Microparticles of phosphonate-functionalized copolymers and their composites with CdTe nanocrystals prepared by sonication-precipitation†
Ioannis
Kanelidis
a,
Ozcan
Altintas‡
a,
Jan-Christoph
Gasse
b,
Ronald
Frahm
b,
Alexander
Eychmüller
c and
Elisabeth
Holder
*ad
aFunctional Polymers Group and Institute of Polymer Technology, University of Wuppertal, Gaußstr. 20, D-42097, Wuppertal, Germany. E-mail: holder@uni-wuppertal.de; Fax: +49-202-439-3880; Tel: +49-202-439-3879
bExperimental Physics, University of Wuppertal, Gaußstr. 20, D-42097, Wuppertal, Germany
cPhysical Chemistry/Electrochemistry, TU Dresden, Bergstr. 66b, D-01062, Dresden, Germany
dDutch Polymer Institute (DPI), P.O. Box 513, NL-5600 AX, Eindhoven, The Netherlands
First published on 8th September 2011
Abstract
Two phosphonate-functionalized, side-chain modified copolymers were synthesized by utilizing either a Yamamoto or a Suzuki protocol, correspondingly. The copolymers' backbones were comprised of 2,7-dibromo-9,9-bis(6-bromohexyl)-9H-fluorene and 2,7-dibromo-9,9-dioctyl-9H-fluorene or 2,2′-(9,9-dioctyl-9H-fluorene-2,7-diyl)bis(1,3,2-dioxaborinane), respectively. These precursor polymers were subjected to a post-functionalization using triethyl phosphite and the resulting target copolymers were fully characterized. The optical classification parameters were determined in solutions and in films as well. The copolymers revealed blue light emission with wide optical bandgaps Egopt of 2.93 eV and quantum yields of 0.54 in chloroform solutions. After following a precipitation method, facilitated by means of an ultrasonic bath, stable dispersions of copolymer microparticles in water were achieved. These particles revealed bright and stable emission with colors ranging from blue to white, only depending on the catalysis method applied. Note the quantum yields beneficially increase up to 0.84 due to the particle formation. Additionally, the combination of the copolymers with semiconductor CdTe nanocrystals, applying again the ultrasonic bath-precipitation method, allowed the formation of copolymer-CdTe composites, furthermore usefully tuning their emission intensity and color differing from the original colors of the involved partners.
Introduction
Nanoparticles (NPs) consisting of conjugated polymers have emerged as important fluorescent labels with a variety of potentially useful optical properties. The polymer nanoparticles can be prepared using a variety of methods, including nano-precipitation,1–7 mini-emulsion dispersion,8mini-emulsion polymerization,9–12 and use of preformed polymers1 or dendrimer-based strategies.13 The nano-precipitation method provided nanoparticles with controlled sizes up to 50 nm, exhibiting high fluorescence brightness as compared to other fluorescent nanoparticles of similar size.2,3 Additionally, conjugated polymer nanoparticles exhibit extreme high two-photon excitation cross-sections, which are promising for applications in two-photon excited fluorescence imaging.4 Two-photon imaging holds great hope for the field of in vivo microscopic physiological studies like neurobiology, immunology and tissue engineering.14–16 A limiting factor in such cellular-imaging applications is the hydrophobicity originating from the π-conjugated backbone. Ultrafiltration techniques could overcome the issue by preparation of stable conjugated polymer nanoparticles of 80–100 nm in size.17 Live cells could then uptake the nanoparticles with no noticeable inhibition of cellular viability, even after seven days of incubation.17 For biological labeling applications, quantum dots with a diameter threshold of 5 nm are proposed only to be acceptable, as renal clearance from the body would not be possible in any other case.18Quantum dots can be synthesized with such small diameters but are typically composed of toxic materials, such as cadmium. Their toxicity can be circumvented when encapsulated in biocompatible materials,19–22 which procedure however, increases the hydrodynamic diameter of the particles. A new trend for the synthesis of semiconducting polymer nanospheres is the use of well-studied polymers, known for their benign properties in combination with biocompatible quantum dots. Chen and Li for example, reported the synthesis of water-soluble fluorescent nanoparticles containing fluorescent polymers with molecules possessing both hydrophilic and hydrophobic properties (amphiphile).23 The resulting nano-/microparticles varied between sizes of 10 nm and 10 microns, revealing features useful for cell imaging and similar biological applications. Poly(fluorene)s (PF)s constitute a promising class of fluorescent conjugated polymers, exhibiting good processability and the advantage of facile side-chain modification.24–27 Poly(fluorene)s with sulfonic-,28amine-,29–32phenol-33 or phosphonate-34 side-chain functionalized groups have been designed without affecting the main-chain conjugation. The phosphonate-groups are of particular interest not only due to their amphiphilic character35 but also due to their use as precursors for the synthesis of polymers with phosphonic acid-groups in the backbone.36,37 The interaction of the phosphonate-groups with inorganic nanocrystals, deriving from their strong chemical affinity to the quantum dots,38 allows their surface functionalization and opens-up the possibility for the preparation of hybrid systems. A further aspect of the phosphonate backbone incorporation is the possibility to design polymers that are highly soluble in polar solvents, even soluble in water.39 These properties are highly demanded in biological systems but also in processing modern multi-layer40 or large area devices.41–45 Very recently, an ethanol-soluble phosphonate-functionalized poly(fluorene) has been used as electron injection layer (EIL) in polymer white-light emitting diodes (PWLEDs).46 The polymer is believed to contribute to the electroluminescence performance enhancement through the more efficient electron injection and the protecting effect from the diffusion of Al atoms into the emissive layer. A further phosphonate-functionalized poly(fluorene) namely poly(9,9-bis-(6′-diethoxylphosphorylhexyl)fluorene was used as an electron injection layer to fabricate efficient multilayer polymer light emitting diodes (PLEDs) with Al cathode.34,47,48 Both, the favorable interfacial dipoles of the phosphonate-groups and the intense coordination interaction between the phosphonate-groups and Al at the polymer/Al interface are responsible for the efficient electron injection, resulting in superior performance compared to PLEDs with Ba or Ca cathode.In this work, we introduce the synthesis and characterization of two novel phosphonate-functionalized fluorene-based copolymers by a post-treatment of side-chain bromo-functionalized precursors. By means of a precipitation-sonication method, water-stable polymer microparticles of altered optical properties compared to their linear analogues are prepared, while their combination with CdTe nanocrystals gives particulate composite systems with exceptional optical properties and long term stability.
Results and discussion
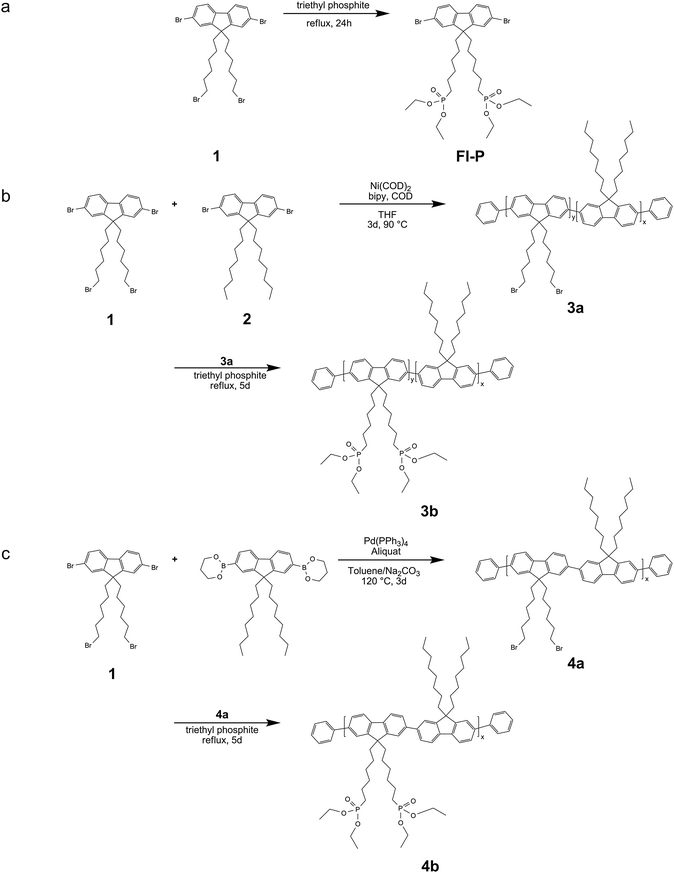
Random49 and alternating50 copolymers were designed with 2,7-dibromo-9,9-bis(6-bromohexyl)-9H-fluorene (1) and 2,7-dibromo-9,9-dioctyl-9H-fluorene as building blocks, correspondingly. 2,7-Dibromo-9,9-bis(6-bromohexyl)-9H-fluorene (1) was synthesized by a base-mediated alkylation, using a phase-transfer catalyst (PTC)51 and was utilized for the synthesis of two side-chain functionalized copolymers obtained by means of Ni(0) and Pd(0) catalyzed step-growth polymerizations according to Yamamoto and Suzuki. Yamamoto protocols normally lead to high molecular weights. On the other hand, Suzuki demands dibromo- and diboronic acid ester- functionalities, which render an exact 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the starting materials essential and makes a control over the backbone constitution possible. The different reactivity of the comonomers, however, can be a reason for lower molecular weights in case of the Suzuki catalysis and on the other hand, for a ranging comonomer content in case of the Yamamoto catalysis. 6-(2,7-Dibromo-9H-fluoren-9-yl)hexylphosphonate (Fl-P) was obtained by a typical Arbuzov protocol, refluxing compound 1 in triethyl phosphite for 24 h (Scheme 1a).52 For the copolymerization according to a Ni(0)-mediated Yamamoto C–C coupling protocol,532,7-dibromo-9,9-bis(6-bromohexyl)-9H-fluorene (1) and 2,7-dibromo-9,9-dioctyl-9H-fluorene (2)54 were used as building blocks and the step-growth reaction was conducted in THF by means of Ni(COD)2 and 2,2′-bipyridine as the organometallic counterpart (Scheme 1b). For the Suzuki step-growth polymerization,552,7-dibromo-9,9-bis(6-bromohexyl)-9H-fluorene (1) and 2,2-(9,9-dioctyl-9H-fluorene-2,7-diyl)bis(1,3,2-dioxaborinane) were applied by means of catalyst tetrakis(triphenyl)phosphine palladium(0) and aliquat 336 as phase-transfer agent (Scheme 1c). The random copolymer 3a was obtained by using an equal molar feed-ratio of the two comonomers 1 and 2 and a 1:1 stoichiometry of comonomer 1 to 2,2′-(9,9-dioctyl-9H-fluorene-2,7-diyl)bis(1,3,2-dioxaborinane) was the case in the synthesis of the alternating copolymer 4a as well. In order to remove impurities and to obtain unimodal molecular-weight distributions the copolymers were exhaustively purified by continuous extractions with ethanol, isopropanol and chloroform over intervals of 1 to 3 days by means of a Soxhlet apparatus. The copolymers were well-soluble in solvents of medium to high polarity like chloroform, dichloromethane, or tetrahydrofuran. Ongoing, the copolymers were post-functionalized in order to tether phosphonate-groups onto the side-chains of the backbone. The post-functionalization was conducted in triethyl phosphite at 165 °C reaction temperature and acquisition of the polymers by precipitation from n-hexane (Scheme 1b,c). The obtained polymers exhibited exceptional solubility in even more polar solvents like dimethylformamide. Fig. 1 shows the 1H-NMR spectra (exemplarily for polymers 3a,b) recorded in deuterated chloroform (CDCl3), comparing the side-chain bromo-functionalized copolymers to the phosphonate-functionalized monomer and respective copolymers. The resonance at 3.32 ppm (copolymers 3,4a) is assigned to the protons of the carbon atom with the attached bromine (C5H10CH2-Br), and is evidence for the success of the applied protocols (Fig. 1a). The achievement of the applied post-functionalization procedure is verified by the new signals monitored at 1.64 and 4.04 ppm in the spectra of the final polymers 3,4b (Fig. 1b). The resonance at 4.04 ppm is assigned to the protons of the carbon atoms linked to the oxygen atom of the phosphonate-group [(P–(O–CH2)2], while the signal at 1.64 ppm is the high field-shifted resonance of the protons attached to the carbon bond next to the phosphor atom (C5H10CH2-P). Both resonances can be found in the spectrum of the monomer as well (Fig. 1c).31 P-NMR spectroscopy is a further proof for the success of the post-functionalization with the signal arising at 32.48 ppm, which is typical for phosphor incorporated in the phosphonate-group. Comparing this value to the chemical shift of the corresponding monomer Fl-P (32.41 ppm) an excellent coincidence is observed. Moreover, 1H-NMR spectroscopy was used for the determination of the actual percentages of each building block in polymers 3,4a. As expected, copolymer 4a revealed an alternating backbone constitution of 44% of the 9,9-bis(6-bromohexyl)-9H-fluorene derivative 1 and 56% of the 9,9-dioctyl-9H-fluorene moiety 2, while in copolymer 3a the 9,9-dioctyl-9H-fluorene part predominates exhibiting a 76% backbone incorporation (Table 1).
:1 stoichiometry of the starting materials essential and makes a control over the backbone constitution possible. The different reactivity of the comonomers, however, can be a reason for lower molecular weights in case of the Suzuki catalysis and on the other hand, for a ranging comonomer content in case of the Yamamoto catalysis. 6-(2,7-Dibromo-9H-fluoren-9-yl)hexylphosphonate (Fl-P) was obtained by a typical Arbuzov protocol, refluxing compound 1 in triethyl phosphite for 24 h (Scheme 1a).52 For the copolymerization according to a Ni(0)-mediated Yamamoto C–C coupling protocol,532,7-dibromo-9,9-bis(6-bromohexyl)-9H-fluorene (1) and 2,7-dibromo-9,9-dioctyl-9H-fluorene (2)54 were used as building blocks and the step-growth reaction was conducted in THF by means of Ni(COD)2 and 2,2′-bipyridine as the organometallic counterpart (Scheme 1b). For the Suzuki step-growth polymerization,552,7-dibromo-9,9-bis(6-bromohexyl)-9H-fluorene (1) and 2,2-(9,9-dioctyl-9H-fluorene-2,7-diyl)bis(1,3,2-dioxaborinane) were applied by means of catalyst tetrakis(triphenyl)phosphine palladium(0) and aliquat 336 as phase-transfer agent (Scheme 1c). The random copolymer 3a was obtained by using an equal molar feed-ratio of the two comonomers 1 and 2 and a 1:1 stoichiometry of comonomer 1 to 2,2′-(9,9-dioctyl-9H-fluorene-2,7-diyl)bis(1,3,2-dioxaborinane) was the case in the synthesis of the alternating copolymer 4a as well. In order to remove impurities and to obtain unimodal molecular-weight distributions the copolymers were exhaustively purified by continuous extractions with ethanol, isopropanol and chloroform over intervals of 1 to 3 days by means of a Soxhlet apparatus. The copolymers were well-soluble in solvents of medium to high polarity like chloroform, dichloromethane, or tetrahydrofuran. Ongoing, the copolymers were post-functionalized in order to tether phosphonate-groups onto the side-chains of the backbone. The post-functionalization was conducted in triethyl phosphite at 165 °C reaction temperature and acquisition of the polymers by precipitation from n-hexane (Scheme 1b,c). The obtained polymers exhibited exceptional solubility in even more polar solvents like dimethylformamide. Fig. 1 shows the 1H-NMR spectra (exemplarily for polymers 3a,b) recorded in deuterated chloroform (CDCl3), comparing the side-chain bromo-functionalized copolymers to the phosphonate-functionalized monomer and respective copolymers. The resonance at 3.32 ppm (copolymers 3,4a) is assigned to the protons of the carbon atom with the attached bromine (C5H10CH2-Br), and is evidence for the success of the applied protocols (Fig. 1a). The achievement of the applied post-functionalization procedure is verified by the new signals monitored at 1.64 and 4.04 ppm in the spectra of the final polymers 3,4b (Fig. 1b). The resonance at 4.04 ppm is assigned to the protons of the carbon atoms linked to the oxygen atom of the phosphonate-group [(P–(O–CH2)2], while the signal at 1.64 ppm is the high field-shifted resonance of the protons attached to the carbon bond next to the phosphor atom (C5H10CH2-P). Both resonances can be found in the spectrum of the monomer as well (Fig. 1c).31 P-NMR spectroscopy is a further proof for the success of the post-functionalization with the signal arising at 32.48 ppm, which is typical for phosphor incorporated in the phosphonate-group. Comparing this value to the chemical shift of the corresponding monomer Fl-P (32.41 ppm) an excellent coincidence is observed. Moreover, 1H-NMR spectroscopy was used for the determination of the actual percentages of each building block in polymers 3,4a. As expected, copolymer 4a revealed an alternating backbone constitution of 44% of the 9,9-bis(6-bromohexyl)-9H-fluorene derivative 1 and 56% of the 9,9-dioctyl-9H-fluorene moiety 2, while in copolymer 3a the 9,9-dioctyl-9H-fluorene part predominates exhibiting a 76% backbone incorporation (Table 1).
 | ||
| Fig. 1 1H-NMR spectra of the precursor polymer 3a (a), polymer 3b (b) and the phosphonate-functionalized monomer Fl-P (c) recorded in CDCl3 (CHCl3 signal from remaining CHCl3 in CDCl3). | ||
| Polymer | Monomer 2 | Monomer 1 | Fl-P |
|---|---|---|---|
| x (%) | y (%) | y (%) | |
| 3a | 76 | 24 | — |
| 4a | 56 | 44 | — |
| 3b | 71 | — | 29 |
| 4b | 54 | — | 46 |
Suzuki-synthesized polymers like 4a tend to favor an alternating sequence with one diboron-ester functionality more in the backbone perhaps due to insufficient reactivity of the bromo-functionalities.29,56 Obstinately, the predomination of one specific comonomer in the polymer backbone of 3a is not unusual for Yamamoto-based polymers32 where deviations from the actual feed-ratios can come about, most probably due to the better solubility of the dioctyl-fluorene blocks in the tested reaction medium. As the inherent reactivity of the bromine-functional groups in the two fluorene cores should be equivalent, the preference for monomer 2 could be attributed to solubility matters of the hexyl side-chains of monomer 1 compared to the octyl side-chains. Calculations of the content of the post-functionalized polymers resulted in case of 3b in a ratio-content of dioctyl-fluorene of 71%. The Suzuki-based post-functionalized copolymer 4b follows a similar alternating backbone pattern as the precursor 4a and the building blocks 2 and Fl-P are integrated into the backbone by a 54:46% ratio, correspondingly.
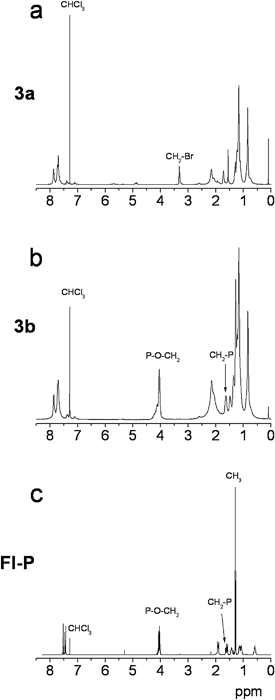
Copolymers 3,4a and their post-functionalized counterparts 3,4b were further characterized by means of gel permeation chromatography (GPC). The number-average molecular weights of copolymers 3,4b are 15100 g mol−1 (PDI 2.28) and 9100 g mol−1 (PDI 2.26), correspondingly, while the unimodal elugrams of the precursors in Fig. 2 illustrate the homogeneity of the isolated polymers. Deviations in the molecular weights of precursor and final polymers arise due to their quite different dissolution behaviour. The post-functionalized polymers are, due to the high polarity of their phosphonate-groups, well-soluble in dimethylformamide (DMF). This solvent was moreover utilized as eluent for the performance of the GPC measurements. The limited solubility of 3,4a originates from the bromine-atoms, which make chains with higher molecular weights less soluble.57 On the other hand, the enhanced polarity of the phosphonate-groups arising through the presence of the electronegative oxygens is responsible for the extremely good dissolution of polymers 3,4b. A further aspect considered, is the different work-up methods of the precursor and final polymers.58 The extraction of the former with chloroform may result in a partial fractionation, leading to the aforementioned less soluble high molecular weight chains, which is not the case when the final polymers are purified by precipitation from n-hexane. Considering that the post-functionalization is performed almost quantitatively and regarding that the molecular weight values and polydispersity indices should be treated as rough estimates due to the differences in hydrodynamic volumes between the conjugated polymers and the poly(methyl methacrylate) standards, the amphiphilic interaction of the phosphonate-groups with DMF and their larger total weight compared to the former bromine-functionalities, might be the key-reason for the enhancement of the molecular weights.
 | ||
| Fig. 2 Gel permeation chromatography elugrams of copolymers 3,4a. | ||
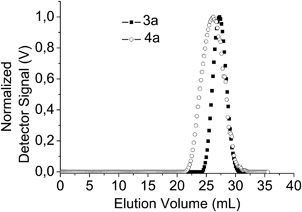
The optical properties of the copolymers 3,4a and 3,4b were additionally determined. Fig. 3 illustrates the optical properties of the final copolymers in solution (a) and films (b), while Table 2 lists the information regarding the optical properties of all polymers in solution and films. The final products 3,4b revealed blue light emission in chloroform solutions with an emission maximum at 416 nm and quantum yields up to 0.54. An advantageous precipitation method allows the shift of their blue emission to white by preparation of water-stable dispersions. Moreover, very high quantum yields of up to 0.84 were obtained (Table 2).
 | ||
| Fig. 3 Normalized emission and absorption (inset) spectra of copolymers 3,4b in solution (chloroform, 10−6 mol L−1) (a), films (drop cast from chloroform, 1 mg mL−1) (b) and as polymer microparticles prepared via a precipitation-sonication procedure (water, 10−4 mol L−1) (c). | ||
| Abssola(lgε)e [nm] | Absfilm [nm] | Emsola,b [nm] | Emfilmb [nm] | Egsola,c [eV] | Egfilmc [eV] | Φsola,d | |
|---|---|---|---|---|---|---|---|
| a In chloroform solution (10−6 mol L−1) for polymers 3,4a and 3,4b and in water solutions (10−4 mol L−1) for polymer particles 3,4bMPs. b λexc. 390 [nm]. c Calculated from the absorption band edge. d Determined according to Demas and Crosby62 by using PF as reference. e Logε in L mol−1 cm−1. | |||||||
| 3a | 382 (5.44) | 380 | 418/439 | 425/446 | 2.92 | 2.89 | 0.33 |
| 4a | 380 (5.20) | 381 | 417/440 | 429/449 | 2.95 | 2.88 | 0.16 |
| 3b | 381 (4.98) | 388 | 416/440 | 431/448 | 2.93 | 2.85 | 0.54 |
| 4b | 382 (5.71) | 411 | 416/441 | 432/453 | 2.93 | 2.58 | 0.52 |
| 3b MPs | 380 (2.69) | 397 | 432/455/530 | — | 2.86 | 2.59 | 0.61 |
| 4b MPs | 382 (2.45) | 400 | 436/464/493 | — | 2.83 | 2.60 | 0.84 |
This precipitation procedure, involves the dissolution of the polymers in tetrahydrofuran and their injection in water, while sonicating for 1 h at room temperature (Scheme 2a). By using this method, in principle, we are dealing with a solvent exchange method, in which the target polymers are dissolved in a ‘good’ solvent (THF) and rapidly mixed by means of sonication with an excess of a ‘poor’ solvent (water). Due to the discrepancy in the solubility of the polymer in the two solvents and the miscibility of the two media, a collapse of the polymer chains is induced, resulting in the formation of hydrophobic polymer particles.1–55
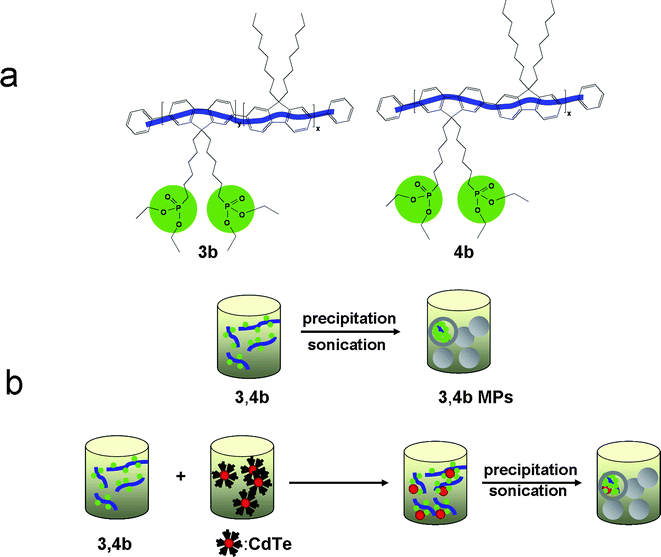 | ||
| Scheme 2 Schematic representation of the formation of the polymer microparticles 3,4b (a) and their composites with CdTe nanocrystals (b) via the precipitation-sonication method. | ||
The sudden change of solvent quality leads to the densely packed structures of particulate spheres through the aggregation of the organic molecules. The resulting particulate material is designated as microparticles (MPs) and this is the case for the polymer-CdTe systems deriving from the application of the precipitation-sonication process as well. The differentiation of the optical properties of the achieved polymer microparticles compared to the linear copolymers can be seen in Fig. 3a–c and constitutes one interesting outcome of this study as microparticles of the 3bpolymer exhibit a different behavior regarding their optical properties compared to the 4bMPs.
The differentiation is attributed to the Yamamoto-mediated step-growth mechanism applied for copolymer 3b being thus catalysis-dependent. The pronounced impact in case of 3bMPs where an additional band at 530 nm arises and complements the blue-light emitting band could be ascribed to increased interchain interactions, resulting in a small fraction of red-shifted aggregate species (Fig. 3c).59 A white-light emitting dispersion (Fig. 4a), which remains stable for over a period of three weeks is the result, a behavior not observed in the case of the 4bMPs, where the out coming bands are sharper compared to 4b, extend however, on the same wavelengths (Fig. 3c). The absence of a pronounced band at the green region of the linear visible spectrum leads, in case of 4b, to a blue-light emitting solution (Fig. 4b). The copolymers in form of their microparticles seem to possess electronic states, which might be in common with a supposed ketodefect phenomenon. A comparison of the polymer microparticles with fluorenone-containing poly(fluorene)s (PFOs) showed an additional peak at about 530–560 nm for PFOs in different surroundings,60 thus not clearly excluding the possibility of backbone oxidation of the polymers during the Yamamoto catalysis. The photophysical properties of the microparticles of copolymer 3b may moreover be affected from solubility matters and the structural configuration of the backbone, as the alternating configuration for polymer 4b microparticles does not provide a similar color-shift. This shifting in case of 3bMPs can therefore be attributed to intrachain energy-transfer or interchain excimer emission, which is both facilitated by the closer get-together of the polymer chains.61 Allying the phosphonate-functionalized copolymers with CdTe nanocrystals using the sonication method resulted in microparticulate dispersions, which remained stable without signs of deposition or illuminating power diminution. Experiments performed eliminating the sonication step resulted in solutions with a blue tinge under UV-vis irradiation, even after bringing green or red-light emitting CdTe nanocrystals into play (Fig. 5a,b). The used nanocrystals are water-stable and represent two different species, both end-capped with thioglycolic acid (TGA) and exhibit emission maxima at 534 (green CdTe) and 631 nm (red CdTe). Increasing the concentration of the red CdTe nanocrystals (from 1 × 10−5 mol L−1 to 6 × 10−5 mol L−1), a color change can indeed be observed but non-sonicated blends miss luminous intensity. As the optical properties of the nanocrystals remain unaltered when injecting them in sonicating water, the combination of the CdTe nanocrystals with the copolymers allows an intensification of the polymer-nanocrystal composite emission color when both counterparts are subjected to the sonication procedure (Fig. 5c).
 | ||
| Fig. 4 Photographs of the polymer microparticles 3b (a) and 4b (b), both illuminated with UV-vis irradiation under an excitation wavelength of λexc.: 366 nm. | ||
 | ||
| Fig. 5 Photographs of 3b, 3b + CdTegreen, 3b + CdTered, 3b + CdTegreen+red, CdTered, CdTegreen, CdTegreen+red (a) 4b, 4b + CdTegreen, 4b + CdTered, 4b + CdTegreen+red, CdTered, CdTegreen, CdTegreen+red (b) 3b MPs, 3b MPs + CdTered, 4b MPs, 4b MPs + CdTered, CdTered (c) (in that order left to right, under illumination of UV light, λexc.: 366 nm). Dispersions in 5c are prepared by the precipitation-sonication method, not the case for the solutions in 5a,b. | ||
In Scheme 2b, the bringing-together of the copolymers and the nanocrystals forming microparticulate composites via the precipitation-sonication procedure is illustratively depicted. To the best of our knowledge, this report is the first time describing such systems. The stability of these composites is tested through centrifugation experiments of their dispersions followed by fluorescence emission measurements of the resulting supernates and precipitates. Independently from the time and velocity of the centrifugation, coexistence of polymers and nanocrystals is evidenced through the fluorescence emission of both counterparts in the supernates, indicating the composite configuration of the two components (Figures S1 and S2 in the ESI). Dynamic light scattering (DLS) measurements, even qualify the aforementioned composites for the class of hybrid materials as their unimodal and symmetric particle distributions reveal (Figure S4 in the ESI). Only by reducing the CdTe concentration by a factor of 6, the phenomenon of polymer leaching in the supernate and nanocrystal sedimentation in the precipitate is observed as can be seen in Figure S3.
Fig. 5a,b show that the addition of CdTe in the non-sonicated aqueous polymer solutions does not cause color changes while a pronounced color changeover is the case only for the sonicated systems (Fig. 5c). The color transitions achieved in these systems can render them applicable in a broad field like in multi-layer colored or white OLED devices as well as in biological or environmental sensing applications.
In terms of UV-vis and fluorescence spectroscopy, the reason for the different behavior of the solutions is visualized in Fig. 6. All UV-vis spectra are dominated by the CdTe absorption pattern. The higher absorbance of the non-sonicated systems can be attributed to scattering effects of larger nanocrystal aggregates, which cover the polymer absorption band while the absorption pattern of the sonicated systems can be considered as a further indication of their composite-like nature due to the fact that polymer microparticles and nanocrystals seem both to participate and establish optical properties of the supposed composite material. The predomination of the nanocrystal absorption pattern is also connected to their higher electron affinity compared to conjugated polymers.63 Regarding the emission spectra, pronounced CdTe peaks (at 645 nm) come into sight only when applying sonication. The absence of these peaks in the non-sonicated systems is not due to concentration effects. Sonication seems to contribute to a closer coming-together of polymers and nanocrystals (evidenced by AFM, Fig. 7) and may thus support energy-transfer due to exciton diffusion from 3b to the inorganic dye. The inadequate transfer in case of 4b can be an issue of marginal inorganic dye concentration for this kind of copolymer. The fact that polymer 4b exhibits a moderate tendency for aggregation compared to the high aggregation of 3b as dynamic light scattering measurements elucidated, could lead to inter- and intra-forces between nanocrystals and polymers, which are less pronounced for this kind of polymer. As an excimer-like state is responsible for the observed photoluminescence behavior, weaker inter-chain interactions are expected in case of 4b, due to the moderate folding and packing of the chains and this might be the reason for the intensity differentiation.63
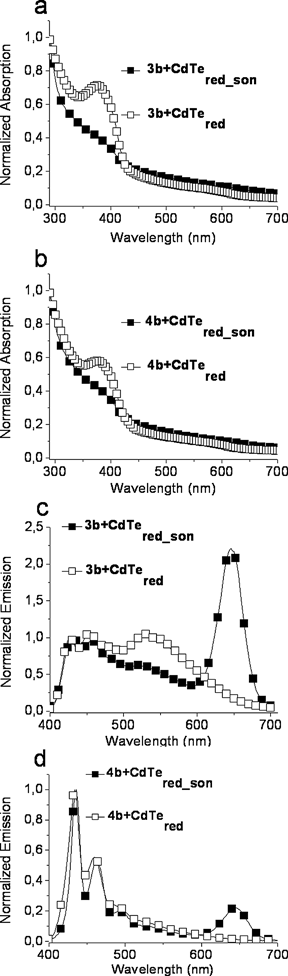 | ||
| Fig. 6 Normalized UV-vis (a,b) and fluorescence (c,d) spectra of the polymer-CdTe water dispersions prepared via the precipitation-sonication process and compared to non-sonicated systems. Fluorescence spectra were normalized regarding the polymer emission maximum (λexc: 390 nm, C3,4b: 10−4 mol L−1 and CCdTe: 6 × 10−5 mol L−1). | ||
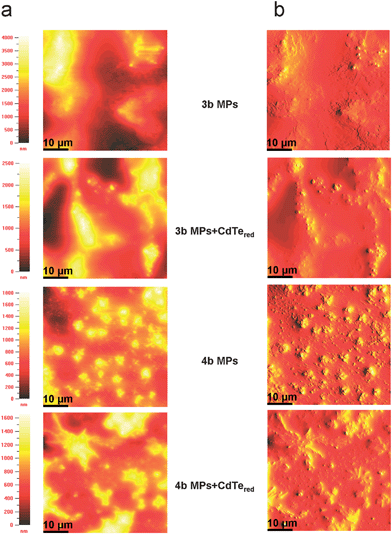 | ||
| Fig. 7 AFM images of the copolymer microparticles 3,4b and the copolymer-CdTered composites prepared from water dispersions via the precipitation-sonication method. (a) False color representation of the raw height data acquired in the tapping mode. (b) Contour plot calculated from the raw data with slope shading. The films were prepared by drop casting copolymer microparticles (10−2 mol L−1) and microparticulate copolymer-CdTered composites (10−2 mol L−1 and 10−3 mol L−1) from water on glass substrates. The AFM images have a scale of 50 × 50 μm. | ||
The surface morphology of the systems was exemplarily investigated for polymer microparticles solely and in combination to red-light emitting CdTe nanocrystals by means of atomic force microscopy (AFM). In Fig. 7a the AFM images are recorded in tapping mode and show the raw height data in top view and false color representation. In order to improve the contour visualization and acquire more information about the surface morphology, the recorded AFM images are also presented with slope shading (contour plot, Fig. 7b). This option is a calculation of the raw data, which adds a perspective illumination from the right to the images.64–66
Images of polymer 4b illustrate the tendency of formation of well-defined homogeneous microparticle aggregates (500 nm in diameter), which agglomerate in even larger clusters (2500 nm in size). Microparticle formation occurs less pronounced in case of polymer 3b, where they can be partly seen building microparticle clusters (900 nm in size, top-right side of the image). Dynamic light scattering (DLS) measurements complement the picture of the size distribution profile of the polymer microparticles in solution. Microparticles of 3b revealed a particle distribution of 540.1 nm in diameter and a standard deviation of 343.6 while the 4bMPs dispersion contains particles of 324.5 nm in diameter with a standard deviation of 143.3. The size differences are probably due to the different molecular weights and the occurrence of interchain interactions.59 The findings of the particle sizes in solution (DLS) are reasonably comparable to the AFM values taking the respective standard deviations into consideration and keeping in mind that the preparation conditions of the samples differ for the two applied methods whereas the solid state measurements result in tightly packed structures due to aggregation.
Furthermore by the AFM measurements in case of the 4b + CdTeredred composite system, an alignment of the nanocrystals around the polymer-chains can be supposed, whereas formation of nanocrystal aggregates, which stand out from the polymer surface can be conjectured for the 3b + CdTeredred system. The average surface roughness (RMS deviation) of the polymer microparticles 4b (264 nm) is by a factor of 3 lower compared to the average surface roughness of the 3bMPs (817 nm). This can be a further reason for the differentiation in the appearance of the two polymer-microparticles on the AFM images. On the other hand, their combination with the CdTe nanocrystals smooths the surface in case of 3b + CdTeredred (499 nm surface roughness) but does not affect immense the surface characteristic for the 4b + CdTeredred system (278 nm surface roughness) allowing thus the observation of the presumable coming-together of the nanocrystals around the polymer chains.
Experimental
Instrumentation
The elemental analyses were performed by means of the Vario Elemental EL analyzer. Ultraviolet-visible measurements (UV-vis) were performed on a Jasco V-550 spectrophotometer (1 cm cuvettes, chloroform) at concentration of 2.5 × 10−5 mol L−1 for compounds 1 and FI-P, 2.01 × 10−6 and 5.22 × 10−6 mol L−1 for the precursor polymers 3,4a, 9.41 and 1.1 × 10−6 mol L−1 for the post-functionalized ones 3,4b, 5.1 and 8.4 × 10−4 mol L−1 (1 cm cuvettes, water) for the polymer microparticles 3,4bMPs. Fluorescence spectroscopic measurements were done on a Cary Eclipse Fluorescence Spectrophotometer (1 cm cuvettes, chloroform) at concentration of 5 × 10−5 mol L−1 for compounds 1 and FI-P and the aforementioned concentrations and solvents for the precursor, the post-functionalized polymers and the polymer microparticles respectively. 1H- and 13C-NMR spectra were recorded in deuterated chloroform (CDCl3) with TMS as internal standard on a Bruker ARX 600 Fourier Transform Nuclear Resonance Spectrometer. The chemical shift δ is given in ppm. Infrared studies were conducted on a JASCO FT/IR-4200 Fourier-Transform-Spectrometer. Mass spectra were obtained using a Bruker micrOTOF instrument equipped with an electrospray ionization source (ESI-MS). For the electron impact ionization (EI) mass spectra, the device MAT 311A from Varian was utilized. Gel permeation chromatography analysis of the polymers was performed by means of a Jasco AS2055 Plus apparatus using Jasco UV/VIS-2070/75 and Jasco RI-2031 as detectors (GRAM columns, dimethylformamide (DMF) as eluent with ammonium hexafluorophosphate 5 mM as salt, 40 °C). For the determination of the molecular weights, a calibration based on poly(methyl methacrylate) standards was applied. Atomic force microscopy was performed on a Q-Scope™ 250 (Quesant Instrument Corporation). A scan head was utilized of 1 nm lateral resolution, 9 μm maximal vertical range, 0.1 nm vertical resolution and a four-quadrant photodiode. Samples were prepared by drop casting the water dispersions on glass substrates. The concentrations used were 10−2 mol L−1 for the polymers and 10−3 mol L−1 for the CdTe nanocrystals. Dynamic light scattering (DLS) measurements were carried out on a Delsa Nano C Particle Analyzer of the Beckman Coulter Company by means of the software Delsa Nano Beckman Coulter Inc. The polymer concentration used was 10−3 mol L−1 in water and 10−4 mol L−1 was the nanocrystal concentration. For the purification of the monomers by means of column chromatography silica gel of particle size 50–200 mesh was utilized as the stationary phase.Materials
All reactions were carried out under inert conditions (argon). N-Bromosuccinimide, tetrakis(triphenylphosphine)palladium(0) were purchased from ABCR, 1,6-dibromohexane, tetrabutylammonium bromide, bis(1,5-cyclooctadiene)nickel(0), 2,2′-bipyridine and triethyl phosphite from Acros, fluorene, aliquat 336, 2,2′-(9,9-dioctyl-9H-fluorene-2,7-diyl)bis(1,3,2-dioxaborinane) and 1,5-cyclooctadiene from Aldrich and bromobenzene from Merck. 2,7-Dibromo-9,9-bis(6-bromohexyl)-9H-fluorene (1),512,7-dibromo-9,9-dioctyl-9H-fluorene (2),54 and 6-(2,7-dibromo-9H-fluoren-9-yl)hexylphosphonate (Fl-P)52 were synthesized according to literature procedures. Polymerizations were performed in extra dry THF and toluene/Na2CO3 1:1 mixture according to literature known procedures.49,50,53,55CdTe nanocrystals were synthesized following a standard aqueous synthetic approach.67 The required chemicals, cadmium perchlorate hexahydrate, aluminum telluride lumps, thioglycolic acid (TGA) and solvent Milli-Q water were purchased from Alfa Aesar, Cerac Inc. Merck and Millipore, correspondingly.
Synthesis of the monomers
 ), 39.95, 33.72, 32.59, 28.92, 27.72, 23.45 (aliph.); IR (vmax/cm−1): 2925 (m, CH2, stretching), 2850 (m, CH2, stretching), 1568 (w, C
), 39.95, 33.72, 32.59, 28.92, 27.72, 23.45 (aliph.); IR (vmax/cm−1): 2925 (m, CH2, stretching), 2850 (m, CH2, stretching), 1568 (w, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, stretching), 1460 (m, CH2, scissor), 1446 (s, C–H, deformation), 810 (s, aromatic C–H, out-of-plane deformation); ESI-MSm/z Calcd. C25H30AgBr4 758; Found 758.8.
O), 1.82–1.85 (m, 4H, Ar-CH2), 3.95–3.99 (m, 8H, OCH2CH3), 7.34–7.35 (d, 2H, arom.), 7.38–7.39 (m, 2H, arom.), 7.44–7.45 (d, 2H, arom.); 13C-NMR (CDCl3, 600 MHz): δ (ppm): 152.24, 139.05, 130.28, 126.07, 121.53, 121.21 (arom.), 61.36 (CH2–O), 55.56 (
C, stretching), 1460 (m, CH2, scissor), 1446 (s, C–H, deformation), 810 (s, aromatic C–H, out-of-plane deformation); ESI-MSm/z Calcd. C25H30AgBr4 758; Found 758.8.
O), 1.82–1.85 (m, 4H, Ar-CH2), 3.95–3.99 (m, 8H, OCH2CH3), 7.34–7.35 (d, 2H, arom.), 7.38–7.39 (m, 2H, arom.), 7.44–7.45 (d, 2H, arom.); 13C-NMR (CDCl3, 600 MHz): δ (ppm): 152.24, 139.05, 130.28, 126.07, 121.53, 121.21 (arom.), 61.36 (CH2–O), 55.56 (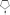 ), 40.16, 30.28 (CH2–P), 30.17, 29.34, 25.99, 25.06, 16.48 (aliph.); 31P-NMR (CDCl3, 600 MHz): δ (ppm): 32.41; IR (vmax/cm−1): 3461 (w, PO, stretching overtone), 2980 (w, CH2, stretching), 2930 (m, CH2, stretching), 2855 (w, CH2, stretching), 1648 (w, CC, stretching), 1569, (w, CC, stretching), 1451 (m, CH3, deformation), 1393 (m, O–CH2, wagging), 1232 (s, PO, stretching), 1024 (s, P–O/=C–H, stretching/in-plane deformation), 951 (s, P–O/=C–H, bending/out-of-plane deformation); GC-MS (m/z): Calcd C22H28Br2NO3P: 545.24; Found 545.03.
), 40.16, 30.28 (CH2–P), 30.17, 29.34, 25.99, 25.06, 16.48 (aliph.); 31P-NMR (CDCl3, 600 MHz): δ (ppm): 32.41; IR (vmax/cm−1): 3461 (w, PO, stretching overtone), 2980 (w, CH2, stretching), 2930 (m, CH2, stretching), 2855 (w, CH2, stretching), 1648 (w, CC, stretching), 1569, (w, CC, stretching), 1451 (m, CH3, deformation), 1393 (m, O–CH2, wagging), 1232 (s, PO, stretching), 1024 (s, P–O/=C–H, stretching/in-plane deformation), 951 (s, P–O/=C–H, bending/out-of-plane deformation); GC-MS (m/z): Calcd C22H28Br2NO3P: 545.24; Found 545.03.
Synthesis of the copolymers – Yamamoto procedure
1, 2, Ni(COD)2, 2,2′-bipyridine and 1,5-cyclooctadiene were added together in a Schlenk tube. Subsequently, THF (10 mL) was added to the reaction system and stirring was allowed for 3 days at 80 °C. 3 Hours before stopping the reaction, 0.05 mL of bromobenzene were added and after cooling down to room temperature the reaction solution was taken with chloroform and washed with 2 N HCl (2 × 100 mL), saturated NaHCO3 solution (1 × 100 mL) and water (2 × 100 mL). The organic phase was dried over Na2SO4 and the solvent removed by reduced pressure. The residue was dissolved in chloroform (1–2 mL), precipitated from methanol (300 mL) and the yellow solid further extracted with ethanol, isopropanol and chloroform. The chloroform fraction was concentrated under vacuum and precipitated from acetone.Synthesis of the copolymers – Suzuki procedure
1, 2,2′-(9,9-dioctyl-9H-fluorene-2,7-diyl)bis(1,3,2-dioxaborinane), tetrakis(triphenylphosphine)palladium(0) and aliquat 336 were added in a Schlenk tube. 2M Na2CO3 solution and toluene were subsequently added in a 1:1 mixture and the system was allowed to stir for 3 days at 120 °C. 3 Hours before stopping the reaction, bromobenzene and phenylboronic acid were added and after cooling down to room temperature the reaction solution was taken with chloroform and washed with 2 N HCl (2 × 100 mL), saturated NaHCO3 solution (1 × 100 mL) and water (2 × 100 mL). The organic phase was dried over Na2SO4 and the solvent removed by reduced pressure. The residue was dissolved in chloroform (1–2 mL), precipitated from methanol (300 mL) and the brown solid further extracted with ethanol, isopropanol and chloroform. The chloroform fraction was evaporated under vacuum and reprecipitated from acetone.
C, stretching), 1605 (w, CC, stretching), 1451 (m, C–H, deformation), 1249 (m, C–C, skeletal), 1024 (m, =C–H, in-plane deformation), 811 (s, =C–H, out-of-plane deformation); GPC (g mol−1): Mn: 7700; Mw: 12600; PDI: 1.64.
Synthesis of the post-functionalized polymers – general procedure
Precursors were dissolved in triethyl phosphite (10 mL) and vigorously stirred for 5 days under reflux. Solvent was removed by distillation, the residue dissolved in CHCl3 (1 mL) and precipitated from cold n-hexane (300 mL).O, stretching), 1453 (m, C–H/P–OC2H5, deformation/stretching), 1397 (w, P–OC2H5, wagging), 1233 (m, PO, stretching), 1020 (s, P–OC2H5/=C–H, stretching/in-plane deformation), 953 (s, P–OC2H5/=C–H, bending/out-of-plane deformation), 812 (s, =C–H, out-of-plane deformation); GPC (g mol−1): Mn: 15100; Mw: 34400; PDI: 2.28.
O, stretching), 1453 (m, C–H/P–OC2H5, deformation/stretching), 1396 (w, P–OC2H5, wagging), 1229 (m, PO, stretching), 1160 (w, =C–H/P–OC2H5, in-plane deformation/rocking), 1025 (s, P–OC2H5/=C–H, stretching/in-plane deformation), 954 (s, P–OC2H5/=C–H, bending/out-of-plane deformation), 814 (m, =C–H, out-of-plane deformation); GPC (g mol−1): Mn: 9100; Mw: 20600; PDI: 2.26.
Preparation of polymer-CdTe dispersions
THF solutions of polymers 3,4b (0.1 mg mL−1) were prepared and filtered through a micropore syringe filter (0.45 μm). 2 mL of the stock solutions were injected in 50 mL distilled water, THF was removed under vacuum and the dispersions filtered through a paper filter (90 mm of diameter). CdTe water solutions (100 μL) of 5.58 × 10−5 mol L−1 concentration for green-light emitting nanocrystals and 10−5 to 6 × 10−5 mol L−1 concentration for red-light emitting species were injected in the polymer dispersions (10 mL, 10−4 mol L−1polymer concentration). The fluid systems were sonicated for 1 h at room temperature. As a control experiment, nanocrystals or polymers solely with the aforementioned concentrations were just injected in water and sonicated for 1 h at room temperature. For solutions, which were not subjected to the ultrasonic-bath procedure, concentrations of 10−4 mol L−1 for the copolymers (10 mL), 1 × 10−5 mol L−1 for the red CdTe (100 μl) and 5.58 × 10−5 mol L−1 for the green CdTe (100 μl) nanocrystals were used, respectively (Fig. 5a,b).Conclusion
In summary, we prepared two bromo-functionalized, fluorene-based copolymers using nickel(0)- and palladium(0)-mediated polymerizations. The copolymers were post-functionalized in terms of their bromine end-groups and substituted by diethyl phosphonate ones. The final copolymers can be processed from polar solvents such as tetrahydrofuran or dimethylformamide and therefore be incorporated in a precipitation-sonication procedure, building water-stable dispersions with very high quantum yields of up to 0.84. It is interesting to find that the implemented procedure alters the optical properties catalysis-dependent from blue in the organic solvents to even white emission in the ‘green chemistry’ solvent water and generates microparticulate material of 500 to 900 nm in size in the solid state (AFM measurements) while microparticles of 234.5 to 540.1 nm in diameter were formed in the water solutions (DLS measurements). Combining the copolymers with semiconducting CdTe nanocrystals a worthwhile shifting of the emission color could be achieved whereby the dispersions maintained their stability and their intact emission brightness as evidenced by centrifugation experiments and the subsequent fluorescence emission measurements of the supernates. Well endowed with the aforementioned properties and an uncontroversial harmless solvent like water, these systems can become a further useful option in the direction of the ‘multi-layer light-emitting devices’ evolution. Moreover, these highly emissive water-stable systems could probably recommend themselves for biological or environmental sensing applications. For example, they could offer great potential and prospective promising chances in the fields of disease diagnostics (two photon microscopy imaging), environmentally compatible water-based deposition techniques (ink-jet printing) or in the area of sensor technology by the detection of heavy metals through the emission color changeover of the polymer microparticles upon addition of nanocrystals.Acknowledgements
The Deutsche Forschungsgemeinschaft (DFG) is acknowledged by E. H., I. K. and O. A. for financial support within the grant application HO3911/2-1: “Hybrid polymer/nanocrystals structures: fabrication and studies of energy transfer, charge generation and transport”. A. E. further acknowledges the European Union (E. U.) for financial support within the grant applications “Network of Excellence nanophotonics4energy” and “Innovative Materials for Future Generation Excitonic Solar Cells' (INNOVASOL)”. Vladimir Lesnyak is acknowledged for synthesizing and providing the CdTe nanocrystals and Jan Poppe for supporting and assisting in the DLS measurements. Prof. Ullrich Scherf is acknowledged for granting access to the tools of the Macromolecular Chemistry.Notes and references
- C. Szymanski, C. F. Wu, J. Hooper, M. A. Salazar, A. Perdomo, A. Dukes and J. McNeill, J. Phys. Chem. B, 2005, 109, 8543 CrossRef CAS.
- C. F. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415 CrossRef CAS.
- J. B. Yu, C. F. Wu, S. P. Sahu, L. P. Fernando, C. Szymanski and J. McNeill, J. Am. Chem. Soc., 2009, 131, 18410 CrossRef CAS.
- C. F. Wu, C. Szymanski, Z. Cain and J. McNeill, J. Am. Chem. Soc., 2007, 129, 12904 CrossRef CAS.
- C. F. Wu, H. S. Peng, Y. F. Jiang and J. McNeill, J. Phys. Chem. B, 2006, 110, 14148 CrossRef CAS.
- C. F. Wu, Y. L. Zheng, C. Szymanski and J. McNeill, J. Phys. Chem. C, 2008, 112, 1772 CAS.
- C. F. Wu, B. Bull, K. Christensen and J. McNeill, Angew. Chem., Int. Ed., 2009, 48, 2741 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488 CrossRef CAS.
- M. C. Baier, J. Huber and S. Mecking, J. Am. Chem. Soc., 2009, 131, 14267 CrossRef CAS.
- K. Müller, M. Klapper and K. Müllen, Macromol. Rapid Commun., 2006, 27, 586 CrossRef.
- M. Green, P. Howes, C. Berry, O. Argyros and M. Thanou, Proc. R. Soc. London, Ser. A, 2009, 465, 2751 CrossRef CAS.
- Z. Q. Yang, W. T. S. Huck, S. M. Clarke, A. R. Tajbakhsh and E. M. Terentjev, Nat. Mater., 2005, 4, 486 CrossRef CAS.
- P. Taranekar, J. Y. Park, D. Patton, T. Fulghum, G. J. Ramon and R. Advincula, Adv. Mater., 2006, 18, 2461 CrossRef CAS.
- W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73 CAS.
- M. J. Miller, S. H. Wei, I. Parker and M. D. Cahalan, Science, 2002, 296, 1869 CrossRef CAS.
- A. Agarwal, M. L. Coleno, V. P. Wallace, W. Y. Wu, C. H. Sun, B. J. Tromberg and S. C. George, Tissue Eng., 2001, 7, 191 CrossRef CAS.
- J. H. Moon, W. McDaniel, P. MacLean and L. E. Hancock, Angew. Chem., Int. Ed., 2007, 46, 8223 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS.
- C. Medina, M. J. Santos-Martinez, A. Radomski, O. I. Corrigan and M. W. Radomski, Br. J. Pharmacol., 2009, 150, 552 CrossRef.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11 CrossRef CAS.
- A. Hoshino, K. Fujioka, T. Oku, M. Suga, Y. F. Sasaki, T. Ohta, M. Yasuhara, K. Suzuki and K. Yamamoto, Nano Lett., 2004, 4, 2163 CrossRef CAS.
- R. Hardman, Environ. Health Perspect., 2006, 114, 165 CrossRef.
- Z. Chen, X. Li, WO 2007/027159 A1, 2007.
- M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867 CrossRef CAS.
- W. Y. Wong, Coord. Chem. Rev., 2005, 249, 971 CrossRef CAS.
- W. L. Yu, J. Pei, W. Huang and A. J. Heeger, Adv. Mater., 2000, 12, 828 CrossRef CAS.
- J. Lee, H. J. Cho, B. J. Jung, N. S. Cho and H. K. Shim, Macromolecules, 2004, 37, 8523 CrossRef CAS.
- C. Fan, K. W. Plaxco and A. J. Heeger, J. Am. Chem. Soc., 2002, 124, 5642 CrossRef CAS.
- I. Kanelidis, Y. Ren, V. Lesnyak, J. C. Gasse, R. Frahm, A. Eychmüller and E. Holder, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 392 CrossRef CAS.
- I. Kanelidis, A. Vaneski, D. Lenkeit, S. Pelz, V. Elsner, R. M. Stewart, J. Rodríguez-Fernández, A. A. Lutich, A. S. Susha, R. Theissmann, S. Adamczyk, A. L. Rogach and E. Holder, J. Mater. Chem., 2011, 21, 2656 RSC.
- V. Marin, E. Holder, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 793 CrossRef CAS.
- I. Kanelidis, V. Elsner, M. Bötzer, M. Butz, V. Lesnyak, A. Eychmüller and E. Holder, Polymer, 2010, 51, 5669 CrossRef CAS.
- G. Zhou, Y. Cheng, L. Wang, X. Jing and F. Wang, Macromolecules, 2005, 38, 2148 CrossRef CAS.
- G. Zhou, G. Qian, L. Ma, Y. Cheng, Z. Xie, L. Wang, X. Jing and F. Wang, Macromolecules, 2005, 38, 5416 CrossRef CAS.
- C. Edder and J. M. J. Frechet, Org. Lett., 2003, 5, 1879 CrossRef CAS.
- M. R. Pinto, J. R. Reynolds and K. S. Schanze, Polym. Prepr., 2002, 43, 139 CAS.
- K. Stokes, K. Heuze and R. McCullough, Macromolecules, 2003, 36, 7114 CrossRef CAS.
- X. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019 CrossRef CAS.
- C. J. Qin, H. Tong and L. X. Wand, Sci. China, Ser. B: Chem., 2009, 52, 833 CrossRef CAS.
- N. Tian, A. Thiessen, R. Schiewek, O. J. Schmitz, D. Hertel, K. Meerholz and E. Holder, J. Org. Chem., 2009, 74, 2718 CrossRef CAS.
- E. Tekin, E. Holder, D. Kozodaev and U. S. Schubert, Adv. Funct. Mater., 2007, 17, 277 CrossRef CAS.
- E. Tekin, H. Wijlaars, E. Holder, D. A. M. Egbe and U. S. Schubert, J. Mater. Chem., 2006, 16, 4294 RSC.
- V. Marin, E. Holder, M. M. Wienk, E. Tekin, D. Kozodaev and U. S. Schubert, Macromol. Rapid Commun., 2005, 26, 319 CrossRef CAS.
- V. Marin, E. Holder, R. Hoogenboom, E. Tekin and U. S. Schubert, Dalton Trans., 2006, 1636 RSC.
- E. Tekin, E. Holder, V. Marin, B.-J. de Gans and U. S. Schubert, Macromol. Rapid Commun., 2005, 26, 293 CrossRef CAS.
- B. Zhang, C. Qin, J. Ding, L. Chen, Z. Xie, Y. Cheng and L. Wang, Adv. Funct. Mater., 2010, 20, 2951 CrossRef CAS.
- X. Niu, C. Qin, B. Zhang, J. Yang, Z. Xie, Y. Cheng and L. Wang, Appl. Phys. Lett., 2007, 90, 203513 CrossRef.
- B. Zhang, C. Qin, X. Niu, Z. Xie, Y. Cheng, L. Wang and X. Li, Appl. Phys. Lett., 2010, 97, 043506 CrossRef.
- A. Tsami, X.-H. Yang, F. Galbrecht, T. Farrell, H. Li, S. Adamczyk, R. Heiderhoff, L. J. Balk, D. Neher and E. Holder, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4773 CrossRef CAS.
- A. Tsami, X.-H. Yang, T. Farrell, D. Neher and E. Holder, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7794 CrossRef CAS.
- E. P. Woo, M. Inbasekaran, W. R. Shiang, G. R. Roof. WO//05184, 1997.
- C. Qin, Y. Cheng, L. Wang, X. Jing and F. Wang, Macromolecules, 2008, 41, 7798 CrossRef CAS.
- L. Kinder, J. Kanicki and P. Petroff, Synth. Met., 2004, 146, 181 CrossRef CAS.
- B. Liu, B. S. Gaylord and G. C. Bazan, J. Am. Chem. Soc., 2003, 125, 6705 CrossRef CAS.
- U. Giovanella, M. Pasini, S. Destri, W. Porzio and C. Botta, Synth. Met., 2008, 158, 113 CrossRef CAS.
- J. Langecker and M. Rehahn, Macromol. Chem. Phys., 2008, 209, 258 CrossRef CAS.
- M. Svensson, F. Zhang, S. C. Veenstra, W. J. H. Verhees, J. C. Hummelen, J. M. Kroon, O. Inganäs and M. R. Andersoon, Adv. Mater., 2003, 15, 988 CrossRef CAS.
- R. Gütner, U. Asawapirom, M. Forster, C. Schmitt, B. Stiller, B. Tiersch, A. Falcou, H.-G. Nothofer and U. Scherf, Thin Solid Films, 2002, 417, 1 CrossRef.
- C. Wu and J. McNeill, Langmuir, 2008, 24, 5855 CrossRef CAS.
- S. Shekhar, E. Aharon, N. Tian, F. Galbrecht, U. Scherf, E. Holder and G. L. Frey, ChemPhysChem, 2009, 10, 576 CrossRef CAS.
- Q. Pei and Y. Yang, J. Am. Chem. Soc., 1996, 118, 7416 CrossRef CAS.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- F. Guo and P. Xie, Chin. J. Chem., 2009, 27, 1427 CrossRef CAS.
- E. J. Houser, D. B. Chrisey, M. Bercu, N. D. Scarisoreanu, A. Purice, D. Colceag, C. Constantinescu, A. Moldovan and M. Dinescu, Appl. Surf. Sci., 2006, 252, 4871 CrossRef CAS.
- H. L. Hing, S. J. Shee, M. A Kaswandi, A. H. Abd Aziz and A. Z. Sahalan, Proc. MST Conf., 2010, 12 Search PubMed.
- S. S. Stewart-Clark and Y. M. Lvov, J. Coat. Technol. Res., 2010, 8, 275 Search PubMed.
- A. L. Rogach, T. Franzl, T. A. Klar, J. Feldmann, N. Gaponik, V. Lesnyak, A. Shavel, A. Eychmüller, Y. P. Rakovich and J. F. Donegan, J. Phys. Chem. C, 2007, 111, 14628 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00337b |
| ‡ Current address: Preparative Macromolecular Chemistry, Institute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, D-76128 Karlsruhe, Germany. |
| This journal is © The Royal Society of Chemistry 2011 |