
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUiO(Zr)-based MOF catalysts for light-driven aqueous pollutant degradation†
Samuel C.
Moore‡
 a,
Isabella L.
Hubble‡
b,
Alyssa L.
Ritchie
b,
Jeffrey E.
Barzach
b and
Michele L.
Sarazen
*ab
a,
Isabella L.
Hubble‡
b,
Alyssa L.
Ritchie
b,
Jeffrey E.
Barzach
b and
Michele L.
Sarazen
*ab
aDepartment of Chemical and Biological Engineering, Princeton University, 41 Olden Street, Princeton, New Jersey 08544, USA. E-mail: msarazen@princeton.edu
bDepartment of Chemistry, Frick Chemistry Laboratory, Princeton University, Princeton, New Jersey 08544, USA
First published on 12th June 2024
Abstract
Zirconium-carboxylate metal–organic frameworks (MOFs) of isoreticular crystal morphologies and contrasting pore sizes are examined to understand the relative influence of linker size (UiO-67 vs. UiO-68) and secondary metal incorporation in photocatalytic aqueous pollutant degradation. Here, iron (Fe) is chosen given its prevalence in wastewater treatment literature and applications, resulting from its low toxicity and ability to activate benign oxidants. UiO-67 with Fe incorporated (Fe-UiO-67) via incipient wetness impregnation demonstrates reduced band gap energy relative to the UiO-67 parent and higher apparent photocatalytic degradation under UV light toward methylene blue dye using hydrogen peroxide (H2O2), with catalyst mass-normalized pseudo-first order rate constants of 6.8 ± 0.5 g−1 ks−1 and 2.0 ± 0.3 g−1 ks−1, respectively. While structural characterization via X-ray diffraction remains unperturbed for Fe-UiO-67 before and after reaction, some Fe leaching is evident, as indicated by recharge experiments in the filtrate. Synthesized UiO-68, which possesses increased pore size, also has reduced band gap energy resulting in higher UV-light activation than UiO-67 (pseudo-first order rate constant of 3.5 ± 0.4 g−1 ks−1). Further, UiO-68 demonstrates high stability and exhibits a higher productive H2O2 utilization fraction than either of the UiO-67 catalysts. Together, this work clarifies the relative influence of linker modulation and active metal incorporation in UiO-MOFs for pollutant degradation and aqueous applications broadly.
Introduction
Rising industrial activity has led to significant pollution of natural water resources, posing serious long-term threats to both human health and ecosystems.1 By 2050, the global demand for freshwater is projected to increase by 30%, while concomitant spread of pollutants will exacerbate ecosystem and biodiversity loss.1,2 Continued pursuit of effective wastewater treatment methodologies can help mitigate this impact. Advanced oxidation processes (AOPs) are one such practice of focus for recalcitrant pollutants, such as aromatic compounds found in industrial effluents with harmful effects. For instance, Fenton-based applications use ferrous (and some ferric) salts to break down an oxidant, specifically hydrogen peroxide (H2O2), to generate radicals that can non-selectively degrade aqueous pollutants. Although these salts are cheap relative to other metal sources, Fenton reactions have strict pH limitations, which impair catalyst activity and can precipitate amorphous ferric oxyhydroxides.1,3,4 Heterogeneous catalysts, including metal–organic frameworks (MOFs), have shown potential as an alternative class of materials for AOPs, and photocatalytic activation of these structures could further enhance degradative performance.5,6 With iron (Fe)-containing solid materials in particular, the benefits of Fenton-like chemistry are achievable while simultaneously mitigating unfavorable deactivation via precipitation.3,7–11MOFs, consisting of networks of metal-containing nodes connected by organic linkers, constitute a diverse class of materials with tunable porosity and chemical functionalities, making them attractive for a variety of applications.12,13 MOFs constructed from non-toxic, earth-abundant metals are especially attractive as adsorbent materials or for catalytic degradation of organic pollutants.6,14–16 Additionally, many MOFs exhibit semiconductor-like behavior, allowing light at or above the band gap energy (BGE) of the material to excite electrons and form holes. These can interact directly to degrade toxic organic pollutants or create highly reactive, transient radical species (particularly in the presence of oxidants) that can then break down the pollutant moieties. Significant attention is devoted to Fe-based MOFs, such as the Fe-MIL series, that function as heterogeneous analogs to homogeneous Fenton catalysts with BGEs in the visible light range (for instance, 2.4 eV for MIL-101(Fe)) enabling electron transfer between the node and linker sites of the MOF.17–22 However, they suffer from significant stability limitations in aqueous environments, causing a seemingly enhanced catalyst performance through the conflated impact of leached Fe and solid reactivity.6,23–25
From this perspective, zirconium (Zr)-based UiO series MOFs are more attractive alternatives, given their high chemical and thermal stability derived from the strength of the Zr–oxygen (O) bonding in their Zr6O8 metal nodes.26–29 However, Zr does not intrinsically promote thermal H2O2 activation in the manner of Fe, since it does not possess characteristics of other variable valence state metals that facilitate a redox cycle; instead, any low H2O2 activation observed is typically attributed to peroxidase-like activity of Zr–OH(H2O) nodal moieties found in many Zr-MOFs.30,31 Given this limited intrinsic reactivity for pollutant oxidation, the semiconductor-like behavior of UiO structures is paramount to inducing significant degradation. UiO-66 and UiO-67, formed using linear dicarboxylate linkers benzene 1,4-dicarboxylate (BDC) and 4,4-biphenyl-dicarboxylate (BPDC; Fig. 1), respectively, are well-studied but have higher experimentally-measured BGEs than typical Fe-MIL-series MOFs (UiO-66: 4.0 eV and UiO-67: 3.6 eV), requiring high energy UV light to induce photocatalytic effects.32,33 Employing strategies to modulate band gap—such as by altering linker identity/functionality, introducing alternative metals to the structure, and adjusting material defect density—will reduce energy-intensity of their use in photocatalysis.32–35
One option explored herein is use of a longer, terphenyl-4,4″-dicarboxylate (TPDC; Fig. 1) linker to form UiO-68, characterized by a lower theoretical BGE of 2.9 eV, with its longer, more conjugated linker that better promotes electron transfer due to a greater density of carbon states that raise the valence band maximum.32,36 Larger UiO linker sizes correspond to higher porosity materials—theoretically 0.45 cm3 g−1, 1.05 cm3 g−1, and 1.82 cm3 g−1, for UiO-66, UiO-67, and UiO-68, respectively—with similar surface area trends (970 m2 g−1, 1580 m2 g−1, and 2820 m2 g−1 reported experimental BET surface areas, respectively).36,37 The longer linkers may also influence adsorption of pollutants by promoting π–π interactions, in addition to modulating photocatalytic reactivity.
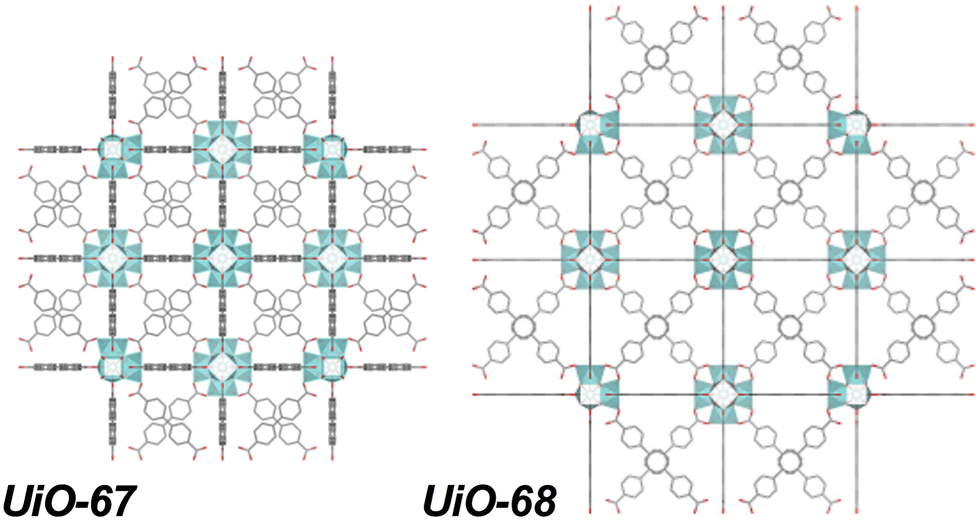 | ||
| Fig. 1 Zr-based UiO-67 and UiO-68 MOF structures based on BPDC and TPDC linkers, respectively. | ||
Structural modifications through secondary metal incorporation could further influence the reactivity of the material, as well as impact pollutant adsorption, with many doping approaches for modifying UiO structures reported. For instance, titanium (Ti) substitution for Zr after synthesis in aminated UiO-66 (2-aminoterephthalate linker) decreased its BGE, shifting the photoabsorption edge to the visible light range (though this is partially attributable to the increased number of missing linkers resulting from Ti introduction).38 Indium (In) doping in place of Zr during synthesis of UiO-66 likewise reduced the BGE, from 4.0 to 2.8 eV, while also improving ligand to metal charge transfer.39
Nanoparticle incorporation in lieu of nodal metal exchange represents another promising path toward more effective wastewater treatment and BGE modulation. For instance, growth of cadmium sulfide (CdS) nanoparticles on UiO-66 reduced the BGE of the composite to intermediate values between the two parent materials and demonstrated enhanced photocatalytic reactivity above the level of physical mixtures of CdS and UiO-66.40 Integration of magnetic Fe3O4 during the synthesis of Cu-based HKUST-1 showed improved persulfate-mediated degradation of tetracycline compared to equivalent mass loading of Fe3O4 or HKUST-1 without light.5 Another study supported Fe3O4 onto UiO-66/67 after synthesis and showed enhanced Fenton-like performance for BPA degradation in the absence of light over the parent structures.11 Similarly, recent work synthesized a Fe3O4 composite with UiO-66-NH2 (and functionalized graphene oxide), which showed effective adsorption of lead and dye molecules.16 Although adsorption and Fenton-like behavior have been demonstrated previously, studies interrogating Fe oxide loaded UiO structures in photocatalytic pollutant degradation remain limited, and contextualizing results in material stability is underemphasized.
In this work, supported Fe oxide nanoparticles and dispersed Fe moieties are incorporated into UiO-67 through an incipient wetness impregnation (IWI) procedure. The relative performance of Fe-loaded UiO-67 (herein termed Fe-UiO-67) is compared against synthesized parent UiO-67 and UiO-68 in the degradation of methylene blue (MB) under catalytic and photocatalytic (visible and UV light) conditions using excess H2O2. The dye MB is chosen as the organic pollutant for study because it is a suitable probe for understanding the degradation of aromatic compounds pertinent to industrial waste effluent.41,42 Reactivity differences, inferred from extracted pseudo-first order kinetic rate constants, demonstrate the enhanced degradation of MB by UiO-68 compared to UiO-67, suggesting the positive impact of linker length extension. Moreover, Fe-UiO-67 reactivity exceeds that of UiO-68, confirming the positive role of metal nanoparticle incorporation. However, catalyst stability investigations via leach tests and post-reaction characterizations emphasize the nuanced balance between apparent initial performance and catalyst recoverability and reuse, with UiO-68 demonstrating a higher productive oxidant utilization. Collectively, these results inform the importance of contextualizing reactivity observations with structural changes to understand and improve MOFs for aqueous applications.
Experimental
Materials
Zr4+ chloride (ZrCl4; Thermo Scientific, 99.5+%), 4,4-biphenyl dicarboxylic acid (H2BPDC; Chemimpex, 99.1%), [1,1′:4′,1″-terphenyl]-4,4″-dicarboxylic acid (H2TPDC; Ambeed, 95%), acetic acid (VWR Chemicals, 99.7+%), hydrochloric acid (HCl; EMD Millipore Corporation and Ward's Scientific, 37% in water), trifluoroacetic acid (TFA; Chem-Impex, 99.9%), Fe3+ chloride hexahydrate (FeCl3·6H2O; Acrōs Organics, 99+%), and N,N-dimethylformamide (DMF; Fisher Chemical, 99+%), were used for catalyst synthesis. Acetone (Fisher Chemical, 99.5+%) and methanol (Fisher Chemical, 99.8%) were used as washing solvents, along with DMF. Tetrahydrofuran (THF; Alfa Aesar, 99% stabilized with 250–350 ppm BHT), ethanol (Decon Labs, 99.9+%), and acetone were used in catalyst recovery after reaction. Methylene blue trihydrate (MB; Fisher Chemical, 99%) and hydrogen peroxide (H2O2; Fisher Chemical, 29+% v/v) were used in reactivity tests. Potassium bromide (KBr; Acrōs Organics, 99+%) and barium sulfate (BaSO4; Sigma, 99%) were used to make dilute FT-IR and DR-UV-vis mixtures for analysis, respectively. Potassium permanganate (KMnO4; Aldrich Chemical, 99+%) and sulfuric acid (H2SO4; Fisher Chemical, 93–98%) were used in H2O2 titration experiments. All starting materials were utilized as received without additional purification.MOF synthesis
UiO-67 was synthesized following a procedure reported in literature by mixing 1 mmol of ZrCl4, 1 mmol of H2BPDC, 10 mmol of acetic acid, and 2 mmol HCl in 30 mL DMF and sonicating for 20 minutes.43 The opaque white solution was sealed with a glass stopper and keck clip in a 50 mL Erlenmeyer flask and heated at 393 K for 48 hours. After cooling to ambient temperature, the product was isolated through centrifugation (7250 RPM, 10 minutes) and washed three times with 50 mL DMF. The material was soaked in 50 mL DMF overnight to remove any free H2BPDC linker, then washed three times with 50 mL acetone and dried under vacuum (∼70 kPa) at 373 K for 24 hours, producing a white product.Fe-UiO-67 was synthesized adapting an incipient wetness impregnation (IWI) procedure reported for platinum incorporation into UiO-67 in literature.44 An Fe-containing precursor solution (34 mM) was made by adding 0.067 mmol of FeCl3·6H2O to 2 mL of acetone and sonicating for 10 minutes. This amount of precursor was determined by targeting nominal 8 wt% Fe oxide, assuming complete metal incorporation.45 The precursor solution was added dropwise to 100 mg UiO-67 in 50 μL increments and stirred manually until the MOF was saturated and had a clay-like texture (300–350 μL). This mixture was heated in a convection oven at 338 K for 20 minutes. These two steps were repeated until complete incorporation of precursor solution. The resultant material was dried overnight under vacuum (∼70 kPa) at 383 K. Afterward, the product was washed twice with 45 mL of acetone, twice with 45 mL methanol, and once with 45 mL acetone, centrifuging (7000 RPM, 10 minutes) between washes. The resulting Fe-UiO-67 was dried overnight at 383 K, forming a light-yellow powder.
UiO-68 was synthesized adapting a procedure reported in literature for an isoreticular structure with a modified linker by mixing 0.20 mmol ZrCl4, 0.17 mmol H2TPDC, and 1 mL TFA in 40 mL DMF and sonicating for 3 minutes.46 The opaque white solution was sealed with a glass stopper and keck clip in a 50 mL Erlenmeyer flask and heated at 393 K for 96 h. After cooling to ambient temperature, the product was isolated through centrifugation (7800 RPM, 5 minutes) and washed three times with 25 mL DMF and three times with 25 mL methanol. Washed MOF was dried overnight under vacuum (∼70 kPa) at 353 K, forming a fine white powder.
Material characterization
Long-range order was examined via X-ray diffraction (XRD) patterns obtained on a Bruker D8 Discover X-ray diffractometer with Cu-Kα radiation (λ = 0.15418 nm) operating at 40 kV and 40 mA. Chemical functionalities were examined via ex situ Fourier-transform infrared spectroscopy (FT-IR) on a Nicolet iS50 infrared spectrometer. MOF samples were diluted in a mixture of KBr at 1.3 wt% loading and were pressed into 13 mm pellets using a Carver hydraulic press at 4500 kg for 5 min. Thermogravimetric analysis (TGA) was performed on a Perkin Elmer TGA 8000 by heating 3–10 mg of catalyst under argon to 383 K followed by a temperature ramp from 383–1173 K (or to 873 K for spent material) under zero air to induce burnoff. Water and organic species removal from the material were separated in the resulting TGA profiles, with the formation of Zr oxide (and Fe oxide) at the end obtained to calculate framework atomic composition. MOF crystal images were taken using a Quanta 200 field emission gun environmental scanning electron microscope (SEM). Fe-UiO-67 images were taken on a Philips Talos F200X scanning/transmission electron microscope (TEM) with a four detector, Super-X energy dispersive X-ray (EDX) to observe Fe nanoparticles. Diffuse reflectance UV-vis spectroscopy (DR UV-vis) measurements used to calculate BGEs were obtained on an Agilent Technologies Cary 60 UV-vis spectrophotometer with a custom stage to hold a Harrick praying mantis diffuse-reflectance reaction chamber. Samples were thoroughly mixed with BaSO4 at 1% weight-loading for analysis. N2 physisorption isotherms (77 K) were obtained on a Micromeritics 3Flex physisorption instrument after 383 K outgassing under vacuum for 24 h. Pore size distributions were determined based on a N2-Tarazona non-local density functional theory (NLDFT) model assuming cylindrical pore geometry. Surface areas were determined using Brunauer–Emmett–Teller (BET) theory with P/Po values selected to satisfy the Rouquerol consistency criteria based on BET surface identification software.47Photocatalytic experiments
Catalytic experiments were performed at ambient temperature and pressure in a tin foil-wrapped 250 mL round bottom flask. In a typical experiment, 0.025 g MOF was added to 100 mL of a 0.031 mM MB stirred stock solution. H2O2 was added at 3.6 ks to produce a solution concentration of 4.0 mM, and light was introduced to the reaction. For visible light driven reactions, a fiber optic cable connected to a halogen lamp (Dolan-Jenner Model Mi-150 Fiber Optic Illuminator, 150 W halogen, 487 mW cm−2 1 cm from the lamp) was placed inside the flask 5.75 cm above the reaction broth. For UV light reactions, the tin foil was removed, and the flask was placed between two 370 nm, 44 W UV light lamps set to 25% intensity (Kessil PR160L, 370 nm 44 W LED, second generation with a built-in fan, typically 399 mW cm−2 1 cm from the lamp at full power), with the flask centered 10 cm from each lamp. The reaction environment was further shielded by cardboard and plastic coverings to prevent unintentional light exposure. Aliquots were removed from solution and quenched with known quantities of methanol prior to absorbance measurement using a Cary 60 UV-vis. MB concentrations were determined via the Beer–Lambert law based on the molar absorptivity obtained through calibration on the instrument. Conversion was calculated either through normalization to the initial bulk concentration or by renormalizing to the bulk concentration in solution at the time of light introduction/oxidant addition. Rate constants were fit over the first 3.6 ks of exposure to oxidant/light and normalized based on the catalyst quantity present upon their introduction.Leach testing was performed at the end of typical reactions (or at truncated times) by recovering 50 mL supernatant fluid from solution with centrifugation (7000 RPM, 10 minutes) and recharging it with MB to initial concentration conditions (assuming complete conversion). The flask was reintroduced to the pertinent light condition and aliquots were taken periodically. Separate trials were performed in which H2O2 was recharged to initial concentration conditions, concurrently with MB. Recovered solid catalysts were dried in air overnight or immersed in 30 mL of solvent (acetone, ethanol, or THF) overnight before drying under thermal vacuum activations. Water stability tests were performed by loading reaction level concentrations of MOF into the batch reactor at 50 mL scale in the absence of dye, oxidant, or light. Unproductive H2O2 decomposition tests were performed by exposing 50 mL solution with reaction level concentrations of catalyst and H2O2 to pertinent light conditions and tracking the concentration through time via acidic KMnO4 titrations of removed aliquots.
Error bars for each temporal conversion profile are determined by propagating observed standard deviation in UV-vis absorbance with uncertainty from volumetric measurements as applicable through calculation of concentration and conversion. Data for control and most activation conditions are single experiments. Data for degradation of MB by MOFs with oxidant and UV light (and their associated leach tests) represent the averages of duplicate trials with error bars reflecting their spread. Uncertainty associated with pseudo-first order rate constants is taken as the extracted standard deviation of the slope from linearized plots, which are averaged to represent data presented of duplicate trials and propagated appropriately to reflect normalization. Extracted rate constants represent the lumped, total pseudo-first order rate constant for MB degradation in the system except where normalized by H2O2 concentration, wherein control rate constant is subtracted.
Results and discussion
Material characterization
Successful synthesis of the catalysts was confirmed using several standard characterization techniques. Primary low angle diffraction peaks in the XRD patterns (Fig. 2a for (Fe-)UiO-67, Fig. S1a† for UiO-68) for all three UiO-MOFs synthesized match simulations for a defect-free crystal constructed using RIETAN-FP in VESTA and agree with literature reports.43,46,48–52 Mild peak attenuation is observed in the Fe-loaded MOF relative to the UiO-67 parent measured under similar sample loading conditions, consistent with some pore occlusion via the impregnation procedure applied in synthesis and processing. Likewise, FT-IR spectra for both the parent UiO-67 and Fe-UiO-67 are similar to one another (Fig. 2b, full spectrum in Fig. S2†), indicating the retention of the MOF structure after Fe incorporation procedures. The band at 1540 cm−1 is assigned to C–C ring stretches in the linker,53 while bands at 1420 cm−1 and 1600 cm−1 represent the symmetric and asymmetric carboxylate stretches, respectively, in the linker, connecting each node.43,53 Nodes are indicated by bands centered at 770 cm−1 and 660 cm−1, which have been attributed to the transverse and longitudinal vibrational modes of Zr–O bonds, respectively.54,55 Additional bands include adsorbed molecules: 1690 cm−1 and 1100 cm−1 bands are consistent with the C–O vibration of residual DMF (other DMF bands are likely obscured by larger functionalities from the MOF),53 and bands in the 2800–3600 cm−1 region are indicative of water and hydroxyl groups present in the structure. Similar framework functionalities are observed on the UiO-68 MOF (Fig. S1b†).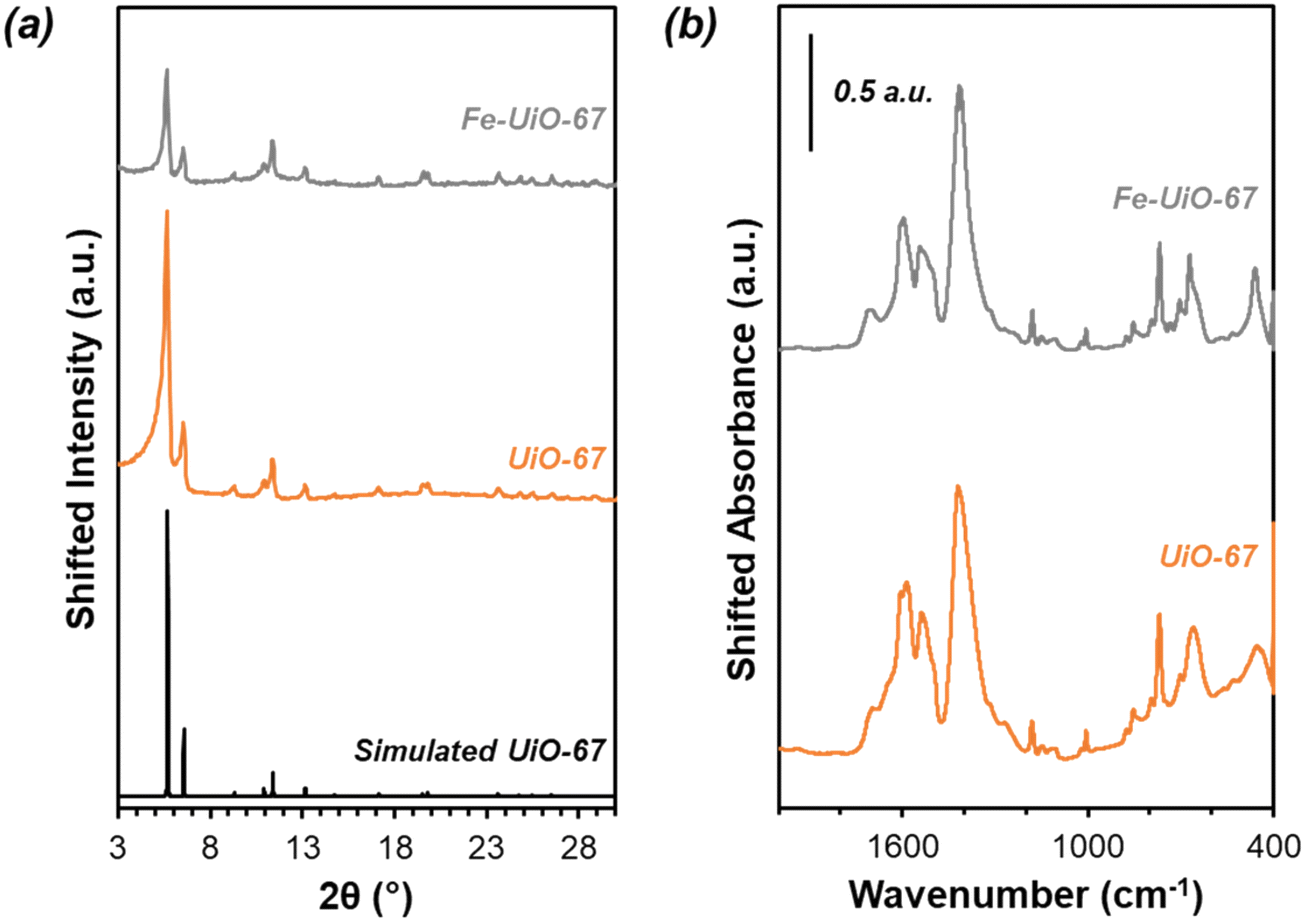 | ||
| Fig. 2 (a) UiO-67 and Fe-UiO-67 experimental (orange and gray, respectively) and simulated (black) XRD patterns. Fe-UiO-67 intensity scaled up 2.5×. (b) Ex situ FT-IR absorbance spectra for synthesized UiO-67 and Fe-UiO-67. Fe-UiO-67 absorbance scaled down 0.35×. | ||
Both isoreticular structures demonstrate comparable octahedral crystal morphologies based on SEM imaging, although some inhomogeneity exists due to choice of acid modulator (Fig. 3a–c).56 Particle sizes for the MOFs are similar to one another, although notably UiO-67 crystallites, measuring 0.5 ± 0.3 μm along their longest dimension, are larger than those of Fe-UiO-67, at 0.4 ± 0.3 μm (Fig. S3a and b†). Mild attenuation in average size is attributed to the synthetic workup for the Fe-incorporation. UiO-68 features crystals of a similar scale, with a wider distribution, at 1.1 ± 0.8 μm (Fig. S3c†). The presence of isolated or clustered Fe moieties distributed throughout Fe-UiO-67 is confirmed through EDX mapping of agglomerated particles (Fig. S4†). However, scanning TEM (STEM) of Fe-UiO-67 also suggests Fe-containing nanoparticles, as indicated by the darker areas on the high-angle annular dark-field (HAADF) image (Fig. 4a; where Fe will show up darker compared to higher atomic number Zr). EDX images in Fig. 4b–d demonstrate one such ∼10 nm diameter region on a particle surface that exhibits Fe and O presence, but little Zr content. The occurrence of these nanoparticles confirm the existence of a Fe/O domain, which is expected to be Fe2O3 based on the synthetic procedure applied.45 Notably, peaks corresponding to (any) Fe oxide are not present in the Fe-UiO-67 XRD pattern, indicating they must be small and limited in quantity (Fig. S5†).
 | ||
| Fig. 3 SEM images of (a) UiO-67, (b) Fe-UiO-67, and (c) UiO-68. | ||
 | ||
| Fig. 4 (a) STEM HAADF image of Fe-UiO-67 with accompanying EDX mapping (b) Zr, (c) O, and (d) Fe elements (yellow, green, and orange, respectively). | ||
Elemental contents (Table 1) are further assessed from normalized combustion TGA curves (Fig. S6†) with distinct regions of water loss, organic combustion, and final metal oxide formation. Parent UiO-67 exhibits less water normalized by dry mass of Zr-MOF (2.7 mmol H2O per gMOF) compared with Fe-UiO-67 and UiO-68 (3.2 mmol H2O per gMOF and 4.0 mmol H2O per gMOF, respectively). This water loss has been correlated with defect density,57 suggesting mild increases in defects accruing from Fe incorporation methods. Regardless, Fe content in Fe-UiO-67, determined based on the mass loss profile of parent UiO-67 material, is calculated as 3.8 wt%. Collectively, the compositional and structural characterizations for UiO-67 and UiO-68 are consistent with their successful syntheses and confirm the integration of oxidized Fe nanoparticles and dispersed moieties in the Fe-UiO-67 material.
| MOF | Zr (wt%) | H (wt%) | O (wt%) | C (wt%) | Fe (wt%) |
|---|---|---|---|---|---|
| Ideal UiO-67 | 25.8 | 2.5 | 24.1 | 47.6 | — |
| Experimental UiO-67 | 29.1 | 2.7 | 26.8 | 41.4 | — |
| Experimental Fe-UiO-67 | 29.2 | 2.6 | 27.2 | 37.2 | 3.8 |
| Ideal UiO-68 | 21.2 | 3.0 | 19.9 | 55.9 | — |
| Experimental UiO-68 | 21.6 | 3.5 | 24.6 | 50.4 | — |
Photocatalytic reactivity
UiO-67 and UiO-68 both demonstrate sorptive MB uptake from bulk aqueous solution (∼20% of loaded MB) in absence of light or oxidant (Fig. 5a). The dye interaction with the MOF is likely in a site-specific manner, with MB binding to available external and internal metal nodes via heteroatoms or to structure linkers through positive π–π interactions. Lack of enhanced removal by UiO-68 compared to UiO-67 is consistent with adsorptive removal of dye from solution, as the higher pore size and BET surface area of UiO-68 compared to UiO-67 (measured at 3010 m2 g−1 and 1600 m2 g−1, respectively) would be expected to enhance removal if absorption plays a dominant role (Fig. S7†). Uptake is more limited for Fe-UiO-67, likely owing to reduced adsorption site availability caused by Fe presence and binding to the MOF structure rather than significant changes to the porosity and internal site access in the material. This is consistent with a slight reduction in mass normalized adsorption isotherm, differential pore volume, and BET surface area for Fe-UiO-67 as compared with its parent (1450 m2 g−1vs. 1600 m2 g−1 total surface areas, respectively) (Fig. S7†).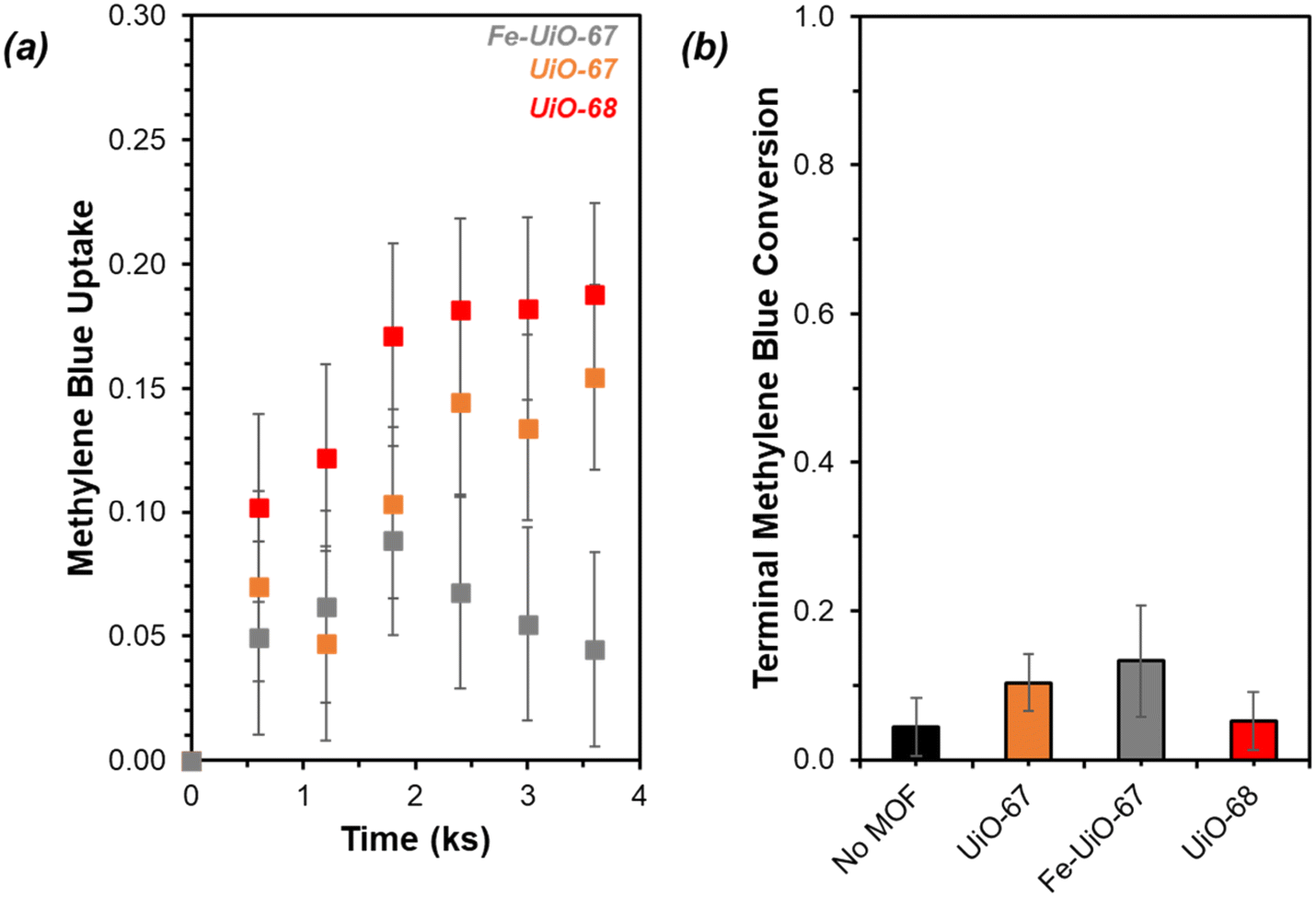 | ||
| Fig. 5 (a) MB uptake in absence of light or oxidant by Fe-UiO-67 (gray), UiO-67 (orange), and UiO-68 (red) (initial charge of 0.25 g L−1 MOF, 0.031 mM MB, 0 mM H2O2, 100 mL). (b) Terminal conversion of MB with visible light activation and oxidant present (Fe-UiO-67 (gray), UiO-67 (orange), UiO-68 (red), no MOF (black)) normalized to the bulk concentration remaining after a 3.6 ks period of interaction between the dye and MOF (initial charge of 0.25 g L−1 MOF, 0.031 mM MB, 4.0 mM H2O2, 150 W halogen lamp, 100 mL). | ||
Notably, addition of H2O2 and exposure of visible or near-UV light to MB solutions in the absence of MOFs does not result in significant conversion in the timescale considered (9.9 ks exposure, Fig. S12†). Slight enhancement is achieved by activating H2O2 with visible light in the absence of MOF, with MB conversion of 0.04 ± 0.04 by after 9.9 ks (Fig. 5b, temporal reaction profile in S13 with pseudo-first order model values in Table S1†). Here, conversion is calculated based on the bulk concentration present at 3.6 ks after dark adsorptive uptake has occurred. Identical catalyst mass loading of UiO-67 and Fe-UiO-67 shows mild enhancement above the control with MB conversions of 0.10 ± 0.04 and 0.13 ± 0.07, respectively, while UiO-68 performs closer to the control at 0.05 ± 0.04. Application of a pseudo-first order model over the first 3.6 ks post-oxidant introduction results in rate constants that have similar trends (Fig. S13† fitted profiles; lumping H2O2 concentration into this value, as it is assumed to be constant). Regardless, the rate constants and conversions by MOFs with visible light and oxidant present are all low and within the uncertainty of the no MOF control, suggesting higher energy drivers are needed to induce significant dye degradation.
This motivates the use of UV light, which leads to a more marked difference in MB breakdown across the MOFs considered. Observed conversion at 9.9 ks by UiO-67 of 0.40 ± 0.02 is not enhanced relative to the UV-H2O2 control value of 0.45 ± 0.03 after consideration of error (Fig. 6, extended profile in Fig. S14†). Meanwhile, conversion increases for UiO-68 and Fe-UiO-67, with final values of 0.56 ± 0.02 and 0.70 ± 0.03, respectively. Notably, this increase is not due to a mild temperature increase over the course of reaction (1–3 K) caused by the UV-lamp, as it is roughly the same across all trials, including the control. Application of a pseudo-first order reaction model to early temporal conversion data results in mass normalized rate constants of 3.5 ± 0.4 g−1 ks−1 (1.5 ± 0.2 mmolZr−1 ks−1) and 6.8 ± 0.5 g−1 ks−1 (2.1 ± 0.2 mmolZr−1 ks−1) for UiO-68 and Fe-UiO-67, respectively, consistent with their trend in terminal conversion.
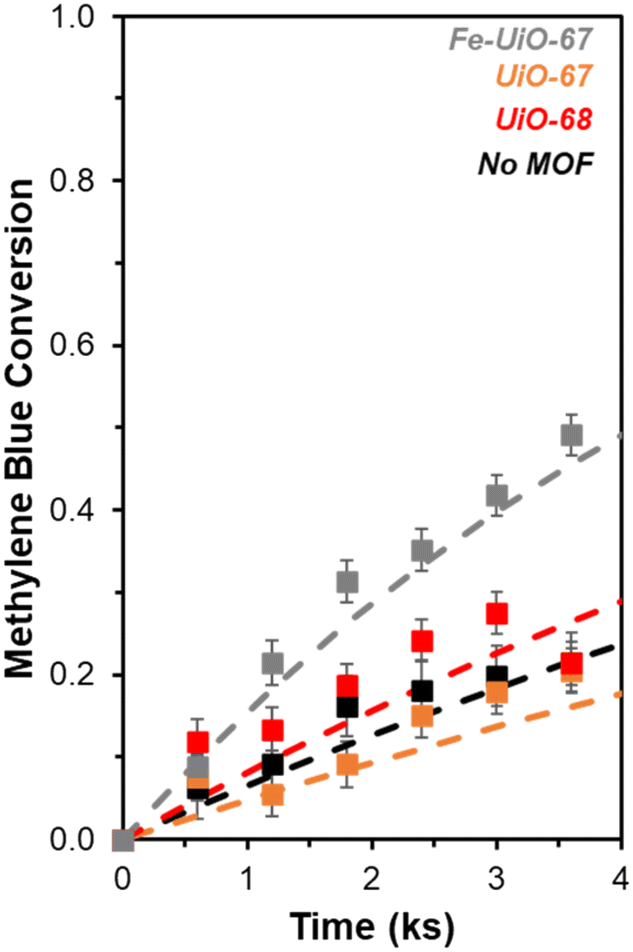 | ||
| Fig. 6 Temporal conversion profiles of MB with UV light activation and oxidant present (Fe-UiO-67 (gray), UiO-67 (orange), UiO-68 (red), no MOF (black)) renormalized to the bulk concentration remaining after a 3.6 ks interaction period between the dye and MOF (initial charge of 0.25 g L−1 MOF, 0.031 mM MB, 4.0 mM H2O2, 370 nm UV lamp, 100 mL, 0 ks corresponds to oxidant/light addition). Dashed lines represent modeled pseudo-first order kinetic profiles. | ||
In contrast, UiO-67 has a first order rate constant of 2.0 ± 0.3 g−1 ks−1 (0.6 ± 0.1 mmolZr−1 ks−1), which is similar to the unnormalized value for the no MOF control (0.049 ± 0.007 ks−1vs. 0.068 ± 0.007 ks−1, respectively). A slight offset of the UiO-67 model from data at increasing conversion may be a result of averaging the extracted rate constants from independent trials to produce the fit and comparing them to the average conversion. Regardless, these values for the three catalysts and their associated rates are interpreted as kinetic in nature based on the Weisz–Prater criterion, considering the size of particles observed via SEM (details presented in ESI†), though notably this does not clarify the extent of light penetration into crystal structures or electron–hole lifetimes.58 The increased rate constant for Fe-UiO-67 compared to UiO-67 suggests added conversion from photo-Fenton based chemistry. However, similar rate constants when normalized per Zr for UiO-68 and Fe-UiO-67 indicate another property of these two materials may be similar.
DR-UV-vis is used to determine how the series of MOFs interact with light to explain apparent trends in reactivity. Transforming the reflectance data for each MOF (Fig. S8†) using Kubelka–Munk (K–M) theory results in absorption features that have distinct slopes (Fig. 7, extended absorbance profiles in S9†). The x-intercept of these slopes, determined after systematically applying a four-point averaging method to the data, reveal that the measured BGEs for UiO-67, UiO-68, and Fe-UiO-67 are 3.6 eV, 3.3 eV, and 3.4 eV, respectively. Because the UV lamp used emits light centered at (and distributed around) 370 nm, it is best suited to activate electrons in materials with BGEs around 3.35 eV. Therefore, both Fe-UiO-67 and UiO-68 can facilitate enhanced formation of electron–hole pairs that may interact with oxidant species to activate radicals or dye directly to induce MB conversion, but UiO-67 cannot. Additive photocatalytic reactivity of Fe oxide is ruled out because the BGE of Fe oxides typically lie in the visible light range, yet Fe-UiO-67 exhibits no enhanced improvement over UiO-67 under visible light conditions (Fig. 5b and S13†). Likewise, dispersed Fe moieties in the structure do not contribute to reactivity significantly via traditional, Fenton-type chemical reactions, or similar enhancement would be seen under visible light activation. Nevertheless, Fe-UiO-67 exhibits a higher mass-normalized pseudo-first order rate constant than UiO-68 under identical total mass loading and lighting conditions despite its marginally higher measured BGE. This could potentially derive from longer electron–hole lifetimes in the Fe-loaded structure or more efficient light utilization resulting from its smaller crystallites. Regardless, the enhanced degradation from these two catalysts under UV-light warrants investigation into their stability, particularly for Fe-UiO-67, because Fe-containing MOFs have demonstrated leaching in aqueous pollutant degradation systems that can conflate observed reactivity.23
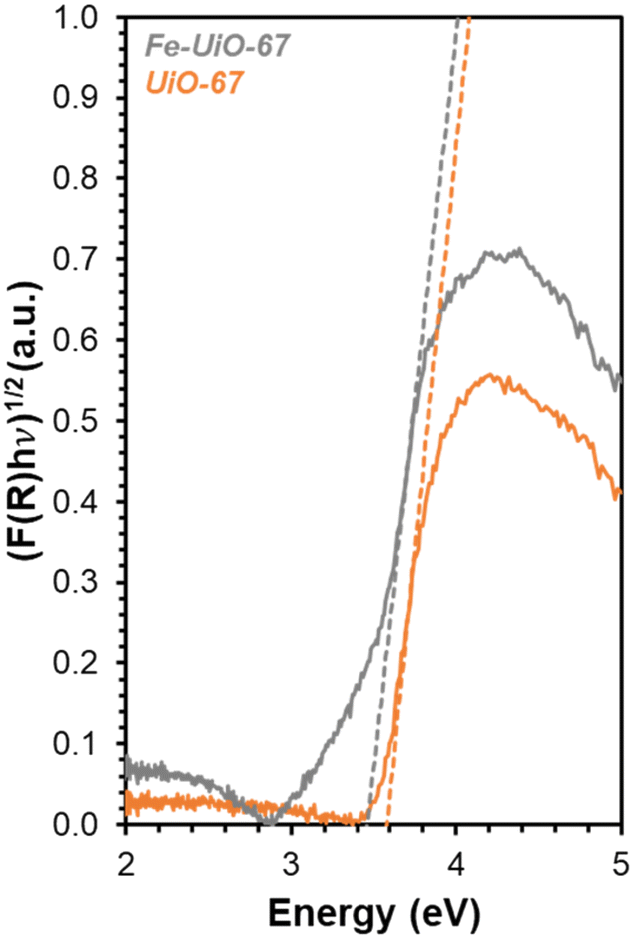 | ||
| Fig. 7 DR-UV-vis data for Fe-UiO-67 (gray) and UiO-67 (orange) with K–M transformation applied. Dashed lines represent tangent lines to the rise in absorption peak for BGE calculation. | ||
Catalyst stability
Given results of reportedly-water stable MOF instability under aqueous oxidation reactions,23–25 investigation into stability of the materials from this work is probed. Recharging recovered supernatant fluid with MB after removing catalyst results in only mild conversion under visible light conditions, suggesting limited leaching of metal. However, it is larger on Fe-UiO-67 than on UiO-67 or UiO-68, with pseudo-first order rate constants of 0.03 ± 0.01 ks−1, 0.009 ± 0.002 ks−1, and 0.003 ± 0.005 ks−1, respectively (Fig. S15†). The larger value for Fe-UiO-67 is likely the result of mild Fe-loss to solution by the end of reaction, in the form of Fe-species that are active for Fenton chemistry. Supernatant fluid reactivities for UiO-67 and UiO-68 systems are like the no MOF control reaction (0.003 ± 0.002 ks−1). Also, supernatant conversion for UiO-67 and UiO-68 under UV light activation is similar to the control, with pseudo-first order rate constants of 0.073 ± 0.011 ks−1 and 0.077 ± 0.003 ks−1, respectively, vs. 0.068 ± 0.007 ks−1 without MOF (Fig. 8a, extended profile in S16a†). This indicates no significant quantity of active leached species are present for either UiO-67 or UiO-68 under either lighting condition. While Zr salts are not known for their activation of H2O2, the lack of enhancement for UiO-68 confirms the methodology applied removes solid material from solution that could otherwise induce photocatalytic reactivity. | ||
| Fig. 8 Temporal conversion profiles of MB with UV light activation and oxidant present (Fe-UiO-67 (gray), UiO-67 (orange), UiO-68 (red), no MOF (black)) (a) in recovered supernatant fluid with additional dye added (0.0016 mmol MB added to 50 mL supernatant, 370 nm UV lamp) and (b) with additional dye and oxidant added (0.0016 mmol MB and 0.19 mmol H2O2 added to 50 mL supernatant, 370 nm UV lamp). Dashed lines represent modeled pseudo-first order kinetic profiles. | ||
In contrast, the filtrate for the Fe-UiO-67 material exhibits lower conversion and extracted rate constant in the UV system, at 0.018 ± 0.005 ks−1 (Fig. 8a). Here, the depressed conversion suggests the H2O2 concentration is reduced significantly despite its high initial excess in comparison to MB, altering the initial conditions in the leach test for the Fe-UiO-67 material relative to UiO-67 and UiO-68. Indeed, KMnO4 titrations of aliquots in the absence of dye demonstrate limited unproductive H2O2 degradation in the UiO-68 and no MOF control trials with UV light exposure, with conversions by 3.6 ks of 0.05 ± 0.03 and 0.05 ± 0.04, respectively, justifying the application of a pseudo-first order model to these systems (Fig. S17†). However, oxidant conversion of 0.74 ± 0.03 over this same period for Fe-UiO-67, culminating in a final conversion of 0.95 ± 0.03 at the end of the typical reaction timescale, indicates the H2O2 concentration is not approximately constant in this setup. Although the extracted rate constant on this structure (6.8 ± 0.5 g−1 ks−1) is an appropriate model for the system at early conversions, deviations at longer reaction times are consistent with this decrease in oxidant concentration (Fig. S14b†).
When H2O2 is recharged alongside MB to the Fe-UiO-67 recovered supernatant fluid to initial reaction concentrations, the apparent degradation significantly exceeds the no MOF control (Fig. 8b, extended profile in S16b†). The resulting extracted 0.50 ± 0.05 ks−1 rate constant suggests the presence of active leached species in solution, likely in the form of an Fe complex, which may exhibit higher reactivity than the supported metal form. These species conflate observed MB degradation driven by solid and homogeneous species during reaction because of the propensity for Fe to participate in Fenton-like chemistry, partially explaining the apparent increase in degradation of Fe-UiO-67 relative to UiO-68, despite their similar BGEs. Notably, the apparent conversion by leached species from the UV activated system is significantly higher than in the visible light activated system (extracted pseudo-first order rate constant of 0.50 ± 0.04 ks−1vs. 0.03 ± 0.01 ks−1). While solvent interactions with water may contribute, the significant difference in conversion is consistent with UV light interaction or oxidant turnovers driving observed leaching. By truncating the reaction at intermediate time points and performing supernatant tests, the turnover dependence of leaching is explored. In separate trials truncated at 2.4 ks after oxidant and light exposure—one performed with typical reaction conditions, one without adding dye to the initial reaction—similar filtrate conversion profiles are obtained with consistent unnormalized pseudo-first order rate constants of 0.31 ± 0.01 ks−1 and 0.30 ± 0.01 ks−1, respectively (Fig. S18†). Assuming similar levels of H2O2 consumption in these systems, the overlap in value between the reactions with and without dye present highlights the minimal role MB presence and breakdown plays in leaching under these conditions. Moreover, similar pseudo-first order rate constants across filtrates are obtained for systems truncated at 2.4 ks and terminated at the typical reaction timescale after correcting for difference in H2O2 presence (0.13 ± 0.01 ks−1 mMH2O2−1, 0.13 ± 0.02 ks−1 mMH2O2−1, and 0.12 ± 0.02 ks−1 mMH2O2−1 for the truncated reaction, the truncated timescale no-dye exposure, and the full reaction system supernatant tests, respectively). Collectively these data highlight the importance of UV light interaction or initial UV light-MOF mediated H2O2 turnovers in inducing species leaching in the Fe-UiO-67 material.
Fe species in solution may also contribute to observed selectivity toward higher, unproductive use of H2O2 in the Fe-UiO-67 system. KMnO4 titrations reveal contrasting H2O2 utilization efficiencies across the materials studied by demonstrating the capacity for each MOF to unproductively consume oxidant through disproportionation reaction to form water and oxygen. Fe-UiO-67 only uses 9.2 ± 0.4% productively (assuming 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O2 used to MB consumed and complete combustion for calculation) compared to the no MOF control at 23 ± 4% (Fig. 9). In contrast, UiO-68 exhibits a higher efficiency of 36 ± 7%, suggesting it reacts more effectively with H2O2 despite doing so at lower rates than Fe-UiO-67.
:1 H2O2 used to MB consumed and complete combustion for calculation) compared to the no MOF control at 23 ± 4% (Fig. 9). In contrast, UiO-68 exhibits a higher efficiency of 36 ± 7%, suggesting it reacts more effectively with H2O2 despite doing so at lower rates than Fe-UiO-67.
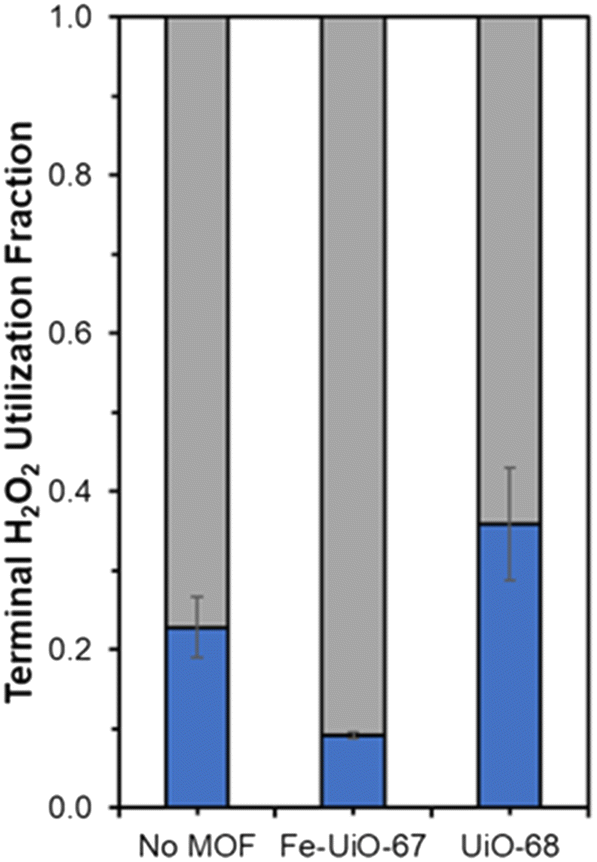 | ||
| Fig. 9 Terminal H2O2 productive (blue) and unproductive (gray) conversion fractions for Fe-UiO-67, UiO-68, and the no MOF control under UV light activation. | ||
Despite apparent leaching and greater unproductive H2O2 turnovers relative to UiO-68, recovered Fe-UiO-67 material continues to demonstrate reactivity under identical initial reactant and catalyst concentration conditions with UV light exposure (Fig. S19a and b†). The unnormalized rate constant in the spent material is 0.12 ± 0.02 ks−1, only slightly lower than that of the fresh material, at 0.17 ± 0.01 ks−1. This reduction is partially explained by the spent catalyst (blue in color) containing residual dye and intermediate breakdown products despite its solvent exchange in acetone prior to drying and reuse. TGA combustion of spent material indicates consistent total organic content with fresh material, but the profile shape during organic loss changes in agreement with residual dye presence (Fig. S10†). Buildup of dye and breakdown product species on the surface may contribute to deviations in reactivity from the pseudo-first order model for fresh material at longer time points, in addition to reduced oxidant presence (Fig. S14†). The lower reactivity of the spent sample at identical catalyst mass concentration loading to fresh MOF is at least in part caused by reduced MOF content in the spent sample. Indeed, TGA data suggests there is ∼50% less Fe incorporated in the MOF structure by the end of the reaction, consistent with decreased MOF presence with residual dye incorporation, though it could also be driven by Fe content reduction after leaching. Additionally, the remaining Fe species in the structure still appear susceptible to leaching based on the continued reactivity of supernatant fluid recovered in the spent Fe-UiO-67 trial (Fig. S19c†). This suggests the observed catalyst leaching may be tied to oxidant turnovers or interaction with UV light and is not merely due to residual weakly chemisorbed or physiosorbed Fe species present after synthesis, which would have been removed after the first trial. Taken together, the degradative reactivity of the spent material indicates continued functionality of the catalyst despite mild leaching, at least over a limited period of cycling.
This explanation is consistent with XRD data indicating catalyst bulk structure appears intact. After solvent exchanging with acetone and drying at 343 K, Fe-UiO-67 used in the UV light reactions still exhibits clear peaks at low angles akin to fresh material (Fig. 10). Notably, solvent exchanging with a lower surface tension solvent than water is necessary prior to drying to ensure crystalline material is recovered as water can collapse the pores in UiO-MOFs via capillary interactions during evaporation.59,60 Taking UiO-67 as a test case, the material shows reduced peak intensity when ambient drying occurs after water exposure, even when subsequently treated with another solvent (Fig. S11a†). Exchanging with THF, acetone, and ethanol prior to drying all allow for recovery of crystalline material under heating conditions ranging from 343 K to 473 K. Similar results are observed when UiO-67 is exposed to water alone and then exchanged with these lower surface tension solvents, confirming the role of water in dictating this process, as opposed to H2O2 or MB interaction (Fig. S11b†). Mild peak broadening on the spent Fe-UiO-67 material may be attributable in part to MB presence on or within the structures (indicated by the blue color of the material). Thus, Fe-UiO-67 continues to demonstrate reactivity upon recovery and washing and retains long range order, although it does leach active species and features lower productive H2O2 utilization than UiO-68.
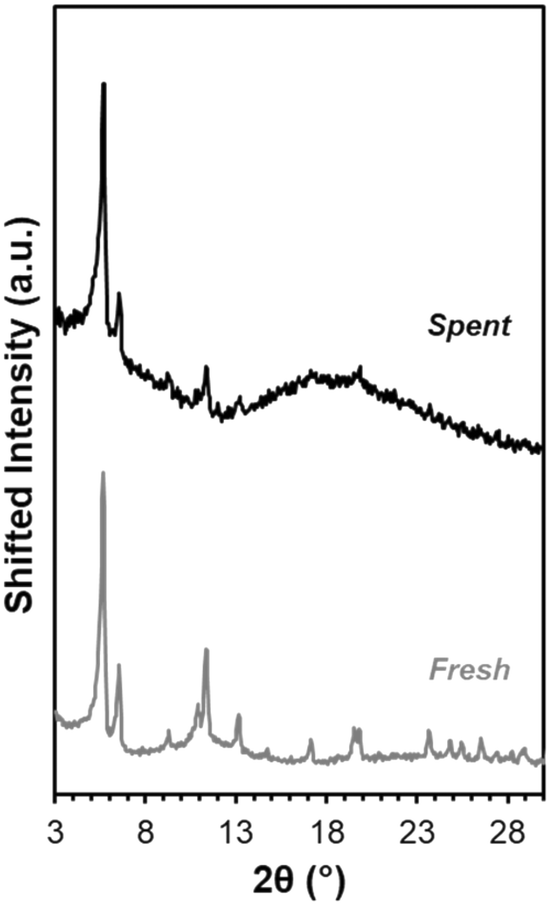 | ||
| Fig. 10 XRD patterns for fresh Fe-UiO-67 (gray) and Fe-UiO-67 recovered from reaction with oxidant and UV light and solvent exchanged with acetone prior to 343 K heating (black, scaled up 1.6×). | ||
Conclusions
Degradation of MB by UiO-family MOFs is investigated to elucidate the impact of contrasting linker and alternative base-metal incorporation for photocatalytic pollutant degradation. While Fe-UiO-67 demonstrates enhanced MB oxidation with a pseudo-first order rate constant of 6.8 ± 0.5 g−1 ks−1 under UV light activation with excess H2O2, parent UiO-67 hardly exceeds controls in the absence of MOF, with a rate constant of 2.0 ± 0.3 g−1 ks−1. This difference is consistent with the lower BGE of Fe-UiO-67 as compared with UiO-67, leading to its activation under the 370 nm UV lamp applied (3.4 eV vs. 3.6 eV, respectively). Using a longer, triphenyl linker in the case of UiO-68 also enhances reactivity, with a pseudo-first order rate constant of 3.5 ± 0.4 g−1 ks−1 (BGE 3.3 eV). However, leach tests reveal the presence of active, Fe-containing species in solution for Fe-UiO-67, potentially conflating homogeneous and heterogeneous reactivity as well as leading to more unproductive H2O2 conversion. Nevertheless, the long-range order of Fe-UiO-67 is largely maintained. The catalyst can be reused upon solvent exchange and drying, enabling potential in this and other aqueous applications. While both secondary metal incorporation and linker modulation can induce promotional effects in the context of pollutant oxidation, reduced risk of leaching and concomitant reduction in BGE in UiO-68, alongside its higher effective H2O2 use, may make it a more advantageous material than Fe-UiO-67. Broadly, linker modulation on the aqueously stable UiO series of MOF shows significant promise toward improvement of oxidant usage in water-based applications.Data availability statement
Data available upon request from the authors.Author contributions
Samuel Moore: methodology (equal), investigation (equal), formal analysis (equal), validation (lead), writing – original draft (equal), project administration (supporting). Isabella Hubble: investigation (equal), formal analysis (equal), writing – original draft (equal). Alyssa Ritchie: investigation (equal), formal analysis (supporting), validation (supporting). Jeffrey Barzach: investigation (supporting), formal analysis (supporting). Michele Sarazen: conceptualization (lead), methodology (equal), resources, writing – review and editing (lead), project administration (lead), supervision (lead), funding acquisition (lead).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the High Meadows Environmental Institute (HMEI) at Princeton University through the Mary and Randall Hack '69 Research Fund and Water and the Environment Grand Challenge Award. Additionally, the authors recognize HMEI, the Princeton Department of Chemistry, and the Princeton Office of Undergraduate Research for their support of the Summer Undergraduate Research Fellows in Chemistry Program and thesis funding. The authors acknowledge the National Science Foundation through Princeton University's Materials Research Science and Engineering Center (MRSEC) Princeton Center for Complex Materials (PCCM) for seed funding (DMR-1420541) and the use of Princeton's Imaging and Analysis Center (IAC, DMR-2011750). The authors thank Marco R. Ivancevic and Dr. Yueh-Lin Loo for preliminary UV lamp use at project inception and James L. Trettin Dr. Michael R. Smith, and Dr. Bruce E. Koel for assistance with the DRIFTS and DR-UV-vis measurements. Lastly, the authors especially thank Dr. Rachel A. Yang for her insights and support in conversations and interpretation surrounding this project.References
- X. Li, B. Wang, Y. Cao, S. Zhao, H. Wang, X. Feng, J. Zhou and X. Ma, ACS Sustainable Chem. Eng., 2019, 7, 4548–4563 CrossRef CAS.
- A. Boretti and L. Rosa, npj Clean Water, 2019, 2, 1–15 CrossRef.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- N. Wang, T. Zheng, G. Zhang and P. Wang, J. Environ. Chem. Eng., 2016, 4, 762–787 CrossRef CAS.
- T. Wu, W. Gao, J. Zhang, M. Hao, S. Zhang and H. Tao, React. Chem. Eng., 2024, 9, 728–739 RSC.
- X. Liu, Y. Zhou, J. Zhang, L. Tang, L. Luo and G. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 20255–20275 CrossRef CAS PubMed.
- Y. Gao, S. Li, Y. Li, L. Yao and H. Zhang, Appl. Catal., B, 2017, 202, 165–174 CrossRef CAS.
- H. Zhang, S. Chen, H. Zhang, X. Fan, C. Gao, H. Yu and X. Quan, Front. Environ. Sci. Eng., 2019, 13, 1–18 CrossRef CAS.
- K. G. M. Laurier, F. Vermoortele, R. Ameloot, D. E. De Vos, J. Hofkens and M. B. J. Roeffaers, J. Am. Chem. Soc., 2013, 135, 14488–14491 CrossRef CAS PubMed.
- L. Ai, C. Zhang, L. Li and J. Jiang, Appl. Catal., B, 2014, 148–149, 191–200 CrossRef CAS.
- Y. Yin, Y. Ren, J. Lu, W. Zhang, C. Shan, M. Hua, L. Lv and B. Pan, Appl. Catal., B, 2021, 286, 1–8 CrossRef.
- G. A. Udourioh, M. M. Solomon, C. O. Matthews-Amune, E. I. Epelle, J. A. Okolie, V. E. Agbazue and U. Onyenze, React. Chem. Eng., 2023, 8, 278–310 RSC.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. Zhou, Adv. Mater., 2018, 30, 1–35 Search PubMed.
- Y. Yang, Y. Xue, J. Li, H. Xia and M. Zhou, React. Chem. Eng., 2023, 8, 2195–2210 RSC.
- S. Dhaka, R. Kumar, A. Deep, M. B. Kurade, S. Ji and B. Jeon, Coord. Chem. Rev., 2019, 380, 330–352 CrossRef CAS.
- M. Cao and W. Yang, React. Chem. Eng., 2023, 9, 317–338 RSC.
- Y. Liu, Y. Xie, M. Dai, Q. Gong and Z. Dang, Materials, 2019, 12, 1–20 Search PubMed.
- H. Fu, X. X. Song, L. Wu, C. Zhao, P. Wang and C. C. Wang, Mater. Res. Bull., 2020, 125, 110806 CrossRef CAS.
- H. Hu, H. Zhang, Y. Chen and H. Ou, Environ. Sci. Pollut. Res., 2019, 26, 24720–24732 CrossRef CAS PubMed.
- H. Hu, H. Zhang, Y. Chen, Y. Chen, L. Zhuang and H. Ou, Chem. Eng. J., 2019, 368, 273–284 CrossRef CAS.
- Q. Zhao, L. Zhang, X. Wang, X. Jia, P. Xu, M. Zhao and R. Dai, Transition Met. Chem., 2019, 44, 789–797 CrossRef CAS.
- R. S. Bhise, Y. A. Patil and G. S. Shankarling, React. Chem. Eng., 2023, 8, 3046–3059 RSC.
- S. C. Moore and M. L. Sarazen, AIChE J., 2023, 69, 1–11 CrossRef.
- I. Bezverkhyy, G. Weber and J. P. Bellat, Microporous Mesoporous Mater., 2016, 219, 117–124 CrossRef CAS.
- A. M. Chávez, A. Rey, J. López, P. M. Álvarez and F. J. Beltrán, Sep. Purif. Technol., 2021, 255, 117660 CrossRef.
- Y. Liu, Y. Zhao, Z. Zhou, Y. Shen and L. Jiang, Colloids Surf., A, 2023, 674, 131907 CrossRef CAS.
- A. H. Vahabi, F. Norouzi, E. Sheibani and M. Rahimi-Nasrabadi, Coord. Chem. Rev., 2021, 445, 214050 CrossRef CAS.
- B. Wang, X. L. Lv, D. Feng, L. H. Xie, J. Zhang, M. Li, Y. Xie, J. R. Li and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed.
- M. Cedrún-Morales, M. Ceballos, E. Polo, P. del Pino and B. Pelaz, Chem. Commun., 2023, 59, 2869–2887 RSC.
- Y. Yin, R. Lv, X. Li, L. Lv and W. Zhang, Appl. Catal., B, 2021, 299, 1–8 CrossRef.
- H. Q. Zheng, Y. N. Zeng, J. Chen, R. G. Lin, W. E. Zhuang, R. Cao and Z. J. Lin, Inorg. Chem., 2019, 58, 6983–6992 CrossRef CAS PubMed.
- E. Flage-Larsen, A. Røyset, J. H. Cavka and K. Thorshaug, J. Phys. Chem. C, 2013, 117, 20610–20616 CrossRef CAS.
- R. M. Cedeno, R. Cedeno, M. A. Gapol, T. Lerdwiriyanupap, S. Impeng, A. Flood and S. Bureekaew, Inorg. Chem., 2021, 60, 8908–8916 CrossRef CAS PubMed.
- S. C. Moore, M. R. Smith, J. L. Trettin, R. A. Yang and M. L. Sarazen, ACS Energy Lett., 2023, 8, 1397–1407 CrossRef CAS.
- M. A. Syzgantseva, C. P. Ireland, F. M. Ebrahim, B. Smit and O. A. Syzgantseva, J. Am. Chem. Soc., 2019, 141, 6271–6278 CrossRef CAS PubMed.
- X. Ye and D. Liu, Cryst. Growth Des., 2021, 21, 4780–4804 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- Y. Feng, Q. Chen, M. Cao, N. Ling and J. Yao, ACS Appl. Nano Mater., 2019, 2, 5973–5980 CrossRef CAS.
- X. Su, T. Xu, R. Ye, C. Guo, S. M. Wabaidur, D. L. Chen, S. Aftab, Y. Zhong and Y. Hu, J. Colloid Interface Sci., 2023, 646, 129–140 CrossRef CAS PubMed.
- J. J. Zhou, R. Wang, X. L. Liu, F. M. Peng, C. H. Li, F. Teng and Y. P. Yuan, Appl. Surf. Sci., 2015, 346, 278–283 CrossRef CAS.
- M. N. Nimbalkar and B. R. Bhat, J. Environ. Chem. Eng., 2021, 9, 106216 CrossRef CAS.
- S. E. H. Etaiw and M. M. El-bendary, Appl. Catal., B, 2012, 126, 326–333 CrossRef CAS.
- X. Zhu, B. Li, J. Yang, Y. Li, W. Zhao, J. Shi and J. Gu, ACS Appl. Mater. Interfaces, 2015, 7, 223–231 CrossRef CAS PubMed.
- R. Vakili, E. K. Gibson, S. Chansai, S. Xu, N. Al-Janabi, P. P. Wells, C. Hardacre, A. Walton and X. Fan, ChemCatChem, 2018, 10, 4238–4242 CrossRef CAS PubMed.
- M. L. Rache, A. R. García, H. R. Zea, A. M. T. Silva, L. M. Madeira and J. H. Ramírez, Appl. Catal., B, 2014, 146, 192–200 CrossRef CAS.
- M. C. Lawrence and M. J. Katz, J. Phys. Chem. C, 2022, 126, 1107–1114 CrossRef CAS.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F. Coudert, Y. Cui, B. Hou, D. M. D'Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. Veen, D. Rega, V. Van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14, 5370–5372 CrossRef.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- B. Lerma-Berlanga, C. R. Ganivet, N. Almora-Barrios, S. Tatay, Y. Peng, J. Albero, O. Fabelo, J. González-Platas, H. García, N. M. Padial and C. Martí-Gastaldo, J. Am. Chem. Soc., 2021, 143, 1798–1806 CrossRef CAS PubMed.
- M. Deng, X. Bo and L. Guo, J. Electroanal. Chem., 2018, 815, 198–209 CrossRef CAS.
- S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626 RSC.
- Y. Wu, X. Wang, K. O. Kirlikovali, X. Gong, A. Atilgan, K. Ma, N. M. Schweitzer, N. C. Gianneschi, Z. Li, X. Zhang and O. K. Farha, Angew. Chem., 2022, 134, 1–8 Search PubMed.
- Q. Zhao, Q. Du, Y. Yang, Z. Zhao, J. Cheng, F. Bi, X. Shi, J. Xu and X. Zhang, Chem. Eng. J., 2022, 433, 1–12 Search PubMed.
- X. Zhang, X. Shi, Q. Zhao, Y. Li, J. Wang, Y. Yang, F. Bi, J. Xu and N. Liu, Chem. Eng. J., 2022, 427, 1–14 Search PubMed.
- R. A. Yang and M. L. Sarazen, ACS Catal., 2022, 12, 14476–14491 CrossRef CAS.
- P. B. Weisz and C. D. Prater, Adv. Catal., 1954, 6, 143–196 CAS.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef CAS.
- G. Ayoub, T. Islamoglu, S. Goswami, T. Friščić and O. K. Farha, ACS Appl. Mater. Interfaces, 2019, 11, 15788–15794 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00172a |
| ‡ Samuel Moore and Isabella Hubble have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |