
Disorder-to-order transition of regiorandom P3HT upon electrochemical doping†
Seth R.
Jackson
 ,
Garrett W.
Collins
,
Rand L.
Kingsford
,
Perry W.
Martin
,
Jolene N.
Keller
and
Connor G.
Bischak
*
,
Garrett W.
Collins
,
Rand L.
Kingsford
,
Perry W.
Martin
,
Jolene N.
Keller
and
Connor G.
Bischak
*
Department of Chemistry, University of Utah, Salt Lake City, UT 84112, USA. E-mail: connor.bischak@utah.edu
First published on 10th June 2024
Abstract
The microstructure of organic mixed ionic–electronic conductors (OMIECs) plays a crucial role in their functionality. OMIECs are generally polycrystalline conjugated polymers consisting of a complex arrangement of disordered and ordered regions. While considerable research has focused on structural transformations in the ordered regions of OMIECs upon ion insertion, the evolution of disordered regions is difficult to probe. Here, we explore changes in the disordered conjugated polymer regiorandom (RRa) poly(3-hexylthiophene) (P3HT), focusing on its response to repeated electrochemical doping/dedoping cycles and thermal annealing. By performing a series of cyclic voltammograms (CVs) followed by grazing incidence wide angle X-ray scattering (GIWAXS), we show how the molecular packing and microstructure of RRa P3HT change. Additionally, we employ nanoscale infrared imaging via photoinduced force microscopy (PiFM) to visualize the changes in polymer morphology and the distribution of ions within the polymer matrix. Our results indicate that RRa P3HT experiences significant structural reorganization after repeated doping/dedoping cycles. These large structural rearrangements lead to partially irreversible doping due to ion trapping in electronically disconnected polymer regions. Moreover, we observe that electrochemical cycling followed by thermal annealing promotes crystallinity in RRa P3HT, yielding GIWAXS diffractograms akin to those of polycrystalline regioregular (RR) P3HT. Our research conclusively shows that disordered conjugated polymers can undergo substantial structural and morphological transformations through repeated electrochemical doping, leading to enhanced ordering and ion trapping within the polymer matrix.
 Connor G. Bischak | Dr Connor Bischak is an Assistant Professor in the Department of Chemistry at the University of Utah. He received his PhD from UC Berkeley (2017) with Prof. Naomi Ginsberg. He then carried out postdoctoral research at the University of Washington with Prof. David Ginger as a Washington Research Foundation Postdoctoral Fellow and a Cottrell Postdoctoral Fellow. His research interests include organic and hybrid semiconductors, mixed conductors, structural phase transitions, nanoscale imaging, and self-driving chemical laboratories. |
Introduction
Organic mixed ionic–electronic conductors (OMIECs) have emerged as a versatile materials class due to their unique capability to transport both electronic charge carriers and ions.1–6 This dual transport capability renders OMIECs highly suitable for a diverse array of applications, such as organic electrochemical transistors (OECTs) for bioelectronics and biosensing,7–10 neuromorphic computing,11–13 and thermoelectrics.14–16 Typically, OMIECs are conjugated polymers that undergo electrochemical doping. In electrochemical doping, electrons or holes are injected into the polymer from an underlying electrode. These injected charges are balanced by ions from an adjacent liquid electrolyte, creating polaron–ion pairs.1,17 The process of electrochemical doping is complex, involving couplings between ion motion, electron transport, and structural changes within the polymer, which depend on many factors, including the electrolyte identity,9,18 the chemistry of the polymer side chains,7,8,19 and the nanoscale morphology of the conjugated polymer OMIEC.6,20,21 While the impact of electrochemical doping on the crystalline regions of OMIECs has been extensively characterized,6,22,23 the behavior and evolution of the amorphous regions remain less explored.Few studies have investigated changes in disordered conjugated polymers upon electrochemical doping. Notably, Neusser et al. showed a significant increase in the conductivity of regiorandom (RRa) poly(3-hexylthiophene) (P3HT) of over six orders of magnitude upon electrochemical doping.24 More recently, Cavassin et al. employed various spectroelectrochemistry techniques to reveal that different degrees of polaronic and bipolaronic species form in regioregular (RR) P3HT versus RRa P3HT with a propensity for bipolaron formation in disordered regions.25 Baustert et al. showed that while the anion identity has only a minimal impact on the oxidation potential of RRa P3HT, large ions are effectively excluded from the crystalline domains of RR P3HT.26 Additional studies indicate that disordered regions of conjugated polymers are advantageous for ion transport.18,20,27,28 However, the specifics of how disordered regions evolve upon electrochemical doping remains largely unexplored.
Several studies have shown that chemical doping can induce ordering in disordered conjugated polymers. For instance, Lim et al. investigated structural changes in blends of RRa P3HT and RR P3HT using 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) as the chemical dopant. Their findings revealed that vapor phase doping enhances the organization of disordered regions of P3HT thin films and improves the long-range connectivity of the polymer.29 In a complementary study, Yee, P. et al. further investigated disorder-to-order transitions in RRa P3HT, uncovering that doping with F4TCNQ and FeCl3 not only orders the structure of the film but does so through a charge transfer mechanism.30 Adding a similar small molecule non-dopant, tetracyanoquinodimethane (TCNQ), to RRa P3HT does not induce a significant structural change. Although these studies highlight the capacity of chemical doping to transform disordered RRa P3HT into a more ordered structure, the question remains whether electrochemical doping achieves a similar effect in enhancing polymer order.
In this work, we investigate the disorder-to-order transition of RRa P3HT upon electrochemical doping. Initially, upon applying a continuous doping potential to the RRa P3HT thin film in an aqueous electrolyte, we detect minimal changes in the structure of the polymer. However, upon performing a series of sequential cyclic voltammograms (CVs), we observe an expansion of the lamellar spacing and, upon thermal annealing, a significant ordering of RRa P3HT and a GIWAXS diffractogram that resembles ordered RR P3HT. We also observe irreversible ion trapping in the polymer film, coinciding with significant morphological changes due to repeated electrochemical doping. Employing nanoscale infrared imaging with photoinduced force microscopy (PiFM),31,32 an atomic force microscopy (AFM) based method previously used to map ion locations in OMIECs,18,20,23,33 and energy dispersive spectroscopy (EDS) we find that wrinkles form that are rich in aggregated salts and that irreversible ion trapping occurs throughout the thin film. Our findings conclusively demonstrate that a combination of repeated electrochemical doping/dedoping cycles and thermal annealing facilitates a disorder-to-order transition in RRa P3HT.
Results and discussion
Initially, we dope RRa P3HT at a constant potential to determine if electrochemical doping induces a disorder-to-order transition in the polymer. Fig. 1(A) shows the chemical structures of RRa P3HT, the electrolyte species potassium trifluoromethanesulfonimide (KTFSI), and the chemical dopant F4TCNQ.29,30 To determine whether electrochemical doping induces ordering in RRa P3HT, we subject a thin film of RRa P3HT (53 ± 5 nm thick) to a doping potential of 0.7 V vs. Ag/AgCl for 20 minutes in a 100 mM aqueous KTFSI electrolyte. We chose 0.7 V vs. Ag/AgCl as the doping bias to avoid potential water oxidation and minimize damage to the polymer (Fig. S1, ESI†). Fig. 1(B) shows ex situ 2D GIWAXS diffractograms of three RRa P3HT thin films: a RRa P3HT thin film immersed in a 100 mM KTFSI solution for 20 min without applying a bias, a RRa P3HT thin film post electrochemical doping (0.7 V vs. Ag/AgCl for 20 min), and a RRa P3HT sample immersed in a 10 mg mL−1 F4TCNQ acetonitrile (ACN) solution for the same duration. Compared to the immersed RRa P3HT thin film, the doped RRa P3HT thin film exhibits enhanced intensity of the ring corresponding to the lamellar side chain stacking peak (100) of the polymer at q = 0.39 Å−1 (15.9 Å). Contrastingly, the RRa P3HT thin film immersed in the F4TCNQ solution exhibits a notable expansion of the lamellar spacing (q = 0.36 Å−1, 17.5 Å) as well as a narrowing of the corresponding diffraction peak, aligning with previous observations of induced ordering of RRa P3HT when doped with F4TCNQ.29,30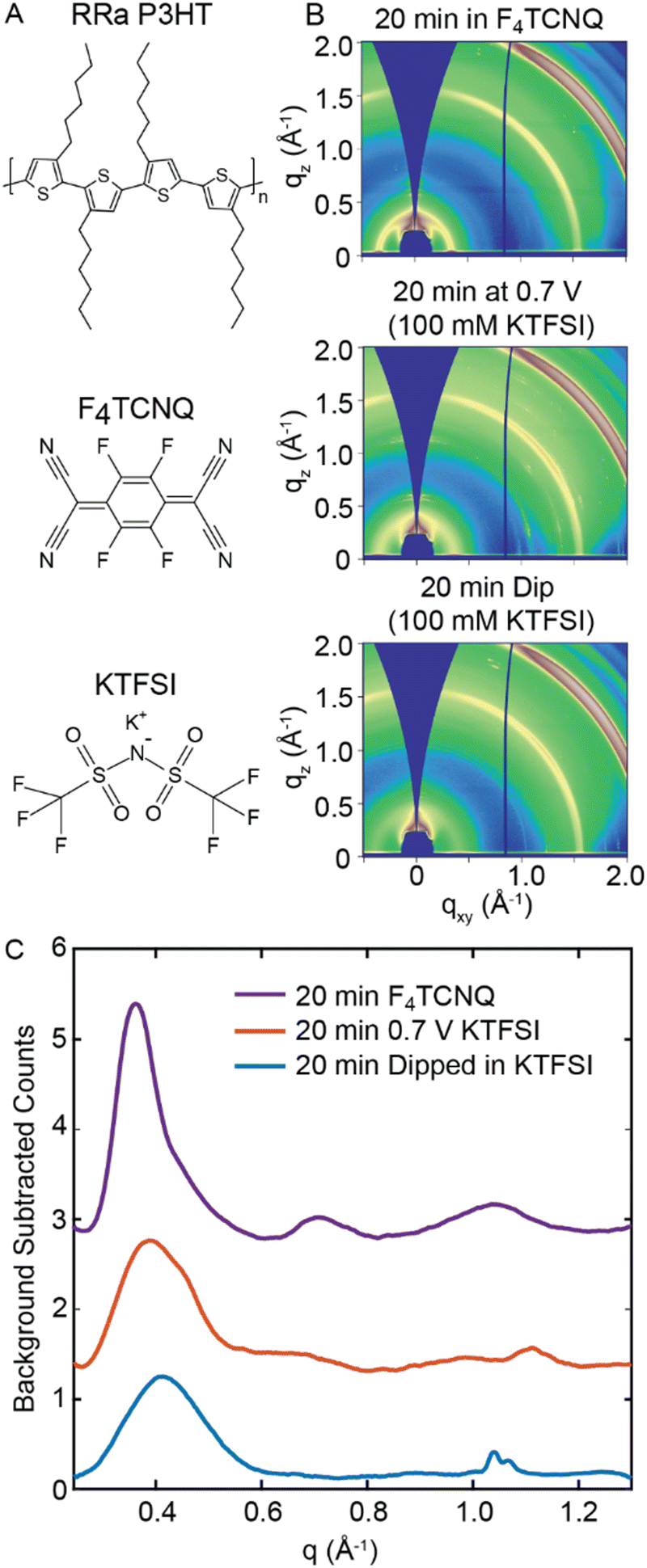 | ||
| Fig. 1 2D GIWAXS diffractograms and radially integrated diffractograms of RRa P3HT under different conditions. (A) Chemical structures of RRa P3HT, F4TCNQ, and KTFSI. (B) GIWAXS diffractograms of a RRa P3HT thin film immersed in 10 mg mL−1 F4TCNQ for 20 min (top), doped at 0.7 V vs. Ag/AgCl for 20 min (middle), and immersed in an aqueous 100 mM KTFSI electrolyte for 20 min (bottom). (C) Radially integrated and background subtracted GIWAXS diffractograms of the RRa P3HT thin films. | ||
Fig. 1(C) shows background subtracted radially integrated diffractograms, integrated from an angle of 30° to 55° to minimize contributions from the indium tin oxide (ITO) background, for the immersed RRa P3HT thin film, the electrochemically doped thin film, and the thin film doped with F4TCNQ. The thin film doped with F4TCNQ shows the characteristic (100) lamellar peak, along with the higher order (200) and (300) lamellar peaks, showcasing a diffractogram that closely resembles the one typically observed for RR P3HT.29,30 Compared to the RRa P3HT thin film immersed in the KTFSI electrolyte without a bias applied, the electrochemically-doped thin film exhibits a slight expansion of the lamellar spacing from 15.5 Å to 15.9 Å. Overall, chemical doping with F4TCNQ results in a much higher degree of ordering of RRa P3HT compared to electrochemical doping.
We next examine how electrochemical doping of RRa P3HT changes its optical properties. Fig. S2 (ESI†) shows UV/Vis absorption spectra of undoped RRa P3HT, RRa P3HT electrochemically doped in KTFSI at 0.7 V vs. Ag/AgCl, RRa P3HT electrochemically in KTFSI at 0.7 V and then dedoped at −0.5 V vs. Ag/AgCl, RRa P3HT doped with F4TCNQ, and RRa P3HT dedoped by thermal annealing. Notably, the UV/Vis absorption spectrum of RRa P3HT upon electrochemical doping and then dedoping shows a slight red shift, suggesting an increase in the ordering of RRa P3HT upon electrochemical doping and subsequent dedoping. We also observe irreversible polaron formation, indicating that not all ions leave the polymer matrix upon applying a dedoping potential. Fig. S3 (ESI†) shows images of the undoped, doped, and doped/dedoped RRa P3HT thin films, indicating that the higher absorbance is not a result of increased scattering following doping and dedoping. The F4TCNQ doped film exhibits absorption peaks appearing around 800 and 400 nm appeared that indicate doping.34–36 Overall, the GIWAXS diffractograms show minimal differences between the electrochemically doped RRa P3HT film and the RRa P3HT thin film immersed in the electrolyte with no applied bias, indicating the absence of induced ordering under constant biasing when compared to F4TCNQ doped films. The spectroelectrochemistry results, however, demonstrate irreversible electrochemical doping of RRa P3HT.
To examine changes in the morphology of RRa P3HT and the distribution of trapped TFSI− ions at the nanoscale, we perform nanoscale infrared imaging with PiFM. We acquire PiFM images at 1200 cm−1, which corresponds to the C–F stretching vibrational mode of TFSI−.37Fig. 2(A)–(C) show the AFM topography and PiFM images of three samples: an undoped RRa P3HT thin film, a doped RRa thin film (0.7 V vs. Ag/AgCl in 100 mM KTFSI for 20 min followed by 45 s at −0.5 V), and RRa P3HT doped with F4TCNQ, respectively. The undoped RRa P3HT exhibits a very smooth surface with a 0.36 nm root mean squared (RMS) roughness. Following electrochemical doping in KTFSI, the film develops elevated features that contain a high concentration of TFSI−. Fig. S4 (ESI†) shows topography and PiFM images of a larger field-of-view (50 × 50 μm), highlighting the trapping of TFSI− in the electrochemically doped films. Comparatively, the topography image of the F4TCNQ doped thin film shows an increase in roughness compared to the undoped RRa P3HT thin film with a 0.55 nm RMS surface roughness. This increase in surface roughness upon chemical doping is consistent with prior studies.38,39 PiFM and Fourier transform infrared spectroscopy (FTIR) spectra, shown in Fig. S5 (ESI†), confirm the presence of F4TCNQ in the RRa P3HT matrix. While the application of a constant potential to RRa P3HT indeed alters the morphology of the polymer and leads to the irreversible trapping of ions, it does not significantly induce ordering.
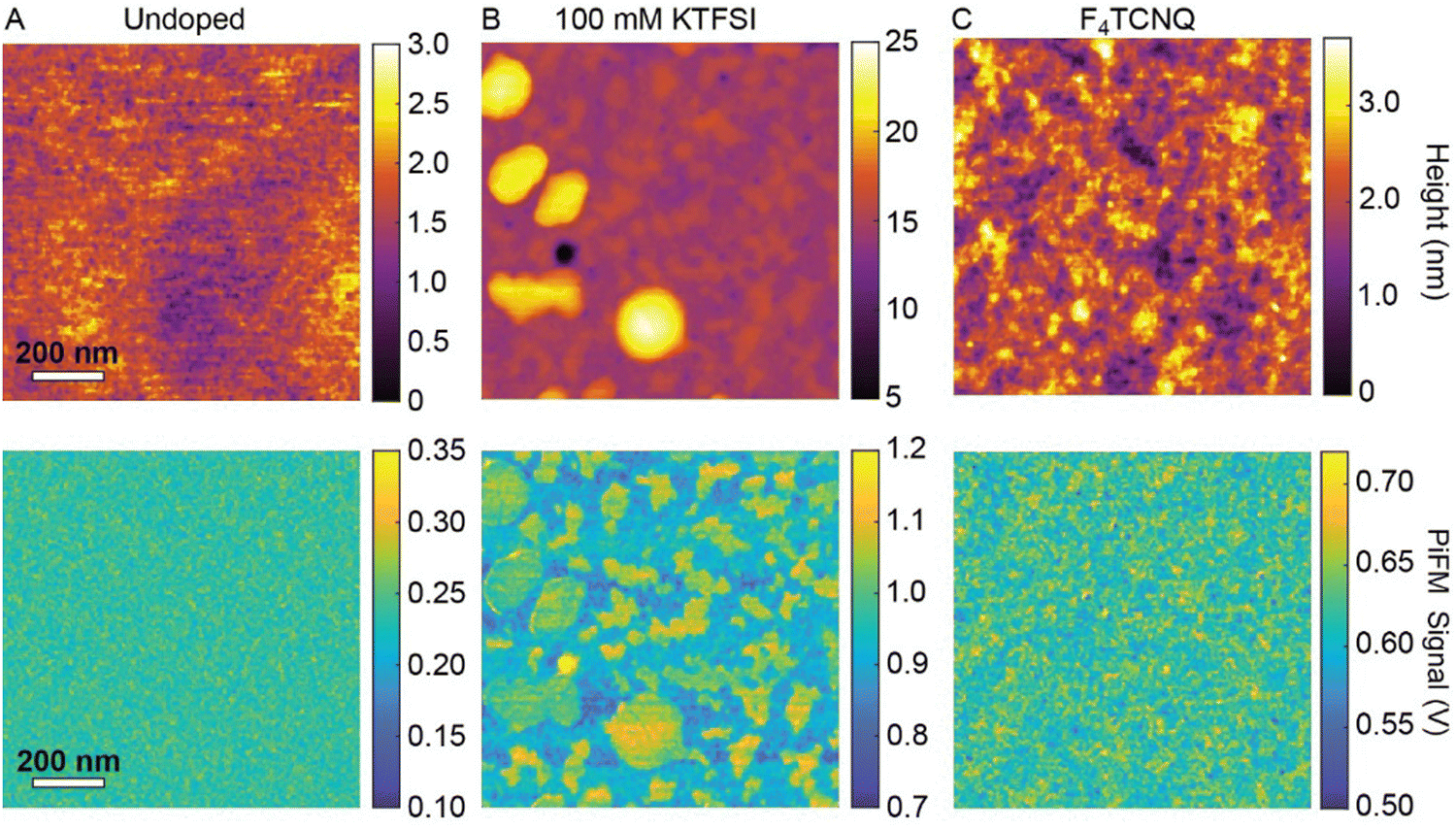 | ||
| Fig. 2 PiFM images of RRa P3HT films that underwent electrochemical and chemical doping. AFM topography and PiFM nanoscale infrared images of (A) an undoped RRa P3HT film, (B) a RRa P3HT thin film doped for 20 min in an aqueous 100 mM KTFSI electrolyte, and (C) a RRa P3HT film doped in a F4TCNQ solution for 20 min. | ||
Rather than applying a constant potential, we next investigate whether repeated electrochemical doping/dedoping cycles induces a disorder-to-order transition in RRa P3HT. Fig. 3(A) shows 50 consecutive CVs performed on a RRa P3HT thin film (−0.5 to 0.9 V vs. Ag/AgCl at a scan rate of 0.1 V s−1). As we acquire more CVs, the oxidation potential of RRa P3HT shifts towards lower voltage, potentially indicating ordering of the RRa P3HT thin film, as more ordered P3HT thin films exhibit a lower oxidation potential.20,26Fig. 3(B) shows a series of UV/Vis absorption spectra acquired after every five CVs after applying a dedoping bias of −0.5 V for 45 s. The repeated doping and dedoping cycles lead to a redshift in the π–π* absorption peak of RRa P3HT, further indicating an increase in the ordering of the polymer like in F4TCNQ doped films.30 Furthermore, the persistence of the polaron absorption peak, even after dedoping, signifies the irreversible trapping of ions within the RRa P3HT structure. Irreversible ion trapping is also supported by electrochemical quartz crystal microbalance (EQCM) measurements, showing an irreversible increase in the mass and accumulated charge of the RRa P3HT thin film upon performing a series of 50 CVs (Fig. S6, ESI†). A series of thickness measurements upon an increasing number of CVs agrees with the EQCM data, as the film thickness increases from 53 ± 5 nm at 0 CVs to 105 ± 12 nm at 50 CVs (Fig. S7, ESI†). These observations provide evidence of changes in structural ordering in RRa P3HT upon repeated electrochemical doping/dedoping.
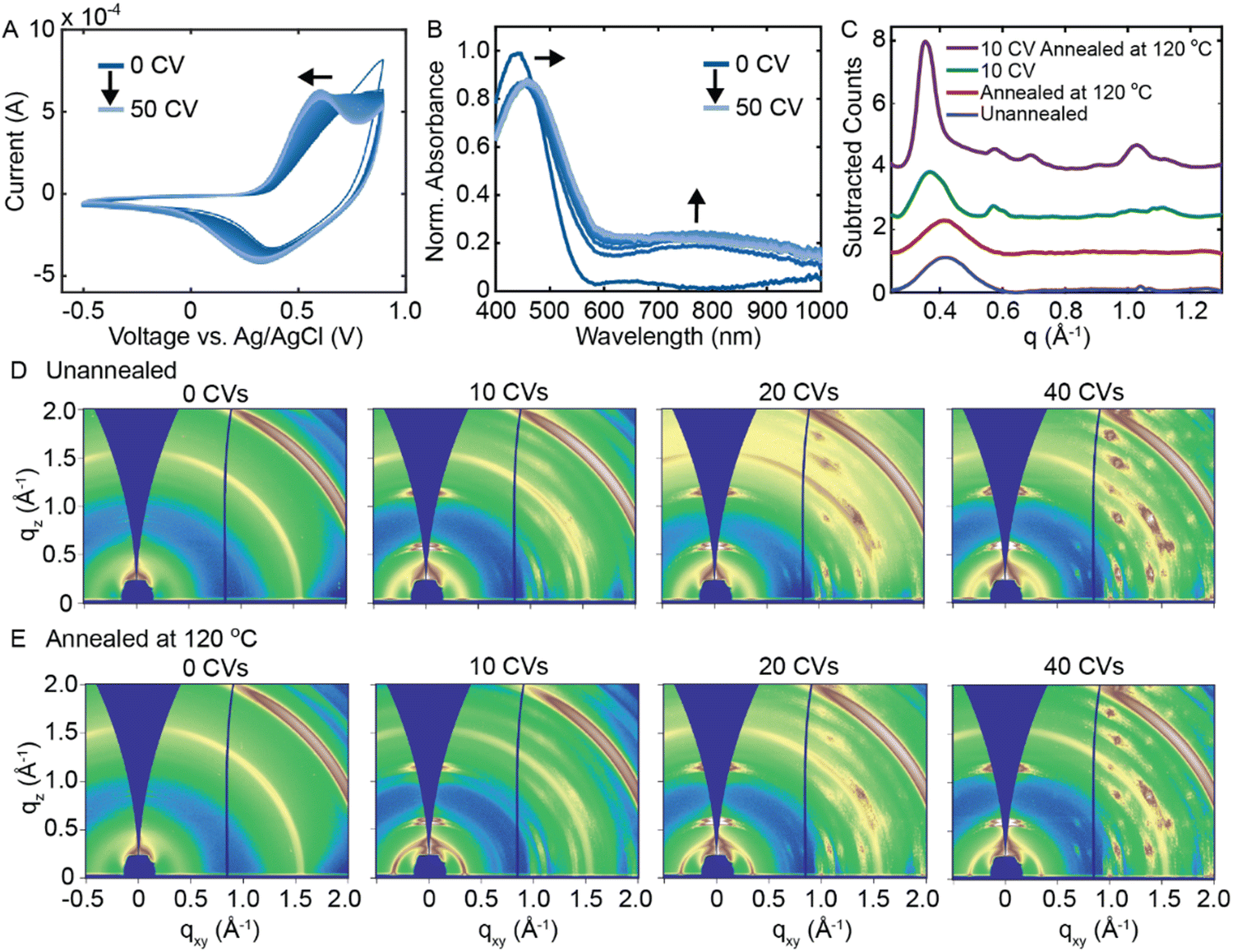 | ||
| Fig. 3 Impact of performing a series of CVs on the structure of RRa P3HT thin films. (A) A series of 50 CVs acquired of RRa P3HT from −0.5 to 0.9 V at a scan rate of 0.1 V s−1. (B) UV/Vis spectra collected every 5 CVs after a 45 s dedoping step at −0.5 V vs. Ag/AgCl. (C) Background subtracted radially integrated GIWAXS diffractograms of four RRa P3HT thin films: not annealed, annealed at 120 °C for 20 min, after performing 10 CVs in aqueous 100 mM KTFSI, and after performing 10 CVs followed by a 20 minute 120 °C annealing step. (D) A series of 2D GIWAXS diffractograms of RRa P3HT after 0, 10, 20, and 40 CVs. (E) A series of 2D GIWAXS diffractograms of RRa P3HT after 0, 10, 20, and 40 CVs followed by thermal annealing at 120 °C for 20 min. | ||
Based on the observed changes in the acquired CVs and absorption spectra with repeated electrochemical doping, we delve deeper into the potential induced structural ordering of RRa P3HT by analyzing GIWAXS diffractograms as a function of the number of electrochemical doping/dedoping cycles. Fig. 3(C) shows the background subtracted radially integrated GIWAXS diffractograms of four different RRa P3HT thin films: an unannealed RRa P3HT thin film, an annealed RRa P3HT thin film, a RRa P3HT thin film that underwent 10 CVs, and a RRa P3HT thin film that underwent 10 CVs followed by a 20 minute 120 °C annealing step. Fig. 3(D) shows 2D GIWAXS diffractograms of a series of RRa P3HT thin films after 0, 10, 20, and 40 CVs and Fig. 3(E) shows 2D GIWAXS diffractograms of the same series of films followed by a thermal annealing step at 120 °C for 20 min. Both the radially integrated diffractograms and 2D GIWAXS diffractograms show a shift in the lamellar spacing of RRa P3HT corresponding to a lattice expansion of 14.9 Å to 17.1 Å after 10 CVs. Upon thermal annealing at 120 °C for 20 min, the intensity of the (100) peak increases significantly and the higher-order (200) and (300) peaks become visible, indicating significantly enhanced ordering in RRa P3HT upon performing a series of CVs followed by thermal annealing. Notably, annealing the RRa P3HT without first acquiring CVs does not show a significant change in the GIWAXS diffractogram, indicating that both repeated electrochemical doping/dedoping and thermal annealing are required for the disorder-to-order transition to occur. In addition to the increased ordering observed in the RRa P3HT thin film, additional peaks appear in the GIWAXS diffractograms with an increasing number of CVs that we attribute to the aggregation of KTFSI in the polymer matrix. Fig. S8 (ESI†) shows a 2D GIWAXS diffractogram of a dropcast KTFSI film that matches the GIWAXS diffractogram that emerges with an increasing number of CVs. The observed KTFSI aggregation upon repeated electrochemical doping/dedoping cycles also occurs in RR P3HT to a lesser degree (Fig. S9, ESI†).
To characterize the morphological changes in RRa P3HT as well as ion trapping, we acquire AFM topography images and PiFM images as a function of the number of CVs performed. Before acquiring each topography and PiFM image, we dedope the film for 20 minutes to focus only on the remaining trapped ions. Fig. 4 shows a set of 50 × 50 μm topography and PiFM images acquired at 1200 cm−1 of 0, 10, 30, and 50 CVs on RRa P3HT films. Initially, the RRa P3HT film, without any CVs performed, has a low surface roughness with only background PiFM signal, indicating the lack of TFSI−. After 10 CVs, we observe the formation of small wrinkles on the samples that contain high concentrations of TFSI−. With an increasing number of CVs, the wrinkles become larger and the PiFM signal increases, indicating higher ion concentrations in the wrinkled regions. We note that we observe no significant changes in the morphology of the thin films upon annealing at 120 °C for 20 min (Fig. S10, ESI†). The presence of high concentrations of TFSI− in the wrinkled regions is further supported by nanoscale PiFM spectra (Fig. S11, ESI†), which highlight the increased concentration of TFSI− ions in the wrinkles (Fig. S12, ESI†). To determine if the TFSI− signal detected by PiFM originates from trapped TFSI− or KTFSI aggregates, we perform energy dispersive spectroscopy (EDS) in a scanning electron microscope (SEM). Our results support that a significant amount of KTFSI aggregates form in the wrinkled regions. The TFSI− PiFM signal in the non-wrinkled regions most likely comes from a combination of small KTFSI aggregates and trapped TFSI− (Fig. S13, ESI†).
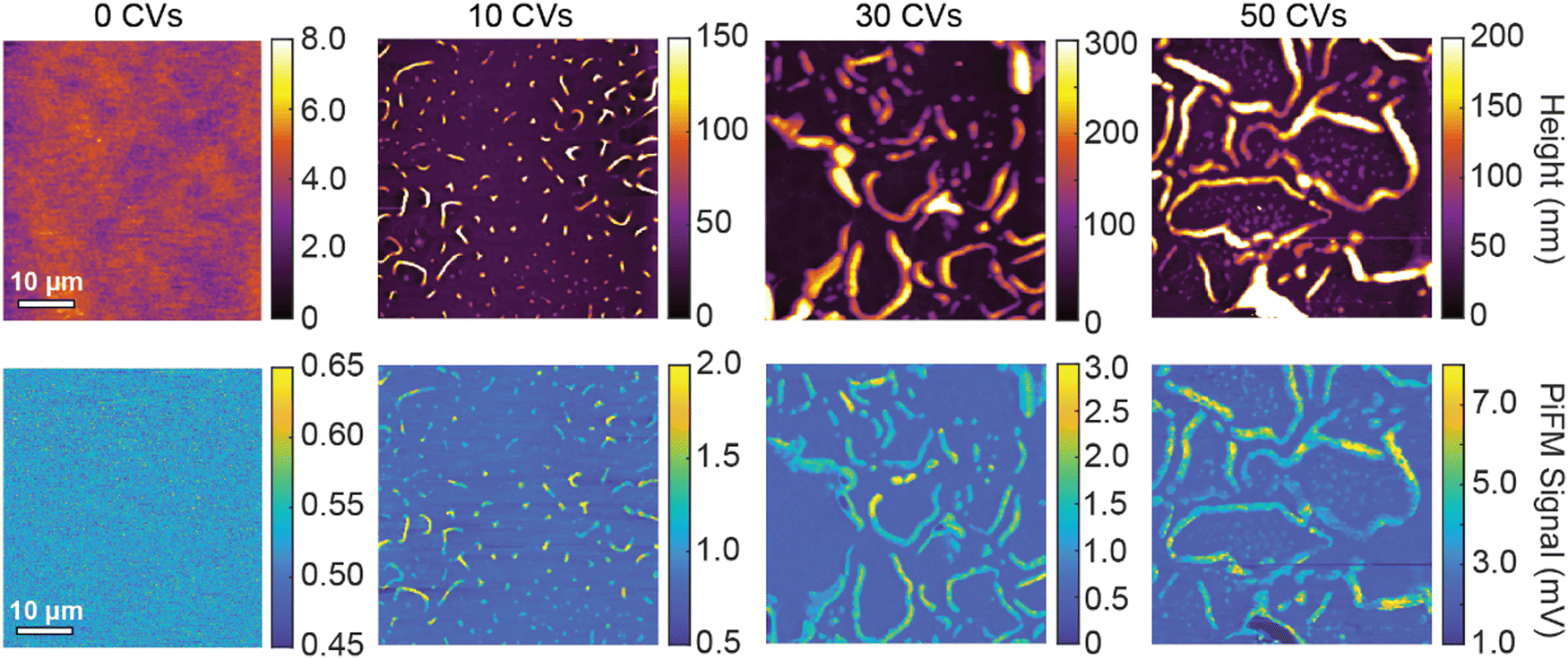 | ||
| Fig. 4 AFM topography and PiFM images of a RRa P3HT thin film upon increasing numbers of CVs performed in aqueous 100 mM KTFSI after a 40 s −0.5 V (vs. Ag/AgCl) dedoping step. 50 × 50 μm AFM topography and PiFM images are collected of an undoped RRa P3HT thin film, as well as RRa P3HT thin films that underwent 0, 10, 30, and 50 CVs. | ||
We next aim to understand the role of the electrolyte on the disorder-to-order transition of RRa P3HT. To interrogate the role of the electrolyte solvent, we performed 50 CVs in water, acetonitrile (ACN) and propylene carbonate (PC) with 100 mM KTFSI. We then probed changes in polymer structure with ex situ GIWAXS and UV/Vis spectroelectrochemistry. Initially, we doped RRa P3HT thin films in the three solvents at 0.7 V, 1.4 V, and 1.2 V (vs. Ag/AgCl) for 20 min in water, ACN, and PC, respectively. The resulting GIWAXS diffractograms show minimal changes after 20 min of doping in each electrolyte (Fig. S14, ESI†). The UV/Vis absorption spectra show irreversible ion trapping in aqueous and ACN electrolytes but not in PC (Fig. S15, ESI†). AFM topography and PiFM images of doped RRa P3HT films show significant changes in the morphology of films doped in the water and PC electrolytes, yet the RRa P3HT thin film doped in ACN retains a smooth surface after doping (Fig. S16, ESI†).
We next performed a series of CVs in each electrolyte to probe structural changes upon repeated electrochemical doping/dedoping cycles. Fig. 5(A) displays a CV performed in water, ACN, and PC (100 mM KTFSI). We adjusted the electrochemical window for each electrolyte to ensure RRa P3HT oxidation. Fig. S17 (ESI†) shows the full 50 CVs for each solvent, demonstrating changes in the oxidation onset with an increasing number of CVs, which is most significant for the aqueous electrolyte. Fig. S18 (ESI†) displays the spectral changes, and the shift in the peak absorption wavelength as a function of the number of CVs applied, with neither ACN nor PC KTFSI resulting in a significant red shift. The 2D GIWAXS diffractograms for each solvent in Fig. 5(B) show a distinct difference in the amount of ion trapping with water showing the highest amount of trapping and ACN showing the lowest. Interestingly, the linecuts in Fig. 5(C) indicate that there is a slightly greater degree of ordering in PC KTFSI than in the other solvents. These results demonstrate that the choice of electrolyte solvent has a major impact on the morphology of RRa P3HT.
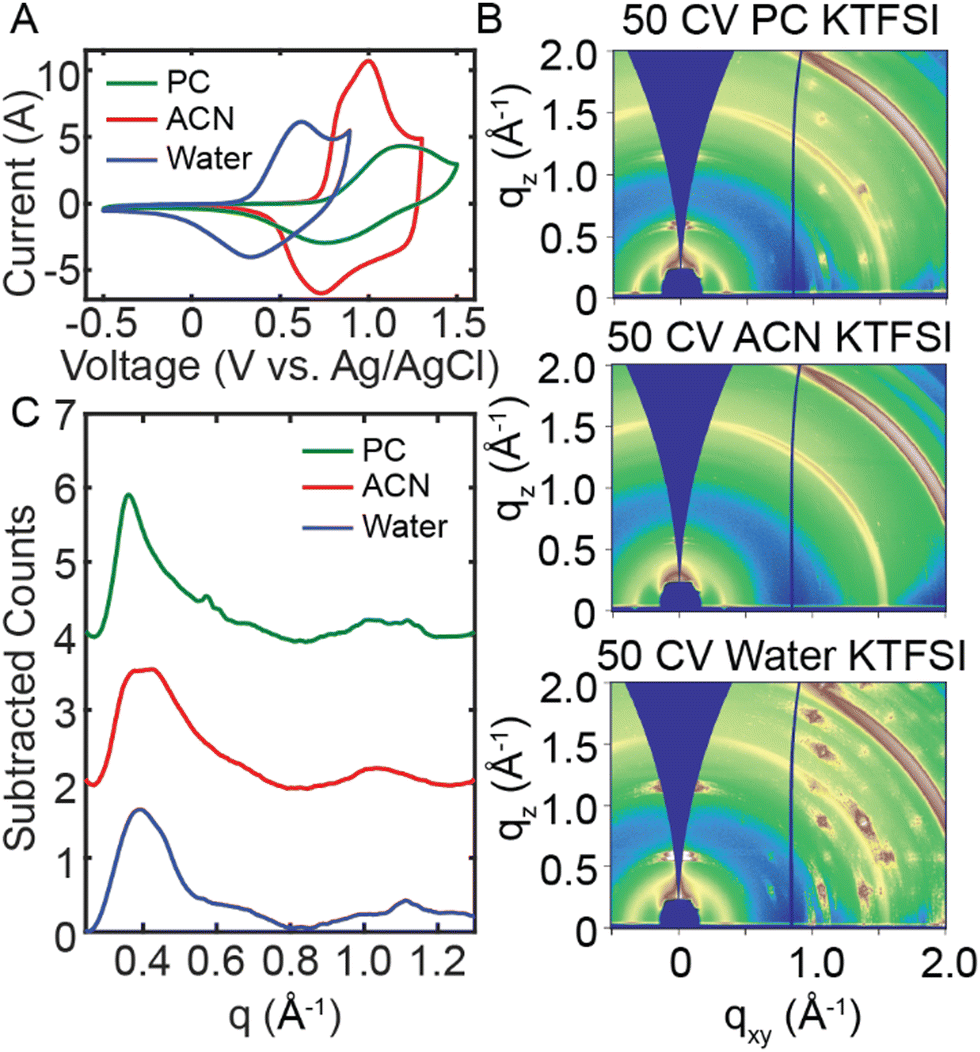 | ||
| Fig. 5 Comparison between using different electrolyte solvents. (A) CVs of RRa P3HT thin films in water, acetonitrile, and propylene carbonate (100 mM KTFSI). (B) GIWAXS diffractograms of P3HT thin films after undergoing 50 CVs in water (bottom), acetonitrile (middle) and propylene carbonate (top) (100 mM KTFSI). (C) ITO background subtracted linecuts of each thin film after undergoing 50 CVs. | ||
In addition to the electrolyte solvent, we also consider the effect of the anion identity and investigated the impact of using potassium perchlorate (KClO4), potassium hexafluorophosphate (KPF6), and potassium trifluoromethanesulfonate (KOTf) as the electrolyte salt. Fig. S19 (ESI†) shows the 50 CV sequences in each aqueous electrolyte. We do not observe as significant a shift in the oxidation onset with these other ions. Fig. S20 (ESI†) shows GIWAXS diffractograms of RRa P3HT using each of these electrolytes after 50 CVs. Here, we observe no significant difference between the GIWAXS patterns, yet the UV/Vis spectroelectrochemistry data show differences in the magnitude of the redshift of the absorption spectra as a function of the number of CVs performed (Fig. S21, ESI†). Overall, the choice of electrolyte has a minor impact on the structural ordering of RRa P3HT upon repeated doping/dedoping cycles.
Conclusions
Our results suggest that repeated electrochemical doping/dedoping has a significant impact on the crystallinity as well as the microstructure of disordered conjugated polymers. In the case of RRa P3HT in an aqueous electrolyte, electrochemical doping of the polymer does not lead to enhanced order, yet a disorder-to-order transition is observed upon performing consecutive CVs followed by thermal annealing. The GIWAXS data shows that the initial electrochemical doping/dedoping cycles expand the characteristic lamellar spacing, allowing the polymer to reorganize and become more ordered upon thermal annealing. Yet, we find that these structural transformations depend on the choice of ion and choice of electrolyte solvent, making these parameters important factors in using OMIECs in operational devices.The spectroelectrochemistry and PiFM studies show that irreversible ion trapping and doping is a major consequence of the observed structural rearrangements upon repeated doping/dedoping cycles. The observed irreversible ion trapping could lead to electronically disconnected regions of the conjugated polymer on repeated doping could have significant implications on OMIEC-based technologies, as these regions could reduce the overall volumetric capacitance of the polymer. Overall, understanding transformations in disordered conjugated polymers, especially in electrochemical settings, could be the key to developing new structure–property relationships for efficient and stable OMIECs.
Experimental section
Materials
Regiorandom P3HT was purchased from Rieke Metals (Lot BLS26-15, Mw = 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1). Regioregular P3HT (M1010 97.3% Regioregular, Mw = 74000 g mol−1)_and 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) were purchased from Ossila. Potassium trifluoromethanesulfonimide (KTFSI) was purchased from TCI America. Potassium hexafluorophosphate (KPF6), potassium perchlorate (KClO4), potassium trifluoromethanesulfonate (KOTf), anhydrous chlorobenzene, anhydrous acetonitrile, propylene carbonate, acetone, and isopropyl alcohol were purchased from Millipore Sigma. Acetonitrile (non-anhydrous) was purchased from Fischer Chemical.
000 g mol−1). Regioregular P3HT (M1010 97.3% Regioregular, Mw = 74000 g mol−1)_and 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) were purchased from Ossila. Potassium trifluoromethanesulfonimide (KTFSI) was purchased from TCI America. Potassium hexafluorophosphate (KPF6), potassium perchlorate (KClO4), potassium trifluoromethanesulfonate (KOTf), anhydrous chlorobenzene, anhydrous acetonitrile, propylene carbonate, acetone, and isopropyl alcohol were purchased from Millipore Sigma. Acetonitrile (non-anhydrous) was purchased from Fischer Chemical.
Preparation of regiorandom and regioregular P3HT solutions
All solutions were prepared in a N2 glovebox. P3HT was dissolved in anhydrous chlorobenzene in a 3 mL amber vial for a final concentration of 20 mg mL−1. P3HT solutions were stirred overnight at 60 °C and filtered through a 0.2 μL syringe filter (Acrodisk) before processing.Preparation of electrolyte solutions
Each electrolyte was prepared using milliQ water and volumetric glassware to achieve a concentration of 100 mM. The 100 mM KTFSI solutions made in acetonitrile and propylene carbonate were prepared the same way. Before each electrochemistry measurement, the electrolyte solutions were degassed by bubbling N2 for 10 minutes.Preparation of F4TCNQ solutions
F4TCNQ was dissolved in anhydrous acetonitrile to achieve a concentration of 10 mg mL−1 inside the glovebox. The solution was stirred for 30 minutes then filtered through a 0.2 μL syringe filter (Acrodisk) before used for doping RRa P3HT.Preparation of P3HT thin films
Glass coated with indium tin oxide (ITO) (MSE Supplies Inc.) were diced to 0.75 cm × 5 cm for spectroelectrochemistry measurements and 1.5 × 2 cm for GIWAXS and AFM measurements. The substrates were then cleaned in an ultrasonicator for 10 minutes in a 1% Alconox aqueous solution, milliQ water, acetone, and isopropyl alcohol. The ITO glass substrates were then dried with nitrogen and placed in a plasma cleaner (Harrick Basic Plasma Cleaner) on high with ambient air for 10 minutes before spin-coating. A spin coater (Ossila) was used to fabricate thin films on the ITO-coated glass substrates spinning at 500 rpm for 5 seconds then 2000 rpm for 45 seconds. 50 μL of P3HT solution was deposited on the substrates before spin-coating. After spin-coating, the P3HT thin films were annealed on a hotplate at 160 °C for 10 minutes.Preparation of electrochemically doped P3HT films
The P3HT thin film on ITO-coated glass was half-submerged in 100 mM KTFSI in an electrochemical cell with an Ag/AgCl reference electrode (A–M Systems) and a Pt mesh counter electrode (Millipore Sigma). The ITO-coated glass substrate served as the working electrode. A potential was applied using a Metrohm Autolab PGSTAT302N potentiostat for 45 s. The sample was then washed with MilliQ water and dried with N2.Preparation of chemically doped P3HT films
The P3HT thin film on ITO-coated glass was dipped in 10 mg mL−1 F4TCNQ dissolved in anhydrous acetonitrile for 20 minutes inside the glovebox. The film was washed with anhydrous acetonitrile after the 20 minutes, then the film was taken out of the glovebox.Preparation of EQCM gold substrates
Au EQCM substrates (SRS QCM-200 Crystals, Phillip Technologies) were washed with acetone and isopropyl alcohol then plasma cleaned for 10 minutes. A P3HT thin film was deposited (70 μL) through spin coating using the same procedure as described above. 50 CV cycles were applied to each EQCM substrate sample from −0.5 to 0.9 V at a scan rate of 0.1 V s−1. The CVs as well as the voltages, current, integrated charge, and the change in mass as a function of time for the RRa P3HT samples were collected on the NOVA program. All data analysis was done on MATLAB.40 The mass calculations were obtained using the Sauerbrey equation (eqn (1)):| Δf = −Cf × Δm | (1) |
GIWAXS measurements
Ex situ GIWAXS measurements were performed on an Anton Paar SAX/WAXS/GISAXS/RheoSAXS laboratory beamline at the University of Utah Nanofab using the GISAXS 2.0 stage. All measurements were performed using extreme WAXS mode with an alpha angle of 0.2° and an exposure time of 2 minutes. High-resolution ex situ GIWAXS measurements were collected at the Advanced Light Source (ALS) at the Lawrence Berkeley National Labs on beamline 7.3.3 with an alpha angle of 0.2° and an exposure time of 10 seconds.41 All diffraction images and data reductions were processed on Igor Pro using the Nika package in Igor Pro 9 and WAXTools42 software packages and MATLAB.40GIWAXS ITO and X-ray noise background subtractions
With each linecut, peaks were selected based on where their lamellar spacing peaks/rings were at (1st, 2nd, and 3rd degree peaks) and any TFSI− trapping peaks (1st and 2nd degree peaks). The width of each of those peaks were removed from any polynomial fit (5th and 6th degree polynomial fits were used in all linecuts). After fitting the curves without the peaks to a polynomial fit, the original data is subtracted by that fit to obtain flattened linecuts for each film used.AFM measurements
Photoinduced force microscopy (PiFM) measurements were collected using a molecular vista one atomic force microscope (AFM) and NCH-PtIr PiFM cantilevers (molecular vista). We prepared the OMIEC films using the same method as the GIWAXS samples. Nanoscale infrared images were acquired at 1200 cm−1 using a scan rate of 0.1 lines per s and image sizes of 256 × 256 pixels. Smaller images in the 20 min continuous doped scans were 1 × 1 μm and the large CV sequence scans were 50 × 50 μm. Nanoscale IR spectra were collected on both undoped and doped P3HT regions with a range of 1800 to 790 cm−1 with an exposure time of 10–15 seconds. All images were processed on SurfaceWorks software (Molecular Vista) and MATLAB.40 PiFM images were flattened and filtered to remove line to line variations in intensity.Spectroelectrochemistry measurements
All electrochemistry and spectroelectrochemistry measurements were performed using the Metrohm Autolab PGSTAT302N potentiostat and Autolab Spectrophotometer UV/Vis/NIR kit. The working electrode is an ITO-coated glass substrate, the reference electrode was an Ag/AgCl puck electrode (A–M Systems), and the counter electrode was a Pt mesh (Millipore Sigma). For the continuous doping measurements, each sample in water was doped for 20 min at 0.7 V followed by 20 min at −0.5 V, each sample in acetonitrile was doped for 20 min at 1.4 V then dedoped for 20 min at −0.1 V, and each sample in propylene carbonate was doped for 20 min at 1.2 V then dedoped for 20 min at −0.3 V. The F4TCNQ doped and dedoped samples were placed in the cuvette with no solvent and quick scans were taken. For the CV cycling measurements in water, a 45 s −0.5 V step was applied to get a background dedoped spectrum, then CV cycles were applied to the sample from −0.5 to 0.9 V at a scan rate of 0.1 V s−1 for 50 CVs. A 45 s −0.5 V dedoping step at the end of every 5 CVs, then a spectrum is taken. The same for acetonitrile and propylene carbonate except with ranges of −0.1 to 1.4 V and −0.5 to 1.5 V, respectively. This repeat occurs until 50 CVs is reached. Continuous doping and CV cycling absorbance measurements were collected with the NOVA 2.1.5 program and the data was processed in MATLAB.40EQCM measurements
EQCM measurements were performed using a Stanford QCM200 5 MHz Quartz Crystal Microbalance and the Metrohm Autolab PGSTAT302N potentiostat. 50 CV cycles were applied to each EQCM substrate sample from −0.5 to 0.9 V at a scan rate of 0.1 V s−1. CV, voltage, mass change, current, and charge as a function of time for each of the P3HT films were collected with the NOVA program. The resulting data was processed in MATLAB.40FTIR measurements
FTIR measurements were performed on a Thermo Scientific Nicolet iS-50 FTIR between 400 and 4000 cm−1 using attenuated total reflection (ATR) mode. The thin films were prepared by spin coating a P3HT solution on ITO-coated glass, starting at a rate of 500 rpm for 5 seconds, then 2000 rpm for 45 seconds. A blank ITO-coated glass substrate was used as a background. FTIR measurements were processed in MATLAB.40Energy dispersive spectroscopy (EDS) measurements
EDS measurements were performed using the FEI Teneo scanning electron microscope (SEM) at the Utah Nanofab Electron Microscopy and Surface Analysis Lab. The EDS attachment used was the EDAX Octane Elite EDS System. RRa P3HT 50 CV thin film were prepared the same as the GIWAXS measurements except using 100 mM NaTFSI instead of 100 mM KTFSI as the electrolyte. An SEM accelerating voltage of 10 kV and beam current of 6.4 μA was used in selecting the field of view for EDS analysis. The magnification was 5000×. EDS elemental maps were collected with a dwell time of 50 μs (512 × 512 pixels) for a total of 32 frames, which were summed to form the final EDS map.Author contributions
C. G. B. and S. R. J. conceived of the project and designed the experiments. S. R. J. performed the experiments, performed the data analysis, and prepared the manuscript. R. L. K., G. W. C. and J. N. K. aided with synchrotron experiments at the Advanced Light Source. P. W. M. aided with the EDS experiments at the University of Utah Electron Microscopy and Surface Analysis Lab. All authors contributed to writing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (CHE-2304613). C. G. B. and S. R. J. acknowledge support from the Camille and Henry Dreyfus Foundation Machine Learning in the Chemical Sciences and Engineering Award. Beamline 7.3.3 of the Advanced Light Source is supported by the Director of the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. This work made use of University of Utah shared facilities of the Micron Technology Foundation Inc. Microscopy Suite sponsored by the College of Engineering, Health Sciences Center, Office of the Vice President for Research, and the Utah Science Technology and Research (USTAR) initiative of the State of Utah. GIWAXS measurements at the University of Utah were supported by the National Science Foundation under grant number: CBET 2018413. C. G. B., S. R. J., R. L. K., G. W. C., P. W. M. and J. N. K. also acknowledge the Nanofab- Electron Microscopy and Surface Analysis Lab (EMSAL) and helpful discussions with Brian van Devener, J. Paulo Perez, and Randy Polson. We acknowledge partial financial support from MathWorks.References
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Organic Mixed Ionic–Electronic Conductors, Nat. Mater., 2020, 19(1), 13–26, DOI:10.1038/s41563-019-0435-z.
- D. Ohayon, V. Druet and S. Inal, A Guide for the Characterization of Organic Electrochemical Transistors and Channel Materials, Chem. Soc. Rev., 2023, 52(3), 1001–1023, 10.1039/D2CS00920J.
- A. Marks, S. Griggs, N. Gasparini and M. Moser, Organic Electrochemical Transistors: An Emerging Technology for Biosensing, Adv. Mater. Interfaces, 2022, 9(6), 2102039, DOI:10.1002/admi.202102039.
- S. Inal, J. Rivnay, A.-O. Suiu, G. G. Malliaras and I. McCulloch, Conjugated Polymers in Bioelectronics, Acc. Chem. Res., 2018, 51(6), 1368–1376, DOI:10.1021/acs.accounts.7b00624.
- H. Kim, Y. Won, H. W. Song, Y. Kwon, M. Jun and J. H. Oh, Organic Mixed Ionic–Electronic Conductors for Bioelectronic Sensors: Materials and Operation Mechanisms, Adv. Sci., 2023, 2306191, DOI:10.1002/advs.202306191.
- R. Wu, M. Matta, B. D. Paulsen and J. Rivnay, Operando Characterization of Organic Mixed Ionic/Electronic Conducting Materials, Chem. Rev., 2022, 122(4), 4493–4551, DOI:10.1021/acs.chemrev.1c00597.
- A. Giovannitti, D.-T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanifi, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Controlling the Mode of Operation of Organic Transistors through Side-Chain Engineering, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(43), 12017–12022, DOI:10.1073/pnas.1608780113.
- B. T. DiTullio, L. R. Savagian, O. Bardagot, M. De Keersmaecker, A. M. Österholm, N. Banerji and J. R. Reynolds, Effects of Side-Chain Length and Functionality on Polar Poly(Dioxythiophene)s for Saline-Based Organic Electrochemical Transistors, J. Am. Chem. Soc., 2023, 145(1), 122–134, DOI:10.1021/jacs.2c08850.
- L. Q. Flagg, R. Giridharagopal, J. Guo and D. S. Ginger, Anion-Dependent Doping and Charge Transport in Organic Electrochemical Transistors, Chem. Mater., 2018, 30(15), 5380–5389, DOI:10.1021/acs.chemmater.8b02220.
- M. Sophocleous, L. Contat-Rodrigo, E. Garcia-Breijo and J. Georgiou, Organic Electrochemical Transistors as an Emerging Platform for Bio-Sensing Applications: A Review, IEEE Sens. J., 2021, 21(4), 3977–4006, DOI:10.1109/JSEN.2020.3033283.
- Q. Liu, Y. Liu, J. Li, C. Lau, F. Wu, A. Zhang, Z. Li, M. Chen, H. Fu, J. Draper, X. Cao and C. Zhou, Fully Printed All-Solid-State Organic Flexible Artificial Synapse for Neuromorphic Computing, ACS Appl. Mater. Interfaces, 2019, 11(18), 16749–16757, DOI:10.1021/acsami.9b00226.
- P. C. Harikesh, C.-Y. Yang, H.-Y. Wu, S. Zhang, M. J. Donahue, A. S. Caravaca, J.-D. Huang, P. S. Olofsson, M. Berggren, D. Tu and S. Fabiano, Ion-Tunable Antiambipolarity in Mixed Ion–Electron Conducting Polymers Enables Biorealistic Organic Electrochemical Neurons, Nat. Mater., 2023, 22, 242–248, DOI:10.1038/s41563-022-01450-8.
- Y. Van De Burgt, E. Lubberman, E. J. Fuller, S. T. Keene, G. C. Faria, S. Agarwal, M. J. Marinella, A. Alec Talin and A. Salleo, A Non-Volatile Organic Electrochemical Device as a Low-Voltage Artificial Synapse for Neuromorphic Computing, Nat. Mater., 2017, 16(4), 414–418, DOI:10.1038/nmat4856.
- L. M. Cowen, J. Atoyo, M. J. Carnie, D. Baran and B. C. Schroeder, Review—Organic Materials for Thermoelectric Energy Generation, ECS J. Solid State Sci. Technol., 2017, 6(3), N3080–N3088, DOI:10.1149/2.0121703jss.
- S. Kee, M. A. Haque, Y. Lee, T. L. Nguyen, D. Rosas Villalva, J. Troughton, A.-H. Emwas, H. N. Alshareef, H. Y. Woo and D. Baran, A Highly Conductive Conjugated Polyelectrolyte for Flexible Organic Thermoelectrics, ACS Appl. Energy Mater., 2020, 3(9), 8667–8675, DOI:10.1021/acsaem.0c01213.
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, Enhanced N-Doping Efficiency of a Naphthalenediimide-Based Copolymer through Polar Side Chains for Organic Thermoelectrics, ACS Energy Lett., 2018, 3(2), 278–285, DOI:10.1021/acsenergylett.7b01146.
- R. Wu, B. D. Paulsen, Q. Ma and J. Rivnay, Mass and Charge Transport Kinetics in an Organic Mixed Ionic–Electronic Conductor, Chem. Mater., 2022, 34(21), 9699–9710, DOI:10.1021/acs.chemmater.2c02476.
- L. Q. Flagg, C. G. Bischak, J. W. Onorato, R. B. Rashid, C. K. Luscombe and D. S. Ginger, Polymer Crystallinity Controls Water Uptake in Glycol Side-Chain Polymer Organic Electrochemical Transistors, J. Am. Chem. Soc., 2019, 141(10), 4345–4354, DOI:10.1021/jacs.8b12640.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, The Role of the Side Chain on the Performance of N-Type Conjugated Polymers in Aqueous Electrolytes, Chem. Mater., 2018, 30(9), 2945–2953, DOI:10.1021/acs.chemmater.8b00321.
- S. R. Jackson, R. L. Kingsford, G. W. Collins and C. G. Bischak, Crystallinity Determines Ion Injection Kinetics and Local Ion Density in Organic Mixed Conductors, Chem. Mater., 2023, 35(14), 5392–5400, DOI:10.1021/acs.chemmater.3c00657.
- R. Steyrleuthner, R. Di Pietro, B. A. Collins, F. Polzer, S. Himmelberger, M. Schubert, Z. Chen, S. Zhang, A. Salleo, H. Ade, A. Facchetti and D. Neher, The Role of Regioregularity, Crystallinity, and Chain Orientation on Electron Transport in a High-Mobility n-Type Copolymer, J. Am. Chem. Soc., 2014, 136(11), 4245–4256, DOI:10.1021/ja4118736.
- B. D. Paulsen, A. Giovannitti, R. Wu, J. Strzalka, Q. Zhang, J. Rivnay and C. J. Takacs, Electrochemistry of Thin Films with In Situ/Operando Grazing Incidence X-Ray Scattering: Bypassing Electrolyte Scattering for High Fidelity Time Resolved Studies, Small, 2021, 17(42), 2103213, DOI:10.1002/smll.202103213.
- C. G. Bischak, L. Q. Flagg, K. Yan, T. Rehman, D. W. Davies, R. J. Quezada, J. W. Onorato, C. K. Luscombe, Y. Diao, C.-Z. Li and D. S. Ginger, A Reversible Structural Phase Transition by Electrochemically-Driven Ion Injection into a Conjugated Polymer, J. Am. Chem. Soc., 2020, 142(16), 7434–7442, DOI:10.1021/jacs.9b12769.
- D. Neusser, C. Malacrida, M. Kern, Y. M. Gross, J. van Slageren and S. Ludwigs, High Conductivities of Disordered P3HT Films by an Electrochemical Doping Strategy, Chem. Mater., 2020, 32(14), 6003–6013, DOI:10.1021/acs.chemmater.0c01293.
- P. Cavassin, I. Holzer, D. Tsokkou, O. Bardagot, J. Réhault and N. Banerji, Electrochemical Doping in Ordered and Disordered Domains of Organic Mixed Ionic–Electronic Conductors, Adv. Mater., 2023, 2300308, DOI:10.1002/adma.202300308.
- K. N. Baustert, A. Abtahi, A. N. Ayyash and K. R. Graham, Impact of the Anion on Electrochemically Doped Regioregular and Regiorandom Poly(3-Hexylthiophene), J. Polym. Sci., 2022, 60(3), 602–609, DOI:10.1002/pol.20210699.
- R. Giridharagopal, L. Q. Flagg, J. S. Harrison, M. E. Ziffer, J. Onorato, C. K. Luscombe and D. S. Ginger, Electrochemical Strain Microscopy Probes Morphology-Induced Variations in Ion Uptake and Performance in Organic Electrochemical Transistors, Nat. Mater., 2017, 16(7), 737–742, DOI:10.1038/nmat4918.
- J. O. Guardado and A. Salleo, Structural Effects of Gating Poly(3-hexylthiophene) through an Ionic Liquid, Adv. Funct. Mater., 2017, 27(32), 1701791, DOI:10.1002/adfm.201701791.
- E. Lim, A. M. Glaudell, R. Miller and M. L. Chabinyc, The Role of Ordering on the Thermoelectric Properties of Blends of Regioregular and Regiorandom Poly(3-hexylthiophene), Adv. Electron. Mater., 2019, 5(11), 1800915, DOI:10.1002/aelm.201800915.
- P. Y. Yee, D. T. Scholes, B. J. Schwartz and S. H. Tolbert, Dopant-Induced Ordering of Amorphous Regions in Regiorandom P3HT, J. Phys. Chem. Lett., 2019, 10(17), 4929–4934, DOI:10.1021/acs.jpclett.9b02070.
- D. Nowak, W. Morrison, H. K. Wickramasinghe, J. Jahng, E. Potma, L. Wan, R. Ruiz, T. R. Albrecht, K. Schmidt, J. Frommer, D. P. Sanders and S. Park, Nanoscale Chemical Imaging by Photoinduced Force Microscopy, Sci. Adv., 2016, 2(3), e1501571, DOI:10.1126/sciadv.1501571.
- R. A. Murdick, W. Morrison, D. Nowak, T. R. Albrecht, J. Jahng and S. Park, Photoinduced Force Microscopy: A Technique for Hyperspectral Nanochemical Mapping, Jpn. J. Appl. Phys., 2017, 56(8S1), 08LA04, DOI:10.7567/JJAP.56.08LA04.
- R. Giridharagopal, J. Guo, J. Kong and D. S. Ginger, Nanowire Architectures Improve Ion Uptake Kinetics in Conjugated Polymer Electrochemical Transistors, ACS Appl. Mater. Interfaces, 2021, 13(29), 34616–34624, DOI:10.1021/acsami.1c08176.
- Y. Wu, Q. M. Duong, A. F. Simafranca, C. Z. Salamat, B. J. Schwartz and S. H. Tolbert, Crystal Structure Control of the Energetics of Chemical Doping in Rub-Aligned P3HT Films, ACS Mater. Lett., 2024, 6(2), 489–497, DOI:10.1021/acsmaterialslett.3c01543.
- V. Untilova, H. Zeng, P. Durand, L. Herrmann, N. Leclerc and M. Brinkmann, Intercalation and Ordering of F 6 TCNNQ and F 4 TCNQ Dopants in Regioregular Poly(3-Hexylthiophene) Crystals: Impact on Anisotropic Thermoelectric Properties of Oriented Thin Films, Macromolecules, 2021, 54(13), 6073–6084, DOI:10.1021/acs.macromol.1c00554.
- A. Hamidi-Sakr, L. Biniek, J. Bantignies, D. Maurin, L. Herrmann, N. Leclerc, P. Lévêque, V. Vijayakumar, N. Zimmermann and M. Brinkmann, A Versatile Method to Fabricate Highly In-Plane Aligned Conducting Polymer Films with Anisotropic Charge Transport and Thermoelectric Properties: The Key Role of Alkyl Side Chain Layers on the Doping Mechanism, Adv. Funct. Mater., 2017, 27(25), 1700173, DOI:10.1002/adfm.201700173.
- I. Rey, P. Johansson, J. Lindgren, J. C. Lassègues, J. Grondin and L. Servant, Spectroscopic and Theoretical Study of (CF3SO2)2N− (TFSI−) and (CF3SO2)2NH (HTFSI), J. Phys. Chem. A, 1998, 102(19), 3249–3258, DOI:10.1021/jp980375v.
- I. D. V. Ingram, D. J. Tate, A. V. S. Parry, R. Sebastian Sprick and M. L. Turner, A Simple Method for Controllable Solution Doping of Complete Polymer Field-Effect Transistors, Appl. Phys. Lett., 2014, 104(15), 153304, DOI:10.1063/1.4871096.
- X. Han, Z. Wu and B. Sun, Enhanced Performance of Inverted Organic Solar Cell by a Solution-Based Fluorinated Acceptor Doped P3HT:PCBM Layer, Org. Electron., 2013, 14(4), 1116–1121, DOI:10.1016/j.orgel.2013.01.031.
- MATLAB Version 9.13.0 (R2022b), The Mathworks Inc., 2022, https://www.mathworks.com Search PubMed.
- A. Hexemer, W. Bras, J. Glossinger, E. Schaible, E. Gann, R. Kirian, A. MacDowell, M. Church, B. Rude and H. Padmore, A SAXS/WAXS/GISAXS Beamline with Multilayer Monochromator, J. Phys. Conf. Ser., 2010, 247, 012007, DOI:10.1088/1742-6596/247/1/012007.
- S. D. Oosterhout, V. Savikhin, J. Zhang, Y. Zhang, M. A. Burgers, S. R. Marder, G. C. Bazan and M. F. Toney, Mixing Behavior in Small Molecule:Fullerene Organic Photovoltaics, Chem. Mater., 2017, 29(7), 3062–3069, DOI:10.1021/acs.chemmater.7b00067.
Footnote |
| † Electronic supplementary information (ESI) available: Continuous doping and dedoping absorption spectra in 100 mM KTFSI (all solvents) and F4TCNQ, 50 × 50 μm PiFM images of the continuous doping steps, FTIR for F4TCNQ and CV cycling samples, Electrochemical quartz crystal microbalance data of CV cycling, additional GIWAXS data, additional PiFM images, full 50 CVs for each solvent and electrolyte used, and spectroelectrochemistry wavelength comparisons between each solvent and each electrolyte. (PDF). See DOI: https://doi.org/10.1039/d4tc01029a |
| This journal is © The Royal Society of Chemistry 2024 |