
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-tandem catalysis of fast Mg2+ desolvation and sulfur conversions for ultrahigh-performance Mg–S batteries via serially-assembled atomic reactors†
Qinghua
Guan‡
ac,
Jian
Wang‡
 *abi,
Quan
Zhuang‡
e,
Jing
Zhang
f,
Linge
Li
ac,
Lujie
Jia
a,
Yongzheng
Zhang
g,
Hongfei
Hu
ac,
Huimin
Hu
a,
Shuang
Cheng
ac,
Huang
Zhang
h,
Huihua
Li
h,
Meinan
Liu
ac,
Shuangyin
Wang
*d and
Hongzhen
Lin
*ac
*abi,
Quan
Zhuang‡
e,
Jing
Zhang
f,
Linge
Li
ac,
Lujie
Jia
a,
Yongzheng
Zhang
g,
Hongfei
Hu
ac,
Huimin
Hu
a,
Shuang
Cheng
ac,
Huang
Zhang
h,
Huihua
Li
h,
Meinan
Liu
ac,
Shuangyin
Wang
*d and
Hongzhen
Lin
*ac
ai-Lab & CAS Key Laboratory of Nanophotonic Materials and Devices, Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences, Suzhou 215123, China. E-mail: wangjian2014@sinano.ac.cn; jian.wang@kit.edu; hzlin2010@sinano.ac.cn
bHelmholtz Institute Ulm (HIU), Ulm D89081, Germany
cSchool of Nano-Tech and Nano-Bionics, University of Science and Technology of China, Hefei 230026, China
dState Key Laboratory of Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China. E-mail: shuangyinwang@hnu.edu.cn
eInner Mongolia Key Laboratory of Carbon Nanomaterials, Nano Innovation Institute (NII), College of Chemistry and Materials Science, Inner Mongolia Minzu University, Tongliao 028000, China
fSchool of Materials Science and Engineering, Xi’an University of Technology, Xi’an 710048, China
gState Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China
hKey Laboratory of Engineering Dielectric and Applications (Ministry of Education), School of Electrical and Electronic Engineering, Harbin University of Science and Technology, Harbin 150080, China
iKarlsruhe Institute of Technology (KIT), Karlsruhe D-76021, Germany
First published on 20th March 2024
Abstract
Rechargeable magnesium–sulfur (Mg–S) batteries are famous for their high volumetric energy density and much improved safety without possible dendrite formation. However, the performances of Mg–S batteries are severely hindered by the sluggish and dissatisfactory electrochemical kinetics of interfacial Mg2+ desolvation and successive sulfur species conversions. Herein, a self-tandem catalysis for achieving fast Mg2+ desolvation and sulfur conversions is pioneered and the serially-assembled train-like atomic reactors of zinc atomic catalysts employed in long-conductive nitrogen-doped nanocarbons are designed (STAR@LCNC), serving as electrochemical kinetic promotors. Benefitting from these, the STAR@LCNC provides the capability in propelling dissociation kinetics of bridged MgCl(THF)x+ to form more Mg2+, and then guarantees high-speed electron exchange and highly active catalytic capability in catalyzing sulfur species at the interface, as thoroughly demonstrated from experimental and in situ spectroscopical characterizations i.e. sum frequency generation spectroscopy to theoretical calculations. The so-fabricated sulfur cathode with STAR@LCNC delivers a long lifespan (400 cycles at 0.5C) and the highest rate performance (612 mA h g−1 at 2C) in the Mg–S battery systems reported so far. Also, the high sulfur loading cathode (4 mg cm−2) is capable of stabilizing at 2.92 mA h cm−2 for 50 cycles, indicating a bright future for tandem catalysis in practical batteries.
Broader contextAn essential problem in secondary batteries based on conversion-type electrodes is that: for the redox conversion reactions to take place smoothly and efficiently, a prerequisite fundamental step, often overlooked in the physical models described in the current literature, is the rapid desolvation of the charge carriers. The desolvation process has typical features of a chemical reaction (or a combination of a series of chemical steps) with substantially high energy barriers, which can be comparable or even larger than that of the subsequent conversion step. In the particular systems of magnesium sulfur (Mg–S) batteries in our contribution, this problem can even be severe. Furthermore, the ion desolvation issue has never been considered in these previous reports. In this work, self-tandem catalysis for fast Mg2+ desolvation and sulfur/polysulfide conversion is proposed by serial train-like atom reactors on long conductive nanocarbon (STAR@LCNC), as revealed by various characterizations, opening new avenues for achieving faster, long-lasting, higher energy density batteries. |
Introduction
The daily increasing pursuit of high volumetric energy densities in smart reversible energy storages for portable devices and electrical vehicles has resulted in an urgent need to develop economical but safe rechargeable batteries.1–5 In comparison to popular lithium–sulfur batteries, magnesium–sulfur (Mg–S) batteries possess a higher theoretical volumetric specific energy density (3221 W h L−1) and benefit from the natural abundance of magnesium and sulfur in the earth.6–11 Importantly, metallic Mg offers much improved safety without possible dendrite formation in comparison to Li metal electrodes.7,12–15 However, the state-of-art developments of Mg–S batteries are still in their early stage with miserable cycling stability and depressing rate performance.12 They are restricted by the electronic/ionic insulation nature of S/MgS, the large volumetric changes, and the formation and shuttling of soluble polysulfides (MgSx, 4 ≤ x < 8). Specifically, kinetic problems of the sluggish interfacial Mg2+ desolvation and redox conversion rates of sulfur species, and high decomposition barrier of insulative MgS in high mass loading cathodes prevent the performance improvement.12,16–19Strategies in dealing with the above-mentioned challenges are mainly centered on: (i) constructing conductive porous carbon/sulfur or organic polymer/sulfur hybrid composites i.e. polyacrylonitrile/sulfur, improving the integral conductivity and accommodating volume changes;2,20–24 (ii) designing and introducing polar sites to adsorb or block the outward polysulfides physically or chemically, restricting MgSx shuttling;25–29 (iii) synthesizing various compatible electrolytes.23,30–34 However, none of these strategies involve interfacial ion diffusion and conversion kinetics, which are responsible for the redox kinetics of sulfur reduction reactions (SRRs) and sulfide oxidation reactions (SORs).12,35,36 As described in Fig. 1A, pristine carbon only transports electrons rapidly to reach sulfur, while it cannot effectively adsorb polysulfides chemically and accelerate the polysulfide conversions, so the redox conversion kinetics are too slow since it lacks active sites, exhibiting dissatisfactory performance and stability. Existing studies have concluded that the state of the carrier such as Li+ and Mg2+ depends on the successive reactions.37 Always, the desolvation steps take place at the electrode/electrolyte interface before reacting with sulfur. Generally, Mg2+ together with solvents will form a large solvation structure in the electrolyte, which hinders the mobility of Mg2+ and generates huge steric effect, thus resulting in slow reaction kinetics. As highlighted, the performance of the sulfur cathode is closely related to the free insertion/extraction amount of metal ions.38–40 Prior to magnesiation of sulfur, it is inevitable to dissociate the MgCl(THF)x+ cluster structure to form free Mg2+ at the electrode/electrolyte interface. Afterwards, the free Mg2+ is involved in the successive polysulfide conversions, displaying two-step electrochemical processes. In other words, two successive procedures occur during the sulfur cathode reaction: the first procedure is the desolvation of the MgCl(THF)x+ complex to produce free Mg2+, and the second procedure is the free Mg2+ being involved in the conversion of the sulfur and polysulfides. Common strategies of modulating solvation shells are achieved by electrolyte additives or pore morphology chemistry. In spite of these, our group had also proposed an electrocatalytic method with pore morphology to promote the desolvation process of solvated Li+ at the electrode/electrolyte interface to uniformize Li plating or accelerate sulfur conversions.37–40 Therefore, the ideal strategy is to seek stable and highly active electrocatalysts and simultaneously regulate the solvation behavior of Mg2+ and promote sulfur/polysulfide conversions, aiming at reducing the desolvation and reaction energy barriers of MgSx conversions and MgS decomposition along with more free Mg2+ formation.
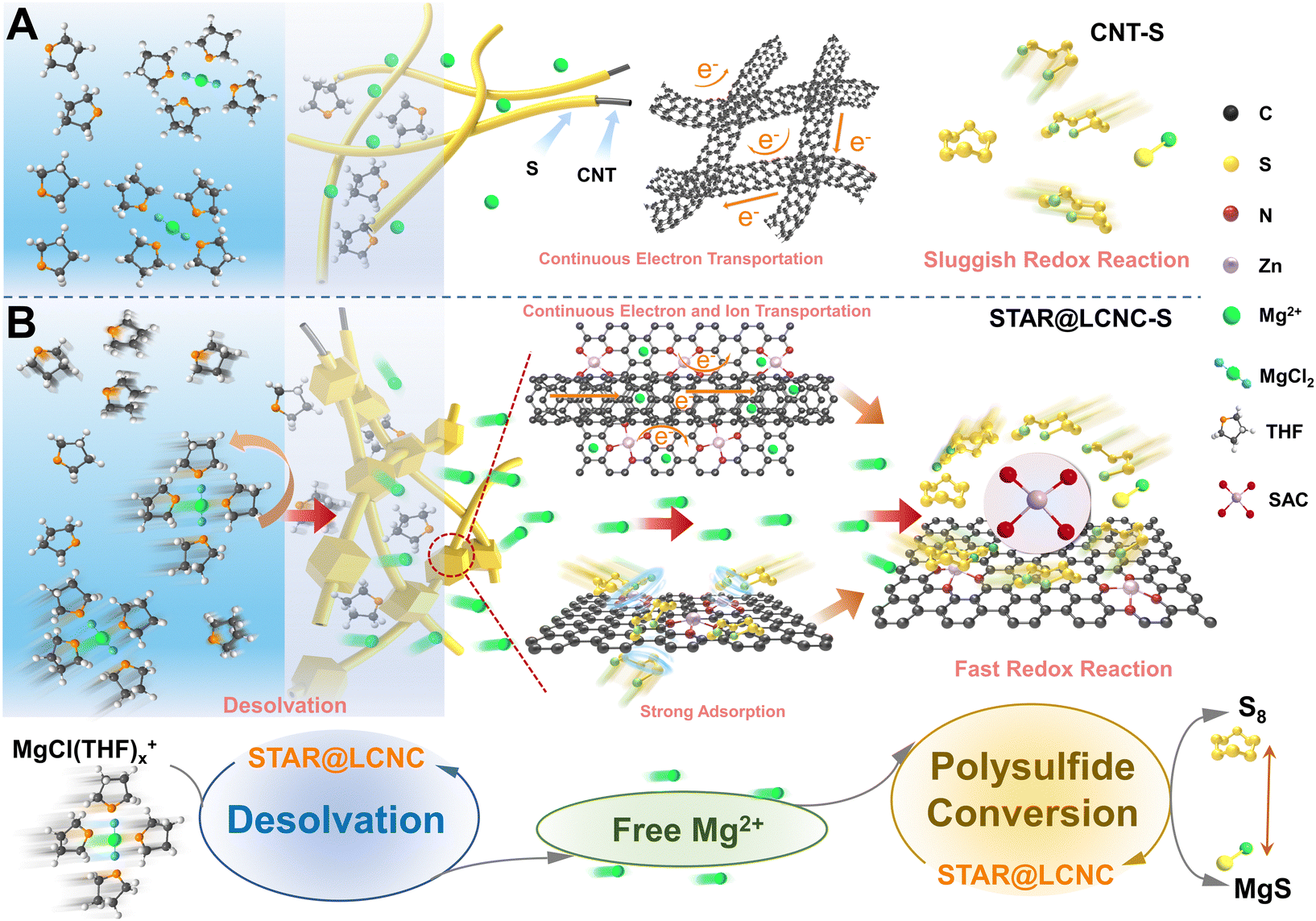 | ||
| Fig. 1 Schematic illustrations of atomic reactors in triphasic interfaces. The schematic illustrations of (A) electronic transport along the cross-linked CNT and (B) fast electronic transport, strong chemisorption, and rapid conversion kinetics of magnesium polysulfides at the solid–liquid–solid ternary interfaces with the catalysis of atomic reactors. | ||
Different from common metal-based compounds, isolated atomic reactors such as single-atom catalysts (SACs) anchored on conductive supports are endowed with a high surface area and the highest specific activity with ideally high atomic exposure.41–46 While our and other research groups have demonstrated the superiority of SACs in Li–S battery systems,47–51 no one yet knows how SACs perform in conversion-based Mg–S batteries, and the underlying working mechanism of SACs after cycling has not been explored until now.19 What is worse, current progresses of Mg–S batteries are evaluated under a low mass loading (<0.5 mg cm−2) and low current rate (<0.1C), which are far from practical requirements. Therefore, the catalytic efficiency will be maximized if the atomic reactor sites are delicately located at the sulfur/matrix/electrolyte triphasic interface, which can effectively accelerate the dissolution of the solvated structure of Mg2+, release more free Mg2+ to participate in the conversions of sulfur and polysulfides, and improve the sulfur utilization.52
Herein, a self-tandem catalysis strategy for achieving fast Mg2+ desolvation and sulfur conversions is pioneered and the protocol serially-assembled train-like atomic reactors of zinc atomic catalysts employed in long-conductive nanocarbons (STAR@LCNC) are designed (Fig. 1B), serving as electrochemical redox kinetic promoters in Mg–S batteries. In contrast to pristine conductive carbon, the STAR@LCNC on the cross-linked electron network can effectively accelerate the dissociation of MgCl(THF)x+, releasing a large amount of free Mg2+ and further involving the transformation reaction of polysulfides (Fig. 1B, bottom). At the same time, the robust structure can ensure the rapid transport of Mg2+ at the electrode/electrolyte interface, and the STAR domains provide the triphasic local catalytic environment to allow high efficiency to promote the electrochemical kinetics of SRRs and SORs (Fig. 1B, below). Consequently, the modified sulfur cathode with STAR@LCNC is capable of delivering a capacity of 1012 mA h g−1 at 0.5C. A high-rate performance up to 2C (612 mA h g−1 at 2C) and a long-term cycling lifespan of 400 cycles are achieved under the assistance of zinc atomic catalysts. More impressively, the sulfur cathode with 4.0 mg cm−2 stabilizes the areal capacity of 2.92 mA h cm−2 after 50 cycles, indicating fast electrochemical conversion kinetics without polysulfide shuttling.
Results and discussion
Systematic self-tandem catalysis of STAR@LCNC revealed by theoretical simulations
Initially, theoretical simulations were performed to understand the dissociation of MgCl(THF)x+ on different substrates. Typical MgCl2(THF)2 species are selected to imitate the solvation shell behaviors at the electrode/electrolyte interface. Its decomposition into MgCl2·THF and THF molecule is described on STAR@LCNC, corresponding to a dissociation energy barrier of ∼2.7 kJ mol−1, which is lower than that in the electrolyte (Fig. 2A, B and Fig. S1, ESI†). Meanwhile, the optimized configurations and electrostatic potential (ESP) results in Fig. 2C show the desolvation process of MgCl2(THF)2 on STAR@LCNC had lower electronegativity than that on nitrogen-doped carbon (NC), which means that the interaction between STAR@LCNC and MgCl2(THF)2 is weaker and more conducive to dissociating and forming free Mg2+. The interactions and catalytic functions of STAR@LCNC in sulfur species adsorption and decomposition are displayed in Fig. 2D, E and Fig. S2 (ESI†).53–55 All structures and free energies of the S8 and various MgSx (x = 1, 2, 4, 6, 8) species were optimized on various carbon matrix with/without STAR. As summarized in Fig. 2E, the adsorption energies of S-related species on the STAR@LCNC surface are the highest, much stronger than other systems. For example, the STAR@LCNC displays the improved binding energy of 2.66 eV to MgS, about 3.3 times higher than that on pristine carbon, indicating the sulfiophilic effect induced by STAR@LCNC. Furthermore, the sulfur–matrix interaction (SMI) mechanism was revealed by the charge density difference (CDD) and Bader charge analysis (Fig. 2F).56 Taking the MgS4 intermediator as an example, in contrast to pristine carbon and NC, the strong bonding and charge transfer interactions of Zn–S and N–Mg were realized along with an obvious charge transfer (0.1889e−), much higher than the controlled ones (0.0296e− for NC and 0.0115e− for pristine carbon). In the projected density of states (PDOS) spectra (Fig. 2G and Fig. S3–S5, ESI†), the obvious hybridizations between Zn 3d and S 2p, and N 2p and Mg 2p are formed with the introduction of zinc atom reactor into the system, demonstrating chemical interactions between STAR and polysulfides. Compared with the occupation of C 2p for both valence bands and conduction bands in pristine carbon, the top of the valence bands is dominated by the hybridized states of both C 2p and N 2p, while the bottom of the conduction bands is contributed by C 2p for NC and C–N hybridized states for STAR@LCNC. Furthermore, the gap between the conduction and valence bands shows a decreasing tendency (band gap: pristine carbon > NC > STAR@LCNC; conductivity: pristine carbon < NC < STAR@LCNC), indicating the enhanced electronic transportation in STAR@LCNC, which is consistent with the above charge transfer analysis. However, the SMI is rather weak without zinc atomic catalysts, which might be attributed to the physical van der Waals (vdW) interaction. In spite of the adsorption interactions, the reversibility and decomposition capability of the insulative MgS are more significant for evaluating the oxidation kinetics. The initial demagnesium step in the solid-phase MgS plays a decisive role and has strong impacts on the successive conversion reactions, which determine the later cycling performances and utilizations.54,57 A higher energy needs to be overcome to make one Mg atom move away from the Mg2S2 on pristine carbon and the energy barrier is as high as 4.1 eV (Fig. 2H). Thanks to the high catalytic capability of single atomic Zn, the decomposition barrier is sharply reduced to 0.45 eV, about ten times lower than that of pristine MgS, clearly confirming the superior electrocatalytic effect of STAR@LCNC accelerating MgS phase transformation and improving sulfur utilization.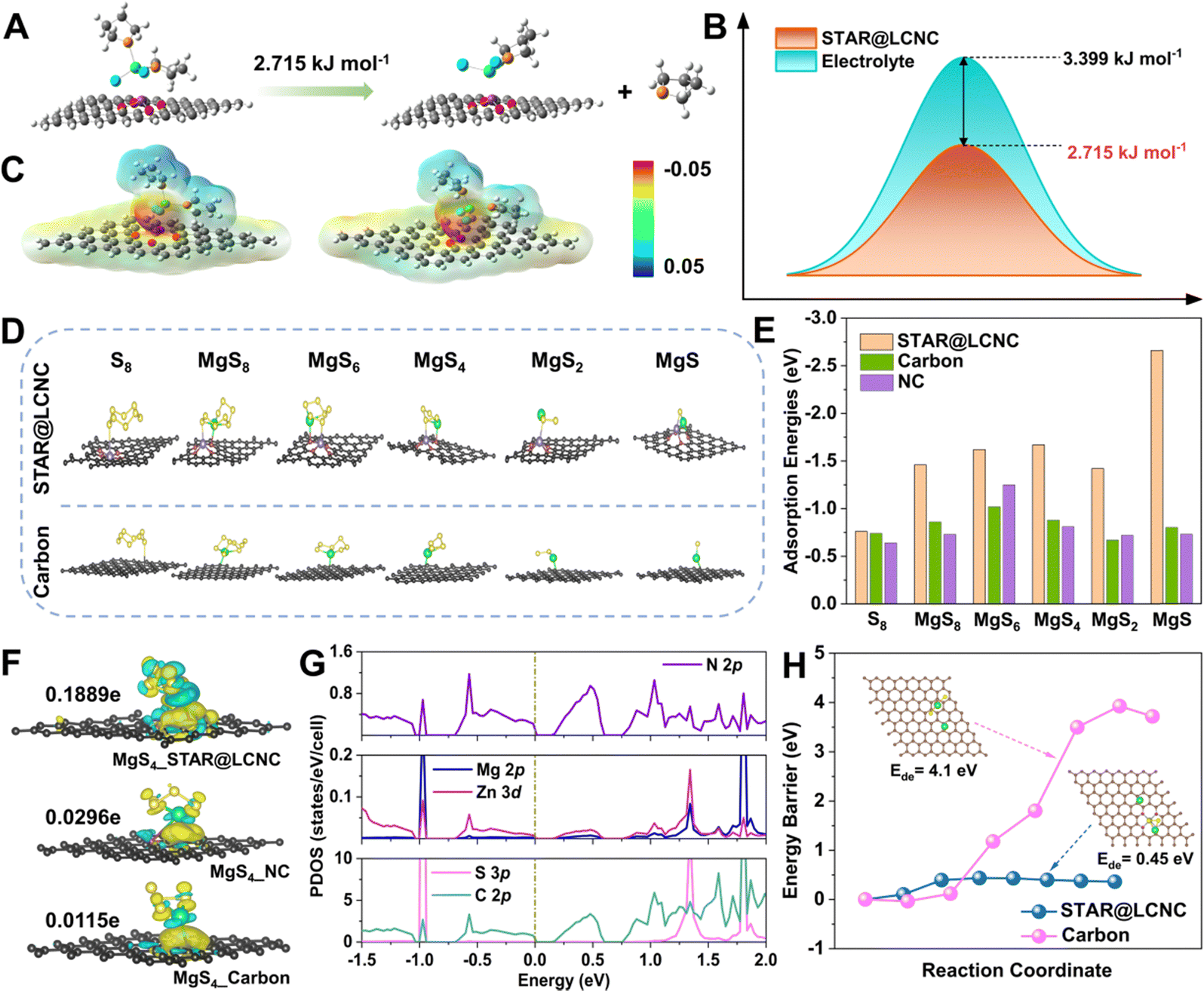 | ||
| Fig. 2 Systematic illustrations of atomic reactors in triphasic interfaces revealed by theoretical simulations. (A) Desolvation barrier of MgCl2(THF)2 into MgCl2·THF and THF on STAR@LCNC. (B) Desolvation barrier of MgCl2(THF)2 into MgCl2·THF and THF on STAR@LCNC or in liquid electrolyte, respectively. (C) The electrostatic potential for STAR@LCNC (left) and NC (right). (D) The corresponding structure illustrations of different S8, MgS8, MgS6, MgS4, MgS2 and MgS on STAR@LCNC and Carbon substrates. (E) Comparison of adsorption binding energies between sulfur species and various matrix (see Table S1 for details, ESI†). (F) Charge density difference between MgS4 and various carbon substrates with/without zinc atomic catalysts. (G) The projected density of states (PDOS) for the adsorbed configuration of MgS4 on the substrate with zinc atomic catalysts. (H) Comparison of decomposition energy barriers of the Mg2S2 cluster on STAR@LCNC and carbon, respectively. | ||
Synthesis and characterizations of the STAR@LCNC
According to the above findings, a schematic illustration of the synthesis process of STAR@LCNC is depicted in Fig. 3A. Briefly, the 2-MeIm and Zn2+ precursors are physically adsorbed on the uniform CNT suspension via van der Waals interaction forces.58 After stirring, filtration, and successive heating reaction under a NH3 atmosphere, the formed precursors (ZIF-8) on CNT composites were converted into the STAR@LCNC assemblies, generating abundant atomic reactor sites (Fig. S6, ESI†). Similar to the structure of traditional Chinese red lanterns (Fig. 3B), scanning electron microscopy (SEM) shows the synthesized ZIF-8 (analogy to lanterns) are uniformly grown on the long-length conductive CNTs (analogy to the conductive wire), exhibiting an obvious serially-assembled train-like structure (Fig. 3C). Even after carbonization, most of the serially-assembled train-like structures are reserved (Fig. 3D). The STAR@LCNC shows a larger surface area (1024.6 m2 g−1) and richer porous structure than the pristine CNT assemblies (Fig. S7, ESI†), which are beneficial for potentially sieving solvated Mg2+ and adsorbing polysulfides. Fig. 3E shows that the elemental carbon, nitrogen and zinc are homogenously distributed throughout the composite without any aggregation. To better reveal the presence and distribution of atomic Zn, high-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was performed (Fig. 3F and G).46–48 No apparent aggregates (clusters or particles) of Zn exist and the isolated Zn atoms could be clearly observed as the bright spots in the magnified image (highlighted with red circles), strongly demonstrating the formation of STAR on the nanocarbon during the NH3 heat treatment (Fig. 3F). Meanwhile, the chemical bonding states of the zinc and nitrogen elements in the STAR@LCNC were investigated using X-ray photoelectronic spectroscopy (XPS). As shown in Fig. 3H, two peaks located at 1021.5 and 1044.7 eV assigned to the Zn 2p3/2 and Zn 2p1/2 could be found in the high-resolution Zn 2p spectrum, indicating the valence value (m) of Znm+ is between 0 and +2 without the formation of Zn–Zn or Zn–O bonds. Except for the typical peaks assigned to pyrrolic and pyridinic N groups for adsorbing polysulfides, another peak located at 399.3 eV appeared in high-resolution N 1s spectrum (Fig. 3I), attributed to the formation of a N–Zn bond to anchor zinc atomic catalysts.49 According to the XPS results, the content of zinc atoms is estimated to be 5.04 at% in the STAR@LCNC.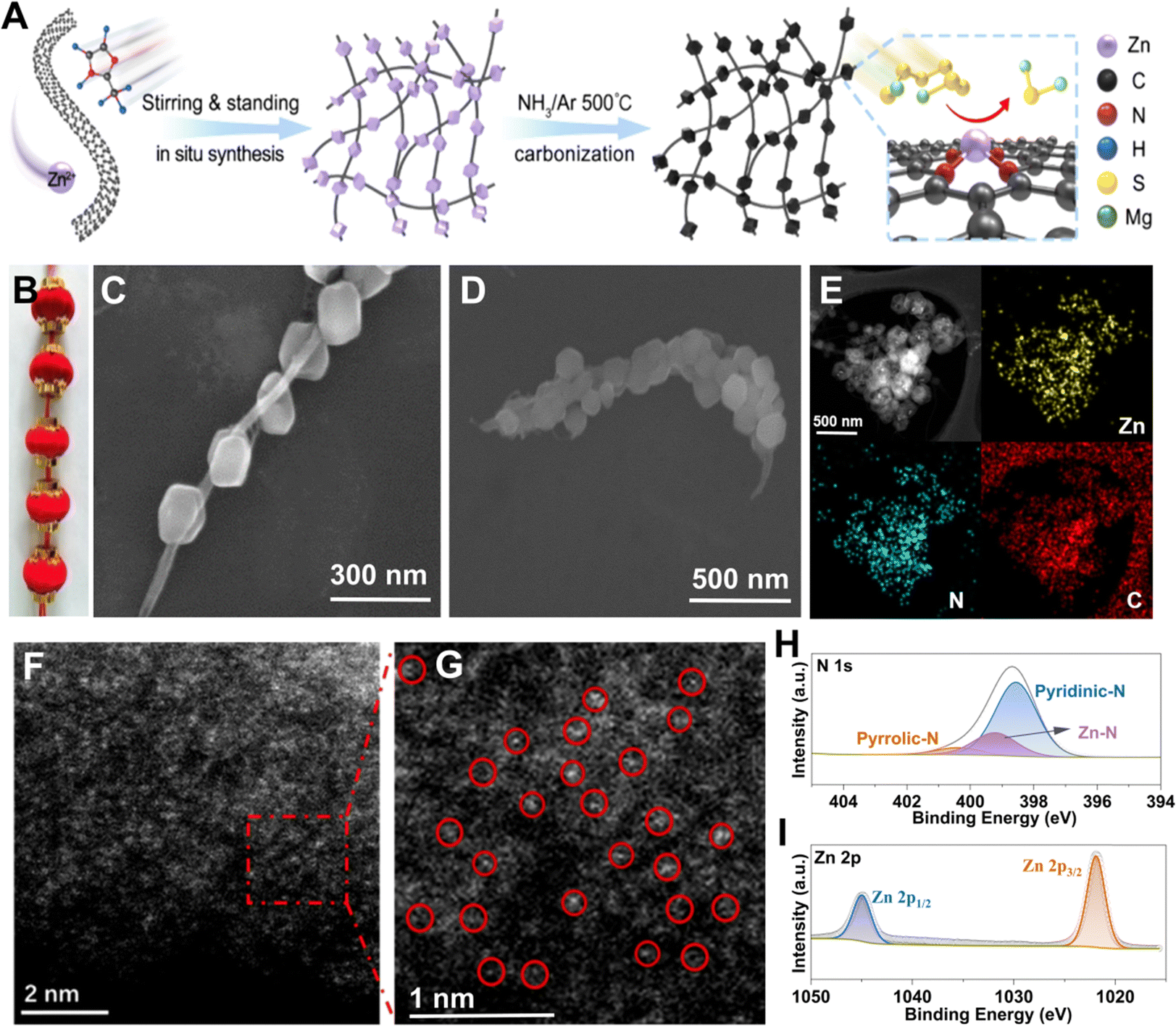 | ||
| Fig. 3 Synthesis and characterizations of the STAR@LCNC catalyst. (A) A schematic synthesis illustration of the STAR@LCNC. (B) An optical image of a Chinese lantern. SEM images of (C) ZIF-8/CNT and (D) STAR@LCNC samples. (E) The energy dispersed X-ray elemental mappings of C, Zn and N in STAR@LCNC. (F) and (G) HAADF-STEM images of the single zinc atoms in the STAR@LCNC composite (highlighted by circles in (G)). High-resolution XPS spectra of (H) N 1s and (I) Zn 2p of STAR@LCNC. | ||
Interfacial desolvation catalyzed by STAR@LCNC for enhancing redox kinetics
Catalytically dissociating the MgCl(THF)x+ complex to release free Mg2+ more efficiently at the electrode/electrolyte interface is crucial for increasing the Mg2+ mobility and reactivity in the subsequent polysulfide redox conversion. In general, the interfacial arrangement of cations and anions can be described by the double-layer model with an inner Helmholtz layer and an outer diffuse layer.59 As one would expect the charged species in the diffuse layer are dominated by solvated ions, and the ions in the inner layer can exist as solvated, partially solvated, or free ones without any solvent shell, depending on the electrode surface properties and local environments. Meanwhile, a certain amount of free solvent molecules or solvent clusters may also enter the inner layer and get directly bounded to the electrode surface. Probing the interconversion between several types of interfacial solvent molecules will be very helpful to characterize the solvation/desolvation processes at the interface. As illustrated in Fig. 4A and Fig. S8 (ESI†), the configuration of in situ interface-sensitive sum frequency generation (SFG) spectroscopy was constructed to monitor the desolvation behaviors of the solvated Mg2+ with/without bias voltage. The C–H stretching vibrations of the solvent THF were investigated in the 2700–3000 cm−1 range at the electrode/electrolyte interface by SFG spectroscopy with ssp polarizations. Two different electrodes were applied, one made from pristine CNT without any catalyst, and the other from the catalytic STAR@LCNC. Identical MgCl2–LiCl/THF electrolyte was used in both cases, and control samples with pure THF were also investigated for comparison. For the CNT/pure THF interface, two characteristic peaks at 2845 and 2930 cm−1 were observed (Fig. S9, ESI†), which can be readily assigned to the symmetric stretching (ss) and its Fermi resonance (FR) mode of CH2, respectively.60 As illustrated in Fig. 4B, three peaks at around 2800, 2890, and 2945 cm−1 can be identified for the CNT/electrolyte interface. While the 2890 and 2945 cm−1 peaks can be attributed to other FR modes of CH2 symmetric stretching in correspondence to strong intermolecular interaction,61 the 2800 cm−1 peak is seldom reported in the literature for THF or other CH2-containing molecules. We found that the 2800 cm−1 peak was dramatically enhanced as a bias voltage of 100 mV was applied, and meanwhile the 2945 cm−1 peak was weakened, a phenomenon not observable for the contact interface with neat THF. Therefore, it must be related to the presence of ionic species in the electrolyte solution and their redistribution at the interface upon applying an external electric field. In other words, the 2800 cm−1 peak comes from the solvent molecules in the solvation shell of ions. The strong intermolecular interaction in the solvent shell results in strong coupling to raise the FR modes and the disappearance of a basic mode at ∼2845 cm−1. The central ion polarizes the surrounding solvent molecules and decreases the electron density of the C–H bonds and thus lowers the vibration frequency. The bias voltage drives the accumulation of more solvated ions at the interface, leading to enhancement of the 2800 cm−1 peak. The STAR@LCNC/pure THF displays SFG peaks at 2830 and 2875 cm−1, redshifted in comparison to CNT/pure THF due to stronger binding of THF to the modified surface. At the STAR@LCNC/electrolyte interface, the SFG intensities are significantly lower than that at the CNT electrolyte interface, while the two electrodes in contact with pure THF shows a similar intensity level, indicating a substantial fraction of the STAR@LCNC/electrolyte interface is covered by non-solvated Mg ions or partially solvated ions with a low coordination number of solvents. Moreover, the THF-related SFG resonance of the STAR@LCNC/electrolyte interface can be obviously divided into two groups, with one group (2826 and 2860 cm−1) having a similar feature to pure THF, and the other group (2778 and 2916 cm−1) in correspondence to THF in the solvation shell of ions. Importantly, upon applying a bias voltage of 100 mV, the signal of the solvation shell THF almost disappears, while the maintained peaks (2840 and 2938 cm−1) should be assigned to free, non-specifically adsorbed THF molecules, similar to that observed for the CNT/pure THF interface. The above results cogently reveal that the catalyst can accelerate the dissociation of MgCl(THF)x+ structures, and release a large number of free Mg2+ at the interface. As a result, the Mg2+ transference number is greatly enhanced from 0.14 for CNT to 0.22 for STAR@LCNC (Fig. 4C and Fig. S10, ESI†), which clearly shows that STAR@LCNC can significantly improve the ion diffusion and transport kinetics by promoting the Mg2+-solvent dissociation. Fig. 4D and Fig. S11 (ESI†) schematically depict the above model that STAR@LCNC catalyzes the release of Mg2+ from the solvation structure and facilitates their spreading over the cathode surface, entering into the active cathode phase, and participating in the sulfur conversion redox.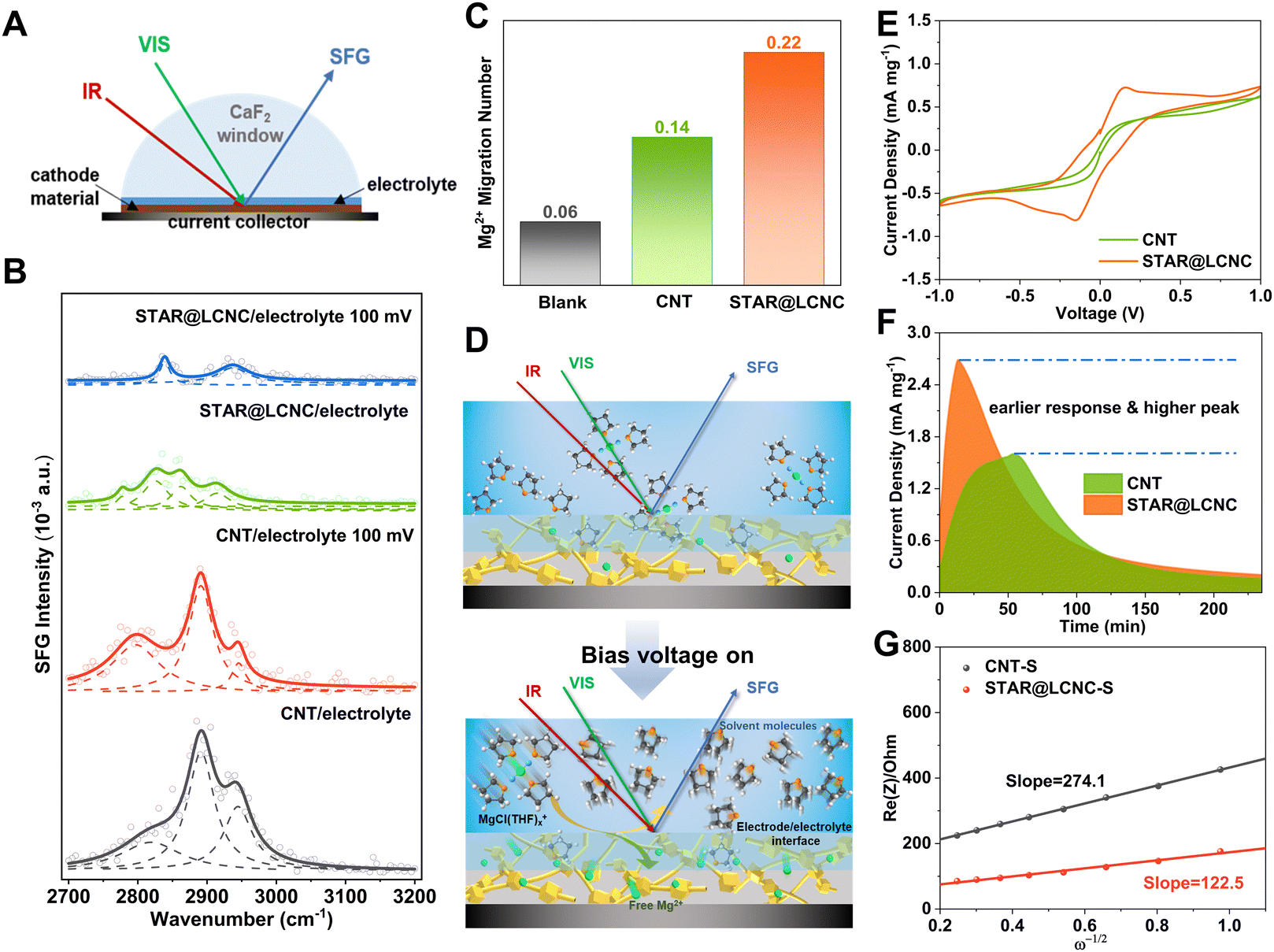 | ||
| Fig. 4 Experimental of interfacial desolvation and sulfur species kinetics catalyzed by STAR@LCNC. (A) Schematic illustration of the in situ SFG probing the electrolyte/catalyst interface. (B) The SFG spectra of different adsorption states of the Mg2+ solvation structure in MgCl2-LiCl/THF electrolyte. (C) Magnesium ion migration number for blank magnesium foil, CNT and STAR@LCNC materials. (D) The molecular states of the Mg2+ solvation structure in the STAR@LCNC electrode/electrolyte interface before and after turning the bias voltage on. (E) Comparisons of magnesium polysulfide conversions in a symmetric cell based on the STAR@LCNC and the CNT electrodes, respectively. (F) Comparison of MgS precipitation kinetics on the surface between CNT and STAR@LCNC at a constant potential of 0.7 V, corresponding to a liquid/solid phase transition. (G) The Mg2+ diffusion comparison derived from EIS by the linear relationship between Re(Z) and reciprocal exponential (−1/2) at low frequency. | ||
As is well known, the reversibility and performance of Mg–S batteries depend on the kinetics of the MgCl(THF)x+ desolvation and the redox conversion of sulfur species in inhibiting polysulfide migration, which are influenced by huge desolvation/reaction barriers.12 Compared with the initial dark yellow (Fig. S12A, ESI†), the remaining solution with STAR@LCNC became much lighter after reacting for 24 h. Correspondingly, the peak at 420 nm ascribed to the short-chain S42− species decreased significantly in the UV-vis spectroscopy (Fig. S12B, ESI†). And the corresponding remaining polysulfide concentration was reduced to 13.69% for the STAR@LCNC system, while it was 23.70% for the CNT one, indicating fast SMI interactions after catalysis.62 The reported magnesium chloride and lithium chloride in tetrahydrofuran (MgCl2–LiCl/THF) is chosen as the electrolyte system with the upper voltage window up to 3.5 V (vs. Mg/Mg2+) (Fig. S13, ESI†).34 In order to directly investigate the capability of STAR on catalyzing polysulfide conversions electrochemically, symmetric cells based on STAR@LCNC and CNT electrodes (without sulfur loading) were assembled by adding MgSx cathodelyte.63 The STAR@LCNC electrodes exhibit the characteristic redox peaks of MgSx at 0.2 mV s−1. The increased redox capability is reflected in the area of the wrapped curve and peak current, of which the STAR@LCNC cell is significantly larger than those of the CNT one (Fig. 4E), which indicates the abundant MgSx catalyzed by STAR@LCNC.47,64 Furthermore, Fig. 4F displays a quick response to the formation of MgS within 15 min on the STAR@LCNC surface, implying the fast conversion of MgSx to MgS precipitates with the aid of STAR.65 The higher peak current and larger peak area for the STAR@LCNC also demonstrate more MgS were generated and deposited during the same time, indicating the high conversion utilization.
After sulfur loading, the sulfur was observed to be well-distributed over the catalytic STAR@LCNC host matrix with a weight ratio of ∼71 wt% (Fig. S6 and S14–S16, ESI†). To evaluate the practical catalysis ability of STAR@LCNC, a variety of electrochemical tests were performed on the Mg–S batteries. In the fresh state (Fig. S17A, ESI†), the two cells exhibit relatively high internal and charge transfer resistances (4556 and 3476 Ohm) in the electrochemical impedance spectroscopy (EIS), attributed to the presence of a passivation layer on the commercial Mg anode.23 During the 30 cycles, the STAR@LCNC-S cathode displays much smaller charge transfer resistance than the controlled CNT-S cathode, implying the fast electron transfer stimulated by the STAR (Fig. S17, ESI†). In addition, the magnesium ion diffusion rate is determined according to the reported diffusion equation 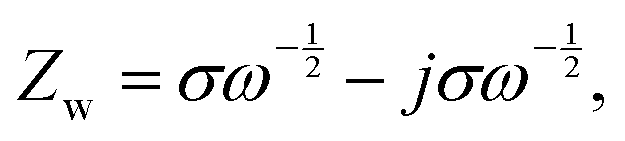 where ω represents frequency. The magnesium ion diffusion rate is determined by the slope in Fig. 4G according to the formula of
where ω represents frequency. The magnesium ion diffusion rate is determined by the slope in Fig. 4G according to the formula of 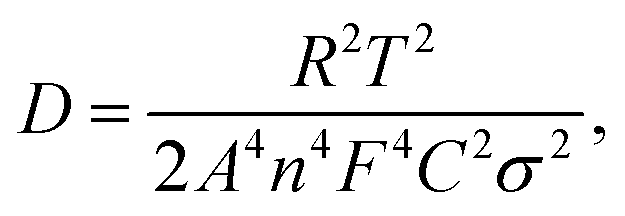 where R (8.3145 J K−1 mol−1), T (room temperature), and F (96
where R (8.3145 J K−1 mol−1), T (room temperature), and F (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1) are the constants and A, n, C, σ and D represent the area of the electrode, the number of transfer electron, the concentration of magnesium ions, the fitting slope and the magnesium ion diffusion rate, respectively.66 As we can see, the STAR@LCNC-S cathode exhibits a smaller slope derived from EIS (122 vs. 274) (Fig. 4G), indicating the higher Mg ion transport kinetics across the interface with the aid of the highly active zinc atomic catalysts. Fig. S18A–C (ESI†) illustrate the multi-step redox reactions of sulfur species. Apparently, the STAR@LCNC-S cathode displays the first oxidation peak at 1.643 V, lower than that of CNT-S (1.677 V), indicating the lower demagnesium barrier to be overcome.65 In the successive cycling, the oxidation peak in the STAR@LCNC-S cathode moves to the lower potential range (Fig. S18A, ESI†). The obtained higher redox peaks and lower oxidation potential peak strongly demonstrate the reduced barriers for interfacial desolvation, the decomposition of MgS and polysulfide conversions in the presence of STAR. The galvanostatic voltage profiles in Fig. S19 (ESI†) show the decomposition potential barrier is decreased from 1.58 to 1.45 V for STAR@LCNC-S and it delivers a specific capacity of 1107 mA h g−1 at 0.05C (1C = 1672 mA g−1, based on S weight), which means a lower overpotential and activation energy barrier needs to be overcome to reach high utilization.
485 C mol−1) are the constants and A, n, C, σ and D represent the area of the electrode, the number of transfer electron, the concentration of magnesium ions, the fitting slope and the magnesium ion diffusion rate, respectively.66 As we can see, the STAR@LCNC-S cathode exhibits a smaller slope derived from EIS (122 vs. 274) (Fig. 4G), indicating the higher Mg ion transport kinetics across the interface with the aid of the highly active zinc atomic catalysts. Fig. S18A–C (ESI†) illustrate the multi-step redox reactions of sulfur species. Apparently, the STAR@LCNC-S cathode displays the first oxidation peak at 1.643 V, lower than that of CNT-S (1.677 V), indicating the lower demagnesium barrier to be overcome.65 In the successive cycling, the oxidation peak in the STAR@LCNC-S cathode moves to the lower potential range (Fig. S18A, ESI†). The obtained higher redox peaks and lower oxidation potential peak strongly demonstrate the reduced barriers for interfacial desolvation, the decomposition of MgS and polysulfide conversions in the presence of STAR. The galvanostatic voltage profiles in Fig. S19 (ESI†) show the decomposition potential barrier is decreased from 1.58 to 1.45 V for STAR@LCNC-S and it delivers a specific capacity of 1107 mA h g−1 at 0.05C (1C = 1672 mA g−1, based on S weight), which means a lower overpotential and activation energy barrier needs to be overcome to reach high utilization.
High electrochemical performances of Mg–S cells with STAR@LCNC catalyst
The catalytic effects of STAR@LCNC were also demonstrated in the rate and cycling performances. The STAR@LCNC-S electrode delivers outstanding rate performances of 925, 900, 860 and 610 mA h g−1 at 0.1, 0.5, 1 and 2C, respectively (Fig. 5A). It recovers to 866 mA h g−1 when the current density is switched back to 1C, displaying robust reversibility. As highlighted in Fig. 5B, the rate performances of our cell are superior to the other Mg–S results reported so far, especially cycled at a high rate, verifying the validity of the single-atom catalysts in propelling conversion kinetics for high power density. In the various voltage profiles (Fig. S20A, ESI†), obvious plateaus are still available even under 2C, implying the much faster conversion kinetics assisted by STAR. In the long-term cycles (Fig. 5C), the specific capacities of STAR@LCNC-S and CNT-S electrodes increase slowly in the initial ten cycles at 0.5C, which might be ascribed to the sufficient immersion of electrolyte and the activation and removal of the MgO passivation layer on the anode side. After 400 cycles, the capacity of the STAR@LCNC-S cell stabilizes at approximately 577 mA h g−1 and possesses low overpotentials (Fig. S20B, ESI†), much better than the CNT one (260 mA h g−1). It should be mentioned that the STAR@LCNC matrix itself only contributes to 10 mA h g−1 (Fig. S21, ESI†), which is negligible in the entire capacity. As far as we know, the lifespans in the previous reports are limited within 100 cycles, and we are delighted to claim that our results in performance and lifespan are the best one (Fig. 5D), thanks to the accelerated conversion kinetics.2,22,67–69 Furthermore, enhancing the mass loading to 4 mg cm−2, the cell could power a series of star lights (Fig. 5F) and display an initial capacity of 964 mA h g−1 at 0.1C. After 50 cycles, the high areal loading cathode still exhibits an areal capacity of 2.92 mA h cm−2 with a capacity retention of 75.6% (Fig. 5E), which has never been reached for low/high loading cells in earlier reports (Fig. 5F). These results strongly prove that the STAR propels the dissociation of MgCl(THF)x+, and the kinetics of Mg2+ and polysulfide conversions for higher rate and longer lifespan.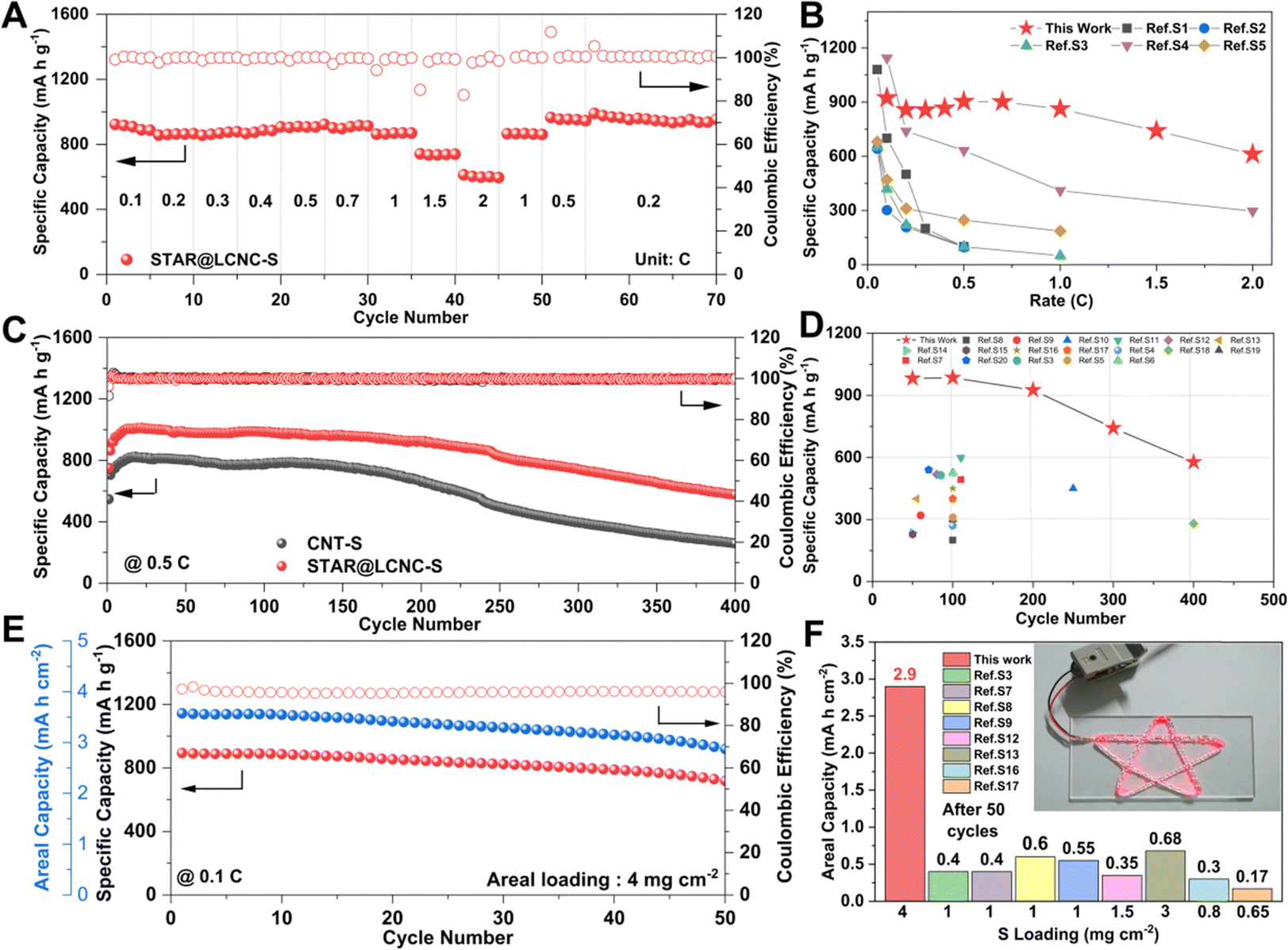 | ||
| Fig. 5 High electrochemical performances of Mg–S cells with STAR@LCNC catalyst. (A) Rate performance for the STAR@LCNC-S electrode from 0.05 to 2C. (B) Comparison of rate performances between the STAR@LCNC-S electrode and ever reported S electrodes in Mg–S batteries (see Table S2 for details, ESI†). (C) Cycling performance of the STAR@LCNC-S and CNT-S electrodes for 400 cycles at 0.5C. (D) Comparison of the electrochemical properties between the STAR@LCNC-S electrode and previously reported S electrodes in Mg–S batteries (see Table S3 for details, ESI†). (E) The high mass loading STAR@LCNC-S electrode cycled at 0.1C. (F) Comparison of the areal capacity after 50 cycles between the STAR@LCNC-S electrode and previously reported S electrodes in Mg–S batteries (see Table S4 for details, ESI†) and the high-loading cell with the STAR@LCNC-S electrode powers a series of star-shaped lights. | ||
Catalytic mechanism analysis of STAR@LCNC after cycling
To gain deeper understandings of the catalytic mechanism of STAR, ex situ SEM, time-of-flight secondary-ion-mass-spectrometry (TOF-SIMS), XPS, and HAADF-STEM measurements were carried out, respectively. As displayed in Fig. S22 (ESI†), the surface of the coupled Mg anode after cycling was also investigated. As shown, the Mg anode from the STAR@LCNC-S full cell is much smoother with lower sulfur content on the surface (0.3% vs. 1%), which indicates that most of the polysulfides are transformed under the help of STAR@LCNC and little polysulfides shuttle to the metallic Mg anode.23 As shown in the TOF-SIMS image after 50 cycles (under the fully charged state) (Fig. 6A and B), uniform distribution of MgS is achieved both in the interior and on the top surface under the electrochemical modulation of STAR@LCNC, while more MgS aggregates are found in the CNT-S electrode. The evolution curve of the S− species intensity shows an opposite trend to that of the MgS− one at the beginning sputter time (Fig. 6C and D), indicating the surface of the CNT-S is surrounded and covered by soluble MgSx.70,71 In contrast, both the MgS− and S− species intensities for the cycled STAR@LCNC-S display a gradually increasing tendency, indicating less polysulfide species on the cathode surface and most of the discharged product was transformed into sulfur in the bulk cathode interior catalyzed by STAR. The cycled electrode was subjected to HAADF-STEM characterization. As shown in Fig. 6E and F, the single Zn atoms still maintained the atomic morphology after 50 cycles and no aggregation of Zn atoms was observed, implying the stability and robustness of the STAR in the present system for catalyzing the redox reaction kinetics. Compared with the pristine STAR@LCNC, little changes of XPS peaks are observed after discharging or charging (Fig. 6G). Meanwhile, the peak located at 399.6 eV is assigned to the formation of Zn–N (Fig. S23A, ESI†), suggesting the stability of the atomic Zn catalyst after cycling in the STAR@LCNC-S cathode.2,49 The slight shift of the Zn 2p1/2 peak toward lower binding energy after cycling might be ascribed to the interaction or catalyst effect between STAR and the formed polysulfide intermediator (Fig. S23B, ESI†). Above all, the designed STAR@LCNC still keeps the atomic presence and works as an atomic catalyst well during the charge/discharge process. Therefore, compared with the pristine CNT-S cell with a severe shuttling effect (Fig. 6H), the STAR@LCNC modified Mg–S cell exhibits a cross-linked conductive network, strong adsorption toward MgSx, and fast conversion kinetics with decreased barriers by introducing atomic reactor promoters (Fig. 6I), contributing to the high-rate performance and lifespan in low or high mass loading cells.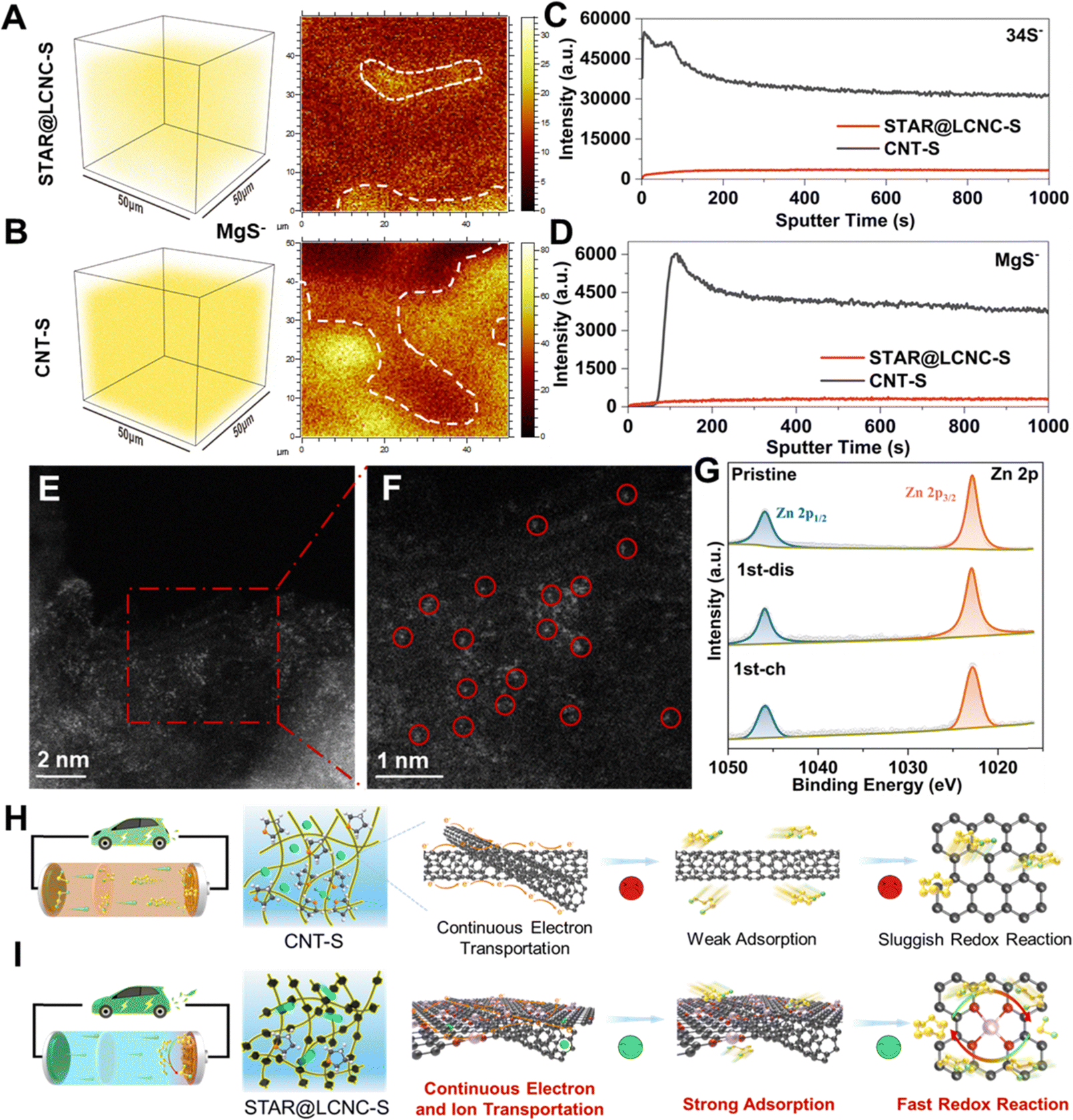 | ||
| Fig. 6 Catalytic mechanism analysis of STAR@LCNC after cycling. The 3D reconstructions and top-view surface mapping of MgS− species in cycled (A) STAR@LCNC-S cathode and (B) CNT-S cathode after 50 cycles at 0.5C via TOF-SIMS. The scalebar is 50 μm. The signal intensity comparisons of (C) S− species and (D) MgS− species in cycled STAR@LCNC-S and CNT-S cathodes, respectively. The white dashed line indicates the aggregation location of MgS− species. (E) and (F) HAADF-STEM images of the single zinc atoms in the cycled STAR@LCNC-S cathode composite after 50 cycles at 0.5C (highlighted by red circles in (F)). (G) The high-resolution XPS of Zn 2p for pristine, fully charged, and fully discharged STAR@LCNC-S electrodes. The proposed catalytic mechanism without atomic reactors in (H) CNT-S or with (I) STAR@LCNC-S in Mg–S batteries. | ||
Conclusion
In summary, a self-tandem catalysis for achieving fast Mg2+ desolvation and sulfur conversions is pioneered to deal with the sluggish reaction kinetics in polysulfide conversions and decomposition of MgS for the first time. As a demo, the serially-assembled train-like atomic reactors of zinc atomic catalysts employed in long-conductive nanocarbons (STAR@LCNC) are fabricated by locating the Zn atoms at the porous beads linked on long-chain CNT. The crucial catalytic roles and working mechanism of STAR@LCNC in tailoring the solvation structure, accelerating Mg2+ transport and catalyzing polysulfide conversion reactions by lowering the barriers are comprehensively demonstrated by theoretical simulations, static polysulfide adsorption, electrochemical analysis, and in situ spectroscopical and electron characterizations. Consequently, the STAR@LCNC can effectively promote the dissolution of MgCl(THF)x+ structure, release a large amount of free Mg2+, and then accelerate the transfer of Mg2+ to the sulfur cathode to participate in the conversion of sulfur and polysulfide, effectively improving the utilization of sulfur. The as-fabricated STAR@LCNC-S cathodes show the best ever rate performances up to 2C and with the so-far longest lifespan of 400 cycles. More impressively, the cell with a high mass loading of 4.0 mg cm−2 is capable of maintaining a high capacity of 729 mA h g−1 after 50 cycles, corresponding to an areal capacity of 2.92 mA h cm−2, which has never been achieved in previous Mg–S batteries. These results open up new routes to design and fabricate highly active catalysts with favorable interface environments to develop faster, longer-lasting, higher-energy density Mg–S batteries.Author contributions
J. Wang, S. Wang, and H. Lin conceived the idea. Q. Guan and J. Wang designed and conducted the experiments; L. Li, J. Zhang, L. Jia, H. Hu, Y. Zhang, H. Hu, S. Cheng, H. Zhang and H. Li helped to characterize the samples. Q. Zhuang carried out the theoretical simulation. Q. Guan, J. Wang, M. Liu, H. Lin, and S. Wang co-wrote and revised the manuscript. All authors were involved in the discussion of all the results and the revision of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge the National Key R&D Program of China (2021YFA1201503), the National Natural Science Foundation of China (No. 21972164, 22279161, 12264038, 22309144), the Natural Science Foundation of Jiangsu Province (BK. 20210130), Innovative and Entrepreneurial Doctor in Jiangsu Province (JSSCBS20211428), China Postdoctoral Science Foundation (2023M732561, 2023M731084), Shanxi Natural Science Basic Research Plan (No. 2022JQ-137) as well as the technical support from Nano-X, Suzhou Institute of Nano-tech and Nano-bionics, Chinese Academy of Sciences. Dr. J. Wang is thankful for the funding fellowship provided by the Alexander von Humboldt Foundation and the Helmholtz Association. We also thanks Peng Cheng for the help with the schematic illustration.References
- A. Gurung, K. M. Reza, S. Mabrouk, B. Bahrami, R. Pathak, B. S. Lamsal, S. I. Rahman, N. Ghimire, R. S. Bobba, K. Chen, J. Pokharel, A. Baniya, M. A. R. Laskar, M. Liang, W. Zhang, W. H. Zhang, S. Yang, K. Xu and Q. Qiao, Adv. Funct. Mater., 2020, 30, 2001865 CrossRef CAS.
- X. Zhou, J. Tian, J. Hu and C. Li, Adv. Mater., 2018, 30, 1704166 CrossRef PubMed.
- R. F. Service, Science, 2018, 359, 1080–1081 CrossRef CAS PubMed.
- N. Wang, X. Zhang, Z. Ju, X. Yu, Y. Wang, Y. Du, Z. Bai, S. Dou and G. Yu, Nat. Commun., 2021, 12, 4519 CrossRef CAS PubMed.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- W. Cao, J. Zhang and H. Li, Energy Storage Mater., 2020, 26, 46–55 CrossRef.
- R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Joule, 2019, 3, 27–52 CrossRef CAS.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- Y. Lu, C. Wang, Q. Liu, X. Li, X. Zhao and Z. Guo, Small Methods, 2021, 5, 2001303 CrossRef CAS PubMed.
- J. Zhang, R. He, L. Jia, C. You, Y. Zhang, M. Liu, N. Tian, H. Lin and J. Wang, Adv. Funct. Mater., 2023, 33, 2305674 CrossRef CAS.
- P. Wang and M. R. Buchmeiser, Adv. Funct. Mater., 2019, 29, 1905248 CrossRef CAS.
- L. Kong, C. Yan, J.-Q. Huang, M.-Q. Zhao, M.-M. Titirici, R. Xiang and Q. Zhang, Energy Environ. Mater., 2018, 1, 100–112 CrossRef CAS.
- J. Wang, L. Li, H. Hu, H. Hu, Q. Guan, M. Huang, L. Jia, H. Adenusi, K. V. Tian, J. Zhang, S. Passerini and H. Lin, ACS Nano, 2022, 16, 17729–17760 CrossRef CAS PubMed.
- H. Zhang, L. Qiao and M. Armand, Angew. Chem., Int. Ed., 2022, 61, e202214054 CrossRef CAS PubMed.
- S. H. Chung and A. Manthiram, Adv. Mater., 2019, 31, 1901125 CrossRef PubMed.
- J. Zhang, C. You, H. Lin and J. Wang, Energy Environ. Mater., 2022, 5, 731–750 CrossRef CAS.
- B. P. Vinayan, Z. Zhao-Karger, T. Diemant, V. S. Chakravadhanula, N. I. Schwarzburger, M. A. Cambaz, R. J. Behm, C. Kubel and M. Fichtner, Nanoscale, 2016, 8, 3296–3306 RSC.
- A. Robba, A. Vizintin, J. Bitenc, G. Mali, I. Arčon, M. Kavčič, M. Žitnik, K. Bučar, G. Aquilanti, C. Martineau-Corcos, A. Randon-Vitanova and R. Dominko, Chem. Mater., 2017, 29, 9555–9564 CrossRef CAS.
- D. Muthuraj, M. Pandey, M. Krishna, A. Ghosh, R. Sen, P. Johari and S. Mitra, J. Power Sources, 2021, 486, 229326 CrossRef CAS.
- B. P. Vinayan, H. Euchner, Z. Zhao-Karger, M. A. Cambaz, Z. Li, T. Diemant, R. J. Behm, A. Gross and M. Fichtner, J. Mater. Chem. A, 2019, 7, 25490–25502 RSC.
- Z. Zhang, Z. Cui, L. Qiao, J. Guan, H. Xu, X. Wang, P. Hu, H. Du, S. Li, X. Zhou, S. Dong, Z. Liu, G. Cui and L. Chen, Adv. Energy Mater., 2017, 7, 1602055 CrossRef.
- S. Zhang, W. Ren, Y. NuLi, B. Wang, J. Yang and J. Wang, Chem. Eng. J., 2022, 427, 130902 CrossRef CAS.
- P. Wang, K. Küster, U. Starke, C. Liang, R. Niewa and M. R. Buchmeiser, J. Power Sources, 2021, 515, 230604 CrossRef CAS.
- Z. Ye, P. Li, W. Wei, C. Huang, L. Mi, J. Zhang and J. Zhang, Adv. Sci., 2022, 9, 2200067 CrossRef CAS PubMed.
- X. Yu and A. Manthiram, ACS Energy Lett., 2016, 1, 431–437 CrossRef CAS.
- P. Wang, J. Trück, J. Häcker, A. Schlosser, K. Küster, U. Starke, L. Reinders and M. R. Buchmeiser, Energy Storage Mater., 2022, 49, 509–517 CrossRef.
- Z. Zhou, B. Chen, T. Fang, Y. Li, Z. Zhou, Q. Wang, J. Zhang and Y. Zhao, Adv. Energy Mater., 2019, 10, 1902023 CrossRef.
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47, 8804–8841 RSC.
- H. Fan, X. Zhang, J. Xiao, Y. Lin, S. Ren, Y. Zhao, H. Yuan, L. Pan, Q. Lin, H. Liu, Y. Su, Y. Su, Y. Liu and Y. Zhang, Energy Storage Mater., 2022, 51, 873–881 CrossRef.
- Q. Zou, Y. Sun, Z. Liang, W. Wang and Y. C. Lu, Adv. Energy Mater., 2021, 11, 2101552 CrossRef CAS.
- L. Sheng, Z. Hao, J. Feng, W. Du, M. Gong, L. Kang, P. R. Shearing, D. J. L. Brett, Y. Huang and F. R. Wang, Nano Energy, 2021, 83, 105832 CrossRef CAS.
- H. Fan, Y. Zhao, J. Xiao, J. Zhang, M. Wang and Y. Zhang, Nano Res., 2020, 13, 2749–2754 CrossRef CAS.
- H. Fan, Z. Zheng, L. Zhao, W. Li, J. Wang, M. Dai, Y. Zhao, J. Xiao, G. Wang, X. Ding, H. Xiao, J. Li, Y. Wu and Y. Zhang, Adv. Funct. Mater., 2019, 30, 1909370 CrossRef.
- H. Dong, O. Tutusaus, Y. Liang, Y. Zhang, Z. Lebens-Higgins, W. Yang, R. Mohtadi and Y. Yao, Nat. Energy, 2020, 5, 1043–1050 CrossRef CAS.
- J. Zhang, C. You, H. Lin and J. Wang, Energy Environ. Mater., 2022, 5, 731–750 CrossRef CAS.
- J. Wang, J. Zhang, J. Wu, M. Huang, L. Jia, L. Li, Y. Zhang, H. Hu, F. Liu, Q. Guan, M. Liu, H. Adenusi, H. Lin and S. Passerini, Adv. Mater., 2023, 35, e2302828 CrossRef PubMed.
- X. Li, Q. Guan, Z. Zhuang, Y. Zhang, Y. Lin, J. Wang, C. Shen, H. Lin, Y. Wang, L. Zhan and L. Ling, ACS Nano, 2023, 17, 1653–1662 CrossRef CAS PubMed.
- L. Li, H. Tu, J. Wang, M. Wang, W. Li, X. Li, F. Ye, Q. Guan, F. Zhu, Y. Zhang, Y. Hu, C. Yan, H. Lin and M. Liu, Adv. Funct. Mater., 2023, 33, 2212499 CrossRef.
- X. Zhang, X. Li, Y. Zhang, X. Li, Q. Guan, J. Wang, Z. Zhuang, Q. Zhuang, X. Cheng, H. Liu, J. Zhang, C. Shen, H. Lin, Y. Wang, L. Zhan and L. Ling, Adv. Funct. Mater., 2023, 2302624 CrossRef CAS.
- Z. Liang, J. Shen, X. Xu, F. Li, J. Liu, B. Yuan, Y. Yu and M. Zhu, Adv. Mater., 2022, 34, 2200102 CrossRef CAS PubMed.
- C. Lu, R. Fang and X. Chen, Adv. Mater., 2020, 32, 1906548 CrossRef CAS PubMed.
- W. Chen, B. Wu, Y. Wang, W. Zhou, Y. Li, T. Liu, C. Xie, L. Xu, S. Du, M. Song, D. Wang, Y. liu, Y. Li, J. Liu, Y. Zou, R. Chen, C. Chen, J. Zheng, Y. Li, J. Chen and S. Wang, Energy Environ. Sci., 2021, 14, 6428–6440 RSC.
- W. Ni, Z. Liu, Y. Zhang, C. Ma, H. Deng, S. Zhang and S. Wang, Adv. Mater., 2021, 33, e2003238 CrossRef PubMed.
- J. Wang, J. Zhang, S. Cheng, J. Yang, Y. Xi, X. Hou, Q. Xiao and H. Lin, Nano Lett., 2021, 21, 3245–3253 CrossRef CAS PubMed.
- J. Wang, J. Zhang, S. Duan, L. Jia, Q. Xiao, H. Liu, H. Hu, S. Cheng, Z. Zhang, L. Li, W. Duan, Y. Zhang and H. Lin, Nano Lett., 2022, 22, 8008–8017 CrossRef CAS PubMed.
- J. Wang, L. Jia, S. Duan, H. Liu, Q. Xiao, T. Li, H. Fan, K. Feng, J. Yang, Q. Wang, M. Liu, J. Zhong, W. Duan, H. Lin and Y. Zhang, Energy Storage Mater., 2020, 28, 375–382 CrossRef.
- J. Wang, L. J. Jia, J. Zhong, Q. B. Xiao, C. Wang, K. T. Zang, H. T. Lin, H. C. Zheng, J. Luo, J. Yang, H. Y. Fan, W. H. Duan, Y. Wu, H. Z. Lin and Y. G. Zhang, Energy Storage Mater., 2019, 18, 246–252 CrossRef.
- R. Wang, R. Wu, X. Yan, D. Liu, P. Guo, W. Li and H. Pan, Adv. Funct. Mater., 2022, 32, 2200424 CrossRef CAS.
- J. Wang, W. Qiu, G. Li, J. Liu, D. Luo, Y. Zhang, Y. Zhao, G. Zhou, L. Shui, X. Wang and Z. Chen, Energy Storage Mater., 2022, 46, 269–277 CrossRef.
- X. Fan, S. Chen, W. Gong, X. Meng, Y. Jia, Y. Wang, S. Hong, L. Zheng, L. Zheng, C. W. Bielawski and J. Geng, Energy Storage Mater., 2021, 41, 14–23 CrossRef.
- J. Wang, L. Jia, H. Lin and Y. Zhang, ChemSusChem, 2020, 13, 3404–3411 CrossRef CAS PubMed.
- L. Wang, P. Jankowski, C. Njel, W. Bauer, Z. Li, Z. Meng, B. Dasari, T. Vegge, J. M. G. Lastra, Z. Zhao-Karger and M. Fichtner, Adv. Sci., 2022, 9, e2104605 CrossRef PubMed.
- Y. Xu, Y. Ye, S. Zhao, J. Feng, J. Li, H. Chen, A. Yang, F. Shi, L. Jia, Y. Wu, X. Yu, P. A. Glans-Suzuki, Y. Cui, J. Guo and Y. Zhang, Nano Lett., 2019, 19, 2928–2934 CrossRef CAS PubMed.
- Q. Zhao, R. Wang, Y. Zhang, G. Huang, B. Jiang, C. Xu and F. Pan, J. Magnesium Alloys, 2021, 9, 78–89 CrossRef CAS.
- X. Song, M. Zhang, M. Yao, C. Hao and J. Qiu, ACS Appl. Mater. Interfaces, 2018, 10, 43896–43903 CrossRef CAS PubMed.
- Y. Xu, Y. Zhao, S. Zhao, J. Zhang, J. Li, J. Guo and Y. Zhang, Energy Storage Mater., 2021, 42, 513–516 CrossRef.
- F. Fu, B. Zheng, L.-H. Xie, H. Du, S. Du and Z. Dong, Crystals, 2018, 8, 367 CrossRef.
- Y. Zhou, M. Su, X. Yu, Y. Zhang, J.-G. Wang, X. Ren, R. Cao, W. Xu, D. R. Baer, Y. Du, O. Borodin, Y. Wang, X.-L. Wang, K. Xu, Z. Xu, C. Wang and Z. Zhu, Nat. Nanotechnol., 2020, 15, 224–230 CrossRef CAS PubMed.
- C. J. Kliewer, C. Aliaga, M. Bieri, W. Huang, C.-K. Tsung, J. B. Wood, K. Komvopoulos and G. A. Somorjai, J. Am. Chem. Soc., 2010, 132, 13088–13095 CrossRef CAS PubMed.
- W. Wang and S. Ye, Phys. Chem. Chem. Phys., 2017, 19, 4488–4493 RSC.
- L. Hu, C. Dai, H. Liu, Y. Li, B. Shen, Y. Chen, S.-J. Bao and M. Xu, Adv. Energy Mater., 2018, 8, 1800709 CrossRef.
- R. Zhang, C. Cui, R. Xiao, R. Li, T. Mu, H. Huo, Y. Ma, G. Yin and P. Zuo, Chem. Eng. J., 2023, 451, 138663 CrossRef CAS.
- H. Lin, L. Yang, X. Jiang, G. Li, T. Zhang, Q. Yao, G. W. Zheng and J. Y. Lee, Energy Environ. Sci., 2017, 10, 1476–1486 RSC.
- S. Cheng, J. Wang, S. Duan, J. Zhang, Q. Wang, Y. Zhang, L. Li, H. Liu, Q. Xiao and H. Lin, Chem. Eng. J., 2021, 417, 128172 CrossRef CAS.
- J. Wang, J. Zhang, S. Duan, T. Li, L. Jia, H. Liu, L. Li, S. Cheng, H. Hu, M. Huang, H. Hu, S. Zhang, Q. Xiao and H. Lin, Chem. Eng. J., 2022, 429, 132352 CrossRef CAS.
- Y. Xu, W. Li, G. Zhou, Z. Pan and Y. Zhang, Energy Storage Mater., 2018, 14, 253–257 CrossRef.
- Y. Yang, Y. Qiu, Y. NuLi, W. Wang, J. Yang and J. Wang, J. Mater. Chem. A, 2019, 7, 18295–18303 RSC.
- Y. Ji, X. Liu-Théato, Y. Xiu, S. Indris, C. Njel, J. Maibach, H. Ehrenberg, M. Fichtner and Z. Zhao-Karger, Adv. Funct. Mater., 2021, 31, 2100868 CrossRef CAS.
- C. Zhao, A. Daali, I. Hwang, T. Li, X. Huang, D. Robertson, Z. Yang, S. Trask, W. Xu, C. J. Sun, G. L. Xu and K. Amine, Angew. Chem., Int. Ed., 2022, 61, e202203466 CrossRef CAS PubMed.
- R. Fang, H. Xu, B. Xu, X. Li, Y. Li and J. B. Goodenough, Adv. Funct. Mater., 2020, 31, 2001812 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee04028c |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2024 |