Design and synthesis of new anionic “polymeric ionic liquids” with high charge delocalization
A. S.
Shaplov
*a,
P. S.
Vlasov
ab,
M.
Armand
c,
E. I.
Lozinskaya
a,
D. O.
Ponkratov
a,
I. A.
Malyshkina
d,
F.
Vidal
e,
O. V.
Okatova
f,
G. M.
Pavlov
g,
C.
Wandrey
h,
I. A.
Godovikov
a and
Y. S.
Vygodskii
a
aA.N. Nesmeyanov Institute of Organoelement Compounds Russian Academy of Sciences (INEOS RAS), Vavilov str. 28, 119991, GSP-1, Moscow, Russia. E-mail: zipper@ineos.ac.ru; Fax: +7 499 1355085; Tel: +7 499 1359244
bDepartment of Macromolecular Chemistry, Saint-Petersburg State University, Universitetsky pr. 26, 198504, Saint-Petersburg, Russia
cLaboratoire de Réactivité et Chimie des Solides (LRCS), University de Picardie Jules Verne, UMR 6007 CNRS, 33 rue de Saint-Leu, 80039, Amiens, France
dDepartment of Physics, Moscow State University, Vorob'evy gory, 119992, Moscow, Russia
eLaboratoire de Physico-chimie des Polymères et des Interfaces (LPPI), Universite' de Cergy-Pontoise, 5 mail Gay-Lussac, 95031, Cergy-Pontoise, Cedex, France
fInstitute of Macromolecular Compounds Russian Academy of Sciences (IMC RAS), Bolshoy av. 31, Saint-Petersburg, 199004, Russia
gInstitute of Physics, Saint-Petersburg State University, Ulyanovskaya str. 1, Petrodvorets, 198504, Saint-Petersburg, Russia
hLaboratoire de Médecine Régénérative et de Pharmacobiologie (LMRP), Ecole Polytechnique Fédérale de Lausanne (EPFL), station 15, CH-1015, Lausanne, Switzerland
First published on 25th August 2011
Abstract
Three novel ionic monomers having highly delocalized anions and electrochemically stable mobile cations, namely, 1-butyl-1-methylpyrrolidinium 1-[3-(methacryloyloxy)propylsulfonyl]-1-(trifluoromethane-sulfonyl)imide, 1-butyl-1-methylpyrrolidinium 1,1-dicyano-1-[(3-(methacryloyloxy)propylsulfonyl)]methanide and 1-butyl-1-methylpyrrolidinium 1-cyano-1-[(3-(methacryloyloxy)propylsulfonyl)]imide were synthesized and characterized. The structure of these monomers was designed to be a mimic of the most highly conductive bis(trifluoromethylsulfonyl)imide, tricyanomethanide and dicyanamide anions. By radical polymerization procedure a series of new anionic “polymeric ionic liquids” (PILs) were prepared. The solubility of these linear PILs, thermal stability, glass transition temperatures, molar masses and ionic conductivities were estimated. An advantage of the novel PILs was demonstrated by the comparison of their ionic conductivity at 25 °C (2.0 × 10−8 ÷ 1.6 × 10−7 S cm−1) with the unmodified poly(1-ethyl-1-methylpyrrolidinium 3-(methacryloyloxy)propane-1-sulfonate) analog. The increase in ionic conductivity is as high as three orders of magnitude and was found to depend on the size of the attached anion. The new ionic monomers were subsequently copolymerized with poly(ethylene glycol) dimethacrylate and poly(ethylene glycol) methyl ether methacrylate. The investigation of the copolymers properties revealed further improvement of the conductivity in approximately two orders of magnitude and the achievement of σ = 4.8 ÷ 6.8 × 10−6 S cm−1) at 40 °C.
Introduction
Rechargeable lithium batteries have been investigated as an attractive alternative power source for a wide variety of applications.1 Owing to the growing needs for battery safety, ionic liquids (ILs) have attracted considerable attention as new type of non-volatile conductive liquid electrolyte.2,3 They possess potentially useful physicochemical properties, including high ion conductivity, wide electrochemical window, chemical and thermal stability, non-flammability, and nearly absence of vapor pressure.4 Despite the advantages offered by ILs, the possibility of their leakage from the battery during the mechanical damage still remains a possibility.More recently, interest has turned to the polymers based on ionic liquids and their potential application as a new class of polymers that combine all beneficial properties of ILs with those of classical polyelectrolytes.5–9 The use of “polymeric ionic liquids” (PILs) as advanced polymer electrolytes was suggested by Ohno.7,10 As in the case of ILs, one of the important advantage of PILs is their diverse chemical composition, structure, and hence properties, that can be readily changed by pairing any of a variety of organic cations with a wide range of either inorganic or organic anions. The final challenge for PILs is to achieve a high level of ion dissociation, eliminating ion pairs or aggregates, so that the conductivity becomes high enough; besides the transport number of the active ion should be the highest possible. To date lithium batteries demand solid polyelectrolyte membranes having ionic conductivity in the range of 10−5–10−4 S cm−1 at 25 °C.1 However, the most promising trend is the utilization of Li batteries in conjunction with other energy sources. Thus, as a part of energy system, Li batteries are allowed to work at higher temperatures than r.t. and at 25 °C can possess conductivity lower then mentioned one. Nevertheless, the general tendency is to have as high conductivity as possible.1,3,6,10
Taking into account that the privileged charge carrier in modern batteries is the cation (Li+ in particular) the required PILs or their copolymers should be of the polyanion type.6,11 To enhance the ion conductivity and the ion dissociation, the anionic species should be only weakly basic and preferably have a diffuse charge, as, for example, in bis(trifluoromethylsulfonyl)imide (TFSI),12–15PF6 or ClO4 anions.4,16 In its turn, ionic liquids with pyrrolidinium cations have emerged as one of the most promising ionic liquids for application in lithium batteries due to their better electrochemical stability with respect to the more frequently investigated imidazolium ILs.2,3 To our knowledge, till now only one example of utilization of PILs as solid separators in Li batteries has been published by group of D. Mecerreyes.17
Very recently, we reported the preparation and study of the conductive and mechanically stable polymer films based on (3-sulfopropyl) methacrylate polyanion and quaternized (aprotic) pyrrolidinium or imidazolium counter-ions.18 The conductivity of these PILs is greatly affected by the humidity and can increase up to 220 times while transferring from dry conditions to 20% relative humidity. Although the conductivity results at ambient temperature and humidity were satisfactory, the ionic conductivity in dry conditions was too low.
Another anionic PIL published by our group lately19 possessed structure shown on Scheme 1. The reported bulk ionic conductivity (6.5 × 10−4 S cm−1) was measured at ambient conditions, i.e. at ∼40% relative humidity. Originally, it was designed for actuator application and therefore contained less electrochemically stable imidazolium cation and –CH2–N–SO2CF3 anion having low stability towards metal lithium.
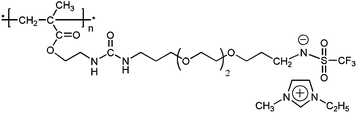 | ||
| Scheme 1 | ||
The main goal of this work is to report the synthesis and investigation of new family of PILs not prone to water take-up and having highly delocalized anions in the polymer backbone and quaternized pyrrolidinium cations as mobile counterions. For the first time the ionic liquid like monomers (ILMs) with –SO2–C(CN)2 and –SO2–N–CN negatively charged groups as a mimic of the most highly conductive tricyanomethanide and dicyanamide anions have been designed and prepared.
Experimental
Materials
Poly(ethylene glycol) dimethacrylate (PEGDM, Mw = 750 g mol−1, Aldrich), poly(ethylene glycol) methyl ether methacrylate (PEGM, Mw = 475 g mol−1, Aldrich), 4-methoxyphenol (99%, Acros), cyanamide (99%, Aldrich), trifluoromethanesulfonamide (97%, ABCR), cyclohexanone (99.8%, Acros), benzophenone (99%, Acros), phosphorus pentoxide (98%, Alfa Aesar), ethanol (HPLC grade, Carlo Erba), methanol (Carlo Erba), 1,1,1,3,3,3-hexafluoroisopropanol (HFIP, ≥99%, Aldrich) were used without further purification. 2,2′-Azobisisobutyronitrile (AIBN, initiator, 98%, Acros) was recrystallized from methanol before use. 1-Bromobutane (98%, Acros) and N-methylpyrrolidine (98%, Acros) were distilled under inert atmosphere over CaH2. Thionyl chloride (>99%, Aldrich) was distilled over linseed oil prior to use. Malononitrile (99%, Aldrich) was distilled under reduced pressure. Tetrahydrofuran (THF, 99+%, Acros) was dehydrated by distillation over sodium and benzophenone radical anion. Reagent-grade chloroform, acetonitrile, N,N-dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), hexamethylphosphoramide (HMPA), dimethyl sulfoxide (DMSO), dichloromethane, ethyl acetate and diethyl ether were obtained from Aldrich, Acros or Merck and were purified by vacuum distillation over P2O5. Potassium 3-(methacryloyloxy)propane-1-sulfonate (SPMA, 98%, Aldrich) was carefully dried under vacuum (<1 mm Hg) at 25 °C for 2 h prior to use.Characterization
NMR spectra were obtained on Bruker AMX-300, Bruker AMX-400 and Bruker AMX-600 spectrometers at 25 °C in the indicated deuterated solvent and are listed in ppm. The signal corresponding to the residual protons of the deuterated solvent was used as an internal standard (1H, 13C relative to tetramethylsilane) and CHCl2F was used as an internal standard for 19F NMR. A Nicolet Magna-750 Fourier IR-spectrometer was used to record IR spectra at a resolution of 2 cm−1 and with the scan number equal to 128 (KBr pellets).Inherent viscosities (ηinh) were measured using an Ostwald capillary viscometer (0.05 g of polymer in 10.0 mL of solvent at 25.0 °C), while the intrinsic viscosities [η] were determined with an Ubbelohde type capillary viscometer at 25.0 °C.
The average MsD polymer molecular masses were determined at 25.0 °C in 0.25M 1-butyl-1-methylpyrrolidinium bromide ([1-Bu-1-MePyrr]Br) solution in DMF for poly-9 and poly-10 by sedimentation-diffusion analysis. The density and viscosity of the 0.25M [1-Bu-1-MePyrr]Br solution in DMF were determined as ρo = 0.9593 g cm−3 and ηo = 0.9437 cPa. Velocity sedimentation was performed in a Beckman XLI analytical ultracentrifuge (ProteomeLab XLI Protein Characterization System, Beckman Coulter Inc., USA) at a rotor speed of 40 × 103 rpm. The buoyancy factor (1 − υρ0) and the partial specific volumes υ (0.733 and 0.817 mL g−1 for poly-9 and poly-10, correspondingly) were estimated from the solution densities measured by the DMA-4000 densitometer (Anton Paar GmbH, Austria). The dn/dc values for poly-9 and poly-10 were determined as 0.039 and 0.076 g mL−1, respectively.
The glass transition temperatures (Tg) of the linear PILs were determined by thermomechanical analysis (TMA) using a UIP-70M (Russia) thermomechanical analyzer at a heating rate of 2.5 °C min−1 and a constant load of 0.08 MPa. Dynamic Mechanical Thermal Analysis (DMTA) measurements were carried out on copolymer (coPILs) films (typically length × width × thickness = 15 × 8 × 0.5 (mm)) with a Q800 model (TA Instruments, USA) operating in tension mode (strain between 0.05 and 0.07%, pretension: 10−2 N). Experiments were performed at 1 Hz frequency with a heating rate of 3 °C min−1 from −90 to +150 °C. The set up provided the storage and loss modules (E′ and E′′). The damping parameter or loss factor (tanδ) was defined as the ratio tanδ = E′′/E′. Thermogravimetric analysis (TGA) was performed in air on a Q50 model (TA Instruments, USA) applying a heating rate of 5 °C/min.
The efficiency of the cross-linking reactions between the ILMs, PEGDM and PEGM was estimated by the extraction of the copolymer films in a Soxhlet extractor with dichloromethane for 10 h with subsequent drying in a vacuum at 80 °C over night.
Dielectric spectroscopy studies of monomers, PILs and coPIL's films were run on a Novocontrol Broadband Dielectric Spectrometer equipped with an Alpha analyzer and a Quatro temperature controller (Novocontrol GmbH, Germany). The films were preliminarily dried in a vacuum oven containing a Petri dish with P2O5 at 80 °C/1 mm Hg and then sandwiched between the gold-coated brass electrodes. The experiments were carried out at 25 °C in the 10−1–107 Hz frequency range in air. Conductivities were duplicated by complex impedance analysis using impedance analyzer Autolab PGSTAT30 (Autolab Frequency Response Analyser System, Metrohm Autolab B.V., The Netherlands). The runs were performed in air, varying the frequency from 10−2 to 108 Hz at 25 °C. A polymer film was put in a cell with pressure contact stainless steel electrodes.
3-(Chlorosulfonyl)propyl methacrylate (2)
Freshly dried potassium 3-(methacryloyloxy)propane-1-sulfonate (1) (15.0 g, 0.061 mol) was suspended in 25 mL of anhydrous THF under inert atmosphere. 1.7 mL of DMF as a catalyst was injected via syringe. The reaction flask was cooled to 0–1 °C and an excess of the thionyl chloride (39.9 g, 0.335 mol) was added dropwise under stirring. The reaction proceeded at 0–1 °C for 1 h, and then at room temperature for 12 h. The obtained suspension was carefully poured into the ice-water (200 mL). The upper aqueous layer was decanted and the lower organic oily layer was diluted with dichloromethane (80 mL). The CH2Cl2 solution was washed with water (6 × 25 mL) and then dried over anhydrous magnesium sulfate. MgSO4 was filtered off and the dichloromethane was gently evaporated under reduced pressure at temperature ≤ 30 °C. The residual colorless or slightly yellow transparent oil was finally dried at 25 °C/12 mm Hg for 2 h and additionally at 25 °C/1–2 mm Hg for 8 h with a special flask filled with P2O5 and introduced into the vacuum line. Yield: 11.3 g (81%); Anal. Calcd for C7H11ClO4S (226.68): C, 37.09%; H, 4.89%; Cl, 15.64%; Found: C, 37.11%; H, 4.99%; Cl, 15.69%; 1H NMR (600.22 MHz, CDCl3): δ = 6.05 (s, 1H,![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2 = C(CH3)–), 5.55 (s, 1H, 2 = C(CH3)–), 4.25 (t, 2H, JHH = 6.2 Hz, CO–O–2–), 3.76 (m, 2H, –2–SO2Cl), 2.35 (m, 2H, CO–O–CH2–2–), 1.87 (s, 3H,CH2 = C(3)–); 13C NMR (150.93 MHz, CDCl3): δ = 166.6 (C
2 = C(CH3)–), 5.55 (s, 1H, 2 = C(CH3)–), 4.25 (t, 2H, JHH = 6.2 Hz, CO–O–2–), 3.76 (m, 2H, –2–SO2Cl), 2.35 (m, 2H, CO–O–CH2–2–), 1.87 (s, 3H,CH2 = C(3)–); 13C NMR (150.93 MHz, CDCl3): δ = 166.6 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 135.4, 126.0, 61.9, 61.0, 23.9, 17.9 (CH2 = C(3)–); IR (KBr pellet): 2968 (m, νC–H), 2929 (m, νC–H), 1719 (vs, νCO), 1638 (m, νCC), 1512 (w), 1454 (m), 1375 (vs, νasSO2), 1321 (s), 1298 (s), 1238 (w), 1162 (νsSO2), 1065 (w), 1027 (m), 947 (m), 894 (w), 816 (m), 770 (w), 741 (w), 699 (w), 655 (w), 594 (m), 548 (m), 531 (s), 509 (m) cm−1.
O), 135.4, 126.0, 61.9, 61.0, 23.9, 17.9 (CH2 = C(3)–); IR (KBr pellet): 2968 (m, νC–H), 2929 (m, νC–H), 1719 (vs, νCO), 1638 (m, νCC), 1512 (w), 1454 (m), 1375 (vs, νasSO2), 1321 (s), 1298 (s), 1238 (w), 1162 (νsSO2), 1065 (w), 1027 (m), 947 (m), 894 (w), 816 (m), 770 (w), 741 (w), 699 (w), 655 (w), 594 (m), 548 (m), 531 (s), 509 (m) cm−1.
Triethyl ammonium 1-[3-(methacryloyloxy)propylsulfonyl]-1-(trifluoromethane-sulfonyl)imide (3)
Trifluoromethanesulfonamide (7.3 g, 0.049 mol) was loaded into the flask and flushed with argon. Freshly distilled anhydrous triethylamine (10.9 g, 0.107 mol) was added under stirring. The reaction mixture was diluted with 40 mL of anhydrous THF and the obtained colorless solution was cooled down to 0 °C on an ice bath. The solution of 3-(chlorosulfonyl)propyl methacrylate (11.1 g, 0.049 mol) in 15 mL of anhydrous THF was added dropwise under inert atmosphere to the cooled THF solution of NH2SO2CF3 and N(C2H5)3. The reaction proceeded at 0 °C for 1 h, then for 1 h at r.t. The resulted precipitate was removed by filtration under inert atmosphere and the filtrate was gently evaporated at r.t. The residual slightly yellow oil was redissolved in 90 mL of dichloromethane. The CH2Cl2 solution was washed with water (4 × 35 mL) and dried over anhydrous magnesium sulfate. MgSO4 was filtered off, a catalytic amount of 4-methoxyphenol (inhibitor) was added and the dichloromethane was gently evaporated under reduced pressure at temperature ≤30 °C. The residual slightly yellow transparent oil was finally dried at 35 °C/12 mm Hg for 1 h and additionally at 35 °C/1–2 mm Hg for 3 h with a special flask filled with P2O5 and introduced into the vacuum line. Yield: 16.7 g (78%); Found: C, 38.14%; H, 6.32%; N, 6.37%; Calc. for C14H27F3N2O6S2 (440.50): C, 38.17%; H, 6.18%; N, 6.36%; 1H NMR (400.13 MHz, CDCl3): δ = 7.76 (bs, 1H,–N(C2H5)3), 6.02 (s, 1H, 2=C(CH3)–), 5.51 (t, 1H, JHH = 1.6 Hz, 2=C(CH3)–), 4.19 (t, 2H, JHH = 6.4 Hz, CO–O–2–), 3.19−3.10 (m, 8H, H–N(2CH3)3 + –2–SO2–N–), 2.19 − 2.12 (m, 2H, O–CH2–2–CH2–), 1.85(s, 3H,CH2 = C(3)–), 1.16 (t, 9H, JHH = 7.2 Hz, H–N(CH23)3); 13C NMR (100.61 MHz, CDCl3): δ = 167.1 (CO), 135.9, 125.8, 124.9 − 114.6 (q, JCF = 322 Hz, N–SO2-![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif) 3), 62.6, 51.9, 46.8, 23.7, 18.2, 8.6; 19F NMR (282.40 MHz, CDCl3): δ = −78.2 (s, CF3); IR (KBr pellet): 3080 (w), 2985 (m, νC–H), 2958 (w, νC–H), 2810 (w), 2705 (w), 1718 (s, νCO), 1637 (m, νCC), 1475 (m), 1456 (m), 1402 (w), 1319 (vs, νasSO2), 1299 (s, νasSO2), 1278 (m), 1181 (vs, νCF), 1123 (vs, νsSO2), 1056 (s, νCF), 945 (w), 835 (m), 818 (m), 702 (w), 623 (m), 608 (m), 557 (m), 514 (m) cm−1.
3), 62.6, 51.9, 46.8, 23.7, 18.2, 8.6; 19F NMR (282.40 MHz, CDCl3): δ = −78.2 (s, CF3); IR (KBr pellet): 3080 (w), 2985 (m, νC–H), 2958 (w, νC–H), 2810 (w), 2705 (w), 1718 (s, νCO), 1637 (m, νCC), 1475 (m), 1456 (m), 1402 (w), 1319 (vs, νasSO2), 1299 (s, νasSO2), 1278 (m), 1181 (vs, νCF), 1123 (vs, νsSO2), 1056 (s, νCF), 945 (w), 835 (m), 818 (m), 702 (w), 623 (m), 608 (m), 557 (m), 514 (m) cm−1.
Triethyl ammonium 1,1-dicyano-1-[(3-(methacryloyloxy)propylsulfonyl)]methanide (4)
The same procedure was used as indicated for 3 with the exception of the utilization of malononitrile instead of trifluoromethanesulfonamide. Yield: 70%; Found: C, 53.74%; H, 7.64%; N, 11.97%; Calc. for C16H27N3O4S (357.47): C, 53.76%; H, 7.61%; N, 11.75%; 1H NMR (400.13 MHz, CDCl3): δ = 7.86 (bs, 1H,–N(C2H5)3), 5.97 (s, 1H, 2=C(CH3)–), 5.47 (s, 1H, 2=C(CH3)–), 4.13 (t, 2H, JHH = 6.4 Hz, CO–O–2–), 3.11 − 3.03 (m, 8H, H–N(2CH3)3 + -2-SO2-C–), 2.11−2.06 (m, 2H, O–CH2–2–CH2–), 1.79 (s, 3H, CH2 = C(3)–), 1.23 (t, 9H, JHH = 7.2 Hz, H–N(CH23)3); 13C NMR (100.61 MHz, CDCl3): δ = 167.1 (CO), 135.9, 125.9, 120.0 (C≡N), 62.5, 54.4, 47.2, 39.2, 23.4, 18.2, 8.9; IR (KBr pellet): 3037 (w), 2986 (m, νC–H), 2807 (w), 2726 (w), 2496 (w), 2188 (s, νsC≡N), 2161 (vs, νasC≡N), 1717 (s, νCO), 1638 (m, νCC), 1474 (m), 1456 (m), 1400 (m), 1299 (s, νasSO2), 1169 (s), 1130 (s, νsSO2), 1078 (m), 1019 (m), 947 (w), 905 (w), 888 (w), 810 (w), 788 (m), 722 (w), 659 (w), 584 (m), 571 (m) cm−1.
Triethyl ammonium 1-cyano-1-[(3-(methacryloyloxy)propylsulfonyl)]imide (5)
The same procedure was used as for 3 with the substitution of trifluoromethanesulfonamide for cyanamide. Yield: 53%; Found: C, 50.47%; H, 8.29%; N, 12.51%; Calc. for C14H27N3O4S (333.45): C, 50.43%; H, 8.16%; N, 12.60%; 1H NMR (600.22 MHz, CDCl3): δ = 8.95 (bs, 1H,–N(C2H5)3), 5.88 (s, 1H, 2=C(CH3)–), 5.37 (s, 1H, 2=C(CH3)–), 4.06 (t, 2H, JHH = 6.3 Hz, CO–O–2–), 2.99−2.98 (m, 6H, H–N(2CH3)3), 2.93 (t, 2H, JHH = 7.2 Hz, 2-SO2-N–), 2.04−2.01 (m, 2H, O-CH2-2-CH2–), 1.72 (s, 3H, CH2 = C(3)–), 1.16 (t, 9H, JHH = 7.2 Hz, H–N(CH23)3); 13C NMR (150.93 MHz, CDCl3): δ = 166.5 (CO), 135.5, 125.0 (m, C≡N), 118.6, 62.2, 50.3, 46.2, 23.5 (m), 17.7 (m), 8.3 (m); IR (KBr pellet): 2981 (m, νC–H), 2936 (m, νC–H), 2736 (w), 2676 (m), 2492 (w), 2168 (vs, νC≡N), 1716 (vs, νCO), 1636 (m, νCC), 1476 (m), 1454 (m), 1398 (m), 1320 (s, νasSO2), 1297 (vs, νasSO2), 1264 (s), 1170 (vs), 1123 (s, νsSO2), 1061 (w), 1019 (m), 947 (w), 897 (w), 835 (s), 636 (w), 611 (w), 576 (m), 476 (w) cm−1.
1-Butyl-1-methylpyrrolidinium 1-[3-(methacryloyloxy)propylsulfonyl]-(trifluoromethane-sulfonyl)imide (9)
A suspension of lithium hydride (0.26 g, 0.033 mol) in 15 mL of anhydrous THF was added dropwise to the solution of 3 (9.8 g, 0.022 mol) in 30 mL of anhydrous THF at 0 °C under inert atmosphere. The ice bath was removed and the reaction was allowed to warm up to room temperature. The stirring was then continued at 25 °C for 2 h, whereupon the unreacted LiH was removed by filtration under inert atmosphere. The filtrate was concentrated under reduced pressure and the obtained oil was washed with hexane (3 × 20 mL) under vigorous stirring. After the decantation of the last portion of hexane and drying at 25 °C/1–2 mm Hg for 1 h, the intermediate product 6 was recovered in quantitative yield and represented slightly yellow very viscous oil, which can be recrystallized from dry dichloromethane in the form of white crystals. Further on the 6 (7.59 g, 0.022 mol) was dissolved in 50 mL of distilled water and added dropwise to the solution of 1-butyl-1-methylpyrrolidinium bromide (6.84 g, 0.031 mol) in 20 mL of H2O at ambient temperature. The mixture was stirred 1 h at 25 °C and the precipitation of an oil was observed. The upper aqueous layer was decanted and the residual oil was redissolved in 50 mL of dichloromethane. The CH2Cl2 solution was washed with water (3 × 30 mL) and dried over anhydrous MgSO4. The magnesium sulfate was filtered off, a catalytic amount of 4-methoxyphenol was added as an inhibitor and the dichloromethane was stripped off under the reduced pressure at temperature ≤30 °C. The 9 was obtained as colorless transparent fluid oil, which was finally dried at 30 °C/1–2 mm Hg for 1 h. Yield: 6.3 g (59%); Found: C, 42.62%; H, 6.57%; N, 5.69%; Calc. for C17H31F3N2O6S2 (480.56): C, 42.49%; H, 6.50%; N, 5.83%; 1H NMR (400.13 MHz, CDCl3): δ = 5.95 (s, 1H,2=C(CH3)–), 5.45 (s, 1H, 2=C(CH3)–), 4.12 (t, 2H, JHH = 6.4 Hz, CO–O–2–), 3.42 − 3.41 (m, 4H, (Pyrr)), 3.24 − 3.21 (m, 2H, Pyrr, = N(CH3)-2-CH2-CH2-CH3), 3.09 − 3.06 (m, 2H, -2-SO2-N-SO2-CF3), 2.93 (s, 3H, Pyrr, = N(3)-CH2-CH2-CH2-CH3), 2.12 − 2.09 (m, 6H, (Pyrr) + CO–O–CH2-2–), 1.79 (s, 3H, CH2 = C(3)–), 1.65−1.57 (m, 2H, Pyrr, =N(CH3)-CH2-2-CH2-CH3), 1.32 − 1.23 (m, 2H, Pyrr, =N(CH3)-CH2-CH2-2-CH3), 0.84 (t, 3H, JHH = 7.2 Hz, Pyrr, = N(CH3)-CH2-CH2-CH2-3); 13C NMR (100.61 MHz, CDCl3): δ = 166.8 (CO), 135.6, 125.4, 124.7−115.5 (q, JCF = 322 Hz, N-SO2-3), 114.3, 63.9, 62.5, 51.6, 47.8, 25.3, 23.4, 21.1, 19.2, 17.8, 13.1; 19F NMR (376.50 MHz, CDCl3): δ = −78.3 (s, CF3); IR (KBr pellet): 2967 (m, νC–H), 2883 (w, νC–H), 1717 (s, νCO), 1634 (m, νCC), 1472 (m), 1453 (m), 1322 (vs, νasSO2), 1298 (s, νasSO2), 1222 (s), 1184 (vs, νCF), 1123 (vs, νsSO2), 1053 (s, νCF), 935 (m), 822 (m), 750 (w), 705 (w), 622 (m), 608 (m), 574 (m), 555 (w), 514 (m) cm−1.
1-Butyl-1-methylpyrrolidinium 1,1-dicyano-1-[(3-(methacryloyloxy)propylsulfonyl)] methanide (10)
The same procedure was used as for 9. After the isolation and subsequent drying 10 represents a transparent light brown fluid liquid. Yield: 61%; Found: C, 57.16%; H, 7.94%; N, 10.39%; Calc. for C19H31N3O4S (397.53): C, 57.40%; H, 7.86%; N, 10.57%; 1H NMR (400.13 MHz, CDCl3): δ = 5.97 (s, 1H,2=C(CH3)–), 5.46 (s, 1H, 2=C(CH3)–), 4.14 (t, 2H, JHH = 6.4 Hz, CO–O–2–), 3.52−3.51 (m, 4H, (Pyrr)), 3.34−3.29 (m, 2H, Pyrr, =N(CH3)–2-CH2–CH2–CH3), 3.03 (s, 3H, Pyrr, =N(3)–CH2–CH2–CH2–CH3), 3.03−2.98 (m, 2H, –2–SO2–C–(CN)2), 2.17 (m, 4H, (Pyrr)), 2.15−2.08 (m, 2H, –CO–O–CH2–2–), 1.81 (s, 3H, CH2 = C(3)–), 1.70−1.62 (m, 2H, Pyrr, =N(CH3)–CH2–2–CH2–CH3), 1.37−1.28 (m, 2H, Pyrr, =N(CH3)–CH2–CH2–2–CH3), 0.88 (t, 3H, JHH = 7.2 Hz, Pyrr, =N(CH3)–CH2–CH2–CH2–3); 13C NMR (150.93 MHz, CDCl3): δ = 167.1 (CO), 135.9, 125.5 (C≡N), 64.3, 64.2, 62.8, 50.5, 48.3 (m), 25.7, 23.9, 21.5, 19.5, 18.1, 13.4; IR (KBr pellet): 2964 (m, νC–H), 2877 (w, νC–H), 2186 (s, νsC≡N), 2160 (vs, νasC≡N), 1716 (s, νCO), 1636 (m, νCC), 1466 (s), 1321 (s, νasSO2), 1298 (s, νasSO2), 1170 (s), 1129 (s, νsSO2), 1078 (m), 1023 (w), 932 (w), 889 (w), 810 (w), 784 (w), 725 (w), 662 (w), 584 (m), 570 (m) cm−1.
1-Butyl-1-methylpyrrolidinium 1-cyano-1-[(3-(methacryloyloxy)propylsulfonyl)]imide (11)
The same procedure was used as for 9 with the exception that 11 during the last stage was not precipitating from aqueous solution, however it was extracted from it with dichloromethane. The CH2Cl2 solution was dried over anhydrous MgSO4, filtered off and the dichloromethane was stripped off under the reduced pressure at temperature ≤30 °C. After drying at 30 °C/1–2 mm Hg for 1 h 11 was isolated as slightly yellow transparent fluid oil. Yield: 77%; Found: C, 54.57%; H, 8.61%; N, 11.04%; Calc. for C17H31N3O4S (373.51): C, 54.67%; H, 8.37%; N, 11.25%; 1H NMR (400.13 MHz, CDCl3): δ = 6.03 (s, 1H,2=C(CH3)–), 5.50 (s, 1H, 2=C(CH3)–), 4.19 (t, 2H, JHH = 6.4 Hz, CO–O–2–), 3.58 − 3.57 (m, 4H, (Pyrr)), 3.39 − 3.36 (m, 2H, Pyrr, =N(CH3)–2–CH2–CH2-CH3), 3.09 (s, 3H, Pyrr, =N(3)–CH2–CH2–CH2–CH3), 3.09 − 3.05 (m, 2H, –2–SO2–N–CN), 2.23 (m, 4H, (Pyrr)), 2.20 − 2.15 (m, 2H, –CO–O–CH2–2–), 1.86 (s, 3H, CH2 = C(3)–), 1.75 − 1.67 (m, 2H, Pyrr, =N(CH3)–CH2–2–CH2–CH3), 1.43−1.33 (m, 2H, Pyrr, =N(CH3)–CH2–CH2–2–CH3), 0.93 (t, 3H, JHH = 7.2 Hz, Pyrr, =N(CH3)–CH2–CH2–CH2–3); 13C NMR (150.93 MHz, CDCl3): δ = 167.1 (CO), 135.9, 125.5 (C≡N), 64.3, 64.2, 62.8, 50.4, 48.3 (m), 25.7, 23.9, 21.5, 19.5, 18.1, 13.4; IR (KBr pellet): 3024 (w), 2963 (s, νC–H), 2934 (s, νC–H), 2877 (m, νC–H), 2161 (vs, νC≡N), 1715 (vs, νCO), 1637 (m, νCC), 1467 (s), 1321 (s, νasSO2), 1297 (s, νasSO2), 1265 (s), 1200 (s), 1171 (vs), 1119 (vs, νsSO2), 1061 (w), 1019 (m), 933 (m), 898 (w), 831 (s), 637 (w), 611 (w), 575 (m), 476 (m) cm−1.
Polymerization procedure
The following example describes the procedure used for the synthesis of the linear polymer poly-9. Other polymers were synthesized accordingly, with the ratio of monomer to initiator, the solvent and the monomer concentration varied. 9 (0.40 g, 0.83 mmol), DMF (0.40 g) and AIBN (0.0040 g, 1 wt%) were gently mixed in a flask at ambient temperature. The solution was transferred into a glass ampoule. After triple freeze-thaw-pump cycles the ampoule was sealed under vacuum and heated to 60 °C. The polymerization was carried out at this temperature for 6 h. The resulting transparent highly viscous polymer solution was slightly diluted with acetone (acetonitrile for poly-10 and poly-11) and precipitated into dichloromethane. Poly-9 was then thoroughly washed with dichloromethane and dried at 80 °C under vacuum (1 mm Hg) for 48 h. Yield: 0.33 g (82%); ηinh DMF = 3.17 dL g−1 (25.0 °C).General procedure for the copolymerization of 9–11 with PEGDM
The crosslinked polymer films of 9–11 with PEGDM were prepared as follows: 0.114 g of PEGDM and 0.228 g of the corresponding ionic monomer were dissolved in 0.4 mL of ethanol at ambient temperature under inert gas. 0.0103 g of AIBN (3 wt%) was then added and the mixture was thoroughly stirred until the formation of a clear solution, which was poured into a mould made from two glass plates clamped together and sealed with a 500 mm thick Teflon® gasket. Afterwards, the mould was kept at 60 °C for 10 h. The resultant film was taken out from the mould at ambient temperature, dried at 60 °C for 3 h and finally for 10 h at 80 °C/1 mm Hg.General procedure for the terpolymerization of 9–11 with PEGDM and PEGM
The formation of the crosslinked films via terpolymerization was accomplished in a full accordance with the procedure given for the copolymerization of 9, 10 or 11 and PEGDM with the exception that the reaction solution was composed of 0.076 g of PEGDM, 0.038 g of PEGM, 0.0103 g of AIBN (3 wt%), 0.228 g of the corresponding ionic monomer and 0.4 mL of ethanol.Film casting from linear PILs
Poly-9, poly-10 and poly-11 films were cast from 5 wt% polymer solutions in acetonitrile directly on the gold electrode's surface. The solvent was slowly evaporated at 50 °C. The obtained thin polymer coatings together with the electrodes were dried at 80 °C/1 mm Hg for 24 h.Results and discussions
Synthesis and characterization of ionic liquid like monomers (ILMs) 9–11
The design of the proposed ILMs 9, 10 and 11 (Scheme 2) was guided by the goal to create highly delocalized anion in the polymer backbone and by the previously established relationships between the monomer structure and the ionic conductivity of the respective polymer.6,7 Negatively charged –SO2–N–SO2–CF3, –SO2–C(CN)2 and –SO2–N–CN groups were selected as a mimic of the most highly conductive bis(trifluoromethylsulfonyl)amide, tricyanomethanide and dicyanamide anions.3 The reactive methacrylate functional group and the delocalized anion were separated by the flexible spacer containing three CH2 groups. The quaternized (aprotic) pyrrolidinium cation was chosen as a mobile counterion due to its exceptional electrochemical stability in comparison with respective protic cations20 or imidazolium ones.21![Synthetic route for the preparation of monomers 9, 10 and 11. Reagents and conditions: (i) SOCl2, THF/DMF, 0 °C → RT; (iia) NH2SO2CF3, THF, NEt3, 0 °C → RT; (iib) CH2(CN)2, THF, NEt3, 0 °C → RT; (iic) NH2CN, THF, NEt3, 0 °C → RT; (iii) LiH, THF, 10 °C → RT; (iv) 1.4 [1-Bu-1-Me Pyrr]Br, H2O, RT.](/image/article/2011/PY/c1py00282a/c1py00282a-s2.gif) | ||
| Scheme 2 Synthetic route for the preparation of monomers 9, 10 and 11. Reagents and conditions: (i) SOCl2, THF/DMF, 0 °C → RT; (iia) NH2SO2CF3, THF, NEt3, 0 °C → RT; (iib) CH2(CN)2, THF, NEt3, 0 °C → RT; (iic) NH2CN, THF, NEt3, 0 °C → RT; (iii) LiH, THF, 10 °C → RT; (iv) 1.4 [1-Bu-1-Me Pyrr]Br, H2O, RT. | ||
For the preparation of monomers 9–11 the synthetic method consisting of four steps (Scheme 2) was developed. On the first stage potassium 3-(methacryloyloxy)propane-1-sulfonate was converted into the sulfonyl chloride derivative by reaction with thionyl chloride in the presence of DMF as a catalyst. This reaction can be carried with/without anhydrous THF as a co solvent. The presence of THF as a solvent was influencing only the yield of the reaction, i.e. slightly increasing it to 80%. Due to the hydrophobic nature of 2 as well as its relative stability towards cold water in comparison with carboxylic acid chlorides,22 it was isolated from the reaction mixture by pouring into the ice-water. The subsequent extraction with methylene chloride and drying over magnesium sulfate resulted in the obtaining of 2 in high purity.
The second stage consisted in the reaction of 2 with trifluoromethanesulfonamide (Scheme 2, 3), malononitrile (Scheme 2, 4) or cyanamide (Scheme 2, 5) in the presence of a 2.2 molar excess of triethylamine. When the reaction was carried out in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio between the 2 and triethylamine, the triethylammonium hydrochloride was isolated only in 50% yield and the inseparable mixture of 3 and its acid form (in 1:1 ratio according to NMR) was obtained. Thus, it was found that the desired monomeric acids can be successfully isolated only in the form of their triethylammonium salts (Scheme 2, 3, 4 or 5). Further studies revealed that triethylamine appeared to be the best reagent for this reaction. Its substitution with pyridine, a weaker base, did not result in the isolation of the target compound. According to22,23 the similar reaction of aliphatic sulfonyl halides with nucleophiles proceeds via one of the two competing pathways: nucleophilic substitution (nucleophilic reaction and general-base mechanism) or elimination-addition mechanism (formation of sulfenes R–CH = SO2). The realization of the specific mechanism was found to be dependent on the reaction conditions including the nature of the base.23 As far as the nitrogen atom in triethylamine is less nucleophilic and more hindered than in pyridine, the sulfene mechanism may predominate. The application of stronger bases, such as NaH and EtONa, was complicated by various side reactions of cyano groups and resulted in intensively colored and insoluble products. The third stage of the suggested synthetic path was concluded in the interaction of 3–5 with lithium hydride in anhydrous THF (Scheme 2). To exclude the possibility of an ionic polymerization, the reaction was started at temperatures < −10 °C. However, probably due to the limited solubility of LiH in the anhydrous THF, this reaction proceeded smoothly only at 10 °C, that was monitored by the visible evolution of hydrogen. After the removal of the excess of lithium hydride by filtration, 6–8 were recovered as highly viscous colored oils in a virtually quantitative yield. It was further revealed that 6 and 8 can be additionally recrystallized from anhydrous methylene chloride as white needle like crystals.
:1 molar ratio between the 2 and triethylamine, the triethylammonium hydrochloride was isolated only in 50% yield and the inseparable mixture of 3 and its acid form (in 1:1 ratio according to NMR) was obtained. Thus, it was found that the desired monomeric acids can be successfully isolated only in the form of their triethylammonium salts (Scheme 2, 3, 4 or 5). Further studies revealed that triethylamine appeared to be the best reagent for this reaction. Its substitution with pyridine, a weaker base, did not result in the isolation of the target compound. According to22,23 the similar reaction of aliphatic sulfonyl halides with nucleophiles proceeds via one of the two competing pathways: nucleophilic substitution (nucleophilic reaction and general-base mechanism) or elimination-addition mechanism (formation of sulfenes R–CH = SO2). The realization of the specific mechanism was found to be dependent on the reaction conditions including the nature of the base.23 As far as the nitrogen atom in triethylamine is less nucleophilic and more hindered than in pyridine, the sulfene mechanism may predominate. The application of stronger bases, such as NaH and EtONa, was complicated by various side reactions of cyano groups and resulted in intensively colored and insoluble products. The third stage of the suggested synthetic path was concluded in the interaction of 3–5 with lithium hydride in anhydrous THF (Scheme 2). To exclude the possibility of an ionic polymerization, the reaction was started at temperatures < −10 °C. However, probably due to the limited solubility of LiH in the anhydrous THF, this reaction proceeded smoothly only at 10 °C, that was monitored by the visible evolution of hydrogen. After the removal of the excess of lithium hydride by filtration, 6–8 were recovered as highly viscous colored oils in a virtually quantitative yield. It was further revealed that 6 and 8 can be additionally recrystallized from anhydrous methylene chloride as white needle like crystals.
The final step represented a metathesis reaction between the lithium salts of the desired monomers (Scheme 2, 6, 7 and 8) and the respective pyrrolidinium bromide ionic liquid. As in the synthesis of bis(trifluoromethylsulfonyl)imide containing ILs,24 the reaction leads to the preparation of clean products. The hydrophobic 9 and 10 precipitated from the aqueous medium and can be easily isolated. In contrast, the cyanamide derivative 11 appeared to be somewhat hydrophilic and water miscible. However, it was possible to extract it with methylene chloride and recover after the solvent evaporation in 78% yield. It should be noted that monomers 9–11 after their isolation need to be stored at low temperatures in the presence of dissolved 4-methoxyphenol (inhibitor).
The structure and purity of 9–11 were proved by 1H and 13C NMR, IR spectroscopy and elemental analysis. All three ILMs represent viscous liquids at r.t. To study their thermal behavior the differential scanning calorimetry (DSC) was used. The DSC traces performed with a slow rate of 2 °C min−1 confirmed the absence of any crystallization or melting processes for the studied ionic compounds. The monomers showed only glass transition Tg at −71.0, −69.9 and −75.5 °C for 9, 10 and 11, respectively. As expected, ionic monomer 11 having the smallest delocalized anion with little rotational barrier of S–N bond demonstrates the lowest Tg.
The ionic conductivity measured for cited monomers resulted in the values of 1.4 − 3.1 × 10−4 S cm−1 at 25 °C. In spite of the difference in the nature of the attached anion, 9–11 visibly seemed to have similar viscosity and as a result demonstrated nearly equal conductivity. Evidently the large size of the anionic part imparts high viscosity for the investigated ILMs, that in its turn reduces the ion mobility and the ionic conductivity in comparison with the analogous 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide ionic liquid (σ = 2.2 × 10−3 S cm−1).14
Radical polymerization of ionic monomers 9–11
Various methods of the polymerization of 9–11 were investigated. AIBN was used as initiator.18 The polymerization of 9–11 was firstly accomplished in bulk and resulted in insoluble polymers probably due to side curing reactions.6,18 In contrast, the solution polymerization allowed for the synthesis of soluble poly-9–poly-11. The determined optimal conditions providing high molar mass polyelectrolytes were found to be: monomer concentration [ILM]:[solvent] = 1:1 by wt.; initiator concentration [AIBN] = 1 wt%; reaction duration 6 h. Among tested solvents, DMF was shown to produce polymers with the highest inherent viscosities equal to 3.17, 5.29 and 3.20 dL g−1 for poly-9, poly-10 and poly-11, respectively (Table 1).
| No | Polymer | η inh (dL g−1)a | [η] (dL g−1)b | MsD (g mol−1)c | Tg (°C)d | σDC (S cm−1) e | T d (°C)f | |
|---|---|---|---|---|---|---|---|---|
| a For the solution of 0.05 g of polymer in 10.0 mL of DMF at 25.0 °C. b For the solution of polymer in 0.25 M [1-Bu-1-MePyrr]Br in DMF at 25.0 °C. c Determined by sedimentation-diffusion analysis in 0.25M [1-Bu-1-MePyrr]Br in DMF. d Tg was determined by TMA. e Conductivity of polymer film at 25 °C. f Onset loss temperature according to TGA. | ||||||||
| 1 |

|
poly-9 | 3.17 | 0.77 | 0.5 × 106 | 14 | 2.0 × 10−8 | 285 |
| 2 |

|
poly-10 | 5.29 | 2.00 | 1.7 × 106 | 38 | 3.1 × 10−8 | 235 |
| 3 |
|
poly-11 | 3.20 | 0.71 | — | 11 | 1.6 × 10−7 | 245 |
The determination of [η] in DMF was complicated by the insufficient electrostatic screening. To suppress it, 1-butyl-1-methylpyrrolidinium bromide was added to DMF. The addition of pyrrolidinium based IL instead of standard salt electrolyte (LiCl, Li(CF3SO2)2N, etc.) was driven by the aim to exclude the ion exchange between the mobile pyrrolidinium cation of the polyelectrolyte and the metal ions. The use of 0.25 M solution of 1-butyl-1-methylpyrrolidinium bromide in DMF as a solvent yielded the intrinsic viscosities 0.77, 2.00 and 0.71 dL g−1 for poly-9, poly-10 and poly-11, respectively (Table 1).
The sedimentation-diffusion analysis was applied for investigation of poly-9 and poly-10 molar masses (Table 1). 0.25 M solution of 1-butyl-1-methylpyrrolidinium bromide in DMF was used as a solvent for the study of the sedimentation velocity and the translational diffusion. The molar mass (MsD) determined by this method was equal to 500 × 103 and 1700 × 103 g mol−1 for poly-9 and poly-10, respectively.
In spite of the apparent similarity of monomer structures, the solubility of poly-9–poly-11 are different (Table 2). The most pronounced disparity concluded in the hydrophobic nature of poly-9 and poly-10 and hydrophilicity of poly-11, soluble in water and aq. 0.5 M NaCl. While in DMF all three studied polymers were soluble, DMSO was capable to dissolve only poly-9 and poly-10. Poly-9 and poly-10 were soluble in tetrachloroethane/phenol mixture. The solubility of poly-9 in acetone and poly-10 in methanol deserves to be noted. In contrast to the majority of hydrophobic PILs, the solubility of poly-10 in methanol was found to be its distinctive peculiarity. At the same time the uncommon solubility of poly-9 in acetone can be attributed to the fluorinated nature of the attached anion. As well as ILs, poly-9–poly-11 were soluble in acetonitrile and insoluble in diethyl ether.
| Solvent | Polymer | ||
|---|---|---|---|
| poly-9 | poly-10 | poly-11 | |
| a Solubility: (+) soluble at ambient temperature, (+t) soluble on heating, (±) partly soluble on heating, (–) insoluble even on heating. | |||
| DMF | + | + | + |
| 0.5 M Li(CF3SO2)2N in DMF | + | + | — |
| DMSO | + | + | — |
| H2O | — | — | +t |
| 0.5 M NaCl in H2O | — | — | +t |
| Methanol | — | + | — |
| HFIP | ± | ± | ± |
| Acetone | + | — | — |
| Acetonitrile | + | + | + |
| CH2Cl2 | — | — | — |
| Chloroform | — | — | — |
| Cyclohexanone | — | — | — |
| Diethyl ether | — | — | — |
|
Symm-Tetrachloroethane/phenol (3:1, by wt) |
+ | + | ± |
Thermal stability of novel PILs (onset temperature of polymer degradation, Td) and their heat resistance (Tg) were estimated (Table 1). Poly-9 demonstrated the highest Td among the investigated polymers. The thermal stability of poly-10 and poly-11 was lower by 40–50 °C and equal to 235 and 245 °C, respectively. Tgs of poly-9 and poly-11 are in the order: poly-11 < poly-9 < rt < poly-10 (Table 1). Upon consideration of poly-9–poly-11 as polymeric analogs of 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide, tricyanomethanide and dicyanamide ILs, respectively, it was of interest to compare their thermal properties. It is of common knowledge, that bis(trifluoromethylsulfonyl)amide anion usually imparts high thermal stability to the respective ILs, increasing their Td over 420 °C.4 The thermal decomposition temperatures for the tricyanomethanide ILs vary between 290 and 335 °C25 that are quite similar to the dicyanamide salts.26 According to the onset temperature of decomposition ILs follow the general stability order: [1-Bu-1-MePyrr]N(CF3SO2)2 > [1-Bu-1-MePyrr]C(CN)3 ≥ [1-Bu-1-MePyrr]N(CN)2. It is noteworthy, although the thermal stability of poly-9–poly-11 is lower in comparison with the corresponding ILs, their thermal properties (both Td and Tg) follow the same general tendencies.
According to the values of ionic conductivity at 25 °C the investigated polyelectrolytes can be arranged in the following order: poly-11 > poly-10 > poly-9 (Table 1). Such sequence is in a full agreement with the conductivity of the ILs with dicyanamide, tricyanomethanide and bis(trifluoromethylsulfonyl)imide mobile anions.3,27 The higher conductivity of poly-11 can be attributed to the smallest size of the –SO2–N–CN anion.
A clear advantage of the achievement of high delocalization in the attached anion can be demonstrated by comparison of poly-9–poly-11 and unmodified poly(1-ethyl-1-methylpyrrolidinium 3-(methacryloyloxy)propane-1-sulfonate) (Scheme 3, poly-12). The ionic conductivity of poly-9–poly-11 at 25 °C (2.0 × 10−8 ÷ 1.6 × 10−7 S cm−1) was found to be two or three orders of magnitude higher than that of poly-12 (3.3 × 10−10 S cm−118). At the same time Tg of poly-9–poly-11 (11 ÷ 38 °C) were close to r.t. and lower than the Tg of poly-12 (63 °C18). Thus, PILs containing weakly nucleophilic attached anions are more conductive and demonstrate lower Tgs. Another interesting comparison can be performed on poly-9 and structurally similar poly-13 (Scheme 3). The latter polycation serves as a good reference as it has the same polymer backbone and the same length of the alkyl spacer. Tg of poly-13 is higher (55 °C18) while its conductivity (7.8 × 10−9 S cm−116) is lower in comparison with anionic poly-9. Probably, at similar degree of delocalization of the imide anions in polycation and polyanion, the presence of mobile bulky 1-butyl-1-methylpyrrolidinium cation in poly-9 leads to the loosening of the polymer packing and subsequent decrease of the polymerTg and an increase in ions mobility.
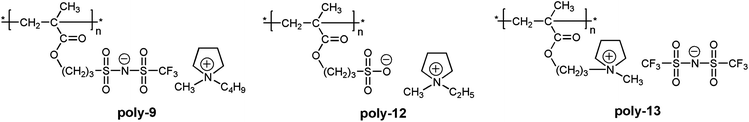 | ||
| Scheme 3 Chemical structures of poly-9, unmodified poly-12 and structurally similar polycation poly-13. | ||
Copolymerization of 9–11 with PEGDM and PEGM
The 9–11 were copolymerized with PEGDM using the previously determined optimal ratio (Table 3).28,29 In contrast to rubber like linear poly-9–poly-11 the copolymerization yielded cross-linked tough films and according to the DMTA (Fig. 1) resulted in a 10–20 °C decrease in the Tg (Tα more correctly). At this, the order of the copolymers according to their Tgs was found to be identical to that for linear polyelectrolytes: copoly-11/PEGDM < copoly-9/PEGDM < copoly-10/PEGDM (Table 3, entries 1, 3 and 5). The estimation of the ionic conductivity revealed the enhancement in more than 10 times with the average value of 5 × 10−7 S cm−1 at 25 °C. The improvement was more pronounced in the case of polymer networks based on 9 and 10 (Table 3, entries 1 and 3). Probably, the increase of ionic conductivity can be attributed to the lowering of the copolymersTg and relevant increase of the ions mobility.5–7,29,30| No | Composition | T α (°C)a | σDC, (S cm−1)b | T d (°C)c | ||
|---|---|---|---|---|---|---|
| ILM | PEGDM (wt%) | PEGM (wt%) | ||||
| a T α was determined by DMTA. b Conductivity of polymer film at 25 °C. c Onset loss temperature according to TGA. | ||||||
| 1 |
|
30 | — | 5.5 | 5.5 × 10−7 | 230 |
| 2 | 20 | 10 | −1.4 | 2.0 × 10−6 | 215 | |
| 3 |

|
30 | — | 15.0 | 6.7 × 10−7 | 230 |
| 4 | 20 | 10 | 0.6 | 2.7 × 10−6 | 230 | |
| 5 |

|
30 | — | 4.4 | 2.9 × 10−7 | 230 |
| 6 | 20 | 10 | −2.1 | 1.5 × 10−6 | 215 | |
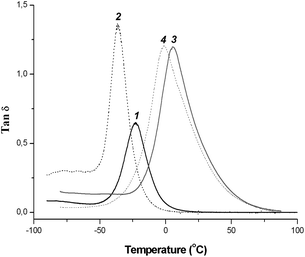 | ||
| Fig. 1 DMTA analysis for PEGDM film (11) and copolymer films: coPEGDM/PEGM (22), copoly-9/PEGDM (33) and copoly-9/PEGDM/PEGM (44). | ||
The copolymerization of the 9–11 was further performed with both PEGDM and PEGM (Table 3, entries 2, 4 and 6). The tercopolymerization resulted in the next decrease of the Tg to the region close to or less than 0 °C. In turn, the simultaneous reduction of the Tg and the increase of the degrees of freedom (“dangling chains” effect1) led to the significant increase of the bulk ionic conductivity. It is noticeable that all three copolymer films demonstrated at 25 °C practically equal conductivity which moved to the 10−6 S cm−1 region (Table 3, entries 2, 4 and 6).
To check the copolymerization reaction completion and the presence of any unpolymerized ionic monomers or oligomers which can influence PIL's properties, the obtained films were extracted with CH2Cl2. It was found that the soluble fraction content is quite low (0.8–4.2 wt%) which proves the high efficiency of the cross-linking reaction. Additionally, the tan δ vs. temperature dependence for all obtained copolymers showed single narrow relaxation peaks (Fig. 1).
Fig. 2 shows the temperature dependence of the ionic conductivity for tercopolymers of 9 and 10. The observed dependence was found to be nonlinear. However, in accordance with the general tendency, the ionic conductivity rises significantly with temperature. Thus, at 40 °C the σ values were two times higher than at r.t. and were equal to 4.8 × 10−6 and 6.8 × 10−6 S cm−1 for poly-9/PEGDM/PEGM and poly-10/PEGDM/PEGM, respectively. Reaching 10−5 S cm−1 was observed already at 45–48 °C.
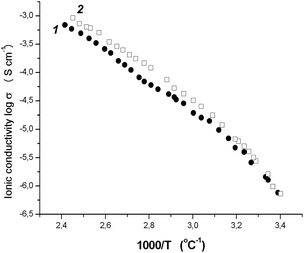 | ||
| Fig. 2 Temperature dependence of ionic conductivity for copoly-9/PEGDM/PEGM (11) and copoly-10/PEGDM/PEGM (22) films. | ||
TGA of the obtained copolymers revealed almost identical (∼230 °C) onset loss temperature (Table 3). This can be explained by the fact that the thermal stability of these films is mainly governed now by the decomposition of the PEO fragments.
Conclusions
Three novel ionic monomers with highly delocalized anions and mobile aprotic pyrrolidinium cations were synthesized. The structure of these ILMs was designed to be a mimic of the most highly conductive bis(trifluoromethylsulfonyl)imide, tricyanomethanide and dicyanamide anions. The performed free radical polymerization of such monomers resulted in the preparation of the new “polymeric ionic liquids” having high molar masses (MsD up to 1.7 × 106 g mol−1) and conductivities in the range of 2.0 × 10−8 ÷ 1.6 × 10−7 S cm−1 at 25 °C. As previously observed for the other families of PILs, their properties, such as solubility, viscosity, thermal stability, Tg and ionic conductivity are affected mainly by the constitution of the ILMs. At this, the lowest Tg and the highest conductivity were demonstrated by the polymer with the smallest size of the –SO2–N–CN anion. The suggested modification of the –SO3group allows for up to three orders of magnitude improvement in polymer ionic conductivity.The subsequent copolymerization of ILMs with PEGDM and PEGM leads to the preparation of novel solid polymer electrolytes with enhanced ionic conductivity. The conductivity of polymer films achieved at 40 °C was 4.8÷6.8 × 10−6 S cm−1, while already at 45–48 °C, σ reached 10−5 S cm−1. These findings along with polymer good thermal stability make them attractive candidates for the application as materials in various electrochemical devices.
Acknowledgements
This work was supported by the Russian Foundation for Basic Researches (Grant no. 10-03-00391-a) and by Swiss National Science Foundation (SCOPES grant no. IZ73Z0_128071/1). Associate Prof. Dr Shaplov is grateful to the University of Cergy-Pontoise for the sponsorship of his visit and work in the LPPI laboratory. Authors would like to thank Prof. L. I. Komarova for IR spectra and Dr T. M. G. Nguyen for her assistance.Notes and references
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- A. Lewandowski and A. Swiderska-Mocek, J. Power Sources, 2009, 194, 601–609 CrossRef CAS.
- H. Ohno, Ed., Electrochemical Aspects of Ionic Liquids, Wiley-Interscience, Hoboken N.J., 2005 Search PubMed.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, 2nd ed., Wiley-VCH, Weinheim, 2007 Search PubMed.
- H. Ohno and M. Yoshizawa, in Ionic Liquids IIIB: Fundamentals, Progress, Challenges, and Opportunities: Transformations and Processes, American Chemical Society, Washington, DC, 2005, pp. 159–170 Search PubMed.
- A. S. Shaplov, E. I. Lozinskaya, and Y. S. Vygodskii, Chapter 9 “Polymer Ionic Liquids: Synthesis, Design and Application in Electrochemistry as Ion Conducting Materials” in Electrochemical Properties and Applications of Ionic Liquids, Novapublishers, New York, 2010 Search PubMed.
- H. Ohno, Macromol. Symp., 2007, 249–250, 551–556 CrossRef.
- O. Green, S. Grubjesic, S. Lee and M. A. Firestone, Polym. Rev., 2009, 49, 339 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011 DOI:10.1016/j.progpolymsci.2011.05.007.
- H. Ohno and K. Ito, Chem. Lett., 1998, 27, 751–752 CrossRef.
- J. Sun, D. R. MacFarlane and M. Forsyth, Solid State Ionics, 2002, 147, 333–339 CrossRef CAS.
- M. Armand, M. Gauthier and D. Muller, US Pat., 4 851 307, 1989.
- P. Johansson, S. P. Gejji, J. Tegenfeldt and J. Lindgren, Electrochim. Acta, 1998, 43, 1375–1379 CrossRef CAS.
- D. R. McFarlane, J. Sun, J. Golding, P. Meakin and M. Forsyth, Electrochim. Acta, 2000, 45, 1271–1278 CrossRef CAS.
- Y. U. Paulechka, G. J. Kabo, A. V. Blokhin, A. S. Shaplov, E. I. Lozinskaya, D. G. Golovanov, K. A. Lyssenko, A. A. Korlyukov and Y. S. Vygodskii, J. Phys. Chem. B, 2009, 113, 9538–9546 CrossRef CAS.
- F. M. Gray, Ed., Solid polymer electrolytes: fundamentals and technological applications., Wiley-VCH, New York, 1991 Search PubMed.
- G. B. Appetecchi, G.-T. Kim, M. Montanino, M. Carewska, R. Marcilla, D. Mecerreyes and I. De Meatza, J. Power Sources, 2010, 195, 3668–3675 CrossRef CAS.
- A. S. Shaplov, P. S. Vlasov, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, O. V. Okatova, G. M. Pavlov, C. Wandrey, A. Bhide, M. Schönhoff and Y. S. Vygodskii, Macromolecules, 2011 Search PubMed , accepted.
- J. Juger, F. Meyer, F. Vidal, C. Chevrot and D. Teyssié, Tetrahedron Lett., 2009, 50, 128–131 CrossRef CAS.
- H. Matsumoto, H. Sakaebe and K. Tatsumi, J. Power Sources, 2005, 146, 45–50 CrossRef CAS.
- M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef CAS.
- J. F. King, J. Y. L. Lam and S. Skonieczny, J. Am. Chem. Soc., 1992, 114, 1743–1749 CrossRef CAS.
- S. N. Lyashchuk, Y. G. Skrypnik and V. P. Besrodnyi, J. Chem. Soc., Perkin Trans., 1993, 2, 1153–1159 Search PubMed.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram, M. Grätzel and M. Armand, Inorg. Chem., 1996, 35, 1168–1178 CrossRef.
- S. A. Forsyth, S. R. Batten, Q. Dai and D. R. MacFarlane, Aust. J. Chem., 2004, 57, 121–124 CrossRef CAS.
- D. R. MacFarlane, S. A. Forsyth, J. Golding and G. B. Deacon, Green Chem., 2002, 4, 444–448 RSC.
- Y. Yoshida, K. Muroi, A. Otsuka, G. Saito, M. Takahashi and T. Yoko, Inorg. Chem., 2004, 43, 1458–1462 CrossRef CAS.
- A. S. Shaplov, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, P.-H. Aubert, O. V. Okatova, G. M. Pavlov, C. Wandrey and Y. S. Vygodskii, Electrochim. Acta, 2011 DOI:10.1016/j.electacta.2011.06.041.
- A. S. Shaplov, L. Goujon, F. Vidal, E. I. Lozinskaya, F. Meyer, I. A. Malyshkina, C. Chevrot, D. Teyssié, I. L. Odinets and Y. S. Vygodskii, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4245–4266 CrossRef CAS.
- Y. S. Vygodskii, A. S. Shaplov, E. I. Lozinskaya, K. A. Lyssenko, D. G. Golovanov, I. A. Malyshkina, N. D. Gavrilova and M. R. Buchmeiser, Macromol. Chem. Phys., 2008, 209, 40–51 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |