
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultichannel nitrogen-doped carbon fiber confined Fe3C nanoparticles for efficient electroreduction of nitrate†
Fangzhou
Zhang
a,
Zhangsheng
Shi
 c,
Junliang
Chen
a,
Hongxia
Luo
a,
Jun
Chen
*b and
Jianping
Yang
*a
c,
Junliang
Chen
a,
Hongxia
Luo
a,
Jun
Chen
*b and
Jianping
Yang
*a
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, P. R. China. E-mail: jianpingyang@dhu.edu.cn
bIntelligent Polymer Research Institute, Australian Institute of Innovative Materials, Faculty of Engineering and Information Sciences, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: junc@uow.edu.au
cDepartment of Chemistry, City University of Hong Kong, Hong Kong SAR, China
First published on 5th February 2024
Abstract
Electrochemical conversion of nitrate into benign dinitrogen is a promising solution for water purification and environmental remediation. The development of environmentally friendly electrocatalysts possessing excellent catalytic activity and stability has attracted increasing attention. Herein, a 1D hierarchical architecture with uniformly dispersed Fe3C nanoparticles confined in multichannel nitrogen-doped carbon fibers (Fe3C/MNCFs) is reported as a highly efficient NO3RR electrocatalyst. Fe3C/MNCFs-800 demonstrates a nitrate conversion of 90.9%, an N2 selectivity of 99.53%, and up to 15 cycles of electrocatalytic stability. The excellent electrocatalytic activity is proposed to be mainly due to the multichannel fibrous architecture beneficial for exposing more active sites and facilitating mass diffusion. Moreover, the strong interaction between active species and fibrous support guarantees the chemical stability and long cycle life. This work provides a reference for the development of high-performance noble-metal-free electrocatalysts for eco-friendly nitrate reduction.
Broader contextNitrate is widely distributed in industrial wastewater, nuclear wastewater, and contaminated groundwater, which has increasingly provoked ecological and environmental concerns. The electrochemical nitrate reduction reaction (NO3RR) has emerged as a promising approach for converting NO3− into harmless product N2. It is indispensable to develop electrocatalysts with higher catalytic activity and selectivity for the NO3RR. In this study, we report a novel 1D hierarchical architecture with uniformly dispersed Fe3C nanoparticles confined in multichannel nitrogen-doped carbon fibers (Fe3C/MNCFs) as an efficient NO3RR electrocatalyst. Fe3C/MNCFs-800 demonstrates a nitrate conversion of 90.9%, an N2 selectivity of 99.53%, and up to 15 cycles of electrocatalytic stability. This work provides new insights into the design of cost-effective NO3RR catalysts for water purification and environmental remediation. |
Introduction
Anthropogenic activities such as fossil-fuel combustion, overfertilization, and discharge of wastewater have caused an imbalance in the global nitrogen cycle, posing a major threat to the environment and public health.1–3 Therefore, the World Health Organization (WHO) has stated that the nitrate concentration in public water systems should not exceed 10 mg L−1.4 Engineered reduction of excessive NO3− to benign dinitrogen (N2) is becoming progressively essential. Among the current state-of-the-art denitrification methods, the electrocatalytic nitrate reduction reaction (NO3RR) has been considered a promising way because of the high energy efficiency, moderate operating conditions, and no chemical input.5–8Catalytic reduction capacity and selectivity for N2 generation are vital factors for the NO3RR. Many studies suggest that low-cost and nontoxic iron-based electrocatalysts exhibit high reduction ability in the conversion of NO3−–N to NH4+, which could be further converted to N2 through the electrochlorination process around the anode.9 A series of iron-based materials have been found to be active towards the NO3RR, such as zero-valent iron, iron nitride, carbon-supported Fe–Nx/C compounds, iron-based bimetallic alloys, etc.10–12 Recently, a new type of iron-based catalyst Fe3C, whose lattice contains carbon atoms located in trigonal prismatic interstices among close-packed iron atoms with space group C(4c) (a = 5.032 Å, b = 6.708 Å, and c = 4.477 Å), has been reported as an ideal NO3RR catalyst.13,14 Such a crystalline structure leads to high activation of iron atoms on the carbide surface, making it an efficient catalyst toward accelerating the conversion of NO3−.15 Furthermore, it has been confirmed that the metallic Fe3C possesses small work functions, which are favorable for the charge transfer kinetics and beneficial for the NO3RR performances.16
Nevertheless, phase-pure Fe3C nanoparticles (NPs) inevitably undergo agglomeration and polydispersion during catalytic reactions, leading to the low catalytic capacity and durability. It has been confirmed that the NPs can be dispersed onto various solid substrates to enhance the accessibility of active sites. Numerous heterogeneous substrates, such as mesoporous carbon, graphene, carbon nanotubes, and carbon fibers, have been used to disperse NPs and regulate their particle size to create more active sites.17,18 Compared with other carbon-based materials, N-doped carbon fibers are more attractive since the one-dimensional (1D) nanostructures could fully expose active-sites in the radial direction and promote charge transfer along the axial dimension in microns.19,20 Furthermore, the uniform dissolution of the metal precursor in the electrospun solution enables good dispersion of metal species in the fiber matrix.21 Previous studies have found that N-doping facilitates the adsorption of nitrate, and the continuous fibrous structure could enhance the stability of the catalyst and prevent the corrosion of the active sites.22 However, the lack of interconnected pores leads to the poor exposure of NPs and thus results in lower catalytic activity. Therefore, constructing effective fiber-based electrocatalysts with tailored void space is considered vital for maximizing available active sites and facilitating mass transport.
Herein, we demonstrate the synthesis of uniformly dispersed Fe3C NPs confined in multichannel nitrogen-doped carbon fibers (Fe3C/MNCFs) via electrospinning and subsequent pyrolysis. The 1D hierarchical structure with highly parallel channels ensures good electrical contact between the catalyst and support, abundant active sites, and sufficient space for mass transfer. Moreover, the strong interaction between Fe3C NPs and nitrogen-doped carbon fibers helps to stabilize Fe3C NPs without aggregation or detachment. When used as the NO3RR electrocatalyst, the Fe3C/MNCFs demonstrate ultrahigh nitrate conversion of 90.9% and nitrogen selectivity of 99.53%, and robust stability in neutral media. This strategy for the fabrication of a unique fibrous structure will open a new chapter in the development of highly efficient electrocatalysts for the NO3RR.
Experimental
Preparation of electrospun nanofibers (PAN/PS/Fe(acac)3)
In a typical synthesis, 600 mg PAN and 600 mg PS were added into 7 mL DMF with stirring overnight at room temperature to form a homogeneously dispersed solution. Then 200 mg of Fe(acac)3 was added into the above solution with vigorous stirring for 6 h. Then, PAN/PS/Fe(acac)3 nanofibers were electrospun on an Al foil collector from the precursor solution. The voltage, feeding rate, and the distance between the syringe and collector are fixed at 16 kV, 0.8 mL h−1, 16 cm, respectively. The obtained electrospun nanofibers were dried at 60 °C overnight in a vacuum. Similarly, Fe(acac)3 was also replaced with Co(acac)2 and Ni(acac)2 to synthesize a series of nanofibers, defined as PAN/PS/Co(acac)2 and PAN/PS/Ni(acac)2.Synthesis of Fe3C nanoparticle anchored multichannel carbon fibers (Fe3C/MNCFs)
The as-obtained PAN/PS/Fe(acac)3 nanofiber film was preoxidated in air at 200 °C for 4 h at a heating rate of 5 °C min−1. After that, the PAN/PS/Fe(acac)3 nanofiber film was annealed at 700, 800, 900 °C for 2 h under a H2/Ar atmosphere with a ramp rate of 2 °C min−1 to form the Fe3C/MNCFs.Materials characterization
Scanning electron microscopy (SEM) images were taken using a TESCAN/MAIA3 (Czech Republic). Transmission electron microscopy (TEM) images and elemental mappings were obtained at 200 kV using a Talos F200S. X-ray diffraction (XRD) patterns were recorded using a Rigaku D/Max-2550 PC. Thermogravimetric analysis (TGA) was performed using NETZSCH STA449F5 apparatus under an air atmosphere. Raman spectra were recorded using a Dilor LabRam-1B Raman spectrometer with an excitation wavelength of 633 nm. X-ray photoelectron spectroscopy (XPS) was performed on an Escalab 250 Xi. The N2 isothermal adsorption/desorption was recorded using a Quantachrome Autosorb-Iq. DMPO spin-trapping ESR spectra were obtained using a EMXnano (Germany). Thermogravimetric analysis (TGA) was performed using a NETZSCH STA449F5 system under an air atmosphere.Electrocatalytic measurements
Electrochemical measurements were carried out by using a Bio-Logic VMP3 electrochemical analyzer system in a three-electrode cell. The platinum and saturated calomel electrodes were used as the counter and reference electrodes, respectively. To prepare the working electrode, 4 mg catalysts and 0.5 mg carbon black were dispersed in 50 μL, 10 wt% polyvinylidene fluoride, and sonicated for 10 min to form a homogenous ink. Then the ink was drop-cast on the nickel foam (1 × 1 cm) and dried at 70 °C under vacuum for 12 h.Before the nitrate electroreduction test, linear sweep voltammetry (LSV) was performed until the polarization curves achieved a steady state at a rate of 10 mV s−1 from −0.5 to −2 V. Then, the potentiostatic test was carried out under different conditions, including applied potentials (−1.1, −1.2, −1.3, −1.4, and −1.5 V), electrolyte, and NO3−–N concentrations (25, 50, 100, 150, and 200 mg L−1).
Determination of ion concentration
An ultraviolet-visible (UV-Vis) spectrophotometer was used to detect the ion concentration of pre- and post-test electrolytes after diluting to appropriate concentration to match the range of calibration curves. The specific detection methods from a previous work by our group are employed in this study.Calculation of the nitrate conversion and N2 selectivity
The nitrate conversion and N2 selectivity were evaluated using the following equations: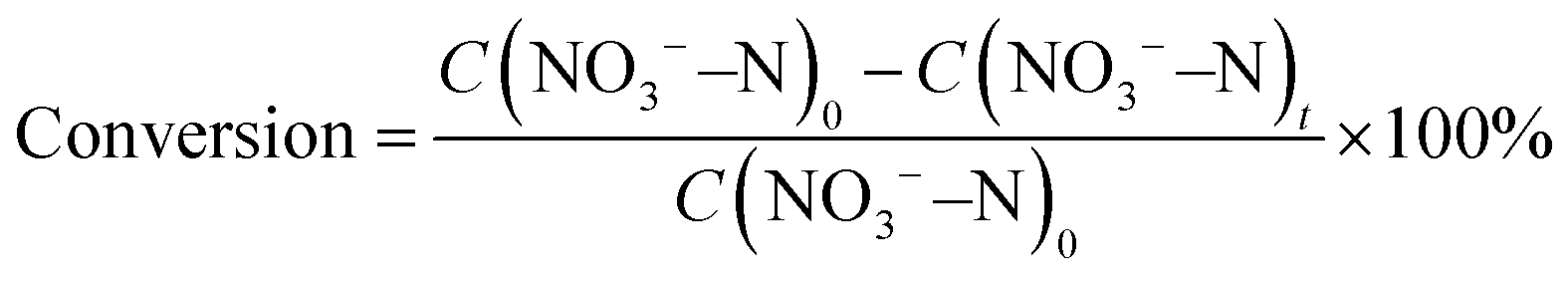
where C(NO3−–N)0 is the initial concentration (mg L−1) of NO3−–N, C(NO3−–N)t is the concentration (mg L−1) of NO3−–N at time t, and ΔC(NO3−–N), ΔC(NO2−–N), and ΔC(NH4+–N) are the differences of NO3−–N, NO2−–N, and NH4+–N concentrations before and after the reaction, respectively. Nitrate removal capacities (Q) were calculated using the following equation: Q = [C(NO3−–N)0 − C(NO3−–N)t]V/mFe, where V (L) is the volume of nitrate solution and mFe (g) is the mass of iron in the composite coated on the nickel foam.
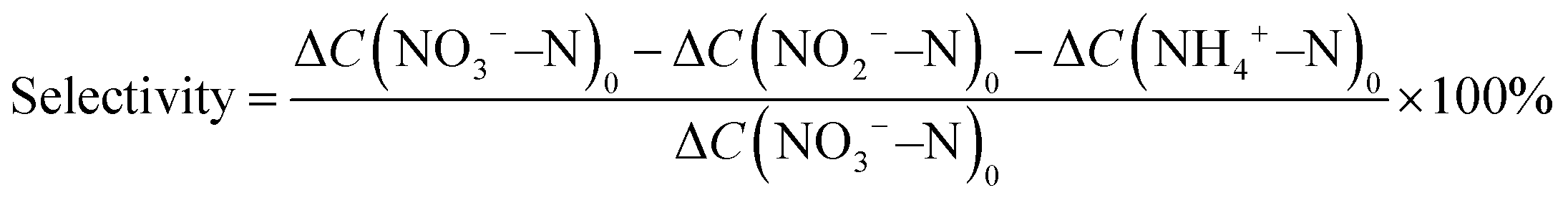
Computational methods
All the spin-polarized DFT calculations were performed with the Vienna ab initio simulation package (VASP) using the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA). The projected augmented wave (PAW) potentials were used to describe the ionic cores. A plane wave cut-off energy of 500 eV was applied for all the calculations. The criteria for force and electronic energy convergence are set to 0.01 eV Å−1 and 10−5 eV, respectively, during all the calculations. A Monkhorst–Pack mesh of 5 × 4 × 5 was applied for structural relaxation of bulk Fe3C. The Gaussian smearing with a smearing width was set as 0.10 eV.The optimized lattice parameters of the bulk Fe3C unit cell were a = 5.011 Å, b = 7.040 Å, and c = 4.672 Å, respectively. According to the high-resolution TEM image, the Fe3C (210) surface was selected as the model, and a slab of five layers using a (1 × 3) supercell was cut from the bulk. The bottom two-layers were fixed for all calculations. A vacuum slab of 20 Å was built to avoid the interactions between periodic images. The dipole correction was adopted to remove the dipole interaction along the z direction. The DFT-D3 empirical correction method was used to describe van der Waals interactions. A Monkhorst–Pack mesh of 1 × 1 × 1 was used for slab calculations. More computational details were followed from previous work.
Results and discussion
Synthesis and structural characterization of Fe3C/MNCFs
The construction of Fe3C/MNCFs was achieved by a facile electrospinning-calcination process, as shown in Fig. 1a. First, homogeneous solutions containing appropriate amounts of polyacrylonitrile (PAN), polystyrene (PS), and metal salts (Fe3+, Co2+, and Ni2+) were used to synthesize the 1D fibrous structures through electrospinning. The PS could be stretched into nanoscaled wires in the PAN fibers during the electrospinning process, and then decomposed to generate nanochannels during the pyrolysis process. Meanwhile, the metal precursors were reduced in situ to form nanoparticles embedded or anchored on the fiber skeleton. By adjusting the pyrolysis temperature from 700 to 900 °C, the 1D hierarchical structure with different skeleton structures and particle morphologies can be obtained.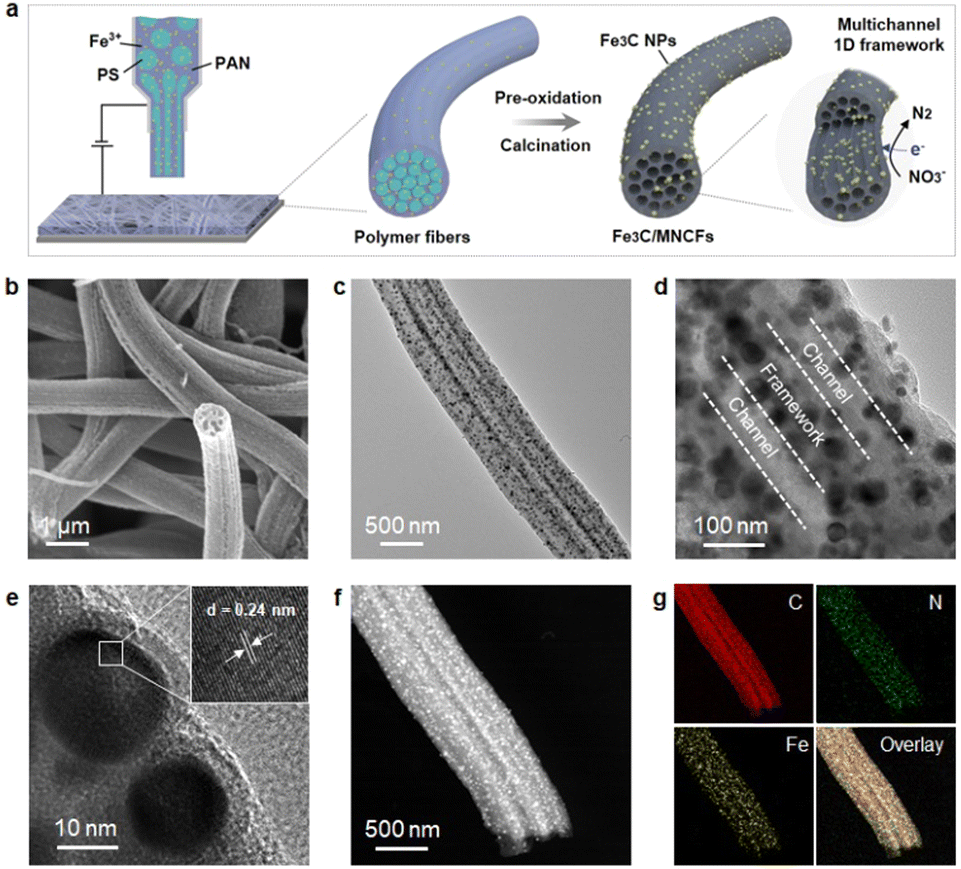 | ||
| Fig. 1 (a) Illustration for the preparation of Fe3C/MNCFs-800 via the electrospinning method and the subsequent pyrolysis. (b) SEM image of Fe3C/MNCFs-800. (c) and (d) TEM images of Fe3C/MNCFs-800. (e) HR-TEM image of Fe3C/MNCFs-800. (f) HAADF-STEM image and (g) elemental mapping of Fe3C/MNCFs-800. | ||
The morphology and structure of Fe3C/MNCFs were investigated by electron microscopy. The scanning electron microscopy (SEM) image showed that the obtained Fe3C/MNCFs-800 maintained a continuous fibrous structure without obvious agglomeration or structural collapse (Fig. S1, ESI†). The cross-sectional SEM image demonstrates that Fe3C NPs are homogenously anchored on the fiber skeleton with well-defined channels (Fig. 1b). The transmission electron microscopy (TEM) image further confirmed the 1D hierarchical structure of Fe3C/MNCFs-800, consisting of plenty of well-dispersed Fe3C NCs anchored on the hollow fiber inner wall and matrix (Fig. 1c and d). The HRTEM image in Fig. 1e demonstrated that the NPs were enclosed by carbon shells and the visible lattice fringe was 0.24 nm, corresponding to the (210) planes of the Fe3C phase. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and elemental mapping images in Fig. 1f and g suggest that C, N and, Fe are evenly distributed within the entire Fe3C/MNCFs-800. The energy-dispersive X-ray spectroscopy (EDS) line scans further verified that the Fe3C NCs are distributed on the parallel channel walls and the fiber matrix (Fig. S2, ESI†).
Typical X-ray diffraction (XRD) patterns of the Fe3C/MNCFs-x are shown in Fig. 2a. Apart from the broad diffraction hump at about 25° assigned to the (002) plane of graphitic carbon, the remaining sharp diffraction peaks can be indexed to metallic Fe3C (JCPDS card no. 35-0772). Apparently, the intensity of Fe3C peaks increases with temperature, which can be attributed to the higher annealing temperature leading to the deeply reduced products with higher crystallinity. Raman analysis was carried out to determine the nature of the carbon fiber matrix (Fig. 2b). The two broad peaks of D and G bands at 1350 cm−1 and 1596 cm−1 correspond to the disordered defective carbon and the orderly graphitic carbon, respectively. The ID/IG ratio decreased with the increase of annealing temperature, indicating that high temperature can increase the degree of graphitization, which will contribute to better electronic conduction between the fiber skeleton and Fe3C NPs.23 N2 adsorption–desorption analysis was performed to obtain the specific surface area and average pore size distribution of Fe3C/MNCFs-x.24 The specific surface area of Fe3C/MNCFs-700, Fe3C/MNCFs-800, and Fe3C/MNCFs-900 is 37.5 m2 g−1, 138.8 m2 g−1, and 210.1 m2 g−1, respectively (Fig. 2c). This significant increment of the specific surface area is due to the increase of the nanopores. As shown in Fig. 2d, the average pore size of Fe3C/MNCFs-800 is 4.84 nm, which is smaller than that of the Fe3C/MNCFs-900 (6.40 nm). This indicated that high annealing temperature will accelerate the nucleation of Fe3C NCs, thus leaving numerous nanopores in the fiber skeleton.
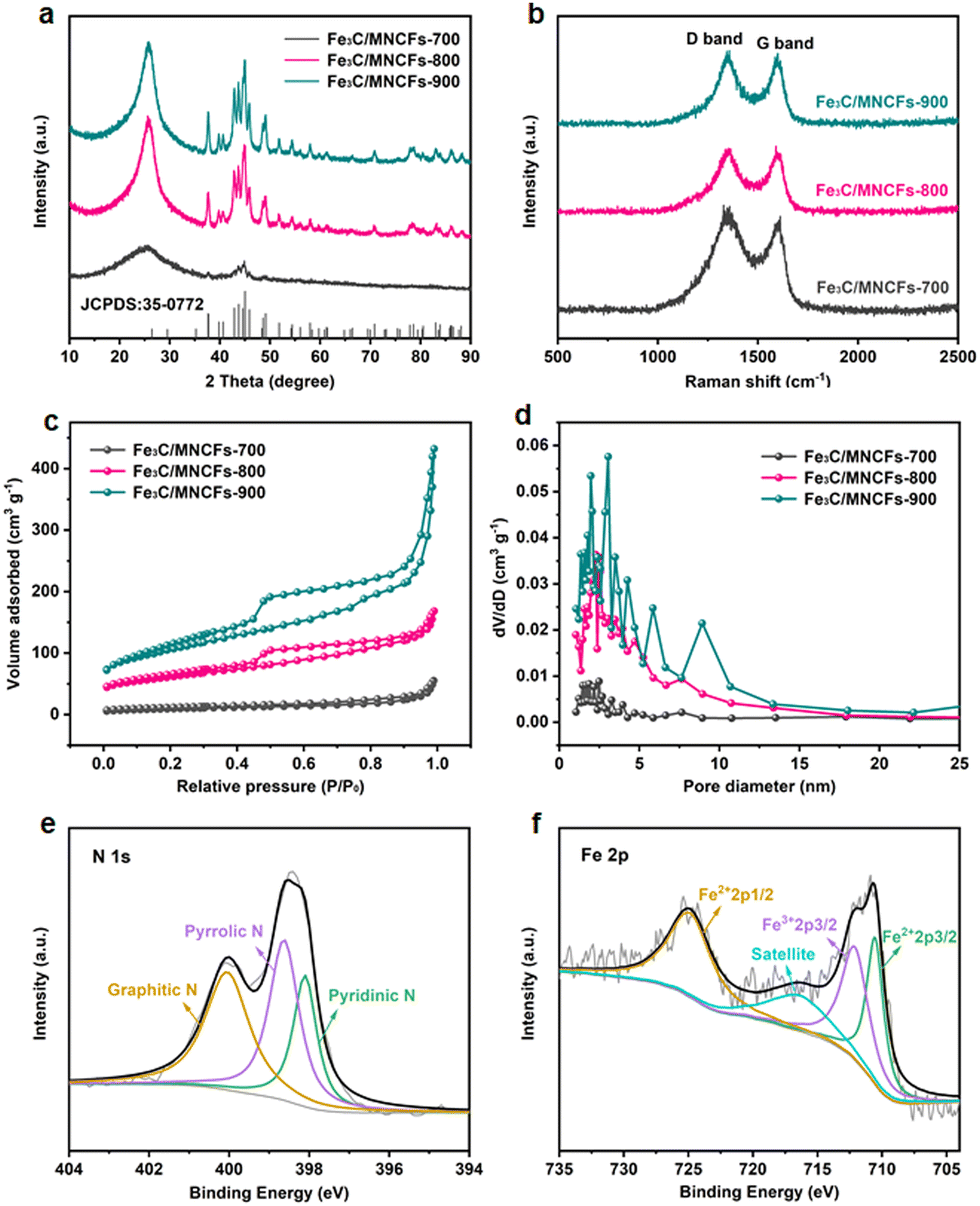 | ||
| Fig. 2 (a) XRD patterns, (b) Raman spectra, (c) nitrogen adsorption–desorption isotherms, and (d) pore diameter of Fe3C/MNCFs with pyrolysis temperatures at 700 °C, 800 °C, and 900 °C. (e) N 1s and (f) Fe 2p spectra of Fe3C/MNCFs-800. | ||
X-ray photoelectron spectroscopy (XPS) was used to elucidate the surface composition and binding structure of Fe3C/MNCFs-800. The survey XPS spectra confirmed the co-existence of C, N, O, and Fe elements in Fe3C/MNCFs-800 (Fig. S3a, ESI†). The high-resolution C 1s spectrum displays two deconvoluted peaks at 286.2 and 284.8 eV, corresponding to the C–N/C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species, respectively (Fig. S3b, ESI†).25 The N 1s spectrum in Fig. 2e shows three peaks that are assigned to pyridinic-N (398.2 eV), pyrrolic-N (398.8 eV), and graphitic N (400.5 eV), confirming the successful doping of nitrogen into carbon fibers.26 The high-resolution Fe 2p spectra show that all the iron can be attributed to Fe2+/Fe3+ ionic species, confirming the existence of Fe3C in the prepared Fe3C/MNCFs-800 (Fig. 2f).11
O species, respectively (Fig. S3b, ESI†).25 The N 1s spectrum in Fig. 2e shows three peaks that are assigned to pyridinic-N (398.2 eV), pyrrolic-N (398.8 eV), and graphitic N (400.5 eV), confirming the successful doping of nitrogen into carbon fibers.26 The high-resolution Fe 2p spectra show that all the iron can be attributed to Fe2+/Fe3+ ionic species, confirming the existence of Fe3C in the prepared Fe3C/MNCFs-800 (Fig. 2f).11
The structures of Fe3C/MNCFs-800 can be easily controlled by changing the applied voltage and weight ratio of PAN, PS, and Fe(acae)3 (Fig. S4, ESI†). When the applied voltage increases, the Fe3C/MNCFs exhibit a bead like structure (Fig. S4a and d, ESI†). As shown in Fig. S4b and e (ESI†), the uniformity of the fibrous structure deteriorates with the increase of Fe(acae)3 content. An ideal support material should provide large internal space for high catalyst loading, so we attempted to change the weight ratio of PAN to PS to increase the number of inner channels. However, with the increase of the PS content, the decomposition of PS makes it impossible for PAN to form a continuous one-dimensional fiber skeleton (Fig. S4c and f, ESI†). Hence, all Fe3C/MNCFs used in this work are fabricated according to the optimized PAN/PS/Fe(acae)3 ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1. Furthermore, the thermal carbonization temperature plays an important role in controlling the composition and morphology on the composite fibers. As the pyrolysis temperature increases from 700 to 900 °C, the particle size of Fe3C increases gradually, leaving nanoholes in the fiber skeleton due to transformation of metal salts (Fig. S5, ESI†).
:3:1. Furthermore, the thermal carbonization temperature plays an important role in controlling the composition and morphology on the composite fibers. As the pyrolysis temperature increases from 700 to 900 °C, the particle size of Fe3C increases gradually, leaving nanoholes in the fiber skeleton due to transformation of metal salts (Fig. S5, ESI†).
To demonstrate the adaptability of the approach, other metal salts (Co2+ and Ni2+) were used to synthesize the 1D hierarchical structure. SEM and TEM images confirmed that the multichannel 1D structure we observed in Fe3C/MNCFs could be well maintained when the Fe3C was changed to Co (Fig. S6a and b, ESI†) and Ni (Fig. S6d and e, ESI†). Their XRD patterns clearly identify the consistent crystalline structure of Co/MNCFs-800 (JCPDS card no. 15-0806) and Ni/MNCFs-800 (JCPDS card no. 87-0712), as shown in Fig. S7 (ESI†). The uniform dispersion of different metals was further confirmed by STEM and elemental mapping images, suggesting the versatility of this method in synthesizing different metal-based channel-rich fibrous structures (Fig. S6c and f, ESI†).
Electrocatalytic performance toward the NO3RR
The NO3RR electrocatalytic activity of the Fe3C/MNCFs was evaluated using a three-electrode system. The generated products were detected using ultraviolet-visible (UV/Vis) absorbance spectra (Fig. S8, ESI†). At −1.3 V, which was selected as the reaction potential, the Fe3C/MNCFs-800 displayed superior NO3RR performance (NO3− conversion: 90.9% and N2 selectivity: 99.53%) to Fe3C/MNCFs-700 (NO3− conversion: 46.3% and N2 selectivity: 86.7%) and Fe3C/MNCFs-900 (NO3− conversion: 85.2% and N2 selectivity: 97.77%) (Fig. 3a). Obviously, both the crystalline nature and well-defined multichannel fibrous structure synergistically promoted the catalytic performance of Fe3C/MNCFs-800. Even though the Fe3C/MNCFs-900 possess abundant nanopores for mass diffusion, the grain coarsening caused by high annealing temperature inevitably leads to insufficient exposure of active sites. Moreover, the NO3RR performance of Fe3C/MNCFs-800 is significantly higher than that of Co/MNCFs-800 (NO3− conversion: 86.9% and N2 selectivity: 88.98%) and Ni/MNCFs-800 (NO3− conversion: 58.6% and N2 selectivity: 84.62%). Fig. 3b depicts the LSV linear sweep voltammetry (LSV) curves of different electrocatalysts. The Fe3C/MNCFs-800 exhibited the strongest current density under reaction potential compared with Fe3C/MNCFs-700 and Fe3C/MNCFs-900, indicating obvious electroactivity of Fe3C/MNCFs-800 for the NO3RR.27 The Fe3C/MNCFs-800 displays significantly higher current density in 100 ppm of NO3− solution than in NO3− free solution, suggesting the occurrence of nitrate reduction rather than hydrogen evolution (Fig. S9, ESI†).28 Chronoamperometry measurement testified that the Fe3C/MNCFs-x can maintain a relatively stable current density at −1.3 V for 10 h. The Fe3C/MNCFs-800 showed the highest current density at the same voltage, indicating the best electrocatalytic activity (Fig. S10, ESI†).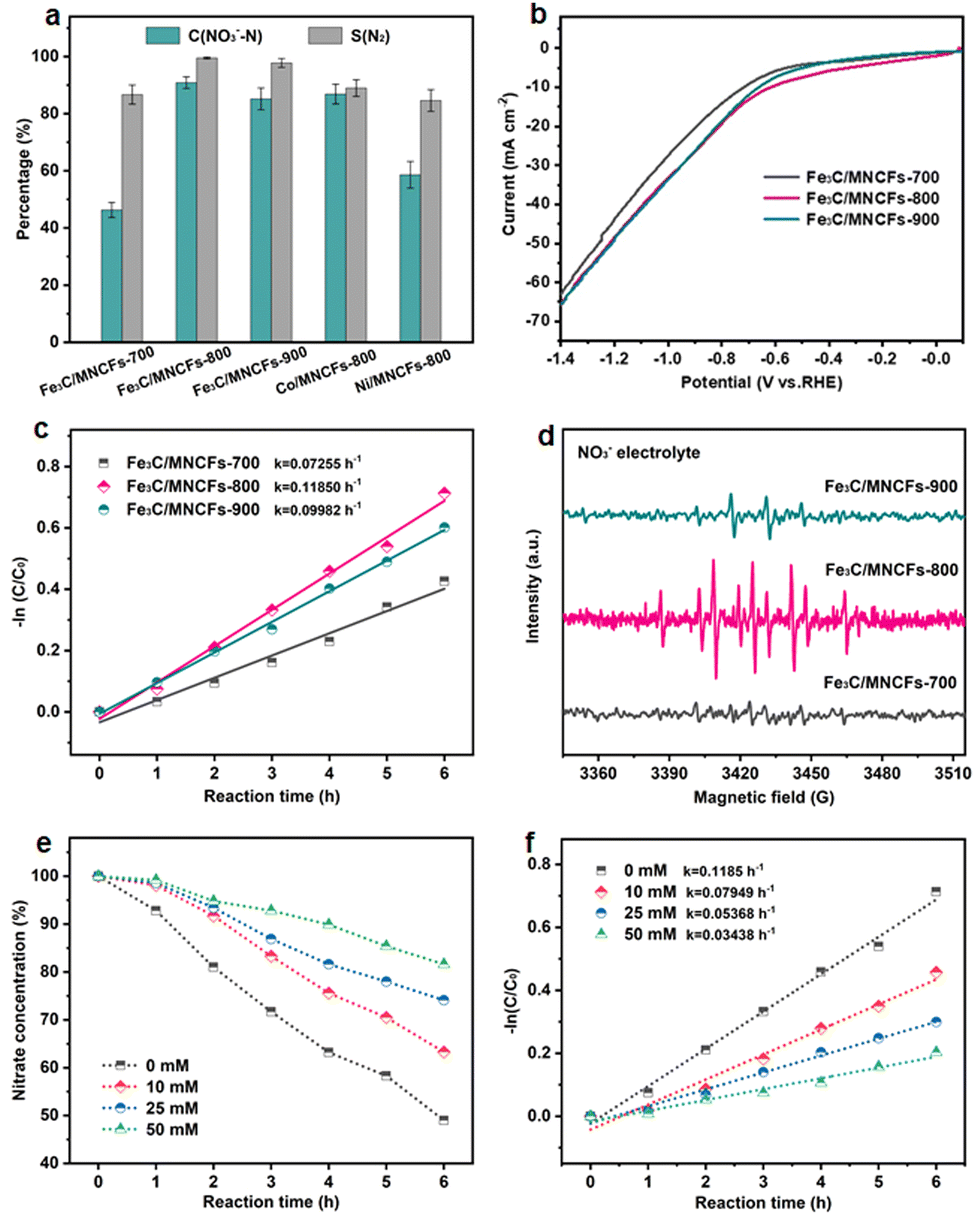 | ||
| Fig. 3 (a) The comparison of the NO3RR performance among different catalysts. (b) LSV curves of Fe3C/MNCFs-x during nitrate electroreduction. (c) The linearized pseudo first-order kinetic profiles of Fe3C/MNCFs-x. (d) DMPO spin-trapping ESR spectra of Fe3C/MNCFs-x. (e) The change of nitrate concentration during the NO3−RR process on Fe3C/MNCFs-800 with different addition concentrations of TBA. (f) The linearized pseudo first-order kinetic profiles of Fe3C/MNCFs-800 with different addition concentrations of TBA. | ||
The kinetic analysis was further performed to reveal the catalytic process. According to the change of nitrate residues during the NO3RR process, it can be seen that the reaction at Fe3C/MNCFs-800 proceeded much faster than at other electrodes (Fig. S11, ESI†). The kinetics followed a pseudo first-order rate law, and the apparent rate constants (kap) can be calculated through the slope of the linear fitting (Fig. 3c). For Fe3C/MNCFs-800, the kap is higher than that of Fe3C/MNCFs-700 and Fe3C/MNCFs-900, indicating a faster reaction rate. Such a high reaction rate implies the existence of abundant available active sites, which make significant contributions to its optimized NO3RR performance. To detect the presence of hydrogen radicals during the reaction process, the electron spin resonance (ESR) experiment was conducted using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a radical trapping reagent.29 The ESR spectra exhibited nine characteristic peaks of DMPO-H with their intensity ratio close to 1:1:2:1:2:1:2:1:1 (Fig. 3d). The strongest peak intensity of DMPO-H for Fe3C/MNCFs-800 demonstrates the high active hydrogen adsorption capacity.30
Moreover, the tert-butyl alcohol (TBA), a specific quenching agent of hydrogen radicals, was used to identify the role of active hydrogen in the catalytic process.31 As shown in Fig. 3e, the nitrate residue increases with the increase of TBA concentration. The kap of nitrate conversion decreased gradually when the TBA increased from 0 to 50 mM, indicating that the addition of TBA has a negative impact on nitrate reduction (Fig. 3f). The results showed that the active hydrogen plays a key role in catalytic reactions, and the high active hydrogen adsorption capacity of Fe3C/MNCFs-800 is the main reason for its high activity.
The NO3RR performance of Fe3C/MNCFs-800 was evaluated under a variety of applied potential from −1.0 V to −1.5 V vs. SCE (Fig. 4a). The NO3− conversion rate increased with the increase of cathode potential, while the N2 selectivity exhibited a volcanic trend. The increased NH4+ selectivity was observed when the cathode potential exceeds −1.3 V, which is probably attributed to the Cl− in the electrolyte not being sufficient to consume the excessive NH* generated around the electrode.
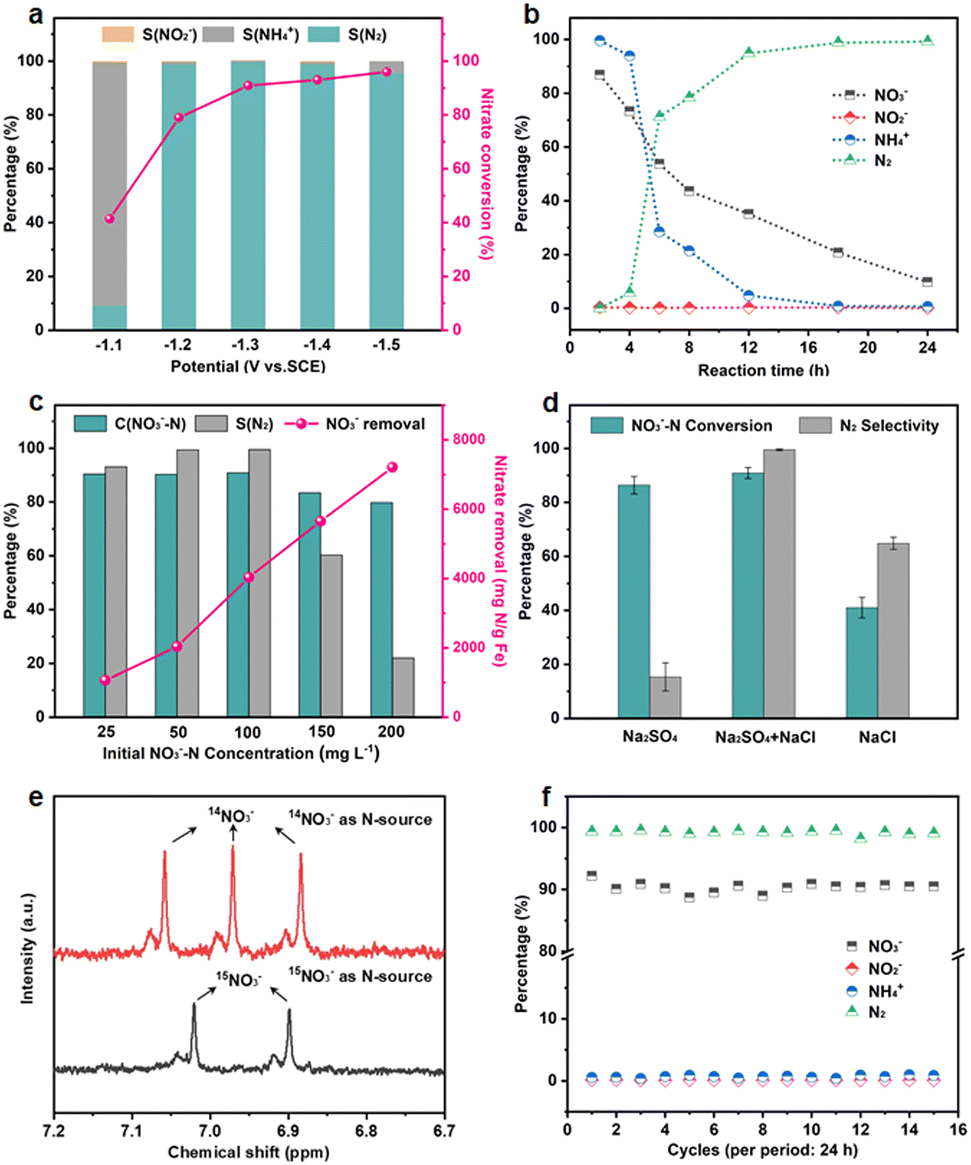 | ||
| Fig. 4 (a) Nitrate conversion and product selectivity of Fe3C/MNCFs-800 at different given potentials. (b) The evolution of NO3−, NO2−, NH4+, and N2 of Fe3C/MNCFs-800 for different reaction times. (c) Nitrate conversion, N2 selectivity, and nitrate removal capacity of Fe3C/MNCFs-800 at different initial NO3−–N concentrations. (d) Nitrate conversion and N2 selectivity of Fe3C/MNCFs-800 at different electrolytes. (e) 1H NMR spectra of the electrolyte after the electrocatalytic NO3RR using 14NO3− and 15NO3− as the nitrogen source. (f) Nitrate conversion and N2 selectivity of Fe3C/MNCFs-800 during 15 consecutive cycles. Reaction conditions: 0.1 M Na2SO4, 0.02 M NaCl, and −1.3 V, 24 h. | ||
The effect of the evolution of NO3− and intermediate products (NO2−, NH4+, and N2) on the performance of Fe3C/MNCFs-800 was investigated to reveal the sequence of catalytic reactions over a reaction time of 24 h (Fig. 4b). The NO3− concentration decreased from the initial 100 ppm to the final 9.8 ppm, achieving a conversion efficiency of 90.2%. The NO2− was almost below the detection limit (0.001 ppm) during 24 h of the reaction, indicating that NO2− as an intermediate can be rapidly reduced to N2 or NH4+. The NH4+ rapidly decreases within 4 to 6 h, and the residual amount is only 0.47% after 24 hours. The selectivity of N2 was calculated to be 99.53% after 24 h on the basis of the N mass balance. The effect of the nitrate concentration on the catalytic performance was evaluated (Fig. 4c). The NO3− removal capacity was calculated based on the proportion of Fe3C (14.55 wt%, according to the TGA result) (Fig. S12, ESI†). The NO3− removal capacity enhanced from 1064 to 7207 mg N g−1 Fe3C as the initial concentration of NO3−–N increased from 25 to 200 ppm. The electrolyte showed significant influence on the conductivity and electrochemical reactions. To the best of our knowledge, the Na2SO4 can improve the NO3− conversion, while NaCl contributes to the selectivity of N2. The iron-based electrocatalyst exhibited high reduction ability in the conversion of NO3−–N to NH4+, and the generated NH4+–N can be converted into N2 through the electrochlorination processes on the anode. In the Na2SO4 system, NO3−–N conversion reached 86.4% after 24 h of the reaction, while the N2 selectivity was only 15.4% (Fig. 4d). In NaCl electrolyte, the N2 selectivity (64.8%) is much higher than that in the Na2SO4 system, since NH4+ is oxidized to N2 (2NH4+ + 3HClO → N2 + 3H2O + 3Cl− + 5H+) by the generated HClO (Cl2 + H2O → HClO + Cl− + H+).32 In the mixed system, NO3−–N conversion is much higher than that of the single electrolyte, and the N2 selectivity reaches almost 100%. These results indicated that the Na2SO4 accelerated NO3− conversion while NaCl promoted the oxidation of NH4+ on the anode, leading to the simultaneously improved NO3− removal efficiency and N2 selectivity.33 To verify the origin of ammonia and eliminate the possible interference from the external environment, a isotope-labeling test was conducted (Fig. 4e). It can be seen that the formation of ammonia originated from the electroreduction of nitrate.
The long-term stability was measured in the Na2SO4–NaCl system, and the NO3−–N conversion and N2 selectivity almost remain unchanged during 15 test cycles, indicating the outstanding electrocatalytic stability of the Fe3C/MNCFs-800 (Fig. 4f). After consecutive electrocatalysis, the TEM images of the Fe3C/MNCFs-800 exhibit the retained fibrous structure and well-dispersed nanoparticles (Fig. S13, ESI†). The strong anchoring effect enables the NPs stabilized on the fiber matrix without detachment and aggregation, while the thin carbon layer wrapped around Fe3C NPs can prevent corrosion and leakage of active ingredients. The chemical states of Fe3C/MNCFs-800 after the electrocatalytic nitrate reduction were identified by XPS. In Fig. S14 (ESI†), the Fe 2p spectrum shows that the new peak ascribed to Fe(0) appeared. The Fe(I) in Fe3C could be reduced to Fe(0) by the electrons and active hydrogen from the cathode, thus facilitating electron transfer and improving the reaction activity.
The reaction pathways of the NO3RR and the free energy of each intermediate over Fe3C/MNCFs were calculated using density functional theory (DFT) calculations (Fig. 5). Nitrate ions are first adsorbed on Fe3C/MNCFs with a formation free energy change of −0.68 eV. After that, the N–O bond is continuously cleaved by proton-coupled electron transfer to form *NO2 and *NO. Notably, the reduction of nitrate may occur in two cases after the production of *NO, that is, the *NOH pathway and *N pathway, respectively. From *NO to *NOH, the protonation of *NO → *NOH requires an energy uphill (ΔG = 0.16 eV). In contrast, the formation of *N species is more favourable with a free energy of −0.88 eV. Thus, pathway I and II are the favorable pathways for the nitrate reduction reaction. In pathway I, the *N undergoes a hydrogenation process to generate *NH3, and the hypochlorite selectively oxidizes ammonia to N2 through electrochlorination processes. Pathway II is more favorable in terms of the free energy change (ΔG = −2.40 eV) for N2 production. This is highly consistent with the high selectivity of N2 since the pathway II is thermodynamically barrierless compared with the other two paths.
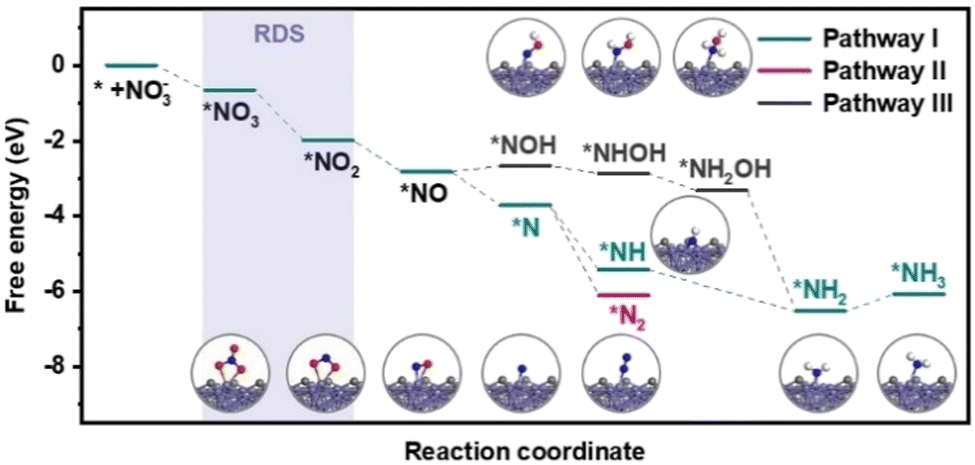 | ||
| Fig. 5 Gibbs free energy diagrams for different NO3RR pathways on Fe3C/MNCFs. | ||
Based on the electrochemical behavior analysis, the high catalytic performance of Fe3C/MNCFs-800 can be attributed to not only the high activity of Fe3C NPs but also the well-designed 1D architecture. The multi-channel 1D fiber support promotes the permeation of active species in the electrolyte as well as the mass diffusion, contributing to accelerating the reaction kinetics. Furthermore, the tight connection between the Fe3C NPs and conductive MNCFs guarantees the structural stability and effective electronic pathways. Compared with the previous NO3RR studies, Fe3C/MNCFs-800 exhibits a high conversion efficiency and N2 selectivity that are superior or comparable to the performances of alloy or noble metal catalysts (Table S1, ESI†).34–40
Conclusions
In summary, multichannel nitrogen-doped carbon fiber confined Fe3C NPs have been rationally designed and synthesized through a facile electrospinning method and a pyrolysis process. The hierarchical 1D architecture helps to maximize the utilization of effective active sites and to promote efficient electron transfer. Benefiting from the synergy between the multichannel fibrous structure and the strong interfacial interaction between the active species and supports, the obtained catalysts delivered a high nitrate conversion of 90.9% and nitrogen selectivity of 99.53%, as well as long-term stability. This work may provide new insights into the design of cost-effective NO3RR catalysts for water purification and environmental remediation.Author contributions
F.-Z. Z., and J.-P. Y. designed the studies. F.-Z. Z. synthesized the catalysts and performed the catalytic tests. Z.-S. S. performed the density functional theory calculations. F.-Z. Z., J.-L. C., and H.-X. L. conducted SEM, TEM, XRD, XPS, and TGA measurements. F.-Z. Z. wrote the paper with the help of J.-P. Y. and J. C. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 52172291, 52122312, and 92163121), and the “Shuguang Program” supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (22SG31).References
- X. Zhang, B. B. Ward and D. M. Sigman, Chem. Rev., 2020, 120, 5308–5351 CrossRef CAS PubMed.
- Z. Chang, G. Meng, Y. Chen, C. Chen, S. Han, P. Wu, L. Zhu, H. Tian, F. Kong, M. Wang, X. Cui and J. Shi, Adv. Mater., 2023, 35, 2304508 CrossRef CAS PubMed.
- Y. Lv, S.-W. Ke, Y. Gu, B. Tian, L. Tang, P. Ran, Y. Zhao, J. Ma, J.-L. Zuo and M. Ding, Angew. Chem., Int. Ed., 2023, 62, e202305246 CrossRef CAS PubMed.
- A. S. Fajardo, P. Westerhoff, C. M. Sanchez-Sanchez and S. Garcia-Segura, Appl. Catal., B, 2021, 281, 119465 CrossRef CAS.
- K. Chu, W. Zong, G. Xue, H. Guo, J. Qin, H. Zhu, N. Zhang, Z. Tian, H. Dong, Y.-E. Miao, M. B. J. Roeffaers, J. Hofkens, F. Lai and T. Liu, J. Am. Chem. Soc., 2023, 145, 21387–21396 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, L. Han, S. Wang, T. Cui, Y. Yan, M. Xu, H. Duan, Y. Kuang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202213711 CrossRef CAS PubMed.
- H. Xu, Y. Ma, J. Chen, W.-X. Zhang and J. Yang, Chem. Soc. Rev., 2022, 51, 2710–2758 RSC.
- X. Zhao, G. Hu, G.-F. Chen, H. Zhang, S. Zhang and H. Wang, Adv. Mater., 2021, 33, 2007650 CrossRef CAS PubMed.
- H. Yu, S. Qu, P.-R. Chen, K.-Q. Ou, J.-Y. Lin, Z.-H. Guo, L. Zheng, J.-K. Li, S. Huang, Y. Teng, L. Zou and J.-L. Song, J. Hazard. Mater., 2022, 430, 128351 CrossRef CAS PubMed.
- W. Teng, N. Bai, Y. Liu, Y. Liu, J. Fan and W.-X. Zhang, Environ. Sci. Technol., 2018, 52, 230–236 CrossRef CAS PubMed.
- W. Duan, G. Li, Z. Lei, T. Zhu, Y. Xue, C. Wei and C. Feng, Water Res., 2019, 161, 126–135 CrossRef CAS PubMed.
- Y. Liu and J. Wang, Sci. Total Environ., 2019, 671, 388–403 CrossRef CAS PubMed.
- M. Ma, S. You, W. Wang, G. Liu, D. Qi, X. Chen, J. Qu and N. Ren, ACS Appl. Mater. Interfaces, 2016, 8, 32307–32316 CrossRef CAS PubMed.
- W. Zong, H. Gao, Y. Ouyang, K. Chu, H. Guo, L. Zhang, W. Zhang, R. Chen, Y. Dai, F. Guo, J. Zhu, Z. Zhang, C. Ye, Y.-E. Miao, J. Hofkens, F. Lai and T. Liu, Angew. Chem., Int. Ed., 2023, 62, e202218122 CrossRef CAS PubMed.
- X. Fan, Z. Peng, R. Ye, H. Zhou and X. Guo, ACS Nano, 2015, 9, 7407–7418 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, Y. Niu, D. Fang, J. Wang, Q. Su and C. Wang, Green Chem., 2021, 23, 7594–7608 RSC.
- X. Ma, J. Zhong, W. Huang, R. Wang, S. Li, Z. Zhou and C. Li, Chem. Eng. J., 2023, 474, 145721 CrossRef CAS.
- J. Wang, W. Teng, L. Ling, J. Fan, W.-X. Zhang and Z. Deng, Environ. Sci.: Nano, 2020, 7, 1496–1506 RSC.
- F. Zhang, J. Luo, J. Chen, H. Luo, M. Jiang, C. Yang, H. Zhang, J. Chen, A. Dong and J. Yang, Angew. Chem., Int. Ed., 2023, 62, e202310383 CrossRef CAS PubMed.
- X. Cao, L. Zhao, B. Wulan, D. Tan, Q. Chen, J. Ma and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202113918 CrossRef CAS PubMed.
- F. Zhang, J. Chen, G. G. Wallace and J. Yang, Prog. Mater. Sci., 2023, 133, 101069 CrossRef CAS.
- Y. Lan, H. Luo, Y. Ma, Y. Hua, T. Liao and J. Yang, Nanoscale, 2021, 13, 10108–10115 RSC.
- S.-H. Bae, J.-E. Kim, H. Randriamahazaka, S.-Y. Moon, J.-Y. Park and I.-K. Oh, Adv. Energy Mater., 2017, 7, 1601492 CrossRef.
- J. Zheng, X. Chen, X. Zhong, S. Li, T. Liu, G. Zhuang, X. Li, S. Deng, D. Mei and J.-G. Wang, Adv. Funct. Mater., 2017, 27, 1704169 CrossRef.
- H. Li, T. A. Ha, S. Jiang, C. Pozo-Gonzalo, X. Wang, J. Fang, P. C. Howlett and X. Wang, Electrochim. Acta, 2021, 377, 138089 CrossRef CAS.
- J. Wang, Z. Deng, T. Feng, J. Fan and W.-X. Zhang, Chem. Eng. J., 2021, 417, 129160 CrossRef CAS.
- Z.-X. Ge, T.-J. Wang, Y. Ding, S.-B. Yin, F.-M. Li, P. Chen and Y. Chen, Adv. Energy Mater., 2022, 12, 2103916 CrossRef CAS.
- H. Liu, X. Lang, C. Zhu, J. Timoshenko, M. Rüscher, L. Bai, N. Guijarro, H. Yin, Y. Peng, J. Li, Z. Liu, W. Wang, B. R. Cuenya and J. Luo, Angew. Chem., Int. Ed., 2022, 61, e202202556 CrossRef CAS PubMed.
- J. Lu, X. Liu, H. Zhang, M. Fu, H. Zheng, Q. Chen and J. Zhou, Chem. Eng. J., 2022, 433, 134586 CrossRef CAS.
- C. Feng, W. Zhou, H. Wu, Q. Huo, J. Shao, X. Li, H. Yang, Q. Hu and C. He, Appl. Catal., B, 2024, 341, 123280 CrossRef CAS.
- Y. Wang, H. Li, W. Zhou, X. Zhang, B. Zhang and Y. Yu, Angew. Chem., Int. Ed., 2022, 61, e202202604 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhao, Z. Chen, L. Wang, P. Wu and F. Wang, Electrochim. Acta, 2018, 291, 151–160 CrossRef CAS.
- J. Sun, W. Gao, H. Fei and G. Zhao, Appl. Catal., B, 2022, 301, 120829 CrossRef CAS.
- H. Zhang, C. Wang, H. Luo, J. Chen, M. Kuang and J. Yang, Angew. Chem., Int. Ed., 2023, 62, e202217071 CrossRef CAS PubMed.
- X. Chen, T. Zhang, M. Kan, D. Song, J. Jia, Y. Zhao and X. Qian, Environ. Sci. Technol., 2020, 54, 13344–13353 CrossRef CAS PubMed.
- Y. Lan, J. Chen, H. Zhang, W.-X. Zhang and J. Yang, J. Mater. Chem. A, 2020, 8, 15853–15863 RSC.
- H. Luo, C. Wang, J. Wang, Y. Ma and J. Yang, Inorg. Chem. Front., 2023, 10, 4526–4533 RSC.
- F. Ni, Y. Ma, J. Chen, W. Luo and J. Yang, Chin. Chem. Lett., 2021, 32, 2073–2078 CrossRef CAS.
- L. Su, D. Han, G. Zhu, H. Xu, W. Luo, L. Wang, W. Jiang, A. Dong and J. Yang, Nano Lett., 2019, 19, 5423–5430 CrossRef CAS PubMed.
- H. Luo, S. Li, Z. Wu, Y. Liu, W. Luo, W. Li, D. Zhang, J. Chen and J. Yang, Adv. Mater., 2023, 35, 2304695 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00016a |
| This journal is © The Royal Society of Chemistry 2024 |