
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into performances of magnetic and bio-based doped-nanohydroxyapatites as water decontamination agents
Maria Laura
Tummino
 a,
Giuliana
Magnacca
b,
Monica
Rigoletto
b,
Mery
Malandrino
b,
Claudia
Vineis
a and
Enzo
Laurenti
*b
a,
Giuliana
Magnacca
b,
Monica
Rigoletto
b,
Mery
Malandrino
b,
Claudia
Vineis
a and
Enzo
Laurenti
*b
aInstitute of Intelligent Industrial Technologies and Systems for Advanced Manufacturing, National Research Council of Italy (CNR-STIIMA), Corso G. Pella 16, 13900 Biella, Italy
bDepartment of Chemistry, University of Torino, Via P. Giuria 7, 10125 Torino, Italy. E-mail: enzo.laurenti@unito.it
First published on 29th May 2024
Abstract
Hydroxyapatite, a mineral from the apatite group, is widely distributed in living organisms and largely studied because of its many properties, including the adsorption of many different substances. In this work, two functionalized nanohydroxyapatites were synthesized starting from their precursors (calcium hydroxide and phosphoric acid) in the presence of Fe(II)/(III) ions and bio-based substances (BBS) isolated from green compost. The products were characterized with different techniques (nitrogen adsorption/desorption, ATR-FTIR, XRD, TGA and ζ-potential measurements) and compared to nanohydroxyapatite obtained without further functionalization. The ability of these materials to remove different water pollutants by adsorption was tested using two organic dyes (crystal violet and methyl orange) and four inorganic ions, Al(III), Cr(III), Ni(II) and As(V), characterized by different ionic charges, dimensions and nature. Moreover, for the same purpose, the antibacterial properties of iron- and iron/BBS-added materials were also tested. The result showed the effective adsorption capability of the materials, in particular with respect to crystal violet, Al(III) and Cr(III), and an enhancement of adsorption capacity with respect to all the adsorbates after functionalization. Finally, the tests towards Staphylococcus aureus and Escherichia coli showed high antimicrobial activity for the bare nanohydroxyapatite samples, whereas the doping with iron and BBS or the high-temperature treatment remarkably impacted this capacity depending on the bacterial strain to eliminate.
1. Introduction
Hydroxyapatite, Ca10(PO4)6(OH)2, is a mineral from the apatite group and has calcium and phosphates as main constituents to form hexagonal crystalline structures.1 The arrangement of atoms in crystalline units generates a tunnel structure with walls composed of CaO6 and PO4 polyhedra joined at the vertices; in these tunnels, the hydroxyl units are inserted.2 Apatites naturally exist in many minerals3 and in the bone tissue of vertebrates;4 in fact, 99% of the calcium present in the human body is stored in the bone tissue in the form of hydroxyapatite,5 making these materials considered biocompatible, and also in nanometric size.6 These materials are indeed the subject of various studies in the biomedical field for the production of bioceramics and as drug carriers,7 but they are promising materials as environmental decontamination tools, as well.8,9Hydroxyapatites and nanohydroxyapatites have been proven to be excellent adsorbents for the removal and sequestration of heavy metals, actinides, radionuclides, halogen ions, and organics from wastewater thanks to their high stability under both oxidizing and reducing conditions and easy accessibility thanks to methods for industrial-scale production, which have significantly reduced the cost.10–12 In addition to the use in aqueous solutions, the addition of hydroxyapatite nanoparticles (nAp) as a soil improver reduced the mobilization of heavy metals and increased their geochemical stability in metal-contaminated soils.13 Possible sorption mechanisms include adsorption, ion exchange, surface complexation and precipitation (or dissolution–precipitation) of metal phosphates.12,14
The morphological and chemical properties of nanohydroxyapatites can be modified if the synthesis occurs in the presence of inorganic and organic additives.15 For example, the incorporation of iron into the hydroxyapatite structure is of great interest for the possibility of forming nanoparticles with magnetic properties and developing efficient adsorbent materials and catalysts that can be removed by applying a magnetic field.16,17 The use of organic substances as templates or, in general, modifiers in the synthesis of nanohydroxyapatites can significantly influence the morphology and surface chemistry of the final materials, causing a significant improvement in sorbent capacity.18,19 These strategies have been exploited by Mercado et al.,20,21 who synthesized paramagnetic iron-containing hydroxyapatite nanoparticles and paramagnetic materials further mixed with soluble bioorganic substances (SBOs, in particular, the so-called CVT230) derived from 230 day-composted urban waste as synthesis precursors. SBOs are mixtures of substances with different molecular weights, formed by long aliphatic C chains substituted by aromatic rings and COOH, NCO, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, PhOH, O-alkyl, O-aryl, OCO, OMe, and alkyl amine functional groups.22,23 In the hydroxyapatite system, SBO introduction was found to increase Cu(II) and Pb(II) sorbent capacity.20,21
O, PhOH, O-alkyl, O-aryl, OCO, OMe, and alkyl amine functional groups.22,23 In the hydroxyapatite system, SBO introduction was found to increase Cu(II) and Pb(II) sorbent capacity.20,21
In this research, we deeply investigated the performance of similar materials through the adsorption of anionic and cationic dyes and other inorganic species, characterized by different ionic charges, radii and nature (metallic and semi-metallic), namely Al(III), Cr(III), Ni(II) and As(V). Indeed, it is essential to widen the application field of adsorbents and evaluate their decontamination capability in differently polluted wastewater.24–26 For the same purpose, we also investigated the antibacterial properties of iron- and iron/SBO-added materials, considering previous studies reporting such antimicrobial ability for different hydroxyapatites.27–33 The hydroxyapatite-based materials produced here are inspired by the studies of Mercado et al.,20,21 but the SBOs used as templates are compounds labeled bio-based substances (BBS), obtained by a slightly different procedure to CVT230 (lab-scale).22,34 The materials were characterized by a multi-analytical approach and the features after UV treatment and calcination were also assessed.
2. Materials and methods
Following a well-established methodology, the bio-based substances (BBS) were isolated from green compost sampled from the Polo Ecologico process line of the ACEA Pinerolese Industriale S.p.A. waste treatment plant in Pinerolo, Italy.34 Green compost was obtained from composting urban public park trimmings and home gardening residues for more than 180 days. Briefly, for the isolation of BBS, the green compost was treated with a water solution of 6 M NaOH (Sigma Aldrich) (4 hours/60 °C). The liquid phase, separated from the reaction mix by centrifugation, was concentrated and different fractions were obtained through a lab-scale ultrafiltration unit equipped with a membrane (molar mass cut-off of 5 kDa). The retentate fraction was then dried at 60 °C for 24 h.All the other chemical reagents were used as provided by the suppliers without further purification. In the experiments, if not otherwise specified, distilled water was used.
2.1 Synthesis of nanohydroxyapatite powders
Following the procedure of Mercado et al.20,21 for the synthesis of the reference nanohydroxyapatite (nAp), a suspension of 5 g of Ca(OH)2 (95%, Sigma Aldrich) in 40 mL of H2O was mechanically stirred at 80 °C. Afterward, a solution of 4.44 g of H3PO4 (85%, Sigma Aldrich) in 60 mL of H2O was added dropwise into the suspension of calcium hydroxide for 2 hours at constant temperature and stirred to obtain the suspension of nAp. Finally, the precipitate was separated from the supernatant by centrifugation at 5000 rpm, washed several times with deionized water and dried in an oven at 80 °C for 24 hours.A similar process was used to synthesize nanohydroxyapatite with iron (nAp–Fe). A mixture of FeCl2·4H2O (1.27 g, >99 wt%, Sigma Aldrich), FeCl3·6H2O (1.79 g, 97 wt%, Sigma Aldrich) and phosphoric acid (4.44 g in 60 mL of H2O) was left under stirring for approximately 4 hours at a temperature of 60 °C to favor the complete dissolution of the iron salts. At the end of the dissolution process, the prepared solution was added dropwise to a suspension of calcium hydroxide (5 g in 40 mL of H2O) over a 2 h period at 80 °C under stirring. The reaction products were kept under stirring for 24 hours at room temperature and then centrifuged, and the solid product was washed and dried, as done for the plain nAp sample.
For the synthesis of iron-containing hydroxyapatite and in the presence of bio-based substances (nAp–Fe–BBS), 1.00 g of BBS and 5 g of Ca(OH)2 were added to 40 mL of water. The resulting suspension was left under stirring for 24 hours before starting the synthesis. 1.27 g of FeCl2·4H2O and 1.79 g of FeCl3·6H2O were added to a solution of 60 mL of water and 4.44 g of phosphoric acid and placed under stirring at 60 °C for 4 hours until complete dissolution of the salts. Subsequently, this solution was added dropwise to the suspension of Ca(OH)2 and BBS over 2 hours under stirring at a constant temperature of 80 °C and then kept for 24 hours at room temperature. After this step, the precipitate was separated from the supernatant by centrifugation, washed several times with deionized water, and finally dried in an oven at 80 °C for 24 hours. In Fig. 1a, images of the powders of the three samples are shown.
 | ||
| Fig. 1 As-prepared nAp, nAp–Fe and nAp–Fe–BBS photographs (a) and preliminary characterization, N2 adsorption isotherms (b), BJH pore size distribution curves (c) and X-ray diffractograms (d). | ||
A portion of the samples was subjected to UV-C irradiation for 2 hours for sterilization purposes with a lamp emitting UV rays at 253.7 nm and an ultraviolet output of 13.4 W (Sankyo Denki, Japan). The indication “UV” was added to the labels of the so-treated samples.
Another treatment the samples underwent was calcination at 600 °C for 3 hours in a muffle furnace. In this case, the materials were identified as nAp 600, nAp–Fe 600 and nAp–Fe–BBS 600.
2.2 Characterization methods
In order to study the reproducibility of the materials, especially considering the fact that a slightly different type of bio-based substance (BBS instead of CVT230) was used, X-ray diffraction (XRD) measurements and N2 adsorption experiments were performed.The diffractometer used for XRD was a PW3040/60 X'Pert PRO MPD X-ray diffractometer of PANalytical (Malvern, UK), with a Bragg Brentano geometry. Qualitative data processing took place with PANalytical X'pert High Score software and the diffractograms of the samples were compared with those provided in the ICDD-PDF-2 database. An evaluation of the crystalline domain was calculated employing the Scherrer equation relating the size of submicrometre particles in a solid to the broadening of diffraction pattern main peaks and considering k = 0.89 as the shape factor and λ = 0.154056 nm as the wavelength of the X-rays for Cu Kα1 radiation. Full width at half-maximum (FWHM) determination was performed with Origin23 software.
Nitrogen gas-volumetric adsorption experiments for textural property determination were performed at 77 K through an ASAP2020 (Micromeritics, Norcross, GA, USA). Before each measurement, the samples were outgassed overnight at 80 °C at a residual pressure of about 10−2 mbar to guarantee a good cleaning of the sample surface. The specific surface area of the samples was calculated by the Brunauer–Emmett–Teller (BET) model,35 whereas the total pore volume and the pore size distribution were evaluated by applying the Barrett–Joyner–Halenda (BJH) equation36 to the desorption branch of the isotherms, typically employed for mesoporous materials.
ζ-Potential measurements were carried out on a Zetasizer (Malvern Instrument, Malvern, UK). The ζ-potential values were measured using principles of laser Doppler velocimetry and phase analysis light scattering (M3-PALS technique). All the suspensions were prepared by dispersing 10 mg of powder in 20 mL of double distilled water. The pH values were adjusted in a range of 5.5–10 by adding the necessary volume of 0.1 M HCl or 0.1 M NaOH aqueous solutions. The analysis could not be conducted at pH values lower than 5.5, due to the higher solubility of the hydroxyapatite under such conditions.
The vibrational features of the materials were investigated through attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy. The spectra were recorded with a Thermo Nicolet iZ10 spectrometer (Milan, Italy) equipped with a Smart Endurance TM (ZnSe crystal) in the range of 4000–650 cm−1 with 32 scans and 4 cm−1 band resolution.
The thermal behavior of the samples was investigated by thermogravimetric analysis (TGA). For these analyses (TGA 1 Star System of Mettler Toledo, Schwerzenbach, Switzerland), ca. 10–15 mg of samples were heated from 30 °C to 800 °C at a rate of 20 °C min−1 in a 70 mL min−1 N2 flow. The first derivative of TGA (DTG) was utilized to identify the temperature of maximum mass-loss rates.
2.3 Adsorption tests
A previously reported procedure34,37 was slightly modified and applied for the adsorption experiments of inorganic ions in the presence of 200 mg L−1 nanohydroxyapatite samples prepared in this work. Single-ion aqueous solutions of aluminum, nickel, chromium and arsenic (1 × 10−4 M), prepared by concentrated commercial standards Tritisol® in MilliQ® water (Al(NO3)3·9H2O, NiCl2, CrCl3, As2O5), were put in contact at 25 °C with samples in a beaker and left under mechanical stirring at 500 rpm throughout the measurement. During the experiment, the pH, which was set at 7, and temperature were continuously monitored by means of a pH electrode and a thermometer introduced in the beaker. The experiment duration was one hour, sufficient to evaluate the adsorption process from a kinetic point of view. At different times, 10 mL of suspension was withdrawn and filtered with a cellulose filter (0.45 μm, Minisart, Sartorius, Göttingen, Germany) supported on syringes with plungers devoid of rubbery parts (BD Discardit™) to remove the adsorbent powder. After filtration, 10 μL of ultrapure HNO3 (65%, Suprapur®, Merck) was added to each sample and the solutions were stored at 4 °C until further analysis. The ion concentration was determined using an inductively coupled plasma optical emission spectrometer (ICP-OES) Optima 7000 DV (Perkin Elmer, Waltham, MA, USA), equipped with a cross-flow nebulizer, a Scott spray chamber and a double monochromator (prism and Echelle grating). The instrumental conditions were: plasma power 1.3 kW, sample aspiration rate 1.5 mL min−1, argon nebulizer flow 0.8 L min−1, argon auxiliary flow 0.2 L min−1 and argon plasma flow 15 L min−1.Desorption tests of Al and Cr were conducted in ad hoc tests, after 1-hour adsorption. The suspension was, then, centrifuged and the solid was separated and mildly washed with MilliQ® water to remove the metal ions in excess on the surface (but not adsorbed). This material was left to dry. In the desorption process, the spent powder was suspended in acidified water (nitric acid, pH 5.5) for 1 hour, and the desorbed ions were quantified by ICP-OES.
Moreover, to understand the effect of the nanohydroxyapatite leaching, the release of Ca, Fe, Al, Ni and Cr ions was monitored (200 mg L−1 of each sample in MilliQ® water at a circumneutral pH). After 1 h stirring, the powders were separated by centrifugation and the amount of metal ions in the remaining supernatant was determined by ICP-OES.
Nanohydroxyapatite powders (200 mg L−1) were also used to adsorb 5 mg L−1 dye solutions (methyl orange, 85%, Sigma Aldrich and crystal violet, >90%, Sigma Aldrich) adjusted with NaOH to reach neutral pH. The dye bleaching was monitored using a double-beam UV-visible spectrophotometer CARY 100 SCAN (Varian, Palo Alto, CA, USA). A sample quartz cell of 1 cm path length was used. The maximum absorbance of the dyes was monitored to determine the adsorption percentage (464 nm for methyl orange and 590 nm for crystal violet). The adsorption isotherm at 25 °C was constructed using different concentrations of crystal violet (CV) in the range of 0–40 mg L−1 at pH 7, keeping the materials under stirring in the dye solution for 24 hours to ensure equilibration. The Langmuir model was used to describe the dye adsorption from the solution, whose equation is given below (eqn (1)):
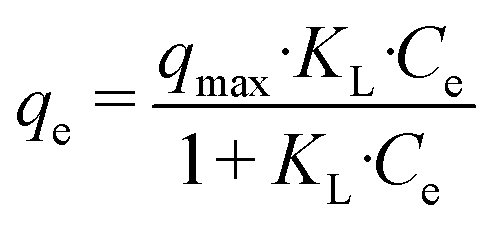 | (1) |
Crystal violet desorption tests were carried out by re-suspending 750 mg L−1 spent powder (after filtration and drying) in distilled water and acidified water (nitric acid, pH 5.5) for 1 hour and 24 hours.
2.4 Antibacterial tests
The antimicrobial activity was evaluated according to the quantitative ASTM E 2149-2013 procedure “Standard test method for determining the antimicrobial activity of immobilized antimicrobial agents under dynamic contact conditions”.38 The bacteria were Staphylococcus aureus ATCC 6538 (Gram-positive) and Escherichia coli ATCC 11229 (Gram-negative).The bacteria were grown in a suitable nutrient broth (buffered peptone water for microbiology, VWR Chemicals) for 24 h at 37 °C. The bacteria concentration was measured with a spectrophotometer (following the absorbance at 475 nm) and diluted into a sterile buffer to obtain a 1.5–3.0 × 105 CFU ml−1 working dilution. This bacterial inoculum was put in contact with the antibacterial agent under shaking at room temperature for 1 hour with a nanohydroxyapatite/inoculum ratio of 1 g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ml. After this time, 1 ml of inoculum was diluted 1000 times and plated in Petri dishes with yeast extract agar (Sigma Aldrich). The Petri dishes were incubated for 24 hours at 37 °C and then, the surviving bacteria colonies were counted and compared to the initial bacteria concentration of the inoculum to calculate the % bacterial reduction using this equation (eqn (2)).
:50 ml. After this time, 1 ml of inoculum was diluted 1000 times and plated in Petri dishes with yeast extract agar (Sigma Aldrich). The Petri dishes were incubated for 24 hours at 37 °C and then, the surviving bacteria colonies were counted and compared to the initial bacteria concentration of the inoculum to calculate the % bacterial reduction using this equation (eqn (2)).
 | (2) |
The materials employed were nAp, nAp–Fe, nAp–Fe–BBS after UV sterilization and the calcined powders.
3. Results and discussion
3.1 Characterization
As regards texture analyses, the main data are reported in Fig. 1b and c and Table 1. The isotherms are of type IV and show a hysteresis loop, albeit rather limited, typical of mesoporous systems (pore diameter between 2 and 50 nm).39| Sample | Specific surface area, BET (m2 g−1) | Cumulative pore volume, BJH (cm3 g−1) |
|---|---|---|
| nAp | 34 | 0.39 |
| nAp–Fe | 83 | 0.41 |
| nAp–Fe–BBS | 90 | 0.21 |
It is observed that the co-precipitation of iron oxide produced particles of reduced size, causing an increase in specific surface area from 34 to 83 m2 g−1. In parallel, the porosity of the samples increased as a result of the presence of interparticle spaces. By also adding the BBS, the surface area seems to have increased further, although not significantly (since the error to be attributed to the model is ±5%) and the total porosity has halved, indicating a system of particles denser than the previous one.
The XRD results for nAp, nAp–Fe and nAp–Fe–BBS in Fig. 1d resembled those reported elsewhere,20 confirming that the incorporation of iron and BBS did not modify the nanohydroxyapatite structure, except for the crystallite size, which resulted in smaller dimensions in the presence of Fe and/or BBS. In fact, as determined by the Scherrer method, applied to the principal reflection of hydroxyapatite, the 20–35 nm crystallites of nAp decreased in size by 30 up to 50% for nAp–Fe and nAp–Fe–BBS.
Such change is also confirmed by BET surface area results (increasing in the order n-Ap < n-Ap–Fe < n-Ap–Fe–BBS), suggesting that crystallite and particle sizes go hand in hand, thus excluding the presence of additional amorphous phases. Among the peaks at 2θ = 35.5°, 30.2° and 62.5° that have been previously attributed to the presence of iron oxides, herein, only the reflection at 35.5° as a single, independent peak is easily detectable. However, the formation of magnetite/maghemite can be found in the magnetic activity of the samples. The signals at 2θ = approximately 13°, 26.5° and 30° visible on the nAp sample are attributable to impurities of other phases of calcium phosphates (i.e., β-tricalcium phosphate and monetite),40,41 which are not found in the modified nanohydroxyapatites, probably since their formation is not favored in the presence of other substances (metallic or organic).
Regarding ζ-potential, it can be observed in Fig. 2 that from pH 5.5, all the samples presented a negatively charged surface that progressively became more negative, with both increasing the pH and adding the different doping compounds (magnetite/maghemite and BBS). This latter evidence is explicable by considering that magnetite/maghemite alone42–44 and BBS-containing materials follow similar trends; in particular, for BBS, this fact is attributable to the wide number of dissociable carboxylic groups present in their macromolecular structure.34
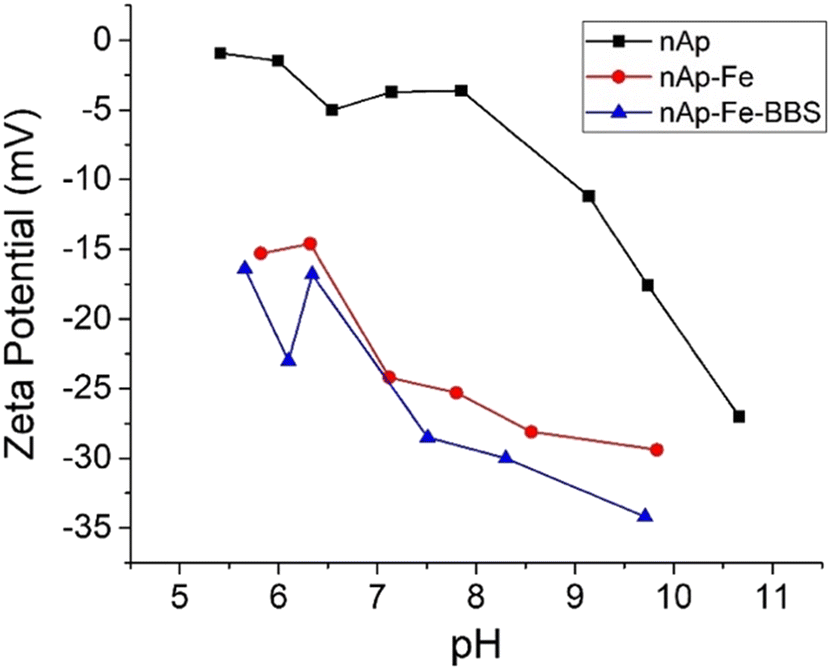 | ||
| Fig. 2 Zeta potential trend as a function of pH values for the different samples of nanohydroxyapatites. | ||
In the infrared spectra of Fig. 3, it is possible to distinguish the fundamental vibration modes and the related signals of the phosphate groups of the apatites (960 and 1022 cm−1), which are sharper for the nAp specimens (Fig. 3a). The residual carbonates derived from the synthesis process and the interaction with atmospheric CO2 are visible at 875, 1415 and 1455 cm−1 due to the substitution of PO43− by CO3−2,45–47 whereas the hydroxyls of physisorbed water typically present on the surface of solids fall under the signals centered at 1640 and 3430 cm−1 (bending and stretching, respectively).48 Only in the nAp spectrum, the peak at 3570 cm−1 was distinguishable (although weak), representing the structural OH ions of the hydroxyapatite lattice,48,49 which was previously correlated to a high crystallinity degree.20 For the Fe-modified nanohydroxyapatites (Fig. 3b), the main characteristic peaks of the iron oxides were not identifiable, since, given their low concentration, they could have been covered by the hydroxyapatite signals. Still, a higher presence of hydroxyl groups has increased the intensity of the bands around 3430 and 1640 cm−1. As already found for other magnetite-containing systems, this event may be ascribed to the formation of iron hydroxyl functional groups in the form of Fe(OH)2, Fe(OH)3 and FeO(OH).50 Following this trend, as a consequence of the addition of BBS (Fig. 3c), more intense absorptions could be found in the same zone, due to the OH groups of phenols, alcohols and carboxyl groups, as well as by the CO groups of carboxyls and carbonyls, abundantly present in BBS.22 Regarding the influence of post-synthesis treatments, on the one hand, the vibrational modes seemed not to be affected by UV exposure (photostability51). On the other hand, the calcination changed different spectral features. First, the region of OH suffered from a remarkable intensity decrease as a result of surface water desorption and, in the case of nAp–Fe–BBS, the combustion of organic substances. Moreover, by looking at the zoomed-in region in Fig. 3d, it is possible to make some considerations about the carbonate-related signals. As mentioned, signals at 875 (out-of-plane bending vibrations), 1415 and 1455 cm−1 (asymmetric stretching vibrations) indicate the substitution of carbonates for phosphate, which defines the B-type carbonates.52,53 Instead, when the substitution occurs at the expense of hydroxides, A-type carbonates are formed and are characterized by the shift of the peaks to 878, 1450 and 1545 cm−1; the relative A- and B-type presence in apatites is known to influence their structural and functional properties.52–55 After calcination at 600 °C, aided by the high temperature and by the fact that the hydroxyl group amount has been evidently reduced, the materials nAp–Fe and nAp–Fe–BBS were progressively transformed into AB-type carbonated hydroxyapatites.52,56,57 For nAp–Fe–BBS, this phenomenon could have been accentuated due to the availability of organic molecules that could have locally created a CO2-richer environment.
 | ||
| Fig. 3 ATR-FTIR spectra of different samples of as-prepared, UV-treated and calcined nAp (a), nAp–Fe (b) and nAp–Fe–BBS (c); in (d), the zoomed-in region of 1900–1200 cm−1 is reported for all the samples. | ||
From Fig. 4, TGA outputs can be examined. First, the as-prepared and UV-subjected samples followed substantially the same trend and this was also verified by DTG peaks (data not shown), confirming the stability to the UV treatment, already seen in the FTIR outcomes. The mass loss related to adsorbed water (centered at 150 °C, according to DTG) for nAp and nAp UV was very limited (<1 wt%), whereas for modified nanohydroxyapatites, the value was comprised between 4 and 8 wt%. The dehydration of hydroxides (380–500 °C) and the elimination of surface carbonate-like groups (500–800 °C) were found to be the subsequent steps of thermal degradation and, for the BBS-added samples, organic matter pyrolysis occurred.20 The steeper phenomenon visible at about 440 °C for nAp(UV) has been correlated to the reaction of two molecules of calcium monohydrogen phosphate, which drove the formation of pyrophosphate and water.58 The DTG peaks present at 670 °C for nAp–Fe(UV) and 705 °C for nAp–Fe-BBS(UV) (broader), corresponding to changes in the slope in the TGA curves, were instead relatable to decarboxylation.58 The residues at 800 °C were approximately 5, 12 and 20 wt% for nAp(UV), nAp–Fe(UV) and nAp–Fe–BBS(UV), respectively. The BBS contribution to the TGA residue is due to the complex composition, which includes the presence of about 30% of inorganic compounds constituted by oxides and carbonates of Ca, Si, Fe, K, Na, Al, and Mg, plus other microelements.22
 | ||
| Fig. 4 TGA curves for the samples prepared in this work and the related UV- and heat-treated ones. | ||
Regarding the calcined samples, the nAp 600 residue was lower than 2 wt%, while nAp–Fe 600 and nAp–Fe–BBS 600 reached 3.5 wt%. If the DTG of nAp 600 was very flat throughout the whole temperature range, for nAp–Fe 600 and nAp–Fe–BBS 600, the peaks related to carbonate decomposition were both centered at 690 °C. For the BBS-containing sample, the phenomenon was broader, probably indicating that multiple events were still subtended to the peak.
3.2 Adsorption tests
As observable in Fig. 5, the materials are especially performing towards Al(III) and Cr(III) since high abatement values were achieved in a short time, but in both cases, the addition of BBS did not lead to any improvement. In contrast, for Ni(II), the addition of BBS brought a doubling of the observed adsorption values, which, however, remained rather limited (maximum reduction shown by the nAp–Fe–BBS sample equal to 30%). A similar trend was observed in the case of As(V), with a maximum reduction shown by the nAp–Fe and nAp–Fe–BBS samples equal to approximately 20%. Al(III) and Cr(III), in general, reached outstanding removal in a few minutes since their adsorption on a negatively charged surface was promoted by their high positive charge and, for aluminum, by small ion dimensions.34,37 Ni(II) adsorption on the BBS-containing sample could be enhanced by the surface texture characteristics (higher specific surface area) and the complexation effect guided by the multiple functionalities of BBS.34 As(V), present mainly as an oxyanion at pH 7,59,60 was not favorably attracted to the powder surface through electrostatic interaction. The explanation for the greater adsorption by BBS can be the same given for nickel, whereas, in the case of nAp–Fe, it stands in the very well-known affinity of iron with arsenic.61 These results can be integrated with the determination of the intrinsic leaching of the different nanohydroxyapatites in water. None of the selected metal adsorbates were released in solution, excluding any competition between leaching and adsorption for the same type of ion. However, for all the samples, a portion of calcium was found in the solution (38 mg g−1 for nAp, 14 mg g−1 for nAp–Fe and 7 mg g−1 for nAp–Fe–BBS). Small amounts of iron were released from nAp–Fe (ca. 1 mg g−1) and from nAp–Fe–BBS (ca. 0.4 mg g−1). These data highlight that, in the presence of BBS, the structure is less subjectable to structural ion leaching, probably due to the BBS complexing effect. | ||
| Fig. 5 Adsorption 1 h kinetics for nAp, nAp–Fe, and nAp–Fe–BBS (200 mg L−1) in contact with solutions of Al(III), Cr(III), Ni(II) and As(V) (1 × 10−4 M) at pH 7. | ||
Regarding the desorption tests of Al and Cr, it was negligible, indicating a strong interaction with the nanohydroxyapatites, as already observed with the irreversible adsorption of Pb(II).20,21
Regarding the adsorption performances on dyes that have been tested here for the first time for these doped systems, methyl orange was not removed from the solution by any of the tested materials, due to the non-advantageous electrostatic interaction between the adsorbent negative surface and the methyl orange anionic molecule. Crystal violet (cationic), instead, was captured, but only by the nAp–Fe–BBS powder (Fig. 6a), reaching the 60% adsorption level in 1 hour and thus confirming the affinity of this dye with BBS.16,23,62Qmax was estimated through isotherm fitting with the Langmuir model (R2 = 0.92) to be 52 mg g−1 (see Fig. 6b), which is in line with the average results of other hydroxyapatite-based systems and, in general, other materials already described in the literature.63–66 Under the tested conditions, the CV desorption was limited to 1–2%.
 | ||
| Fig. 6 (a) Adsorption 1 h kinetics for nAp–Fe–BBS (200 mg L−1) with 5 mg L−1 CV solutions; (b) isotherm at 25 °C for CV adsorption and fitting with the Langmuir model (red line). | ||
3.3 Antibacterial activity
Based on the previous findings of Lamkhao et al.,28 who found a higher antibacterial activity for calcined hydroxyapatites, both the UV and heat-treated series were tested: the outcomes of the antibacterial trials against Staphylococcus aureus and Escherichia coli are reported in Table 2.| Sample | S. aureus bacterial reduction (%) | E. coli bacterial reduction (%) |
|---|---|---|
| nAp UV | 87 | 95 |
| nAp–Fe UV | 91 | 0 |
| nAp–Fe–BBS UV | 14 | 15 |
| nAp 600 | 65 | 95 |
| nAp–Fe 600 | 39 | 31 |
| nAp–Fe–BBS 600 | 0 | 48 |
The antibacterial activities of nanohydroxyapatites have already been proven32 and confirmed herein against both the analyzed bacterial strains. The nano-size of the particles can be itself an important characteristic in destabilizing the bacterial cell membrane and enhancing the penetration ability.30,67,68 Moreover, positive charges (surface Ca2+)69 of nAp can react with the negative charges of the bacterial cell membrane, and also reactive oxygen species (ROS) able to inhibit the growth of bacteria can be formed, resulting from the optical properties of nAp that can capture visible light and generate hydroxyl radicals (·OH).32 Hariani et al.,30 who tested both Staphylococcus aureus and Escherichia coli, attributed the activity of a nanohydroxyapatite produced from snakehead fish bone to the action of the OH group, which can destroy the cell wall and the structure of bacterial proteins through different mechanisms. In the literature, antibacterial properties were also attributed to iron oxide nanoparticles.68
Looking at the results in Table 2 regarding the S. aureus decontamination process, BBS addition caused a significant decrease in antibacterial activity and, for all the materials, calcination negatively affected the performances.
The loss of antibacterial activity for the BBS-containing material could not be attributed only to a matter of surface charge change, which became significantly more negative than nAp (around −25 mV vs. −4 mV at pH 7, Fig. 2). Indeed, it is recognized that the higher the positive charges, the higher the efficiency in puncturing bacteria cell membranes can be obtained,70,71 but, in the present research, nAp–Fe showed a very similar zeta potential trend with respect to nAp–Fe–BBS, without losing its activity. One of the reasons on the basis of nAp–Fe–BBS UV behavior can be related to its microstructure, given the reduction of the crystalline domain and the formation of more agglomerated particles72 (this latter consideration is also valid for nAp 600, nAp–Fe 600 and nAp–Fe–BBS 600, considering the agglomeration as an effect of calcination20).
However, what also seems interesting to investigate is the modification of surface chemical reactivity. Actually, the increment in modified nAp of OH groups, detected by ATR-FTIR and previously by X-ray photoemission spectrometry,20 did not bring any positive effect, leading to the hypothesis of a more important role of calcium ions, which may have been, instead, firstly substituted by Fe2+/Fe3+ and then complexed by BBS, hindering their function. Similarly, the alteration of surface species can be the key to the activity diminishing after calcination. Based on the ATR-FTIR interpretation with the new arrangements of the carbonate species, it is worth taking into account that the exchange of phosphate ions (−3) with carbonate ions (−2) has been reported to lead to the creation of OH− and/or Ca2+ vacancies.31,73 The extent of this process may have been limited after calcination with the partial substitution of B-type with A-type carbonates, whose charge compensation involves the removal of two OH− ions per CO32− moiety.73 Moreover, if OH− ions could have had an effect on the antibacterial properties in nanohydroxyapatites, their lessening after heating was deleterious in the S. aureus reduction. Lastly, as anticipated, BBS, due to their inherent characteristics, brought about inorganic compounds, which, remaining as ashes after organic matter decomposition at 600 °C, could be another factor of property modification.
When the levels of E. coli reduction are examined (Table 2), the scenario changes. Except for nAp, which maintained excellent antimicrobial performance even after calcination, the overall situation seemed inverted: both nAp–Fe UV and nAp–Fe–BBS UV showed an irrelevant activity, whereas calcination enhanced the antibacterial features, although limited. This fact is attributable to the difference in the cell membranes between Gram-negative and Gram-positive bacteria and their susceptibility to the composition and active species of antibacterial agents.70 Briefly, Gram-negative bacteria cell walls are composed of thin peptidoglycan layers, surrounded by an outer membrane containing lipopolysaccharides. Gram-positive bacteria, instead, lack an outer membrane but are surrounded by layers of peptidoglycan that are significantly thicker than those present in Gram-negative bacteria.74 In the case of nAp–Fe UV and nAp–Fe–BBS UV, the most impactful characteristics preventing bacterial reduction could be the strongly negative surface charge that can cause intense charge repulsion and, although somehow contradictory with respect to some literature studies, the relevant presence of OH− species (indeed, nAp and nAp 600 samples with a low OH FTIR signal retained their antimicrobial properties).75 Furthermore, the already discussed substitution of carbonates at the expense of hydroxyls, as a consequence of calcination, resulted in being more convenient.
Albeit all these observations, it is not straightforward to correlate the net variation of antibacterial properties to a single specific parameter, but the importance of the presence and exposure of the ionic species (calcium, carbonates, phosphates, iron, hydroxyls) on the material surface clearly emerges, as previously reported.29,54,76,77 Peccati et al.54 studied the remarkable influence of carbonates on water adsorption features of hydroxyapatites by a computational approach. In another study, Ca- and P-deficiency and the incorporation of carbonate anions into phosphate sites for Ag-doped hydroxyapatites played a role in bactericidal properties.29
4. Conclusions
The nanohydroxyapatite materials synthesized in the presence of iron and BBS showed significant changes with respect to plain nanohydroxyapatite, such as a reduction in the crystalline domain, a greater specific surface area, an increase of negative surface charge, and paramagnetism ascribable to the formation of magnetite or maghemite. The nanohydroxyapatites prepared in this work expressed good adsorption potential for Al(III) and Cr(III), while in the presence of Ni(II) the adsorption was limited, but increased in the presence of BBS, and in the case of arsenic, the addition of iron appeared to have a positive effect on the sorbent properties of hydroxyapatites, although rather low compared to other metals. The capture of dyes has proven to be ineffective by hydroxyapatites, except for nAp–Fe–BBS with the cationic dye crystal violet, towards which it demonstrated a good adsorbent capability (Qmax = 52 mg g−1). In general, the sorption properties of these materials could not be explained with a simple electrostatic model, but the nature of adsorbent–adsorbate interactions of these systems can be influenced by the complexing action of the additives in the nanohydroxyapatite structure, in particular in the presence of BBS. Moreover, the desorption of crystal violet, Al and Cr was not significant. The reduction of Staphylococcus aureus operated by the developed nanohydroxyapatite samples with and without iron was about 90%; therefore, a significant antibacterial ability can be attributed to these materials. However, the addition of BBS or the treatment at a high temperature significantly decreased this capacity. Except for nAp, in the case of Escherichia coli, the performance trend was inverted, due to the diverse interaction of cell walls of different bacterial strains with the antibacterial agents. These results lead to the hypothesis that the presence and exposure of the ionic species (calcium, carbonates, iron, phosphates, hydroxyls) represent key factors for the antibacterial features. In conclusion, Fe- and BBS-modified hydroxyapatites showed good potential as environmentally friendly, tunable, and selective materials for wastewater remediation towards different harmful agents, both chemical and biological.Author contributions
Conceptualization: M. L. Tummino, G. Magnacca, E. Laurenti; investigation: M. L. Tummino, G. Magnacca, M. Malandrino, C. Vineis, M. Rigoletto; writing – draft: M. L. Tummino, E. Laurenti; writing – review: G. Magnacca, M. Malandrino, C. Vineis; visualization: M. L. Tummino.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. Magnacca, M. Malandrino, M. Rigoletto and E. Laurenti acknowledge support from the Project CH4.0 under the Italian MUR program “Dipartimenti di Eccellenza 2023-2027” (CUP: D13C22003520001). The study conducted by M. L. Tummino and C. Vineis was carried out within the MICS (Made in Italy—Circular and Sustainable) Extended Partnership and received funding from the European Union Next-GenerationEU [Piano Nazionale di Ripresa e Resilienza (PNRR)—Missione 4 Componente 2, Investimento 1.3—D.D. 1551.11-10-2022, PE00000004].References
- V. Paterlini, M. Bettinelli, R. Rizzi, A. El Khouri, M. Rossi, G. Della Ventura and F. Capitelli, Crystals, 2020, 10, 806 CrossRef CAS.
- X. Lu, H. Zhang, Y. Guo, Y. Wang, X. Ge, Y. Leng and F. Watari, CrystEngComm, 2011, 13, 3741 RSC.
- C. Combes, S. Cazalbou and C. Rey, Minerals, 2016, 6, 34 CrossRef.
- O. Fufă, E. Andronescu, A. M. Grumezescu and D. Rădulescu, in Nanobiomaterials in Hard Tissue Engineering, Elsevier, 2016, pp. 381–412 Search PubMed.
- Ż. Ciosek, K. Kot, D. Kosik-Bogacka, N. Łanocha-Arendarczyk and I. Rotter, Biomolecules, 2021, 11, 506 CrossRef PubMed.
- R. M. Kavasi, C. C. Coelho, V. Platania, P. A. Quadros and M. Chatzinikolaidou, Nanomaterials, 2021, 11, 1–15 CrossRef PubMed.
- R. Verma, S. R. Mishra, V. Gadore and M. Ahmaruzzaman, Adv. Colloid Interface Sci., 2023, 315, 102890 CrossRef CAS PubMed.
- M. Ibrahim, M. Labaki, J.-M. Giraudon and J.-F. Lamonier, J. Hazard. Mater., 2020, 383, 121139 CrossRef CAS PubMed.
- S. Li, Y. Li, W. Shen, Y. Bai and L. Kong, J. Cleaner Prod., 2022, 380, 134961 CrossRef CAS.
- I. Mobasherpour, E. Salahi and M. Pazouki, J. Saudi Chem. Soc., 2011, 15, 105–112 CrossRef CAS.
- A. Veiga, S. Madureira, J. B. Costa, F. Castro, F. Rocha and A. L. Oliveira, Mater. Adv., 2023, 4, 5453–5478 RSC.
- I. L. Balasooriya, J. Chen, S. M. Korale Gedara, Y. Han and M. N. Wickramaratne, Nanomaterials, 2022, 12, 2324 CrossRef CAS PubMed.
- J. Feng and J. Shi, Zhejiang Daxue Xuebao, Lixueban, 2015, 42, 732–738 Search PubMed.
- M. Ferri, S. Campisi, M. Scavini, C. Evangelisti, P. Carniti and A. Gervasini, Appl. Surf. Sci., 2019, 475, 397–409 CrossRef CAS.
- R. Rial, M. González-durruthy, Z. Liu and J. M. Ruso, Molecules, 2021, 26, 1–22 Search PubMed.
- M. L. Tummino, R. Nisticò, F. Franzoso, A. Bianco Prevot, P. Calza, E. Laurenti, M. C. Paganini, D. Scalarone and G. Magnacca, Molecules, 2021, 26, 3361 CrossRef CAS PubMed.
- M. Elkady, H. Shokry and H. Hamad, Chem. Eng. Technol., 2018, 41, 553–562 CrossRef CAS.
- L. Yang, Z. Wei, W. Zhong, J. Cui and W. Wei, Colloids Surf., A, 2016, 490, 9–21 CrossRef CAS.
- J. Ma, M. Xia, C. Belver, Y. Zhang and F. Wang, J. Cleaner Prod., 2022, 361, 132173 CrossRef CAS.
- D. F. Mercado, G. Magnacca, M. Malandrino, A. Rubert, E. Montoneri, L. Celi, A. Bianco Prevot and M. C. Gonzalez, ACS Appl. Mater. Interfaces, 2014, 6, 3937–3946 CrossRef CAS PubMed.
- D. F. Mercado, A. Rubert, G. Magnacca, M. Malandrino, S. Sapino, P. Caregnato, A. B. Prevot and M. C. Gonzalez, J. Nanosci. Nanotechnol., 2017, 17, 9081–9090 CrossRef CAS.
- A. Bianco Prevot, M. L. Testa, E. Laurenti, M. L. Tummino and G. Magnacca, in Materials Science in Photocatalysis, Elsevier, 2021, pp. 589–601 Search PubMed.
- M. L. Tummino, G. Magnacca, D. Cimino, E. Laurenti and R. Nisticò, Int. J. Mol. Sci., 2020, 21, 550 CrossRef CAS PubMed.
- S. Jang, K. Park, S. Song, H. Lee, S. Park, B. Youn and K. Park, Water, 2021, 13, 3302 CrossRef CAS.
- S. Wadhawan, A. Jain, J. Nayyar and S. K. Mehta, J. Water Process Eng., 2020, 33, 101038 CrossRef.
- M. Ahmaruzzaman and Asian J. Water, Environ. Pollut., 2019, 16, 43–53 Search PubMed.
- S. Seyedmajidi, R. Rajabnia and M. Seyedmajidi, J. Lab. Physicians, 2018, 10, 265–270 CrossRef CAS PubMed.
- S. Lamkhao, M. Phaya, C. Jansakun, N. Chandet, K. Thongkorn, G. Rujijanagul, P. Bangrak and C. Randorn, Sci. Rep., 2019, 9, 1–9 CrossRef CAS PubMed.
- O. Gokcekaya, C. Ergun, T. J. Webster, A. Bahadir, K. Ueda, T. Narushima and T. Nakano, Materials, 2021, 14, 3158 CrossRef CAS PubMed.
- P. L. Hariani, M. Muryati, M. Said and S. Salni, Key Eng. Mater., 2020, 840 KEM, 293–299 Search PubMed.
- J. Kolmas, E. Groszyk and D. Kwiatkowska-Rózycka, BioMed Res. Int., 2014, 178123 Search PubMed.
- H. Abed, N. J. Hameed and E. T. Salim, Mater. Res. Express, 2023, 10, 085301 CrossRef.
- S. Chaiarwut, J. Niyompanich, P. Ekabutr, P. Chuysinuan, P. Pavasant and P. Supaphol, Polym. Bull., 2021, 78, 3543–3559 CrossRef CAS.
- M. L. Tummino, M. L. Testa, M. Malandrino, R. Gamberini, A. Bianco Prevot, G. Magnacca and E. Laurenti, Nanomaterials, 2019, 9, 162 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- M. L. Tummino, V. Tolardo, M. Malandrino, R. Sadraei, G. Magnacca and E. Laurenti, Front. Chem., 2020, 8, 763 CrossRef CAS PubMed.
- ASTM E2149–13 Standard Test Method for Determining the Antimicrobial Activity of Immobilized Antimicrobial Agents Under Dynamic Contact Conditions, https://www.astm.org/e2149-13.html Search PubMed.
- N. S. Zaharudin, E. D. Mohamed Isa, H. Ahmad, M. B. Abdul Rahman and K. Jumbri, J. Saudi Chem. Soc., 2020, 24, 289–302 CrossRef CAS.
- R. Karalkeviciene, E. Raudonyte-Svirbutaviciene, A. Zarkov, J.-C. Yang, A. I. Popov and A. Kareiva, Crystals, 2023, 13, 265 CrossRef CAS.
- T. H. A. Corrêa and J. N. F. Holanda, Mater. Res., 2019, 22(suppl. 1), e20190486 CrossRef.
- S. F. Soares, T. Fernandes, T. Trindade and A. L. Daniel-da-Silva, Molecules, 2019, 24, 1958 CrossRef CAS PubMed.
- O. Domingues, D. Remonatto, L. K. dos Santos, J. P. M. Galán, D. L. Flumignan and A. V. de Paula, Appl. Biochem. Biotechnol., 2022, 194, 5419–5442 CrossRef CAS PubMed.
- Y. Liu, K. Xu and J. Cheng, Bull. Environ. Contam. Toxicol., 2020, 104, 477–483 CrossRef CAS PubMed.
- K. C. Vinoth Kumar, T. Jani Subha, K. G. Ahila, B. Ravindran, S. W. Chang, A. H. Mahmoud, O. B. Mohammed and M. A. Rathi, Saudi J. Biol. Sci., 2021, 28, 840–846 CrossRef CAS PubMed.
- S. L. Bee and Z. A. A. Hamid, Composites, Part B, 2019, 163, 562–573 CrossRef CAS.
- M. K. dos S. Horta, C. Westin, D. N. da Rocha, J. B. de Campos, R. F. M. de Souza, M. S. Aguilar and F. J. Moura, Mater. Res., 2023, 26, e20220466 CrossRef CAS.
- X. Liu, Int. J. Nanomed., 2012, 7, 1239 CrossRef CAS PubMed.
- S. Herradi, B. El Bali, M. Khaldi and M. Lachkar, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 186, 012023 Search PubMed.
- C. Muzenda, O. V. Nkwachukwu and O. A. Arotiba, J. Water Process Eng., 2023, 56, 104370 CrossRef.
- C. S. Cunha, P. J. Castro, S. C. Sousa, R. C. Pullar, D. M. Tobaldi, C. Piccirillo and M. M. Pintado, Int. J. Biol. Macromol., 2020, 159, 1177–1185 CrossRef CAS PubMed.
- H. Madupalli, B. Pavan and M. M. J. Tecklenburg, J. Solid State Chem., 2017, 255, 27–35 CrossRef CAS PubMed.
- M. E. Fleet and X. Liu, Biomaterials, 2007, 28, 916–926 CrossRef CAS PubMed.
- F. Peccati, C. Bernocco, P. Ugliengo and M. Corno, J. Phys. Chem. C, 2018, 122, 3934–3944 CrossRef CAS.
- K. Sugimoto, Y. Zhou, T. G. P. Galindo, R. Kimura and M. Tagaya, Biomimetics, 2023, 8(2), 184 CrossRef CAS PubMed.
- I. R. Gibson and W. Bonfield, J. Biomed. Mater. Res., 2002, 59, 697–708 CrossRef CAS PubMed.
- S. M. Barinov, J. V. Rau, S. N. Cesaro, J. Ďurišin, I. V. Fadeeva, D. Ferro, L. Medvecky and G. Trionfetti, J. Mater. Sci.: Mater. Med., 2006, 17, 597–604 CrossRef CAS PubMed.
- M. R. Senra, R. B. de Lima, D. De H. S. Souza, M. De F. V. Marques and S. N. Monteiro, J. Mater. Res. Technol., 2020, 9, 7190–7200 CrossRef CAS.
- P. Smedley and D. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713–764 CrossRef CAS.
- T. G. F. Souza and V. S. T. Ciminelli, Curr. Opin. Environ. Sci. Health, 2023, 33, 100466 CrossRef.
- M. L. Tummino, R. Nisticò, C. Riedo, D. Fabbri, M. Cerruti and G. Magnacca, Chem. – Eur. J., 2021, 27(2), 660–668 CrossRef CAS PubMed.
- A. Jebli, A. El Amri, R. Hsissou, A. Lebkiri, B. Zarrik, F. Z. Bouhassane, E. Mahdi Hbaiz, E. H. Rifi and A. Lebkiri, J. Taiwan Inst. Chem. Eng., 2023, 149, 105006 CrossRef CAS.
- M. Gomaa and A. W. Danial, J. Biol. Eng., 2023, 17, 1–14 CrossRef PubMed.
- A. Ghedjemis, R. Ayeche, M. Kebaili, A. Benouadah and L. F. Gil, Int. J. Appl. Ceram. Technol., 2022, 19, 2124–2134 CrossRef CAS.
- P. L. Homagai, R. Poudel, S. Poudel and A. Bhattarai, Heliyon, 2022, 8, e09261 CrossRef CAS PubMed.
- G. F. El-Said, G. M. El Zokm, A. El-Sikaily and M. M. Ismail, Environ. Nanotechnol., Monit. Manage., 2024, 21, 100908 CAS.
- S. V. Gudkov, D. E. Burmistrov, D. A. Serov, M. B. Rebezov, A. A. Semenova and A. B. Lisitsyn, Antibiotics, 2021, 10, 1–23 CrossRef PubMed.
- V. Bolis, C. Busco, G. Martra, L. Bertinetti, Y. Sakhno, P. Ugliengo, F. Chiatti, M. Corno and N. Roveri, Philos. Trans. R. Soc., A, 2012, 370, 1313–1336 CrossRef CAS PubMed.
- M. L. Tummino, E. Laurenti, P. Bracco, C. Cecone, V. La Parola, C. Vineis and M. L. Testa, Cellulose, 2023, 30, 7805–7824 CrossRef CAS.
- S. Vihodceva, A. Šutka, M. Sihtmäe, M. Rosenberg, M. Otsus, I. Kurvet, K. Smits, L. Bikse, A. Kahru and K. Kasemets, Nanomaterials, 2021, 11, 652 CrossRef CAS PubMed.
- A. M. Díez-Pascual, Int. J. Mol. Sci., 2020, 21, 3563 CrossRef PubMed.
- O. F. Yasar, W. C. Liao, R. Mathew, Y. Yu, B. Stevensson, Y. Liu, Z. Shen and M. Edén, J. Phys. Chem. C, 2021, 125, 10572–10592 CrossRef CAS.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- Z. Li, J. Ma, J. Ruan and X. Zhuang, Nanoscale Res. Lett., 2019, 14, 195 CrossRef CAS PubMed.
- K. Kandori, S. Mizumoto, S. Toshima, M. Fukusumi and Y. Morisada, J. Phys. Chem. B, 2009, 113, 11016–11022 CrossRef CAS PubMed.
- I. Cacciotti, Int. J. Appl. Ceram. Technol., 2019, 16, 1864–1884 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |