
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Post-polymerization functionalization of aliphatic polycarbonates using click chemistry
Mohsin
Hassan
b,
Gulzar A.
Bhat
 *b and
Donald J.
Darensbourg
*a
*b and
Donald J.
Darensbourg
*a
aDepartment of Chemistry, Texas A&M University, College Station, Texas 77843, USA. E-mail: djdarens@chem.tamu.edu
bCentre for Interdisciplinary Research and Innovations, University of Kashmir, Srinagar, Jammu and Kashmir 190006, India. E-mail: gulzarbhat@uok.edu.in
First published on 26th March 2024
Abstract
Presently there is much interest in the synthesis of degradable, more sustainable polymers from resources that minimize the use of petroleum-based feedstocks. A process receiving much attention related to this challenge is the production of polycarbonates from CO2 and epoxides or cyclic carbonates. These polymeric materials generally lack thermal and mechanical properties as well as economic feasibility to compete with the widely used polyolefins and BPA-derived polycarbonates; nevertheless, because of their degradability and biocompatibility, these CO2-derived polymers can play valuable roles in niche applications. This implementation would be greatly enhanced by the functionalization of these polymers. A wide variety of aliphatic polycarbonates are readily synthesized via the copolymerization of epoxides and carbon dioxide. Nevertheless, these polymeric materials have limited utility, in part because of their lack of functionalities. These shortcomings can be modified by way of post-polymerization processes. Prominent among these modifications is click chemistry. Relevant pathways for these materials are thiol–ene and alkyne–azide processes. Herein, we will discuss the functionalization of polymers produced from the copolymerization of CO2 and epoxides and the ring-opening polymerization of six-membered cyclic carbonates. We will also highlight several of the unique applications these novel materials are used for, particularly in biomedical science.
 Mohsin Hassan | Mohsin Hassan was born in Bandipora, Jammu and Kashmir, India, in 1997. He earned his M.Sc. degree in Chemistry from the University of Kashmir. Currently, he is pursuing his Ph.D. under the supervision of Dr. Gulzar Ahmad Bhat at the University of Kashmir, focusing on sustainable polymer chemistry. His research interests lie in the synthesis and functionalization of CO2-based polycarbonates. |
 Gulzar A. Bhat | Gulzar A. Bhat was born in Ganderbal, Jammu and Kashmir, India in 1986. He received his M.Sc. from University of Kashmir in 2011 and his PhD from Indian Institute of Technology Bombay (IITB) in 2018. Dr Bhat worked as a Postdoctoral Research Fellow in the Department of Chemistry at Texas A&M University, Texas USA under the guidance of Prof. Donald J. Darensbourg from 2018-2021. Since July 2021, he has been working as a Ramanujan Fellow in the Centre for Interdisciplinary Research and Innovations University of Kashmir. His research interests lie at the interface of Inorganic and Polymer Chemistry with strong emphasis on development of copolymers from the copolymerization reactions of CO2, COS and epoxides and their functionalization to achieve desired properties in micellar catalysis, drug delivery and as self-healing materials. |
 Donald J. Darensbourg | Donald J. Darensbourg was born in Baton Rouge, LA in 1941 and received his B.S. and Ph.D. degrees from California State University at Los Angeles and the University of Illinois/Urbana, respectively. Following a nine month period at the Texaco Research Center in Beacon, NY, he was on the faculties of State University of New York at Buffalo from 1969 to 1972 and Tulane University from 1973 to 1982. He has been at Texas A&M University since 1982 where he currently is a Distinguished Professor. Among his current interests are the utilization of CO2 as both a monomer and solvent in copolymerization reactions with oxiranes and oxetanes, and the ring-opening polymerization of renewable monomers such as lactides. |
Introduction
Aliphatic polycarbonates represent a promising class of polymeric biomaterials due to their biocompatibility and controlled biodegradability. Additionally, these polymers can be obtained from a variety of resources, including both natural and renewable. These polycarbonates are synthesized via three main routes: (i) polycondensation of aliphatic diols and diallyl carbonates, (ii) ring-opening polymerization of six-membered cyclic carbonates, or (iii) the copolymerization of 3- or 4-membered cyclic ethers and carbon dioxide. Because of their low glass transition temperatures (Tgs), e.g., polypropylene carbonate has a Tg of 40 °C, and often lack of functionality, these copolymers have inferior mechanical properties. For example, BPA polycarbonate has a high Tg value of 140–150 °C, rendering it useful in a wide variety of commercial applications as compared to some aliphatic polycarbonates.Similarly, aliphatic polycarbonates are unlikely to ever be competitive, both in terms of their physical properties and economic feasibility, with the broadly used polyolefins. Nevertheless, since these aliphatic polycarbonates (aPCs) have the potential to be biocompatible and (bio)degradable,1 there are numerous niche applications which greatly benefit from their use, especially in biomedical science. Despite the advantages that aPCs possess, their lack of functionality can greatly hinder their ability to serve as high value-added materials. In efforts to broaden the usability of these polymeric materials, there is a need for enhancing their functionalities. In general, this is most conveniently achieved using a post-polymerization strategy, that is either thiol–ene or alkyne–azide click chemistry. Herein, we will focus on utilizing these approaches for functionalizing aliphatic polycarbonates derived from carbon dioxide (CO2) and cyclic ethers or six-membered cyclic carbonates.
Recently, due to growing environmental concerns, sustainability and development of eco-friendly products are highly desirable. Anastas and Warner developed the “Twelve Principles of Green Chemistry” to represent the objectives of sustainability in the discipline of chemistry. The principles of green chemistry often center around a number of ideas, including the use of renewable feedstocks, employing safe and ecologically friendly materials, and atom economy.2 As carbon dioxide (CO2) is one of the greenhouse gases emitted in large quantities, its emission must be reduced and it must be stored and/or recycled into chemicals in order to maintain its permissible concentration in the atmosphere. In this regard, catalytic coupling of epoxides with CO2 to produce biodegradable polycarbonates can help in recycling of CO2 emissions, hence representing a viable option for utilizing this abundantly available gas for creating value added products. For example, polyether carbonate polyols are presently used on the industrial scale to be employed in the production of polyurethanes. It has been shown by a life-cycle analysis when compared to the production of conventional polyether polyols that polyols containing 20% by weight of CO2 provide a greenhouse gas (GHG) reduction of 11–19%.3 Polycarbonates have diverse applications and have been extensively used as adhesives, ceramic binders, coatings, and packaging materials and in the synthesis of engineering thermoplastics and resins.1,4 The most widely utilized polycarbonate is produced through condensation reactions between bisphenol A (BPA) and phosgene gas (COCl2) in a biphasic NaOH/DCM system (Scheme 1a).5 However, an alternate route for synthesizing BPA polycarbonates via a melt reaction between BPA and diphenyl carbonate has enhanced the eco-friendly nature of its preparation.6 Notwithstanding, BPA remains being toxic and the cause for many other diseases.7
 | ||
| Scheme 1 Comparison of conventional and sustainable approaches for polycarbonate synthesis. (a) Conventional method of synthesis from bisphenol A (BPA) and phosgene.5 (b) Sustainable synthesis of polycarbonates.8,9 (c) Brief description about the synthesis and functionalization of polycarbonates synthesized via the copolymerization of CO2 and epoxides.15 | ||
Currently, there is not a broad-based replacement for BPA-derived polycarbonates; however, the development of environment friendly and biodegradable polycarbonates from renewable sources is an important strategy to reduce the dependency on petroleum-based resources for the synthesis of other useful polycarbonates. In this direction, an important discovery of the copolymerization of propylene oxide (PO) and CO2 catalyzed by an inefficient heterogeneous catalytic system (ZnEt2/H2O) was reported by Inoue and coworkers in 1969 which paved the way for the synthesis of different polycarbonates via this viable approach (Scheme 1b).8,9 This discovery has motivated researchers across the globe to develop well-defined catalytic systems for selective coupling of epoxides with CO2 to copolymers. Furthermore, this approach generally can be carried out in the absence of a solvent as most epoxides are liquids.10 Although the conversion of CO2 into useful products is a challenging process, it can be overcome by reacting CO2 with high energy molecules like oxiranes for the synthesis of polycarbonates or cyclic carbonates.11,12 During this process, formation of several side products like cyclic carbonates via backbiting or formation of a ether linkage due to successive ring opening may also take place.13 Two important epoxides, PO and cyclohexene oxide (CHO), have been widely investigated for the copolymerization process due to their reactivity and product selectivity. In contrast to BPA-based polycarbonates, these polycarbonates are mainly biodegradable. Although the large level of CO2 emissions can't be compensated by utilizing CO2 as a feedstock for chemical synthesis, this copolymerization approach offers a route to the products obtained from renewable and non-toxic resources. Utilizing CO2 is also important with respect to current environmental concerns and the key advantage is that CO2 is inexpensive in nature. On a commercial scale, companies like Covestro, Saudi Aramco and Empower materials are synthesizing different polyols for polyurethane synthesis based on CO2/epoxide copolymerization (vide supra).14 Despite many advantages, the lack of functionalities has limited the use of polycarbonates in several applications.
Fortunately, these polycarbonate materials can be functionalized via pendant or terminal attachments using click chemistry which is the main aim of this review (Scheme 1c).15 For a comprehensive presentation of the current state of metal- and organo-catalysts for the copolymerization of CO2 and epoxides, along with the mechanistic aspects of these processes, see our recent review.16
Synthesis of functional aliphatic polycarbonates
As alluded to earlier, the availability of aliphatic polycarbonates with specific functionalities is essential in advancing the scope of applications open to these polymeric materials. That is, a variety of techniques, including post-polymerization functionalization, are used to change the mechanical properties, manage biodegradation, and extend the chemical and biological properties of polycarbonates.15 In post-polymerization functionalization, the functional groups are anchored in an orthogonal fashion along the main polymeric backbone, hence increasing the side chain length as well. As physical properties like toughness and glass transition temperature (Tg) are attributed to the main chain, therefore, softening of main chain rigidity may occur due to the lengthening of side chains by post-polymerization modification.17 Typically, functional groups, such as aryl, alkyl, alkene, alkyne, and azide, have been used to categorize the methods for post-polymerization modifications. Particularly, post-polymerization alterations of polycarbonates with alkene functionalities have drawn a lot of attention for the synthesis of improved biomaterials.18 There is a need to summarize the methods and approaches that aid in the synthesis of useful functional aliphatic polycarbonates on a large scale. The most notable for this purpose is the class of precise and tunable bio-orthogonal processes known as click chemistry.The term “click chemistry” was first introduced by K. Barry Sharpless, H C. Kolb and M. G. Finn in 2001 through a review article.19 One can simply understand the “click” term by considering the clicking of two pieces of a seat belt. As they were highly impressed by the nature's way of working with substances, the main aim of Sharpless and coworkers was to join small units through a heteroatom linkage and to describe reactions that enable products with high yields and great selectivity.19,26
This approach gave birth to a desire to harness the efficiency of molecular assembly for various applications, hence revolutionizing molecular engineering. For a reaction to be categorized under this approach, it must be “modular, wide in scope, give very high yield, generate only inoffensive byproducts that can be removed by non-chromatographic methods and be stereospecific (not necessarily enantioselective)”. Also the required characteristics for the process include “simple reaction conditions, readily available starting materials, use of no solvent or a solvent that is benign, and simple product isolation.”19,27 It is crucial to understand that click reactions have strong thermodynamic driving forces generally greater than 20 kcal mol−1.19,28 The success of click chemistry has made a huge impact on the research community, as of now two decades later this approach has won Nobel Prizes for making difficult processes easier.29 Various click chemistry reactions that have been reported are summarized in Scheme 2a.20,21
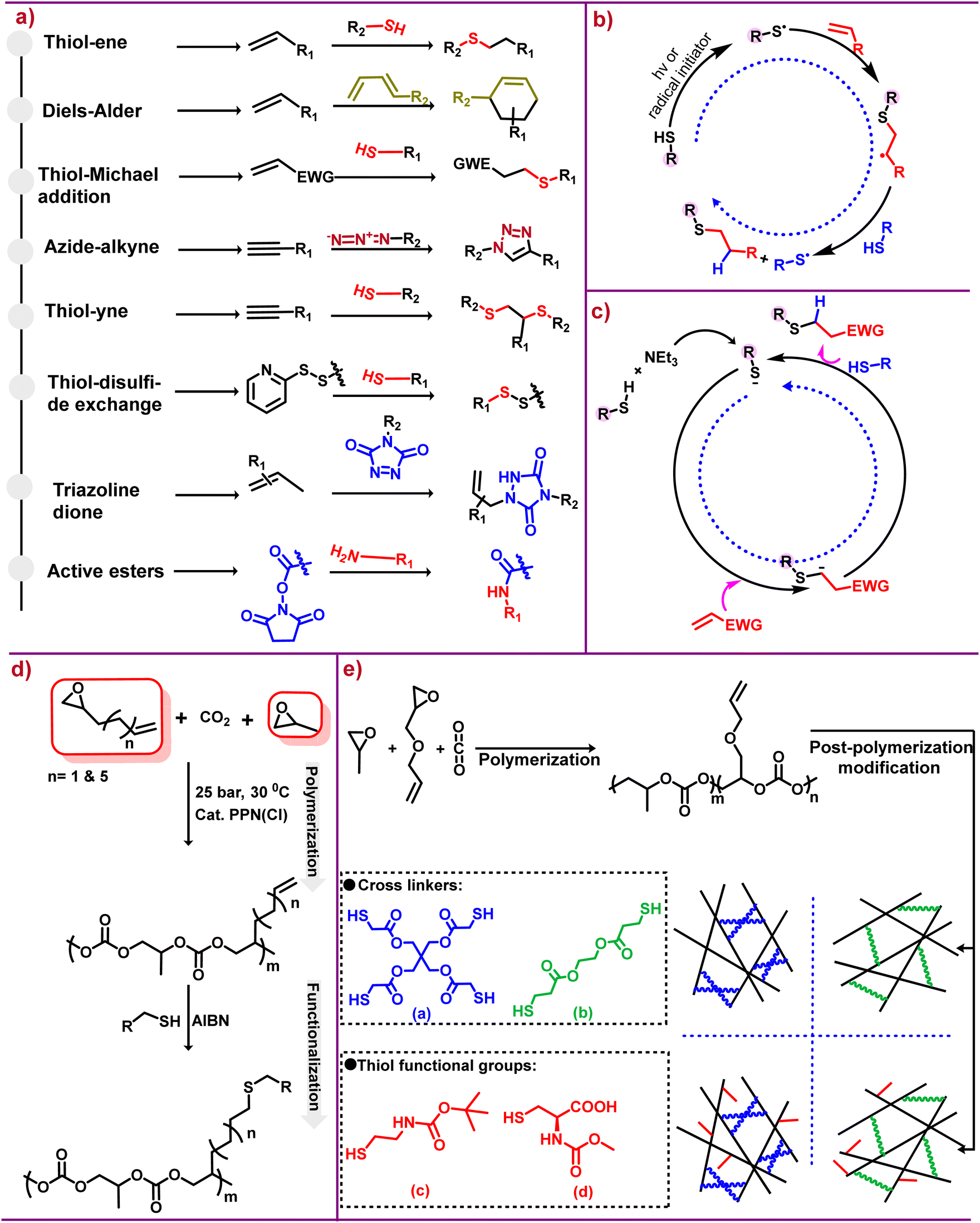 | ||
| Scheme 2 Click chemistry and its applications. (a) Different classes of reactions that can be employed for post-polymerization modification of various polycarbonates.20,21 (b) Mechanistic depiction of the thiol–ene reaction via a radical pathway.22,23 (c) Proposed base-catalyzed mechanism for hydrothiolation.22,23 (d) Terpolymerization followed by post-polymerization modification via thiol–ene click chemistry.24 (e) Polycarbonate synthesis [(a and b) crosslinkers] and [(c and d) thiol functional groups].25 | ||
Of the diverse groups of click reactions depicted in Scheme 2a, thiol–ene click chemistry is the leading technique for functionalizing aliphatic polycarbonates. The word “thiol–ene” is generally used to denote the addition of a thiol to an ene bond; however, thiol–ene is associated with a radical mediated reaction, whereas base/nucleophile-mediated additions of activated enes are described as thiol-Michael addition.30 Although the thiol–ene reaction is simply hydrothiolation of alkene unsaturation, this type of reaction has been widely used for many applications. The key features associated with the thiol–ene reaction are that the process is completed in a fraction of seconds, its dependence on the polarity of the S–H bond, and the S–H bond dissociation energy is less than that of the corresponding O–H bond.31
As stated above, the reaction between thiols and enes proceeds via a radical mechanism called thiol–ene reactions, as shown in Scheme 2b, or through an anionic chain called a thiol-Michael addition, as shown in Scheme 2c, under mild basic or nucleophilic conditions. However, in contrast to the traditional thiol-Michael addition, the use of a weak base like triethylamine (NEt3) is enough for catalysis.22,23 Thiol–ene click reactions, thermally or photochemically induced, occur through an anti-Markovnikov path32 and the rate of free radical thiol–ene chemistry depends upon the structure of the thiol and ene groups.33 The rate dependency on structure is due to steric and electronic effects; additionally the presence of different functional groups can also effect the rate of reaction because of inhibiting or favouring the radical formation.34
In the case of the alkyne–azide click chemistry, the reaction between an alkyne and an azide, generally catalyzed by Cu(I), results in the formation of a five-membered heteroatom ring.35 The widespread employment of this reaction confirms the fact that it fulfills many of the prerequisite conditions of click chemistry. That is, various substituted alkynes and azides are readily available and many others can be synthesized with attached functional groups.36 Nevertheless, due to high reaction yields, readily available materials as well as following the principles of green chemistry such as energy efficiency and use of solvents,33,37 thiol–ene click chemistry is the method of choice for functionalizing aliphatic polycarbonates.38–40 Although post-polymerization modifications result in desired functional group attachments, the addition of one more step for targeted application needs specific requirements like more time and energy. Moreover, each additional step has the potential for the introduction of undesired results. Also to get the preferable product, additional characterization and purification have to be done, therefore adding economic implication to the process. However economic implications can be justified by the utility of the desired functional products.
Thiol–ene click chemistry: an efficient method for functionalization of aliphatic polycarbonates
Post-polymerization functionalization provides an efficient synthetic route to access useful aPCs via a single scaffold. It also enables the addition of groups that if present in monomers will be detrimental in the polymerization process. Although there are several reports that have compiled many aspects of functionalized polycarbonates,14,15,18,41 in this section of the review we will summarize the most recent developments concerning the synthesis of functionalized aPCs via thiol–ene click chemistry. Importantly, in the thiol–ene click approach, pendant alkene groups are the main targets for modification, and therefore no protection/deprotection is needed to provide functionalization. This is true whether aPCs are obtained via coupling of CO2 and epoxides or ring opening of cyclic carbonates. The reaction conditions for this type of post-polymerization process generally involve radical initiators like azobisisobutyronitrile (AIBN), followed by refluxing at appropriate temperatures. A UV light source can also be used for this type of post-polymerization modification.The advantageous features of CO2 and epoxide copolymerization provide a facile method for developing platforms for the synthesis of various active functional polycarbonates. In this regard, Frey and coworkers terpolymerized CO2 with PO and 1,2-epoxy-5-hexene or 1,2-epoxy-9-decene in various feed ratios to provide terpolymers which were functionalized with thiols, mercapto ethanol, thioacetic acid and thioglycolic acid (Scheme 2d).24 This approach paved the way for synthesizing numerous amphiphilic polycarbonates. For example, we synthesized copolymers from 2-vinyl oxirane (VIO) and CO2, as well as terpolymers VIO/PO/CO2. Both were functionalized by thiol–ene click reactions with 2-mercaptoethanol and thioglycolic acid, followed by modification with NH4OH and L-aspartic acid anhydride hydrochloride to yield water soluble polycarbonates.44
Unlike the terpolymer prepared from PO/VIO and CO2, where PO is more reactive than VIO, leading to a more tapered polymer, the terpolymer produced from PO/allylglycidyl ether/CO2 has a random distribution of unsaturated side chains. These terpolymers were subjected to cross linking, as shown in Scheme 2e, using thiol–ene click chemistry.25 Recently, polyols produced from PO/AGE/CO2 were functionalized with mercaptopropionic acid, which afforded polycarbonates with high toughness and tensile strength for synthesizing waterborne polyurethanes.45
This methodology for the schematic synthesis of functional aPCs has provided for the first time polymeric nanoparticles derived from the coupling of CO2/PO/AGE using a (salen) CoTFA/PPNTFA (TFA = trifluoroacetate) binary catalyst system (Scheme 3a).42 In this study, post-polymerization alteration of these triblock polymers with thiols yielded amphiphilic polycarbonates with negative or positive charges, which exhibited self-assembly into spherical nanostructures when added to deionized water. Considering the importance of amphiphilic macromolecules, this research group synthesized another triblock copolymer, poly(phosphate-b-carbonate-phosphate) (PPE-b-PPC-PPE), in the presence of an organocatalyst DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) to afford self-assembled micellar structures in water (Scheme 3b).43 Additionally, this methodology was employed for the synthesis of degradable amphiphilic aPC; following this, the conjugation with gadolinium results in polymeric micelles with efficient magnetic resonance imaging properties for cancer diagnosis and treatement.50
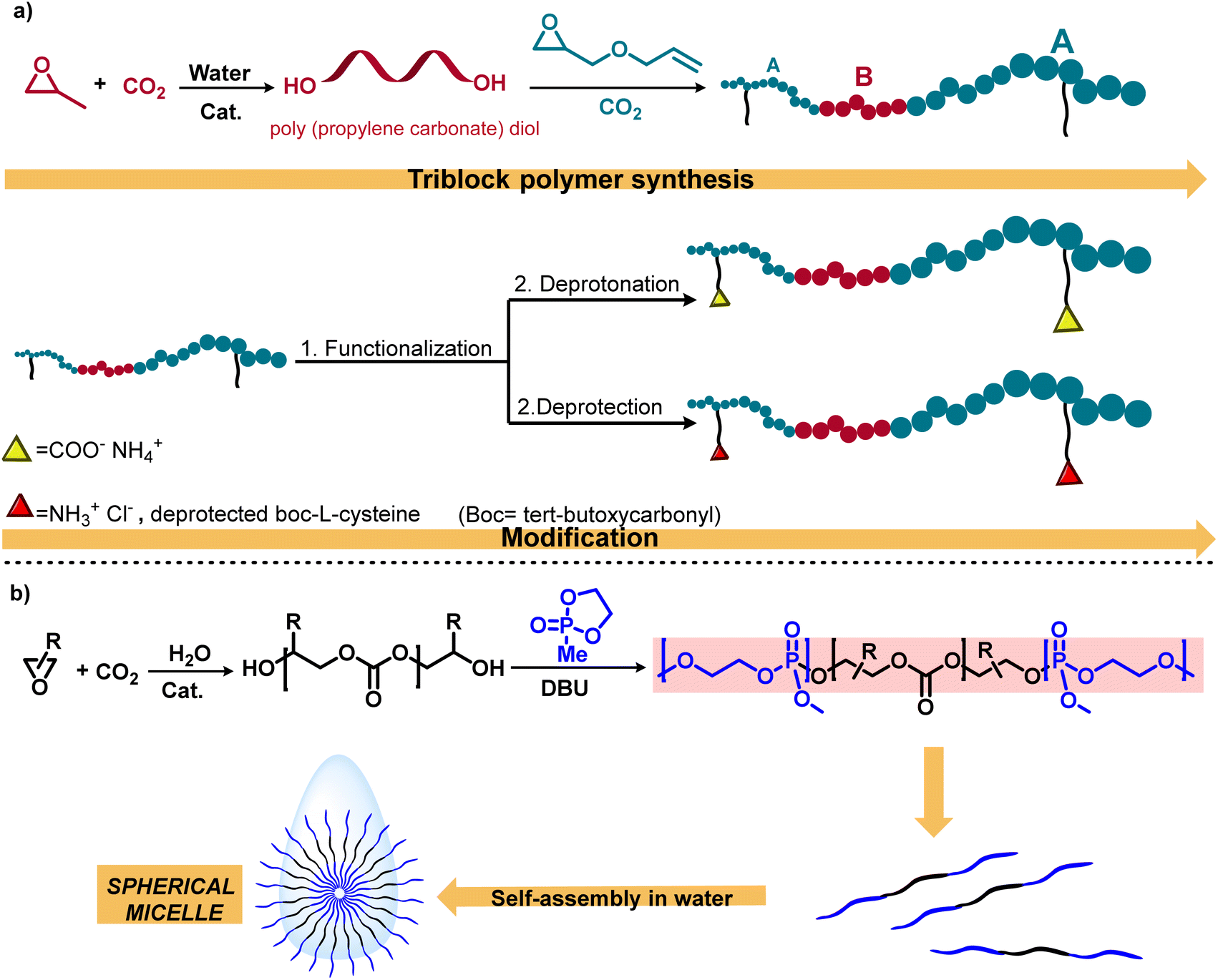 | ||
| Scheme 3 Sequential approach for the synthesis and functionalization of polycarbonates. (a) Water-mediated polycarbonate synthesis and functionalization to achieve amphiphilic polycarbonates.42 (b) Illustration of the mechanistic pathway including metal-catalyzed CO2/epoxide copolymerization and organocatalytic ring-opening polymerization.43 | ||
Subsequent to the successful synthesis of amphiphilic aPCs utilizing chain transfer agents, it became apparent that we could add functionality to the chain transfer agent itself.51,52 Indeed, this behavior was seen upon embedding (bpy) or (bpy)Re(CO)3Br in the hydrophobic part of the backbone chain of an (AGE)(PO)(CO2) triblock polymer employing (2,2′-bipyridine-5,5′-dicarboxylic acid) or its rhenium complex as a CTA prior to its modification with HOOC(CH2)SH via thiol–ene click chemistry and deprotonation with NH4OH (Scheme 4a).46 These anionic amphiphilic polycarbonates containing (bpy)Re(CO)3Br in the backbone chain were shown to self-assemble in water to afford uniform and spherical nanostructures. The rhenium-based metallosurfactant was demonstrated to act as an efficient photocatalyst in the aqueous medium for the selective reduction of CO2 to CO (Scheme 4b).47 In addition to utilizing water as the solvent, the advantages of having the catalyst situated in the hydrophobic nanoreactor over the analogues catalyst in organic solvents are: dimerization of the metal catalyst is retarded by the polymeric backbone; the hydrophobicity of the catalyst avoids photocatalysis of H2O; enhanced solubility of CO2. Other metallosurfactants which have been synthesized in an analogous manner contain a terpyridine ligand for the binding of various metals, as well as bipyridine palladium complexes for carbon–carbon bond forming processes (Scheme 4c).48,49
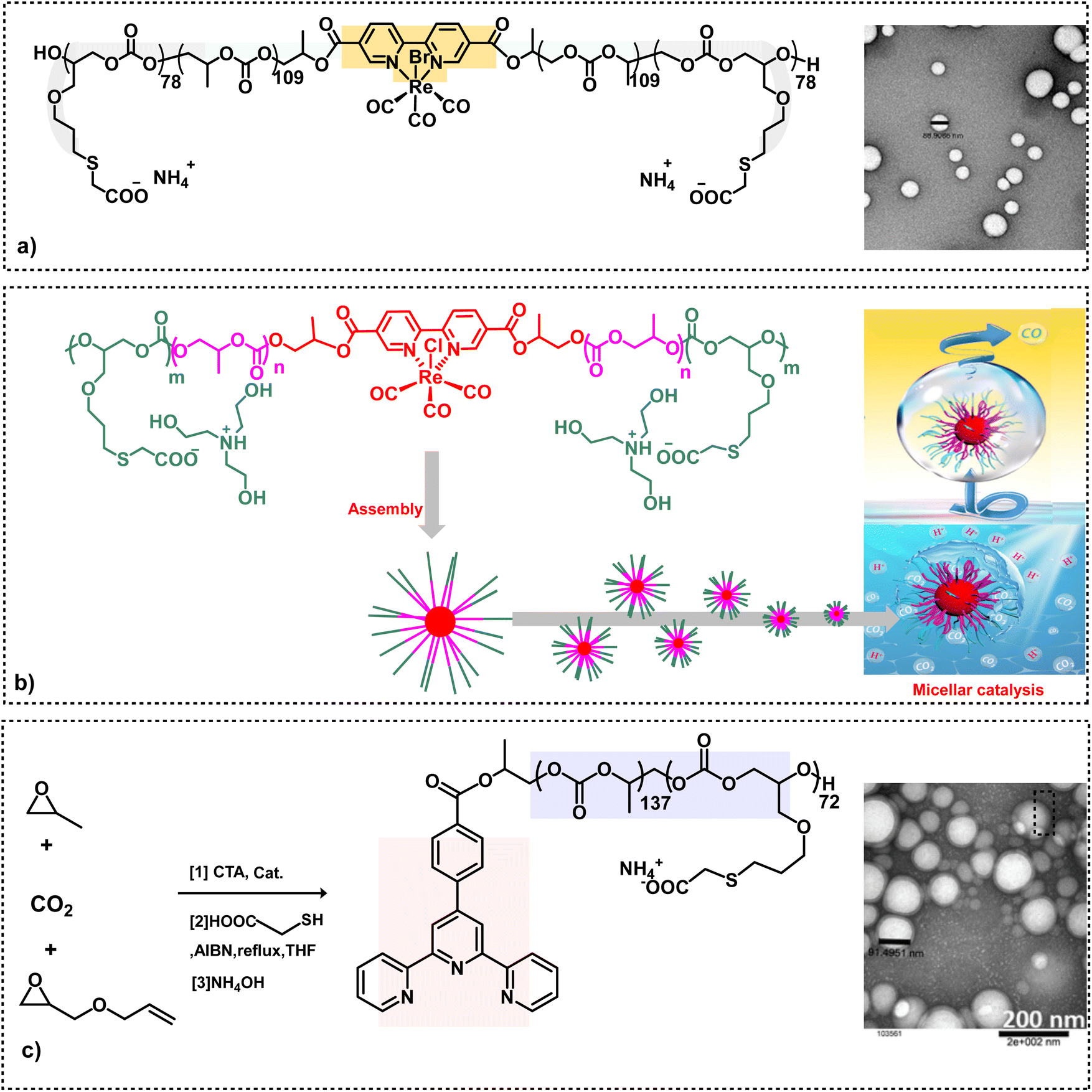 | ||
| Scheme 4 Amphiphilic polycarbonate synthesis. (a) On the left, triblock amphiphilic polymer derived from the copolymerization of CO2 and epoxides containing a bipyridine rhenium complex in its backbone. On the right, TEM image of anionic nano micelles with good uniformity.46 (b) Synthetic route to Re(dcbpy)-based amphiphilic micellar catalysts used for CO2 to reduction in water.47 (c) Synthesis of a triblock polymer attached with a chelating ligand, followed by functionalization. The TEM image shows anionic nanoparticles.48,49 | ||
Derivatives of the commonly employed cyclohexene oxide (CHO) such as vinyl-cyclohexene oxide (VCHO), cyclohexadiene oxide (CHDO), and limonene oxide (LO) have been frequently used for the synthesis of functional polycarbonates. Parenthetically, it should be noted here that these alicyclic epoxides are very selective for copolymer formation from the coupling of CO2 and epoxides. This is due to the higher activation barrier for cyclic carbonate production,53 as well as more favorable thermodynamic stability of the copolymer vs. cyclic carbonates.54,55 In this regard, copolymers from VCHO and CO2 were synthesized and modified employing thiol–ene click chemistry with β-mercaptoethanol to provide side chains with –OH end groups. In a sequential process, ε-caprolactone was grafted onto these –OH groups via ring-opening polymerization to afford degradable well-defined brush polymers.56 In a related alternative process, brush polycarbonates were prepared from the copolymerization of VCHO and CO2 using an organocatalyst, followed by thiol–ene click reactions with mercaptopropionic acid, with subsequent partial deprotonation of the acid side chains to furnish carboxylates for initiating the copolymerization of PO and CO2.57
Use of the epoxide monomer, 1,2-epoxy-4-cyclohexene (1,4-CHDO), for copolymerization with CO2 afforded a completely water-soluble polycarbonate following post-polymerization functionalization using thiol–ene click chemistry with thioglycolic acid and deprotonation, Scheme 5a.58 The glass transition temperature (Tg) of the initially produced polymer was found to be 123 °C or 7 °C higher than that of its saturated analog. Significantly, the 1,4-cyclohexadiene monomer precursor is available from renewable resources.59 The related epoxide, 1,2-epoxy-3-cyclohexene (1,3-CHDO), was shown to be more reactive with CO2 to produce a copolymer with a Tg of 106 °C, much lower than its isomeric counterpart.60 Additionally, the enhanced activity of 1,3-CHDO allows for its terpolymerization with PO and CO2 to afford random terpolymers with a PO to 1,3-CHDO content of 0.81 from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 feed ratio. Under these conditions, a terpolymer with a Tg near 70 °C was produced, which can be further tuned for post-polymerization modifications for a variety of applications. In separate studies, Sugimoto and coworkers have modified in post-polymerization processes the cyclohexene rings of copolymers derived from 1,4-CHDO and CO2 with –Br, –Cl, or –OH groups.17 Other accounts of the synthesis of a terpolymer based on PO/VCHO/CO2 have been reported which provide polymeric materials, which can be functionalized by thiol–ene click chemistry.66
:1 feed ratio. Under these conditions, a terpolymer with a Tg near 70 °C was produced, which can be further tuned for post-polymerization modifications for a variety of applications. In separate studies, Sugimoto and coworkers have modified in post-polymerization processes the cyclohexene rings of copolymers derived from 1,4-CHDO and CO2 with –Br, –Cl, or –OH groups.17 Other accounts of the synthesis of a terpolymer based on PO/VCHO/CO2 have been reported which provide polymeric materials, which can be functionalized by thiol–ene click chemistry.66
 | ||
| Scheme 5 Functionalization by thiol–ene click chemistry. (a) Cyclohexadiene oxide as a biological source for functional polycarbonate synthesis.58 (b) Preparation and functionalization of cationic poly(vinylcyclohexene carbonates) via the copolymerization of VCHO and CO2, also the polycarbonate backbone was blended with polyacrylonitrile (PAN) to form nanofiber meshes (NFMs) via electrospinning.61 (c) Schematic illustration of the formation of CPCHCs/siRNA nanoparticles from functional polycarbonates.62 (d) General approach for synthesizing limonene oxide-based functional and cross-linked polycarbonates. (e) Functionalization of a copolymer with different mercaptans.63 (f) ref. 63, (g) ref. 64, and (h) ref. 65 Special monomers for polycarbonate synthesis. (i) MAC and AOMEC-based functional and cross-linked polycarbonates derived from the ring-opening polymerization of these monomers. (j) Allyl bearing eight-membered cyclic carbonates obtained via a two-step synthesis approach initially, followed by the ring-opening homopolymerization of an allyl bearing eight-membered cyclic carbonate and functionalization via a radical thiol–ene approach.37 | ||
With the aim of synthesizing biodegradable polymeric surfactants, functional poly(cyclohexene carbonate)s have been obtained via thiol–ene click reactions following the copolymerization of VCHO or VCHO/CHO and CO2.61,67 That is, 2-(diethylamino)ethanethiol hydrochloride (DEAET) was anchored onto these polycarbonates by way of a UV-radiated thiol–ene click mechanism with subsequent conversion into nanoparticles via a mini-emulsion interfacial cross-linking technique.67 In this manner, two different DEAET functionalized polycarbonates were examined for their antimicrobial and antitumor activities. The DEAET-modified cationic poly(vinylcyclohexene carbonate)s were found to provide efficient antibacterial activity against Staphylococcus aureus. In order to have additional biomedical applications, the cationic copolymer having a tertiary amine density of 43% relative to the backbone was blended with polyacrylonitrile (PAN) to form nanofiber meshes via electrospinning (Scheme 5b).61 Other related studies utilized terpolymers derived from CHO/VCHO/CO2 functionalized with DEAET to various extent to treat osteosarcoma by acting as efficient synthetic gene vectors. The expression of the polo-like kinase 1 (PLK1) gene in osteosarcoma cells was significantly suppressed when employing polycarbonate/siRNA nanoparticles for treatment. Among the different cationic polycarbonates, ones with 60% amine moieties showed the best results (Scheme 5c).62
Since the development of an effective process for the copolymerization of limonene oxide (LO), a bio-mass-derived monomer and CO2 by Coates and coworkers,68 significant progress has been achieved in the post-polymerization functionalization of this polymeric material.69 Bio-based poly(limonene carbonate)s have been functionalized to provide antibacterial polymers, heat processing-grade polymeric materials, and seawater soluble polymers.70 In these cases, functionalization by thiol–ene click chemistry with 2-mercaptothiol or mercaptoacetic acid enhances hydrophilicity; whereas, addition of butyl-3-mercaptopropionate followed by curing converts the polycarbonate into an elastic rubbery material and treatment with 2-(diethylamino) ethanol, followed by quaternization with benzyl bromide forms a polycarbonate with antibacterial properties. Poly(limonene-8–9-oxide carbonate) has been synthesized via chemo selective coupling of LO and CO2, and was modified with various nucleophiles, including thiols by way of epoxide ring opening.71 Cross-linked polycarbonates derived from renewable resources have received much interest. In this regard, thermoset materials have been synthesized via a thiol–ene approach based on poly(limonene carbonate)s, such as the series of terpolymers obtained from LO/CHO/CO2 with variable limonene carbonate contents which provide polymers with improved thermal properties.72,73 Recently, poly(limonene carbonate) cured with trifunctional thiols afforded poly(thioether-co-carbonate) networks to act as important bio-based coatings.74 In general, a sustainable synthetic pathway for limonene oxide-based functional and cross-linked polycarbonates is shown in Scheme 5d.
Correspondingly, it is possible to synthesize functional polycarbonates via the ring-opening polymerization of 6- or 8-membered cyclic carbonates. This can be achieved either by first synthesizing functional cyclic carbonates, often an onerous task, or by post-polymerization by thiol–ene click chemistry. For example, the cyclic carbonate monomer 5-methyl-5-allyloxy-carbonyl-1,3-dioxan-2-one (MAC) (Scheme 5e and f) and its derivatives are frequently used monomers because of the versatile nature of this cyclic carbonate as demonstrated by Yue et al.63 This pathway was employed by Jing and coworkers to prepare the copolymer poly(LA-co-MAC) which was functionalized by grafting NHS-activated folic acid to the poly(ester carbonate) by thiol–ene click chemistry.75 In another instance, using 2-allyloxymethyl-2-ethyl trimethylene carbonate (AOMEC), Olsen et al. have functionalized poly(AOMEC) via thiol–ene click chemistry using 1-dodecanethiol (Scheme 5g).64 Similarly, using (AOMEC) derived from the easily accessible and cost-efficient precursor trimethylolpropane allyl ether diol (TMAD), polycarbonates with pendant allyl groups were synthesized by way of ring-opening polymerization. The resulting polycarbonates were functionalized using 1-dodecanethiol, 3-mercaptopropionic acid, and benzyl mercaptan to afford polymers with tunable thermal responses.76 The development of a prodrug containing doxorubicin (DOX) through hydrazone bonds based on poly(5-methyl-5-allyloxycarbonyl-1,3-dioxan-2-one)-graft-12-acryloyloxy dodecyl phosphorylcholine (PMAC-graft-ADPC) was obtained by way of ROP of MAC. This prodrug exhibited micellar assembly in aqueous solution and was shown to release DOX in cancer cells. Moreover, this prodrug is biodegradable and can be eliminated from the body following drug release.77 Similarly, 3,4-dihydroxyphenylalanin (DOPA), the naturally occurring amino acid, was anchored on a MAC-based polycarbonate via thiol–ene click chemistry and shown to form self-healing gels in the presence of Fe(III).78 Here, initially pMAC was synthesized, followed by the synthesis of DOPA-thiol, and at the end, DOPA functionalization of the initially synthesized pMAC was obtained via UV initiated thiol–ene coupling between the allyl moiety of the polymer and the thiol group of DOPA-thiol. In other studies, Dove and coworkers have used organocatalysts for the ring-opening polymerization of MAC to provide polycarbonates with narrow molecular weight distributions and high end-group fidelity, with subsequent functionalization of the allyl groups by the radical addition of 1-dodecanethiol.79 Additionally, this group has synthesized a new furan-protected maleimide-containing six-membered cyclic carbonate for ROP to afford homopolymers and copolymers with L-lactide.80 This was followed by activation of the maleimide groups in these polymers via a retro Diels–Alder reaction to provide pendant maleimide moieties for further modification using thiol–ene click chemistry with the thiols, 1-hexanethiol and 6-(ferrocenyl)hexanediol. The latter thiol made accessible a readily discernible NMR probe.
Dove's research group has also synthesized triblock polymers derived from the ROP of MAC and L-lactide, followed by reversible addition–fragmentation chain transfer (RAFT) polymerization of tetrahydropyran.81 This was accomplished by first ROP of a small quantity of MAC (degree of polymerization (DP) = 4) using an organocatalyst and the dual-headed initiator dodecyl 4-(hydroxymethyl) benzyl carbonotrithioate and L-lactide (DP = 32) to provide the diblock polymer PLLA-b-PMAC. Tetrahydropyran acrylate (THPA) was subsequently polymerized from the macro initiator PLLA-b-PMAC by a RAFT polymerization process (DP = 258) to afford the triblock polymer PLLA32-b-PMAC4-b-PTHPA258. Following deprotection of the THPA block, crystallization-driven self-assembly leads to the development of cylindrical micelles where the inner hydrophobic MAC core was shown to undergo click chemistry with the thiols, benzyl mercaptan and 6-(ferrocenyl)hexanethiol. This approach provides a possible novel means for drug delivery. Another synthesis of an amphiphilic polymer utilizing thiol–ene click chemistry for possible drug delivery has been reported by Kuang, et al.82 That is, these researchers have synthesized a mPEG-b-P(LA-co-MAC) polymer backbone, subsequently adding 3-mercapto-1,2-propane diol using click chemistry. The nucleobase, thymine, was grafted onto the hydrophobic segment of these amphiphilic polymers, providing nanoparticles in water, thereby allowing hydrogen bonding of a modified adenine group in the hydrophobic core. In another instance, the polycarbonate synthesized from the cyclic carbonate monomer having pendant unsaturation procured from methyl undec-10-enoate was modified using a cinnamate thiol (Scheme 5h).65 Thus as a result of the ring-opening polymerization of MAC and AOMEC, these cyclic monomers have been used for synthesizing functional and cross-linked polycarbonates, as shown in Scheme 5i. Biologically active functional polycarbonates have also been synthesized via ring opening of cyclic carbonate monomers; these monomers were initially synthesized using 1,1′-carbonyldiimidazole as a key reagent, hence this approach also presents a cost-efficient route for synthesizing customized carbonate monomers.83 Due to the unique properties of boronic acid polymers, access to facile and well-defined boronic acid functional polycarbonates has been presented by utilizing 6-membered cyclic carbonate monomers and by taking advantage of post-polymerization modification.84 Furthermore, a distinctive approach for post-polymerization via transesterification between pentafluorophenyl ester functional polycarbonates and different alcohols has been achieved under milder conditions to afford functional polycarbonates.85
In a recent and topical publication, an amphiphilic sulfonated polycarbonate was synthesized which rapidly inactivates SARS-CoV-2.86 This was achieved utilizing two functional cyclic carbonate monomers derived from 5-methyl-5-carboxyl-1,3-dioxane-2-one (MTC-OH), where one was functionalized with a protected sulfonate prepared via thiol–ene click chemistry and the other with an n-butyl substituent. Several polymers were prepared with varying stoichiometries of hydrophobic n-butyl segments and once deprotected hydrophilic sulfonate pendents, with the copolymer containing 30% sulfonate content showing the highest antiviral activity for SARS-CoV-2 (ancestral wild-type and the delta variant). In addition, this polymer exhibited the least cytotoxicity against mammalian cells.
In contrast to six-membered cyclic carbonates, Alexander and coworkers have employed cyclic eight-membered carbonate monomers to synthesize several functionalized polycarbonates by way of thiol–ene click chemistry (Scheme 5j).37 To broaden the scope of CO2 utilization and to address different concerns, first one-pot synthesis of 7- and 8-menbered cyclic carbonates was performed under ambient conditions using CO2, diols, tosyl chloride and a mild base (triethylamine).87 Concomitant to this, Hedrick and coworkers also reported the synthesis of 6- and 8-membered functional cyclic carbonates followed by ring-opening polymerization to synthesize diverse functional polycarbonates.88 Moreover, considering the sustainability, an innovative strategy was developed involving the depolymerization reaction of a bisphenol A-based polycarbonate in the presence of an organocatalyst along with functional diols to achieve functional 6-membered cyclic carbonate monomers.89
In order to enhance the properties of solid-state batteries for next-generation use, polymers with a specific combination of mechanical, chemical and electrochemical properties could help to overcome the challenges associated with these batteries. In this regard, a block polymer with hard–soft–hard blocks was synthesized. Here, the soft block represents poly(ethylene oxide) for boosting the ionic conductivity and the hard block represents poly(4-vinyl cyclohexene oxide carbonate) for improving oxidative stability and providing mechanical strength. Moreover, the pendent unsaturation in the hard block was functionalized with phosphonic acid via a thiol–ene click chemistry approach for adhesive properties.90
Regarding elastomers, the triblock polymer poly(vinyl-cyclohexene carbonate-b-decalactone-b-vinyl-cyclohexene carbonate) was synthesized in a one pot process. This was followed by UV-initiated attachment of 3-mercaptopropionic acid via thiol–ene click chemistry and coordination with metal centres to afford thermoplastic elastomers.91 Another type of elastomer was synthesized by incorporating copper–thioether coordination within a polycarbonate framework. This was achieved through the copolymerization of cyclohexene oxide, vinyl cyclohexene oxide, and CO2, followed by the functionalization of unsaturated pendant groups using hexanethiol and dodecylthiol via thiol–ene click chemistry. The subsequent introduction of copper enables dynamic, reversible coordination with sulfur, thereby converting the rigid polymer into an elastomeric material.92
In addition to all of these post-polymerization modifications, thiol–ene click chemistry has been utilized for cross-linking various cyclic carbonate-based polycarbonates providing diverse applications such as nanosponges, hydrogels, 3D printed porous materials and elastic polymers.93–96
Functionalization of polycarbonates via azide–alkyne cycloaddition and 1,2,4-triazoline-3,5-dione (TAD)
Although there is much literature utilizing azide–alkyne cycloaddition (AAC) in polymer chemistry, herein, we will solely consider it as an efficient tool for functionalizing polycarbonates. This methodology, in the presence of a Cu(I) catalyst to afford a 1,4-adduct via a click reaction under milder reaction conditions, has expanded the functionalization pathways for polycarbonates. In the absence of a catalyst, this process requires elevated temperatures and leads to a lack of regioselectivity.97 This was first illustrated by Sharpless et al. in 2002 using Cu(I) as a catalyst. The mechanistic details for the reaction can be explained as the catalyst generally (Cu(I)) reacts with a terminal alkyne to form a copper–acetylidine complex. Now the azide moiety coordinates with the copper complex, followed by 1,3-dipolar cycloaddition between the terminal alkyne and the azide group, resulting in the formation of a five-membered ring (1,2,3-triazoline).98 Since that time, other metal catalysts have been employed in this type of click chemistry, including Ru(II) and Ag(I).99 Both alkyne and azide containing polycarbonates have been synthesized, e.g., glycidyl propargyl ether can be utilized in the synthesis of alkyne bearing polycarbonates, whereas cyclic carbonate monomers and polycarbonates can possess azide groups suitable for alkyne–azide click chemistry.100–104 Functional biodegradable aliphatic polycarbonates have been synthesized from 5-methyl-5-propargyloxycarbonyl-1,3-dioxan-2-one (MPC) and L-lactide, resulting in a diblock polymer with orthogonal acetylene moieties for further anchoring with 2-azidoethyl β-D-glucopyranoside or 2-azidoethyl β-lactoside using a Cu(I) catalyst.15,105 Because of its stability and ease of synthesis, MPC has become an efficient monomer for providing polymer modifications.83 These include disulfide,106 azide-modified spiropyran (SP-N3),107 altered dimethylamine,108 and benzylbenzene boronic acid pinacol ester,109 which have been used for the functionalization of aliphatic polycarbonates through alkyne–azide click chemistry for various applications such as controlled release of hydrophobic drugs. Similarly, alkyne-containing cyclic carbonate monomers capable of enhancing the properties of various aliphatic polycarbonates with respect to low cytotoxicity and drug release have been reported. Examples involve 5-methyl-5-propargyl-1,3-dioxan-2-one,110 5,5-di(propargyl)-1,3-dioxan-2-one,111 and methyl-2-O-ethyloxycarbonyl-3-O-propargyloxycarbonyl-4,6-O-carbonyl-α-D-glucopyranoside.112 Alkyne-containing compounds like propargyl alcohol and alkyne-terminated poly(ethylene glycol) can also be employed for the synthesis of functional aliphatic polycarbonates via alkyne–azide click reactions. Utilizing this approach, propargyl palmitate has been successfully grafted onto polycarbonates to generate amphiphilic block–graft copolymers.113 Redox-responsive polymeric micelles having cross-linked cores for stability in aqueous solution with applications in drug loading have been prepared using 2,2-bis(azidomethyl)trimethylene carbonate (ADTC) and 2,2-dimethyltrimethylene carbonate (DTC) with monomethoxy poly(ethylene glycol) (mPEG) as an initiator.114 For the synthesis of an amphiphilic block copolymer, these azide groups aid in cross-linking via a click reaction with a disulfide-containing dialkyne.Cyclic polymers, because of topological effects, have distinct and unique physical/chemical properties, such as high thermodynamic stability compared to their linear counterparts.115 In this direction, a functional amphiphilic cyclic polycarbonate was synthesized via an alkyne azide reaction.116 Following the tremendous work done for the polymerization and post-polymerization functionalization of substituted cyclic and acyclic carbonate monomers, Zhu et al. have shown that alkyne-terminated polyethylene can be anchored onto the azide-containing copolymer of ε-caprolactone (ε-CL) and 5,5-dibromomethyltrimethylene carbonate (DBTC).117,118 Using this pathway, a focus of Ai-Yun Peng's research group was to functionalize the P(BMTC-co-LLA) polymer derived from 2-bromomethyl-2-methyltrimethylene carbonate (BMTC) with L-lactide (LLA). Following the azidation of the polymer, clicking on phenyl acetate provided the functional polycarbonate.119 Another azide polycarbonate 2,2-bis(azidomethyl)trimethylenecarbonate (PADTC-co-PDTC) was anchored with pendant functionalized groups such as alkynyl-terminated poly(ethylene glycol) monomethyl ether (propargyl-PEG), propargyl alcohol, dimethylpropargylamine, and propargyl methacrylate using click chemistry catalyzed by CuBr-Et3N in THF at 35 °C, as shown in Scheme 6a. Interestingly, the PDTC-g-PEG polymer is amphiphilic in nature (PEG acts as a side chain) capable of self-assembling in a particular solvent.120 To circumvent the toxic effects of Cu(I) and to enhance the biomedical applications, functionalization of azide polycarbonate with dibenzocyclooctyne was carried out through copper-free strain-promoted azido-alkyne cycloaddition.121 Also, a method for the in situ generation of Cu(I) has been developed using the precursor copper sulphate (CuSO4) as a feedstock for the generation of Cu(I) using ascorbate for the reduction of Cu(II) to Cu(I).122 Although the copper catalyzed reaction is efficient but due to residual copper, the potential toxicity can't be ignored. In contrast to this, copper-free strain-promoted azido-alkyne cycloaddition relies on the high energy strained cyclooctyne ring to facilitate the reaction. Hence this reaction can be done in biological systems. Thus far, we have described several click reaction pathways for the functionalization of polycarbonates, including thiol–ene, alkyne–azide, and strain promoted azido-alkyne. Recently, the reaction of 1,2,4-triazoline-3,5-dione (TAD)-based derivatives for the functionalization of polycarbonates has received much attention due to the ultra-fast nature of this process.123
 | ||
| Scheme 6 Polycarbonate functionalization using alkyne–azide 1,2,4-triazoline-3,5-dione (TAD) click reactions. (A) Functionalization of polycarbonate bearing a pendant azide group through an alkyne–azide click chemistry approach.120 (B) Pathway depicting the functionalization and formation of network materials.124 | ||
TAD molecules are heterocyclic moieties where the azo group is attached to two carbonyl substituents.125 An important feature of TAD is its electron-withdrawing carbonyl groups and symmetric electron density, giving it an electrophilic nature like that of carbenes or singlet oxygen.126 Due to the presence of the highly electron-deficient N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (nitrogen–nitrogen) double bond, TAD acts as an exceptionally strong electrophile for numerous reactions. TAD can be used as a dienophile for Diels–Alder reactions and as an electrophile for ene-like nucleophilic addition with phenols and indoles.
N (nitrogen–nitrogen) double bond, TAD acts as an exceptionally strong electrophile for numerous reactions. TAD can be used as a dienophile for Diels–Alder reactions and as an electrophile for ene-like nucleophilic addition with phenols and indoles.
Using TAD compounds for the functionalization of aliphatic polycarbonates by click chemistry is a new approach employed initially by Du Prez and coworkers.124 This reaction is a type of Diels–Alder cycloaddition reaction involving 1,2,4-triazoline as a dienophile during the reaction with conjugated dienes; therefore, the mechanism involves a typical cycloaddition pathway. Such a type of click approach generally involves the reaction between TAD and the diene moiety under ambient conditions in an organic solvent. The advantages of TAD click chemistry are no catalyst or initiators are used and no byproducts are formed during the functionalization of the polymer. In a specific example, 5-methyl-5-hexadienoxycarbonyl-1,3-dioxane-2-one (MTC-HD) was copolymerized with a nonfunctional monomer 5-methyl-5-ethyloxycarbonyl-1,3-dioxane-2-one (MEC) using an organocatalyzed ring-opening polymerization (ROP). In order to examine the effect of functionalization on Tg, post-polymerization modifications were carried using 4-butyl-TAD (BuTAD) or 4-phenyl-TAD (PhTAD) (Scheme 6b). To highlight the versatility of this concept, bivalent TAD compounds based on 4,4′-methylene bis(phenyl isocyanate) (MDI-TAD) were used to synthesize cross-linked polycarbonates.
Another type of click reaction that can be used for functionalization of polycarbonates is the thiol–yne click reaction. This reaction proceeds via a two-step path, initially there is the addition of a photo-initiated thiol radical to the alkyne group giving rise to a vinyl sulfide intermediate which is followed by thiol–ene chemistry to give a 1,2-dithioether adduct. This reaction can take place photochemically or thermally. Some research groups have shown that thiol–yne reactions occur with fast kinetics, high efficiency, and at room temperature using sunlight as the renewable source of radiation.127,128 With the aim of developing materials that can be easily degraded, researchers have synthesized functional polycarbonate via thiol–yne click chemistry.129 By designing a tetrafunctional alkyne monomer having an aliphatic polycarbonate backbone, they have managed to synthesize different cross-linked degradable polycarbonates. Interestingly, the ester linkages present in cross-linkers impart fast hydrolytic degradation of the obtained 3D printed shapes in 1 M NaOH solution.
Applications of functionalized aliphatic polycarbonates
Functionalized aliphatic polycarbonates have provided applications for biomedical functionality (including drug delivery vehicles and support structures for hard and soft tissues), polymeric electrolytes, barrier materials, construction materials, electronics, and others (Scheme 7a). A select discussion of some of their implementations is provided here. The important key features of aliphatic polycarbonates compared to their aromatic analogs are their (bio)degradability and good biocompatibility, thereby being preferred for biomedical applications.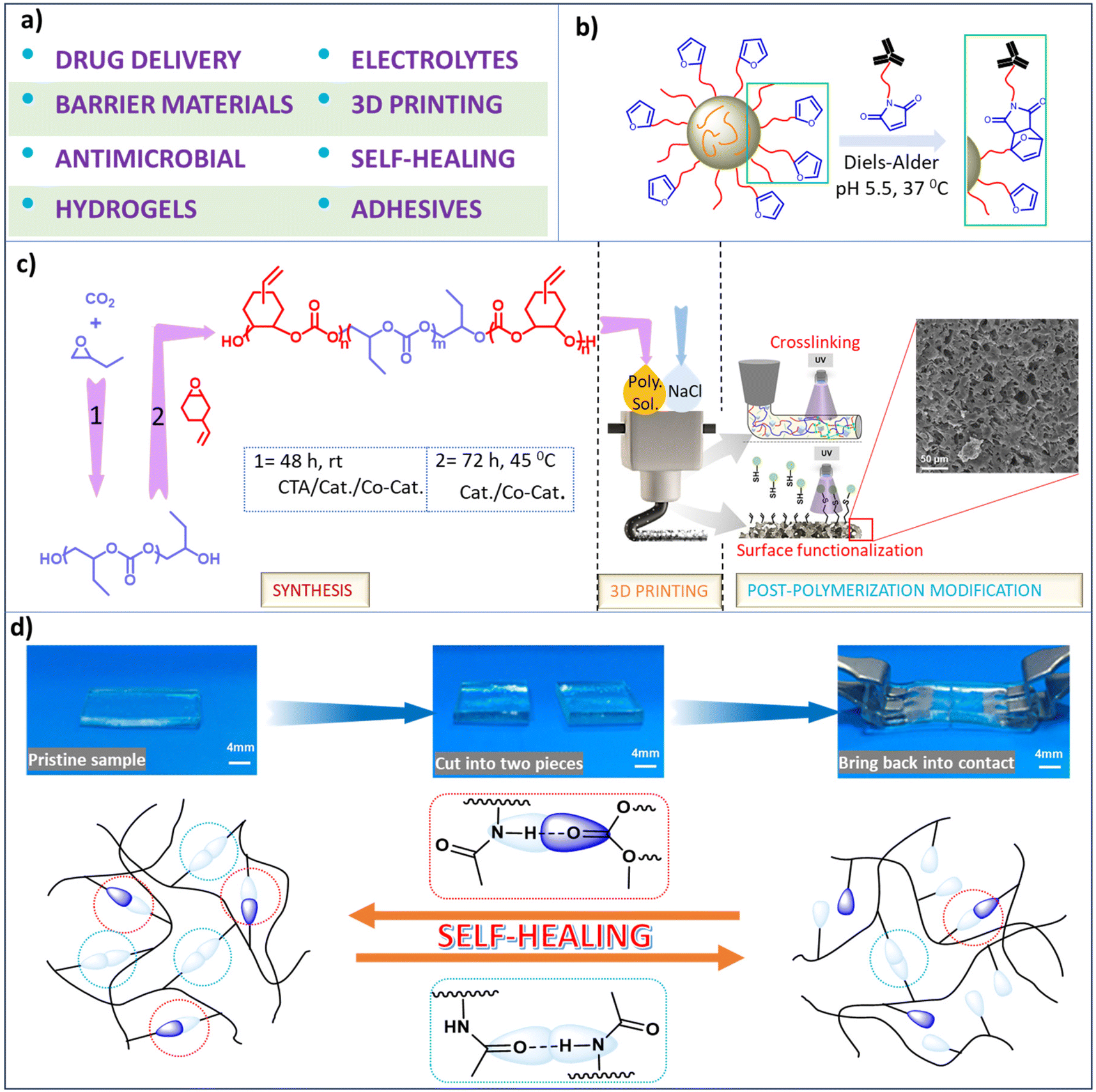 | ||
| Scheme 7 Various applications of synthesized polycarbonates. (a) Depiction of different applications of functional polycarbonates. (b) Representation of the reaction between maleimide-modified antibodies and self-assembled micellar nanoparticles formed from poly(2-methyl-2-carboxytrimethylene carbonate-co-D,L-lactide)-graft-poly(ethylene glycol)-furan.134 (c) Sequential illustration of triblock copolymer synthesis, followed by utilization of the polymer as an ink for 3D printing, and UV-induced cross-linking and surface modification via thiol–ene click chemistry.148 (d) Self-healing material with self-healing ability due to hydrogen bonding.151 | ||
The mechanism of degradation of polycarbonates generally involves end-group activation as opposed to main chain scission, hence polycarbonates can withstand their mechanical properties for a prolonged period of time during degradation.67 Furthermore, the products of degradation of polycarbonates unlike those of polyesters such as polylactides are neutral in nature.130,131 Indeed, several aliphatic polycarbonates are approved for biomedical applications by the US Food and Drug Administration (FDA).44 Functionalized polycarbonates derived from CO2 provides excellent precursor materials for hydrogels which have numerous applications, such as dressings, personal care products, protein microchips, ophthalmic prostheses, and tissue engineering scaffolds, among others that have been summarized by Xu et al.132 Additionally the chemotherapeutic drug, paclitaxel has been reported to be deliverable by polycarbonate-based thiol–ene hydrogels.96 In related efforts, Hedrick and coworkers have synthesized sugar-functionalized polycarbonates that were shown to deliver doxorubicin more efficiently into ASGP-R positive HepG2 cells than in ASGP-R negative HepG2 cells.133 Other studies have shown that functional furan groups located on the polymer surfaces enable the conjugation of biomolecules, e.g., biodegradable graft copolymers of poly(2-methyl-2-carboxytrimethylene carbonate-co-D,L-lactide)-graft-poly(ethylene glycol)-furan(poly(TMCC-co-LA)-g-PEG-furan) were utilized as drug delivery systems because of their ability to self-assemble into micellar immune-polymeric nanoparticles in aqueous solution (Scheme 7b).134 Similarly, self-assembled polymeric micelles of urea-functionalized polycarbonate/PEG, phenylboronic acid-functionalized polycarbonate/PEG, and a 1:1 mixture of both were used for encapsulating the poorly insoluble toxic antifungal drug amphotericin B.135
Other chemotherapeutic investigations involve the high efficiency loading and regulated release of doxorubicin (DOX) into cancer cells via the pendant carboxyl-modified block copolymer based on methoxy poly(ethylene glycol)-b-poly(5-allyloxy-1,3-dioxan-2-one) (mPEG-b-PATMC). Both the carboxyl-modified and unmodified copolymers were shown to self-assemble to produce nano-sized micelles in aqueous solution. It was also revealed that the pendant carboxyl-modified structure in the polymer mPEG-b-PATMC-g-SCH2COOH significantly improves the drug-loading capacity and enhances trapping compared to the unmodified copolymer through electrostatic interactions with amine groups from doxorubicin.136 It was further shown that PEG and N-methylimidazole-functionalized polycarbonate when used in combination with catechol-functionalized polycarbonates for DOX delivery achieved higher drug loading efficiency.137 Taking advantage of biodegradable polymers, the prodrug poly(5-methyl-5-allyloxycarbonyl-1,3-dioxan-2-one)-g-12-acryloyloxy dodecyl phosphorylcholine-co-6-maleimidocaproyl-doxorubicin, Ji et al. have attached DOX to an aliphatic carbonate,77 while another research group attached DOX on an amphiphilic polycarbonate,138 in both cases micelles efficiently release DOX in cancer cells. ε-Caprolactone and 16-membered cyclic decarbonate-derived biodegradable copolymers have also been employed for drug delivery applications for ibuprofen and doxorubicin.139 Relative to tumor detection, aPCs have shown promising results, i.e., researchers have demonstrated the synthesis of aPC micelles accompanied with gadolinium, where these polymeric micelles have shown efficient magnetic resonance imaging (MRI) performance. Moreover the degraded products of these micelles could easily be eliminated from the body via the renal path without any toxic effects.50
The demand for rechargeable lithium-ion batteries for their numerous applications has led to a need for better electrolytes present in them for enhanced charge flow. This has been achieved by the introduction of poly(propylene carbonate) into a gel,140 as well as in solid polymers.141 When poly(vinylidene fluoride)-based gel polymer electrolytes were added to poly(propylene carbonate), the resulting electrolyte displayed a significant increase in ionic conductivity.142 Relative to this objective, Wenhan and coworkers have synthesized a gel polymer electrolyte by functionalizing a terpolymer produced from PO/AGE/CO2 using thiol–ene click chemistry with a thiolated methoxy polyethylene glycol.143 Additionally, a single-ion conducting electrolyte was prepared by the lithiation of the functionalized terpolymer of PO/AGE/CO2. This was realized via thiol–ene click chemistry employing 3-mercaptopropionic acid prior to lithiation. The resulting polymer showed excellent ionic conductance among numerous solid-state single-ion conducting polymer electrolytes.144 In other usages, the functionalization of poly(2-methacryloyloxy)ethyl oleate (PMAEO) using 3-mercaptopropanic acid has provided water-soluble polymers suitable for use as adhesives and paints. Because of the special features of microspheres, i.e., small size, high specific surface area and diffusivity, monodispersed microspheres have been prepared via a step-growth thiol-Michael addition from functionalized monomers, thereby being useful for fluorescence related applications.145–147
Since the mechanical properties of polycarbonates are a function of the nature of the epoxide employed in the copolymerization with CO2, e.g., poly(cyclohexene carbonate) is hard and poly(ethylene carbonate) is soft,1,148 the mechanical behaviour of the polymer can be widely varied by the contents of the epoxide monomers used in its synthesis. In this regard, several research groups have prepared polycarbonates that possess various applications for 3D printing. Recently, Darensbourg and coworkers have synthesized triblock copolymers consisting of soft and hard blocks with pendant alkene groups attached to the hard blocks for cross-linking or thiol–ene click functionalization for their use in 3D printing. This approach allowed for easy tuning of the chemical, mechanical and thermal properties of the printed products (Scheme 7c).148 Similarly, other investigators have prepared cross-linked polycarbonates having alkene substituents for thiol–ene click chemistry, thus providing many clinical applications.149
Finally, synthetically developed materials that mimics nature's ability of self-healing, i.e., having the built-in ability to automatically repair themselves without human intervention are of much interest, and indeed need attention with respect to greener processes from researchers.150 In this regard, thiol–ene click chemistry has been instrumental in providing a self-healing polymeric material based on hydrogen bonding.151 That is, the employment of three different reaction strategies involving propylene oxide hydrolysis to afford a CTA, followed by sequential copolymerization of PO/CO2 and AGE/CO2 to provide a diblock polymer in a one-pot process with the unsaturated block undergoing thiol–ene addition of 2-(acetylamino)ethanethiol. The resulting polycarbonate exhibited autonomous self-healing ability (Scheme 7d), where this attribute was shown to involve both homo- and hetero-hydrogen bonding.
Conclusion and outlook
In the above presentation, we have described the state of the art for functionalizing aliphatic polycarbonates derived from CO2 and epoxides or six- or eight-membered cyclic carbonates. We have attempted to emphasize the main methods for transforming these polymeric materials from those with limited usage to matter with significantly broader applications. In general, modifications of these easily accessible and degradable polycarbonates are best achieved by utilizing post-polymerization click chemistry pathways of thiol–ene or azide–alkyne addition. These are important advancements, given that most polycarbonates obtained by these greener methods lack functionality.The copolymerization of carbon dioxide and epoxides via a chain-growth pathway to provide aliphatic polycarbonates is an effective and greener route compared to that involving polycondensation of diols and carbonates. Indeed, this process can be made to be sustainable when transitioning epoxide monomers from petroleum-based feedstocks to those from natural products. More importantly, these polymeric materials are (bio)degradable, thereby addressing the issues of plastic pollution, as well as providing polymers for biomedical applications. Continuing efforts to advance catalyst development are directed at minimizing the reaction conditions of polymerization temperatures and CO2 pressures, with concomitant product selectivity for copolymers over five-membered cyclic carbonates. Other considerations for improving this technology are understanding polymer degradation and in particular biodegradation, as well as purity requirements of the CO2 monomer.
Nevertheless, the advantageous features which make the above process for synthesizing aliphatic polycarbonates so attractive are particularly negated by these polymeric materials’ lack of functionality. Overcoming this deficiency was indeed the focus of this review article. Researchers have enhanced the usefulness of these polymers by employing epoxide monomers in the copolymerization reactions with CO2 that contain alkene or alkyne groups for their abilities to undergo click reaction chemistry. As we have demonstrated above one of the most effective routes to achieve functionality is utilizing thiol–ene click reactions in post-polymerization pathways. Importantly, these click reactions can be carried out using the photo- or radical-initiated mechanism, for each direction has advantages depending on the nature of the addition. For example, as noted in the use of modifying aPCs following 3D printing, photochemically initiating thiol–ene click chemistry is the method of choice.
Conflicts of interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
D. J. Darensbourg gratefully acknowledges the financial support from the Robert A. Welch Foundation (A-0923) and Gulzar A. Bhat gratefully acknowledges DST-SERB for the Ramanujan fellowship (RJF/2020/000116) and the core research Grant (CRG/2022/003558).References
- S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS.
- P. T. Anastas and J. C. WarnerGreen chemistry: Theory and practice, Oxford University Press, New York, 1998, pp. 10–55 Search PubMed.
- N. Von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- D. A. Czaplewski, J. Kameoka, R. Mathers, G. W. Coates and H. G. Craighead, Appl. Phys. Lett., 2003, 83, 4836–4838 CrossRef CAS.
- T. Laird, Ullmann's encyclopedia of industrial chemistry, VCH, Weinheim, Germany, 1996/1997, vol. 8, pp. 391–392 Search PubMed.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC.
- Y. Ma, H. Liu, J. Wu, L. Yuan, Y. Wang, X. Du, R. Wang, P. W. Marwa, P. Petlulu and X. Chen, Environ. Res., 2019, 176, 108575 CrossRef CAS PubMed.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- S. Inoue, H. Koinuma and T. Tsuruta, Makromol. Chem., 1969, 130, 210–220 CrossRef CAS.
- G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 5007–5034 RSC.
- G. A. Bhat, M. Luo and D. J. Darensbourg, Green Chem., 2020, 22, 7707–7724 RSC.
- M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131, 41141 CrossRef.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC.
- Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- Y. Dai, X. Zhang and F. Xia, Macromol. Rapid Commun., 2017, 38, 1700357 CrossRef PubMed.
- G. A. Bhat and D. J. Darensbourg, Coord. Chem. Rev., 2023, 492, 215277 CrossRef CAS.
- S. Honda, T. Mori, H. Goto and H. Sugimoto, Polymer, 2014, 55, 4832–4836 CrossRef CAS.
- A. W. Thomas and A. P. Dove, Macromol. Biosci., 2016, 16, 1762–1775 CrossRef CAS PubMed.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- A. Sharma and A. Kakkar, Molecules, 2015, 20, 16987–17015 CrossRef CAS PubMed.
- K. A. Günay, P. Theato and H.-A. Klok, Funct. Polym. Post-Polym. Modif., 2013, 1–44 Search PubMed.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- J. Geschwind, F. Wurm and H. Frey, Macromol. Chem. Phys., 2013, 214, 892–901 CrossRef CAS.
- D. J. Darensbourg and Y. Wang, Polym. Chem., 2015, 6, 1768–1776 RSC.
- D. Zeng, W.-P. Deng and X. Jiang, Natl. Sci. Rev., 2023, nwad123 CrossRef CAS PubMed.
- N. K. Devaraj and M. G. Finn, Chem. Rev., 2021, 121, 6697–6698 CrossRef CAS PubMed.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- C. Bertozzi, ACS Central Science, 2022, 9, 558–559 CrossRef PubMed.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- Susanna L. Widicus Weaver and Benjamin J. McCallinCRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Publishing, Boca Raton, FL, 87th edn, 2006 Search PubMed.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS PubMed.
- K.-K. Tremblay-Parrado, C. García-Astrain and L. Avérous, Green Chem., 2021, 23, 4296–4327 RSC.
- S. K. Reddy, N. B. Cramer and C. N. Bowman, Macromolecules, 2006, 39, 3681–3687 CrossRef CAS.
- H. B. El Ayouchia, L. Bahsis, H. Anane, L. R. Domingo and S.-E. Stiriba, RSC Adv., 2018, 8, 7670–7678 RSC.
- P. Cintas, A. Barge, S. Tagliapietra, L. Boffa and G. Cravotto, Nat. Protoc., 2010, 5, 607–616 CrossRef CAS PubMed.
- A. Y. Yuen, A. Bossion, A. Veloso, D. Mecerreyes, J. L. Hedrick, A. P. Dove and H. Sardon, Polym. Chem., 2018, 9, 2458–2467 RSC.
- X. Zhang, J. Liang, Z. Chen, C. Donley, X. Zhang and G. Cheng, Processes, 2020, 8, 498 CrossRef CAS.
- Z. Peng, H. Li, Q. Li and Y. Hu, Colloids Surf., A, 2017, 533, 48–54 CrossRef CAS.
- L. A. Connal, C. R. Kinnane, A. N. Zelikin and F. Caruso, Chem. Mater., 2009, 21, 576–578 CrossRef CAS.
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef.
- Y. Wang, J. Fan and D. J. Darensbourg, Angew. Chem., Int. Ed., 2015, 54, 10206–10210 CrossRef CAS PubMed.
- G.-P. Wu and D. J. Darensbourg, Macromolecules, 2016, 49, 807–814 CrossRef CAS.
- D. J. Darensbourg and F.-T. Tsai, Macromolecules, 2014, 47, 3806–3813 CrossRef CAS.
- J. Wei, Y. Ma, Y. Cai, J. Zheng and H. Fan, Prog. Org. Coat., 2022, 173, 107167 CrossRef CAS.
- G. A. Bhat, A. Z. Rashad, T. M. Folsom and D. J. Darensbourg, Organometallics, 2020, 39, 1612–1618 CrossRef CAS.
- F. Y. Ren, K. Chen, L. Q. Qiu, J. M. Chen, D. J. Darensbourg and L. N. He, Angew. Chem., 2022, 134, e202200751 CrossRef.
- G. A. Bhat, A. Z. Rashad and D. J. Darensbourg, Polym. Chem., 2020, 11, 4699–4705 RSC.
- M. Sengoden, G. A. Bhat, R. J. Rutledge, S. Rashid, A. A. Dar and D. J. Darensbourg, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2312907120 CrossRef CAS PubMed.
- Y. Li, S. Liu, X. Zhao, Y. Wang, J. Liu, X. Wang and L. Lu, Theranostics, 2017, 7, 4689 CrossRef CAS PubMed.
- Z. Huang, Y. Wang, N. Zhang, L. Zhang and D. J. Darensbourg, Macromolecules, 2018, 51, 9122–9130 CrossRef CAS.
- T. M. Folsom, G. A. Bhat, A. Z. Rashad and D. J. Darensbourg, Macromolecules, 2019, 52, 5217–5222 CrossRef CAS.
- D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS PubMed.
- D. J. Darensbourg, Polym. Degrad. Stab., 2018, 149, 45–51 CrossRef CAS.
- F. N. Singer, A. C. Deacy, T. M. McGuire, C. K. Williams and A. Buchard, Angew. Chem., 2022, 134, e202201785 CrossRef.
- J.-F. Zhang, W.-M. Ren, X.-K. Sun, Y. Meng, B.-Y. Du and X.-H. Zhang, Macromolecules, 2011, 44, 9882–9886 CrossRef CAS.
- P. Alagi, G. Zapsas, N. Hadjichristidis, S. C. Hong, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 6144–6152 CrossRef CAS.
- D. J. Darensbourg, W.-C. Chung, C. J. Arp, F.-T. Tsai and S. J. Kyran, Macromolecules, 2014, 47, 7347–7353 CrossRef CAS.
- J. A. Mmongoyo, Q. A. Mgani, S. J. Mdachi, P. J. Pogorzelec and D. J. Cole-Hamilton, Eur. J. Lipid Sci. Technol., 2012, 114, 1183–1192 CrossRef CAS.
- D. J. Darensbourg, W.-C. Chung, A. D. Yeung and M. Luna, Macromolecules, 2015, 48, 1679–1687 CrossRef CAS.
- Z. I. Lin, H. L. Tsai, G. L. Liu, X. H. Lu, P. W. Cheng, P. L. Chi, C. K. Wang, T. H. Tsai, C. C. Wang and J. H. C. Yang, Macromol. Biosci., 2022, 22, 2200178 CrossRef CAS PubMed.
- M. Li, Z. I. Lin, J. Yang, H. Huang, G. L. Liu, Q. Liu, X. Zhang, Y. Zhang, Z. Xu and H. Lin, Adv. Healthcare Mater., 2023, 12, 2201306 CrossRef CAS PubMed.
- J. Yue, X. Li, G. Mo, R. Wang, Y. Huang and X. Jing, Macromolecules, 2010, 43, 9645–9654 CrossRef CAS.
- P. Olsén, K. Odelius and A.-C. Albertsson, Macromolecules, 2014, 47, 6189–6195 CrossRef.
- P.-L. Durand, A. Brège, G. Chollet, E. Grau and H. Cramail, ACS Macro Lett., 2018, 7, 250–254 CrossRef CAS PubMed.
- H. Zhang, B. Liu, H. Ding, J. Chen and Z. Duan, Polymer, 2017, 129, 5–11 CrossRef CAS.
- G.-L. Liu, H.-W. Wu, Z.-I. Lin, M.-G. Liao, Y.-C. Su, C.-K. Chen and B.-T. Ko, Polym. Chem., 2021, 12, 1244–1259 RSC.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- C. Li, S. van Berkel, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2016, 85, 466–477 CrossRef CAS.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed.
- C. Li, R. J. Sablong and C. E. Koning, Angew. Chem., 2016, 128, 11744–11748 CrossRef.
- C. Li, M. Johansson, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2017, 96, 337–349 CrossRef CAS.
- C. Martin and A. W. Kleij, Macromolecules, 2016, 49, 6285–6295 CrossRef CAS.
- C. Li, M. Johansson, P. Buijsen, G. Dijkstra, R. J. Sablong and C. E. Koning, Prog. Org. Coat., 2021, 151, 106073 CrossRef CAS.
- X. Hu, X. Chen, S. Liu, Q. Shi and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1852–1861 CrossRef CAS.
- A. W. Thomas, P. K. Kuroishi, M. M. Pérez-Madrigal, A. K. Whittaker and A. P. Dove, Polym. Chem., 2017, 8, 5082–5090 RSC.
- H. Wang, Y. Wang, Y. Chen, Q. Jin and J. Ji, Polym. Chem., 2014, 5, 854–861 RSC.
- K. Olofsson, M. Malkoch and A. Hult, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2370–2378 CrossRef CAS.
- S. Tempelaar, L. Mespouille, P. Dubois and A. P. Dove, Macromolecules, 2011, 44, 2084–2091 CrossRef CAS.
- S. Onbulak, S. Tempelaar, R. J. Pounder, O. Gok, R. Sanyal, A. P. Dove and A. Sanyal, Macromolecules, 2012, 45, 1715–1722 CrossRef CAS.
- L. Sun, A. Pitto-Barry, A. W. Thomas, M. Inam, K. Doncom, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2016, 7, 2337–2341 RSC.
- H. Kuang, S. Wu, Z. Xie, F. Meng, X. Jing and Y. Huang, Biomacromolecules, 2012, 13, 3004–3012 CrossRef CAS PubMed.
- J. V. Olsson, D. Hult, Y. Cai, S. García-Gallego and M. Malkoch, Polym. Chem., 2014, 5, 6651–6655 RSC.
- N. H. Park, Z. X. Voo, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2017, 6, 252–256 CrossRef CAS PubMed.
- J. M. Chan, H. Sardon, A. C. Engler, J. M. García and J. L. Hedrick, ACS Macro Lett., 2013, 2, 860–864 CrossRef CAS PubMed.
- A. Kuroki, D. J. W. Tay, A. C. Y. Chua, B. S. Y. Tan, N. Li, J. Leong, K. S. Tan, Y. Wang, E. W. P. Tan, L. Rénia, J. L. Hedrick, Y. Y. Yang and G. H. Lee, Macromolecules, 2023, 56, 6003–6009 CrossRef CAS.
- T. M. McGuire, E. M. López-Vidal, G. L. Gregory and A. Buchard, J. CO2 Util., 2018, 27, 283–288 CrossRef CAS.
- J. L. Hedrick, V. Piunova, N. H. Park, T. Erdmann and P. L. Arrechea, ACS Macro Lett., 2022, 11, 368–375 CrossRef CAS PubMed.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, ACS Macro Lett., 2020, 9, 443–447 CrossRef CAS PubMed.
- G. L. Gregory, H. Gao, B. Liu, X. Gao, G. J. Rees, M. Pasta, P. G. Bruce and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 17477–17486 CrossRef CAS PubMed.
- K. C. Poon, G. L. Gregory, G. S. Sulley, F. Vidal and C. K. Williams, Adv. Mater., 2023, 35, 2302825 CrossRef CAS PubMed.
- W. Mo, C. Zhuo, S. Liu, X. Wang and F. Wang, Polym. Chem., 2023, 14, 152–160 RSC.
- D. M. Stevens, S. Tempelaar, A. P. Dove and E. Harth, ACS Macro Lett., 2012, 1, 915–918 CrossRef CAS PubMed.
- V. X. Truong, I. A. Barker, M. Tan, L. Mespouille, P. Dubois and A. P. Dove, J. Mater. Chem. B, 2013, 1, 221–229 RSC.
- I. A. Barker, M. P. Ablett, H. T. Gilbert, S. J. Leigh, J. A. Covington, J. A. Hoyland, S. M. Richardson and A. P. Dove, Biomater. Sci., 2014, 2, 472–475 RSC.
- D. M. Stevens, A. Rahalkar, B. Spears, K. Gilmore, E. Douglas, M. Muthukumar and E. Harth, Polym. Chem., 2015, 6, 1096–1102 RSC.
- Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674–1689 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., 2002, 114, 2708–2711 CrossRef.
- C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS.
- R. J. Ono, S. Q. Liu, S. Venkataraman, W. Chin, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2014, 47, 7725–7731 CrossRef CAS.
- J. Mindemark and T. Bowden, Polym. Chem., 2012, 3, 1399–1401 RSC.
- S. Doran, G. Yilmaz and Y. Yagci, Macromolecules, 2015, 48, 7446–7452 CrossRef CAS.
- M. Basko, M. Bednarek, L. Billiet, P. Kubisa, E. Goethals and F. Du Prez, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1597–1604 CrossRef CAS.
- J. Hilf and H. Frey, Macromol. Rapid Commun., 2013, 34, 1395–1400 CrossRef CAS PubMed.
- C. Lu, Q. Shi, X. Chen, T. Lu, Z. Xie, X. Hu, J. Ma and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3204–3217 CrossRef CAS.
- L. Yu, M. Xie, Z. Li, C. Lin, Z. Zheng, L. Zhou, Y. Su and X. Wang, J. Mater. Chem. B, 2016, 4, 6081–6093 RSC.
- D. Hu, H. Peng, Y. Niu, Y. Li, Y. Xia, L. Li, J. He, X. Liu, X. Xia and Y. Lu, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 750–760 CrossRef CAS.
- L. Yu, C. Lin, Z. Zheng, Z. Li and X. Wang, Colloids Surf., B, 2016, 145, 392–400 CrossRef CAS PubMed.
- W. Zhao, S. Yang, J. Yang, J. Li, J. Zheng, Z. Qing and R. Yang, Anal. Chem., 2016, 88, 10728–10735 CrossRef CAS PubMed.
- X. Yi, D. Zhao, Q. Zhang, J. Xu, G. Yuan, R. Zhuo and F. Li, Polym. Chem., 2016, 7, 5966–5977 RSC.
- M. N. Ganivada, P. Kumar, P. Kanjilal, H. Dinda, J. D. Sarma and R. Shunmugam, Polym. Chem., 2016, 7, 4237–4245 RSC.
- L. Su, S. Khan, J. Fan, Y.-N. Lin, H. Wang, T. P. Gustafson, F. Zhang and K. L. Wooley, Polym. Chem., 2017, 8, 1699–1707 RSC.
- X. Zhang, Z. Zhang, Z. Zhong and R. Zhuo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2687–2696 CrossRef CAS.
- X. Zhang, H. Dong, S. Fu, Z. Zhong and R. Zhuo, Macromol. Rapid Commun., 2016, 37, 993–997 CrossRef CAS PubMed.
- Y. Zhu and N. S. Hosmane, ChemistryOpen, 2015, 4, 408–417 CrossRef CAS PubMed.
- R. J. Williams, A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Macromolecules, 2016, 49, 2802–2813 CrossRef CAS PubMed.
- W. Zhu, Y. Wang, Q. Zhang and Z. Shen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4886–4893 CrossRef CAS.
- Q. J. Zhang, W. P. Zhu and Z. Q. Shen, Chin. Chem. Lett., 2010, 21, 1255–1258 CrossRef CAS.
- L. Li, H. Zhai, T. Wang, X. Qiu, N. Qiang, P. Dong, Y. Bai, A.-Y. Peng and D. Quan, Polymer, 2019, 180, 121705 CrossRef CAS.
- X. Zhang, Z. Zhong and R. Zhuo, Macromolecules, 2011, 44, 1755–1759 CrossRef CAS.
- J. Xu, F. Prifti and J. Song, Macromolecules, 2011, 44, 2660–2667 CrossRef CAS PubMed.
- A. Mandoli, Molecules, 2016, 21, 1174 CrossRef PubMed.
- P. Mondal, P. K. Behera and N. K. Singha, Prog. Polym. Sci., 2021, 113, 101343 CrossRef CAS.
- A. Baroni, L. Vlaminck, L. Mespouille, F. D. Prez, N. Delbosc and B. Blankert, Macromol. Rapid Commun., 2019, 40, 1800743 CrossRef.
- R. Cookson, S. Gilani and I. Stevens, Tetrahedron Lett., 1962, 3, 615–618 CrossRef.
- A. R. Katritzky, Advances in heterocyclic chemistry, Academic press, 1997 Search PubMed.
- R. M. Arnold, N. E. Huddleston and J. Locklin, J. Mater. Chem., 2012, 22, 19357–19365 RSC.
- R. M. Hensarling, V. A. Doughty, J. W. Chan and D. L. Patton, J. Am. Chem. Soc., 2009, 131, 14673–14675 CrossRef CAS PubMed.
- Y. Wu, M. C. Simpson and J. Jin, Macromol. Chem. Phys., 2021, 222, 2000435 CrossRef CAS.
- H. Yan, W. R. Cannon and D. J. Shanefield, Ceram. Int., 1998, 24, 433–439 CrossRef CAS.
- E. Antonakou and D. Achilias, Waste Biomass Valorization, 2013, 4, 9–21 CrossRef CAS.
- J. Xu, E. Feng and J. Song, J. Appl. Polym. Sci., 2014, 131, 39822 CrossRef PubMed.
- F. Suriano, R. Pratt, J. P. Tan, N. Wiradharma, A. Nelson, Y.-Y. Yang, P. Dubois and J. L. Hedrick, Biomaterials, 2010, 31, 2637–2645 CrossRef CAS.
- M. Shi, K. Ho, A. Keating and M. S. Shoichet, Adv. Funct. Mater., 2009, 19, 1689–1696 CrossRef CAS.
- Y. Wang, X. Ke, Z. X. Voo, S. S. L. Yap, C. Yang, S. Gao, S. Liu, S. Venkataraman, S. A. O. Obuobi and J. S. Khara, Acta Biomater., 2016, 46, 211–220 CrossRef CAS PubMed.
- Y.-M. Li, T. Jiang, Y. Lv, Y. Wu, F. He and R.-X. Zhuo, Colloids Surf., B, 2015, 132, 54–61 CrossRef CAS.
- J. M. Chan, J. P. Tan, A. C. Engler, X. Ke, S. Gao, C. Yang, H. Sardon, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2016, 49, 2013–2021 CrossRef CAS.
- T. Jiang, Y.-M. Li, Y. Lv, Y.-J. Cheng, F. He and R.-X. Zhuo, Colloids Surf., B, 2013, 111, 542–548 CrossRef CAS.
- W. Su, H.-f. Wang, J. Feng, X.-h. Luo, X.-z. Zhang and R.-x. Zhuo, J. Mater. Chem., 2011, 21, 6327–6336 RSC.
- X. Huang, J. Huang, J. Wu, X. Yu, Q. Gao, Y. Luo and H. Hu, RSC Adv., 2015, 5, 52978–52984 RSC.
- G. Dukhanin, A. Gaidadin and I. Novakov, Russ. J. Appl. Chem., 2014, 87, 1868–1871 CrossRef CAS.
- X. Huang, S. Zeng, J. Liu, T. He, L. Sun, D. Xu, X. Yu, Y. Luo, W. Zhou and J. Wu, J. Phys. Chem. C, 2015, 119, 27882–27891 CrossRef CAS.
- W. Luo, K. Deng, S. Wang, S. Ren, D. Han, Y. Wang, M. Xiao and Y. Meng, Adv. Polym. Technol., 2020, 2020, 1–12 Search PubMed.
- K. Deng, S. Wang, S. Ren, D. Han, M. Xiao and Y. Meng, ACS Appl. Mater. Interfaces, 2016, 8, 33642–33648 CrossRef CAS PubMed.
- C. Wang, M. Podgórski and C. N. Bowman, Mater. Horiz., 2014, 1, 535–539 RSC.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS.
- H. Kawaguchi, Prog. Polym. Sci., 2000, 25, 1171–1210 CrossRef CAS.
- P. Wei, G. A. Bhat, C. E. Cipriani, H. Mohammad, K. Schoonover, E. B. Pentzer and D. J. Darensbourg, Angew. Chem., Int. Ed., 2022, 61, e202208355 CrossRef CAS PubMed.
- S. L. Brooks, M. C. Arno, A. P. Dove and A. C. Weems, ACS Appl. Polym. Mater., 2022, 4, 5670–5679 CrossRef.
- M. W. Rahman and N. R. Shefa, Adv. Polym. Technol., 2021, 2021, 7848088 CrossRef.
- G.-W. Yang, Y.-Y. Zhang, Y. Wang, G.-P. Wu, Z.-K. Xu and D. J. Darensbourg, Macromolecules, 2018, 51, 1308–1313 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |