
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of NIR light-responsive shape memory composites based on bio-benzoxazine/bio-urethane copolymers reinforced with graphene
Weerapong
Jamnongpak
a,
Sunan
Tiptipakorn
b,
Hariharan
Arumugam
 a,
Krittapas
Charoensuk
a,
Panagiotis
Karagiannidis
c and
Sarawut
Rimdusit
*a
a,
Krittapas
Charoensuk
a,
Panagiotis
Karagiannidis
c and
Sarawut
Rimdusit
*a
aCenter of Excellence in Polymeric Materials for Medical Practice Devices, Department of Chemical Engineering, Faculty of Engineering, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: Sarawut.R@chula.ac.th
bDepartment of Chemistry, Faculty of Liberal Arts and Science, Kasetsart University, Nakhon Pathom 73140, Thailand
cSchool of Engineering, Faculty of Technology, University of Sunderland, Sunderland, SR6 0DD, UK
First published on 18th December 2023
Abstract
In this work, shape memory polymers (SMPs) were developed from a combination of a bio-based benzoxazine (BZ) monomer and polyurethane prepolymer (PU-prepolymer), both derived from bio-based raw materials. The bio-based BZ monomer (V-fa monomer) was synthesized through a Mannich condensation reaction using vanillin, paraformaldehyde, and furfurylamine. The bio-based PU-prepolymer was obtained by reacting palm oil polyol (MW = 1400 Da) and toluene diisocyanate (TDI). To investigate the curing behavior of poly(V-fa/urethane), with a mass ratio of 50/50, differential scanning calorimetry was employed. The structure of the resulting poly(V-fa/urethane) was confirmed using Fourier transform infrared spectroscopy. Furthermore, the synthesized V-fa/urethane copolymers with weight ratios of 70/30, 60/40, 50/50 and 40/60 were observed to exhibit shape memory behaviors induced by near-infrared irradiation (808 nm). Poly(V-fa/urethane), specifically with a mass ratio of 50/50, demonstrated superior shape memory performance. It exhibited a remarkable capacity to retain the temporary shape up to 90%, achieve 99% shape recovery, and exhibit a recovery time of 25 s. The shape memory properties were further improved with the addition of 3 wt% graphene nanoplatelets (GNPs), exhibiting an improvement in the shape fixity value to 94%, and shape recovery time value to 16 s. Moreover, our findings suggest that 60/40 poly(V-fa/urethane) reinforced with 3 wt% GNPs possesses favorable characteristics for applications as multiple SMPs, with shape fixity values of 97% and 94%, and shape recovery values of 96% and 89% for the first and second shapes, respectively.
Introduction
The advancement of new materials and devices holds great promise for enhancing the quality of life. Among these developments, smart materials have emerged as crucial components, exerting a significant influence on advanced technologies due to their distinctive stimulus-response capabilities and autonomous behaviours.1,2 Notably, within the realm of smart materials, two prominent classes, namely piezoelectric materials3 and shape-memory materials4 have attracted considerable attention and are finding widespread applications in diverse fields such as mechanical engineering, materials engineering, and bioengineering. Smart materials possess the outstanding ability to exhibit reversible responses under various environmental conditions such as light, temperature, and electrical, chemical, and mechanical stimuli.5–7 Among these smart materials, thermochromic materials, photochromic materials, hydrogels, shape memory alloys, and shape memory polymers (SMPs) have been extensively studied and employed. SMPs, specifically, belong to a class of smart materials that undergo shape transformations in response to environmental stimuli and subsequently revert to their original form. This characteristic renders SMPs highly applicable in diverse industries including sports equipment (e.g. helmets, gum-shields, and car bumpers), electrical engineering, automotive manufacturing, biomedical engineering, smart textiles, as well as the aerospace sector encompassing aircraft and spacecraft industries.8In recent years, there has been growing research interest in exploring light-responsive shape memory effects, primarily due to the possible potential for applications needing spatially controlled and remotely triggered activations, particularly when considering lower operational temperatures.9 Furthermore, the photo-responsive behaviour can be conveniently paused and resumed as desired by toggling the light source on and off. To achieve such photo-responsive shape memory properties, it is essential to incorporate specific classes of additives, including organic dyes, nanomaterials such as Au nanoparticles and Au nanorods, and carbon materials like carbon nanotubes and graphene nanoplates. These additives possess the capability to convert photo energy into heat, thereby facilitating the photothermal effect within the shape memory polymers. Recently some research publications have explained light-induced changes in the shape of polymers; Li et al. reported a thermal and near-infrared light-induced shape memory film based on polyvinyl alcohol/poly(acrylic acid)–graphene oxide composites.10 These films exhibit the capability to recover from physical damage and restore their shape memory functionality through the utilization of water. Such advancements significantly enhance the reliability and prolong the service life of these materials as shape memory systems. In recent times, our research group has made notable contributions in this field by developing a range of hybrid benzoxazine composites designed for thermal and light-induced SMPs.11
Polybenzoxazines (PBZs) represent a novel category of thermosetting polymers characterized by their exceptional thermal stability, low water absorption, minimal shrinkage upon curing, and robust mechanical properties. Notably, the polymerization reaction of PBZs is devoid of any by-product release. The benzoxazine (BZ) resins are synthesized from phenol, aldehyde, and amine groups through the Mannich condensation reaction. Furthermore, the raw materials of conventional BZ resin are obtained from petroleum resources.12–14 The bio-derived materials are the counter part of petroleum products. In this direction, BZ resin prepared from bio-derived source materials has become a significant attraction for the bio-based raw material research world. Previously, bio-derived phenols (vanillin, eugenol, cardanol, and guaiacol) and amines (furfurylamine, stearylamine, and laurylamine) were successfully utilized for BZ resin preparation.15 PBZ is highly useful for making different composites for various engineering applications. The alloys of PBZ and other thermoset polymers have been investigated for the use of shape memory materials such as benzoxazine–urethane and benzoxazine–epoxy resins.16 In the present work, polyurethane has been taken as a co-material with PBZ, due to its excellent properties. Recently, bio-based SMPs have been applied for making food packaging material, bioactive substances, pharmaceuticals, cosmetics and automotives. Typically, benzoxazine/urethane copolymer will be in the petroleum-based and bio-based BZ/urethane copolymer that has not yet been studied. So, in this work we developed light-induced SMPs based on bio-based BZ/urethane copolymers filled with graphene nanoplates.
In this study, we have undertaken the preparation of bio-based benzoxazine (BZ)/urethane copolymers to explore their applicability as shape memory polymers (SMPs). The bio-based BZ resin (V-fa) was synthesized utilizing bio-derived phenol (vanillin), primary amine (furfurylamine) and paraformaldehyde under appropriate reaction conditions. By varying the ratios, the bio-based BZ/urethane copolymers were synthesized with mass ratios of 70/30, 60/40, 50/50, and 40/60. Additionally, the inclusion of graphene nanoplatelets (GNPs) as photothermal additives facilitated the development of near-infrared (NIR)-responsive nanocomposite SMPs. These resulting SMPs were subjected to a comprehensive analysis of their structural characteristics, photo-thermal effects, thermal properties, dynamic mechanical properties, and shape memory behaviours under NIR stimulation.
Experimental
Chemicals and materials
Vanillin (99%) and furfurylamine (99%) were obtained from Sigma-Aldrich Pte. Ltd (St. Louis, Germany). Paraformaldehyde (AR grade) was purchased from Merck Co., Ltd (Darmstadt, Germany). Palm oil polyol was obtained from Foamtec International Co., Ltd (Thailand). Toluene diisocyanate (TDI) was kindly supplied by Vencorex (Thailand) Co., Ltd, Thailand. GNP grade-H (5–25 μm in diameter, 15 nm in thickness, and surface area of 50–80 m2 g−1) was purchased from XG Sciences (Lansing, United states).Synthesis of BZ resin based vanilin-furfurylamine (V-fa)
The V-fa monomer was prepared using 0.1 mole of vanillin, 0.1 mole of furfurylamine, and 0.2 mole of paraformaldehyde under solvent free conditions by heating at 105 °C for 1 h. After completion of the reaction, the resinous products were placed to cool down at room temperature (RT). The obtained transparent reddish yellow viscous liquid BZ monomer V-fa (Scheme 1) was utilized for further reaction with PU. The ring opening polymerization of V-fa is presented in the same scheme, which can take place by heating at higher temperatures to form homopolymer poly(V-fa).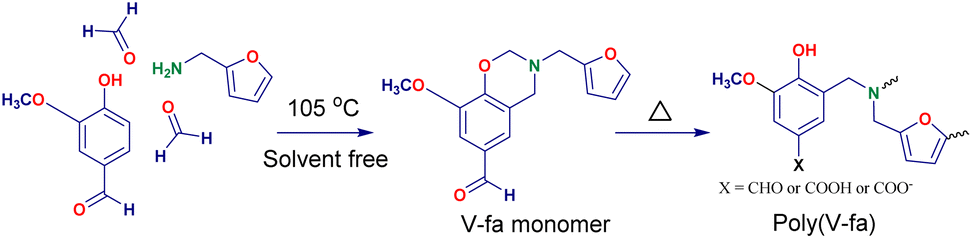 | ||
| Scheme 1 Synthesis of the V-fa monomer and polymerization of poly(V-fa). | ||
Synthesis of the bio-based polyurethane prepolymer (PU-prepolymer)
The synthesis of the bio-based PU-prepolymer involved the reaction between palm oil polyol and toluene diisocyanate (TDI) at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The two reactants were meticulously combined and subjected to continuous stirring at a temperature of 70 °C for a period of 2 h in the presence of a nitrogen atmosphere. The reaction took place in a five-necked round bottom flask, leading to the formation of the PU-prepolymer, which appeared as a transparent yellow viscous liquid (Scheme 2).
:2. The two reactants were meticulously combined and subjected to continuous stirring at a temperature of 70 °C for a period of 2 h in the presence of a nitrogen atmosphere. The reaction took place in a five-necked round bottom flask, leading to the formation of the PU-prepolymer, which appeared as a transparent yellow viscous liquid (Scheme 2).
 | ||
| Scheme 2 Preparation of bio-based poly(V-fa/urethane). | ||
Preparation of the neat V-fa/urethane copolymer and nanocomposites containing GNPs
Neat poly(V-fa/urethane) was synthesized using the V-fa monomer and bio-based PU-prepolymer at different weight ratios of 70/30, 60/40, 50/50, and 40/60. To achieve a homogeneous mixture, the components were heated to 105 °C and continuously stirred. In the case of copolymers filled with GNPs at a content of 3 wt%, the GNPs were gradually introduced into the molten mixtures while stirring until a uniform dispersion was obtained. Subsequently, these mixtures were poured into aluminum molds and kept at 80 °C for 2 h to eliminate any volatile impurities. The temperature was then increased gradually at a rate of 10 °C per hour until 180 °C, where it was held for an additional 2 h. These specific curing conditions were determined based on the results of the curing study conducted using DSC. The reactions occurring during the curing process are illustrated in Scheme 3; upon heating ring open polymerization of BZ takes place, along with the formation of homopolymer poly(V-fa), which then reacts through its hydroxyl groups with the isocyanate groups of the PU-prepolymer (Scheme 1). The prepared samples were brought to RT and were kept for further analysis. | ||
| Scheme 3 Shape memory effect of NIR-responsive poly(V-fa/PU) filled with GNPs. | ||
Characterization
The functional groups of the monomer and polymer network formation were analyzed by the FT-IR spectroscopic technique (Spectrum GX FT-IR spectrometer, PerkinElmer). The curing behaviour of the V-fa/PU-prepolymer blend was analyzed using a differential scanning calorimeter (DSC1 module, Mettler-Toledo, Thailand) at a rate of 10 °C min−1 under N2 purging. The thermal stability of poly(V-fa/urethane) and nanocomposites was investigated using a thermogravimetric analyzer (model TGA1 Module) from Mettler-Toledo (Thailand). A dynamic mechanical analyzer (DMA), DMA1 module, Mettler-Toledo (Thailand) was used to determine the storage modulus (E′) and glass transition temperature (Tg) of the V-fa/urethane copolymers and nanocomposites. The specimen size was designed as 5 mm × 10 mm × 0.3 mm and was tested in tension mode at 1 Hz, a strain amplitude of 10 μm, and from −100 °C to 200 °C at a rate of 2 °C min−1 under an air atmosphere.Specimens of copolymers and nanocomposites were exposed to NIR light with a wavelength of 808 nm (red) for up to 60 s at a distance (between the laser source and specimen) of 60 cm. Photothermal heating of the specimens was carried out by recording the temperature of the irradiated part as a function of time. The temperature of the specimens as a function of time after laser source removal was also recorded. The UV-vis absorption spectra were recorded by using a UV-Vis-NIR spectrometer (Thermo Fisher Scientific G10S) in the wavelength range of 700 to 1000 nm. The distribution of GNPs in polymer matrices was studied using a scanning electron microscope (SEM) JSM-6510A from JEOL Ltd, Tokyo, Japan.
Results and discussion
Curing study by DSC of the BZ (V-fa) monomer and V-fa/urethane copolymer at 50/50 wt/wt
The sample with a mass ratio of 50/50 of the V-fa/PU-prepolymer was chosen as a representative sample for the curing study. The DSC thermograms obtained for these uncured samples are displayed in Fig. 1. A non-symmetrical exothermic peak was recorded, revealing a two-stage curing process, that is, first the thermal ring opening homo-polymerization of the BZ monomer (Scheme 1) and then its copolymerization with the PU-prepolymer (Scheme 2). The initial curing temperature (Tinit) seems to be at about 150–160 °C and the peak temperature at 207 °C. This Tinit indicative of the BZ curing temperature, exhibits a lower value compared to that observed in conventional BZs such as BA-a (Tinit = 190–200 °C), and BA-35X (Tinit = 180–190 °C).17 The carbonyl group of the vanillin moiety seems to reduce the Tinit by catalyzing the ring-opening homopolymerization.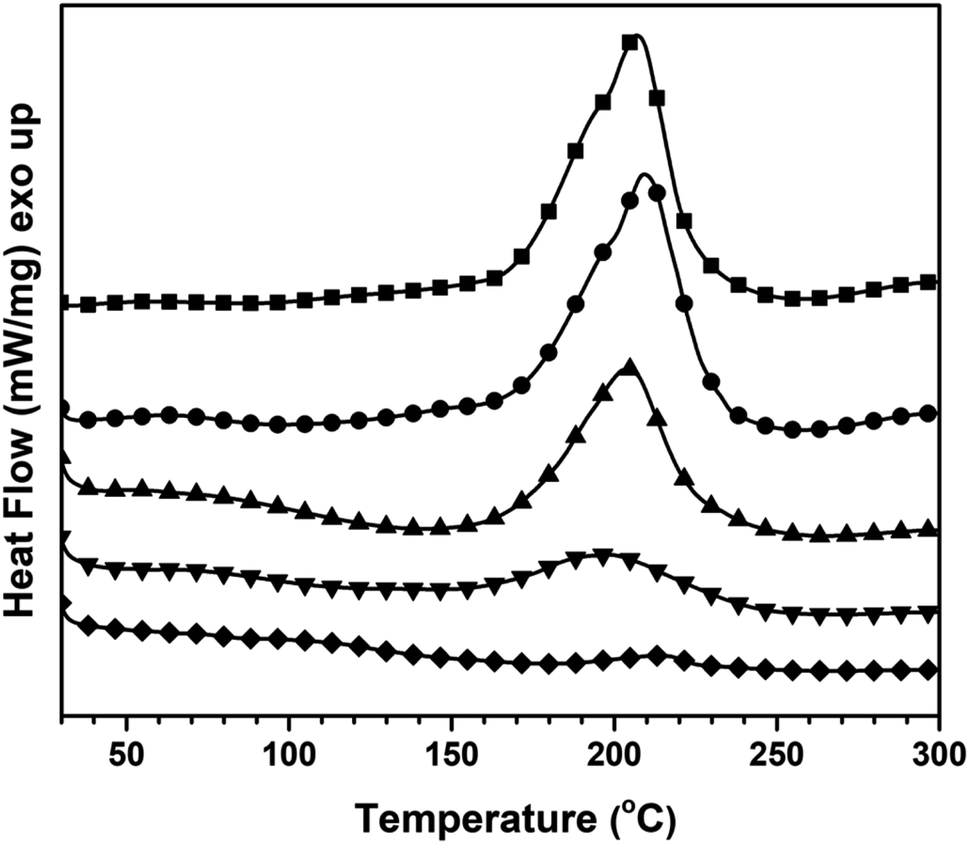 | ||
| Fig. 1 DSC thermograms of 50 wt% of V-fa in poly(V-fa/urethane) (■) uncured, (●) 150 °C/1 h, (▲) 160 °C/1 h, (▼) 170 °C/2 h, and (◆) 180 °C/2 h. | ||
The curing enthalpy (ΔH) obtained from the area under the exothermic peak was 70.2 J g−1. Fig. 1 also shows the curves obtained for the samples of the V-fa/PU-prepolymer binary mixture, which were first heated step wise at 150 °C (1 h), 160 °C (1 h), 170 °C (2 h), and 180 °C (2 h), and then placed in the DSC for further non-isothermal curing. The ΔH values obtained from these curves were 53.8, 35.9, 16.1 and 1.3 J g−1, respectively. The corresponding percentage conversions of poly(V-fa/urethane) were determined to be 23.3%, 48.8%, 77.1%, and 98.1%. A polymerization conversion of 98.1% was preferred for further studies. Kasemsiri et al. reported that bio-based polymers with curing conversions ranging from 80% to 98% demonstrate superior shape memory performances.18
FT-IR investigation
The FT-IR spectra of the V-fa monomer, palm oil polyol-based PU, the uncured V-fa/PU-prepolymer blend (50/50 wt/wt) and cured poly(V-fa/urethane) are presented in Fig. 2(a–d). In Fig. 2(a), the peak at 923 cm−1 is due to the characteristic vibration of the oxazine ring of BZ; and the peak noticed at 1229 cm−1 is associated with the –C–O–C– group of BZ. The presence of a furan group in the BZ structure was confirmed by the peaks at 760, 987, and 1583 cm−1; the peak at 1686 cm−1 corresponds to the carbonyl group (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of vanillin,19 and the characteristic band of hydroxyl groups (–OH) were observed at 3353 cm−1.20,21
O) of vanillin,19 and the characteristic band of hydroxyl groups (–OH) were observed at 3353 cm−1.20,21
 | ||
| Fig. 2 FT-IR spectra of (a) the V-fa monomer, (b) the PU-prepolymer, (c) the uncured V-fa/PU-prepolymer blend 50/50 wt/wt, and (d) cured poly(V-fa/urethane). | ||
The spectrum of the PU-prepolymer (Fig. 2(b)) showed a characteristic absorption peak of –CO stretching vibration at 1740 cm−1. The peaks at 2854 and 2930 cm−1 confirmed the presence of symmetric and asymmetric C–H stretching vibrations in the PU-prepolymer structure, respectively.20 The isocyanate (–NCO) group of the PU-prepolymer appeared at 2276 cm−1,22,23 and the stretching vibration of the ether group (–C–O–C–) appeared at 1063 cm−1.23 A broad peak at 3344 cm−1 was due to stretching vibration of the –NH bond,23,24 while that at 3443 cm−1 corresponded to the –OH stretching.25
The IR spectrum of the uncured V-fa/PU-prepolymer blend is displayed in Fig. 2(c), while the spectrum of the cured poly(V-fa/urethane) is presented in Fig. 2(d). Upon thermal ring-opening polymerization, the V-fa underwent a transformation, resulting in the disappearance of the absorption peak corresponding to the oxazine ring at 923 cm−1.
The distinctive absorption peak associated with the oxazine ring at 923 cm−1 was observed in the spectrum of BZ (Fig. 2(a)) and of the uncured blend V-fa/PU-prepolymer (Fig. 2(c)), while it was not present in the spectrum of the cured poly(V-fa/urethane) (Fig. 2(d)). Also, the broad peak of –NH at around 3344 cm−1 present in the spectrum of the PU-prepolymer (Fig. 2(b)) and that of the uncured V-fa/PU-prepolymer blend (Fig. 2(c)) was drastically decreased in the spectrum of cured poly(V-fa/urethane) (Fig. 2(d)). Furthermore, the characteristic peak of the isocyanate (–NCO) groups at 2276 cm−1 in the infrared spectrum of the PU-prepolymer (Fig. 2(b)) and that of the uncured blend (Fig. 2(c)) completely disappeared. All these results reveal that during the curing process oxazine ring opening takes place with the formation of BZ resin containing free –OH groups, as shown in Scheme 2; these hydroxyl groups are completely consumed then in the reaction with the free isocyanate groups of PU-prepolymer, which are also completely consumed. In a previous study of our research group26 and that of Takeichi et al., it was also found that the phenolic hydroxyl groups in BZ resin could react with the isocyanate group (–NCO) of a PU-prepolymer during curing.22
Thermal stability study of poly(V-fa), PU, poly(V-fa/urethane) and nanocomposites by TGA
The thermal properties of poly(V-fa), PU, and poly(V-fa/urethane) with 40–70 wt% of V-fa and the corresponding composite containing 3 wt% GNPs were investigated using TGA. The TGA thermograms obtained for all samples recorded from 25 °C to 800 °C at a rate of 20 °C min−1 under a N2 atmosphere are shown in Fig. 3(a) and (b). | ||
| Fig. 3 TGA thermograms of (a) neat poly(V-fa/urethane) and (b) poly(V-fa/urethane) nanocomposites with 3 wt% GNPs and V-fa/urethane: (■) 70/30, (●) 60/40, (▲) 50/50, and (▼) 40/60 composites. | ||
These showed that the presence of BZ enhanced the thermal stability of the resulting copolymers, evidenced by the increase in thermal degradation temperature at 10% weight loss (Td10) of the obtained copolymers from 336 °C to 345 °C. The increase in V-fa content resulted in an increase in the thermal stability, owing to the greater amount of aromatic content of BZ compared to that of the aliphatic structure-containing palm oil polyol.
Moreover, the increase in BZ content can also result in a higher degree of crosslinking as confirmed by an increase in E′ in the rubbery plateau of poly(V-fa/urethane) (DMA results). The residual weight of poly(V-fa/urethane) at V-fa contents of 70, 60, 50 and 40 wt% were determined to be 44, 36, 33 and 28 wt%, respectively. The percentage residual weight of poly(V-fa/PU) decreased when the PU mass fraction increased. These results are indicative of the influence of the chemical composition of polyurethane (PU), specifically the palm oil polyol, which exhibits a lower propensity for char formation when compared to the prevalent aromatic benzene rings present in the network structure of BZ.22,27,28
The degradation temperatures at 10% weight loss of poly(V-fa/urethane) at 70/30, 60/40, 50/50, and 40/60 wt/wt filled with 3 wt% GNPs were 352 °C 347 °C 343 °C, and 341 °C, respectively. Furthermore, it's noteworthy that the temperature at which maximum degradation (Tmax) occurs exhibits an upward trend with the progressive increase of V-fa content within the nanocomposites. Similarly, the residual weight of nanocomposites with 70%, 60%, 50% and 40% by weight of V-fa was determined to be 48, 41, 35 and 31 wt%, respectively. Also, Vallés et al. reported that the incorporation of GNPs resulted in a noteworthy enhancement of the thermal stability of the nanocomposites compared to the neat polymer.29 This improvement can be attributed to the establishment of a low flammability network of GNPs within the polymer matrix, which functions as a barrier, effectively impeding the release of decomposition products during combustion.
Dynamic mechanical properties of poly(V-fa/urethane) and nanocomposites
The results obtained from DMA of poly(V-fa/urethane) and poly(V-fa/urethane) nanocomposites are shown in Fig. 4 and 5. In Fig. 4(a), the E′ at the glassy state (−100 °C) of poly(V-fa/urethane) with a V-fa content of 70, 60, 50 and 40 wt% was 2.46, 2.13, 2.06, and 1.85 GPa, respectively. Thus E′ increases on increasing the V-fa content due to the presence of harder V-fa segments in the copolymer. | ||
| Fig. 4 Storage modulus of (a) neat poly(V-fa/urethane) and (b) poly(V-fa/urethane) nanocomposites filled with 3 wt% GNPs and V-fa/urethane: (■) 70/30, (●) 60/40, (▲) 50/50 and (▼) 40/60. | ||
 | ||
| Fig. 5 Loss tangent of (a) neat poly(V-fa/urethane) and (b) poly(V-fa/urethane) nanocomposites filled with 3 wt% GNPs and V-fa/urethane: (■) 70/30, (●) 60/40, (▲) 50/50 and (▼) 40/60. | ||
As depicted in Fig. 4(b), the E′ at −100 °C of poly(V-fa/urethane) nanocomposites with V-fa/urethane ratios of 70/30, 60/40, 50/50 and 40/60 wt/wt was found to be 3.46, 3.09, 2.31, and 2.10 GPa, respectively. This increase in E′ could be explained by the excellent reinforcing action of GNPs which improves the stiffness of these copolymers.
From the loss tangent curve peak maximum as presented in Fig. 5(a) and (b), the Tg values of neat poly(V-fa/urethane) and nanocomposites were determined. The copolymers exhibited two peak maxima, e.g. two Tg; the first one at −44 °C was attributed solely to the PU domains and the second at a higher temperature is due to the contribution of both V-fa and PU domains. The second Tg was found to be at 22 °C, 20 °C, 16 °C and 8 °C for poly(V-fa/urethane) with mass ratios of 70/30, 60/40, 50/50 and 40/60, respectively. This result, i.e. the existence of two Tg suggests that the obtained poly(V-fa/urethane) was partially miscible, resulting in heterogeneous network formation, composed of PU and BZ domains. The PU domain acted as the soft segment or the switch phase, while the BZ domain acted as the hard segment or the stable phase. The Tg value of poly(V-fa/urethane) was increased, with the content of BZ, because BZ has a molecular structure with higher rigidity, due to inter and intra-molecular forces than PU.30,31
The poly(V-fa/urethane) nanocomposites at 70/30 and 40/60 also exhibited two Tg as the non-filled copolymer samples. However, a third peak maximum for nanocomposites with poly(V-fa/urethane) at mass ratios of 60/40 and 50/50 was observed. Additionally, the values of the peak maxima showed a significant shift to higher values compared to their non-filled counterparts.
According to Fig. 5(a) and (b), the first observed peak maximum attributed to the PU domain in the copolymers was approximately −41 °C which increased as the V-fa content increased. As for the second observed peak maximum attributed to poly(V-fa/urethane), the Tg of the poly(V-fa/urethane) nanocomposites was about 41 °C, 30 °C, 22 °C and 12 °C for poly(V-fa/urethane) at mass ratios of 70/30, 60/40, 50/50 and 40/60, respectively. Finally, the third peak maximum was attributed to the restriction of the molecular chain mobility of the copolymer by GNPs. The incorporation of GNPs resulted in an increase in the Tg values. This phenomenon can be attributed to the limitation in molecular chain mobility, subsequently causing a reduction in the free volume within the copolymer matrix.32–35
Shape memory behaviour investigation
All specimens were investigated to evaluate the shape memory behaviours which were reported as shape fixity and shape recovery values. In the first step, the sample was heated in a hot-air oven at 20 °C greater than the Tg value while being subjected to external force, bending its shape by 90°. The sample was then cooled to about −10 °C and then the bending force was disconnected to get a sample with a bending shape of about 90°. The shape fixity (Rf) values for all samples were calculated using eqn (1).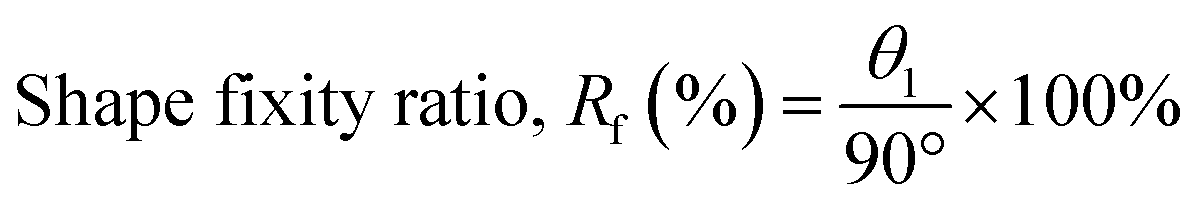 | (1) |
During the shape recovery process, the bending edge of deformed specimen was triggered by NIR light (intensity of 500 mW) at 808 nm in the 60 cm fixed gap of the specimen from the NIR light source. The bent polymer shape was returned to the original shape by NIR light actuation. The irradiated polymer absorbed the light energy which was converted into heat, resulting in an increase in its temperature. The specimens were irradiated until the recorded temperature was above the Tg. The shape recovery (Rr) values were calculated using eqn (2).
 | (2) |
The ability to memorize and retain the temporary shape of poly(V-fa/urethane) was observed as 86%, 90%, 92%, and 95% performance with an increase in V-fa content from 40 to 70 wt% respectively (Fig. 6). In general, the SMP polymers consist of two segments, e.g. hard segments (net points) and soft segments (switch units). The hard segments act as the fixed phase, while the soft segments act as the reversible phase. The shape fixity tends to increase on increasing the V-fa content, due to the higher amount of hard segments which prevent the polymer chains from sliding.
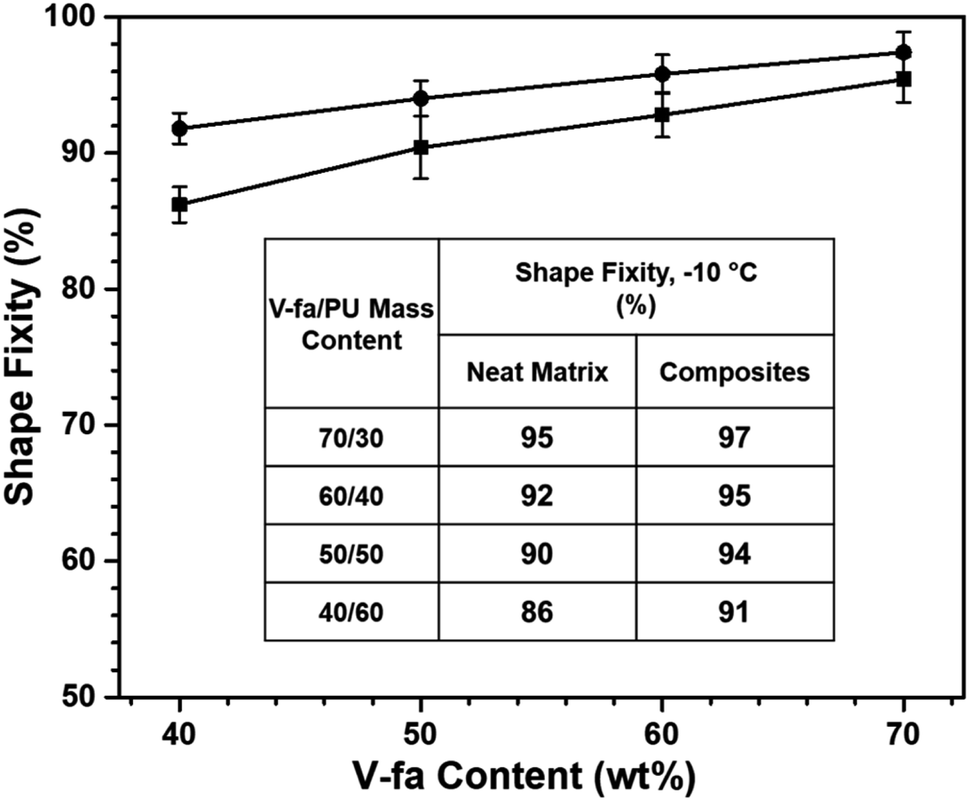 | ||
| Fig. 6 Shape fixity value of (■) neat poly(V-fa/urethane) and (●) poly(V-fa/urethane) nanocomposites filled with 3 wt% GNPs. | ||
The values of shape fixity of poly(V-fa/urethane) nanocomposites with V-fa/urethane of 70/30, 60/40, 50/50, and 40/60 wt/wt, reinforced with 3 wt% GNPs are 97%, 95%, 94%, and 91%, respectively. These values are higher than those of unfilled copolymers, due to the presence of GNPs, which possibly restrict the copolymer chain mobility, thus improving the hard segment in poly(V-fa/urethane). Yadav and Cho reported that hydroxyl groups (–OH) in GNPs can promote the covalent bonding between GNPs and the polymer matrix,36 which could serve as additional net points within the system.
The values of shape recovery (Fig. 7) obtained for poly(V-fa/urethane) with the ratios of 70/30, 60/40, 50/50, and 40/60 wt/wt are 93%, 97%, 99% and 100%, respectively. These values experienced a reduction as the V-fa percentage was increased, primarily due to the decreased proportion of PU within the system. Consequently, the augmentation of PU content, acting as the soft segment within the poly(V-fa/urethane) matrix, led to a notable improvement in the shape recovery performance.
 | ||
| Fig. 7 Shape recovery value of (■) neat poly(V-fa/urethane) and (●) poly(V-fa/urethane) nanocomposites filled with 3 wt% GNPs. | ||
The values of shape recovery of poly(V-fa/urethane) nanocomposites with a V-fa/urethane composition of 70/30, 60/40, 50/50, and 40/60 wt/wt, reinforced with 3 wt% GNPs were observed to be 94%, 98%, 99%, and 100%, respectively; these are slightly higher than those of the corresponding unfilled copolymers. This observed result shows that the restriction of molecular copolymer chain mobility by GNPs did not affect the shape recovery performance of nanocomposites. Furthermore, the nanocomposites showed a rubbery plateau between 0 °C and 150 °C in the E′ curve, characteristic of an elastomer behaviour, and have high molecular chain mobility, resulting in a good recovery performance. The shape recovery values of nanocomposites were enhanced by force absorption and entropic elasticity phenomena. The incorporation of GNPs into the poly(V-fa/urethane) matrix also facilitates the absorption of external forces subsequent to the fixation of the temporary shape. Upon actuation of the fixed temporary shape using NIR light, the stored mechanical energy or stress is released in accordance with the entropy principle. This release enables the specimens to restore their original shape through the application of this force.37
Recovery time (Rt) of poly(V-fa/urethane) and poly(V-fa/urethane) nanocomposites
The recovery time (Rt) of neat poly(V-fa/urethane) reduced from 37 s to 18 s on increasing the PU content. Furthermore, with the incorporation of 3 wt% GNPs within the copolymer matrix, the recovery time was further reduced from 28 s to 13 s, as demonstrated in Fig. 8. | ||
| Fig. 8 Recover time under NIR light actuation of poly(V-fa/urethane): (■) neat matrix, and (●) GNP filled nanocomposites. | ||
This observed phenomenon can be attributed to the ability of GNPs to enhance the thermal conductivity and improve the absorption of NIR. As a result, the photothermal efficiency was increased, leading to improved heat distribution throughout the copolymer matrix, facilitated by an efficient heat transfer process. In the shape recovery process, the GNPs could enhance the recovery force. Therefore, the Rt was related both to the thermal conductivity and recovery force.37 Srisaard et al. and Yang et al. also reported that the addition of GNPs into a matrix could help facilitate improvement in Rt during the shape recovery process.11,38
Furthermore, we conducted a comparative analysis of the shape memory performance of our 50/50 poly(V-fa/PU) nanocomposites filled with 3 wt% GNPs, against closely related systems such as (2) 50/50 poly(V-fa/ECO) filled 3 wt% GNPs,11 (3) 50/50 poly(V-fa/ECO) filled 0.3 wt% MWCNTs,39 (4) 60/40 BA-a/TDI based-PU copolymer,31 and (5) MDI based-PU copolymerized with 17.5 wt% BA-a,40 and the results are summarized in Table 1.
It can be seen that our developed poly(V-fa/PU) nanocomposite exhibits slightly superior shape fixity when compared to those two bio-based V-fa/ECO composite systems, e.g. (2) and (3), which is also better than that of those two synthetic systems e.g. (4) and (5). Moreover, the poly(V-fa/PU) nanocomposite exhibits a lower glass transition temperature, enabling a more efficient shape recovery process at lower temperatures. Consequently, poly(V-fa/PU) and its nanocomposites emerge as promising candidates for applications in the human body. Furthermore, our poly(V-fa/PU) nanocomposites display multiple shape memory behaviors with impressive performance, detailed in Fig. 11–15. It is noteworthy that, in comparison to our work, there are no reported instances of a bio-based system i.e. poly(V-fa/ECO), exhibiting multiple shape memory behaviors. The only closely related system displaying multiple shape memory effects was found in a synthetic SMP based on BA-a/PU copolymer.
Photothermal behaviours
The poly(V-fa/urethane) nanocomposites were tested to study their photothermal behaviours by exposing them to NIR light (808 nm, 500 mW). The temperature of specimens was continuously recorded with a thermocouple. Poly(V-fa/urethane) with 50 wt% of V-fa and 3 wt% of GNPs was taken as a representative sample for NIR exposure at a specific time period and the results are presented in Fig. 9. The temperature of neat poly(V-fa/urethane) increased from RT to 53 °C within 60 s after exposure to NIR light. Furthermore, with the incorporation of GNPs into the polymer matrix, the temperature increased at a higher rate up to a temperature of 65 °C. During the absorption of NIR light by neat poly(V-fa/urethane) and nanocomposites photon energy was converted to thermal energy.22 Therefore, the nanocomposites displayed improved photothermal behaviour compared to the neat poly(V-fa/urethane) matrix. | ||
| Fig. 9 Variation in temperature of poly(V-fa/urethane) (50/50 wt/wt) with irradiation time: (■) neat poly(V-fa/urethane) and (●) 3 wt% of GNP filled poly(V-fa/urethane) nanocomposites. | ||
The investigation of photothermal properties under NIR irradiation is substantiated by the vis-NIR absorption analysis. As depicted in Fig. 10, the vis-NIR absorption characteristics of pure poly(V-fa) were significantly higher than those of the poly(V-fa/urethane) within the wavelength range of 700–1000 nm.
 | ||
| Fig. 10 Vis-NIR absorption spectra of poly(V-fa/urethane) (50/50 wt/wt) with irradiation time: (●) neat poly(V-fa/urethane), (▲) neat poly(V-fa) and (■) 3 wt% of GNP filled poly(V-fa/urethane) nanocomposites. | ||
This observation suggests that the V-fa monomer exhibits remarkable capabilities in enhancing vis-NIR absorption. The NIR light absorption exhibited by poly(V-fa) can be elucidated by the influence of π–π interactions occurring between polymer chains within the V-fa molecules. These interactions facilitate enhanced absorption in the NIR spectrum and improved conversion to thermal energy.41 Moreover, poly(V-fa/urethane) filled with a GNP nanocomposite showed the highest vis-NIR absorption performance, compared with neat poly(V-fa) and neat poly(V-fa/PU). This phenomenon demonstrates that GNPs are an effective photothermal filler for preparing light-induced SMPs.11,38
Poly(V-fa/urethane) and poly(V-fa/urethane) nanocomposites as multiple-SMPs
The range between the onset and endset temperature of the loss tangent curve (Fig. 5), is an important factor for the multiple-shape memory process. Using this temperature range of the copolymer matrix, the deformation to other temporary shapes could be achieved. The loss tangent curve ranges of poly(V-fa/urethane) are presented in Table 2. A copolymer with 60 wt% of V-fa showed the maximum value of 123 °C for the loss tangent curve range. Consequently, both the neat poly(V-fa/urethane) (60/40 wt/wt) and its nanocomposite underwent a designed thermo-programming process as illustrated in Fig. 11, to evaluate their multiple shape memory performance.| V-fa/urethane mass ratio | T g (°C) | Loss tangent curve range (°C) | T g range (°C) |
|---|---|---|---|
| 70/30 | 22 | −12 to 77 | 89 |
| 60/40 | 20 | −24 to 99 | 123 |
| 50/50 | 16 | −15 to 102 | 117 |
| 40/60 | 8 | −18 to 97 | 115 |
 | ||
| Fig. 11 Photographs of multiple-shape memory behaviours of poly(V-fa/urethane) (60/40 wt/wt). | ||
Based on the data presented in Fig. 12, the first temporary shape fixity values of neat poly(V-fa/urethane) (60/40) and of GNP reinforced poly(V-fa/urethane) nanocomposites were measured to be 93% and 97%, respectively. This improvement in shape fixity was attributed to an addition of GNPs into the copolymer matrix. Additionally, the shape fixity value of the second deformed shape also exhibited an increase from 87% to 94%, respectively.
 | ||
| Fig. 12 Shape fixity of (▥) neat and (▤) 3 wt% of GNP reinforced poly(V-fa/urethane) (60/40 wt/wt) nanocomposites for multiple SMP. | ||
Shape recovery (Rr) is also a key point to point out the shape memory performance. Fig. 13 reveals the first- and second-shape recovery performance of neat poly(V-fa/urethane) (60/40) and of the GNP reinforced poly(V-fa/urethane) (60/40) nanocomposite. The shape recoverability of these two systems was found to be 86% and 89%, respectively.
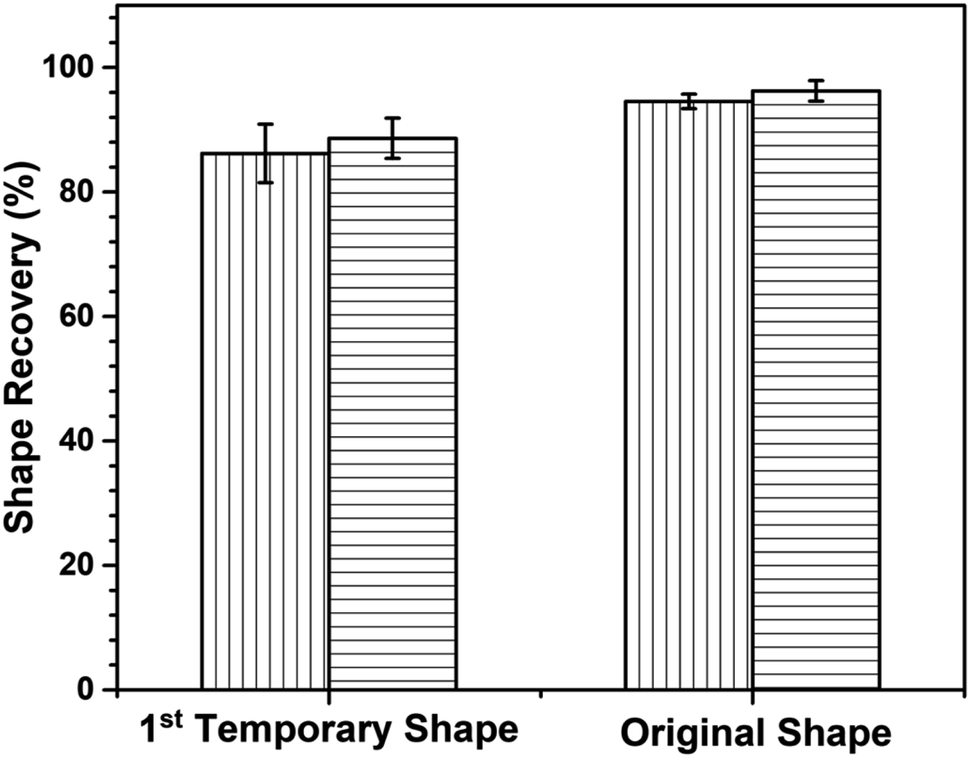 | ||
| Fig. 13 Shape recovery of (▥) neat and (▤) 3 wt% of GNP reinforced poly(V-fa/urethane) (60/40 wt/wt) composites for multiple SMPs. *Rr of the 1st temporary shape refers to the performance of the SMP recovered from the 2nd deformed shape state to the 1st deformed shape state. *Rr of the original shape refers to the performance of the SMP recovered from the 1st deformed shape state to the original shape. | ||
The enhancement in shape recoverability owing to the presence of GNPs in the poly(V-fa/urethane) matrix could enhance the absorption of external force during the shape deformation and entropic elasticity phenomena,37 and after the fixed temporary shape was actuated with NIR light, owing to the entropy principle, the stored mechanical energy or stress was released, allowing the specimens to recover to their initial shape by using this force. Additionally, the shape recoverability of the specimens from the first-deformed shape state to their original shape was also investigated. The poly(V-fa/urethane) nanocomposites also showed a higher shape recovery value than the neat poly(V-fa/urethane), 96% and 94%, respectively.
The number of shape memorization cycles serves as a crucial parameter that gauges the efficacy of materials in maintaining their shape memory performance. Fig. 14 shows the cycle count for both the first- and second-temporary shape fixation of a multiple shape memory polymer, comprising poly(V-fa/urethane) (60/40 wt/wt) nanocomposites reinforced with 3 wt% GNPs. Notably, it is observed that the shape fixity performance of the poly(V-fa/urethane) nanocomposites exhibited a degradation of less than 10% after undergoing deformation in the fourth cycle for the first shape fixation, and in the second cycle for the second shape fixation.
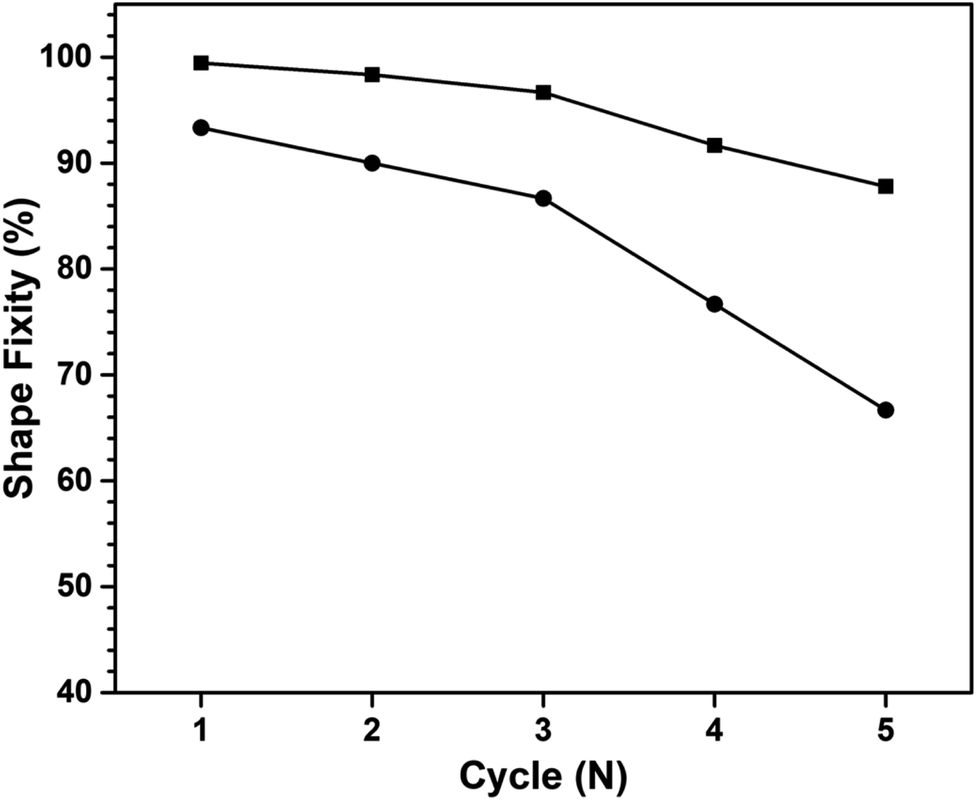 | ||
| Fig. 14 Shape fixity of 3 wt% of GNP reinforced poly(V-fa/urethane) (60/40 wt/wt) nanocomposites: (■) 1st temporary shape and (●) 2nd temporary shape. | ||
In addition, Fig. 15 illustrates the shape recovery performance of these nanocomposites at each subsequent deformation cycle. It is evident that the shape recoverability of poly(V-fa/urethane) nanocomposites decreased as multiple shape memory features decreased with each successive recovery cycle. This decrease can be attributed to the accumulation of internal stress during each subsequent recovery cycle which may contribute to fatigue within the sample, causing debonding or non-entanglement of the bonds, thereby deteriorating the shape memory performance.
 | ||
| Fig. 15 Shape recovery of 3 wt% of GNP reinforced poly(V-fa/urethane) (60/40 wt/wt) nanocomposites: (■) 1st temporary shape, and (●) 2nd temporary shape. | ||
Morphology and filler dispersion
The dispersion of GNPs in the poly(V-fa/urethane) nanocomposites was observed by SEM. The SEM micrographs of neat poly(V-fa/urethane) (60/40 wt/wt) and 3 wt% of GNP reinforced poly(V-fa/urethane) (60/40 wt/wt) are presented in Fig. 16(a) and (b), respectively. The micrographs reveal the well thoroughly distributed PU domains within the poly(V-fa/urethane) matrix, and that of the GNPs were also well dispersed with good adhesion across the V-fa/urethane copolymer matrix. | ||
| Fig. 16 SEM images of (a) neat poly(V-fa/urethane) and (b) 3 wt% of GNP reinforced poly(V-fa/urethane) (60/40 wt/wt) nanocomposites. | ||
Conclusions
The successful synthesis of bio-based copolymers comprising bio-based BZ and bio-based PU was achieved within the range of 40 to 70 wt% for the V-fa monomer and PU, as confirmed through FT-IR spectroscopy analysis. For the development of NIR-responsive shape memory polymers (SMPs), the V-fa/urethane copolymers were further reinforced with 3 wt% of GNPs. The inclusion of GNPs led to an enhancement in the photo-thermal behaviour of the poly(V-fa/urethane) matrix. Remarkably, the incorporation of GNPs notably improved the dynamic mechanical properties and overall performance of the SMPs. The superior shape memory performance of the NIR light-responsive SMP was observed in poly(V-fa/urethane) (50/50 wt/wt) reinforced with 3 wt% GNPs, where a shape fixity of 94%, shape recovery of 99%, and shape recovery time of 16 s were achieved. Additionally, the poly(V-fa/urethane) (60/40 wt/wt) nanocomposites demonstrated the ability to exhibit multiple shape memory behaviours. Furthermore, these prepared copolymers and nanocomposites exhibited capabilities to be remotely activated and achieve site-specific shape recovery under NIR irradiation.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This research endeavour received financial support from the National Research Council of Thailand (NRCT) and Chulalongkorn University (N42A660910), the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (PMU-B) (B05F640086), and Thailand Science Research and Innovation Fund Chulalongkorn University (No. 6641/2566).Notes and references
- F. Guo and Z. Guo, RSC Adv., 2016, 6, 36623–36641 RSC.
- Y. Yin and J. A. Rogers, Chem. Rev., 2022, 122, 4885–4886 CrossRef CAS PubMed.
- S. D. Mahapatra, P. C. Mohapatra, A. I. Aria, G. Christie, Y. K. Mishra, S. Hofmann and V. K. Thakur, Adv. Sci., 2021, 8, e2100864 CrossRef.
- Y. Xia, Y. He, F. Zhang, Y. Liu and J. Leng, Adv. Mater., 2021, 33, e2000713 CrossRef PubMed.
- P. Theato, B. S. Sumerlin, R. K. O'Reilly and T. H. Epps 3rd, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- L. Montero de Espinosa, W. Meesorn, D. Moatsou and C. Weder, Chem. Rev., 2017, 117, 12851–12892 CrossRef CAS PubMed.
- H. Meng and G. Li, Polymer, 2013, 54, 2199–2221 CrossRef CAS.
- F. Pilate, A. Toncheva, P. Dubois and J.-M. Raquez, Eur. Polym. J., 2016, 80, 268–294 CrossRef CAS.
- T. Li, Y. Li, X. Wang, X. Li and J. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 9470–9477 CrossRef CAS.
- S. Srisaard, L. Amornkitbamrung, K. Charoensuk, C. Sapcharoenkun, C. Jubsilp and S. Rimdusit, J. Intell. Mater. Syst. Struct., 2021, 33, 547–557 CrossRef.
- T. Takeichi, T. Kawauchi and T. Agag, Polym. J., 2008, 40, 1121–1131 CrossRef CAS.
- H. Ishida, in Handbook of Benzoxazine Resins, ed. H. Ishida and T. Agag, Elsevier, Amsterdam, 2011, ch. 3–81, pp. 3–81 Search PubMed.
- B. Lochab, M. Monisha, N. Amarnath, P. Sharma, S. Mukherjee and H. Ishida, Polym., 2021, 13(8), 1260 CAS.
- G. Lligadas, A. Tüzün, J. C. Ronda, M. Galià and V. Cádiz, Polym. Chem., 2014, 5, 6636–6644 RSC.
- S. Rimdusit, C. Jubsilp and S. Tiptipakorn, in Alloys and Composites of Polybenzoxazines, Springer Singapore, 1 edn 2013, p. 159 Search PubMed.
- S. Rimdusit, P. Kunopast and I. Dueramae, Polym. Eng. Sci., 2011, 51, 1797–1807 CrossRef CAS.
- P. Kasemsiri, N. Lorwanishpaisarn, U. Pongsa and S. Ando, Polym., 2018, 10, 482 Search PubMed.
- N. K. Sini, J. Bijwe and I. K. Varma, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 7–11 CrossRef CAS.
- T. Parnklang, K. Boonyanuwat, P. Mora, S. Ekgasit and S. Rimdusit, eXPRESS Polym. Lett., 2019, 13, 65–83 CrossRef CAS.
- M. Zeng, T. Pang, J. Chen, Y. Huang, Q. Xu and Y. Gu, J. Mater. Sci.: Mater. Electron., 2018, 29, 5391–5400 CrossRef CAS.
- T. Takeichi, Y. Guo and T. Agag, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4165–4176 CrossRef CAS.
- H. Kalita and N. Karak, J. Appl. Polym. Sci., 2014, 131, 39579 CrossRef.
- E. Głowińska and J. Datta, Cellulose, 2015, 23, 581–592 CrossRef.
- E. F. Assanvo, P. Gogoi, S. K. Dolui and S. D. Baruah, Ind. Crops Prod., 2015, 65, 293–302 CrossRef CAS.
- S. Rimdusit, S. Tiptipakorn, C. Jubsilp and T. Takeichi, React. Funct. Polym., 2013, 73, 369–380 CrossRef CAS.
- T. Takeichi and Y. Guo, Polym. J., 2001, 33, 437–443 CrossRef CAS.
- S. Rimdusit, S. Pirstpindvong, W. Tanthapanichakoon and S. Damrongsakkul, Polym. Eng. Sci., 2005, 45, 288–296 CrossRef CAS.
- C. Vallés, A. M. Abdelkader, R. J. Young and I. A. Kinloch, Compos. Sci. Technol., 2015, 111, 17–22 CrossRef.
- P. Zhao, Q. Zhou, Y. Deng, R. Zhu and Y. Gu, RSC Adv., 2014, 4, 238–242 RSC.
- P. Prathumrat, S. Tiptipakorn and S. Rimdusit, Smart Mater. Struct., 2017, 26, 065025 CrossRef.
- M. Martin-Gallego, R. Verdejo, M. A. Lopez-Manchado and M. Sangermano, Polymer, 2011, 52, 4664–4669 CrossRef CAS.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2010, 20, 1982–1992 RSC.
- T. Ramanathan, S. Stankovich, D. A. Dikin, H. Liu, H. Shen, S. T. Nguyen and L. C. Brinson, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2097–2112 CrossRef CAS.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Compos. Sci. Technol., 2016, 137, 44–51 CrossRef CAS.
- S. K. Yadav and J. W. Cho, Appl. Surf. Sci., 2013, 266, 360–367 CrossRef CAS.
- S. Lashgari, M. Karrabi, I. Ghasemi, H. Azizi, M. Messori and K. Paderni, eXPRESS Polym. Lett., 2016, 10, 349–359 CrossRef CAS.
- Z. Yang, Q. Wang and T. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 21691–21699 CrossRef CAS.
- W. Prasomsin, T. Parnklang, C. Sapcharoenkun, S. Tiptipakorn and S. Rimdusit, Nanomater., 2019, 9, 881 CrossRef.
- N. Erden and S. C. Jana, Macromol. Chem. Phys., 2012, 214, 1225–1237 CrossRef.
- L. Amornkitbamrung, S. Srisaard, C. Jubsilp, C. W. Bielawski, S. H. Um and S. Rimdusit, Polymer, 2020, 209, 122986 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |