
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of a supramolecular spin-crossover tetrahedron†
Hannah H.
Nielsen
 ac,
Pol
Vilariño
a,
Gemma
Rodríguez
a,
Florian
Trepard
a,
Olivier
Roubeau
*d,
Guillem
Aromí
*ab and
David
Aguilà
*ab
ac,
Pol
Vilariño
a,
Gemma
Rodríguez
a,
Florian
Trepard
a,
Olivier
Roubeau
*d,
Guillem
Aromí
*ab and
David
Aguilà
*ab
aDepartament de Química Inorgànica i Orgànica, Secció de Química Inorgànica, Universitat de Barcelona, Diagonal 645, 08028 Barcelona, Spain. E-mail: aromi@ub.edu; david.aguila@ub.edu
bInstitute of Nanoscience and Nanotechnology, University of Barcelona (IN2UB), 08007 Barcelona, Spain
cDepartment of Chemistry, Aarhus University, DK-8000 Aarhus, Denmark
dInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC and Universidad de Zaragoza, Plaza San Francisco s/n, 50009, Zaragoza, Spain. E-mail: roubeau@unizar.es
First published on 20th May 2024
Abstract
A new mononuclear iron(II) SCO compound featuring H-bonding donor and acceptor units has been synthesized and exploited to produce a purely supramolecular switchable [Fe4] tetrahedron. Magnetic and crystallographic measurements evidence a singular magnetic behavior for each of the four Fe(II) centers of the generated architecture and underscore the potential of this strategy to develop novel SCO materials.
Spin-crossover (SCO) compounds are fascinating switchable materials with great potential for the development of novel technological devices.1,2 These coordination complexes exhibit metal ions with two possible electronic configurations (low-spin, LS, and high-spin, HS) which can be toggled using external stimuli such as temperature,1 pressure,3 or light irradiation.4 The different magnetic, optical, and structural features of the two states allow these materials to be exploited for a wide range of applications, such as sensors,5 actuators,6 or for information storage.7 Interestingly, the physical properties of SCO compounds can be tuned by modifying the weak non-covalent interactions exhibited within or in between their molecular entities. In host–guest systems, these interactions offer a versatile tool, for example, for manipulating the transition temperature of encapsulating SCO complexes simply by altering the nature of the supramolecular guest, as shown in dinuclear helicates,8 tetrahedral cages,9–12 or cubic architectures.13 Long range intermolecular interactions can be exploited as well to tune or even to activate/deactivate the SCO behaviour. Such modulation arises from the nature and strength of such interaction, which influence the communication between molecules and thus its cooperativity,14–17 or affect the ligand field exerted by the donor set and therefore the SCO temperature.18 Among the different non-covalent interactions, hydrogen bonds are arguably the most exploited ones for generating these type of supramolecular compounds. Such a strategy has been employed to directly link neighbouring SCO molecules,19 or to produce composite systems using additional anionic or neutral H-bonding organic co-ligands.20,21 Hydrogen bonding can then directly influence the magnetic behaviour, by tuning the transition temperature or even the hysteresis width.22–25 Moreover, the approach opens as well the possibility to produce porous supramolecular architectures, which can be further exploited for gas storage or chemical sensing.
In order to explore the potential of H-bonded SCO materials, we have designed the ligand 2-(5-(pyridin-3-yl)-1H-pyrazol-3-yl)pyridine (HL), which combines the pyrazolylpyridyl chelating unit together with an additional pyridyl moiety (Scheme S1†). The pyrazolylpyridyl fragment is a well-known platform to produce Fe(II)-based SCO compounds, as observed with analogous ligands reported by some of us.26,27 While the N–H group from the pyrazolyl moiety was shown to behave as an H-donor unit, the additional pyridyl group was incorporated to act as an H-acceptor. The synthesis was carried out using the same approach followed for other related ligands. Thus, a 1,3-diketone precursor was first synthesized by a Claisen condensation between ethyl 2-picolinate and 3-acetylpyridine, while hydrazine was used to promote the ring closure (Scheme S1, Fig. S1 and S2†). Coordination of HL with iron(II) was then pursued by mixing three equivalents of the ligand with one equivalent of Fe(BF4)2 in acetonitrile. Slow diffusion of diethyl ether into the resulting orange solution afforded plate shaped orange crystals of [Fe(HL)3](BF4)2 (1) after three weeks. Elemental analysis confirmed the molecular formula revealed by single-crystal X-ray diffraction (SCXRD). The coordination complex crystallizes in the trigonal P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group (Table S1†) featuring one Fe(II) centre encapsulated by the pyrazolylpyridyl pockets of three neutral HL ligands (Fig. 1, top, and Fig. S3†). The electroneutrality of the compound is achieved with the presence of two BF4− anionic units. The ligands provide three N–H donor (pyrazolyl, pz) and three N acceptor (external pyridyl, py) groups, the latter exhibiting the nitrogen atom oriented towards the external part of the molecule. This conformation promotes the interaction with three neighbouring complexes through the formation of six N–H⋯N hydrogen bonds between the corresponding complementary units (Npy⋯Npz = 2.755 Å, Fig. S4 and Table S2†). This framework is extended in the crystal lattice, generating a 2D honeycomb-like supramolecular arrangement (Fig. 1, bottom). At 100 K, the average Fe–N bond distance is 1.985 Å, which rises to 2.110 Å after increasing the temperature to 280 K suggesting the occurrence of a thermally induced SCO (Table S3†). The reversibility of such transition was evaluated by SCXRD following a 300–100–300 K thermal cycle (Table S1†). The average Fe–N bond distance changed from 2.138 Å at 300 K to 1.990 Å at 100 K. Once back to 300 K, a value of 2.140 Å is recovered, thus evidencing a reversible spin crossover (Table S3†). The latter was explored by variable temperature magnetic susceptibility measurements on polycrystalline samples, revealing the switching properties of the compound for which a characteristic full thermal cycle is shown in Fig. 2. At 380 K, the χMT product has a value of 3.05 cm3 mol−1 K, in agreement with the Fe(II) centre laying in the high-spin configuration. On lowering the temperature, χMT gradually decreases, reaching a plateau at around 160 K with a value of 0.27 cm3 mol−1 K that evidences a complete SCO centered at ca. 275 K. A slight change of slope is observed at 275 K, hinting the presence of two steps. Indeed, upon heating, a two-step SCO is clearly observed with a plateau in the 275–320 K range, the second more abrupt step occurring at ca. 328 K, altogether originating a peculiar asymmetric hysteresis. While these properties are globally reproducible, we have found that the height of the plateau and the characteristic temperatures of the 2-step transition upon warming present some scan-rate dependences and annealing phenomena, which are described in more details in the ESI (Fig. S5–S11†). The properties described here are those of a dry/annealed polycrystalline solid. Indeed, we found that exposure of the unsolvated crystals to air results in the uptake of a small amount of water from air moist, as shown through elemental analysis and thermogravimetric measurements (Fig. S12†). The powder X-ray diffraction patterns of the polycrystalline solid is nevertheless in excellent agreement with that derived from the crystal structure (Fig. S13†), implying that the exposure to air does not affect the structural integrity of the material. Slight variations in the packing associated with differences in the degree of hydration could indeed play a role in the original SCO observed. Such variations of the pore content have been observed in a related honeycomb hydrogen bonded framework.28
space group (Table S1†) featuring one Fe(II) centre encapsulated by the pyrazolylpyridyl pockets of three neutral HL ligands (Fig. 1, top, and Fig. S3†). The electroneutrality of the compound is achieved with the presence of two BF4− anionic units. The ligands provide three N–H donor (pyrazolyl, pz) and three N acceptor (external pyridyl, py) groups, the latter exhibiting the nitrogen atom oriented towards the external part of the molecule. This conformation promotes the interaction with three neighbouring complexes through the formation of six N–H⋯N hydrogen bonds between the corresponding complementary units (Npy⋯Npz = 2.755 Å, Fig. S4 and Table S2†). This framework is extended in the crystal lattice, generating a 2D honeycomb-like supramolecular arrangement (Fig. 1, bottom). At 100 K, the average Fe–N bond distance is 1.985 Å, which rises to 2.110 Å after increasing the temperature to 280 K suggesting the occurrence of a thermally induced SCO (Table S3†). The reversibility of such transition was evaluated by SCXRD following a 300–100–300 K thermal cycle (Table S1†). The average Fe–N bond distance changed from 2.138 Å at 300 K to 1.990 Å at 100 K. Once back to 300 K, a value of 2.140 Å is recovered, thus evidencing a reversible spin crossover (Table S3†). The latter was explored by variable temperature magnetic susceptibility measurements on polycrystalline samples, revealing the switching properties of the compound for which a characteristic full thermal cycle is shown in Fig. 2. At 380 K, the χMT product has a value of 3.05 cm3 mol−1 K, in agreement with the Fe(II) centre laying in the high-spin configuration. On lowering the temperature, χMT gradually decreases, reaching a plateau at around 160 K with a value of 0.27 cm3 mol−1 K that evidences a complete SCO centered at ca. 275 K. A slight change of slope is observed at 275 K, hinting the presence of two steps. Indeed, upon heating, a two-step SCO is clearly observed with a plateau in the 275–320 K range, the second more abrupt step occurring at ca. 328 K, altogether originating a peculiar asymmetric hysteresis. While these properties are globally reproducible, we have found that the height of the plateau and the characteristic temperatures of the 2-step transition upon warming present some scan-rate dependences and annealing phenomena, which are described in more details in the ESI (Fig. S5–S11†). The properties described here are those of a dry/annealed polycrystalline solid. Indeed, we found that exposure of the unsolvated crystals to air results in the uptake of a small amount of water from air moist, as shown through elemental analysis and thermogravimetric measurements (Fig. S12†). The powder X-ray diffraction patterns of the polycrystalline solid is nevertheless in excellent agreement with that derived from the crystal structure (Fig. S13†), implying that the exposure to air does not affect the structural integrity of the material. Slight variations in the packing associated with differences in the degree of hydration could indeed play a role in the original SCO observed. Such variations of the pore content have been observed in a related honeycomb hydrogen bonded framework.28
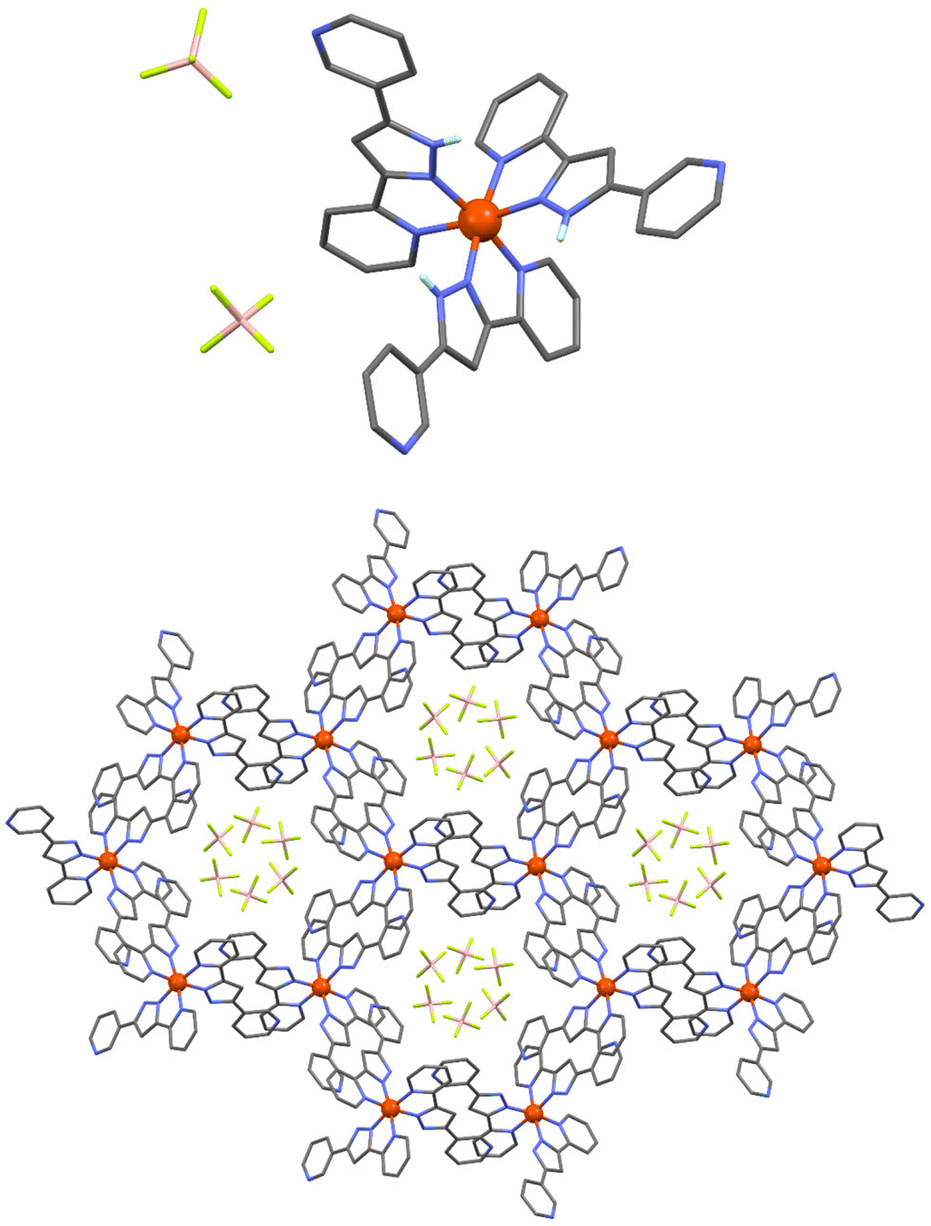 | ||
| Fig. 1 Representation of [Fe(HL)3](BF4)2 (1) (top) and the crystal packing along the c axis (bottom). Fe, N, C, B, F and H atoms are shown in orange, blue, grey, pink, green and white, respectively. Only H atoms from the pyrazolyl NH groups are shown for clarity. | ||
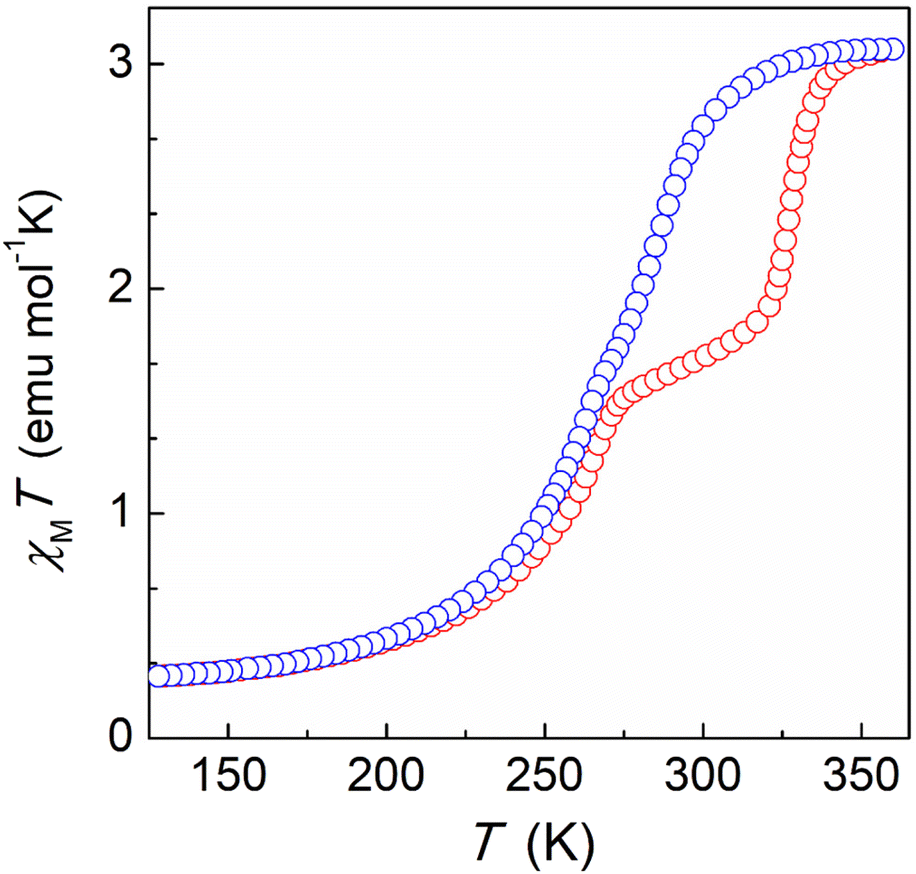 | ||
| Fig. 2 Variable temperature dc magnetic susceptibility (χ = M/H per mole of compound) data for 1 from 125 to 360 K, collected at 3 K min−1 under an applied field of 0.5 T. Cooling and heating modes are shown in blue and red, respectively. | ||
Also, the adsorbed water is easily lost at relatively low temperatures, below 80 °C, but the solid re-absorbs moist very fast. As often in SCO solids, this hydration–dehydration process has consequences on the magnetic properties. This becomes particularly evident in the present case, where the SCO takes place in the vicinity of RT. Thus, the first thermal cycle depends on the history of the sample, which also results in slight differences in the annealed solid (see ESI for details, Fig. S9 and S10†).
After evaluating the switching ability of the [Fe(HL)3]2+ complex in compound 1, we decided to explore the possibility of tuning its supramolecular architecture by introducing a larger anion. Thus, a similar synthetic approach was followed but adding TMA[FeCl4] (TMA = tetramethylammonium) to the reaction solution, using acetone as solvent. The potential of [FeCl4]− to form H-bonding interactions has been observed when acting as a guest within molecular cages, owing to the H-acceptor properties of its halide ligands.29 Slow diffusion of diethyl ether in the orange solution mentioned above afforded red block shaped crystals after one week. SCXRD experiments at 100 K showed the nature of the new compound obtained, featuring both, BF4− and [FeCl4]− anions, which was formulated as [FeCl4]@{[Fe(HL)3]4}[FeCl4]0.5(BF4)6.5·(C3H6O)11.5 (2, Table S4†). Remarkably, the insertion of [FeCl4]− induces the distribution of four [Fe(HL)3]2+ units around it, featuring a tetrahedral topology (Fig. 3 and Fig. S14†). While the latter has been observed in form of tetranuclear molecules based on organic ligands with more than one coordination pocket, the self-assembly of purely H-bonded supramolecular tetrahedrons is very unusual.30–32 The four cationic complexes located at each vertex of compound 2 are crystallographically independent, and produce the supramolecular cage through twelve N–H⋯N hydrogen bonding interactions (Npy⋯Npz = 2.528–2.831 Å, Table S5†). As observed in 1, each [Fe(HL)3]2+ cation interacts with three neighbouring complexes, but shaping in this case a discrete supramolecular system. Such distribution derives from two main factors. First, the role of the tetrahalometallate, which induces additional short interactions between the chloride ligands and the N–H pyrazolyl and ortho C–H pyridyl units of the four iron(II) complexes (Fig. S15 and Table S6†). The latter was already observed in polynuclear tetrahedral Ni(II) complexes encapsulating [FeX4]− (X = Cl, Br).29 Secondly, each external pyridyl group is partially rotated when compared to 1. Such a rotation allows the orientation required to establish the H-bonding network of the discrete supramolecular tetrahedron (Fig. S16†). The positive charge of {[Fe(HL)3]4}8+ is compensated by one [FeCl4]− complex and one BF4− anion, both with 50% occupancy, six further fully occupied BF4− anions and the encapsulated tetrahalometallate. It should be noted that, at this temperature, two of the [Fe(HL)3]2+ cations (Fe3 and Fe4) are disordered over two positions (Table S7†). The Fe–N average bond distances for each of the iron(II) cations are 1.975, 1.983, 1.983 and 1.977 Å for Fe1, Fe2, Fe3 and Fe4, respectively, suggesting a LS configuration for all four of them.
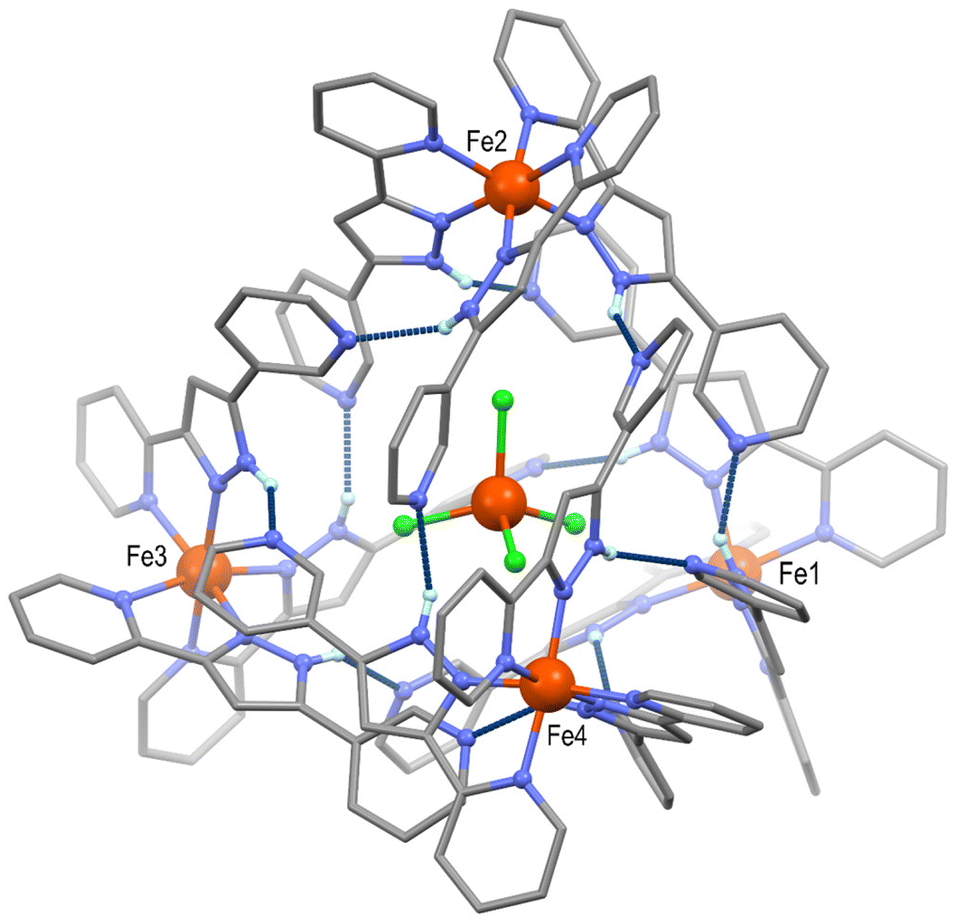 | ||
| Fig. 3 Representation of the cationic supramolecular tetrahedron in compound 2, showing the encapsulated [FeCl4]− moiety. Fe, N, C, and Cl atoms are shown in orange, blue, grey, and green, respectively. H atoms have been omitted for clarity, except these belonging to N–H groups, shown in light blue. H-bonding Npy⋯Npz interactions are represented by dashed blue lines. | ||
The magnetic properties of this supramolecular architecture were then studied by means of temperature dependent magnetic susceptibility measurements. To ensure that the integrity of the compound is guaranteed, crystals were first measured in contact with their acetone mother solution (Fig. S17†). At 100 K, the χMT product shows a value of 7.22 cm3 mol−1 K, which agrees with the presence of the corresponding Fe(III) centres and the four Fe(II) ions in their LS state. Magnetization measurements at variable fields at 5 K also agree with the 1.5 Fe(III) centres per formula and g = 2.06 (Fig. S18†). Upon heating the sample, χMT gradually increases, reaching a value of 13.53 cm3 mol−1 K at 315 K, which evidences a partial transition of the iron centres of the tetrahedron. In addition, a small jump was observed around 230 K, which is reproduced both in the heating and cooling modes (Fig. S18†). In order to explore the response at higher temperatures, magnetic susceptibility was then measured on a dried sample of 2 (Fig. 4, bottom). The behaviour between 100 and 300 K was found to be similar, in this case without the presence of the jump at 230 K. Most likely the observed jump corresponds to a process of localization/diffusion of the lattice acetone molecules, and its suppression is due to the loss of these upon drying. The lack of a plateau and the value of 19.15 cm3 mol−1 K observed at 390 K are consistent with an incomplete transition of the four iron(II) centres, evidencing that higher temperatures are required to further assess the evolution of the magnetic response. Differential scanning calorimetry (DSC) measurements were therefore undertaken on a powder sample of dried 2 in the range 120–450 K (Fig. 5). The calorimetric data revealed very broad anomalies in both the warming and cooling modes covering the range from ca. 200 K to ca. 420 K, in good agreement with the magnetic data. The excess heat capacity associated with the SCO process in 2 was derived after the estimation of a reasonable lattice heat capacity using a polynomial fit of the data below and above the SCO. The excess enthalpy and entropy due to the SCO in 2 can then be derived from integration of ΔCp over T and lnT, respectively, which leads to ΔHSCO = 25.4 kJ mol−1 and ΔSSCO = 79.6 J mol−1 K−1. The corresponding average values per Fe(II) centre, 6.35 kJ mol−1 and 19.9 J mol−1 K−1 respectively, are quite small for a SCO occurring a relatively high temperatures,33–35 the excess entropy being only slightly larger than the purely electronic contribution R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln5 = 13.38 J mol−1 K−1. Clearly, these depict a very gradual SCO process in 2. This is confirmed by the good simulation of the excess heat capacity temperature obtained with the Sorai domain model,34,35 yielding close to 1 (1.07) interacting SCO centres per domain, in line with the regular solution theory. The model also allows defining a SCO temperature T1/2 of 327 K.
ln5 = 13.38 J mol−1 K−1. Clearly, these depict a very gradual SCO process in 2. This is confirmed by the good simulation of the excess heat capacity temperature obtained with the Sorai domain model,34,35 yielding close to 1 (1.07) interacting SCO centres per domain, in line with the regular solution theory. The model also allows defining a SCO temperature T1/2 of 327 K.
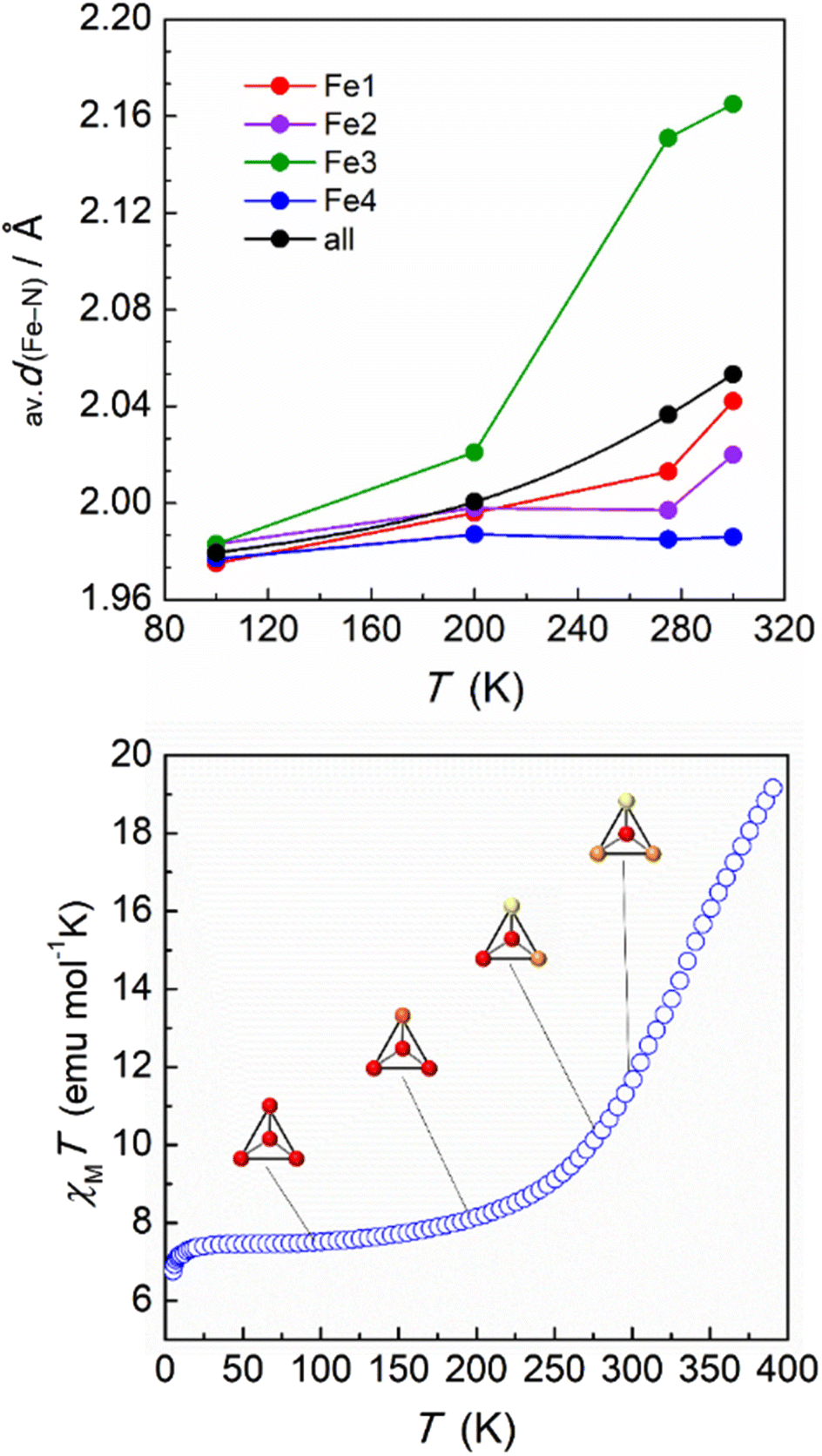 | ||
| Fig. 4 Top: average Fe–N bond lengths for the different iron centres of 2 at selected temperatures. Bottom: variable temperature dc magnetic susceptibility (χ = M/H per mole of compound) data for a dried sample of 2 from 5 to 390 K, collected under an applied field of 0.5 T. A schematic representation of the tetrahedron is shown for the four SCXRD collection temperatures, evidencing a LS (red), HS (yellow) or mixture of both configurations (orange) for each centre. | ||
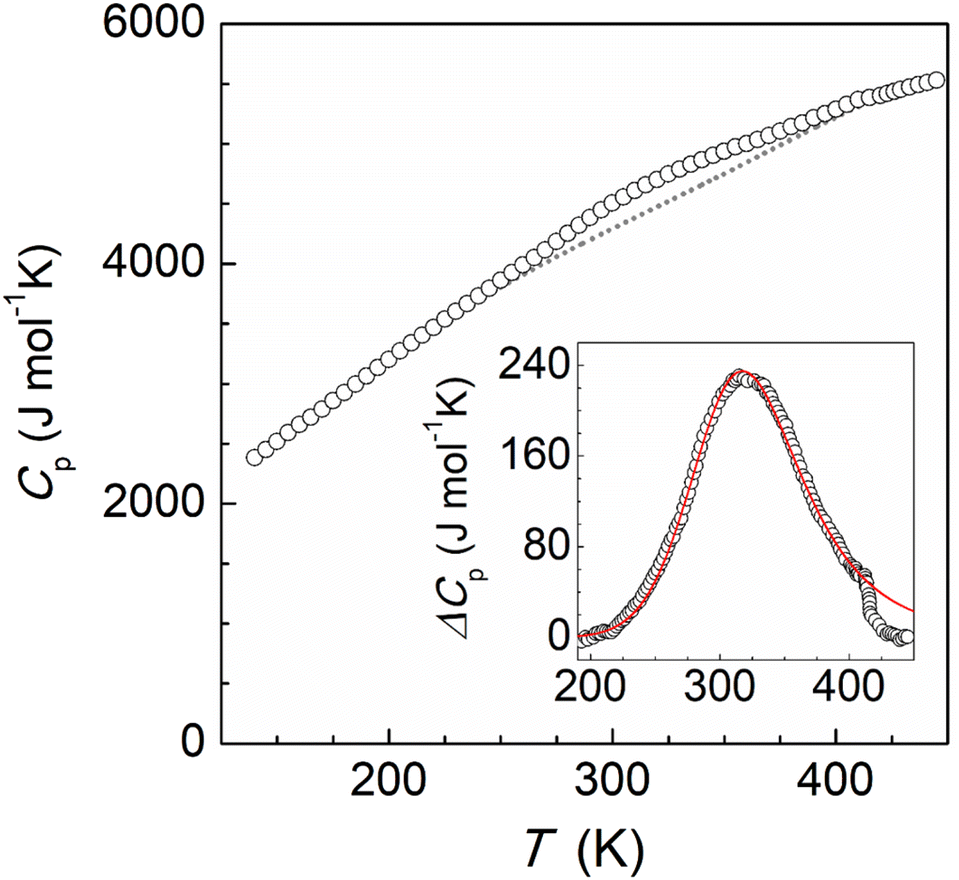 | ||
| Fig. 5 Molar heat capacities of compound 2. The dashed line represents the estimated normal heat capacities used for ΔCp determination. Inset: excess heat capacity and its simulation using Sorai domain model (see text). | ||
The magnetic and DSC experiments performed illustrate the remarkable larger range of temperatures over which the SCO in 2 occurs when compared to 1. In order to assess the magnetic behaviour of each individual iron(II) unit in the supramolecular tetrahedron, the crystal structure was obtained at several higher temperatures (200, 275 and 300 K, Table S4†). The volume and cell axes were found to increase, as expected from the thermal expansion and the SCO process. At 200 K, the average values of the Fe–N bond distances for Fe1, Fe2 and Fe4 do not experience a significant increase with respect to the 100 K values, suggesting still a LS configuration in these centres (Table S7†). In contrast, an increase of this parameter was observed for Fe3 (2.021 Å in average) indicating that, at this temperature, some iron(II) centres in this position acquire the HS state (Fig. 4). This feature agrees with the observed small increase of the χMT product, which shows a value of 8.03 cm3 mol−1 K at 200 K. The trend was then confirmed at 275 K, where an average value of Fe3–N of 2.151 Å was observed, suggesting now a large percentage of the Fe3 ions being in a HS configuration. Interestingly, at this temperature, the Fe–N average values in the other three [Fe(HL)3]2+ moieties of the tetrahedron do not evidence a significant increase, with the exception of Fe1 that slightly grows to 2.013 Å (Table S7† and Fig. 4). Magnetic susceptibility data are in line with these structural observations. The value of 11.17 cm3 mol−1 K observed at 275 K represents an increase of 3.95 cm3 mol−1 K compared to the value at 100 K, which agrees with the Fe3 ions exhibiting a fully HS configuration and Fe1 very partially transformed, leaving the other two iron(II) units in the LS state. It should be noted that, at this temperature, no disorder was observed for Fe3 and Fe4, most probably due to the diffused location of the acetone molecules in the lattice. At 300 K, the average Fe3–N bond distance reaches a value of 2.165 Å, confirming the evolution towards a HS state in this vertex of the tetrahedron (Table S7† and Fig. 4). At this temperature, the Fe1 and Fe2 centres experience as well an enhancement of the Fe–N bond lengths, indicating that the SCO process has also started at these sites. However, the average values suggest that many of these centres are still in their LS configuration (2.042 and 2.020 Å for Fe1 and Fe2, respectively). Interestingly, Fe4 remains in the LS state, as evidenced by the average Fe–N bond length of 1.986 Å. While the setup used for the crystallographic study does not allow a further increase of the temperature (see ESI†), the structural data at 300 K clearly demonstrate the singular behaviour of each [Fe(HL)3]2+ cationic SCO unit in the supramolecular tetrahedron. To further explore the nature of such individual behaviour, the structural parameters of 2 were assessed with Octadist.36 The octahedral nature for each iron(II) centre was evaluated by extracting the stretching (ζ), angular (Σ) and torsional (Θ) distortion parameters (Table S8†). At 100 K, the values observed are similar to these observed for compound 1 in the LS configuration. However, the environment of the Fe3 centres evidences a greater distortion when comparted to its neighbours, with a value of 71.4 and 218.0° for Σ and Θ, respectively. Such a larger distortion could explain the preference of this particular unit for the transition towards a HS configuration, also mirrored by the distortion parameters obtained at higher temperatures. In accordance, the coordination polyhedron around Fe4 was found to be less distorted, as shown by the values obtained both at low and high temperatures (Table S8†).
In conclusion, we have shown the possibility of building a switchable supramolecular tetrahedron constructed purely by H-bonding interactions. The singular structural and magnetic properties observed in each of the [Fe(HL)3]2+ units demonstrate the potential of this synthetic strategy to tune and develop novel SCO materials.
Author contributions
GR and PV synthesized and crystallized compound 1. HHN and FT synthesized and crystallized compound 2. DA made and characterized the organic ligand. DA and OR obtained and solved the SCXRD data. OR performed the magnetic and DSC experiments, and wrote the corresponding discussion. DA wrote the rest of the manuscript, with inputs from all authors. DA and GA organized the whole paper and supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding by the Spanish MCIN through PID2022-137764OB-I00 (GA and DA), TED2021-129214B-I00 (GA and DA), PID2020-118329RB-I00 (OR) and CNS2022-135816 (DA), the Generalitat de Catalunya for the ICREA Academia Prize 2023 (GA) and the Danish Ministry for Higher Education and Science (Q-MAT Lighthouse) (HHN). This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231, and of ALBA synchrotron BL13-XALOC beamline.References
- P. Gütlich and H. A. Goodwin, in Spin Crossover in Transition Metal Compounds I, ed. P. Gütlich and H. A. Goodwin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 1–47. DOI:10.1007/b13527.
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 1703862 CrossRef PubMed.
- A. B. Gaspar, G. Molnár, A. Rotaru and H. J. Shepherd, C. R. Chim., 2018, 21, 1095–1120 CrossRef CAS.
- G. Chastanet, C. Desplanches, C. Baldé, P. Rosa, M. Marchivie and P. Guionneau, Chem. Sq., 2018, 2, 18 Search PubMed.
- H. Li and H. Peng, Curr. Opin. Colloid Interface Sci., 2018, 35, 9–16 CrossRef CAS.
- Y. Zan, M. Piedrahita-Bello, S. E. Alavi, G. Molnár, B. Tondu, L. Salmon and A. Bousseksou, Adv. Intell. Syst., 2023, 5, 2200432 CrossRef.
- A. Bousseksou and G. Molnár, C. R. Chim., 2003, 6, 1175–1183 CrossRef CAS.
- M. Darawsheh, L. A. Barrios, O. Roubeau, S. J. Teat and G. Aromí, Chem. – Eur. J., 2016, 22, 8635–8645 CrossRef CAS PubMed.
- R. A. Bilbeisi, S. Zarra, H. L. C. Feltham, G. N. L. Jameson, J. K. Clegg, S. Brooker and J. R. Nitschke, Chem. – Eur. J., 2013, 19, 8058–8062 CrossRef CAS PubMed.
- W.-K. Han, H.-X. Zhang, Y. Wang, W. Liu, X. Yan, T. Li and Z.-G. Gu, Chem. Commun., 2018, 54, 12646–12649 RSC.
- J. Zheng, L. K. S. von Krbek, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2022, 61, e202212634 CrossRef CAS PubMed.
- A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathonière, R. Clérac and P. E. Kruger, Chem. Commun., 2013, 49, 1597–1599 RSC.
- N. Struch, C. Bannwarth, T. K. Ronson, Y. Lorenz, B. Mienert, N. Wagner, M. Engeser, E. Bill, R. Puttreddy, K. Rissanen, J. Beck, S. Grimme, J. R. Nitschke and A. Lützen, Angew. Chem., Int. Ed., 2017, 56, 4930–4935 CrossRef CAS PubMed.
- C.-Y. Qin, S.-Z. Zhao, H.-Y. Wu, Y.-H. Li, Z.-K. Wang, Z. Wang and S. Wang, Dalton Trans., 2021, 50, 5899–5910 RSC.
- R. Díaz-Torres, W. Phonsri, K. S. Murray, L. Liu, M. Ahmed, S. M. Neville, P. Harding and D. J. Harding, Inorg. Chem., 2020, 59, 13784–13791 CrossRef PubMed.
- K. Hanahara, H. Ono, T. Fujinami, N. Matsumoto and Y. Sunatsuki, Inorg. Chim. Acta, 2015, 429, 93–98 CrossRef CAS.
- O. Roubeau, Chem. – Eur. J., 2012, 18, 15230–15244 CrossRef CAS PubMed.
- G. Lemercier, N. Bréfuel, S. Shova, J. A. Wolny, F. Dahan, M. Verelst, H. Paulsen, A. X. Trautwein and J.-P. Tuchagues, Chem. – Eur. J., 2006, 12, 7421–7432 CrossRef CAS PubMed.
- T. Kanetomo, Z. Ni and M. Enomoto, Dalton Trans., 2022, 51, 5034–5040 RSC.
- E. Coronado, M. C. Giménez-López, C. Giménez-Saiz and F. M. Romero, CrystEngComm, 2009, 11, 2198–2203 RSC.
- L. T. Birchall, G. Truccolo, L. Jackson and H. J. Shepherd, Chem. Sci., 2022, 13, 3176–3186 RSC.
- B. Weber, W. Bauer, T. Pfaffeneder, M. M. Dîrtu, A. D. Naik, A. Rotaru and Y. Garcia, Eur. J. Inorg. Chem., 2011, 2011, 3193–3206 CrossRef CAS.
- I. Nemec, R. Herchel and Z. Trávníček, Dalton Trans., 2015, 44, 4474–4484 RSC.
- L. Salmon, B. Donnadieu, A. Bousseksou and J.-P. Tuchagues, C. R. Acad. Sci., Ser. IIc: Chim., 1999, 2, 305–309 CrossRef CAS.
- H. Zenno, F. Kobayashi, M. Nakamura, Y. Sekine, L. F. Lindoy and S. Hayami, Dalton Trans., 2021, 50, 7843–7853 RSC.
- M. Darawsheh, L. A. Barrios, O. Roubeau, S. J. Teat and G. Aromí, Angew. Chem., Int. Ed., 2018, 57, 13509–13513 CrossRef CAS PubMed.
- D. Y. Aleshin, R. Diego, L. A. Barrios, Y. V. Nelyubina, G. Aromí and V. V. Novikov, Angew. Chem., Int. Ed., 2022, 61, e202110310 CrossRef CAS PubMed.
- J. J. Loughrey, T. P. Comyn, D. C. Apperley, M. A. Little and M. A. Halcrow, Chem. Commun., 2014, 50, 7601–7603 RSC.
- A. J. Scott, J. Vallejo, A. Sarkar, L. Smythe, E. Regincós Martí, G. S. Nichol, W. T. Klooster, S. J. Coles, M. Murrie, G. Rajaraman, S. Piligkos, P. J. Lusby and E. K. Brechin, Chem. Sci., 2021, 12, 5134–5142 RSC.
- Q. Shi, X. Zhou, W. Yuan, X. Su, A. Neniškis, X. Wei, L. Taujenis, G. Snarskis, J. S. Ward, K. Rissanen, J. de Mendoza and E. Orentas, J. Am. Chem. Soc., 2020, 142, 3658–3670 CrossRef CAS PubMed.
- K. Nishi, N. Matsumoto, S. Iijima, M. A. Halcrow, Y. Sunatsuki and M. Kojima, Inorg. Chem., 2011, 50, 11303–11305 CrossRef CAS PubMed.
- K. Nishi, H. Kondo, T. Fujinami, N. Matsumoto, S. Iijima, M. A. Halcrow, Y. Sunatsuki and M. Kojima, Eur. J. Inorg. Chem., 2013, 2013, 927–933 CrossRef CAS.
- O. Roubeau, M. Castro, R. Burriel, J. G. Haasnoot and J. Reedijk, J. Phys. Chem. B, 2011, 115, 3003–3012 CrossRef CAS PubMed.
- M. Sorai, in Spin Crossover in Transition Metal Compounds III, ed. P. Gütlich and H. A. Goodwin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 153–170. DOI:10.1007/b95426.
- M. Sorai, Y. Nakazawa, M. Nakano and Y. Miyazaki, Chem. Rev., 2013, 113, PR41–PR122 CrossRef CAS PubMed.
- R. Ketkaew, Y. Tantirungrotechai, P. Harding, G. Chastanet, P. Guionneau, M. Marchivie and D. J. Harding, Dalton Trans., 2021, 50, 1086–1096 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2334712–2334717 and 2353505–2353507. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00578c |
| This journal is © The Royal Society of Chemistry 2024 |