
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled and sequential single-electron reduction of the uranyl dication†
Tom J. N.
Obey
,
Gary S.
Nichol
 and
Jason B.
Love
*
and
Jason B.
Love
*
EaStCHEM School of Chemistry, Joseph Black Building, University of Edinburgh, Edinburgh EH9 3FJ, UK. E-mail: jason.love@ed.ac.uk
First published on 16th September 2024
Abstract
A flexible tripodal pyrrole-imine ligand (H3L) has been used to facilitate the controlled and sequential single-electron reductions of the uranyl dication from the U(VI) oxidation state to U(V) and further to U(IV), processes that are important to understanding the reduction of uranyl and its environmental remediation. The uranyl(VI) complexes UO2(HL)(sol) (sol = THF, py) were straightforwardly accessed by the transamination reaction of H3L with UO2{N(SiMe3)2}2(THF)2 and adopt ‘hangman’ structures in which one of the pyrrole-imine arms is pendant. While deprotonation of this arm by LiN(SiMe3)2 causes no change in uranyl oxidation state, single-electron reduction of uranyl(VI) to uranyl(V) occurred on addition of two equivalents of KN(SiMe3)2 to UO2(HL)(sol). The potassium cations of this new [UVO2(K2L)]2 dimer were substituted by transmetalation with the appropriate metal chloride salt, forming the new uranyl(V) tetra-heterometallic complexes, [UVO2Zn(L)(py)2]2 and [UVO2Ln(Cl)(L)(py)2]2 (Ln = Y, Sm, Dy). The dimeric uranyl(V)–yttrium complex underwent further reduction and chloride abstraction to form the tetrametallic U(IV) complex [UIVO2YIII(py)]2, so highlighting the adaptability of this ligand to stabilise a variety of different uranium oxidation states.
Introduction
As the least radioactive and most readily available actinyl ion ([AnO2]n+) the uranyl dication [UO2]2+ is by far the most studied. It is the most thermodynamically stable form of uranium found in the environment and is a soluble and problematic environmental contaminant from nuclear waste. It exhibits two linear U–O bonds that are particularly strong due to overlap of the U 5f and 6d orbitals with the O 2p resulting in one σ and two π bonds, giving a formal bond order of three.1,2 This makes the uranyl oxo groups chemically inert and difficult to functionalise. By contrast, analogous transition metal oxo complexes exhibit extensive Lewis-base chemistry and can be used in catalysis and oxo atom-transfer reactions.3–5The single-electron reduction of [UVIO2]2+ is achieved by microbes under anaerobic conditions, but the resulting monocation, [UVO2]+, is unstable in aqueous conditions and disproportionates to uranyl(VI) and uranium(IV) species.6,7 This disproportionation process is thought to proceed through interaction of the oxo groups on adjacent uranyl centres, with this so-called cation–cation interaction (CCI) allowing electron transfer between the metals. These CCIs are also seen in heavier actinyl compounds of neptunium and plutonium, metals that are components of nuclear waste, and disrupt actinide separation processes.8
The U(V) uranyl monocation, [UO2]+, has been found to be stabilised against disproportionation under non-aqueous and anaerobic laboratory conditions by utilising suitable ligands to control the equatorial coordination environment of the uranyl.9–15 The decreased bond order of the U–O bonds accompanied by an increase in the Lewis-basicity of the oxo groups of the 5f1 ion makes the study of these complexes of interest.16,17
Previously, we have reported a macrocyclic ligand with two N4 pyrrole-imine coordination pockets that form “Pacman” shaped complexes (Chart 1, structures A and B) with a uranyl occupying one of the N4 pockets.18 The second pocket is then available for a second metal, such as a lanthanide or transition metal, to coordinate. This second metal is held in a position where it can access one of the uranyl oxo groups, facilitating the single electron-reduction of the uranium and stabilising the U(V) oxidation state.19,20
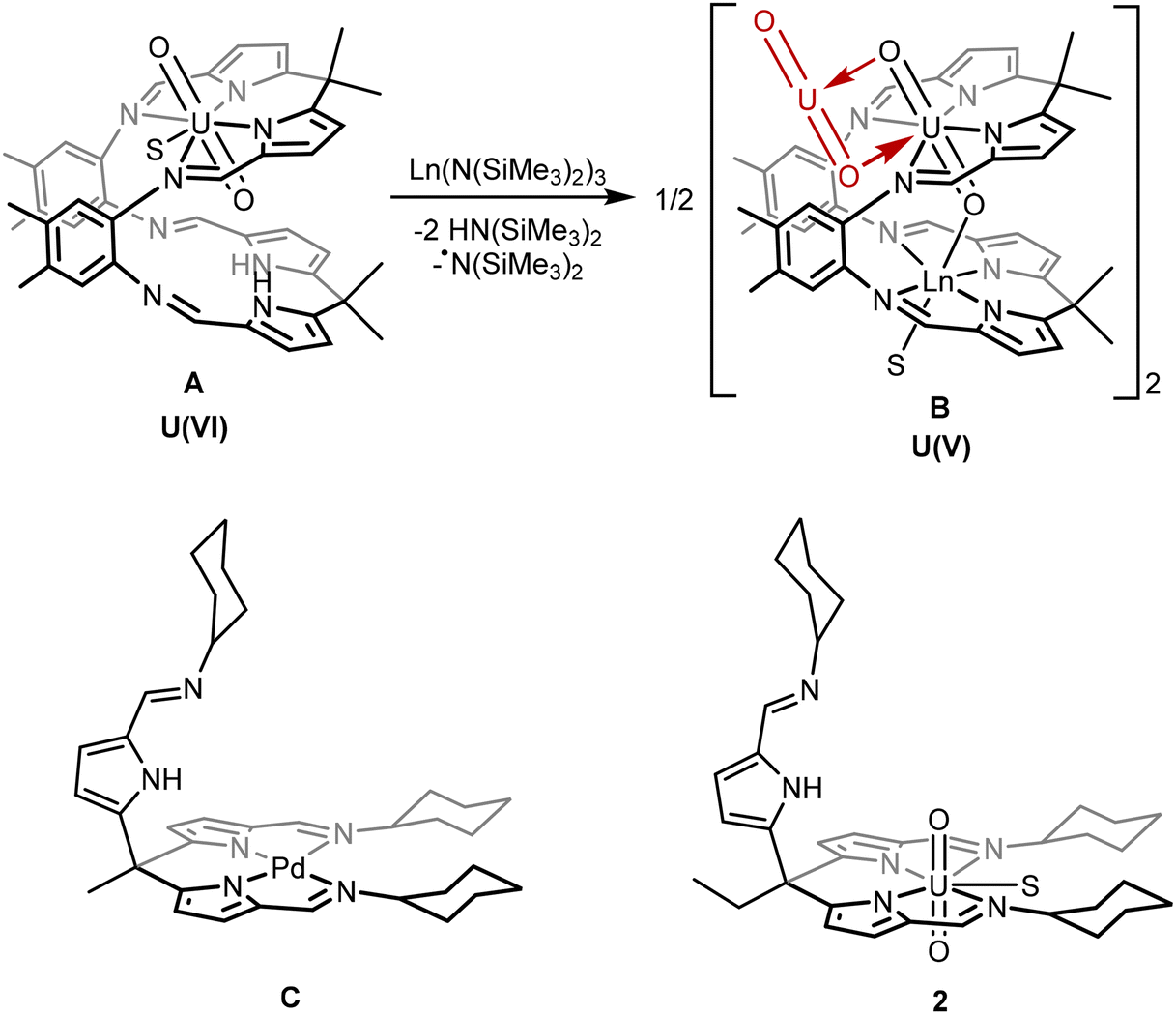 | ||
| Chart 1 Top: Example of the reaction of a uranyl “Pacman” complex (A) and a lanthanide silylamide to form the Ln-functionalised uranyl(V) complex (B) via ligand deprotonation and radical transfer.19,20 Bottom: The “hangman” palladium complex C (previous work) and uranyl complex 2 (this work) in which two ligand “arms” provide a square-planar N4 coordination environment to the metal and the third is pendant. S indicates a THF or pyridine solvent molecule.21,22 | ||
We have also reported a simpler, tripodal ligand system that incorporates similar pyrrole-imine groups as the macrocycle. This ligand can form “hangman” complexes with square-planar transition metals such as in the palladium complex C.21,22 The pendant arm in these complexes has the potential to bind a second metal in a similar way to the Pacman ligand system. However, the formation of multimetallic complexes using this tripodal ligand L has not previously been explored.
The work reported here explores the chemistry of the uranyl complex 2 of the tripodal ligand L in which two of the ligand arms coordinate to the equatorial plane of the uranyl dication leaving the third arm pendant. The pendant arm can in turn coordinate other metals in a position where they can interact with the uranyl oxo groups. Similar to the Pacman complexes A and B, these metals can facilitate the single-electron reduction of the uranyl from U(VI) to U(V) by stabilisation of the U(V) oxidation state, by complexation with alkali-, transition-, and rare-earth metals.19,20 Subsequent reduction of a uranyl(V) complex to uranium(IV) is also achieved, providing a rare example of controlled and sequential reduction of uranyl(VI) to uranium(IV) via uranyl(V) within a single ligand environment.
Results and discussion
In order to form uranyl complexes of the tripodal pyrrole-imine ligand L, the strongly coordinated water molecules associated with L that are generated during the imine condensation reaction needed to be removed.22 This is achieved by the reaction of L(H2O)n with an excess of trimethylsilyl chloride (TMSCl) followed by the addition of freshly sublimed 1,4-diazabicyclo[2.2.2]octane (DABCO) to remove the HCl generated (Scheme 1). The total removal of water from the compound is confirmed by the appearance of the pyrrole NH resonance in the 1H NMR spectrum at 10.6 ppm in d5-pyridine that correctly integrates to 3 protons. Interactions between any bound water and the pyrrole NH proton cause the resonance to shift to lower frequency and broaden significantly (Fig. S1†).23 The dimeric uranyl(VI) complex [UO2(HL)]21 was subsequently synthesised by the reaction of H3L with UO2{N(SiMe3)2}2(THF)2 in toluene under an inert atmosphere (Scheme 1). The silyl amide ligands of the uranyl starting material deprotonate two of the three pyrroles in H3L and the two anionic arms of the ligand coordinate to the uranium centre. The 1H NMR spectrum of 1 shows the loss of the C3 symmetry of the starting H3L, splitting each of the pyrrole, imine and cyclohexyl proton resonances into two with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 integration ratio; only one NH resonance with an integration of 1 is seen at 11.23 ppm. The diamagnetic 1H NMR spectrum (chemical shift range 11.23 to 1.12 ppm) confirms the U(VI) oxidation state along with the infrared spectrum of 1 displaying the O–U–O asymmetric stretch at 910 cm−1, a value typical for the uranyl(VI) dication.24–26
:1 integration ratio; only one NH resonance with an integration of 1 is seen at 11.23 ppm. The diamagnetic 1H NMR spectrum (chemical shift range 11.23 to 1.12 ppm) confirms the U(VI) oxidation state along with the infrared spectrum of 1 displaying the O–U–O asymmetric stretch at 910 cm−1, a value typical for the uranyl(VI) dication.24–26
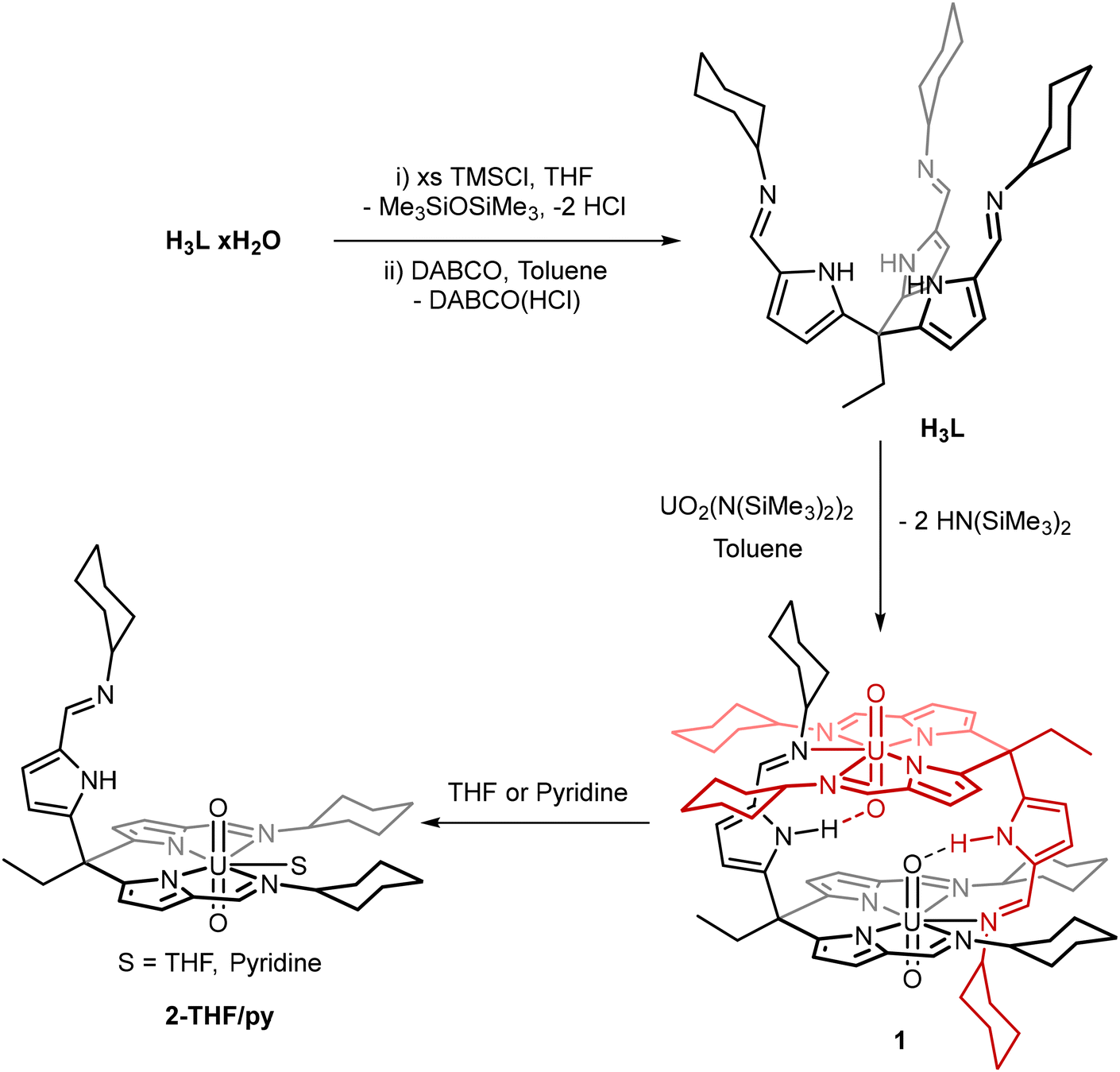 | ||
| Scheme 1 Synthetic procedures to form [UO2(HL)]2 (1) and the solvated [UO2(HL)(THF)] (2-THF) and [UO2(HL)(py)] (2-py) uranyl(VI) complexes. For clarity the separate ligand moieties of 1 have been displayed in black and red. | ||
Single crystals of 1 were obtained by slow evaporation of toluene over several weeks. The X-ray structure shows a dimeric complex in which two of the three ligand arms coordinate to each of the uranyl centres in the equatorial plane (Fig. 1). The third arm remains protonated and coordinates to a second uranyl centre through the imine nitrogen, forming the dimeric solid-state structure. The uranyl dioxo motif is linear and the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distances are similar to those in other uranyl(VI) complexes.27 No significant elongation of U1–O1 is seen, even though a hydrogen-bonding interaction between the O1 oxo group and the N1′ pyrrole hydrogen is evident (O1⋯N1′ 2.793(3) Å). The separation between the adjacent oxo groups O1 and O1′ at 2.877(2) Å is similar to that seen for binuclear, cofacial uranyl Pacman complexes.28 Dissolution of 1 in THF or pyridine and vapour diffusion of hexane results in the formation of single crystals of 2-THF and 2-py, respectively. Analysis of these X-ray crystal structures shows that, in each case, the dimeric structure of 1 is cleaved with the fifth uranyl equatorial site occupied by a THF or pyridine solvent molecule. While there is no notable change to the bonding metrics of the complex due to the identity of the coordinating solvent, the pendant pyrrole-imine arm is now positioned such that no NH-oxo hydrogen bonding occurs, neither within the molecule nor in the extended structure.
O bond distances are similar to those in other uranyl(VI) complexes.27 No significant elongation of U1–O1 is seen, even though a hydrogen-bonding interaction between the O1 oxo group and the N1′ pyrrole hydrogen is evident (O1⋯N1′ 2.793(3) Å). The separation between the adjacent oxo groups O1 and O1′ at 2.877(2) Å is similar to that seen for binuclear, cofacial uranyl Pacman complexes.28 Dissolution of 1 in THF or pyridine and vapour diffusion of hexane results in the formation of single crystals of 2-THF and 2-py, respectively. Analysis of these X-ray crystal structures shows that, in each case, the dimeric structure of 1 is cleaved with the fifth uranyl equatorial site occupied by a THF or pyridine solvent molecule. While there is no notable change to the bonding metrics of the complex due to the identity of the coordinating solvent, the pendant pyrrole-imine arm is now positioned such that no NH-oxo hydrogen bonding occurs, neither within the molecule nor in the extended structure.
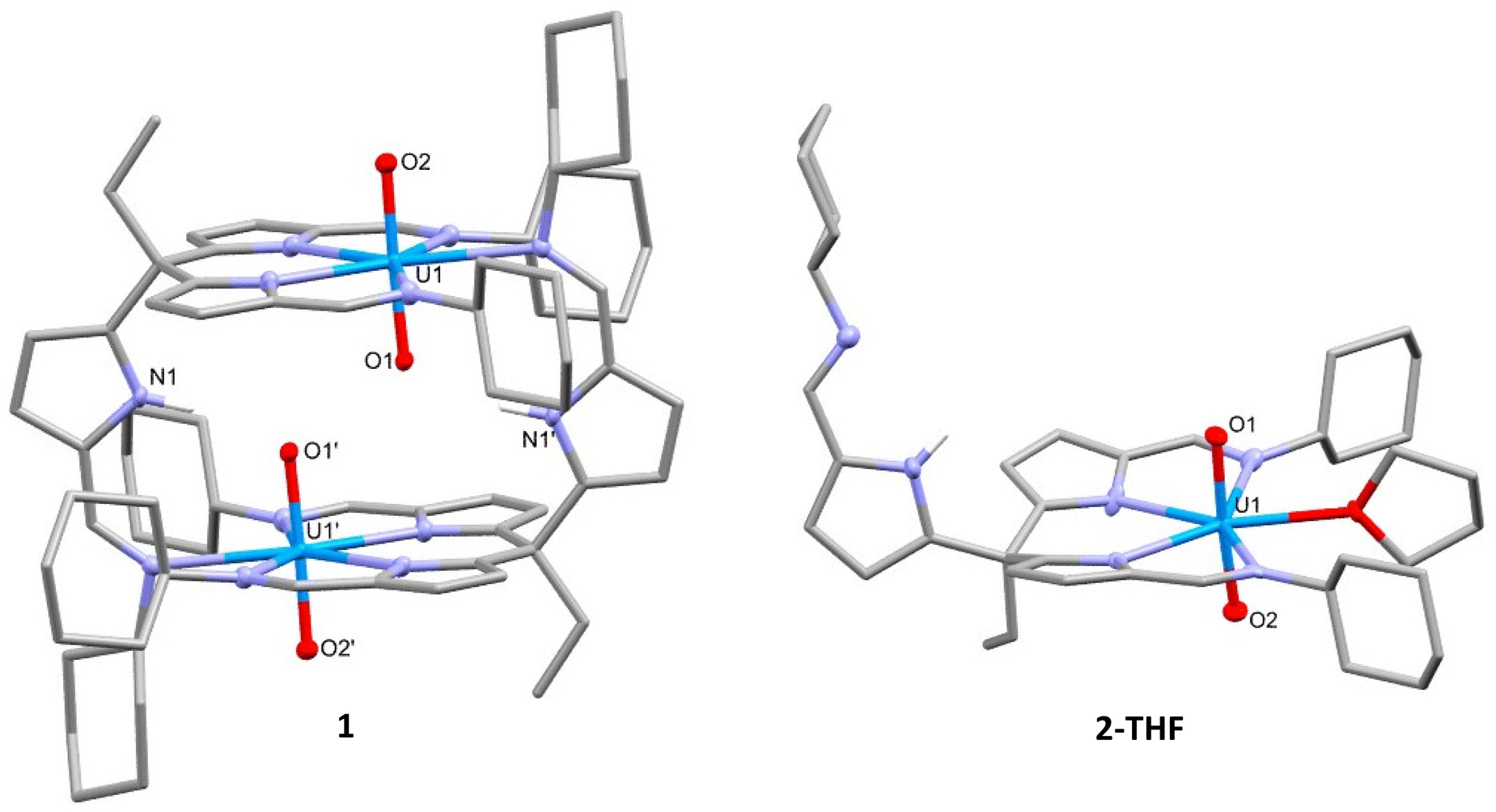 | ||
| Fig. 1 X-ray crystal structures of [UO2(HL)]2 (1, left) and the solvated complex [UO2(HL)(THF)] (2-THF, right). 1. For clarity, all hydrogen atoms except the pyrrole NH are omitted and displacement ellipsoids of the heteroatoms are drawn at 50% probability. Selected bond distances (Å) and angles (°): 1, U1–O1 = 1.774(2), U1–O2 = 1.770(2), N1′–O1 = 2.793(3), O1–O1′ = 2.877(2), O1–U1–O2 = 177.68(8); 2-THF, U1–O1 = 1.764(5), U1–O2 = 1.774(5), O1–U1–O2 = 175.4(2). | ||
Li-functionalised uranyl(VI) complex
The addition of one equivalent of LiN(SiMe3)2 to the uranyl(VI) complex 2-py in pyridine deprotonates the pendant arm of the complex, lithiating the pyrrole and generating [UO2(py){Li(py)2}(L)], 3-Li, and HN(SiMe3)2 (Scheme 2). The resonances in the 1H NMR spectrum of 3-Li appear in the diamagnetic region, confirming that no reduction of the uranyl(VI) centre occurs, and show the disappearance of the pendant NH resonance. Crystals of 3-Li were grown by layering a pyridine solution with hexane and the single-crystal X-ray structure was determined. The solid-state structure shows the uranyl ion coordinated by two arms of the tripodal pyrrole-imine ligand in a similar manner to 2-py with pyridine in the 5th equatorial site and axial and linear oxo groups (Fig. 2). The pendant arm is deprotonated with the lithium cation Li1 coordinated by its pyrrole and imine nitrogen donors along with two molecules of pyridine. This saturation of the coordination sphere of Li1 results in it being directed away from the uranyl centre. It is apparent that the Lewis basicity of the pyridine solvent outcompetes the weak Lewis basicity of the uranyl(VI) oxo group, the result being a compound with no interaction between the lithium ion with the uranyl oxo and therefore no activation of the UO bonding. This is evidenced by the U1–O1 (1.769(3) Å) and U1–O2 (1.772(2) Å) bond distances of 3-Li being almost identical to those in 1, 2, and other [U(VI)O2]2+ complexes.29–31 This is further supported by the observation of the asymmetric O–U–O stretch in the infrared spectrum of 3-Li at 911 cm−1, similar to that seen in 1, and which is typical for uranyl(VI) complexes.
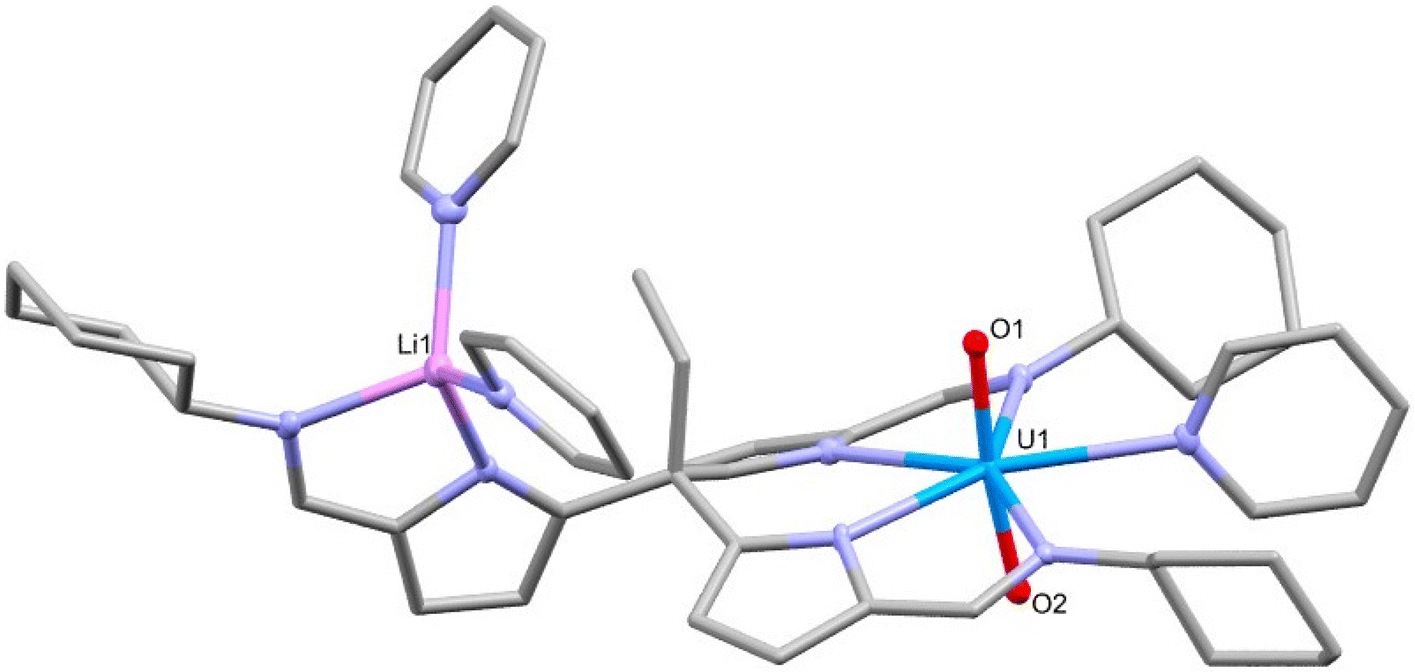 | ||
| Fig. 2 X-ray crystal structure of [UO2(py){Li(py)2}(L)] (3-Li). For clarity, hydrogen atoms are omitted and displacement ellipsoids of the heteroatoms are drawn at 50% probability. Selected bond distances (Å) and angles (°): U1–O1 = 1.769(3), U1–O2 = 1.772(2), O1–U1–O2 = 173.1(1)°. | ||
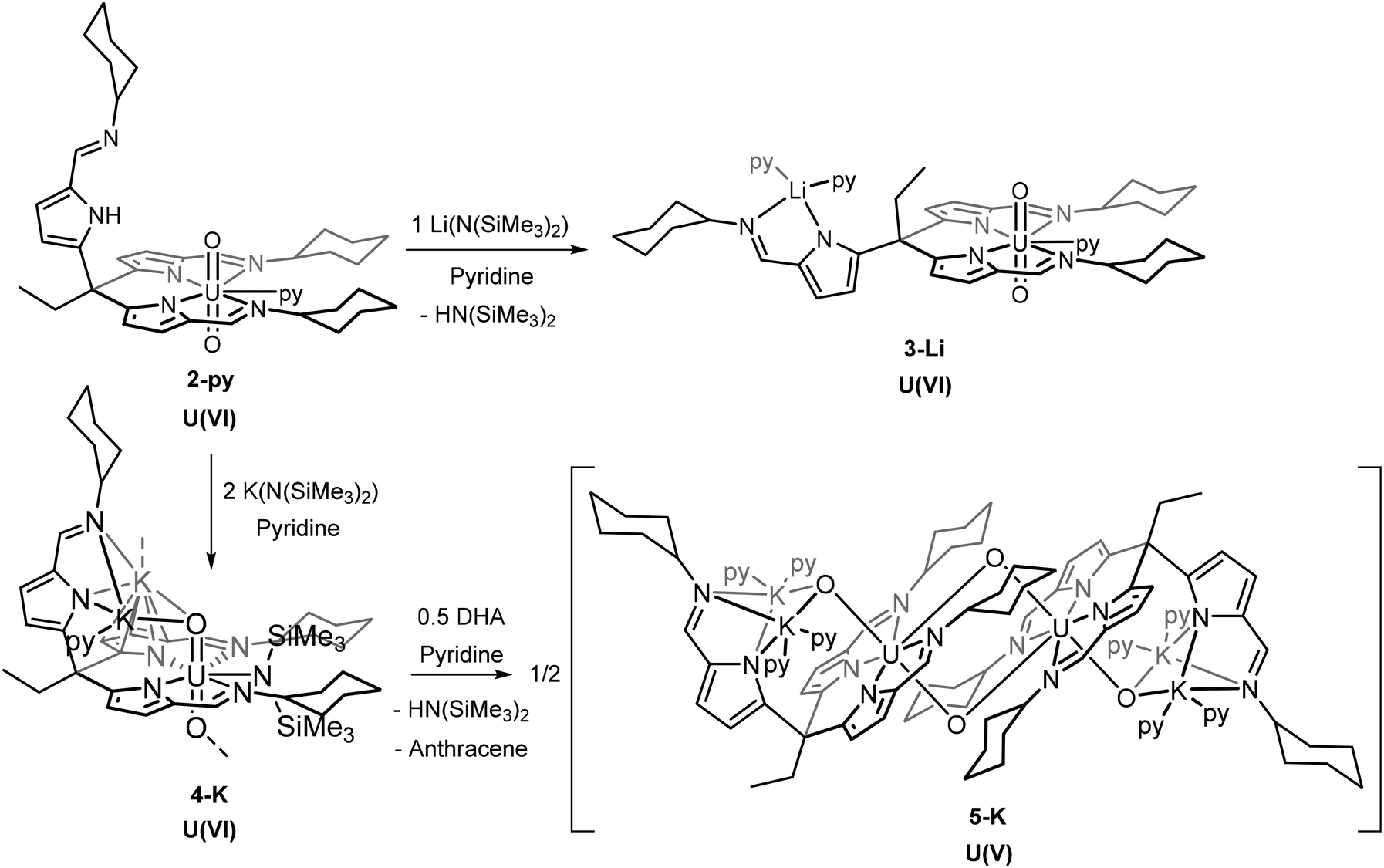 | ||
| Scheme 2 Synthetic procedures to form [UO2(LiL)(py)3] (3-Li), [UO2] and the dimeric, singly-reduced [UO2(K2L)(py)4]2 (5-K). | ||
Reacting 2 with single equivalents of NaN(SiMe3)2 or KN(SiMe3)2 produces 1H NMR spectra that are similar to that from the reaction with LiN(SiMe3)2. The NH resonance disappears, showing deprotonation, and the remaining peaks lie within the diamagnetic region, showing that no uranyl reduction occurs (Fig. S7†). The products of these reactions were not isolated.
Deprotonation coupled with single-electron reduction reactions
The reaction of 2-py with two equivalents of MN(SiMe3)2 (M = Li, Na or K) in pyridine gives mixtures of the diamagnetic complex [M2(py)UO2{N(SiMe3)2}(L)] 4 (e.g., 4-Na/K: 1H NMR chemical shift range 9.36 to 0.68 ppm) and the dimeric paramagnetic complex [M2(py)2UO2(L)]25 (e.g., 5-K: 1H NMR chemical shift range 14.72 to −36.89 ppm) (Scheme 2). X-ray quality crystals of 4-Na/K were isolated from a reaction between 2-py and NaN(SiMe3)2 and the structure was found to be polymeric with bridging potassium cations between the uranyl oxygen atoms of each monomer (Fig. 3). The source of the potassium presumably arises from an impurity in the NaN(SiMe3)2 reagent. As with the structures of 1, 2, and 3, the uranyl centre is coordinated by two of the three iminopyrrolide arms of L but in this case the 5th uranyl equatorial position is occupied by a N(SiMe3)2− anion. The presence of this group is also seen in the 1H NMR spectrum of 4-Na/K as two singlets at 0.68 and 0.72 ppm with integrals of 9H each and in the 29Si NMR spectrum as two peaks at −6.34 and −7.14 ppm. The presence of the N(SiMe3)2− ligand along with the short U–O bond lengths of 1.788(5) Å and 1.798(5) Å show the oxidation state of the uranium has remained unchanged at U(VI). | ||
| Fig. 3 X-ray structures of the U(VI) (4-Na/K), and U(V) (5-K), alkali metal complexes. For clarity, hydrogen atoms are omitted along with the eight pyridine molecules coordinating to the potassium atoms on 5-K; displacement ellipsoids of the heteroatoms are drawn at 50% probability. Selected bond distances (Å) and angles (°): 4-Na/K, U1–O1 = 1.798(5), U1–O2 = 1.788(5), O1–K1 = 2.900(5), O2–K1′ = 2.772(5), O1–U1–O2 = 175.8(2); 5-K, O1–U1 = 1.872(2), O2–U1 = 1.936(2), U1–O2′ = 2.377(2), K1–O1 = 2.927(2), K2–O1 = 2.806(2), U1–U1′ = 3.4993(5), O1–U1–O2 = 177.33(9), O1–U1–O2′ = 110.70(9), O2–U1–O2′ = 71.96(8). | ||
X-ray quality crystals of 5-K were isolated by vapour diffusion of hexane into a pyridine solution and the X-ray structure determined (Fig. 3). In this case, the solid-state structure is a dimeric complex in which the uranium has been reduced to the U(V) oxidation state with oxo groups now bridging the uranium centres in a cation–cation interaction (CCI). The reduction of the uranyl is evidenced by the lengthening of the U–O bonds to 1.872(2) Å and 1.936(2) Å which corresponds to a decrease in bond order associated with the metal reduction and is consistent with other reported uranyl(V) complexes.9,10,30,32 This diamond-shaped dimeric motif also illustrates the propensity of [U(V)O2]+ complexes to form cation–cation interactions due to the increased Lewis basicity of the oxo groups.33,34 The reduction of the uranyl is further evidenced by the asymmetric OUO stretch seen at 716 cm−1 in the IR spectrum of 5-K, which is much lower than in the uranyl(VI) complexes 1 and 3-Li.35,36
In the presence of >0.5 equivalents of 9,10-dihydroanthracene (DHA), the reaction between 2-py and two equivalents of KN(SiMe3)2 in pyridine gives only the uranyl(V) product 5-K and none of the uranyl(VI) product 4-K. Additionally, the formation of anthracene is observed in the 1H NMR spectrum of the reaction mixture. DHA contains weak C–H bonds (bond dissociation energy (BDE) = 79 kcal mol−1) that can undergo sequential H-atom abstractions by a radical to form anthracene. It is therefore likely that a sterically induced homolytic cleavage of the uranium–silylamide bond in 4-K occurs which reduces the uranium and forms the N(SiMe3)2˙ radical that is rapidly quenched by DHA. This mechanism is similar to that proposed for uranyl reduction by lanthanide silylamide complexes.19
Due to the larger radial extension of the 5f orbitals (cf. 4f) providing significant ligand field effects and the presence of large spin–orbit coupling, a simple model of the magnetic moment of these reduced uranyl complexes is difficult to obtain.37 For this reason, determination of oxidation state by room temperature magnetic susceptibility measurements is less straightforward than for transition metals (spin-only approximation, μS) or lanthanides (total angular momentum approximation, μJ). In the current literature, a range of effective magnetic moment (μeff) values have been reported for uranium complexes that shows overlap between the 5+, 4+, and 3+ oxidation states.38 However, preliminary solution state (Evans’ method) susceptibility measurements were undertaken to compare with reported values. The μeff of complex 5-K was determined as a value of 3.36μB (1.68μB per U5+) at 300 K. While this value is below the theoretical value of a free 5f1 ion (2.54μB, 2F5/2), it is within reported ranges for dinuclear U(V) complexes.39–42
Approaches towards heterometallic uranyl complexes
The presence of the alkali metals in the uranyl(VI) and (V) complexes 3–5 makes them attractive synthons to probe the formation of uranyl complexes of transition- and rare-earth metals. In the first instance, transmetalation reactions between 3-Li and a variety of transition metal and rare-earth halides were attempted, but these reactions invariably led to mixtures of intractable products that involve the substitution of the uranyl(VI) cation. As such, attention was focused on transmetalation reactions of the uranyl(V) complexes.A series of metal chlorides (ZnCl2, RECl3 [RE = Y, Sm, Dy]) were used to transmetalate the potassium cations in 5-K. In each case, new hetero-dinuclear dimeric complexes form (6-Zn, 6-Y, 6-Sm and 6-Dy) with a similar overall configuration to 5-K.
On addition of ZnCl2 to a solution of 5-K the new complex 6-Zn forms with KCl precipitating from solution (Scheme 3). The 1H NMR spectrum of 6-Zn shows a similar range in chemical shift values (18.8 to −29.4 ppm) to 5-K indicating that the U(V) oxidation state is retained; this is also evidenced by the O–U–O asymmetric stretch of 660 cm−1 in the IR spectrum. The integration of the resonances in the 1H NMR spectrum corresponds to the number of protons expected. Single crystals of 6-Zn were grown by vapour diffusion of hexane into a pyridine solution and the X-ray crystal structure was determined. The solid-state structure shows elongated U–O bonds (U1–O1, 1.929(2) Å; U1–O2, 1.939(2) Å) and associated CCIs, supporting the U(V) oxidation state (Fig. 4). The U1–O1 bond length in 6-Zn is significantly longer than that seen in 5-K and perhaps reflects an increase in Lewis acidity for Zn2+ compared with two K+; in contrast, the U1–O2 bond lengths which form the CCI are essentially identical. Unfortunately, due to the poor solubility of the crystallised complex, the effective magnetic moment of this complex by Evans’ method could not be measured.
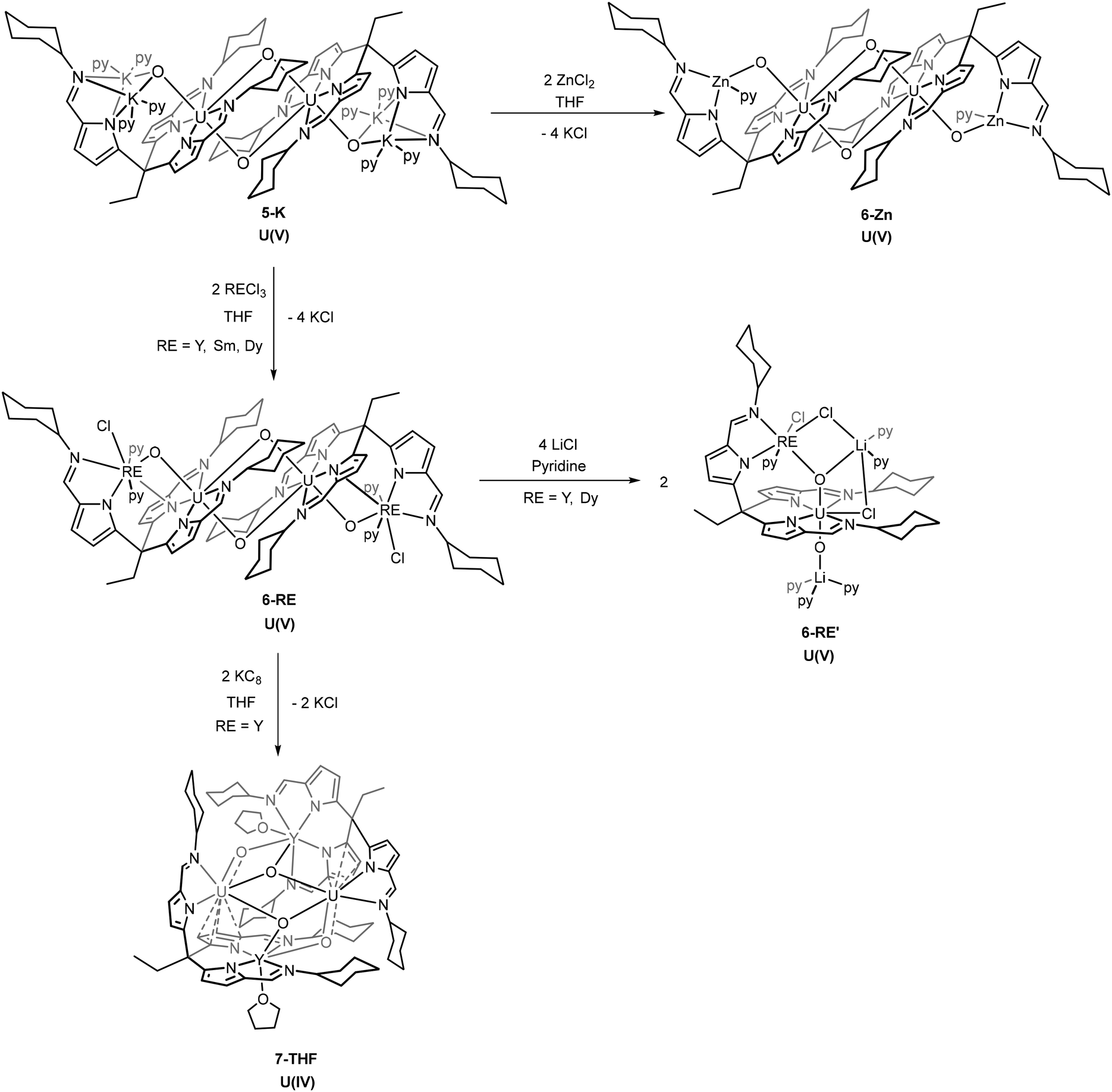 | ||
| Scheme 3 Synthetic procedures to the formation of the U(V) heterobimetallic complexes [UO2Zn(L)]2 (6-Zn), and [UO2RE(L)Cl]2 (6-RE) by reaction of 5-K with the respective metal chloride; [UO2RECl(L)(LiCl)2] (6-RE′) by the treatment of 6-RE with four equivalents of LiCl; and the dimeric U(IV) complex [UO2Y(L)(THF)]2 (7-THF), by the reduction of 6-Y with two equivalents of KC8. | ||
 | ||
| Fig. 4 X-ray structure of 6-Zn. For clarity, hydrogen atoms are omitted and displacement ellipsoids of the heteroatoms are drawn at 50% probability. Selected bond distances (Å) and angles (°): U1–O1 = 1.929(2), U1–O2 = 1.939(2), U1–O2′ = 2.349(1), Zn1–O1 = 1.964(1), Zn1–U1 = 3.4037(5), U1–U1′ = 3.4862(5), O1–U1–O2 = 176.18(6), Zn1–O1–U1 = 121.97(7). Each zinc centre has essentially 1.5 pyridine ligands, one was modelled as half-occupied, consistent with disorder imposed by space group symmetry and, for clarity, is not shown. | ||
The reaction of rare-earth metal chlorides with 5-K results in the formation of the RE–uranyl(V) complexes 6-Y, 6-Sm and 6-Dy (Scheme 3.). The 1H NMR spectrum of 6-Y shows a chemical shift range (17.4 to −30.8 ppm) similar to 5-K and 6-Zn, and which are consistent with a U(V) oxidation state and a diamagnetic Y(III). The 1H NMR spectrum of 6-Dy shows a greater range in its resonances (96.8 to −68.3 ppm) due to the incorporation of the paramagnetic dysprosium ion. NMR spectra of 6-Sm could not be obtained. Single crystals of all of these complexes were obtained by vapour diffusion of hexane into pyridine solutions and were used to determine their solid-state structures by X-ray crystallography (Fig. 5 and Table 1). These complexes adopt similar structures to the potassium and zinc analogues but with a single chloride remaining coordinated to the RE cation. The retention of the U(V) oxidation state is evidenced by the uranyl-oxo bond lengths which are all elongated compared with uranyl(VI) complexes; in this case, the U1–O1 bond lengths increase slightly, but not to the same extent as 6-Zn. One significant difference between 6-RE and 6-Zn is the rare-earth metal interacts through π-bonding with one of the pyrroles coordinated to the uranium centre. This results in the pendant arm being angled to one side for all RE complexes rather than being centrally disposed, as seen in 5-K and 6-Zn. Only small differences in bonding metrics are seen between the complexes of the different rare-earth metals and reflect the small changes in ionic radii on transitioning across the RE series (Table 1).
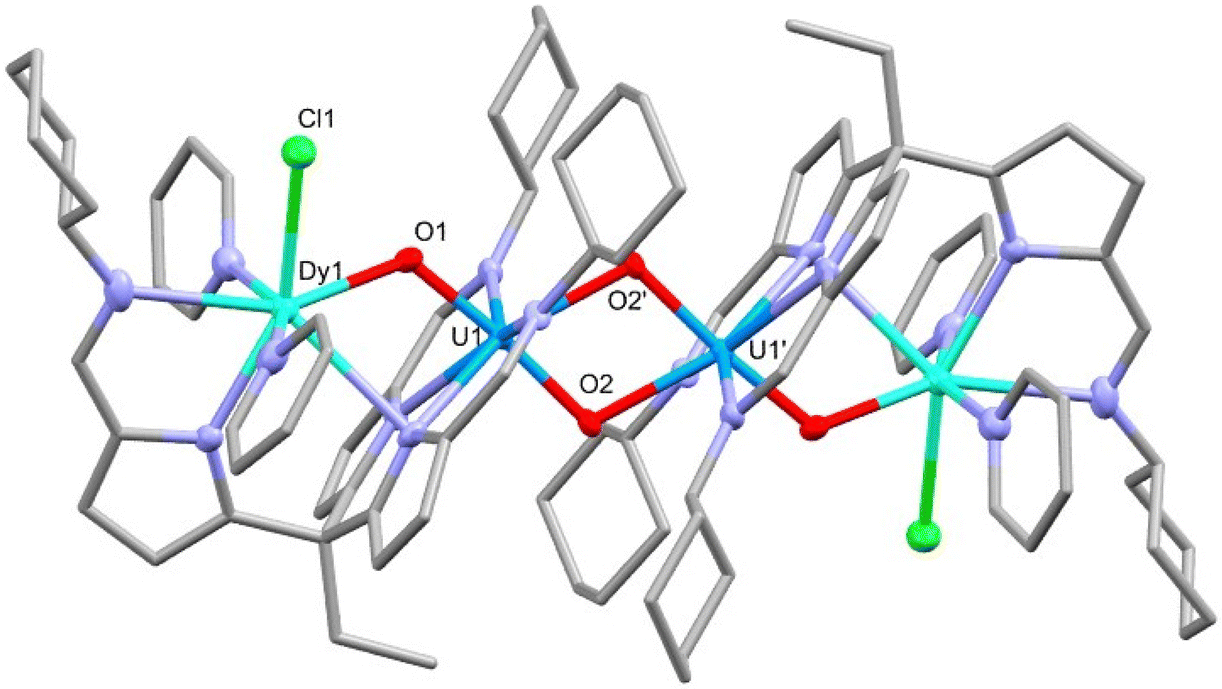 | ||
| Fig. 5 X-ray crystal structure of complex 6-Dy. For clarity, hydrogen atoms are omitted and displacement ellipsoids of the heteroatoms are drawn at 50% probability. Selected bond distances and angles are shown in Table 1. | ||
| Complex | U1–O1 | U1–O2 | U1–O2′ | RE1–O1 | U1–RE1 | U1–U1′ | O1–U1–O2 | U1–O1–RE1 |
|---|---|---|---|---|---|---|---|---|
| 6-Y | 1.903(3) | 1.936(3) | 2.326(2) | 2.231(3) | 3.6236(5) | 3.4426(5) | 174.2(1) | 122.2(1) |
| 6-Sm | 1.904(2) | 1.945(2) | 2.324(2) | 2.296(2) | 3.6564(5) | 3.4416(5) | 175.17(8) | 120.8(1) |
| 6-Dy | 1.899(7) | 1.940(7) | 2.322(5) | 2.244(5) | 3.6255(6) | 3.4461(5) | 174.6(2) | 121.9(3) |
The effective magnetic moments of these uranyl-rare-earth complexes were also measured by Evans’ method at 300 K. The μeff of 6-Y was determined as 2.87μB and 6-Dy as 10.04μB, with the value for 6-Y being similar to the analogous uranyl(V)–yttrium Pacman complex of 2.4μB at 300 K.19,20 The μeff of 6-Sm was calculated as 2.02μB which is lower than expected.
Monomeric hetero-trimetallic uranyl(V) complex
In a similar manner to that seen for dimeric uranyl Pacman complexes, the reaction of these rare-earth U(V) dimeric complexes with 4 equivalents of lithium chloride in pyridine was found to interrupt the CCI and result in the formation of monomeric RE–U(V) complexes 6-RE′ (RE = Y, Dy) (Scheme 3).19 The yttrium complex 6-Y′ shows a much narrower chemical shift range in the 1H NMR spectrum (13.03 to −8.01 ppm) compared with its dimeric analogue 6-Y (13.80 to −30.81 ppm). No satisfactory NMR spectra of 6-Dy′ were acquired due to the paramagnetism of the dysprosium cation. Single crystals of 6-Y′ and 6-Dy′ suitable for X-ray diffraction were obtained by slow diffusion of hexane into pyridine solutions of the complexes. The solid-state structure of 6-Y′ (Fig. 6) shows one pseudo-tetrahedral lithium (Li2) coordinating to the uranyl oxo (O2) with a near linear U1–O2–Li2 angle of 177.3(2)°, and the second five-coordinate lithium (Li1) coordinating to the other uranyl oxo (O1) and bridging between two chlorides (Cl2 and Cl3). One of the newly added chloride ions coordinates in the fifth equatorial position of the uranyl (replacing the bridging oxo in the 6-Y dimer) and the second bridges Li1 and the yttrium ion. The gross structure for 6-Dy′ is essentially identical to that of 6-Y′ with selected bond distances and angles shown in Table 2. The Evans’ method effective magnetic moments were measured at 300 K as 6-Y′ 1.55μB and 6-Dy’ 7.96μB.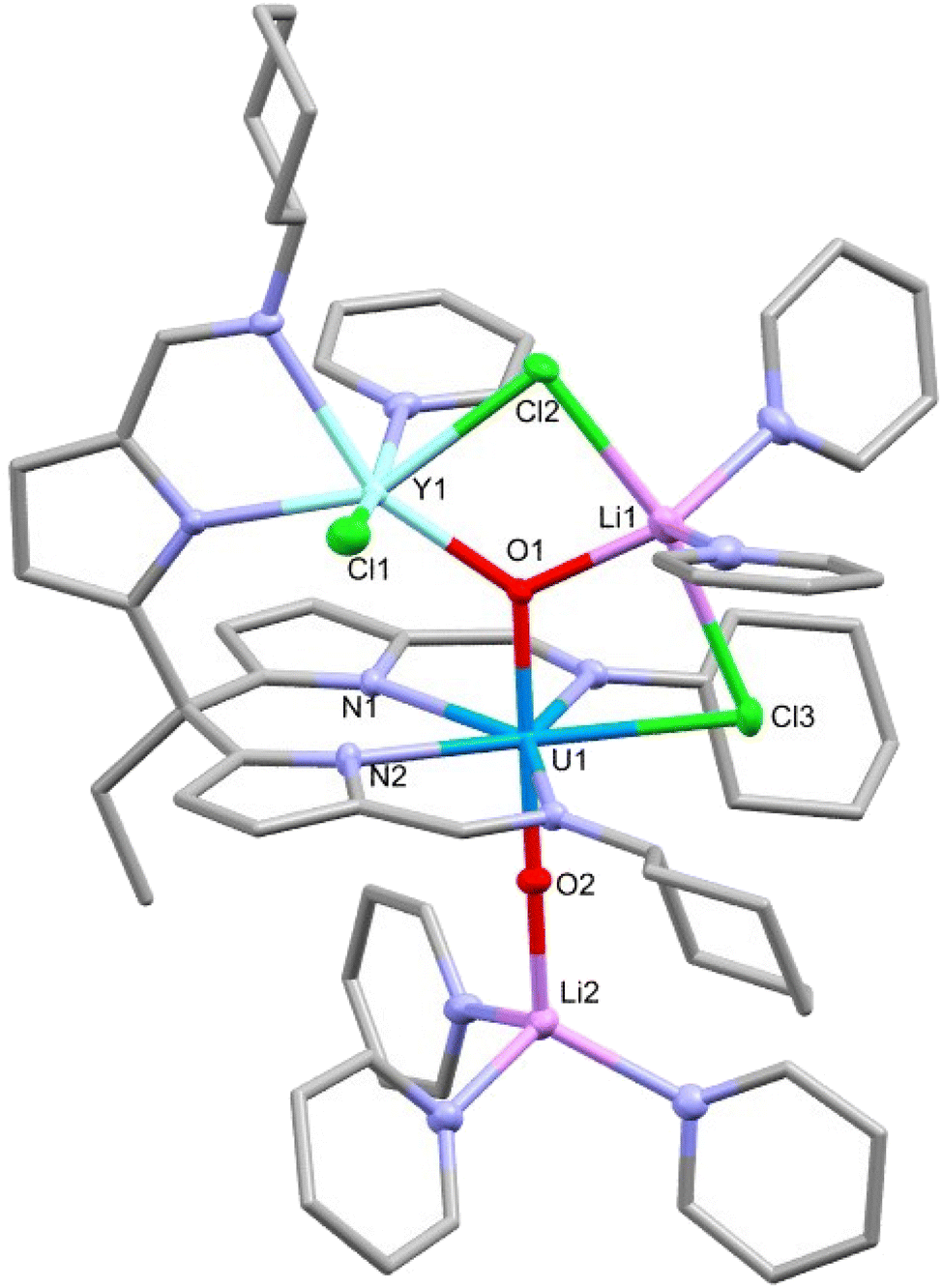 | ||
| Fig. 6 X-ray crystal structure of the monomeric rare-earth-uranium(V) complex 6-Y′. For clarity, hydrogen atoms are omitted and displacement ellipsoids of the heteroatoms are drawn at 50% probability. Selected bond distances and angles are shown in Table 2. | ||
| Complex | U1–O1 | U1–O2 | RE1–O1 | U1–RE1 | O1–U1–O2 | U1–O1–RE1 |
|---|---|---|---|---|---|---|
| 6-Y′ | 1.951(2) | 1.852(2) | 2.242(2) | 3.7979(6) | 179.2(1) | 129.8(1) |
| 6-Dy′ | 1.939(2) | 1.849(3) | 2.268(3) | 3.7876(6) | 179.2(1) | 128.2(2) |
Compared with the dimeric structures 6-Y and 6-Dy, there is a small increase in the U1–O1 bond length and decrease in the U1–O2 bond length when forming the monomers 6-Y′ and 6-Dy′. This is perhaps due to O1 being three-coordinate with a lithium cation which removes electron density from the U–O bond. Interestingly, the U1–O2 bond is significantly shorter in the monomer than in the dimer, likely as a result of the loss of the CCI.
U(V) to U(IV) reduction
To further investigate the reduction chemistry of this uranyl system, the yttrium complex 6-Y was treated with two equivalents of potassium graphite (KC8) (Scheme 3) which results in an increased range (79.29 to −77.65 ppm) of the resonances in the 1H NMR spectrum, suggesting a reduction from the f1 U(V) to an f2 U(IV) complex.27,43–45 Single crystals of the reduced complex 7-THF were isolated by slow evaporation of a THF solution and the X-ray crystal structure was determined (Fig. 7). The solid-state structure shows a dimer in which the loss of a chloride ligand from the yttrium centre has occurred along with the reduction of the uranium centre to U(IV). A rearrangement of the ligands at the two metals is also seen with, in this case, two ligand iminopyrrolide arms coordinating to the yttrium centre with the previously pendant iminopyrrolide arm now bound to a uranium; this coordination motif is opposite to that seen in 6-Y. The uranium centres also engage in a π-interaction with an adjacent pyrrole of one of the ligand arms bound to the yttrium with U–C distances of 2.955(8) to 3.090(10) Å.46,47 The linearity of the oxo groups around the uranium centres has been lost upon reduction with O1–U1–O3 and O2–U2–O4 bond angles of 136.3(2)° and 137.7(2)° respectively. The U–O bond distances increase significantly from the U(V) compound 6-Y with distances elongated to between 2.042(7) to 2.250(5) Å. The combination of these data support the reduction of uranium from 5+ to 4+.45,48,49 The Y–O bond distances of 7-THF (2.203(5)–2.212(6) Å) are very similar to those in 6-Y (2.231(3) Å) and 6-Y′ (2.242(2) Å) supporting the retention of the Y(III) oxidation state. The diamond core of the bridging U–O–U oxo groups (CCI) is somewhat retained but distorted, and all four oxo atoms now coordinating to the yttrium centres. The IR spectrum of 7-THF (Fig. S18†) shows the loss of the asymmetric O–U–O stretch at 672 cm−1 of 6-Y and a new peak at 538 cm−1 that is consistent with a uranium(IV) U–O stretch.49–51 The decrease in wavenumber correlates to a decrease in U–O bond multiplicity arising from the increase in U–O bond distances seen in the X-ray crystal structure.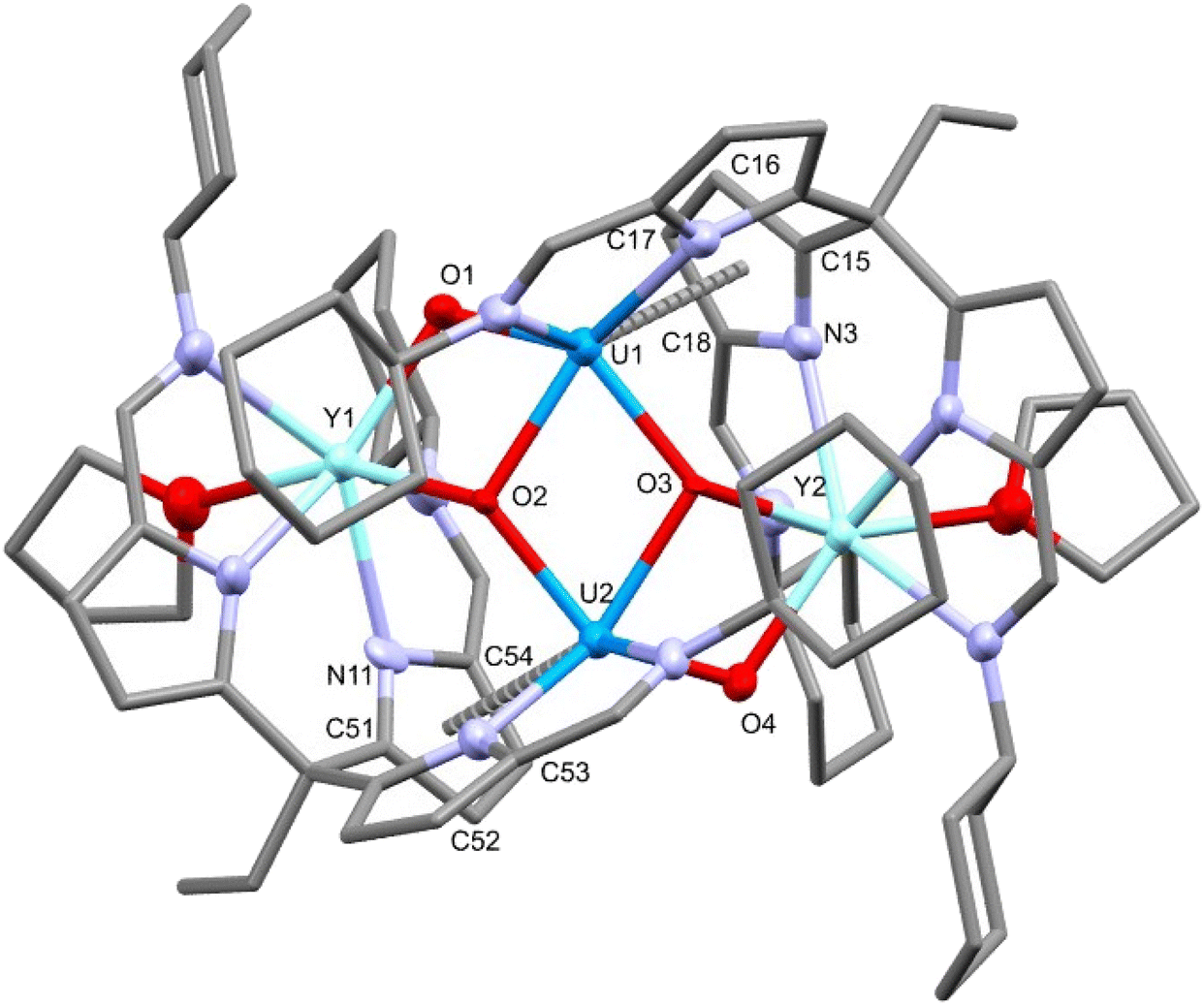 | ||
| Fig. 7 X-ray structure of the U(IV) yttrium–uranium hetero-tetrametallic complex 7-THF. For clarity, hydrogen atoms are omitted and displacement ellipsoids of the heteroatoms are drawn at 50% probability. Selected bond distances (Å) and angles (°): U1–O1 = 2.042(7), U1–O2 = 2.250(5), U1–O3 = 2.175(6), U2–O2 = 2.175(6), U2–O3 = 2.245(5), U2–O4 = 2.044(5), Y1–O1 = 2.218(6), Y1–O2 = 2.204(6), Y2–O3 = 2.203(5), Y2–O4 = 2.212(6), Y1–U1 = 3.469(1), Y2–U2 = 3.4583(8), U1–U2 = 3.5856(5), U1–N3 = 3.091(8), U1–C15 = 3.09(1), U1–C16 = 3.00(1), U1–C17 = 3.011(9), U1–C18 = 3.06(1), U2–N11 = 3.058(8), U2–C51 = 3.05(1), U2–C52 = 2.955(8), U2–C53 = 2.971(8), U2–C54 = 3.009(9), O1–U1–O3 = 136.3(2), O2–U2–O4 = 137.7(2). | ||
Single crystals of 7-py were also grown by vapour diffusion of hexane into a pyridine solution of 7. In this case, the X-ray crystal structure remains largely similar to 7-THF, with pyridine molecules replacing the coordinated THF. However, in contrast, there is an extra pyridine coordinating to one of the two uranium centres which is accompanied by a small change in geometry to accommodate the extra solvent molecule. It is clear from the reactions described above that the flexibility of this ligand design enables significant structural rearrangements in its complexes, which enables the accommodation of multiple metal centres in varying oxidation states. The μeff value was determined for complex 7 to be 3.94μB at 300 K. This value is again lower than the theoretical magnetic moment for two independent 5f2 U(IV) ions (5.06μB),52 but is within the reported range of dinuclear U(IV) metal complexes.41,53–55
Electrochemistry
The cyclic voltammograms (CVs) of 2-THF and 3-Li (Fig. 8) were recorded in a 0.1 M THF solution of [nBu4N][BPh4] electrolyte using a glassy-carbon working electrode, platinum counter electrode and a silver wire pseudo-reference electrode under a nitrogen atmosphere at a scan-rate of 0.1 V s−1.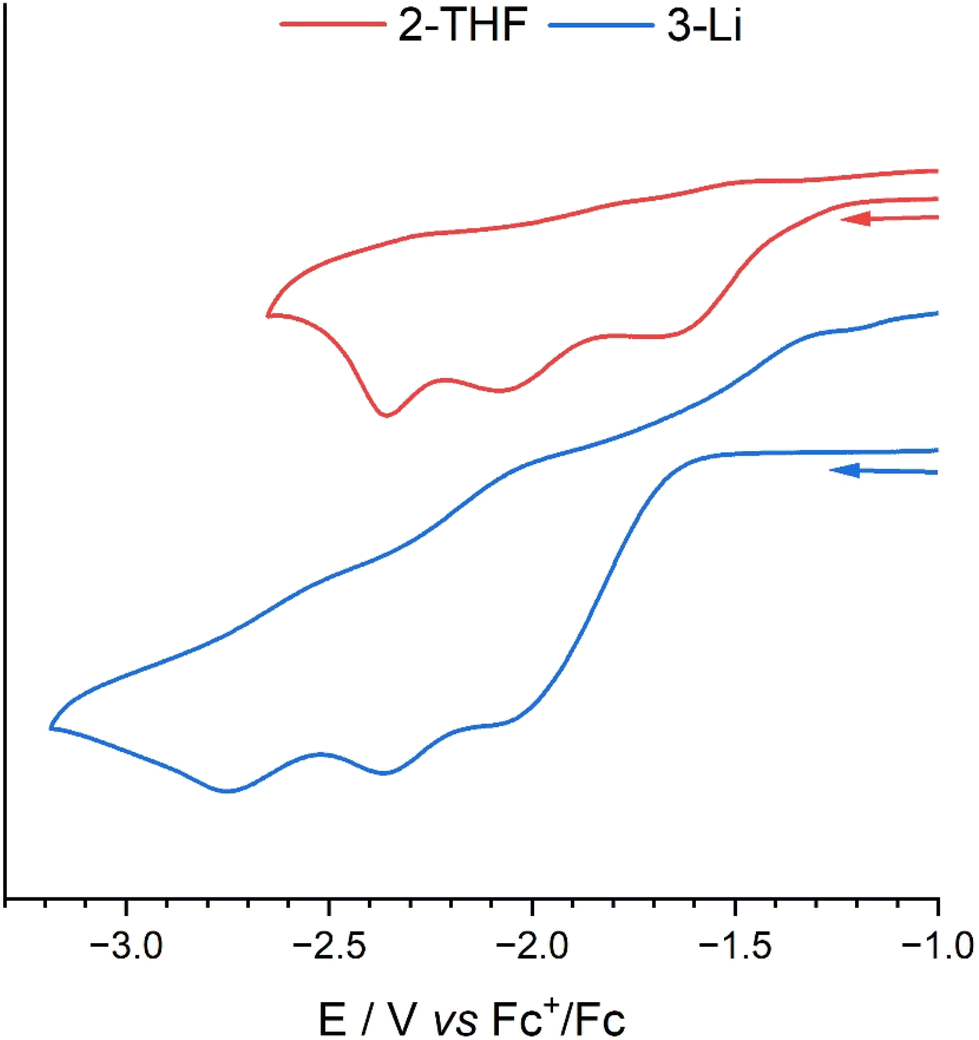 | ||
| Fig. 8 Cyclic voltammogram of complexes 2-THF (RED) and 3-Li (BLUE). A glassy-carbon working electrode, platinum counter electrode and silver wire pseudo-reference electrode were used with a 10 mM solution of analyte in 0.1 M THF solutions of [nBu4N][PF6] electrolyte. Voltammograms were recorded under a dinitrogen atmosphere in a glovebox at a scan-rate of 0.1 V s−1. | ||
The CV of 2-THF features three seemingly irreversible reduction process at Epc = −1.68, −2.08, and −2.36 V vs. Fc+/Fc. The reduction at −1.64 V is attributed to the U(VI)/U(V) single-electron reduction with its irreversibility a result of the formation of a dimeric CCI as seen in the U(V) structures reported above.34,45 This irreversible reduction does not become reversible at increased scan-rates (Fig. S21†). The reduction at EpC = −2.08 V is likely a U(V)/U(IV) single-electron reduction with the following peak being either a second U(V)/U(IV) reduction of the dimeric species as there is likely communication between the uranium centres through the bridging oxo groups, or it is a U(IV)/U(III) reduction. The CV of H3L was also carried out to investigate if these irreversible peaks could be due to hydrogen generation from the acidic protons of 2 (ESI 23†). However, no redox events within the range −1.00 V to −2.70 are seen with only an irreversible reduction at Epc = −3.25 V vs. Fc+/Fc.
The CV of the U(VI)–Li complex 3-Li shows similar redox events to 2-THF, with an irreversible reduction at Epc = −2.12 V that could be attributed to the U(VI)/U(V) reduction that involves the formation of the dimer. There are two more redox events at Epc = −2.37 and −2.75 V that may show two sequential U(V)/U(IV) reductions, due to communication between the two U centres. The irreversible oxidation at Epa = −1.29 V is unrelated to the reduction at EpC = 2.12 V (Fig. S22†) and is likely a result of decomposition of the further reduced products on the electrode.
CVs of 5-K, 6-Zn and 6-Y were also attempted as 1 mM solutions of each complex in 0.1 M THF solutions of [nBu4N][PF6] (ESI 24–26†). Each shows an irreversible reduction at Epc = −2.80, −2.76 and −2.85 V vs. Fc+/Fc for 5-K, 6-Zn and 6-Y, respectively, that are attributable to reduction of the ligand. Other redox events are seen at less negative potentials that may be attributed to the UV/UIV reduction but none are defined enough for formal characterisation. The CVs were also attempted in 0.1 M THF solutions of [nBu4N][BArF4] and [nBu4N][BPh4] but this variation of the electrolyte did not simplify the CVs and so they remain unassigned.
Conclusion
We have shown that the tripodal ligand H3L can accommodate the uranyl cation in both its uranyl(VI) and singly reduced uranyl(V) redox states and that these redox events are facilitated by the presence of the pendant iminopyrrolide arm and its propensity to coordinate to metal ions. The alkali–metal functionalised uranyl(V) complexes act as synthons to uranium–zinc and uranium–rare-earth mixed-metal complexes that retain the U(V) oxidation state. The subsequent reduction of the Y–U complex to form a Y(III)/U(IV) heterobimetallic complex highlights the flexibility of this ligand system which is thus shown to accommodate uranyl(VI), uranyl(V) and uranium(IV) oxidation states without disassembly and to promote the construction of new U–O–M functionalities. In future work, we aim to further exploit this framework to generate new examples of multimetallic f-element complexes, evaluate their magnetic properties, and to probe the redox reactivity of these systems.Data availability
Data are available in the ESI† and on request from the authors.Conflicts of interest
There are no conflicts to declare.References
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- T. W. Hayton, Dalton Trans., 2018, 47, 1003–1009 RSC.
- C. Limberg, Chem. – Eur. J., 2000, 6, 2083–2089 CrossRef CAS PubMed.
- J. L. Smeltz, C. P. Lilly, P. D. Boyle and E. A. Ison, J. Am. Chem. Soc., 2013, 135, 9433–9441 CrossRef CAS PubMed.
- L. Hills, R. Moyano, F. Montilla, A. Pastor, A. Galindo, E. Álvarez, F. Marchetti and C. Pettinari, Eur. J. Inorg. Chem., 2013, 2013, 3352–3361 CrossRef CAS.
- J. C. Renshaw, L. J. C. Butchins, F. R. Livens, I. May, J. M. Charnock and J. R. Lloyd, Environ. Sci. Technol., 2005, 39, 5657–5660 CrossRef CAS PubMed.
- D. R. Lovley, E. J. P. Phillips, Y. A. Gorby and E. R. Landa, Nature, 1991, 350, 413–416 CrossRef CAS.
- H. Steele and R. J. Taylor, Inorg. Chem., 2007, 46, 6311–6318 CrossRef CAS PubMed.
- R. Faizova, R. Scopelliti, A.-S. Chauvin and M. Mazzanti, J. Am. Chem. Soc., 2018, 140, 13554–13557 CrossRef CAS PubMed.
- E. A. Pedrick, G. Wu, N. Kaltsoyannis and T. W. Hayton, Chem. Sci., 2014, 5, 3204–3213 RSC.
- P. L. Arnold, D. Patel, C. Wilson and J. B. Love, Nature, 2008, 451, 315–317 CrossRef CAS PubMed.
- P. L. Arnold, D. Patel, A. J. Blake, C. Wilson and J. B. Love, J. Am. Chem. Soc., 2006, 128, 9610–9611 CrossRef CAS PubMed.
- R. Faizova, S. White, R. Scopelliti and M. Mazzanti, Chem. Sci., 2018, 9, 7520–7527 RSC.
- T. W. Hayton and G. Wu, J. Am. Chem. Soc., 2008, 130, 2005–2014 CrossRef CAS PubMed.
- A. Kumar, D. Lionetti, V. W. Day and J. D. Blakemore, J. Am. Chem. Soc., 2020, 142, 3032–3041 CrossRef CAS PubMed.
- P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS.
- S. Fortier and T. W. Hayton, Coord. Chem. Rev., 2010, 254, 197–214 CrossRef CAS.
- J. B. Love, Chem. Commun., 2009, 3154–3165 RSC.
- P. L. Arnold, E. Hollis, G. S. Nichol, J. B. Love, J.-C. Griveau, R. Caciuffo, N. Magnani, L. Maron, L. Castro, A. Yahia, S. O. Odoh and G. Schreckenbach, J. Am. Chem. Soc., 2013, 135, 3841–3854 CrossRef CAS PubMed.
- P. L. Arnold, E. Hollis, F. J. White, N. Magnani, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed., 2011, 50, 887–890 CrossRef CAS PubMed.
- J. S. Hart, G. S. Nichol and J. B. Love, Dalton Trans., 2012, 41, 5785–5788 RSC.
- J. S. Hart, F. J. White and J. B. Love, Chem. Commun., 2011, 47, 5711–5713 RSC.
- T. A. Babushkina and G. V. Ponomarev, Chem. Heterocycl. Compd., 1998, 34, 474–479 CrossRef CAS.
- N. L. Bell, P. L. Arnold and J. B. Love, Dalton Trans., 2016, 45, 15902–15909 RSC.
- P. J. Cobb, D. J. Moulding, F. Ortu, S. Randall, A. J. Wooles, L. S. Natrajan and S. T. Liddle, Inorg. Chem., 2018, 57, 6571–6583 CrossRef CAS PubMed.
- C. J. Burns, D. L. Clark, R. J. Donohoe, P. B. Duval, B. L. Scott and C. D. Tait, Inorg. Chem., 2000, 39, 5464–5468 CrossRef CAS PubMed.
- B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Chem. Rev., 2019, 119, 10595–10637 CrossRef CAS PubMed.
- P. L. Arnold, G. M. Jones, Q.-J. Pan, G. Schreckenbach and J. B. Love, Dalton Trans., 2012, 41, 6595–6597 RSC.
- M. K. Assefa, E. A. Pedrick, M. E. Wakefield, G. Wu and T. W. Hayton, Inorg. Chem., 2018, 57, 8317–8324 CrossRef CAS PubMed.
- P. L. Arnold, A. F. Pécharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056–1061 CrossRef CAS PubMed.
- S. A. Pattenaude, E. J. Coughlin, T. S. Collins, M. Zeller and S. C. Bart, Inorg. Chem., 2018, 57, 4543–4549 CrossRef CAS PubMed.
- D. P. Mills, O. J. Cooper, F. Tuna, E. J. L. McInnes, E. S. Davies, J. McMaster, F. Moro, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2012, 134, 10047–10054 CrossRef CAS PubMed.
- S.-X. Hu, J. Jian, J. Li and J. K. Gibson, Inorg. Chem., 2019, 58, 10148–10159 CrossRef CAS PubMed.
- K. van Rees, T. Rajeshkumar, L. Maron, S. Sproules and J. B. Love, Inorg. Chem., 2022, 61, 20424–20432 CrossRef CAS PubMed.
- V. Mougel, L. Chatelain, J. Pécaut, R. Caciuffo, E. Colineau, J.-C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef CAS PubMed.
- M. Zegke, G. S. Nichol, P. L. Arnold and J. B. Love, Chem. Commun., 2015, 51, 5876–5879 RSC.
- N. M. Edelstein and G. H. Lander, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J.Fuger, Springer Netherlands, Dordrecht, 2011, pp. 2225–2306 Search PubMed.
- D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865–8882 CrossRef CAS PubMed.
- M. Keener, F. Fadaei-Tirani, R. Scopelliti, I. Zivkovic and M. Mazzanti, Chem. Sci., 2022, 13, 8025–8035 RSC.
- R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588–4590 CrossRef CAS.
- C. Camp, V. Mougel, J. Pécaut, L. Maron and M. Mazzanti, Chem. – Eur. J., 2013, 19, 17528–17540 CrossRef CAS PubMed.
- A.-C. Schmidt, F. W. Heinemann, W. W. Lukens Jr. and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- B. C. Stobbe, D. R. Powell and R. K. Thomson, Dalton Trans., 2017, 46, 4888–4892 RSC.
- C. Deng, J. Liang, R. Sun, Y. Wang, P.-X. Fu, B.-W. Wang, S. Gao and W. Huang, Nat. Commun., 2023, 14, 4657 CrossRef CAS PubMed.
- N. L. Bell, B. Shaw, P. L. Arnold and J. B. Love, J. Am. Chem. Soc., 2018, 140, 3378–3384 CrossRef CAS PubMed.
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Organometallics, 2001, 20, 2552–2559 CrossRef CAS.
- A. Novak, A. J. Blake, C. Wilson and J. B. Love, Chem. Commun., 2002, 2796–2797 RSC.
- M. W. Rosenzweig, J. Hümmer, A. Scheurer, C. A. Lamsfus, F. W. Heinemann, L. Maron, M. Mazzanti and K. Meyer, Dalton Trans., 2019, 48, 10853–10864 RSC.
- J. R. Pankhurst, N. L. Bell, M. Zegke, L. N. Platts, C. A. Lamfsus, L. Maron, L. S. Natrajan, S. Sproules, P. L. Arnold and J. B. Love, Chem. Sci., 2017, 8, 108–116 RSC.
- B. E. Cowie, G. S. Nichol, J. B. Love and P. L. Arnold, Chem. Commun., 2018, 54, 3839–3842 RSC.
- J. L. Brown, C. C. Mokhtarzadeh, J. M. Lever, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 5105–5112 CrossRef CAS PubMed.
- G. Feng, M. Zhang, D. Shao, X. Wang, S. Wang, L. Maron and C. Zhu, Nat. Chem., 2019, 11, 248–253 CrossRef CAS PubMed.
- D. Patel, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2013, 42, 5224–5227 RSC.
- A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed.
- J. Du, D. M. King, L. Chatelain, E. Lu, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2313497–2313510. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02367f |
| This journal is © The Royal Society of Chemistry 2024 |