
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binucleating Jäger-type {(N2O2)2}4− ligands: magnetic and electronic interactions of Fe(II), Ni(II) and Cu(II) across an in-plane TTF-bridge†
Constantin
Schreck
a,
Sophie
Schönfeld‡
a,
Phil
Liebing
 b,
Gerald
Hörner
ab and
Birgit
Weber
*ab
b,
Gerald
Hörner
ab and
Birgit
Weber
*ab
aDepartment of Chemistry, Inorganic Chemistry, University of Bayreuth, 95447 Bayreuth, Germany. E-mail: birgit.weber@uni-jena.de
bNew address: Institute for Inorganic and Analytical Chemistry, Faculty of Chemistry and Earth Sciences, Friedrich-Schiller-University Jena, Germany
First published on 3rd May 2024
Abstract
The simultaneous presence of different electrophores provides an interesting playground for responsive materials. Herein, we present the incorporation of a twice-reversibly oxidizable tetrathiafulvalene (TTF) unit into a binucleating ligand, bridging two metal centers in a fully conjugated plane. A two-step synthesis scheme gave the D2h symmetric Schiff base-like ligand H4L in moderate yields from which the corresponding copper(II) [Cu2L], nickel(II) [Ni2L], [Ni2L(py)4] and iron(II) complexes [Fe2L(py)4], [Fe2L(dmap)4] and [Fe2L(bpee)2]·1 Tol could be obtained. Characterization was performed through 1H-NMR, IR, UV-vis and 57Fe-Mössbauer spectroscopy, SQUID magnetometry and cyclic voltammetry, supported by density functional theory (DFT) calculations. Single crystal X-ray analysis of [Ni2L(py)4] revealed six-coordinate paramagnetic centers, whereas [Ni2L] underwent gradual coordination induced spin state switching (CISSS) in solution. The magnetic independence of both metal centers is echoed by close-to-ideal Curie-plots of the [Cu2L] system and the gradual spin crossover of all iron(II) compounds. By contrast, cyclic voltammetry measurements in solution indicated oxidation-dependent TTF–metal interactions, as well as metal–metal interactions. The reversible TTF-borne events in H4L and [Ni2L] are overlaid with metal-borne events in the case of [Fe2L(py)4], as is corroborated by an analysis of the frontier orbital landscapes and through diagnostic spectral features upon chemical oxidation.
Introduction
The past two decades have witnessed the synthesis and utilization of an increasing number of multifunctional coordination compounds with orthogonal metal-borne and ligand-borne redox-active sites. Of particular interest remain TTF-containing scaffolds (TTF: tetrathiafulvalene), due to the beneficial electrochemical properties of TTF itself. These can lead to interesting combinations of the TTFs redox properties with the properties of the metal center. The degree of interactions between the metal centers and the TTF is adjustable through the method of integration of the TTF moiety into the coordination compound. Several reviews have summarized the state-of-the-art of this field.1 A selection of different topologies with references to recent examples is shown in Scheme 1.2,3–14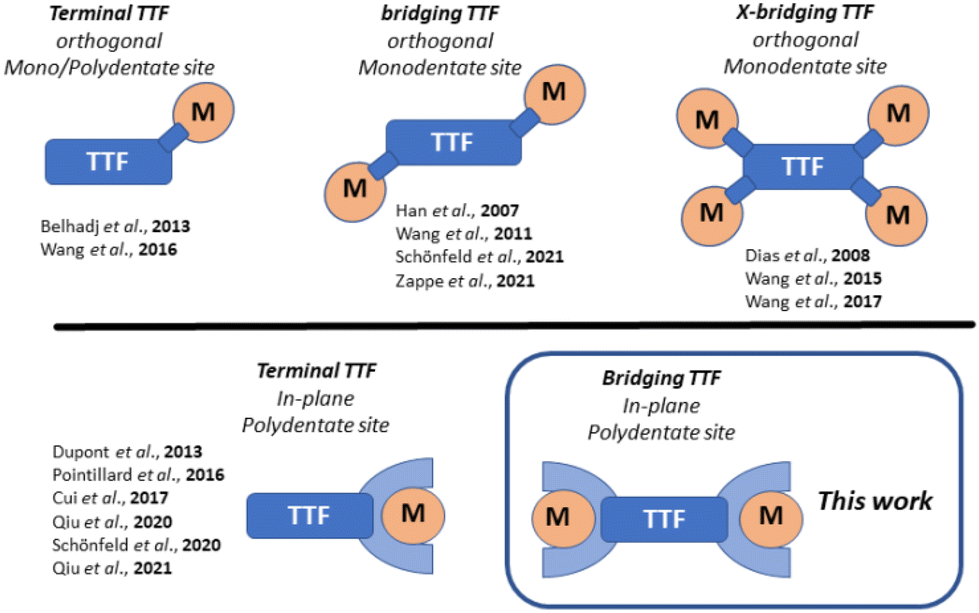 | ||
| Scheme 1 Selected topologies of TTF-containing coordination compounds; orthogonal denotes connection of the electrophores through a single bond; In-plane denotes a topology of merged electrophores within a single conjugated π-system. | ||
Orthogonal arrangements with the TTF and the metal coordination site being connected through a single bond, lead to predominantly or completely decoupled electrophores.15 The implementation of planar, multidentate coordination sites in proximity to the TTF-unit enables stronger interactions.9–11 TTF has also been integrated into bridging ligands, linking multiple metal centers together, in some cases leading to the formation of coordination polymers.3,5–8,16 Due to their type of linkage, both the metal and TTF sites do not interact significantly with each other. In previous work, we had elaborated on the implementation of bridging TTF units in coordination polymers.4 This connection through a single bond led to completely decoupled electrophores. In parallel approaches, we and others merged TTF with the π-backbone of typical Jäger-ligands.12–14
One of these studies found interactions between the TTF unit and iron center, after oxidation of said iron centers from iron(II) to iron(III). A shrinking of the gap between their redox potentials would presumably lead to a greater degree of interaction between the two electrophores. Such an energetic proximity can be achieved through choice of the metal ion and by de-/stabilization of specific oxidation states of the redox centers. A valuable approach is the incorporation of electron donating or electron withdrawing organic groups near the respective redox center. For multinuclear complexes the additional question arises of whether the different metal centers interact with each other e.g., by magnetic coupling.
In this work we present a novel Jäger-type ligand H4L with two (N2O2)2− coordination sites, connected though a planar, fully conjugated tetrathiafulvalene bridge. From this ligand we derived copper(II) [Cu2L], nickel(II) [Ni2L], [Ni2L(py)4] and iron(II) coordination compounds [Fe2L(py)4], [Fe2L(dmap)4], [Fe2L(bpee)2]·1 Tol. Besides their general characterization we focused on potential TTF–metal interactions by determination of the compounds redox processes via cyclic voltammetry (CV) and assigned the observed processes by comparison of the ligand with complexes, as well as literature data. Additionally, we followed the optical properties after chemical oxidation and supported our findings with density functional theory (DFT) calculations. To determine potential metal–metal interactions we used three different approaches. First we studied the coordination induced spin state switching (CISSS) properties for the binuclear nickel(II) complex. Secondly we followed the spin crossover (SCO) properties for the octahedrally coordinated iron(II) compounds by SQUID magnetometry and 57Fe-Mössbauer spectroscopy. Thirdly we determined the coupling constants for the magnetic coupling between the two copper(II) centres in [Cu2L].
Materials and methods
Materials and general procedures
All reagents were of reagent grade and used without further purification. Solvents used for the ligand synthesis were of analytical grade and used without further purification. Dry toluene and pyridine were obtained from ACROS. Ethanol was dried over magnesium and distilled before usage.Synthesis: general procedure
All air and moisture sensitive syntheses were carried out under argon using Schlenk tube techniques. These include all syntheses of iron(II) compounds and the synthesis of [2,2′-bibenzo[d][1,3]dithiolylidene]-5,5′,6,6′-tetraamine 2. The precursor compound 4,5-Diaminobenzole-1,3-dithiol-2-thione 1 was obtained in a two-step synthesis as described in literature.171 H-NMR (300 MHz, DMSO-d6, 300 K): δ [ppm] = 6.55 (s, 4H), 4.67 (s, 8H).
Ligand H4L: Tetraamine 2 (0.53 g, 1.44 mmol) was suspended in EtOH (25 mL) before ethyl-(E)-2-(ethoxymethylene)-4,4,4-trifluoro-3-oxobutanoate (1.74 g, 7.23 mmol) was added. The mixture was heated to reflux for 4 h and, after cooling, a red precipitate was filtered off and washed with EtOH (2 × 5 mL). For further purification the precipitate was recrystallized from EtOH. Yield 0.78 g (47%). Elemental analysis for C42H32F12N4O12S4 (1140.95 g mol−1): found (calc.): C 43.74 (44.21), H 2.84 (2.83), N 5.09 (4.91), S 11.71 (11.24). 1H-NMR (500 MHz, DMSO-d6, 300 K): δ [ppm] = 11.88 (d, J = 13.7 Hz), 11.06 (m), 8.26 (d, J = 14.0 Hz), 8.13 (d, J = 14.0 Hz), 7.92 (s, 4H), 4.21 (m, 8H), 1.24 (t, J = 7.1 Hz, 12H). MS (ESI neg): m/z = 569.01 (H2L2−) FT-IR [cm−1]: ṽ = 2980 (w, ν[–C–H]), 1645 (m, ν[C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]), 1603 (m, n[–CC]), 1417 (m, n[–CC]), 1153 (s, ν[–C–F]).
O]), 1603 (m, n[–CC]), 1417 (m, n[–CC]), 1153 (s, ν[–C–F]).
[Ni2L]: Ligand H4L (0.11 g, 0.10 mmol) was dissolved in EtOH (40 mL) before [Ni(OAc)2]·4H2O (0.06 g, 0.24 mmol) was added. The solution was heated to reflux for 1 h, changing from a red to a dark violet color. The solution was stored at 6 °C overnight and the precipitate was filtered off and washed with cold EtOH (2 × 4 mL). After drying in air, a dark purple solid was obtained. Yield 0.11 g (89%). Elemental analysis for C42H28F12N4Ni2O12S4 (1254.31 g mol−1): found (calc.): C 39.85 (40.22), H 2.59 (2.25), N 4.82 (4.47), S 10.43 (10.22). 1H-NMR (500 MHz, DMSO-d6, 300 K): δ [ppm] = 9.37 (br, 4H), 4.26 (br, 8H), 1.35 (br, 12 H).
[Ni2L(py)4]: [Ni2L] (0.010 g, 0.008 mmol) was dissolved in pyridine (3 mL) by heating to 80 °C. After cooling to room temperature, the solvent was slowly evaporated to obtain single crystals suitable for X-ray structure analysis. Lattice solvent that proved to be present (see below, XRD-structure elucidation), but could not be chemically defined, due to severe disorder.
[Fe2L(py)4]: Ligand H4L (0.57 g, 0.50 mmol) and [Fe(OAc)2] (0.26 g, 1.50 mmol) were dissolved in a mixture of toluene (10 mL) and pyridine (2 mL). The dark brown solution was heated under reflux for 3 h. After keeping the solution at −20 °C overnight, the resulting brown precipitate was filtered off, washed with toluene (2 × 2 mL) and dried under reduced pressure. Yield 0.68 g (86%). Elemental analysis for C62H48F12Fe2N8O12S4 (1565.02 g mol−1): found (calc.): C 47.25 (47.58), H 3.45 (3.09), N 7.26 (7.16), S 7.81 (8.19).
[Fe2L(dmap)4]: Ligand H4L (0.28 g, 0.25 mmol) and [Fe(OAc)2] (0.13 g, 0.75 mmol) were dissolved in EtOH (4 mL) and heated under reflux for 2 h. After storage of the solution at −20 °C, the precipitate was filtered off and dried under reduced pressure. The thus obtained ethanol complex (0.19 g) and 4-(dimethylamino)pyridine (dmap) (0.81 g, 6.6 mmol) were dissolved in EtOH (5 mL) and heated to reflux for 2 h. The solution was stored over night at −20 °C. The precipitate was filtered off, washed with EtOH (2 × 1 mL) and dried under reduced pressure to obtain a brown solid. Yield 0.17 g (40%). Elemental analysis for C70H68F12Fe2N12O12S4 (1737.30 g mol−1): found (calc.): C 48.56 (48.40), H 4.02 (3.95), N 9.87 (9.68), S 7.83 (7.38).
[Fe2L(bpee)2]·1 Tol: [Fe2L(py)4] (0.20 g, 0.13 mmol) and 1,2-bis-(4-pyridyl)ethylene (bpee) (0.23 g, 1.3 mmol) were dissolved in EtOH (20 mL) and heated to reflux for 4 h. All solvent was removed under reduced pressure, before adding EtOH (5 mL) and heating under reflux for 1 h. This last step of drying, solving, and heating was repeated identically for a second time. The solvent was removed under reduced pressure and the obtained solid was re-dispersed in toluene (5 mL). The dispersion was heated under reflux for 1 h and, after cooling, the solid was filtered off, washed with toluene (2 × 2 mL) and dried under reduced pressure to obtain a black solid. Yield 0.14 g (62%). Elemental analysis for C73H56F12Fe2N8O12S4 (1705.21 g mol−1): found (calc.): C 51.51 (51.42), H 3.70 (3.31), N 6.67 (6.57), S 8.31 (7.52).
Physical measurements
Elemental analyses were carried out using a Unicube (Elementar Analysensysteme GmbH, Langenselbold, Germany) with sulfanilamide as standard. NMR spectra were recorded using a Avance III HD 500 MHz spectrometer (Bruker, Ettlingen, Germany) at 25 °C. Mass spectra were recorded using a Thermo Scientific Q Exactive (Life Technologies GmbH, Darmstadt, Germany). In the case of the complexes only strongly fragmented mass spectra were obtained that are therefore not discussed. UV-Vis spectra were recorded using a Cary 60 UV-Vis spectrometer (Agilent, Santa Clara, USA) with a scan rate of 600 nm min−1.X-ray structure analyses were performed on a Stoe StadiVari diffractometer, equipped with a graphite-monochromated Mo-Kα (λ = 0.71073 Å) radiation source and a low-temperature unit from Oxford Cryosystems Ltd. A suitable single crystal was embedded in inert perfluorinated oil (Fomblin® YR-1800) and mounted on a nylon loop before collecting data at the given temperature. Data were corrected for Lorentz and polarization effects; a spherical absorption correction was applied. The structure was solved using OLEX2 software.18 Reflections that were seriously affected by the beamstop were omitted from the final refinement. The crystal was refined as inversion twin and a disordered ethoxy-group was split. Non-coordinate solvent within the crystal structure was treated with the “solvent mask”-routine in OLEX2. The electron density can most likely be attributed to a mixture of pyridine and water. Due to partial release of the solvent upon attempted isolation of the crystals, unambiguous identification by independent analytical techniques was not possible. Furthermore, the wide-angle data were cut off at a resolution of 0.78 Å. Mercury 2022.2.0 was used for structure illustrations/graphical output.19
Magnetic measurements were carried out using a Quantum Design MPMS-XL-5 SQUID magnetometer (Quantum Design, San Diego, USA). The samples were prepared in a gelatine capsule, with a plastic straw as sample holder. A field strength of 0.5–1.5 T was applied, and the samples were measured in a temperature range of 4–400 K to determine the temperature dependency of the magnetism. The temperature range of 20–400 K was measured in sweep mode (5 K min−1) and lower temperatures were measured in settle mode (1 K min−1). The measured values were corrected for diamagnetism of the sample holder and the ligand (tabulated Pascal constants).20 Thermogravimetric analyses were carried out under argon atmosphere with a heat rate of 10 K min−1 using a TG 209 F1 Libra (NETZSCH-Gerätebau GmbH, Selb, Germany). 57Fe Mössbauer spectra were recorded in transmission geometry in a constant-acceleration mode using a Mössbauer spectrometer equipped with a cryostat and a 50 mCi57Co-(Rh) source. The samples were prepared under Argon atmosphere and after insertion of the sample into the cryostat, the sample chamber was evacuated and flushed four times with helium gas. The spectra were fitted with Lorentzian lines using Recoil Mössbauer Analysis Software.21
Redox potentials were determined by cyclic voltammetry using an electrochemical analyser CHI610E (CH Instruments, Inc. Austin, USA). The samples (c = 1.0 × 10−3 M) were measured at room temperature in dichloromethane (DCM) with (Bu)4N(PF6) (c = 0.1 M) as electrolyte, with a saturated calomel reference electrode (SCE) and a scan rate of 50 mV s−1. The potentials are given in reference to the ferrocene/ferrocenium redox couple.
Computational details
Electronic structure calculations on the complexes have been performed through density-functional theory (DFT) methods using the ORCA program package.22 For all optimizations triple-ξ-valence TZVP23 basis sets were used with the generalized gradient approximated functional BP86.24 Grimme's third generation D3 correction of dispersion was used;25 medium effects were approximated in a dielectric continuum approach (COSMO), parameterized for CH3CN.26 Coordinates of the computed structures are assembled in the ESI COORDINATES.† MO energies and pattern of the complexes were extracted from single-point calculations in the optimized positions with the global hybrid functional TPSSh.27Results and discussion
Syntheses and characterization of binucleating ligand H4L
Synthetic access to the binucleating (N2O2)2 ligand H4L and the synthesis of the metal complexes (exemplified for [Cu2L]) are displayed in Scheme 2. The ligand precursor 2 was obtained through a triethyl phosphite-mediated homo-coupling reaction of 1 in toluene. The 1H-NMR spectrum of 2 and signal assignments are displayed in Fig. S1.† Precursor 2 was directly fed into the ligand synthesis without further purification. The ligand H4L was received from tetraamine 2 and the keto–enol ether 3 through a condensation reaction akin to the type invented by Claisen.28 The ligand's identity and purity was confirmed by 1H-NMR (Fig. S2†), mass spectrometry (Fig. S4†) and IR spectroscopy (Fig. S5†), as well as by elemental analysis. 1H-NMR spectra reveal diagnostic characteristics unique to Jäger-type ligands. In ligand H4L, the competing (E)- and (Z)-configurations of the enamine groups give rise to two sets, obvious for the –NH protons at 11.88 ppm and 11.06 ppm, and less pronounced for the other protons. While in most Jäger-type ligands the signals corresponding to the ene sites of the enamines are readily identified through their coupling constants of around 12 Hz,29 the presence of four enamine groups in the H4L adds complexity to the case as all of these enamine groups offer (E)- and (Z)-configuration by themselves. Mutual feedback from one another leads to many more signals in the 1H-NMR-spectrum. The resulting overlap of signals makes the readout of coupling constants impossible in some cases. The aromatic protons in TTF vicinity led to a singlet signal (7.92 ppm) in the same range as seen in the related mono-nucleating Jäger ligand (7.79 ppm) which we have described previously.14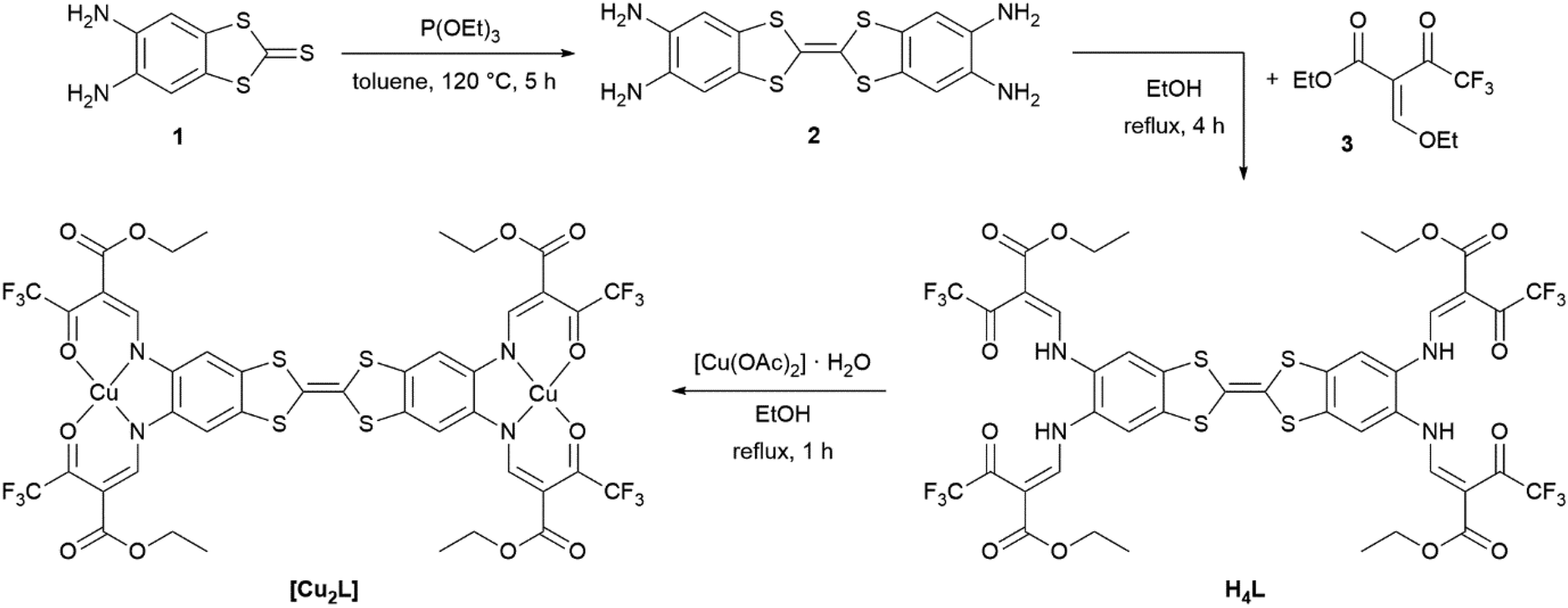 | ||
| Scheme 2 Synthesis of the binucleating ligand H4L, starting from 4,5-diaminobenzole-1,3-dithiol-2-thione 1 and complex synthesis, exemplified for the copper(II) complex [Cu2L]. | ||
Syntheses and characterization of binuclear complexes [M2L]
The copper(II) and nickel(II) complexes [Cu2L] and [Ni2L] were obtained in moderate yields through heating the corresponding metal acetates and the ligand in EtOH. The solid materials afforded as analytical pure microcrystals/powders in most cases indicating solvent-free formulations. In particular, accelerated precipitation of the nickel(II) complex from pyridine solution by rapid cooling results in the non-coordinated complex [Ni2L]. NMR spectroscopic studies of native [Ni2L] proved impossible, due to the very low solubility in non-coordinating solvents. 1H-NMR spectra recorded of dmso-d6 solutions of [Ni2L] reveal massively shifted resonances (Fig. S3†). As was seen previously in related mononuclear systems,30 the axial coordination of one or two solvent molecules to each of the nickel(II) centres renders the complex paramagnetic. The severe line broadening and low solubility of the complex make an assignment of the signals impossible. Independent information on the nature of the paramagnetic nickel complex could be received through single-crystal X-ray crystallography. Crystals suitable for X-ray structure analysis were obtained through slow evaporation of a solution of [Ni2L] in pyridine. These crystals indeed consisted of the pyridine coordinated nickel(II) complex [Ni2L(py)4]. Ni–N and Ni–O bond lengths d > 2.00 clearly point to high-spin configuration of both nickel sites. For a detailed discussion of the solid-state structure, see below.Unfortunately, crystallization efforts were not fruitful for all the iron complexes. The iron(II) complex [Fe2L(py)4] was obtained directly as a brownish powder from a one-pot reaction of ligand H4L, iron acetate and excess pyridine in toluene. For the dmap-coordinated iron(II) complex [Fe2L(dmap)4], in a first step, the EtOH-coordinated complex [Fe2L(EtOH)4] was isolated after reacting ligand H4L with iron acetate in ethanol. In a second step the ethanol ligand was replaced by dmap. Addition of excess dmap served to displace the weakly coordinating alcohol. The coordination polymer [Fe2L(bpee)2]·1 Tol was formed by heating [Fe2L(py)4] in the presence of excess 1,2-bis-(4-pyridyl)ethylene bpee in iterative heating/drying/re-dissolving cycles. To remove dissociated pyridine, the complete solvent was removed under reduced pressure after each cycle before re-adding solvent. This way, analytically pure materials were obtained.
Structures: DFT modeling and X-ray diffraction analysis
The TTF-containing compounds under study largely afforded as microcrystalline materials, akin to our previous experience on related mononuclear complexes14 and coordination polymers.4 In order to receive a consistent set of structural data, we modelled the molecular structures with DFT methods (computational details are given in the Experimental section). Thereby we relied on the GGA functional BP86, which gave very reliable data in related cases.4,5,14,31 Selected metrical data of the optimized complex structures are assembled in Table 1. As could be expected, the binucleating, octadentate ligand offers two identical N2O2 coordination sites, rendering the optimized structures close to ideal D2h symmetric. This pattern is echoed by the solid-state structure of [Ni2L(py)4], where experimental information was accessible (see below).| [Ni2L(py)4] | [Fe2L(py)4] | |||||
|---|---|---|---|---|---|---|
| [Ni2L] | DFT | XRD | [Cu2L] | all-LS | all-HS | |
| a Central bond of the TTF bridge. b Average of the four C–S bond lengths in the TTF bridge. | ||||||
| M(1)–O(1) | 1.861 | 2.052 | 2.020 | 1.945 | 1.929 | 2.011 |
| M(1)–O(2) | 1.857 | 2.051 | 2.065 | 1.947 | 1.932 | 2.025 |
| M(1)–N(1) | 1.849 | 1.998 | 2.008 | 1.938 | 1.900 | 2.080 |
| M(1)–N(2) | 1.850 | 1.993 | 1.991 | 1.944 | 1.894 | 2.082 |
| M(1)–N(ax1) | — | 2.105 | 2.117 | — | 1.962 | 2.180 |
| M(1)–N(ax2) | — | 2.110 | 2.151 | — | 1.991 | 2.232 |
| M(2)–O(3) | 1.862 | 2.054 | 2.039 | 1.946 | 1.931 | 2.006 |
| M(2)–O(4) | 1.858 | 2.049 | 2.010 | 1.942 | 1.932 | 2.003 |
| M(2)–N(3) | 1.849 | 1.994 | 1.961 | 1.943 | 1.898 | 2.074 |
| M(1)–N(4) | 1.849 | 1.990 | 2.009 | 1.937 | 1.893 | 2.069 |
| M(2)–N(ax3) | — | 2.098 | 2.139 | — | 1.988 | 2.179 |
| M(2)–N(ax4) | — | 2.114 | 2.198 | — | 1.972 | 2.243 |
| CCa |
1.354 | 1.352 | 1.345 | 1.353 | 1.352 | 1.353 |
C–Sav![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
1.781 | 1.781 | 1.749 | 1.781 | 1.781 | 1.781 |
| M⋯M | 16.194 | 15.876 | 16.510 | 15.898 | 15.584 | 16.198 |
Largely isotropic Cu–N and Cu–O bond lengths of 1.94 ± 0.01 Å are typical of planar d9 complexes. An overall similar pattern prevails in [Ni2L]; the significant isotropic bond contraction in the latter by ca. 0.1 Å reflects the diminished antibonding character of the diamagnetic d8 configuration at planar nickel(II). By contrast, axial ligation with pyridine renders the six-coordinate nickel in [Ni2L(py)4] paramagnetic, concomitant with significantly elongated bonds. Finally, the respective iron(II) complex was optimized in its all-HS and all-LS state. The derived metrical data are in full agreement with the wealth of experimental data and theoretical experience, which has accumulated for mononuclear congeners.31 That is, all Fe–N bonds experience a strong elongation by ca. 0.2 Å, whereas the Fe–O bond elongation is significantly smaller.
Additional calibration with experimental data could be done in the case of [Ni2L(py)4]. A suitable crystal of [Ni2L(py)4] was obtained from a slow evaporation setup from pyridine solution. The crystallographic data was collected at 180 K; details are given in Table S1.† The complex crystallizes in the tetragonal system in the space group P42 with the asymmetric unit consisting of just one complex molecule (ill-defined electron density points to additional non-bonded solvent molecules, very probably pyridine). The assigned asymmetric unit is given in Fig. 1.
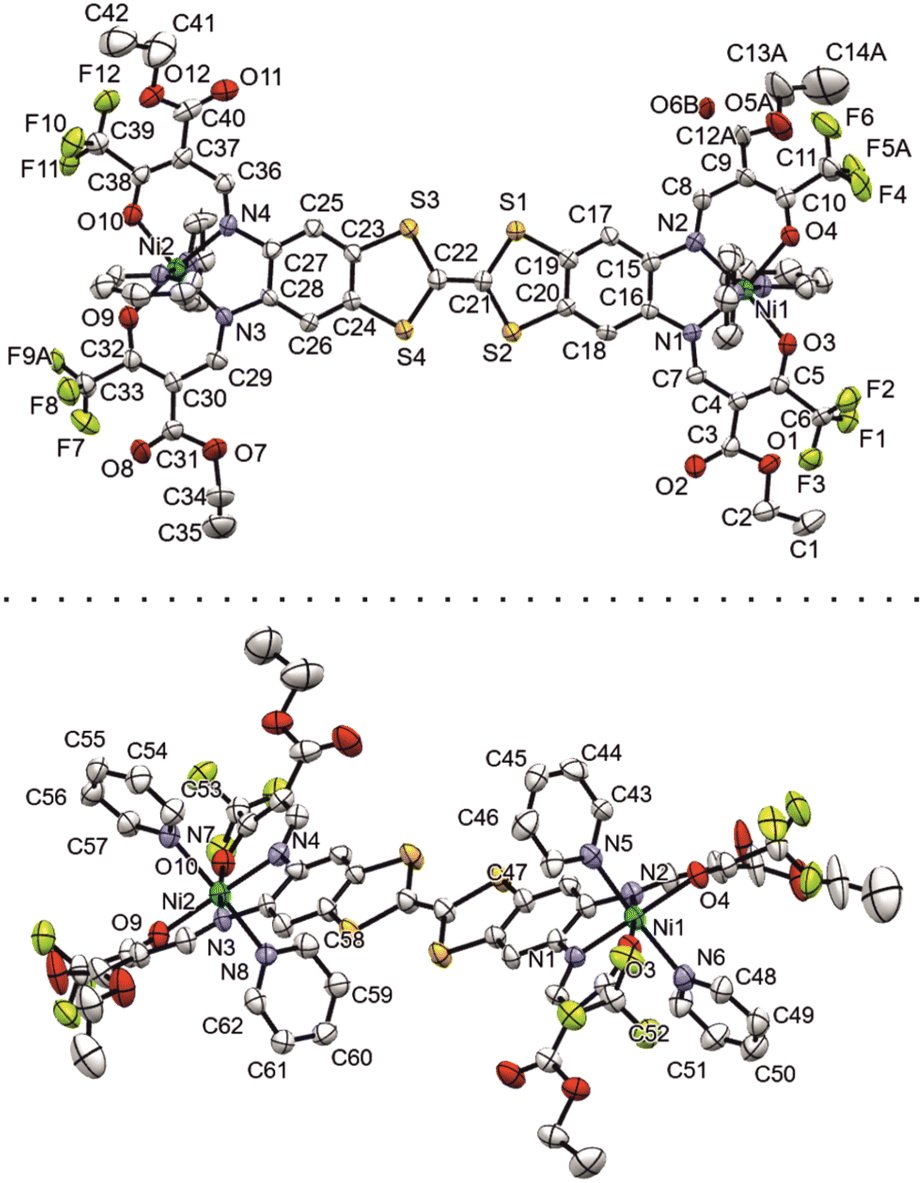 | ||
| Fig. 1 Numbered ORTEP drawing of the asymmetric unit of [Ni2L(py)4], viewed from two different angles. Ellipsoids drawn at 40% probability. | ||
Important bond lengths and angles are given in Table S2.† The nickel(II) centers are in octahedral coordination with the ligand and two axially coordinated pyridine molecules. The bond angles and bond lengths at the nickel centers suggest a slight distortion of the ideal octahedral coordination. As is commonly observed for N2O2 Schiff base-like ligands, the N–Ni bond lengths to the N2O2 chelate are shorter than the N–Ni bond lengths to the axially ligated pyridine. The bridging C–C bond within the TTF-moiety shows a typical double bond length of 1.345(11) Å, confirming the neutral oxidation state of the TTF-moiety. The plains of the axial pyridine molecules are tilted by 90° at both centers. The strand like arrangement of complex molecules within the crystal structure is supported by strong intermolecular interactions (Table S3†) between the terminal CF3 groups, with non-bonded F⋯F distances of 2.867(8) Å, 2.839(7) Å and 2.784(9) Å (Fig. 2, top). The neighboring complex strands are alternatingly rotated by 67°, allowing for aromatic interactions between the ligated pyridine and the TTF-benzene-moieties of neighboring complexes (Fig. 2, bottom). These interactions do not give parallel face-centered stacking, but rather parallel offset. The parameters regarding the aromatic ring interactions are given in Table S4.†
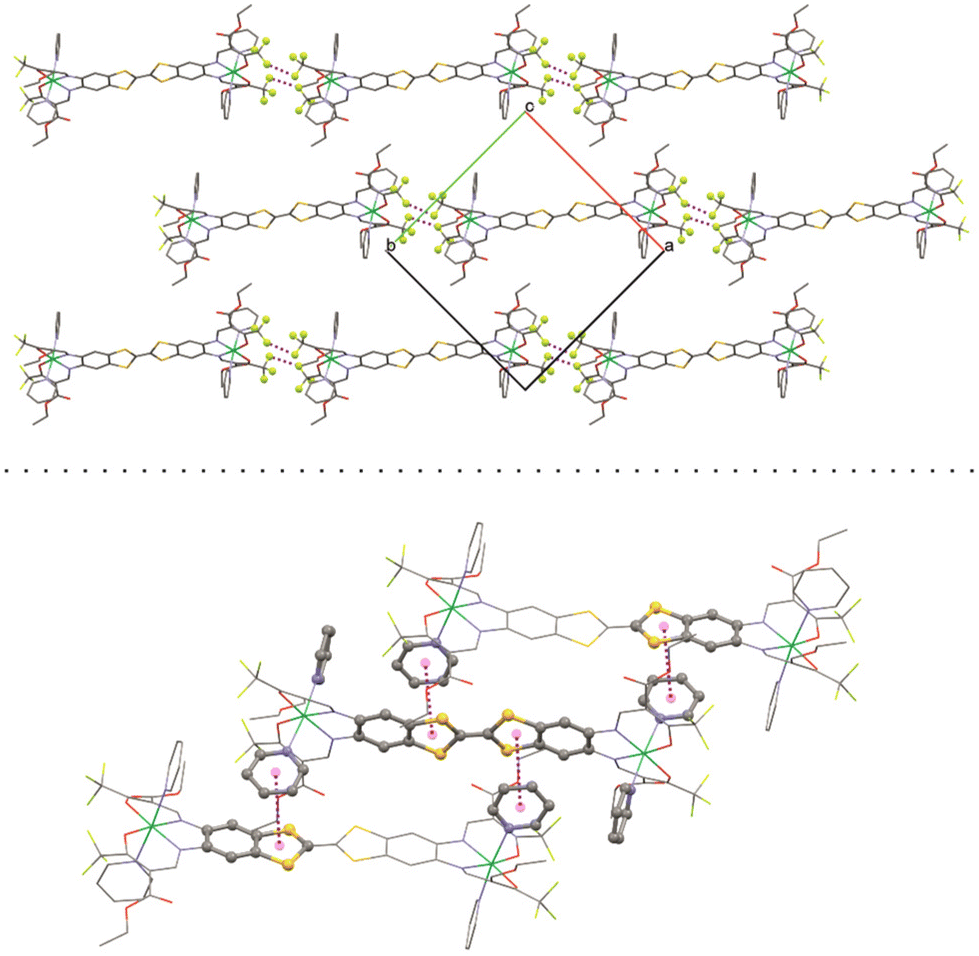 | ||
| Fig. 2 Top: short contact F–F interactions and bottom: aromatic interactions in the crystal structure of [Ni2L(py)4]. | ||
Frontier orbitals and optical properties
TTF-moieties commonly provide potent donor sites. Accordingly, the mononucleating N2O2 ligand with terminal TTF (H2LREF) had revealed an energetically well-isolated HOMO, which was fully localized on the TTF unit,14 whereas the LUMO was localized on the N2O2 chelate. Interestingly, H4L gives an overall similar pattern. Shown in Fig. 4 is the frontier orbital pattern of the ligand H4L (plots of the complexes are shown in Fig. S6 and S7, ESI†). For sake of comparison, the frontier MO energies of H2LREF are re-plotted from ref. 14As we had seen in the mononucleating congener, also the isolated HOMO of the binucleating ligand is localized on the TTF moiety, which is now bridging two chelate units. In more quantitative terms, the bridging function of TTF serves to stabilize the HOMO by ca. 200 meV; clearly also the introduction of CF3 groups will contribute to the stabilization, which shines up as a significant anodic shift of the TTF-waves in CV experiments (see below). More evident is the influence of CF3 in the energies of the unoccupied MOs: the two (N2O2)CF3 chelate sites provide a set of four energetically closely-spaced accepting MOs (LUMO–LUMO+3). Each of them is sharply stabilized by >0.4 eV with respect to the (N2O2)CH3 chelate. The correlated shift of HOMO and LUMO energy makes us expect largely conserved optical properties of the mononucleating and the binucleating ligands.
The UV-Vis absorption spectra of H4L, [Ni2L], [Cu2L] and [Fe2L(py)4] were recorded in diluted DCM solution at ambient temperature and are given in Fig. 3 (c = 1.0 × 10−5M, [Cu2L] was not fully soluble, so that its actual concentration was lower). The ligand H4L retains its yellow color upon dissolution. The UV-spectra show two overlapping, strong absorption bands at λ = 327 and 402 nm. In previous studies on TTF-containing systems, a similar pattern has been observed by others and by us.10,14 The transitions had been previously assigned as an intraligand charge-transfer (ILCT) between the TTF-unit and the ligands’ benzene rings.10 DFT modeling of the ligand spectrum likewise associates these strong bands with CT from the TTF unit and emphasizes the conjugated chelate rings as the most potent acceptor sites (vertical arrows in Fig. 4).
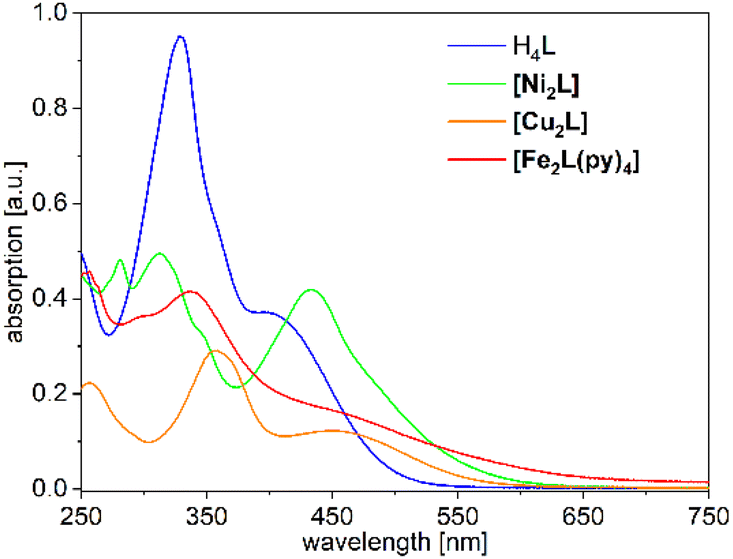 | ||
| Fig. 3 UV-Vis absorption spectra of H4L, [Ni2L], [Cu2L] and [Fe2L(py)4] in DCM (c = 1.0 × 10−5Mfor ligand, Fe and Ni but <1.0 × 10−5 M for Cu). | ||
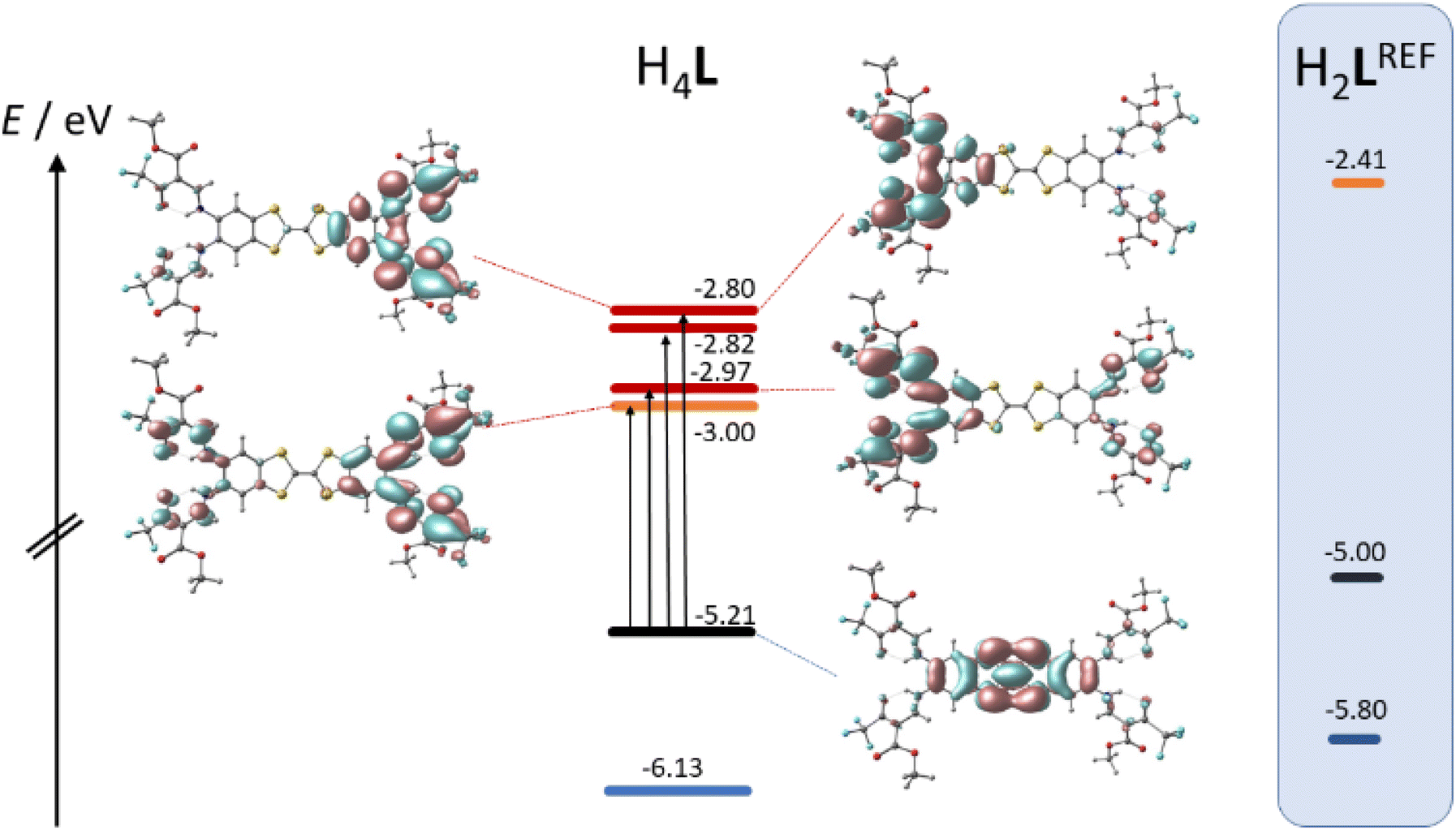 | ||
| Fig. 4 Kohn–Sham frontier MO energy diagram and orbital plots of the binucleating ligand H4L; vertical arrows denote dominant orbital parentage of Vis-transitions at low energy; for sake of comparison, MO energy levels of a related mononucleating ligand H2LREF are re-plotted from ref. 8 | ||
Dissolution of the metal complexes conserves the bulk color as well. In the case of [Cu2L], the overall envelope of the two absorption bands is retained but red-shifted to λ = 357 and 449 nm. The intensity of these leading absorption bands is significantly lowered, an observation which appears to be common to all our complexes. The leading near-UV and Vis transitions of the metal complexes still largely refer to CT from TTF to the chelate. However, the open-shell metal ions obviously offer additional excitation paths, which include the d shell. These differences become most evident from spectral deconvolutions into Gaussian components, which is shown and further discussed in the ESI (Fig. S9†).
The overall broadened appearance of the absorption spectrum of [Fe2L(py)4] can be referred to several additional bands of moderate intensity. The latter are the origin of the low intensity tailing throughout the entire visible spectral range. These bands are typical for iron(II) complexes in similar coordination environment, independent of the presence or absence of TTF-functions.4,14
Magnetic properties
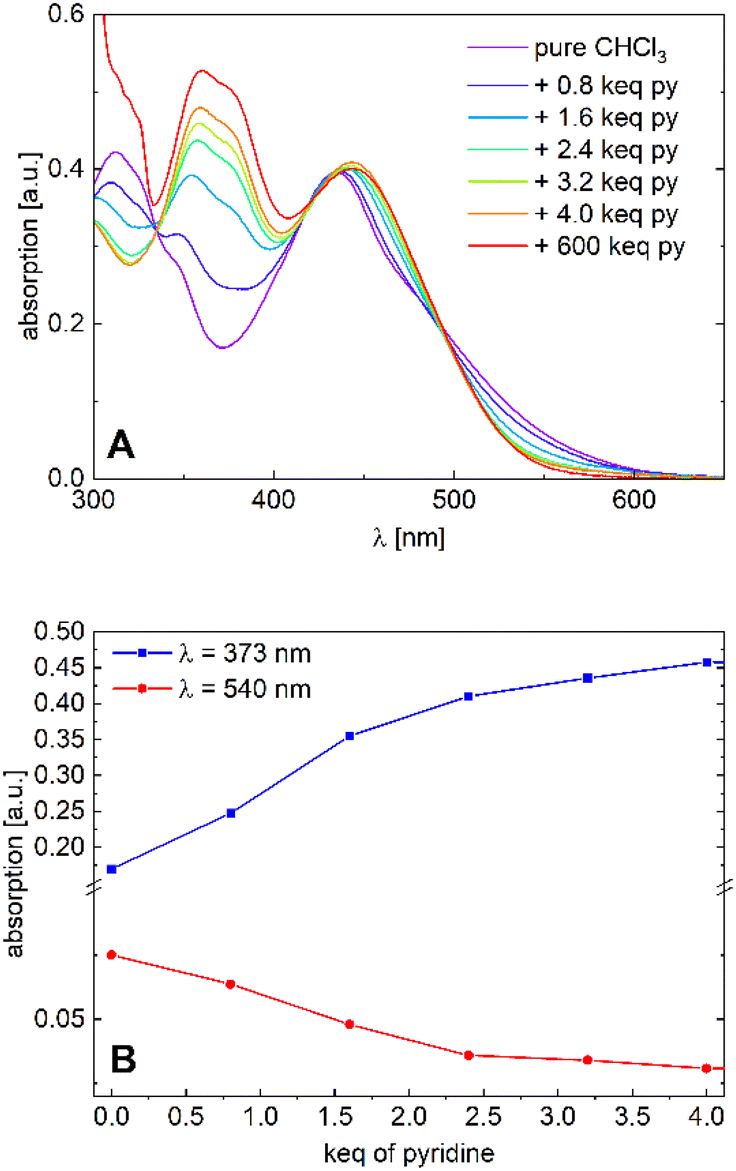 | ||
| Fig. 5 (A) Absorption spectra of [Ni2L] in varying chloroform/pyridine ratios (c = 1.0 × 10−5M) and (B) absorption plots at λ = 373 nm and λ = 530 nm. | ||
We tend to associate the changes in the near-UV range with the in-growth of five- or six-coordinate nickel(II). However, we note that the variation of the Vis absorption bands is rather subtle. In particular, much more pronounced CISSS-related color and spectral changes were observed in a similar mononuclear nickel(II) system.32,33 To follow the spectral evolution, the absorption at the characteristic wavelengths λ = 373 nm and λ = 530 nm was plotted against pyridine concentration. Although a clear distinction between the five- or six-coordinate species does not seem feasible due to spectral overlap, it is safe to assume that at higher pyridine concentrations, six-coordinate nickel(II) is the dominant species. The isosbestic points at λ = 335, 418 and 495 nm, as well as the spectral evolution led to the assumption of a gradual transition from the four-coordinate to the six-coordinate species taking place, with both nickel centers acting as individuals. The complex's high sensitivity toward CISSS (1 keq ≙ 10 mM) can be attributed to the electron withdrawing CF3 groups, which inductively increase the Lewis acidity of the nickel centers.
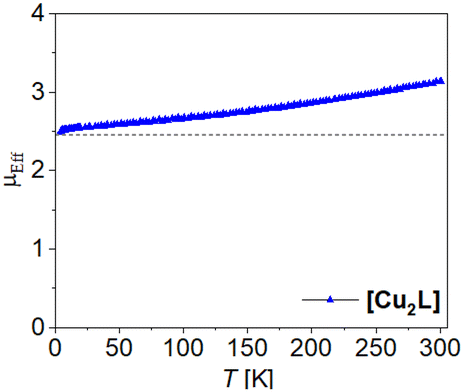 | ||
| Fig. 6 Temperature dependence of μeff of [Cu2L]. | ||
Analogue to this, we expect weak to no coupling between the iron centres in [Fe2L(py)4] However, weak coupling does not necessarily imply the absence of steps during a spin transition. Iron(II) centres in the field of Jäger-type ligands are generally prone to thermal spin crossover. That is, the compounds’ magnetization may vary with temperature in specific ways, typically yielding LS (low spin, S = 0) species at low temperature and HS (high spin, S = 2) species at elevated temperature.34 The emergence of transition steps depends on the relative stability of the mixed HS–LS compared to the stability of a 50:50 mixture of HS and LS states.35 Therefore, the spin state switching behavior of dinuclear complexes attracted the interest of several groups, not only for spin crossover, but also for valence-tautomeric systems.36
Previous NMR studies on binuclear (N2O2)2 variations of the Jäger motif with short, conjugated spacers agree with this notion. It had seen rather uncoupled, individual SCO events at T1/2 ≈ 200 K.37 Different from the previous SQUID magnetometry studies in the solid-state, which found no or only low degrees of SCO completeness in the short-bridge systems,38–41[Fe2L(py)4] and some derivatives do undergo SCO with >80% completeness.
The magnetic properties of the binuclear compounds [Fe2L(py)4], [Fe2L(dmap)4] and [Fe2L(bpee)2]·1 Tol were studied via SQUID magnetometry in a temperature range of 400–4 K. The resulting χMT vs. T plots of the iron(II) complexes with different starting temperatures are given in Fig. 7. At room temperature [Fe2L(dmap)4] (Fig. 7A) shows a χMT value of 6.30 cm3 K mol−1, which is in the typical range for binuclear iron(II) complexes in the HS state.38–41 The χMT products of both, the coordination polymer [Fe2L(bpee)2]·1 Tol (Fig. 7B) and the binuclear complex [Fe2L(py)4] (Fig. 7C) at room temperature are significantly lower (χMT ≈ 4.0 cm3 K mol−1). This deviation can be safely associated with a significant percentage of iron centres remaining in the LS state at this temperature. At 400 K, all iron samples showed χMT values between 6.45 and 6.95 cm3 K mol−1, which can be attributed to all iron(II) centres being in the HS state.
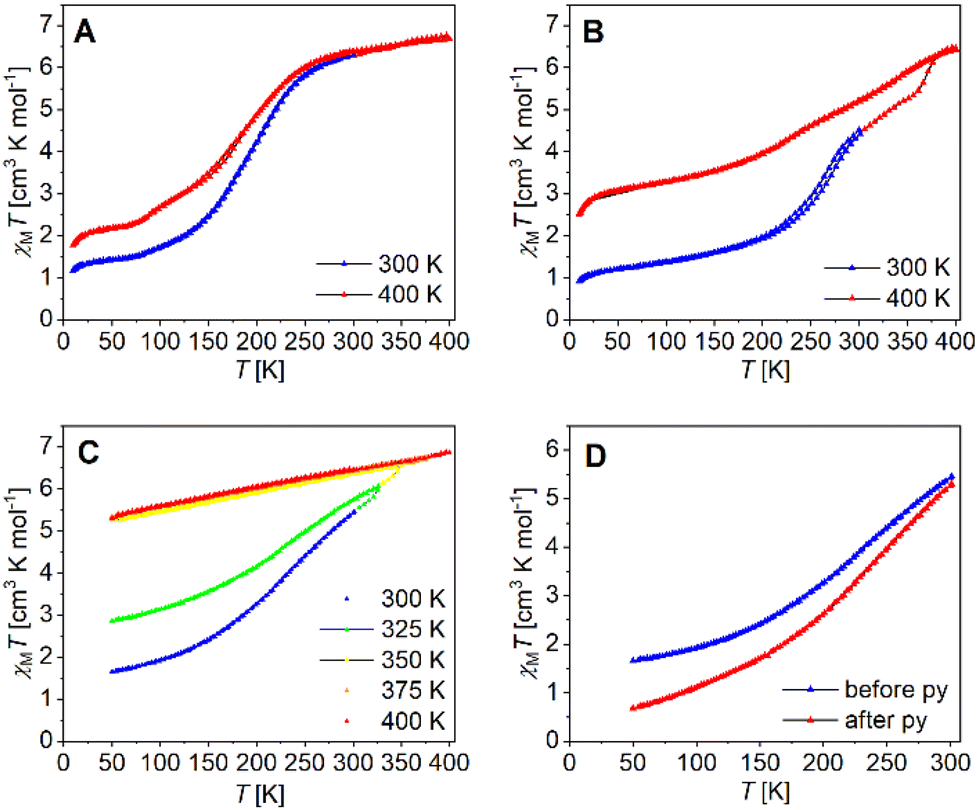 | ||
| Fig. 7 Temperature dependence of χMT of (A) [Fe2L(dmap)4], (B) [Fe2L(bpee)2]·1 Tol, (C) [Fe2L(py)4] in different temperature ranges and (D) [Fe2L(py)4] before and after flushing the sample with pyridine saturated argon. | ||
Upon lowering the temperature, all three compounds undergo gradual spin transitions from the HS to the LS state at different transition temperatures and with varying degrees of completeness. The critical temperature T1/2 where γHS = γLS = 0.50 is the lowest for [Fe2L(dmap)4] with T1/2 = 171 K. The spin transition in [Fe2L(py)4] occurs at higher temperatures with T1/2 = 209 K, whereas in [Fe2L(bpee)2]·1 Tol, T1/2 is already reached at 259 K. All three compounds change their constitution upon heating. That is, iterative cooling of the sample after intermediate heating to 400 K fails to reproduce the initial magnetization curves in all cases, yielding more and more incomplete SCO. In the case of the coordination polymer [Fe2L(bpee)2]·1 Tol the thermal dissociation of the axial ligands as the source of the thermal effect is very unlikely. Similar observations made in related systems point to the effects of lattice solvent loss. Such changes in sample stoichiometry can greatly reduce spin crossover completeness.42 Interestingly, low-temperature magnetization in the pre-heated sample is very close to values expected for a 1:1 HS/LS combination per formula unit.
Volatile pyridine as an axial ligand is known to be susceptible to thermal agitation. For some similar pyridine ligated iron(II) complexes, ligand loss sets in above 350 K,43 while in one case the χMT vs. T plot is largely conserved even after multiple heating cycles up to 400 K.14 Temperature sweeps on [Fe2L(py)4] suggest a loss of ligated pyridine, altering the iron(II) centre's coordination environment and progressively trapping it in the HS state. Iterative cycles with varying starting temperatures greatly reduce spin transition completeness (Fig. 7C). Thermogravimetric analysis of [Fe2L(py)4] (Fig. S8†) reveals continuous mass loss starting at roughly 360 K, equating roughly 1.2 eq. pyridine at 400 K. It is noted that a substantial reduction of spin transition completeness occurs already after heating the SQUID magnetometer to only 325 K, which leads to believe that probe purging and the maintained vacuum in the SQUID probe chamber during measurement, leads to a loss of pyridine already at much lower temperatures. To validate our thesis, we flushed bulk [Fe2L(py)4] with pyridine-saturated argon for one day before preparing a new sample, resulting in a significantly enhanced completeness of the spin transition (Fig. 7D).
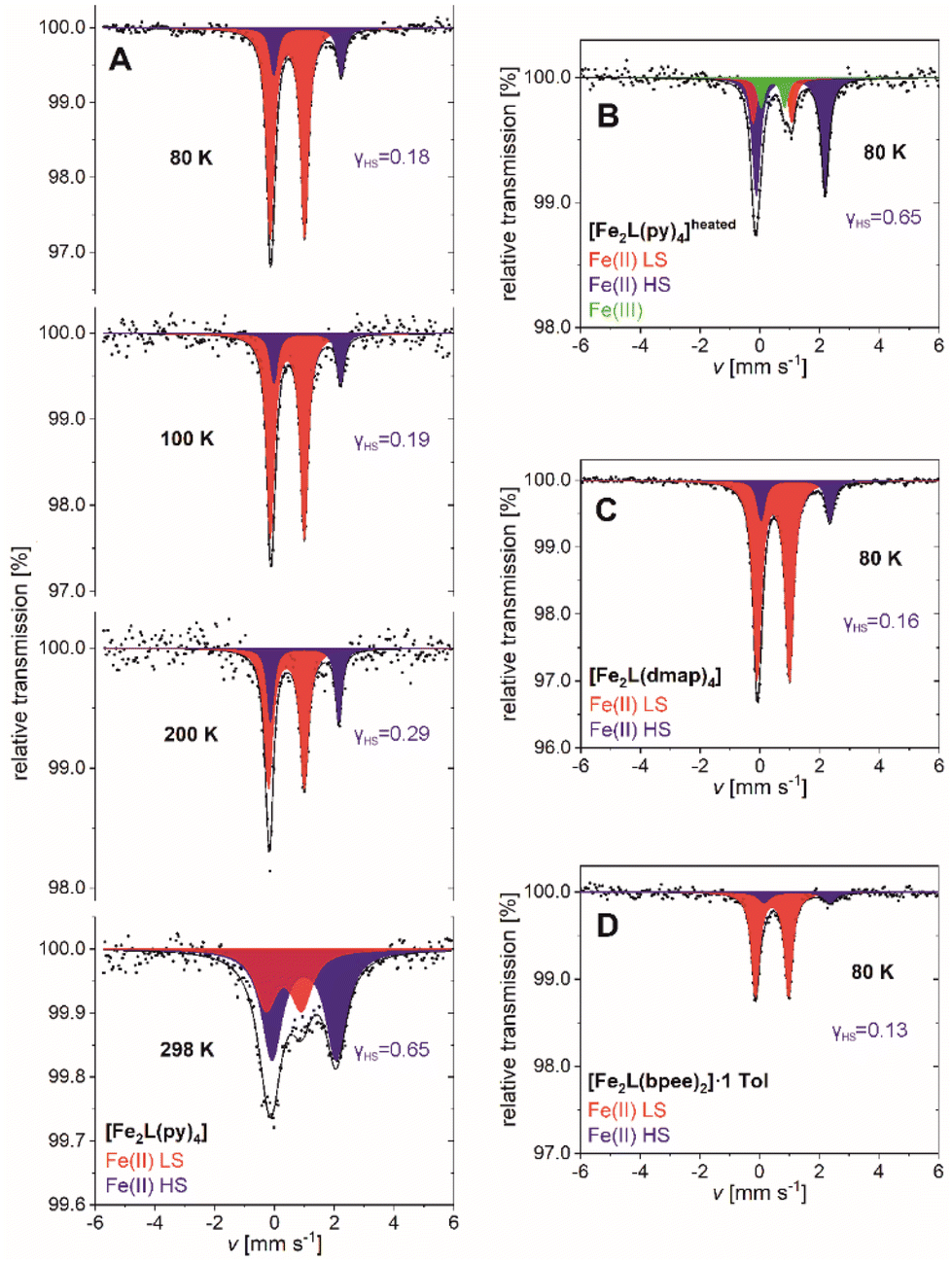 | ||
| Fig. 8 57Fe-Mössbauer spectra of (A) [Fe2L(py)4], (B) [Fe2L(py)4]heated, (C) [Fe2L(dmap)4] and (D) [Fe2L(bpee)2]·1 Tol recorded at various temperatures. | ||
| T [K] | Site | Species | δ [mm s−1] | ΔEQ [mm s−1] | Γ/2 [mm s−1] | Area [%] |
γ
HS
(SQUID) |
|
|---|---|---|---|---|---|---|---|---|
| a Values determined by magnetic susceptibility measurements, with χMT = 6.91 cm3 K mol−1 at 400 K corresponding to γHS = 1.00. | ||||||||
| [Fe2L(py)4] | 298 | D1 | Fe(II) HS | 0.99(4) | 2.15(8) | 0.41(5) | 65(8) | 0.76 |
| D2 | Fe(II) LS | 0.31(7) | 1.19(15) | 0.43(11) | 35(8) | |||
| 200 | D1 | Fe(II) HS | 1.01(4) | 2.29(8) | 0.13(4) | 29(7) | 0.43 | |
| D2 | Fe(II) LS | 0.40(2) | 1.20(5) | 0.17(3) | 71(8) | |||
| 100 | D1 | Fe(II) HS | 1.10(4) | 2.23(8) | 0.14(6) | 19(6) | 0.24 | |
| D2 | Fe(II) LS | 0.430(10) | 1.132(19) | 0.154(14) | 81(5) | |||
| 80 | D1 | Fe(II) HS | 1.104(13) | 2.26(3) | 0.14(2) | 17.7(18) | 0.22 | |
| D2 | Fe(II) LS | 0.435(3) | 1.148(6) | 0.153(4) | 82.3(17) | |||
| [Fe2L(py)4]heated | 80 | D1 | Fe(II) HS | 1.031(13) | 2.31(3) | 0.157(18) | 65(5) | |
| D2 | Fe(II) LS | 0.42(3) | 1.30(8) | 0.14(5) | 22(12) | |||
| D3 | Fe(III) HS | 0.43(4) | 0.78(12) | 0.13(8) | 13(11) | |||
| [Fe2L(dmap)4] | 80 | D1 | Fe(II) HS | 1.189(8) | 2.305(17) | 0.148(13) | 15.5(10) | 0.25 |
| D2 | Fe(II) LS | 0.4455(18) | 1.082(4) | 0.166(26) | 84.5(10) | |||
| [Fe2L(bpee)2]n | 80 | D1 | Fe(II) HS | 1.26(8) | 2.21(16) | 0.23(13) | 13(6) | 0.20 |
| D2 | Fe(II) LS | 0.424(7) | 1.109(13) | 0.156(10) | 87(5) | |||
Spectra of [Fe2L(py)4] can be fitted to two doublets over the full temperature range of 80–298 K. The doublets are attributable to temperature-dependent weights of the iron(II) HS and iron(II) LS site occupation (Fig. 8A). The fit parameters of both doublets are characteristic for iron(II) complexes with Schiff base-like ligands: ΔEQ = 2.26(3) mm s−1 and ΔEQ = 1.148(6) mm s−1 for the quadrupole splitting, δ = 1.104(13) mm s−1 and δ = 0.435(3) mm s−1 for the isomeric shifts of HS and LS, respectively (values at T = 80 K).39,46 At low temperature the HS iron(II) accounts for 17.7(18) % of the sites; higher temperatures see larger HS contributions, in fair quantitative agreement with the aforementioned SQUID data (Table 2). Similar agreement among both techniques holds for the residual HS fractions of [Fe2L(dmap)4] and [Fe2L(bpee)2]·1 Tol at low temperature. The HS molar fraction determined by SQUID magnetometry tends to be slightly higher than the one obtained from Mössbauer spectroscopy but is within the expected error.
SQUID magnetometry had indicated thermal ligand loss in [Fe2L(py)4], leading to increased HS levels of agitated samples. This interpretation is substantiated by Mössbauer spectra. After heating [Fe2L(py)4] to 50 °C under reduced pressure for 3 h, the low temperature Mössbauer spectrum of [Fe2L(py)4]heated retains only traces of the LS species (Fig. 8B). Three doublets are discernible, associated with iron(II) in the HS and LS state, as well as iron(III) in the high spin state. The latter is due to a small portion of iron(II) oxidizing during sample preparation, as the four- and five-coordinate iron(II) centers are even more prone to oxidize then the six-coordinate species. Nevertheless, the molar fraction of LS iron(II) is decreased drastically from γLS = 0.823(18) in the untreated sample to γLS = 0.10(5) after treatment.
Analogous to [Fe2L(py)4], the 80 K Mössbauer spectra for [Fe2L(dmap)4] (Fig. 8C) and [Fe2L(bpee)2]·1 Tol (Fig. 8D) both show two doublets associated with iron(II) in the HS and LS state. HS fractions of γHS = 0.155(10) and γHS = 0.13(6) respectively, confirm the transition of most iron(II) centers to the LS state. These ratios deviate within the scope of expectations from the values obtained through SQUID measurements. The resemblance of determined Mössbauer parameters indicate similar electrochemical environments for all three coordination compounds.
Redox behavior: ligand vs. metal oxidation
The TTF moiety in ligand H4L is a well-known electron donor. TTF and TTF-containing molecules in general can be oxidized to the mono-radical cation TTF˙+ and a dication TTF2+ reversibly and in sequence. To investigate the redox potential of the ligand and its complexes, cyclic voltammograms (CVs) were recorded at room temperature in the non-coordinating solvent DCM. The determined electrochemical parameters of the investigated compounds are given in Table 3. The CV of H4L shows one reversible redox event at E21/2 = 0.321 V which can be associated with the first TTF oxidation to the radical cation TTF˙+ (Fig. 9A).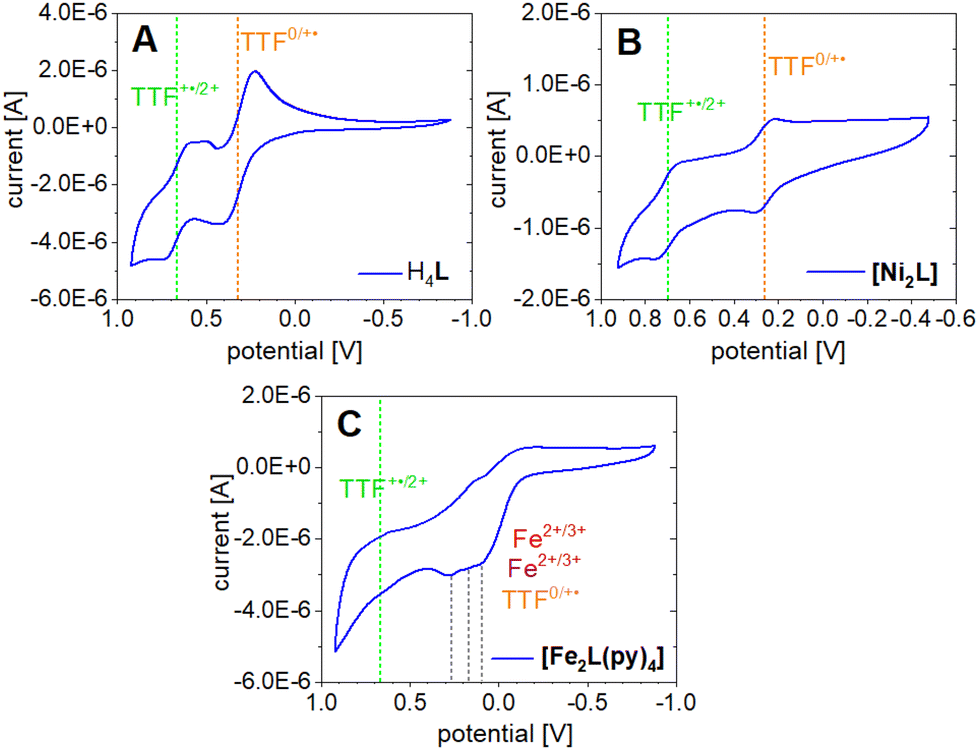 | ||
| Fig. 9 Cyclic voltammograms of H4L (A), [Ni2L] (B) and [Fe2L(py)4] (C) measured in DCM (c = 1.0 × 10−3M) with NBu4PF6 (c = 0.1 M) as electrolyte, on a platinum working and counter electrode with a saturated calomel reference electrode (SCE) and a scan rate of 50 mV s−1. Potentials are reported in reference to Fc/Fc+. | ||
| Sample | Solvent | E 11/2 | E 21/2 | E 31/2 | E 41/2 | E 1ox | E 1red | E 2ox | E 2red | E 3ox | E 3red | E 4ox | E 4red |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Appearance of two reduction peaks. Calculated E1/2 based on the first of two reduction peaks at 0.592 V. | |||||||||||||
| H4L | DCM | 0.665a | 0.321 | 0.737 | 0.592a | 0.414 | 0.228 | ||||||
| 0.504a | |||||||||||||
| [Ni2L] | DCM | 0.697 | 0.263 | 0.740 | 0.653 | 0.296 | 0.229 | ||||||
| [Fe2L(py)4] | DCM | 0.202 | 0.001 | 0.616 | 0.278 | 0.128 | 0.182 | 0.111 | −0.109 | ||||
| ox | DCM | 0.640 | 0.684 | 0.596 | |||||||||
Contrasting our expectations, the second redox event shows splitting of the reduction wave into two separate peaks. This behavior is presumably caused by the formation of the radical cation dimers (TTF˙+) and seems to be solvent dependent, as it is not observed in acetonitrile (Fig. S11†). A half wave potential of E11/2 = 0.665 V is extracted if calculation is based on the first of the two reduction peaks at 0.592 V. While the potential gap between the two redox events lines up well with literature for TTF and its derivatives,47 it is noted that the absolute positions of the waves are shifted anodically with respect to the broad majority of reported data. This is clearly an effect of the electron withdrawing CF3 substituents. For instance, previous CV experiments of a related N2O2 ligand with a terminal TTF unit and a CH3 decorated chelate moiety gave half wave potentials of E1/2 = 0.153 V and E1/2 = 0.368 V.14
The CV of [Ni2L] (Fig. 9B) reveals TTF-centered redox events as well. Two reversible redox events at E21/2 = 0.263 V and E11/2 = 0.697 V correspond to the first and second oxidation of the TTF moiety, respectively. The incorporation of nickel(II) into the ligand leads to a divergence of the TTF's first and second oxidation by almost 100 mV. Predictably, the nickel(II) centers were neither oxidized nor reduced in the investigated potential window. The paucity of metal-borne redox events for nickel can be traced to a conserved frontier MO landscape (Fig. 10), which is dominated by TTF donor orbitals; the same holds true for copper(II) (not soluble enough to be studied with CV). In contrast to nickel(II) and copper(II), the well-purported air-sensitivity of Jäger-type iron(II) complexes made us expect that the iron(II/III) redox couple may fall within the investigated potential window. CVs of mononuclear iron(II) complexes with a TTF moiety in their equatorial ligand had suggested that the iron(II) oxidation takes place at significantly more cathodic potentials than the TTF oxidation.12,14
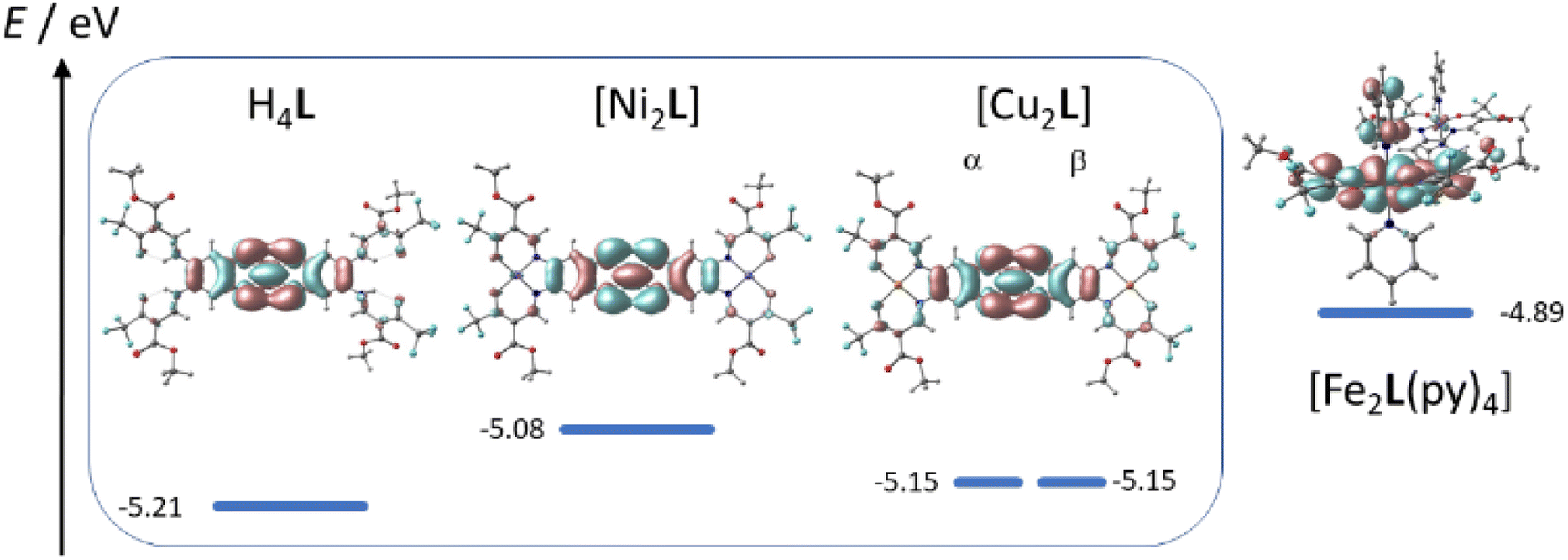 | ||
| Fig. 10 Plot of HOMO energies and orbital parentage. | ||
The idea of redox-active iron(II) is corroborated by the qualitatively different MO order of [Fe2L(py)4] with metal-localized MOs featuring prominently. In fact, close-to-degenerate HOMO and HOMO−1 of [Fe2L(py)4] are largely represented by dxz orbitals of Fe(1) and Fe(2), respectively. TTF-dominated MOs are only found as the HOMO-2 and HOMO-4.
The CV of [Fe2L(py)4] in fact shows four redox events in the studied anodic potential range. Different from the above cases of H4L and [Ni2L] the events are only partially reversible redox processes (Fig. 9C). Additionally, the third and fourth oxidation is convoluted with a decomposition process, inferring with the exact evaluation of its peak potentials. Previous work on mononuclear iron(II) complexes with TTF containing ligands had indicated the presence of largely uncoupled metal-borne and TTF-borne events; that is, the individual half wave potentials could be well matched by reference values taken from single-electrophore models. Comparison with the CVs obtained for the ligand and nickel(II) complex thus suggests that the two redox processes of [Fe2L(py)4] at anodic potentials of ca. 0.67 and 0.20 V still reflect the TTF oxidation. By contrast, the redox processes with oxidation peak potentials at E31/2 = 0.182 V and E41/2 = 0.111 V relate to the iron(II/III) couples. The latter values denote a strong anodic shift of the iron(II/III) redox processes due to the electron withdrawing CF3 groups in the ligand. For comparison, the iron(II/III) wave in the CH3 decorated mononuclear congener was found at E11/2 = −0.41 V.14
In agreement with the above conclusions, step-wise chemical oxidation of H4L and [Fe2L(py)4] clearly addresses qualitatively different molecular sites, that is, the TTF moiety and the iron(II) center(s) of the ligand and the complex, respectively. This difference becomes apparent in UV-Vis absorption spectra of H4L and [Fe2L(py)4] in DCM (c = 2.0 × 10−5M), when recorded before and after addition of aliquots of the strong single-electron oxidant (ox) tris(4-bromophenyl)ammoniumyl tetrakis[3,5-bis(trifluoromethyl)phenyl]borate [N(C6H5Br-4)3]˙+ [{3,5-(CF3)2C6H3}4B]−. This ‘magic blue’ derivative is capable of oxidation of the iron(II) centers and supports the first TTF oxidation event (E1/2(ox) = 0.640 V vs. Fc/Fc+); its absence or presence is readily quantified through the diagnostic absorption band at λ = 750 nm. Formation of TTF-borne oxidation products in turn is evident from the in-growth of TTF˙+-related sub-spectra at λ = 900 nm. Evidently, the first aliquot of ox produces such TTF˙+-related bands in the case of the ligand but fails to produce them in the case of the complex.
The corresponding spectra taken after adding one aliquot ox are shown in Fig. 11. In the case of the ligand H4L the addition of ox leads to new absorption bands at λ = 434 and 462 nm which intensify when a second aliquot ox is added (Fig. S12†). Most diagnostically, the broad absorption band at λ = 880 nm is characteristic for (TTF˙+)2 radical cation dimers, which seem to form in solution.48 Further addition of ox only added the ox's own absorption profile, due to ox's inability to oxidize the TTF unit to the dication (E1/2(ox) = 0.640 V vs. Fc/Fc+). In contrast to the ligand, the first addition of ox to [Fe2L(py)4] fails to add TTF˙+-related features to the spectra. The absorption band at λ = 880 nm only appears after a second addition of ox. This supports our previous assumption of the iron(II) being more easily oxidized than TTF unit. Interestingly, the new absorption band at 880 nm is much broader than the one observed in the ligand and tails to higher wavelengths, suggesting additional absorption bands in the near-infrared range.
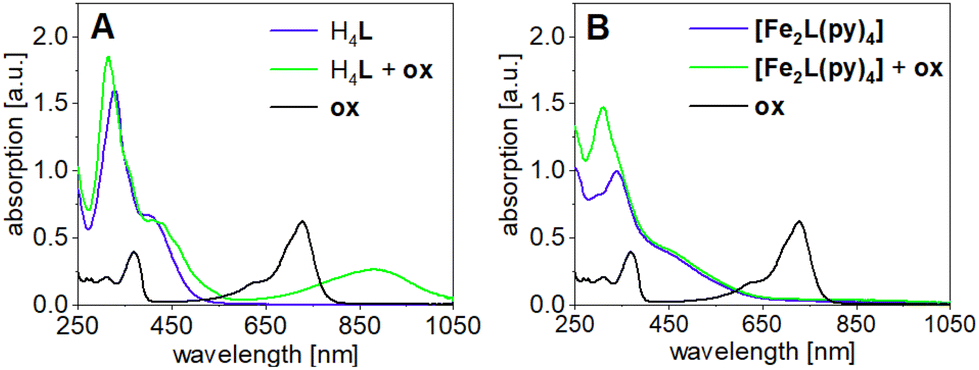 | ||
| Fig. 11 UV-Vis absorption spectra of (A) the ligand H4L and (B) [Fe2L(py)4] in DCM after addition of one aliquot oxidation agent [N(C6H5Br-4)3]˙+ [{3,5-(CF3)2C6H3}4B]−ox; (c = 2.0 × 10−5M). | ||
Conclusions
The combination of redox-active ligand sites with redox-active metal ions gives multiple responsive complexes. Upon stepwise oxidation/reduction a plethora of electronic states can be realized, with tunable interactions between the metal centers and the redox-active site. In this work we introduced a novel binucleating, tetra-anionic ligand L which situates two identical (N2O2)2− chelate sites at both ends of a stiff, π-conjugated backbone. As a redox-active ligand site a TTF unit was incorporated in the π-system. X-ray analysis of the nickel(II) derivative [Ni2L(py)4] revealed the metrics of a reduced TTF unit and bond lengths typical of six-coordinate, paramagnetic nickel(II) complexes.Given the large center-center distance of ca. 16 Å between the coordination sites, consistently seen in the crystal structure and DFT optimizations, direct electronic interaction between the metal centers any binuclear complex [M2L] was expected to be marginal in the case of the reduced ligand. Indeed, this premise was corroborated by all reported complexes of L with copper(II), nickel(II) and iron(II). Vanishing anti-ferromagnetic coupling of the copper(II) ions prevails in [Cu2L] (SQUID) while only very gradual CISSS occurs in solution for the nickel(II) complex [Ni2L] in the presence of pyridine (UV-Vis). Both findings suggest independent metal centers. In keeping with this, all iron(II) coordination compounds [Fe2L(py)4], [Fe2L(dmap)4] and [Fe2L(bpee)2]·1 Tol, exhibited gradual spin crossover behavior (SQUID; 57Fe-Mössbauer).
Electrochemical (cyclic voltammetry) and chemical oxidation (‘magic blue’) of the iron(II) complex [Fe2L(py)4], however, gave evidence of electronic communication of both metal sites across the TTF bridge, after oxidation. Different from the nickel(II) system [Ni2L] and the parent ligand H4L, where two reversible redox events are clearly TTF-localized (diagnostic NIR absorption of TTF˙+), the two iron(II) centers in [Fe2L(py)4] are also prone to oxidation, resulting in four distinguishable, but overlapping oxidation steps in the cyclic voltammograms. The observation of two iron(II/III) waves strongly suggests electronic communication between both metal centers in that the oxidation of the first iron atom impedes the oxidation of the second iron atom. In keeping with the metal-dominated frontier orbital landscape seen in DFT, addition of ‘magic blue’ to a solution of [Fe2L(py)4] did not result in the development of the typical UV-Vis absorption band associated with TTF˙+.
In summary, we have presented a novel TTF containing ligand for binuclear complexes, leading to magnetically independent metal centers for the reduced ligand, while allowing electronic TTF–metal communication, as well as electronic metal–metal interactions if oxidized.
Author contributions
Investigation by C. S. and S. S. (exp.) and G. H. (theor.) Crystallography by P. L. Project conceptualization and supervision by B. W. Original draft written by C. S. and G. H; review and editing by G. H. and B. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. W., G. H. and C. S. acknowledge the German Research Association (DFG) for generous support (Pr.Nr.: 463161096 and 509879467).References
- (a) D. Lorcy, N. Bellec, M. Fourmigué and N. Avarvari, Coord. Chem. Rev., 2009, 253, 1398–1438 CrossRef CAS; (b) H.-Y. Wang, L. Cui, J.-Z. Xie, C. F. Leong, D. M. D'Alessandro and J.-L. Zuo, Coord. Chem. Rev., 2017, 345, 342–361 CrossRef CAS; (c) O. Cador, B. Le Guennic, L. Ouahab and F. Pointillart, Eur. J. Inorg. Chem., 2020, 2020, 148–164 CrossRef CAS.
- (a) Q. Wang, L. Martin, A. J. Blake, P. Day, H. Akutsu and J. D. Wallis, Inorg. Chem., 2016, 55, 8543–8551 CrossRef CAS PubMed; (b) E. Belhadj, A. El-Ghayoury, M. Mazari and M. Sallé, Tetrahedron Lett., 2013, 54, 3051–3054 CrossRef CAS.
- Y.-F. Han, J.-S. Zhang, Y.-J. Lin, J. Dai and G.-X. Jin, J. Organomet. Chem., 2007, 692, 4545–4550 CrossRef CAS.
- S. Schönfeld, G. Hörner, F. W. Heinemann, A. Hofmann, R. Marschall and B. Weber, Z. Anorg. Allg. Chem., 2021, 647, 295–305 CrossRef.
- L. Zappe, S. Schönfeld, G. Hörner, K. A. Zenere, C. F. Leong, C. J. Kepert, D. M. D'Alessandro, B. Weber and S. M. Neville, Chem. Commun., 2020, 56, 10469–10472 RSC.
- S. I. G. Dias, A. I. S. Neves, S. Rabaça, I. C. Santos and M. Almeida, Eur. J. Inorg. Chem., 2008, 2008, 4728–4734 CrossRef.
- H.-Y. Wang, Y. Wu, C. F. Leong, D. M. D'Alessandro and J.-L. Zuo, Inorg. Chem., 2015, 54, 10766–10775 CrossRef CAS PubMed.
- H.-Y. Wang, J.-Y. Ge, C. Hua, C.-Q. Jiao, Y. Wu, C. F. Leong, D. M. D'Alessandro, T. Liu and J.-L. Zuo, Angew. Chem., Int. Ed., 2017, 56, 5465–5470 CrossRef CAS PubMed.
- L. Cui, J.-Y. Ge, C. F. Leong, D. M. D'Alessandro and J.-L. Zuo, Dalton Trans., 2017, 46, 3980–3988 RSC.
- L. K. Keniley, N. Dupont, L. Ray, J. Ding, K. Kovnir, J. M. Hoyt, A. Hauser and M. Shatruk, Inorg. Chem., 2013, 52, 8040–8052 CrossRef CAS PubMed.
- F. Pointillart, X. Liu, M. Kepenekian, B. Le Guennic, S. Golhen, V. Dorcet, T. Roisnel, O. Cador, Z. You, J. Hauser, S. Decurtins, L. Ouahab and S.-X. Liu, Dalton Trans., 2016, 45, 11267–11271 RSC.
- Y.-R. Qiu, L. Cui, P.-Y. Cai, F. Yu, M. Kurmoo, C. F. Leong, D. M. D'Alessandro and J.-L. Zuo, Chem. Sci., 2020, 11, 6229–6235 RSC.
- Y.-R. Qiu, L. Cui, J.-Y. Ge, M. Kurmoo, G. Ma and J. Su, Front. Chem., 2021, 9, 692939 CrossRef CAS PubMed.
- S. Schönfeld, K. Dankhoff, D. Baabe, M.-K. Zaretzke, M. Bröring, K. Schötz, A. Köhler, G. Hörner and B. Weber, Inorg. Chem., 2020, 59, 8320–8333 CrossRef PubMed.
- Q.-Y. Zhu, Y. Liu, Z.-J. Lu, J.-P. Wang, L.-B. Huo, Y.-R. Qin and J. Dai, Synth. Met., 2010, 160, 713–717 CrossRef CAS.
- J. Su, T.-H. Hu, R. Murase, H.-Y. Wang, D. M. D'Alessandro, M. Kurmoo and J.-L. Zuo, Inorg. Chem., 2019, 58, 3698–3706 CrossRef CAS PubMed.
- (a) C. Raby, Ann. Chim. Phys., 1961, 481 CAS; (b) M. Frei, F. Diederich, R. Tremont, T. Rodriguez and L. Echegoyen, Helvetia Chimica Acta, 2006, 89, 2040–2057 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- O. Kahn, Magnetics for Chemists: Molecular Magnetism, VCH, New York, 1993 Search PubMed.
- K. Lagarec and D. G. Rancourt, Mössbauer Spectral Analysis Software for Windows 1.0, Department of Physics, University of Ottawa, Canada, 1998 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. Klamt and G. Schürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- L. Claisen, Ann. Chem., 1897, 1–98 CAS.
- B. Weber and E.-G. Jäger, Eur. J. Inorg. Chem., 2009, 2009, 465–477 CrossRef.
- S. Thies, C. Bornholdt, F. Köhler, F. D. Sönnichsen, C. Näther, F. Tuczek and R. Herges, Chemistry, 2010, 16, 10074–10083 CrossRef CAS PubMed.
- S. Schönfeld, W. Bauer, S. Thallmair, G. Hörner and B. Weber, Z. Anorg. Allg. Chem., 2021, 647, 905–914 CrossRef.
- H. Kurz, K. Schötz, I. Papadopoulos, F. W. Heinemann, H. Maid, D. M. Guldi, A. Köhler, G. Hörner and B. Weber, J. Am. Chem. Soc., 2021, 143, 3466–3480 CrossRef CAS PubMed.
- C. Lochenie, K. G. Wagner, M. Karg and B. Weber, J. Mater. Chem. C, 2015, 3, 7925–7935 RSC.
- (a) I. Šalitroš, N. T. Madhu, R. Boča, J. Pavlik and M. Ruben, Monatsh. Chem., 2009, 140, 695–733 CrossRef; (b) M. A. Halcrow, Spin–Crossover Materials, Wiley, 2013 CrossRef; (c) K. S. Kumar, Y. Bayeh, T. Gebretsadik, F. Elemo, M. Gebrezgiabher, M. Thomas and M. Ruben, Dalton Trans., 2019, 48, 15321–15337 RSC.
- (a) A. Bousseksou, G. Molnár, J. A. Real and K. Tanaka, Coord. Chem. Rev., 2007, 251, 1822–1833 CrossRef CAS; (b) J. Cirera and E. Ruiz, J. Mater. Chem. C, 2015, 3, 7954–7961 RSC; (c) A. B. Gaspar, M. C. Muñoz and J. A. Real, J. Mater. Chem., 2006, 16, 2522–2533 RSC; (d) S. Zein and S. A. Borshch, J. Am. Chem. Soc., 2005, 127, 16197–16201 CrossRef CAS PubMed; (e) K. Nakano, S. Kawata, K. Yoneda, A. Fuyuhiro, T. Yagi, S. Nasu, S. Morimoto and S. Kaizaki, Chem. Commun., 2004, 2892–2893 RSC; (f) V. Ksenofontov, A. B. Gaspar, V. Niel, S. Reiman, J. A. Real and P. Gütlich, Chemistry, 2004, 10, 1291–1298 CrossRef CAS PubMed.
- (a) K. G. Alley, G. Poneti, P. S. D. Robinson, A. Nafady, B. Moubaraki, J. B. Aitken, S. C. Drew, C. Ritchie, B. F. Abrahams, R. K. Hocking, K. S. Murray, A. M. Bond, H. H. Harris, L. Sorace and C. Boskovic, J. Am. Chem. Soc., 2013, 135, 8304–8323 CrossRef CAS PubMed; (b) G. K. Gransbury, B. N. Livesay, J. T. Janetzki, M. A. Hay, R. W. Gable, M. P. Shores, A. Starikova and C. Boskovic, J. Am. Chem. Soc., 2020, 142, 10692–10704 CrossRef CAS PubMed; (c) J. T. Janetzki, M. G. Chegerev, G. K. Gransbury, R. W. Gable, J. K. Clegg, R. J. Mulder, G. N. L. Jameson, A. A. Starikova and C. Boskovic, Inorg. Chem., 2023, 62, 15719–15735 CrossRef CAS PubMed.
- B. Weber and F. A. Walker, Inorg. Chem., 2007, 46, 6794–6803 CrossRef CAS PubMed.
- W. Bauer, S. Schlamp and B. Weber, Chem. Commun., 2012, 48, 10222–10224 RSC.
- B. Weber, E. S. Kaps, J. Obel, K. Achterhold and F. G. Parak, Inorg. Chem., 2008, 47, 10779–10787 CrossRef CAS PubMed.
- W. Bauer and B. Weber, Inorg. Chim. Acta, 2009, 362, 2341–2346 CrossRef CAS.
- B. Weber and E. Kaps, Heteroat. Chem., 2005, 16, 391–397 CrossRef CAS.
- (a) W. Phonsri, D. S. Macedo, C. G. Davies, G. N. L. Jameson, B. Moubaraki and K. S. Murray, Dalton Trans., 2017, 46, 7020–7029 RSC; (b) R. Kulmaczewski, L. J. K. Cook, C. M. Pask, O. Cespedes and M. A. Halcrow, Cryst. Growth Des., 2022, 22, 1960–1971 CrossRef CAS PubMed.
- (a) B. Weber, J. Obel, D. Henner-Vásquez and W. Bauer, Eur. J. Inorg. Chem., 2009, 2009, 5527–5534 CrossRef; (b) S. Schlamp, P. Thoma and B. Weber, Eur. J. Inorg. Chem., 2012, 2012, 2759–2768 CrossRef CAS.
- E. F. Hasty, T. J. Colburn and D. N. Hendrickson, Inorg. Chem., 1973, 12, 2414–2421 CrossRef CAS.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451–465 CAS.
- (a) B. Weber, E. S. Kaps, C. Desplanches, J.-F. Létard, K. Achterhold and F. G. Parak, Eur. J. Inorg. Chem., 2008, 2008, 4891–4898 CrossRef; (b) W. Bauer, T. Pfaffeneder, K. Achterhold and B. Weber, Eur. J. Inorg. Chem., 2011, 2011, 3183–3192 CrossRef CAS; (c) J. Weihermüller, S. Schlamp, W. Milius, F. Puchtler, J. Breu, P. Ramming, S. Hüttner, S. Agarwal, C. Göbel, M. Hund, G. Papastavrou and B. Weber, J. Mater. Chem. C, 2019, 7, 1151–1163 RSC.
- (a) J.-C. Wu, S.-X. Liu, T. D. Keene, A. Neels, V. Mereacre, A. K. Powell and S. Decurtins, Inorg. Chem., 2008, 47, 3452–3459 CrossRef CAS PubMed; (b) S. Oliveira, D. Belo, I. C. Santos, S. Rabaça and M. Almeida, Beilstein J. Org. Chem., 2015, 11, 951–956 CrossRef CAS PubMed.
- (a) J. M. Spruell, A. Coskun, D. C. Friedman, R. S. Forgan, A. A. Sarjeant, A. Trabolsi, A. C. Fahrenbach, G. Barin, W. F. Paxton, S. K. Dey, M. A. Olson, D. Benítez, E. Tkatchouk, M. T. Colvin, R. Carmielli, S. T. Caldwell, G. M. Rosair, S. G. Hewage, F. Duclairoir, J. L. Seymour, A. M. Z. Slawin, W. A. Goddard, M. R. Wasielewski, G. Cooke and J. F. Stoddart, Nat. Chem., 2010, 2, 870–879 CrossRef CAS PubMed; (b) W.-K. Wang, Y.-Y. Chen, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Chem. – Asian J., 2014, 9, 1039–1044 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra; additional structural and analytical data; cyclovoltammograms; crystallographic details. CCDC 2332706. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00479e |
| ‡ Present Address: Fraunhofer Institute for Environmental, Safety and Energy Technology UMSICHT, Institute Branch Sulzbach-Rosenberg, Department Renewable Energy, 92237 Sulzbach Rosenberg. |
| This journal is © The Royal Society of Chemistry 2024 |