
Zinc-doped C4N3/BiOBr S-scheme heterostructured hollow spheres for efficient photocatalytic degradation of tetracycline†
Yaqi
Liu
ab,
Guicheng
Luo
bc,
Yichen
Liu
ab,
Zuozheng
Xu
ab,
Hengxin
Shen
b,
Yuxiang
Sheng
b,
Yuan
Zhu
b,
Shuyi
Wu
*d,
Lizhe
Liu
 *a and
Yun
Shan
*b
*a and
Yun
Shan
*b
aKey Laboratory of Modern Acoustics, MOE, Institute of Acoustics and Collaborative Innovation Center of Advanced Microstructures, National Laboratory of Solid State Microstructures, Nanjing University, Nanjing, 210093, China. E-mail: lzliu@nju.edu.cn
bNanjing Key Laboratory of Advanced Functional Materials, Nanjing Xiaozhuang University, Nanjing, 211171, People's Republic of China. E-mail: yshan@njxzc.edu.cn
cSchool of Chemistry and Chemical Engineering, Nantong University, Nantong, 226019, China
dJiangsu Key Laboratory of Micro and Nano Heat Fluid Flow Technology and Energy Application, School of Physical Science and Technology, Suzhou University of Science and Technology, Suzhou, 215009, People's Republic China. E-mail: wsy@usts.edu.cn
First published on 25th June 2024
Abstract
Photocatalytic degradation of organic pollutants in water is of great significance to the sustainable development of the environment, but encounters limited efficiency when a single compound is used. Thus, there have been enormous efforts to find novel photocatalytic heterostructured composites with high performance. In this work, a novel S-scheme heterostructure is constructed with BiOBr and Zn2+ doped C4N3 (Zn–C4N3) by a solvothermal method for efficient photodegradation of tetracycline (TC), a residual antibiotic difficult to be removed from the aquatic environment. Thanks to Zn2+-doping induced improvement in chemical affinity between Zn–C4N3 and BiOBr, well-formed Zn–C4N3/BiOBr heterostructured hollow spheres are formed. This structure can efficiently suppress fast recombination of photogenerated electron–hole pairs to enhance the photocatalytic activity of BiOBr dramatically. At a room temperature of 25 °C and neutral pH 7, the catalyst can degrade a significant portion of TC pollutants within 30 min under visible light. Also, the Zn–C4N3/BiOBr heterostructure displays good stability after recycling experiments. Free radical capture experiments and ESR tests show that ˙O2− is the main active substance for photocatalytic degradation of TC. This study provides new insights for constructing heterostructures with an intimate interface between the two phases for photocatalytic applications.
1. Introduction
Semiconductor-based heterostructures are considered to be promising photocatalyst candidates due to their ability to separate photogenerated charges efficiently.1 A current attention attracting topic is S-scheme heterojunctions, which are composed of an oxidation type semiconductor and a reduction type semiconductor.2 The formation of S-scheme heterojunctions requires that the conduction band (CB) position and the Fermi level of the reduction photocatalyst (RP) must be higher than those of the oxidation photocatalyst (OP) at the same time.3 When the OP contacts the RP, the electrons in the RP are transferred to the OP through their interfaces and the band bends within the interface region, thus forming a built-in electric field from the RP to the OP.4 In the S-scheme heterojunction, the relatively useless electrons in the CB of the OP and the relatively useless holes in the conduction band (VB) of the RP are recombined and eliminated at the interface after photoexcitation.5 Besides effective separation of photogenerated electrons and holes, they also retain high oxidation and reduction potential which is a great benefit for degradation of aquatic organic pollutants.6 In general, at least one semiconductor in the heterostructure needs to be a good absorber of visible light to take advantage of solar energy efficiently.7 Among several proposed materials to harvest light, BiOBr with a suitable band gap (2.6–2.9 eV) stands out combining prominent optoelectronic properties with excellent stability and low cost.8 Indeed, various morphologies of BiOBr such as nanospheres and nanosheets have been widely used in CO2 reduction,9 photocatalytic H2O2,10 degradation of organic pollutants,11 photocatalytic nitrogen fixation,12etc. In particular, hollow flower-like spherical BiOBr with a large specific surface area is beneficial to the absorption of visible light, which promotes the charge separation and accelerates the catalytic process.13As an oxidation type semiconductor with a low valence band, BiOBr has been connected to many semiconductors with strong reduction ability to construct S-scheme heterostructured photocatalysts. For example, Zan et al.14 prepared CNQDs by an acid etching ultrasonic method and then compounded them on the surface of BiOBr by a hydrothermal method to form a novel 0D/2D CNQDs/BiOBr s-type heterojunction with close interface contact, showing an effective photocatalytic degradation ability for various organic compounds such as rhodamine B (RhB), Congo red (CR) and tetracycline (TC). BiOBr nanosheets were successfully assembled on several layers of the BP surface by liquid-phase ultrasonication combined with solvothermal treatment to construct a layered nanoheterojunction.15 The special structure created favorable conditions for the rapid transfer and separation of photogenerated electron–hole pairs, thereby enhancing the visible light photocatalytic removal of tetracycline (TC) and oxygen evolution activity. Wen et al.2 fabricated S-scheme heterostructured AgBr/BiOBr-OV (ABO) in situ through a simple hydrothermal method, which induced the creation of surface oxygen vacancies and possessed the capacity to photocatalytically degrade sulfonamide antibiotics such as sulfisoxazole (SIZ). Zhang et al.16 constructed a Bi2Sn2O7/BiOBr (BSOB) S-scheme heterojunction by forming a new Bi–O bond through the defect-exposed Bi and O atoms to enhance the built-in electric field, thereby triggering photocatalytic nitrogen reduction efficiently. Most of these pioneering studies, however, focus only on the formation of an intimate interface between BiOBr and the other semiconductor while being unable to control the morphologies of heterostructured composites simultaneously, possibly because of the difficulties in tuning the size and chemical affinity between the two materials.
As a special organic half-metallic material, C4N3 is synthesized by the trimerization reaction of nitrile-functionalized imidazolium ionic liquids, and the scaffold of C4N3 is conjugated by triazine rings connected by carbon atoms.17 The spin polarization of the central carbon atom allows this metal-free half-metal material to possess outstanding charge transfer ability, and the efficient electron–hole separation rate based on singlet-to-triplet conversion contributes to the enhancement of photocatalytic activity.18 Nevertheless, half-metallic C4N3 is seldomly used to construct S-scheme systems largely due to its narrow band gap and poor chemical affinity which makes the direct growth of other semiconductors from the C4N3 surface difficult. Tian et al.19 prepared mesoporous Zn-doped g-C3N4 wrapped CdIn2S4 microsphere nanostructures by a simple hydrothermal method. Zn can partially replace the C element in the g-C3N4 skeleton and enhance the separation of photogenerated electrons and holes. It also plays a key role in exfoliating the stacked layers of g-C3N4 and enhancing the response to light. Deng et al.20 conducted a comprehensive electronic structure analysis through density functional theory (DFT), indicating that the Zn atoms can coordinate with the abundant nitrogen ligands near the vacancies in C4N3 and showing excellent catalytic activity.
Inspired by the above investigations, if a suitable approach is employed to construct intimate heterojunctions between BiOBr and half-metallic C4N3, the obtained nanocomposites can combine the advantages of both components and greatly improve the photocatalytic activity. In this work, Zn2+ ions were introduced into C4N3 by one-step thermal polymerization, and then BiOBr was directly grown on the surface of zinc-doped C4N3 (Zn–C4N3) to form a novel Zn–C4N3/BiOBr S-scheme heterostructure. Owing to Zn2+ doping, the chemical affinity between Zn–C4N3 and BiOBr is effectively improved, endowing the formed heterostructure with an intimate interface. At the same time, the band gap of C4N3 is reduced, thus enhancing the response to light. Moreover, Zn–C4N3 with suitable size and flexibility after facile grinding and ultrasonication allows retaining of the flower-like hollow spherical structure of pristine BiOBr. This special hollow structure endows the photocatalyst with highly exposed reaction sites and elevates photon capture cross-section, greatly boosting the photocatalytic degradation of several common pollutants, such as rhodamine B (RhB), Congo red (CR) and tetracycline (TC). Compared with the original Zn–C4N3 and BiOBr, a significant portion of the TC pollutants can be rapidly decomposed at the Zn–C4N3/BiOBr heterostructure within 30 min.
2. Experimental
2.1. Materials
All the chemicals used in this work were of analytical reagent grade without further treatment. 1-Butyl-3-methylimidazolium chloride ([bmim][Cl], 99%), potassium tricyanomethane (95%), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99%), bismuth nitrate pentahydrate (Bi(NO3)3·5H2O, >99.0%), cetyltrimethylammonium bromide (CTAB, 99%), polyvinylpyrrolidone (PVP, MW = 58000) and ethylene glycol (EG, 99.5%) were purchased from Aladdin Reagent Co., Ltd (Shanghai, China). Ammonium oxalate (AO, 99.99%), 1,4-benzoquinone (BQ, >99.5%), t-butanol (TBA, >99.5%), and L-tryptophan (99.5%) were purchased from Macklin Reagent Co., Ltd (Shanghai, China). Deionized water was used in the experiments.2.2. Preparation of Zn–C4N3
Oily precursor of C4N3 was synthesized following the previously reported procedure.18 0.5 g of the as-prepared oily precursor and 0.01 g of Zn(NO3)2·6H2O were dissolved in 50 mL ethanol under stirring for 4 h at room temperature. The resulting solution was dried overnight at 80 °C to volatilize the ethanol, and the residual mixed oily liquid was then calcined at 450 °C for 1 h in a muffle furnace at a heating rate of 5 C min−1. The obtained Zn-doped C4N3 (Zn–C4N3) samples with half-metallic properties were thoroughly ground and ultrasonicated for subsequent use. The C4N3 samples were prepared via the same procedures, except for the absence of Zn(NO3)2·6H2O in the oily precursor.2.3. Preparation of BiOBr and the Zn–C4N3/BiOBr heterostructure
BiOBr hollow microspheres were fabricated by a solvothermal method using ethylene glycol (EG) as the solvent. Firstly, 1 mmol of Bi(NO3)3·5H2O was dissolved in 5 mL of EG as solution A. 1 mmol of CTAB and 1.125 mg of PVP were dissolved in 20 mL of EG as solution B. After vigorously stirring for 1 h, solution A was slowly added into solution B. Then the mixed solution was transferred into a 50 mL Teflon-lined stainless steel autoclave and heated at 120 °C for 10 h. The final precipitate was washed three times with deionized water and then dried at 60 °C for 10 h. The Zn–C4N3/BiOBr heterostructure was prepared via the same method as BiOBr, except that 0.05 g of Zn–C4N3 was added to solution B under sonication.2.4. Characterization
The morphology and elemental mapping of the samples were characterized by scanning electron microscopy (SEM, Tescan Mira 4) equipped with energy-dispersive X-ray spectroscopy (EDX). Transmission electron microscopy (TEM) images were acquired using an FEI Talos F200X G2 microscope. X-ray diffraction (XRD) patterns were recorded on an X-ray diffractometer (Rigaku, Ultima IV) with Cu Ka radiation (A = 0.15406 nm). Fourier transform infrared (FTIR) spectroscopy was conducted on a PerkinElmer Spectrum Two FT-IR spectrometer. The Raman spectra were acquired on a Raman spectrometer (Horiba HR Evolution) using a 514.5 nm argon ion laser. The surface chemical composition of the samples was determined by X-ray photoelectron spectroscopy (XPS, Thermo Scientific ESCALAB 250Xi) with C ls (284.80 eV) as the reference. The Brunauer–Emmett–Teller (BET) specific surface areas and pore diameter distributions of the samples were measured by nitrogen adsorption–desorption isothermal analysis using a Micromeritics 3Flex at 77 K. The light harvesting capacity was assessed using a Shimadzu UV-3600 Plus spectrometer through diffuse reflectance spectroscopy (DRS). Photoluminescence (PL) emission spectroscopy and transient time-resolved photoluminescence decay measurements were performed on a fluorescence spectrometer (Edinburgh FLS1000, UK).2.5. Photocatalytic tests
The catalytic degradation test was carried out in a 120 mL condensing cup at a constant temperature of 25 °C. The photocatalytic performance was evaluated in a typical photocatalytic system, which contained 100 mL of 25 mg L−1 TC aqueous solution and 20 mg of the photocatalyst. The mixed solution was stirred at room temperature for 30 min to reach adsorption equilibrium in a dark environment. Next, the catalytic activity of the photocatalyst was tested under a 300 W xenon lamp equipped with an AM 1.5 filter. In the process of photocatalytic degradation of TC, 3 mL of solution was filtered with a 0.22 μm filter every 10 min to separate the photocatalyst and the filtrate was detected on a UV-2450 spectrophotometer, especially at the characteristic wavelength of 356 nm.2.6. Active species capturing and ESR tests
Active species capturing experiments were carried out using sacrificial agents AO (ammonium oxalate), BQ (1,4-benzoquinone), TBA (tertiary butyl alcohol) and L-histidine to remove holes (h+), superoxide radicals (˙O2−), hydroxyl radicals (˙OH) and singlet oxygen (1O2), respectively. The electron spin resonance (ESR) was tested on a Bruker EMXplus-6/1 ESR spectrometer, employing 5,5-dimethyl-1-pyrrolidine-N-oxide (DMPO) as a spin trapping reagent under simulated sunlight. Briefly, 10.00 mg of the sample was dispersed in 0.50 mL of deionized water or 0.50 mL of methanol containing 45 μL of DMPO to determine the hydroxyl radical (DMPO–˙OH) or superoxide radical (DMPO–˙O2−).2.7. Photoelectrochemical measurements
10 mg of the sample powder was dispersed in the mixture of 10 μL of 5 wt% Nafion solution and 1 mL of ethanol under ultrasonication for at least 30 min, and then the slurry was evenly dropped on 0.7 × 1.0 cm2 of fluorine-doped tin oxide (FTO) glass. Next, the FTO glass was dried in vacuum at 313 K for 12 h. The electrochemical test was performed on a CHI 660E workstation using 0.1 M Na2SO4 as the electrolyte and a traditional three-electrode configuration, in which the working electrode was sample-coated FTO glass, and a carbon rod and an Ag/AgCl electrode served as the counter and the reference electrode, respectively. Before testing, the electrolytic solution was degassed with argon for 30 min to remove the oxygen. In the photocurrent test, a xenon lamp with an AM 1.5 filter was used as the light source and the power density of light was set to be 100 mW cm−2. Electrochemical impedance spectroscopy (EIS) was conducted on a potentiostat, where the amplitude was set at 10 mV and the frequency range spanned from 0.1 to 100 kHz. The Mott–Schottky plots were computed at frequencies of 800 Hz, 1000 Hz and 1200 Hz, respectively.2.8. Density functional theory
All simulations were performed using the spin-polarized density functional theory (DFT) implemented in the Vienna ab initio simulation package (VASP). The projector augmented wave method is used to describe the ion–electron interaction. The exchange correlation potential is expressed in the Perdew–Burke–Ernzerhof (PBE) form of generalized gradient approximation (GGA). The truncation energy of the plane wave basis set is 450 eV. The energy and force convergence thresholds of the self-consistent field (SCF) iteration are set to 10−5 eV and 0.02 eV Å−1, respectively. In addition, Grimme's DFT-D3 method was also used to consider van der Waals interactions. A 3 × 3 × 1 grid centered at the gamma (Γ) point was used for geometric optimizations and a 5 × 5 × 1 k-point grid for electronic structure calculations. In order to avoid the interaction between two periodic images, a vacuum space of 15 Å in the z-axis direction was used.3. Results and discussion
3.1. Characterization of the Zn–C4N3/BiOBr heterostructure
Fig. 1a and b and Fig. S1 (ESI†) display the representative SEM images of C4N3, Zn–C4N3, BiOBr and the Zn–C4N3/BiOBr heterostructure, respectively. As shown in Fig. S1a (ESI†), pure C4N3 exhibits an aggregated morphology composed of multilayer smooth flakes, which is a typical structural characteristic of bulk C4N3 prepared by the thermal polymerization method.18 Compared with the stacked sheets of C4N3, Zn-doped C4N3 (Zn–C4N3) exhibits an unsmooth plane in Fig. S1b (ESI†). The uneven surface increases its affinity with BiOBr, making it easier for BiOBr to grow in situ on the surface of Zn–C4N3. The BiOBr sample obtained has the morphology of hollow microspheres composed of numerous nanosheets (Fig. 1a). Notably, the distinctive shape of hollow microspheres can give the material a large specific surface area and an enhanced cross-section for scattering photons, which makes it easier to capture and absorb incident irradiation.13 As shown in Fig. 1b, similar hollow microspheres are observed, except for an increase in the size of the nanosheets of the Zn–C4N3/BiOBr heterostructure after Zn–C4N3 loading. Besides, there exist some discernible regions characterized by lighter shades due to the presence of Zn–C4N3. The elemental mapping of the area marked by a yellow box shows a conspicuous outline for the C, N and Zn elements, and Bi, O and Br elements are uniformly distributed. When using Zn-free C4N3 as seeds for the growth of BiOBr, there is an evident impact on the morphology, but no C4N3 can be discerned from the sleek BiOBr surface. Moreover, because C4N3 is too bulky, it is difficult to effectively combine with BiOBr to form heterojunctions. From Fig. S1c and d (ESI†), the difference before and after Zn doping can be clearly seen. These observations suggest that the incorporation of Zn element can lead to an improvement in chemical affinity between BiOBr and C4N3 to make BiOBr grow well from the surface of Zn–C4N3 for the formation of large-sized Zn–C4N3/BiOBr heterostructured nanosheets, which densely pack into a well-defined sphere of about 4 μm diameter (Fig. 1c). As shown in Fig. 1d, the lattice spacing of the (101) crystal plane of BiOBr has a typical value of 0.353 nm,21 indicating that the flexible sheet-like Zn–C4N3 does not affect the growth of BiOBr. More importantly, there exists an intimate interface between well-crystallized BiOBr and amorphous Zn–C4N3,18 which benefits fast charge transfer between them.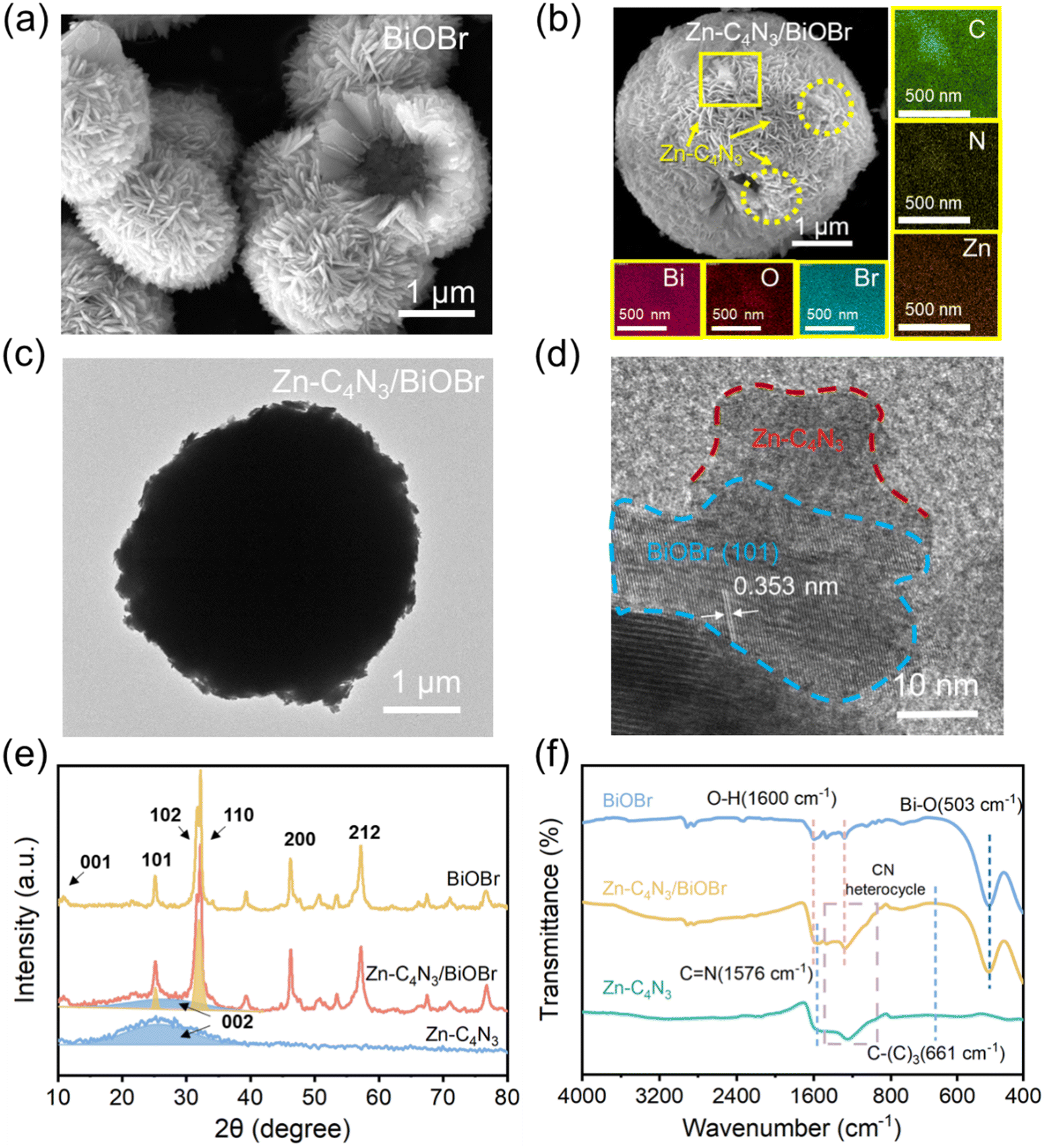 | ||
| Fig. 1 (a) SEM image of BiOBr and (b) SEM image and EDS elemental mapping images of Zn–C4N3/BiOBr. (c) The whole TEM image of Zn–C4N3/BiOBr. (d) HRTEM images of the selected areas of Zn–C4N3/BiOBr. (e) XRD patterns and (f) FTIR spectra of Zn–C4N3, BiOBr and Zn–C4N3/BiOBr. | ||
The powder XRD technique is used to unravel the structure of the as-obtained Zn–C4N3/BiOBr hybrid nanocomposites (Fig. 1e). Zn–C4N3 displays a broad peak at about 26° designated to its (0 0 2) plane, characteristic of an interlayer stacked structure and weak crystallinity in the conjugated aromatic system.17 The addition of Zn(NO3)2·6H2O in the raw material has little effect on the structure of Zn–C4N3, and no relevant diffraction peak is observed. All diffraction peaks of the pure BiOBr sample located at 12.1°, 25.8°, 32.4°, 33.3°, 46.4° and 57.2° correspond to the crystal planes denoted by (0 0 1), (1 0 1), (1 0 2), (1 0 0), (2 0 0) and (2 1 2), respectively (JCPDS No. 09-0393). These distinct peaks corresponding to Zn–C4N3 and BiOBr are all detected in the Zn–C4N3/BiOBr XRD pattern, which indicates the successful preparation of Zn–C4N3/BiOBr hybrid nanocomposites.
Furthermore, the samples are characterized by Raman spectroscopy and the results are shown in Fig. S2 (ESI†). The vibration band at 110 cm−1 in BiOBr is related to the stretching A1g mode of Bi–Br.22 The Raman spectrum of C4N3 can reveal the structural changes before and after Zn doping. In general, the peak located around 1358 cm−1 (D band) belongs to the “disordered” bonds which are related to the breathing mode of the six atom rings, while the peak located around 1560 cm−1 (G band) arises from the symmetric E2g vibration which is caused by vibrational motion of the paired carbon sites.23 The intensity ratio of ID/IG for Zn–C4N3 (1.315) is higher than that of C4N3 (1.125), indicating Zn doping increases the disordering of C4N324 and the zinc element has been successfully doped into the framework of C4N3. The obvious peaks corresponding to Zn–C4N3 and BiOBr are observed in the Raman spectrum of Zn–C4N3/BiOBr, which once again proves the successful preparation of Zn–C4N3/BiOBr hybrid nanocomposites.
The hybridization between Zn–C4N3 and BiOBr is further confirmed by FTIR spectroscopy. As illustrated in Fig. 1f, the pristine BiOBr demonstrates discernible peaks at 503 cm−1 (Bi–O bond25) and 1600 cm−1 (associated with the bending and stretching vibrations of surface-adsorbed water21,26). For Zn–C4N3, the absorption at 661 cm−1 appears due to the stretching vibration of C–(C)3. The bands at 1000–1500 cm−1 and 1576 cm−1 pertain to the CN heterocycle and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations, respectively. These typical characteristic peaks of Zn–C4N3 are all retained after combination. Added to that is the notable absorption peaks at 503 cm−1 and 1600 cm−1 from BiOBr, confirming the successful combination of BiOBr and Zn–C4N3.
N stretching vibrations, respectively. These typical characteristic peaks of Zn–C4N3 are all retained after combination. Added to that is the notable absorption peaks at 503 cm−1 and 1600 cm−1 from BiOBr, confirming the successful combination of BiOBr and Zn–C4N3.
The chemical compositions and valence states of BiOBr, C4N3, Zn–C4N3 and Zn–C4N3/BiOBr are revealed by XPS spectroscopy. Fig. 2a illustrates the C 1s high-resolution spectra of C4N3, Zn–C4N3 and Zn–C4N3/BiOBr. The C 1s spectrum of C4N3 demonstrates three distinct peaks at 284.80 eV, 286.50 eV, and 288.84 eV, which can be ascribed to adventitious carbon, central carbon, and sp2-bonded carbon in triazine rings (N–CN), respectively.27 After Zn doping, the binding energies of center carbon and sp2-bonded carbon (N–CN) shift to lower values of 285.99 eV and 287.26 eV, respectively. Meanwhile, the peaks observed at 399.11 eV and 400.53 eV in the N 1s spectrum of C4N3, originating from the nitrogen atoms present in C–NC and aliphatic CN groups, experience a shift to 398.67 eV and 400.41 eV, respectively (Fig. 2b). As shown in Fig. S3a (ESI†), Zn 2p spectra testify the existence of Zn2+ in Zn–C4N3 with spin–orbit splitting peaks of Zn 2p3/2 and Zn 2p1/2 peaks located at 1022.50 eV and 1045.52 eV, respectively.22 The incorporation of Zn makes the binding energies of C and N peaks shift negatively because the electronegativity of Zn (1.65) is lower than those of C (2.55) and N (3.04). From Raman and XPS analyses, it can be inferred that Zn has been successfully incorporated into the framework of C4N3 and regulates the electronic structure of C4N3. After close contact with BiOBr, the binding energies of center carbon and sp2-bonded carbon (N–CN) shift to lower values of 285.91 eV and 286.68 eV, respectively. Also, two peaks of N 1s experience a negative shift to 398.64 eV and 399.95 eV, respectively. Fig. 2c and d display the high-resolution XPS spectra of Bi 4f and Br 3d, respectively. For pure BiOBr, the binding energies of 159.29 eV and 164.59 eV can be, respectively, identified as Bi 4f7/2 and Bi 4f5/2 with a difference of 5.3 eV,7 confirming the presence of Bi3+. Br 3d5/2 and Br 3d3/2 can be observed at energy levels of 68.33 eV and 69.38 eV, respectively.28 After the formation of the Zn–C4N3/BiOBr heterostructure, the binding energies of Bi 4f and Br 3d both experience an upward shift. In addition, in the high-resolution O 1s spectra of BiOBr and Zn–C4N3/BiOBr, the lattice oxygen (Olatt) and surface oxygen vacancy (Ovac) can be detected at 530.12 eV and 531.44 eV respectively, with a slight increase in binding energy after combination (Fig. S3b, ESI†).29 Note that the n(Ovac)/n(Olatt) ratio of Zn–C4N3/BiOBr is 0.74, obviously higher than 0.67 of BiOBr. Typically, a higher proportion of Ovac tends to enhance the formation of active radicals throughout the photocatalytic process. Moreover, the observed positive shift of Bi 4f, Br 3d and O 1s binding energies and the negative shift of C 1s and N 1s binding energies in Zn–C4N3/BiOBr well verify the strong electronic interaction rather than a physical contact at the interface of Zn–C4N3 and BiOBr in the heterojunction.
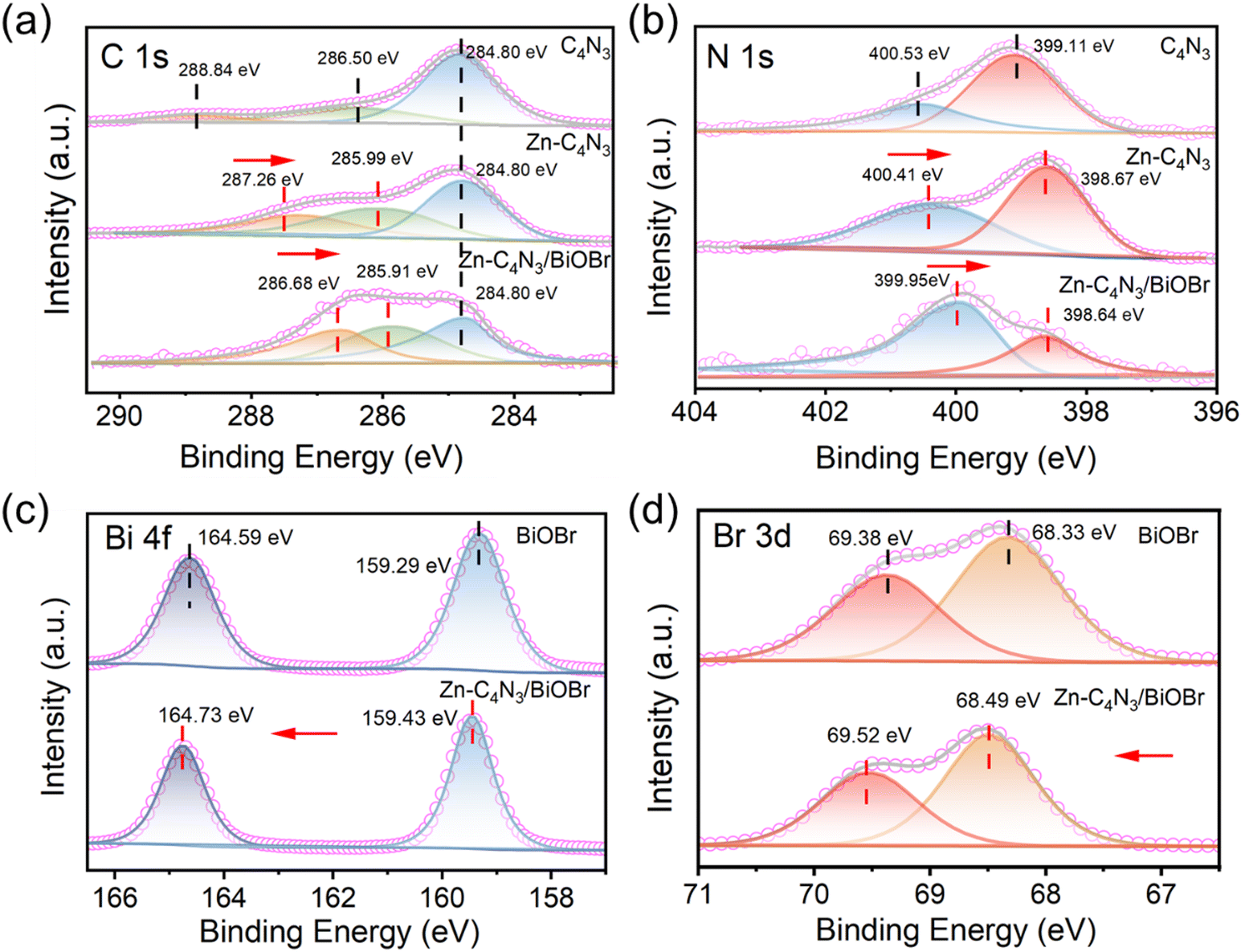 | ||
| Fig. 2 C 1s (a), N 1s (b), Bi 4f (c) and Br 3d (d) XPS spectra of C4N3, Zn–C4N3, BiOBr and Zn–C4N3/BiOBr. | ||
3.2. Light utilization ability and charge transfer features
The Brunauer–Emmett–Teller (BET) specific surface area and pore size distributions of BiOBr and Zn–C4N3/BiOBr are determined using N2 adsorption–desorption isotherms. Based on the IUPAC classification, type IV isotherms and H3-type hysteresis loops of the mesoporous structure are obtained at BiOBr and Zn–C4N3/BiOBr (Fig. 3a), while pure Zn–C4N3 displays no hysteresis loop.29 Notably, Zn–C4N3/BiOBr exhibits a BET specific surface area of 23.90 m2 g−1, which is significantly higher than those of pure BiOBr (12.52 m2 g−1) and pure Zn–C4N3 (1.93 m2 g−1). The pore size distribution curves in Fig. S4 (ESI†) demonstrate two distinct ranges of pore size distribution in BiOBr. Pores with a size smaller than 10 nm are attributed to the compact arrangement of nanoparticles, while pores larger than 10 nm originate from external irregular aggregation. Significantly, there is a decrease in the minimum pore proportion of Zn–C4N3/BiOBr. These observations indicate that the direct growth of BiOBr at Zn–C4N3 helps produce heterostructures with larger size, which self-assemble into nanosheets more loosely.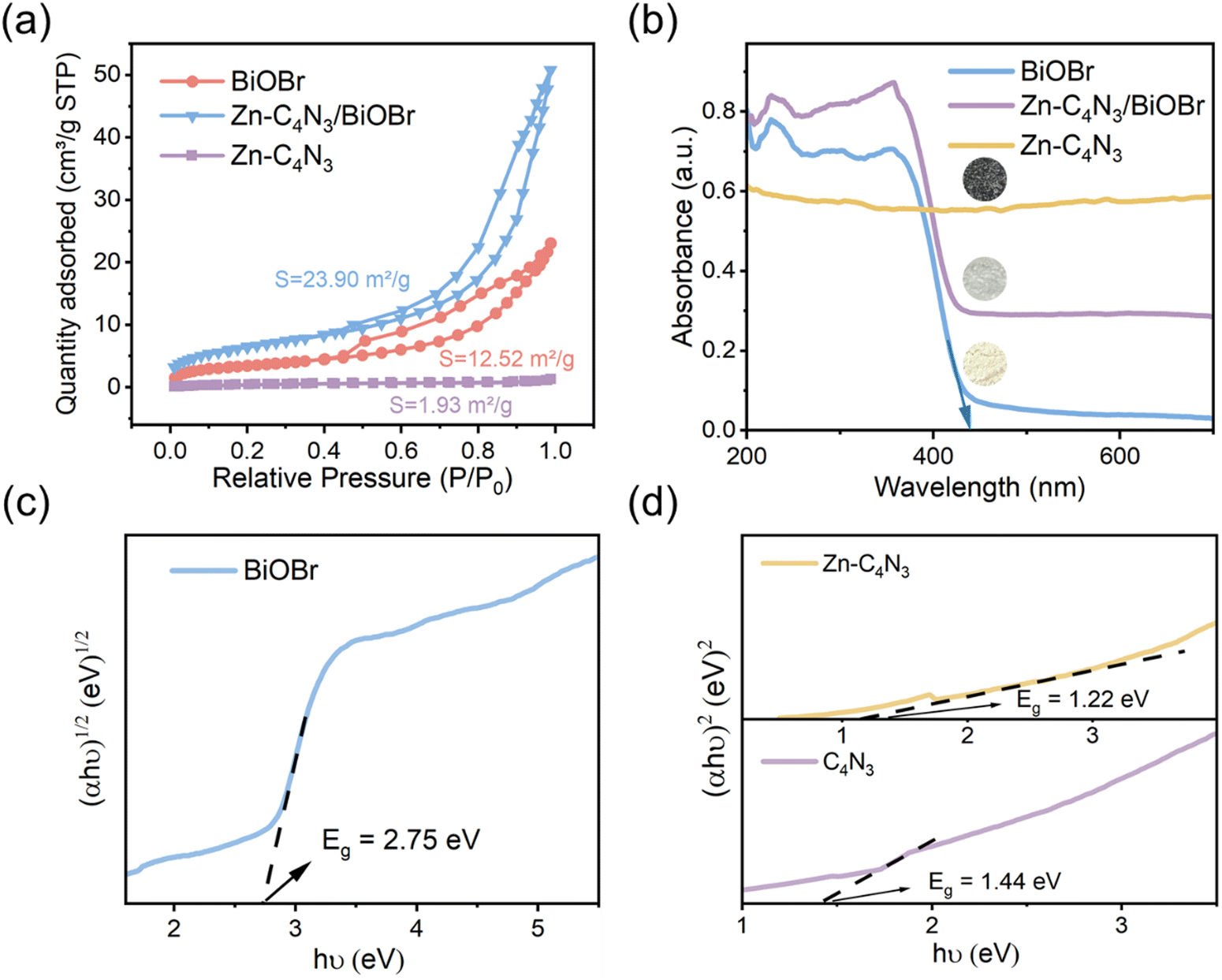 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms of BiOBr and Zn–C4N3/BiOBr. (b) UV-vis absorption spectra of the different catalysts (inset: digital pictures of photocatalysts). (c) and (d) The band gaps of BiOBr, C4N3 and Zn–C4N3. | ||
The optical absorption characteristics of the as-prepared BiOBr, Zn–C4N3, and Zn–C4N3/BiOBr are investigated by UV-vis diffuse reflectance spectroscopy (UV-vis DRS). Pure BiOBr demonstrates significant absorption within the wavelength range of 200 to 420 nm owing to its wide band gap (Fig. 3b). The conspicuous absorption band edge of BiOBr is situated at 441 nm, which corresponds to its pale yellow appearance (inset of Fig. 3b). Compared with C4N3, Zn–C4N3 exhibits an impressive absorption range that stretches to more than two thousand nanometers as a result of its dark appearance (Fig. 3b and Fig. S5, ESI†), covering the entire visible light spectrum. As a result, the heterostructure showcases significantly enhanced light absorption ability in both the visible light region and the ultraviolet region despite a relatively low weight ratio of Zn–C4N3. This improvement can be seen clearly from the distinct alteration in color, confirming the synergistic effect of combining Zn–C4N3 with BiOBr. The excellent light capture ability of Zn–C4N3 in the heterostructure opens up a way to optimize the photocatalytic process, which can absorb a wider spectrum, especially in the visible light range, thus improving the efficiency of the photocatalytic reaction. In addition, the UV-vis DRS spectrum can be used to calculate the band gap energy of BiOBr and Zn–C4N3 according to the formula: αhυ = A (hυ − Eg)n/2, where α, h, υ, A, Eg and n represent the absorption coefficient, Planck constant, light frequency, band gap and transition feature of the semiconductor, respectively (n = 4 for the indirect nature of BiOBr and n = 1 for the direct nature of C4N318,30). As illustrated in Fig. 3c and d, the calculated band gaps for BiOBr, C4N3 and Zn–C4N3 are 2.75 eV, 1.44 eV and 1.22 eV, respectively, which indicates that, due to the addition of Zn element, the band gap of C4N3 is significantly reduced and the light response is enhanced. The narrow band gap of C4N3 is consistent with its half-metallicity feature, contributing to its distinct electronic behavior. Besides, the Mott–Schottky curves of BiOBr and Zn–C4N3 are shown in Fig. S6 (ESI†). The positive slope of the curves suggests that the prepared samples are n-type semiconductors7 and the potential of the conduction band (CB) in n-type semiconductors is proximate to the potential of the flat band (FB).31 The conduction band (CB) positions of BiOBr and Zn–C4N3 are −0.191 eV and −0.679 eV, respectively, in relation to the Ag/AgCl electrode (pH = 7). Meanwhile, according to the conversion formula of the standard hydrogen electrode: 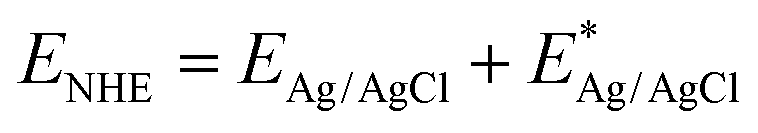 , these positions are 0.006 and −0.482 eV, respectively, in relation to the normal hydrogen electrode (NHE, pH = 7),14 where EAg/AgCl is the experimental potential measured against the Ag/AgCl reference electrode, and
, these positions are 0.006 and −0.482 eV, respectively, in relation to the normal hydrogen electrode (NHE, pH = 7),14 where EAg/AgCl is the experimental potential measured against the Ag/AgCl reference electrode, and  is the standard potential of Ag/AgCl at 298 K (0.197 V). Based on the empirical equation EVB = ECB + Eg, the VB potentials of BiOBr and Zn–C4N3 are 2.756 and 0.738 eV, respectively, where EVB, ECB and Eg are the VB potential, CB potential and the value of band gap, respectively.32
is the standard potential of Ag/AgCl at 298 K (0.197 V). Based on the empirical equation EVB = ECB + Eg, the VB potentials of BiOBr and Zn–C4N3 are 2.756 and 0.738 eV, respectively, where EVB, ECB and Eg are the VB potential, CB potential and the value of band gap, respectively.32
The photogenerated charge carriers in BiOBr can rapidly recombine to emit photons, as shown in Fig. 4a. The strong photoluminescence (PL) emission of BiOBr is composed of two main peaks. One emission band is centered at approximately 450 nm, which can be attributed to its band-to-band emission with energy equivalent to the bandgap of BiOBr (2.75 eV).7 The other peak located at about 570 nm can be attributed to defect emission.33 In contrast, a very weak emission peak is discernible in the PL spectrum of Zn–C4N3, and to some degree it can be considered non-luminescent. As a result, the PL intensity of the Zn–C4N3/BiOBr heterostructure decreases sharply, indicating that the recombination of photogenerated carriers can be effectively inhibited.34 The photogenerated charge transfer dynamics is studied using time-resolved photoluminescence (TRPL) spectroscopy of the ns-level in Fig. 4b. The radiative lifetimes (τ) can be calculated through a biexponential function fitting decay curves (Table S1, ESI†). The fluorescence lifetime of the photogenerated charge carriers in Zn–C4N3/BiOBr, expressed as short (τ1 = 0.62 ns), long (τ2 = 4.43 ns), and average (τav = 3.33 ns), is comparatively shorter than that of pristine BiOBr (τ1 = 1.38 ns, τ2 = 12.06 ns, τav = 7.30 ns). Additionally, the charge transfer rate, which can be determined using the formula k = 1/τav,21 is found to be 0.137 ns−1 for BiOBr and 0.300 ns−1 for Zn–C4N3/BiOBr. The decrease in the fluorescence lifetime of Zn–C4N3/BiOBr indicates that there exists a special electron transfer channel.35–37 The effective charge separation and transfer at the heterojunction interface can reduce the recombination of photogenerated charges, making the photogenerated charges easily captured by the reactants for the subsequent redox reaction.
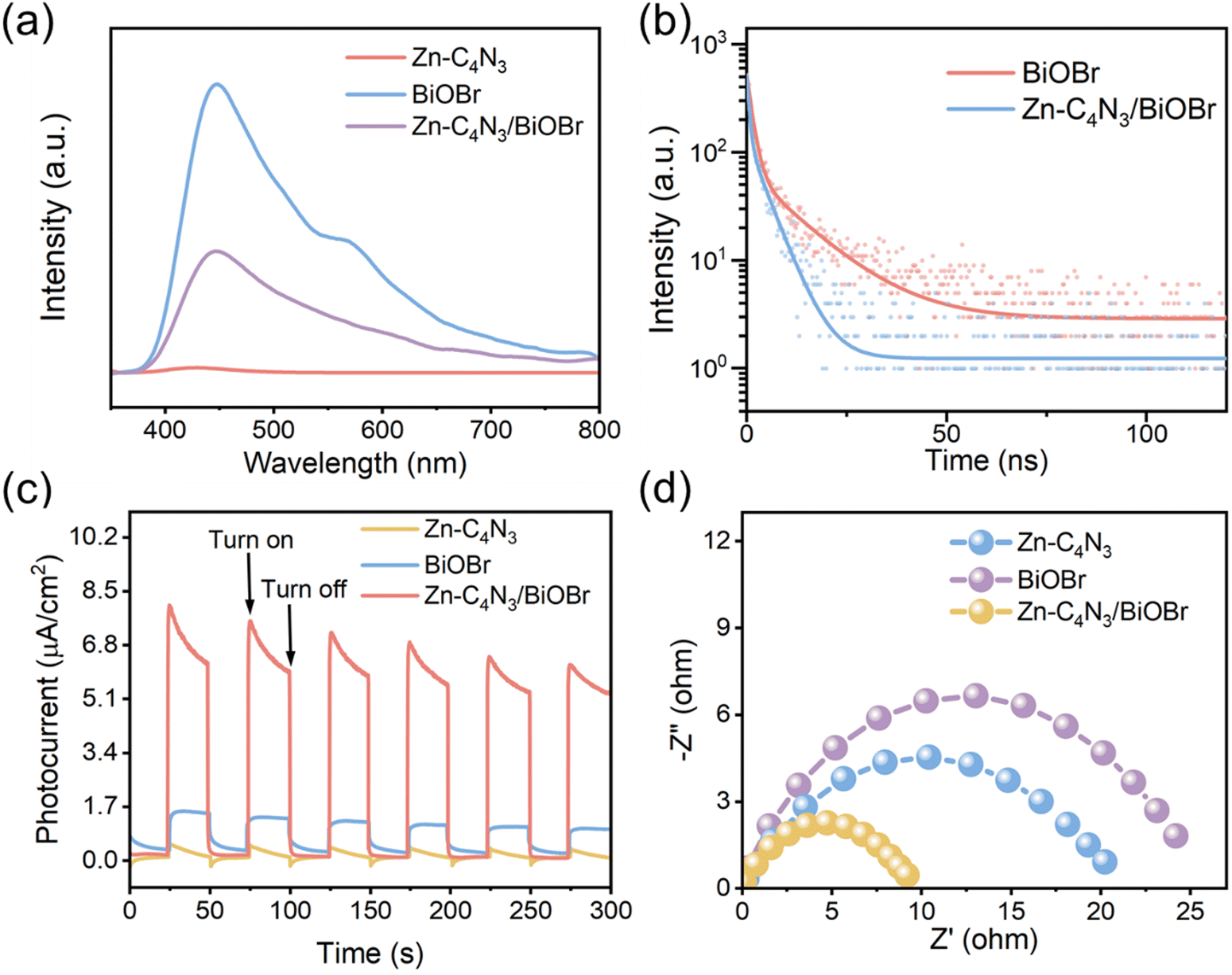 | ||
| Fig. 4 (a) PL spectra (320 nm excitation) of photocatalysts. (b) Time-resolved PL decay curves (monitored at 440 nm) of photocatalysts. (c) Transient photocurrent responses and (d) EIS curves of photocatalysts. | ||
The effective separation of photogenerated electron–holes in the heterostructures is further confirmed by the photocurrent measurements. Fig. 4c illustrates the photocurrent response curves of BiOBr, Zn–C4N3, and Zn–C4N3/BiOBr fabricated on the electrodes by turning the irradiation light on and off for several rounds to collect the I–t curves. Both BiOBr and Zn–C4N3 exhibit a low current intensity upon irradiation due to the lack of an efficient pathway for photogenerated carrier transfer. Impressively, Zn–C4N3/BiOBr shows remarkably improved photocurrent, which is advantageous to photocatalytic degradation. Consistently, the formation of the Zn–C4N3/BiOBr heterostructure greatly reduces the charge transfer resistance, as revealed by the electrochemical impedance spectroscopy (EIS) results in Fig. 4d. This enables fast and effective charge separation, leading to excellent performance in the following photocatalytic degradation.
3.3. Photocatalytic degradation of TC
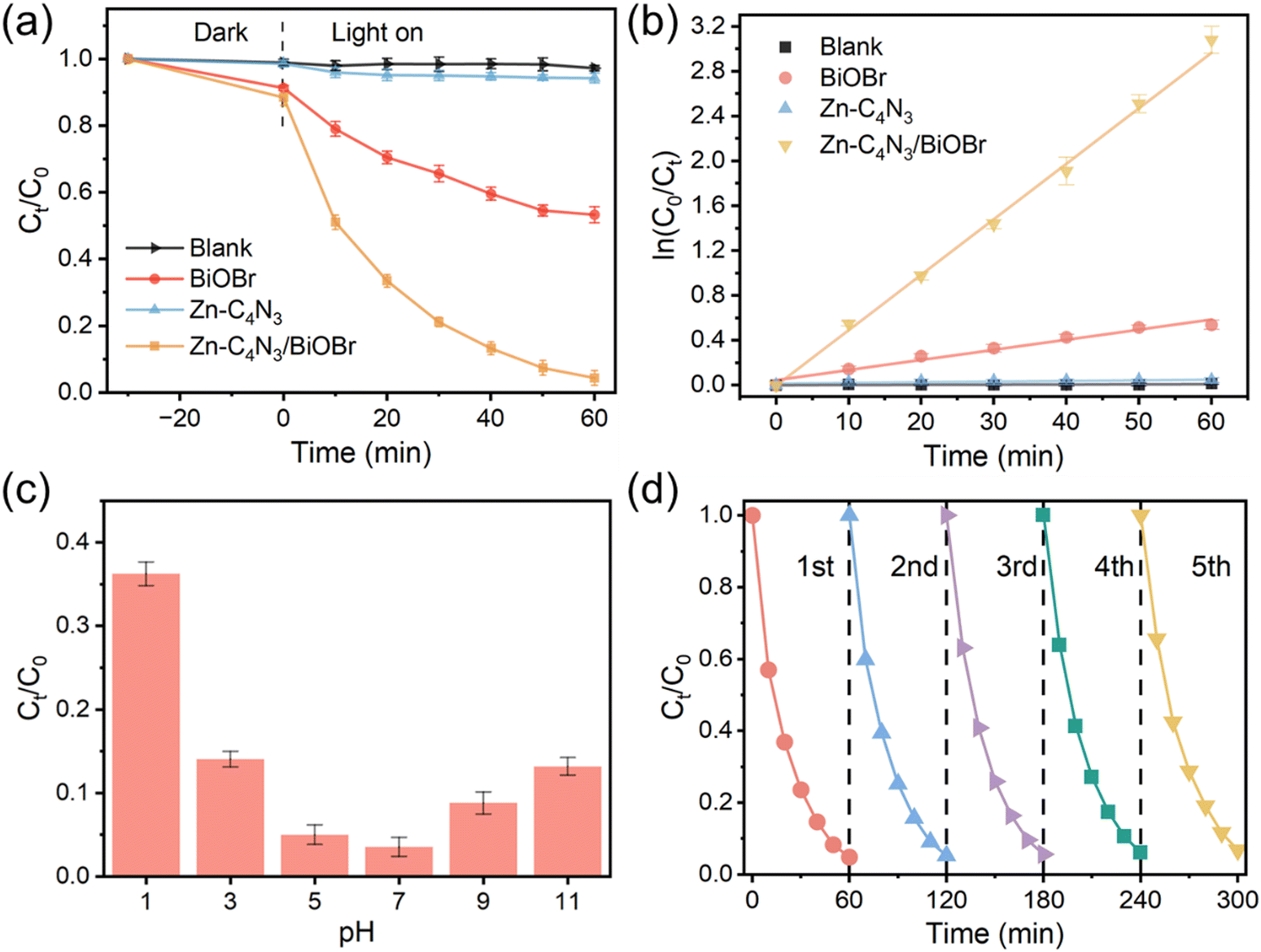 | ||
| Fig. 5 (a) Photodegradation of TC at photocatalysts. (b) Degradation reaction kinetics of TC over photocatalysts. (c) Effect of the initial pH value over Zn–C4N3/BiOBr in degradation of TC. (d) Five recycling runs over Zn–C4N3/BiOBr in degradation of TC. Note that the experiment in Fig. 5a was conducted without pH adjustment. | ||
Due to the variety of pollutants, the ability to degrade other organic pollutants is of great significance for the practical application of Zn–C4N3/BiOBr. Here, the photocatalytic degradation performance of Zn–C4N3/BiOBr on several common pollutants was investigated (Fig. S9a, ESI†). The degradation efficiencies of Congo red (CR), methylene blue (MB) and rhodamine B (RhB) reached 84.45%, 71.71% and 74.57%, respectively, within 60 min light irradiation. These results illustrate the efficient degradation activity of Zn–C4N3/BiOBr for a variety of organic pollutants, which shows its great potential for practical application.
Fig. 5c presents the influence of pH on TC degradation, in which the rate of TC degradation increases with the initial pH rising from 1 to 7 but decreases as the pH further increases from 9 to 11. At low pH, H+ ions participate in trapping of the ˙OH species, thus inhibiting TC degradation.29 Meanwhile, excessively alkaline conditions will also impact its degradation rate. This is likely attributable to the electrostatic repulsion between the charge on the surface of the catalyst and the ions in the solution.39 Therefore, the highest degradation rate of tetracycline occurs in solution close to neutral. The initial unadjusted pH of the TC solution is 6.62. At this pH, TC degradation closely approaches the optimal pH of 7, indicating that Zn–C4N3/BiOBr can be used for practical wastewater treatment without pH adjustment.
At a fixed dosage of the Zn–C4N3/BiOBr photocatalyst, distinct degradation efficiencies were obtained when treating different initial TC concentrations (20–40 mg L−1) (Fig. S9b, ESI†). The highest degradation efficiency is found at a TC concentration of 25 mg L−1. When an appropriate amount of TC solution is adsorbed on the surface of Zn–C4N3/BiOBr, it acts as a mediator for photogenerated electron–hole pairs, hindering their recombination on the surface of the catalyst.40 However, an increased concentration of TC molecules in the reaction solution results in fewer photons entering the reaction solution, thus decreasing the absorption of photons by the Zn–C4N3/BiOBr photocatalyst and directly affecting the degradation rate. Therefore, the optimal concentration of TC is 25 mg L−1 in the following experiments.
The impact of catalyst dosage on the TC degradation rate is also tested with different Zn–C4N3/BiOBr dosages, and the results are shown in Fig. S9c (ESI†). With the dosage of the catalyst increasing from 10 mg to 20 mg, the degradation rate of TC increases, mainly due to the increase of active sites with the increase of catalyst dosage, thus promoting the formation of active radicals. However, further increase to 30 mg will bring about a slight decrease in the degradation rate of TC. This can be attributed to the light scattering caused by excess dosage, resulting in the loss of partial visible light for exciting the photocatalyst.25 Therefore, 20 mg is the best dosage of Zn–C4N3/BiOBr.
Apart from high degradation efficiency, the Zn–C4N3/BiOBr catalyst also has excellent stability (Fig. 5d). After five cycles in the process of degrading TC, Zn–C4N3/BiOBr still maintains high photocatalytic activity. In addition, the morphology of Zn–C4N3/BiOBr remains unchanged and the XRD pattern of Zn–C4N3/BiOBr still exhibits a distinguishable crystal structure after the long-term stability test (Fig. S10, ESI†), indicating that there is no obvious structural deterioration. Moreover, our proposed Zn–C4N3/BiOBr sample is superior to most previously reported catalysts at kinetic rates and degradation efficiency (Table S2, ESI†), exhibiting an obvious advantage in TC degradation.
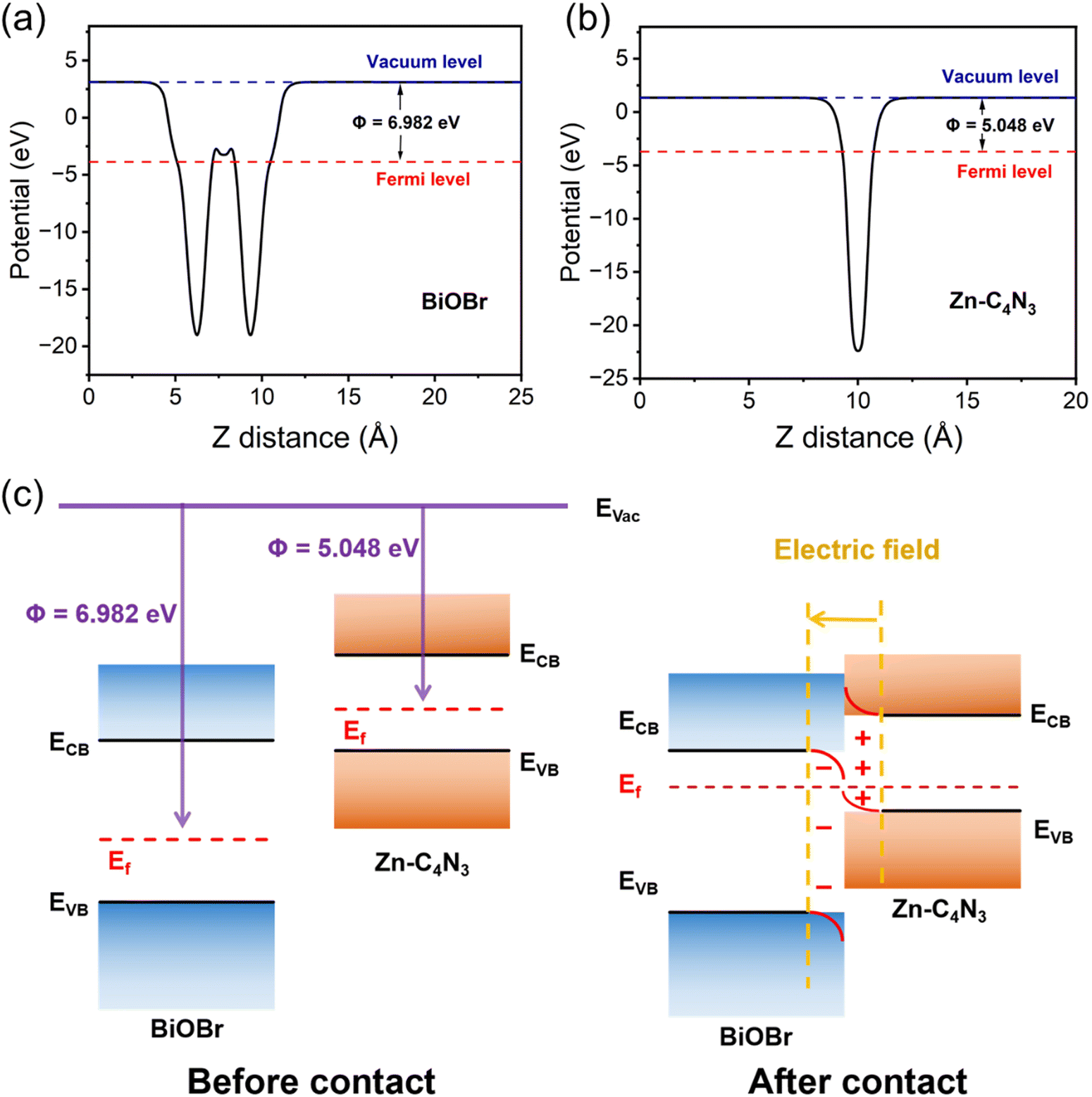 | ||
| Fig. 6 The work function of (a) BiOBr and (b) Zn–C4N3. (c) Schematic illustration of the Zn–C4N3/BiOBr heterostructure: before and after contact in the dark. | ||
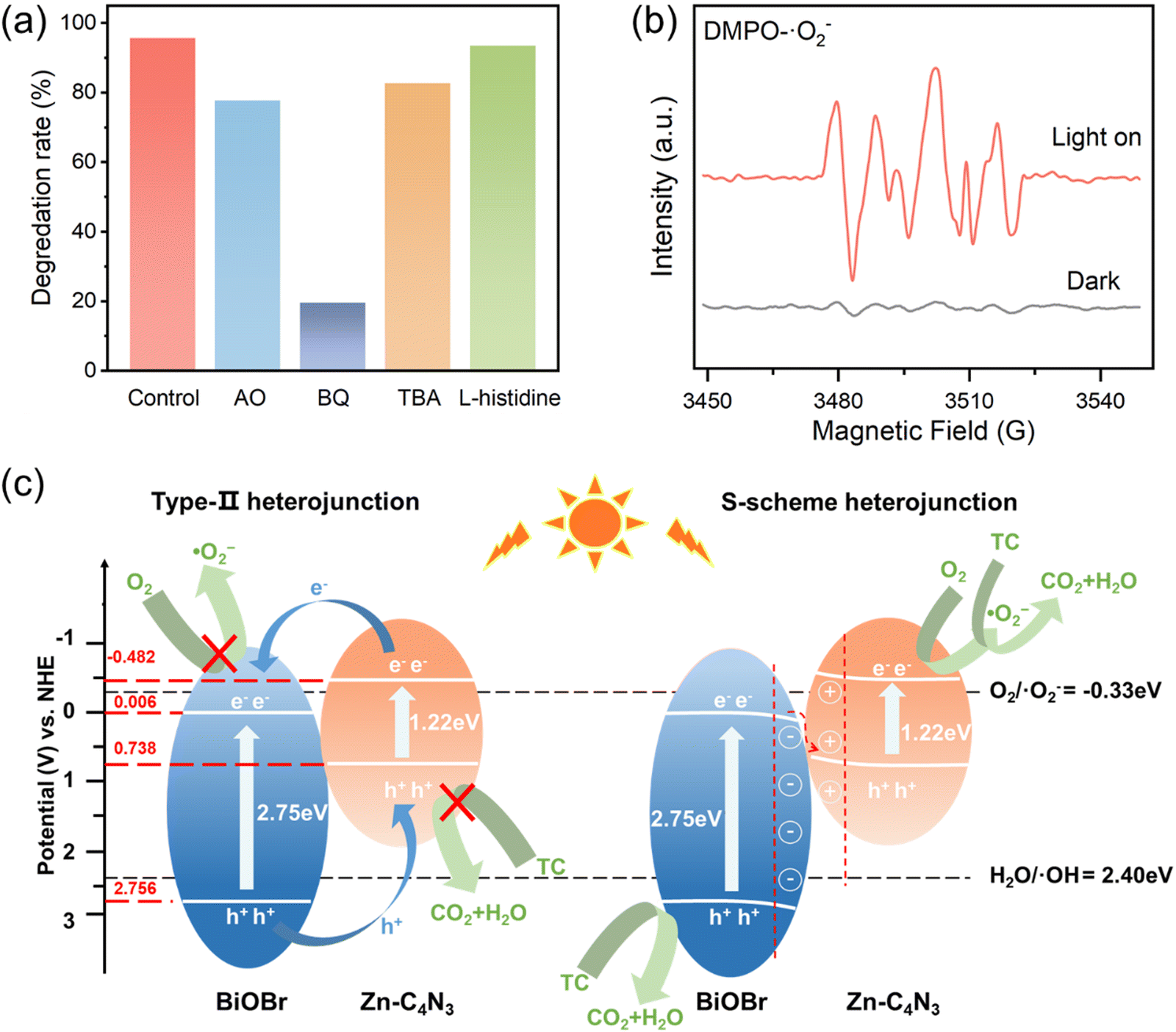
In order to identify the types of reactive oxygen species (ROSs) involved in TC degradation, various scavengers are introduced into the photocatalytic system. With the addition of AO (h+ scavenger), BQ (˙O2− scavenger), TBA (˙OH scavenger), and L-histidine (1O2 scavenger),38 the degradation efficiency of TC decreases significantly from 95.68% to 77.82%, 19.61%, 82.78% and 93.55%, respectively (Fig. 7a). This indicates that the role of ROSs in TC degradation decreases in the following order: ˙O2− > h+ > ˙OH > 1O2.
 | ||
| Fig. 7 (a) Trapping experiments of active species about the photocatalytic degradation of TC. (b) ESR spectra of DMPO–˙O2− in methanol dispersions over Zn–C4N3/BiOBr after irradiation. (c) Possible photocatalytic mechanism for Zn–C4N3/BiOBr. | ||
To further validate the involvement of ˙O2− and ˙OH in the catalytic process, electron spin resonance (ESR) spectroscopy is employed using DMPO as a spin trapping reagent. There exist no ESR signals in the dark but six distinct peaks corresponding to DMPO–˙O2− when the light is on (Fig. 7b), thus effectively verifying that the existence of ˙O2− is not dependent on the self-decomposition of Zn–C4N3/BiOBr in the photocatalytic process.41–43 Upon replacing the solvent methanol with water to capture DMPO–˙OH, a seven-line spectrum is acquired instead of the hydroxyl signal (Fig. S11, ESI†). This phenomenon occurs due to the strong oxidation of Zn–C4N3/BiOBr itself, resulting in the oxidation-induced ring opening of the capture agent DMPO.44,45
According to the results of radical trapping experiments and ESR tests, two possible degradation mechanisms for the degradation of TC at the Zn–C4N3/BiOBr heterostructure can be proposed. The conventional type II heterojunction charge transfer mechanism (Fig. 7c) suggests that Zn–C4N3 and BiOBr are excited by visible light to produce electrons (e−) and holes (h+), respectively. Then, the electrons transfer from the conduction band (CB) of Zn–C4N3 to the conduction band (CB) of BiOBr, and the holes transfer from the valence band (VB) of BiOBr to the valence band (VB) of Zn–C4N3. Nevertheless, the ECB of BiOBr relative to NHE has been calculated to be 0.006 eV using the Mott–Schottky curves in Section 3.2, which is much higher than the O2/˙O2− potential (−0.33 eV).46 In this mechanism, it suggests no ˙O2− generation, which contradicts the results obtained from the active radical species test. Instead, the S-scheme heterojunction supports the charge transfer mechanism better, in which BiOBr and Zn–C4N3 act as the electron donor and electron acceptor, respectively, to guarantee the effective separation of photogenerated electron and hole pairs. Specifically, the effects of the internal electric field (IEF) and bending band ensure that the photogenerated electrons spontaneously flow from BiOBr to Zn–C4N3 to recombine with the holes on the VB of Zn–C4N3 (eqn (1) and (2)). At the same time, electrons remain in the CB of Zn–C4N3 and holes remain in the VB of BiOBr. Because the CB potential of Zn–C4N3 (−0.48 V vs. NHE) is lower than the E(O2/˙O2−) (−0.33 V vs. NHE), the electrons in the CB of Zn–C4N3 can be captured by O2 to produce ˙O2− (eqn (3)), which is consistent with the result that ˙O2− is the primary reactive oxygen species of TC photocatalytic degradation. Simultaneously, one part of holes in the VB of BiOBr can directly react with pollutants, and the other can oxidize H2O to produce ˙OH (eqn (4)) due to the fact that the VB potential of BiOBr (2.76 V vs. NHE) is more negative than the E(H2O/˙OH) (2.40 V vs. NHE).47–49 Finally, numerous photogenerated h+, ˙O2− and ˙OH species trigger the high-efficiency degradation of the adsorbed TC molecules on the surface of Zn–C4N3/BiOBr (eqn (5)). It can be inferred that the S-type heterojunction maintains strong redox ability and is more suitable for the Zn–C4N3/BiOBr system.
| Zn–C4N3/BiOBr + hν → Zn–C4N3/BiOBr (e−, h+) | (1) |
| e− (BiOBr) + h+ (Zn–C4N3) → Recombination | (2) |
| e− (Zn–C4N3) + O2 → ˙O2− | (3) |
| h+ (BiOBr) + H2O → ˙OH + H+ | (4) |
| ˙O2−/˙OH/h+ + TC → CO2 + H2O + small molecules | (5) |
3.4. Degradation pathway of TC
In the conceptual density functional theory framework, Fukui functions are tools used to predict reaction sites in molecules by evaluating electrophilicity and nucleophilicity, providing insights into electronic reactivity. Their application contributes to a deeper understanding of molecular reactivity and offers crucial information for catalyst design and reaction mechanism studies. The Fukui function, denoted as f(γ), is precisely defined as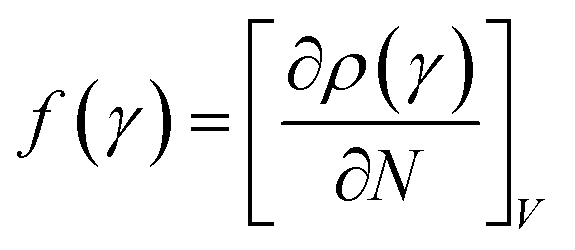 , where ρ(γ) is the electron density at a point γ in space, N is the electron number in the current system, and the constant term V in the partial derivative represents the external potential. In the context of a chemical reaction, the Fukui function (f) can be calculated for electrophilic attack
, where ρ(γ) is the electron density at a point γ in space, N is the electron number in the current system, and the constant term V in the partial derivative represents the external potential. In the context of a chemical reaction, the Fukui function (f) can be calculated for electrophilic attack 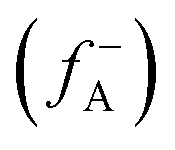 and nucleophilic attack
and nucleophilic attack 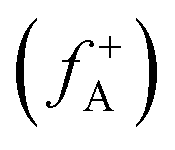 using the atom charges qA of atom A at the corresponding states:
using the atom charges qA of atom A at the corresponding states: 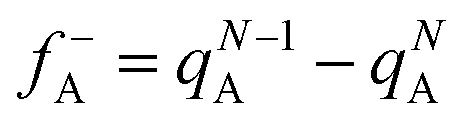 for electrophilic attack and
for electrophilic attack and 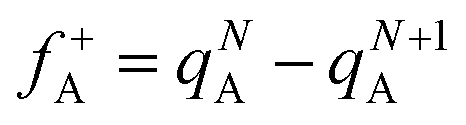 for nucleophilic attack.41 This calculation provides valuable insights into the reactivity of specific atomic sites during a chemical reaction.
for nucleophilic attack.41 This calculation provides valuable insights into the reactivity of specific atomic sites during a chemical reaction.
Herein, natural population analysis (NPA) charges are introduced to investigate the charge distribution of nucleophilic attack (f−) and electrophilic attack (f+) Fukui indices in reactions (Table S3, ESI†). Through NPA, we accurately assess the electron distribution in the reaction system, especially in the interaction between electrophilic and nucleophilic groups. Generally, as electrophilic groups, ˙OH and ˙O2− tend to attack reaction sites that are prone to electron loss. The active reaction region of the TC molecule exhibits larger Fukui indexes. Therefore, atoms such as O20, C10, O26, O22, O21, C16, C14 and O31 exhibit higher reactivity, indicated by their Fukui indices (f0) of 0.0727, 0.0665, 0.034, 0.0402, 0.0409, 0.0454, 0.0457 and 0.0117, respectively (detailed atomic positions are illustrated in Fig. 8b). Furthermore, the electrostatic potential (ESP) mapping (Fig. 8a) reveals that regions marked in red show higher reactivity in the system, which is basically consistent with the atoms with higher f0 values in the Fukui index.
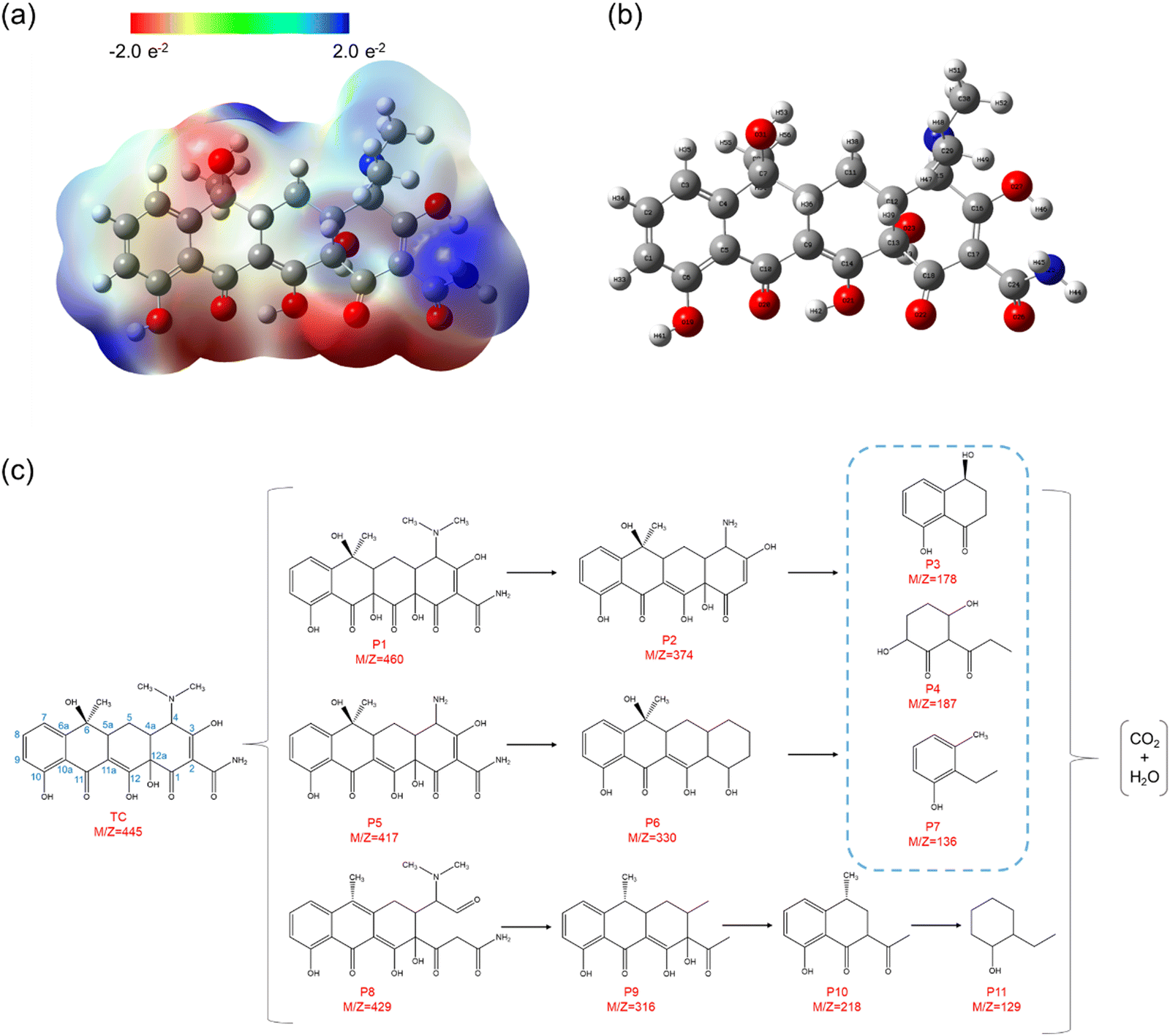 | ||
| Fig. 8 (a) Electrostatic potential (ESP) map. (b) The detailed atomic position of TC. (c) Proposed degradation pathways for TC degradation. | ||
In order to comprehensively explore the proposed degradation pathway of TC, qualitative analysis is conducted using LC–MS techniques based on the mass spectrum. Through experimental analysis, approximately eleven intermediates are detected except for TC, and their details are presented in Table S4 (ESI†). According to the mass spectra of TC and intermediate products obtained after degradation under visible light irradiation (Fig. S12, ESI†), three possible degradation paths can be considered (Fig. 8c). The photoexcited active species (˙O2−, h+ and ˙OH) play a crucial role in attacking the double bonds, aromatic rings, and amino groups during the degradation of tetracycline (TC). This implies that the intermediates primarily result from ring-opening reactions or cleavage of the main carbon bond.41 In the first pathway, a hydroxyl group attacks the double bond at C11a–C12 in the TC molecule to form P1 (m/z = 460) during the hydroxylation process, which is consistent with the previous study.38 Subsequently, P2 (m/z = 374) is formed by the loss of the N-methyl group in the TC molecule at the C4 tertiary amine site due to the low bond energy of the N–C bond.41 In the second pathway, the TC molecule first undergoes N-dealkylation to form P5 (m/z = 417) through a mechanism similar to that of P2 formation. Then, P5 produces P6 (m/z = 330) after the deamination reaction.38 In the above two reaction pathways, P2 and P5 are subsequently decomposed into P3 (m/z = 178), P4 (m/z = 187) and P7 (m/z = 136) under the constantly combined action of ˙O2−, h+ and ˙OH.38 In the third pathway, the hydroxyl group at the C6 position of the TC molecule may be attacked by active species to undergo the dehydration reaction, and the double bond at the C2–C3 position is attacked by superoxide radicals, thus forming P8 (m/z = 429).42 Then, N-dealkylation and deamination reactions occur to form P9 (m/z = 316), and further reaction occurs to form P10 (m/z = 218).43 With the extension of reaction time, the intermediate will decompose into a smaller molecular structure of P11 (m/z = 129) under the joint attack of ˙O2−, h+ and ˙OH. Finally, these small molecular products can be thoroughly decomposed into CO2 and H2O.43
4. Conclusions
In summary, this study has demonstrated that BiOBr can directly grow from the surface of Zn2+ doped C4N3 to construct an S-scheme heterojunction with an intimate interface. More importantly, the original hollow structure of BiOBr is inherited, which provides highly exposed reaction sites and an elevated photon capture cross-section. The synergistic effect between BiOBr and Zn–C4N3, facilitated by Zn2+ doping, significantly boosts the photocatalytic activity of BiOBr, causing a significant portion of TC pollutants to be rapidly removed within 30 minutes. Our findings provide profound insights into the intricacies of advanced photocatalytic materials. This study not only contributes to the sustainable removal of organic pollutants but also introduces a transformative strategy for optimizing the catalytic performance of BiOBr in environmental applications.Data availability
The data underlying this article are available in the article and in its online ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Hebei Natural Science Foundation (B2024205035), National Natural Science Foundation of China (Nos 12174183 and 21872071), and High Performance Computing Centers of Shenzhen.References
- S. Cao, J. Yu, S. Wageh, A. A. Al-Ghamdi, M. Mousavi, J. B. Ghasemi and F. Xu, H2-production and electron-transfer mechanism of a noble-metal-free WO3@ZnIn2S4 S-scheme heterojunction photocatalyst, J. Mater. Chem. A, 2022, 10(33), 17174–17184, 10.1039/d2ta05181h.
- C. Wen, D. Li, J. Zhong, Z. Wang, S. Huang, H. Liu, J. Wu, P. Chen, W. Lv and G. Liu, In situ synthesis of S-scheme AgBr/BiOBr for efficient degradation of sulfonamide antibiotics: Synergistic effects of oxygen vacancies and heterojunctions promote exciton dissociation, Chem. Eng. J., 2022, 450, 138075, DOI:10.1016/j.cej.2022.138075.
- Q. Xu, S. Wageh, A. A. Al-Ghamdi and X. Li, Design principle of S-scheme heterojunction photocatalyst, J. Mater. Sci. Technol., 2022, 124, 171–173, DOI:10.1016/j.jmst.2022.02.016.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Ultrathin 2D/2D WO3/g-C3N4 step-scheme H2-production photocatalyst, Appl. Catal., B, 2019, 243, 556–565, DOI:10.1016/j.apcatb.2018.11.011.
- F. Li, X. Yue, Y. Liao, L. Qiao, K. Lv and Q. Xiang, Understanding the unique S-scheme charge migration in triazine/heptazine crystalline carbon nitride homojunction, Nat. Commun., 2023, 14(1), 3901, DOI:10.1038/s41467-023-39578-z.
- J. Song, J. Zhang, A. Zada, Y. Ma and K. Qi, CoFe2O4/NiFe2O4 S-scheme composite for photocatalytic decomposition of antibiotic contaminants, Ceram. Int., 2023, 49(8), 12327–12333, DOI:10.1016/j.ceramint.2022.12.088.
- X. Ren, M. Gao, Y. Zhang, Z. Zhang, X. Cao, B. Wang and X. Wang, Photocatalytic reduction of CO2 on BiOX: Effect of halogen element type and surface oxygen vacancy mediated mechanism, Appl. Catal., B, 2020, 274, 119063, DOI:10.1016/j.apcatb.2020.119063.
- R. Wang, P. Zhu, M. Duan, J. Xu, M. Liu and D. Luo, Synthesis and characterization of successive Z-scheme CdS/Bi2MoO6/BiOBr heterojunction photocatalyst with efficient performance for antibiotic degradation, J. Alloys Compd., 2021, 870, 159385, DOI:10.1016/j.jallcom.2021.159385.
- M. Yang, Y. Han, J. Ding, Y. Guo, C. Han and X. Gu, Constructing Defective Heterojunctions of BiOBr Nanosheets and Hollow NH2-functionalized MOFs for Visible-light-driven CO2 Reduction with Nearly 100% CO Selectivity by Pure H2O, Chem. – Asian J., 2023, 18(8), e202300033, DOI:10.1002/asia.202300033.
- L. Feng, H. Fu, T. Zhang, Q. Zhang, S. Ren, J. Cheng, Q. Liang and X. Xiao, Synergy of oxygen vacancies and Bi nanoparticles on BiOBr nanosheets for enhanced photocatalytic H2O2 production, New J. Chem., 2024, 48(5), 1998–2007, 10.1039/d3nj03815g.
- W. Zhang, Z. Bian, Y. Peng, H. Tang and H. Wang, Dual-function oxygen vacancy of BiOBr intensifies pollutant adsorption and molecular oxygen activation to remove tetracycline hydrochloride, Chem. Eng. J., 2023, 451, 138731, DOI:10.1016/j.cej.2022.138731.
- Y. Zhang, S. Gu, X. Zhou, K. Gao, K. Sun, D. Wu, J. Xia and X. Wang, Boosted photocatalytic nitrogen fixation by bismuth and oxygen vacancies in Bi2MoO6/BiOBr composite structures, Catal. Sci. Technol., 2021, 11(14), 4783–4792, 10.1039/d1cy00539a.
- J. Zhao, Z. Miao, Y. Zhang, G. Wen, L. Liu, X. Wang, X. Cao and B. Wang, Oxygen vacancy-rich hierarchical BiOBr hollow microspheres with dramatic CO2 photoreduction activity, J. Colloid Interface Sci., 2021, 593, 231–243, DOI:10.1016/j.jcis.2021.02.117.
- Z. Zan, X. Li, X. Gao, J. Huang, Y. Luo and L. Han, 0D/2D Carbon Nitride Quantum Dots (CNQDs)/BiOBr S-Scheme Heterojunction for Robust Photocatalytic Degradation and H2O2 Production, Acta Phys.-Chim. Sin., 2023, 39(6), 2209016, DOI:10.3866/pku.Whxb202209016.
- X. Li, J. Xiong, X. Gao, J. Ma, Z. Chen, B. Kang, J. Liu, H. Li, Z. Feng and J. Huang, Novel BP/BiOBr S-scheme nano-heterojunction for enhanced visible-light photocatalytic tetracycline removal and oxygen evolution activity, J. Hazard. Mater., 2020, 387, 121690, DOI:10.1016/j.jhazmat.2019.121690.
- Y. Zhang, J. Di, X. Zhu, M. Ji, C. Chen, Y. Liu, L. Li, T. Wei, H. Li and J. Xia, Chemical bonding interface in Bi2Sn2O7/BiOBr S-scheme heterojunction triggering efficient N2 photofixation, Appl. Catal., B, 2023, 323, 122148, DOI:10.1016/j.apcatb.2022.122148.
- J. Wu, L. Xiong, Y. Hu, Y. Yang, X. Zhang, T. Wang, Z. Tang, A. Sun, Y. Zhou and J. Shen, et al., Organic half-metal derived erythroid-like BiVO4/hm-C4N3 Z-Scheme photocatalyst: Reduction sites upgrading and rate-determining step modulation for overall CO2 and H2O conversion, Appl. Catal., B, 2021, 295, 120277, DOI:10.1016/j.apcatb.2021.120277.
- G. Zhou, Y. Shan, Y. Hu, X. Xu, L. Long, J. Zhang, J. Dai, J. Guo, J. Shen and S. Li, et al., Half-metallic carbon nitride nanosheets with micro grid mode resonance structure for efficient photocatalytic hydrogen evolution, Nat. Commun., 2018, 9(1), 3366, DOI:10.1038/s41467-018-05590-x.
- S. Tian, H. Ren, W. Sun, Y. Song, H. Ge, A. Yang, W. Zheng and Y. Zhao, CdIn2S4 microspheres embedded with mesoporous Zn-doped g-C3N4 ultrathin nanosheets for efficient photocatalytic hydrogen evolution, New J. Chem., 2024, 48(8), 3695–3706, 10.1039/d3nj05590f.
- H. Deng, D. Deng, S. Jin, Z. Tian and L. M. Yang, Unraveling the Activity and Mechanism of TM@g-C4N3 Electrocatalysts in the Oxygen Reduction Reaction, ACS Appl. Mater. Interfaces, 2024, 16(14), 17617–17625, DOI:10.1021/acsami.4c01342.
- X. Dou, Y. Chen and H. Shi, CuBi2O4/BiOBr composites promoted PMS activation for the degradation of tetracycline: S-scheme mechanism boosted Cu2+/Cu+ cycle, Chem. Eng. J., 2022, 431, 134054, DOI:10.1016/j.cej.2021.134054.
- J. Hao, Y. Zhang, L. Zhang, J. Shen, L. Meng and X. Wang, Restructuring surface frustrated Lewis acid-base pairs of BiOBr through isomorphous Sn doping for boosting photocatalytic CO2 reduction, Chem. Eng. J., 2023, 464, 142536, DOI:10.1016/j.cej.2023.142536.
- H. Guan, Y. Dong, X. Kang, Y. Han, Z. Cheng, L. Han, L. Xie, W. Chen and J. Zhang, Extraordinary electrochemical performance of lithium-sulfur battery with 2D ultrathin BiOBr/rGO sheet as an efficient sulfur host, J. Colloid Interface Sci., 2022, 626, 374–383, DOI:10.1016/j.jcis.2022.06.148.
- F. M. Aghamir, A. R. Momen-Baghdadabad and M. Etminan, Effects of deposition angle on synthesis of amorphous carbon nitride thin films prepared by plasma focus device, Appl. Surf. Sci., 2019, 463, 141–149, DOI:10.1016/j.apsusc.2018.08.154.
- K. Perumal, S. Shanavas, A. Karthigeyan, T. Ahamad, S. M. Alshehri and P. Murugakoothan, Hydrothermal assisted precipitation synthesis of highly stable g-C3N4/BiOBr/CdS photocatalyst with enhanced visible light photocatalytic degradation of tetracycline, Diamond Relat. Mater., 2020, 110, 108091, DOI:10.1016/j.diamond.2020.108091.
- W. D. Oh, S. K. Lua, Z. Dong and T. T. Lim, A novel three-dimensional spherical CuBi2O4 consisting of nanocolumn arrays with persulfate and peroxymonosulfate activation functionalities for 1H-benzotriazole removal, Nanoscale, 2015, 7(17), 8149–8158, 10.1039/c5nr01428j.
- T. Di, B. Zhu, B. Cheng, J. Yu and J. Xu, A direct Z-scheme g-C3N4/SnS2 photocatalyst with superior visible-light CO2 reduction performance, J. Catal., 2017, 352, 532–541, DOI:10.1016/j.jcat.2017.06.006.
- X. Lv, D. Y. S. Yan, F. L.-Y. Lam, Y. H. Ng, S. Yin and A. K. An, Solvothermal synthesis of copper-doped BiOBr microflowers with enhanced adsorption and visible-light driven photocatalytic degradation of norfloxacin, Chem. Eng. J., 2020, 401, 126012, DOI:10.1016/j.cej.2020.126012.
- J. Kang, Y. Tang, M. Wang, C. Jin, J. Liu, S. Li, Z. Li and J. Zhu, The enhanced peroxymonosulfate-assisted photocatalytic degradation of tetracycline under visible light by g-C3N4/Na-BiVO4 heterojunction catalyst and its mechanism, J. Environ. Chem. Eng., 2021, 9, 105524, DOI:10.1016/j.jece.2021.105524.
- X. Ren, J. Li, X. Cao, B. Wang, Y. Zhang and Y. Wei, Synergistic effect of internal electric field and oxygen vacancy on the photocatalytic activity of BiOBrxI1−x with isomorphous fluorine substitution, J. Colloid Interface Sci., 2019, 554, 500–511, DOI:10.1016/j.jcis.2019.07.034.
- S. Wu, X. Tan, J. Lei, H. Chen, L. Wang and J. Zhang, Ga-Doped and Pt-Loaded Porous TiO2-SiO2 for Photocatalytic Nonoxidative Coupling of Methane, J. Am. Chem. Soc., 2019, 141(16), 6592–6600, DOI:10.1021/jacs.8b13858.
- H. Jiang, Q. Wang, P. Chen, H. Zheng, J. Shi, H. Shu and Y. Liu, Photocatalytic degradation of tetracycline by using a regenerable (Bi)BiOBr/rGO composite, J. Cleaner Prod., 2022, 339, 130771, DOI:10.1016/j.jclepro.2022.130771.
- H. Shang, H. Jia, W. Zhang, S. Li, Q. Wang, Q. Yang, C. Zhang, Y. Shi, Y. Wang and P. Li, et al., Surface Hydrogen Bond-Induced Oxygen Vacancies of TiO2 for Two-Electron Molecular Oxygen Activation and Efficient NO Oxidation, Environ. Sci. Technol., 2023, 57(48), 20400–20409, DOI:10.1021/acs.est.3c06593.
- Q. Wu, H. Ma, Y. Wang, J. Chen, J. Dai, X. Xu and X. Wu, Surface Electron Localization in Cu-MOF-Bonded Double-Heterojunction Cu2O Induces Highly Efficient Photocatalytic CO2 Reduction, ACS Appl. Mater. Interfaces, 2022, 14(48), 54328–54337, DOI:10.1021/acsami.2c15278.
- C. Gao, H. Yu, L. Zhang, Y. Zhao, J. Xie, C. Li, K. Cui and J. Yu, Ultrasensitive Paper-Based Photoelectrochemical Sensing Platform Enabled by the Polar Charge Carriers-Created Electric Field, Anal. Chem., 2020, 92(4), 2902–2906, DOI:10.1021/acs.analchem.9b05611.
- Y. Yang, Z. Tang, B. Zhou, J. Shen, H. He, A. Ali, Q. Zhong, Y. Xiong, C. Gao and A. Alsaedi, et al., In situ no-slot joint integration of half-metallic C(CN)3 cocatalyst into g-C3N4 scaffold: An absolute metal-free in-plane heterosystem for efficient and selective photoconversion of CO2 into CO, Appl. Catal., B, 2020, 264, 118470, DOI:10.1016/j.apcatb.2019.118470.
- X. Bao, M. Zhang, Z. Wang, D. Dai, P. Wang, H. Cheng, Y. Liu, Z. Zheng, Y. Dai and B. Huang, Molten-salt assisted synthesis of Cu clusters modified TiO2 with oxygen vacancies for efficient photocatalytic reduction of CO2 to CO, Chem. Eng. J., 2022, 445, 136718, DOI:10.1016/j.cej.2022.136718.
- Y. Lin, X. Wu, Y. Han, C. Yang, Y. Ma, C. Du, Q. Teng, H. Liu and Y. Zhong, Spatial separation of photogenerated carriers and enhanced photocatalytic performance on Ag3PO4 catalysts via coupling with PPy and MWCNTs, Appl. Catal., B, 2019, 258, 117969, DOI:10.1016/j.apcatb.2019.117969.
- S. Wu, Z. Yang, Z. Zhou, X. Li, Y. Lin, J. J. Cheng and C. Yang, Catalytic activity and reaction mechanisms of single-atom metals anchored on nitrogen-doped carbons for peroxymonosulfate activation, J. Hazard. Mater., 2023, 459, 132133, DOI:10.1016/j.jhazmat.2023.132133.
- Y. Chen and K. Liu, Preparation and characterization of nitrogen-doped TiO2/diatomite integrated photocatalytic pellet for the adsorption-degradation of tetracycline hydrochloride using visible light, Chem. Eng. J., 2016, 302, 682–696, DOI:10.1016/j.cej.2016.05.108.
- J. Guo, L. Jiang, J. Liang, W. Xu, H. Yu, J. Zhang, S. Ye, W. Xing and X. Yuan, Photocatalytic degradation of tetracycline antibiotics using delafossite silver ferrite-based Z-scheme photocatalyst: Pathways and mechanism insight, Chemosphere, 2021, 270, 128651, DOI:10.1016/j.chemosphere.2020.128651.
- Y.-Y. Chen, Y.-L. Ma, J. Yang, L.-Q. Wang, J.-M. Lv and C.-J. Ren, Aqueous tetracycline degradation by H2O2 alone: Removal and transformation pathway, Chem. Eng. J., 2017, 307, 15–23, DOI:10.1016/j.cej.2016.08.046.
- X. Jiang, S. Chen, X. Zhang, L. Qu, H. Qi, B. Wang, B. Xu and Z. Huang, Carbon-doped flower-like Bi2WO6 decorated carbon nanosphere nanocomposites with enhanced visible light photocatalytic degradation of tetracycline, Adv. Compos. Hybrid Mater., 2022, 6, 9, DOI:10.1007/s42114-022-00590-4.
- J. Yao, X. Zeng and Z. Wang, Enhanced degradation performance of sulfisoxazole using peroxymonosulfate activated by copper-cobalt oxides in aqueous solution: Kinetic study and products identification, Chem. Eng. J., 2017, 330, 345–354, DOI:10.1016/j.cej.2017.07.155.
- S. Guo, H. Wang, W. Yang, H. Fida, L. You and K. Zhou, Scalable synthesis of Ca-doped α-Fe2O3 with abundant oxygen vacancies for enhanced degradation of organic pollutants through peroxymonosulfate activation, Appl. Catal., B, 2020, 262, 118250, DOI:10.1016/j.apcatb.2019.118250.
- C. Feng, Q. Gu, J. Rong, Q. Liang, M. Zhou, X. Li, S. Xu and Z. Li, Porous dual Z-scheme InOOH/RCN/CoWO4 heterojunction with enhanced photothermal-photocatalytic properties towards norfloxacin degradation, Sep. Purif. Technol., 2023, 308, 122890, DOI:10.1016/j.seppur.2022.122890.
- X. Yao, X. Jiang, D. Zhang, S. Lu, M. Wang, S. Pan, X. Pu, J. Liu and P. Cai, Achieving improved full-spectrum responsive 0D/3D CuWO4/BiOBr:Yb3+,Er3+ photocatalyst with synergetic effects of up-conversion, photothermal effect and direct Z-scheme heterojunction, J. Colloid Interface Sci., 2023, 644, 95–106, DOI:10.1016/j.jcis.2023.04.072.
- X. Fu, J. Tao, Z. He, Y. Gao, Y. Xia and Z. Zhao, Synergy of Z-scheme heterostructure with interfacial S−O bonding in In2S3/BiOBr for efficient tetracycline hydrochloride degradation and Cr(VI) reduction, J. Alloys Compd., 2023, 936, 168202, DOI:10.1016/j.jallcom.2022.168202.
- R. Weng, F. Tian, Z. Yu, J. Ma, Y. Lv and B. Xi, Efficient mineralization of TBBPA via an integrated photocatalytic reduction/oxidation process mediated by MoS2/SnIn4S8 photocatalyst, Chemosphere, 2021, 285, 131542, DOI:10.1016/j.chemosphere.2021.131542.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01043d |
| This journal is © the Owner Societies 2024 |