
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlternative protein encapsulation with MOFs: overcoming the elusive mineralization of HKUST-1 in water†
Jesús Cases
Díaz
and
Mónica
Giménez-Marqués
 *
*
Instituto de Ciencia Molecular (ICMol), Universidad de Valencia, Catedrático José Beltrán 2, 46980 Paterna, Valencia, Spain. E-mail: monica.gimenez-marques@uv.es
First published on 17th November 2023
Abstract
Protein encapsulation by in situ formation of MOFs is a valuable strategy to immobilise and protect these bioentities. However the required biocompatible conditions limits the scope of MOFs under investigation, particularly in the case of hydrolytically unstable MOFs such as HKUST-1. We report alternative synthetic procedures to obtain protein@HKUST-1 biocomposites from related Cu-BTC dense biocomposites. pH dependent dense phase precursors are first obtained and their transformations into HKUST-1 are characterized. Encapsulation efficiency is affected by the protein's nature, and can be modulated by the sequential or simultaneous addition of MOF precursors.
Metal–organic frameworks (MOFs) have been demonstrated as suitable platforms for the immobilization of biomolecules.1 Their versatile functionality and porosity permit fine-tuning of the host–guest interactions, conferring protection against denaturation and allowing the transport of specific substrates.2 Among the developed immobilization approaches,3in situ MOF growth is preferred since avoids biomolecule size limitations and leaching encountered via infiltration or surface immobilization, respectively.4 However, the biocompatible conditions required for mineralization are typically unsuited for the growth of most MOFs. Thus, zeolitic imidazolate frameworks (ZIFs) have been the most exploited materials owing to their optimal stability and facile synthesis under these conditions.5 Very recently, the progress of biocompatible MOF synthesis has permitted incorporating more structures on the list,5,6 offering new chemical scenarios at the MOF-biomolecule interface.
Motivated by the difficulty in obtaining specific MOFs by direct synthesis in water, we propose using alternative routes. One of these elusive MOFs is the archetypal HKUST-1, constituted by Cu2+ cations and benzene 1,3,5-tricarboxylic acid ligands (H3BTC).7 HKUST-1 forms in mixtures of H2O:EtOH,8 a medium that has been employed for the preparation of certain protein@HKUST-1 composites.4,9 However, its in situ growth is prevented in water, forming instead dense Cu-BTC compounds.8 In addition, HKUST-1 is degraded in water or dilute HCl to these layered Cu-BTC compounds with negligible porosity. Some of these degradation products can be transformed back to the initial porous phase upon immersion in EtOH.10 Essentially, the acid degraded material can be converted to HKUST-1 upon EtOH immersion, whereas the water degraded is not transformed. These observations allow us to glimpse an alternative biocompatible HKUST-1 synthetic route for protein encapsulation.
Herein we propose the synthesis of non-porous Cu-BTC materials in water and their later reversible transformation to the porous HKUST-1 phase by immersion in EtOH under specific pH conditions. This biocompatible dense-to-porous reversible transformation enables the formation of elusive protein@HKUST-1 biocomposites in water.
First attempts consisted in the synthesis of Cu-BTC compounds from two aqueous solutions of Na3BTC and CuCl2 (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the presence of different amounts of HCl (0, 0.5 or 1 equivalents) and later incubation in EtOH (Fig. 1). Depending on the acidity of the media, different phases were obtained. In absence of acid (0 eq.), a dense Cu-BTC material that resembles the M3(BTC)2·12H2O (M = Cu, Co, Ni, and Zn)11,13,14 structure is achieved, as deduced from XRPD (Fig. 1A), with negligible N2 sorption capacity (Fig. 1B). This phase, termed as H0, cannot transform to HKUST-1 upon overnight exposure to EtOH, but can be converted upon acidification with acetic acid (HAc), HCl or the acid ligand H3BTC (Fig. S1, ESI†). Addition of HCl (0.5 eq.) leads to a coexistence of the previous H0 phase with a new one denoted as H1, with the latter being able to transform into HKUST-1 upon immersion in EtOH (Fig. 1C), with the expected gain in sorption capacity (Fig. 1D). When more HCl is added (1 eq.), only the ethanol transformable phase H1 is obtained as deduced by XRPD (Fig. 1E) and the expected gain in N2 sorption capacity (Fig. 1F). This H1 structure with formula Cu(HBTC)·3H2O,12,15 corresponds to the one obtained by G. Majano et al. after degradation of HKUST-1 in water, which is reversibly reconstructed to the starting material upon exposure to EtOH.10 As a result, two dense layered structures of Cu-BTC, H0 and H1 can be obtained from aqueous solutions depending on the pH. These two phases can be inter-converted (Fig. S2, ESI†) and lately transformed into HKUST-1 upon immersion in EtOH as depicted in Fig. 2. The transformed HKUST-1 samples from H0 or H1 are respectively termed HK0 and HK1. Morphological and size particle differences are observed upon transformation in both cases (Fig. S3, ESI†). It was found that under these biocompatible conditions, the phase transformations can be reversibly repeated (Fig. S4 and S5, ESI†) with a good recovery of the sorption capacity (Fig. S6, ESI†).
:1) in the presence of different amounts of HCl (0, 0.5 or 1 equivalents) and later incubation in EtOH (Fig. 1). Depending on the acidity of the media, different phases were obtained. In absence of acid (0 eq.), a dense Cu-BTC material that resembles the M3(BTC)2·12H2O (M = Cu, Co, Ni, and Zn)11,13,14 structure is achieved, as deduced from XRPD (Fig. 1A), with negligible N2 sorption capacity (Fig. 1B). This phase, termed as H0, cannot transform to HKUST-1 upon overnight exposure to EtOH, but can be converted upon acidification with acetic acid (HAc), HCl or the acid ligand H3BTC (Fig. S1, ESI†). Addition of HCl (0.5 eq.) leads to a coexistence of the previous H0 phase with a new one denoted as H1, with the latter being able to transform into HKUST-1 upon immersion in EtOH (Fig. 1C), with the expected gain in sorption capacity (Fig. 1D). When more HCl is added (1 eq.), only the ethanol transformable phase H1 is obtained as deduced by XRPD (Fig. 1E) and the expected gain in N2 sorption capacity (Fig. 1F). This H1 structure with formula Cu(HBTC)·3H2O,12,15 corresponds to the one obtained by G. Majano et al. after degradation of HKUST-1 in water, which is reversibly reconstructed to the starting material upon exposure to EtOH.10 As a result, two dense layered structures of Cu-BTC, H0 and H1 can be obtained from aqueous solutions depending on the pH. These two phases can be inter-converted (Fig. S2, ESI†) and lately transformed into HKUST-1 upon immersion in EtOH as depicted in Fig. 2. The transformed HKUST-1 samples from H0 or H1 are respectively termed HK0 and HK1. Morphological and size particle differences are observed upon transformation in both cases (Fig. S3, ESI†). It was found that under these biocompatible conditions, the phase transformations can be reversibly repeated (Fig. S4 and S5, ESI†) with a good recovery of the sorption capacity (Fig. S6, ESI†).
The possibility of in situ protein encapsulation using the alternative HKUST-1 synthesis via transformation of dense Cu-BTC phases was explored. With this aim, a variety of proteins including casein (Cas), bovine serum albumin (BSA), subtilisin Carlsberg (SubC), bovine cytochrome c (CytC), trypsin (Try) and lysozyme (Ly) were selected, covering a broad range of isoelectric points (pI) (Fig. 3A and Table S1, ESI†), an electrostatic parameter that has been demonstrated to highly determine the occurrence of surface interactions that promote effective encapsulation.16 First encapsulation attempts consisted of mixing the protein with a Na3BTC aqueous solution, and in a second step, adding the CuCl2 aqueous solution on top (see ESI†). Reasonably good encapsulation efficiencies (∼81–63%) were obtained for the low pI proteins, with a significant drop evidenced for the proteins with higher pI (∼42–33%) (Fig. 3A). These results are in good agreement with the previously postulated encapsulation mechanism,4,16,17 which states that MOF formation around the biomolecule depends on the effective interaction between the constituent metal cations and the functional groups on the biomolecule surface, which turns into nucleation points for the MOF growth. These groups are mainly carboxylates in the low pI proteins, or amine and guanidinium groups in the high pI ones, which effectively bind to metal cations, or hamper their interaction, respectively. Following this principle, we have recently established the possibility to dominate these surface interactions by employing hard Lewis acid cations as MOF precursor, obtaining optimal encapsulation efficiencies irrespective of the protein nature for a variety of MIL-100(Fe) biocomposites.18 As expected, the soft Lewis acid cation used in this work, Cu2+, results in a compromised encapsulation of basic proteins (Fig. 3A). In this situation, it was hypothesized that the carboxylates from the BTC−3 binding to the available Cu2+ may strongly hamper an effective interaction at the biomolecule interface especially in the high pI proteins, which is then reflected in a reduced encapsulation efficiency (Fig. 3B).
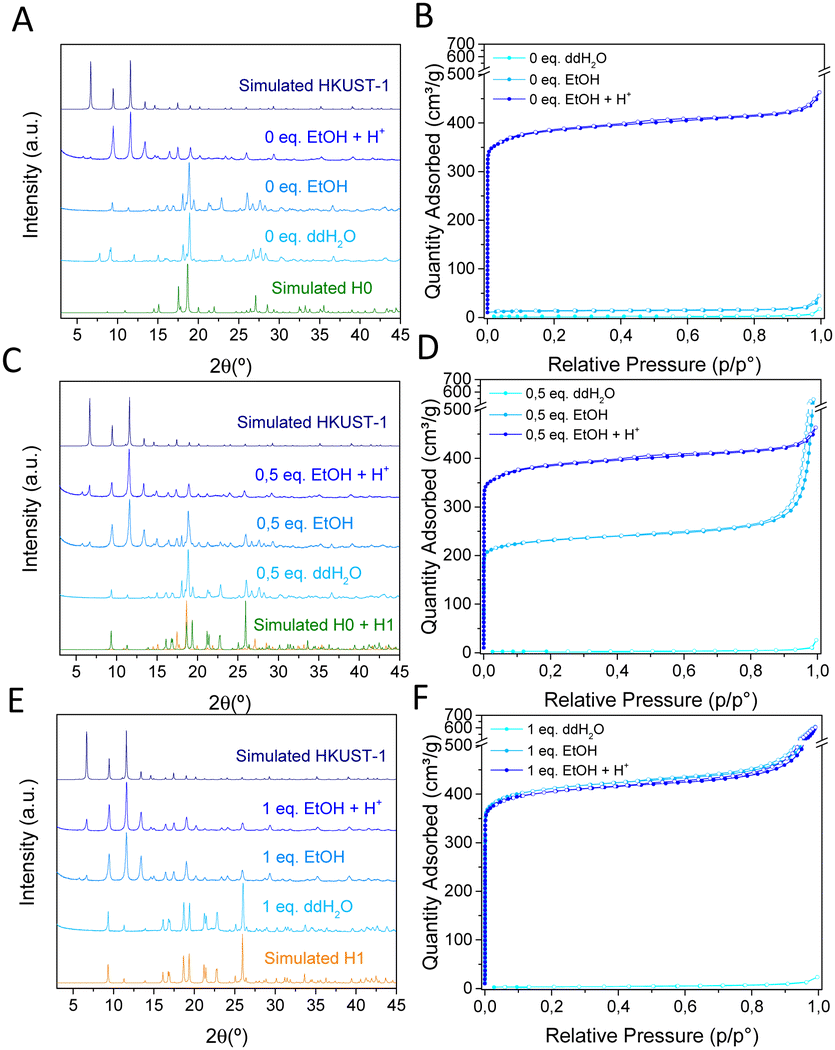 | ||
| Fig. 1 XRPD patterns (left) and N2 sorption isotherms (right) of Cu-BTC materials synthesized with (A, B) 0 equivalents, (C, D) 0.5 equivalents and (E, F) 1 equivalent of HCl and the corresponding products washed with water (ddH2O), ethanol (EtOH) or ethanol and acid (EtOH + H+). H0: CCDC 609754,11 H1: CCDC 1288676.12 | ||
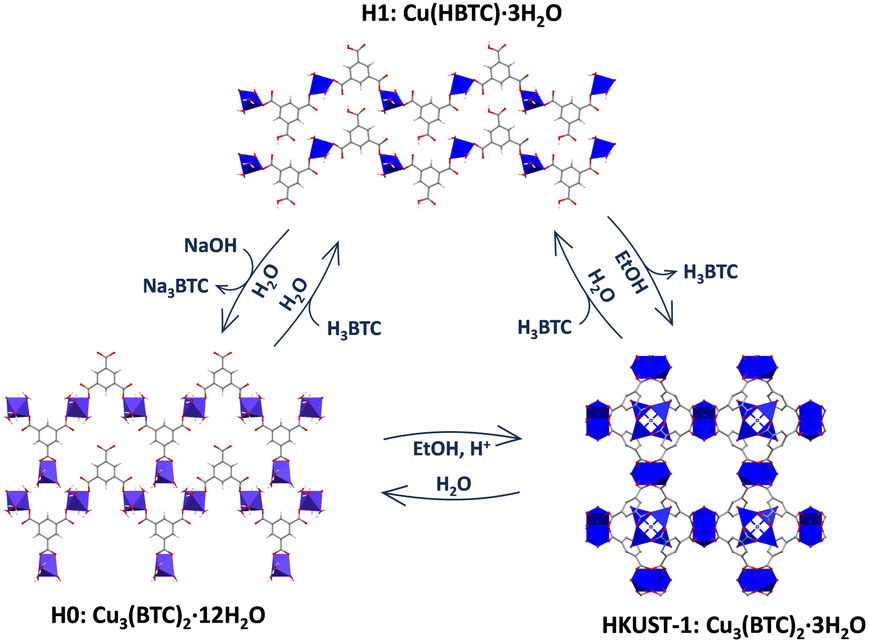 | ||
| Fig. 2 Schematic representation of the reversible transformation processes among the different Cu-BTC phases. | ||
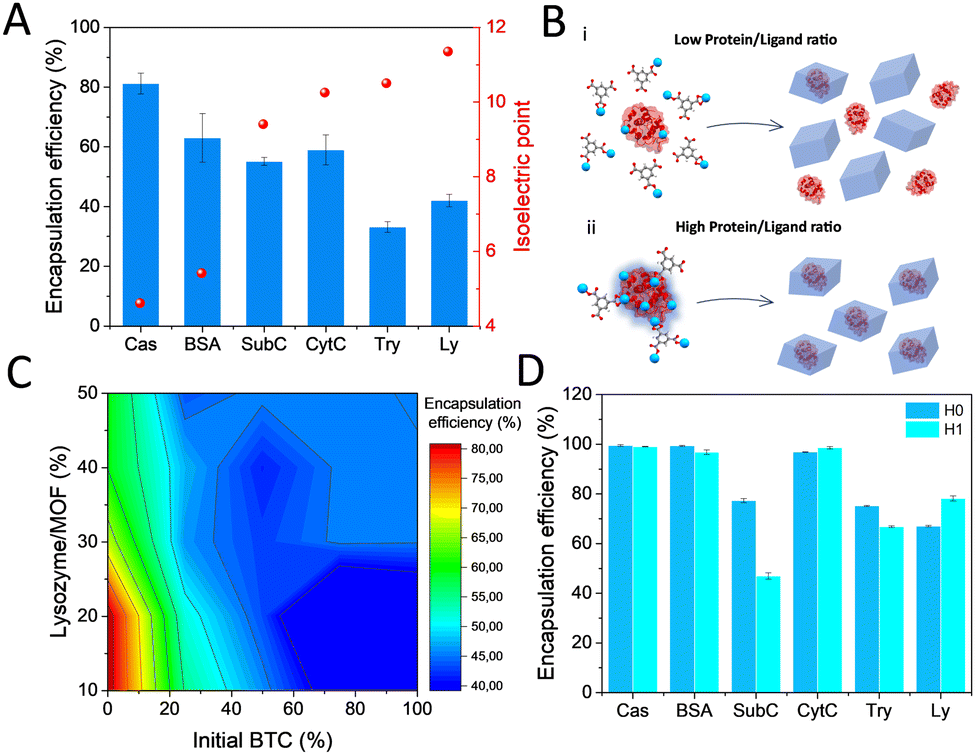 | ||
| Fig. 3 (A) Encapsulation efficiency obtained for the different proteins with H0 (pI indicated in right axis). (B) Representation of the interactions at the biomolecule surface in presence of (i) an excess or (ii) limited ligand salt, resulting respectively in moderate or significant encapsulation efficiency (metal cations represented as blue spheres). (C) Encapsulation efficiency of lysozyme in H0 as a function of the initial amount of protein and BTC. (D) Encapsulation efficiency of the optimized synthesis for H0 and H1 with the set of proteins. | ||
To gain more insight into the actual interactions at the biomolecule-MOF interface, we thoroughly analyzed the hindered biocomposite formation using the highly basic Ly (pI 11.35)19 protein as model. A systematic study was carried out by adding the corresponding metal and ligand precursor aqueous solutions (1:1 ratio) on top of a Ly aqueous solution with increasing amounts of protein (see ESI† for details). CuCl2 aqueous solution (50 mM) was added at a fixed rate, whereas a varying percentage of the BTC solution (50 mM) was added beforehand. The rest of the BTC solution was added in the same timelapse than the CuCl2. With these conditions, it is possible to cover the situations where BTC is added in different amounts prior to the synthesis. The results depicted in Fig. 3C show that the encapsulation efficiency rapidly decays as more BTC is added before the synthesis, with the highest encapsulation efficiency (∼85%) reached when BTC and CuCl2 precursor solutions are added simultaneously on top of a pure protein aqueous solution. In addition, a limited uptake capacity is shown since the encapsulation efficiency decays with the amount of added protein. The optimized procedure was extended for the rest of proteins with H0 and H1 dense phases. As a general trend, higher encapsulation efficiencies were found in all cases, with the exception of SubC (Fig. 3D). The Cu-BTC phases obtained in presence of the proteins are identical to the ones obtained in its absence for both H0 and H1 (Fig. 4B and C), even though some of the H0 samples, like Cas and Ly biocomposites exhibit additional peaks at 7.7°, 9.2° and 12.0°. This pattern has been previously reported as a humidity and temperature dependent degradation product of HKUST-1.20
Essentially, the optimized synthetic scheme to obtain protein@HKUST-1 biocomposites consist in the encapsulation of proteins in H0 or H1, and their subsequent transformation to the porous HK0 or HK1 by immersion in ethanol solutions (Fig. 4A). The in situ synthesis of the dense phases in presence of the proteins does not alter their transformation process, yielding in all cases HKUST-1 (Fig. 4D and E). This transformation was also confirmed by ATR-FTIR spectroscopy, (Fig. S7, ESI†) where the initial H0 and H1 phases with clearly different spectra, lead to similar HKUST-1 spectra after transformation. Protein loadings were estimated from TGA analysis (Fig. S8, ESI†), resulting in values ranging from ∼7 to ∼13%, except for SubC@HK1, with a lower encapsulation efficiency and a lower loading (∼5%) (Tables S2 and S3, ESI†). N2 sorption studies were conducted to monitor the changes in specific surface. In all cases, a lower sorption capacity is obtained for the composites (Fig. S9 and Table S4, ESI†) as compared to the control empty material, with BET values dropping from 1529 to ∼1100–1350 m2 g−1 respectively for HK0 and the corresponding protein@HK0 materials, and from 1611 to ∼1150–1350 m2 g−1 respectively for HK1 and the protein@HK1 materials. Morphological characterization by TEM revealed the polydisperse nature of the samples (Fig. S10 and S11, ESI†).
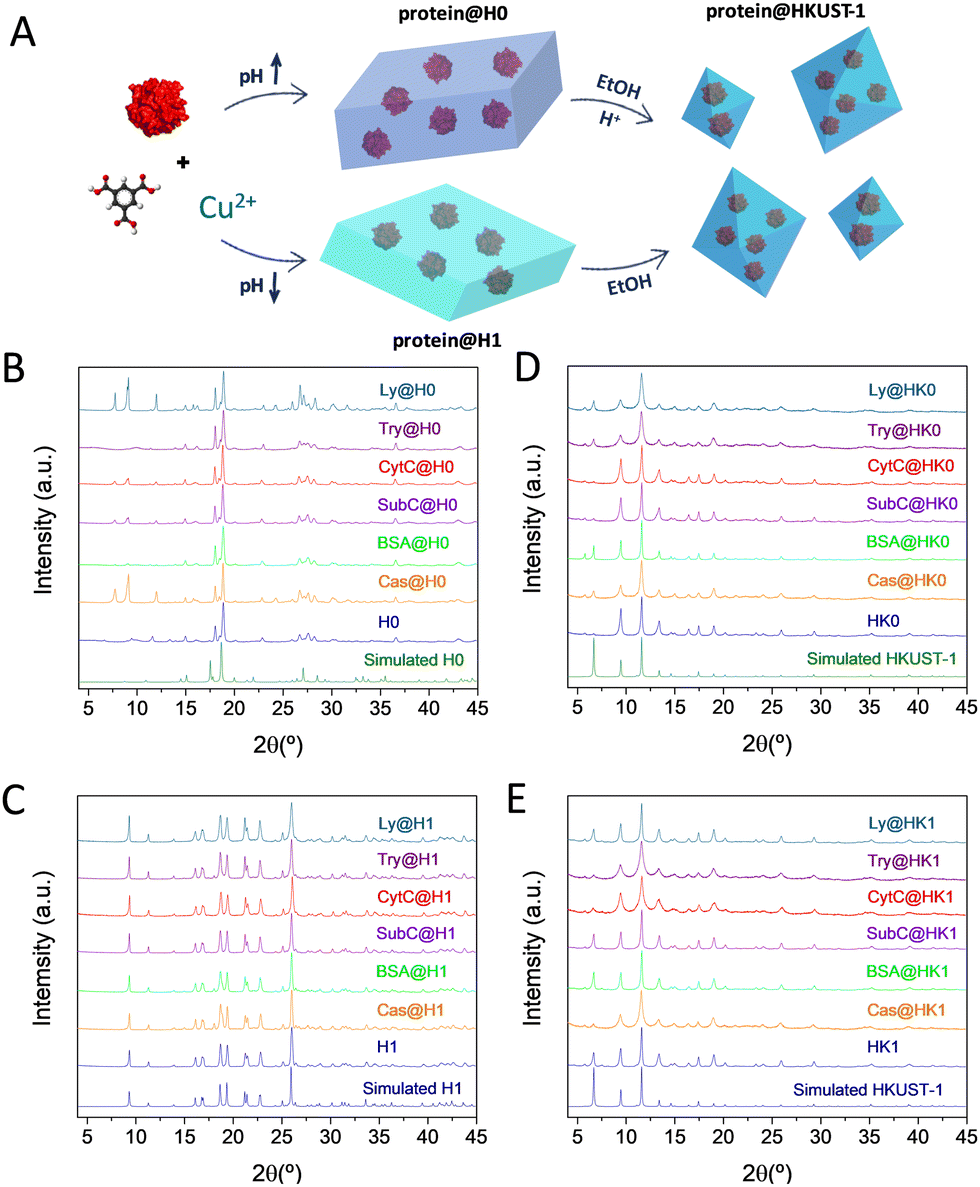 | ||
| Fig. 4 (A) Schematic representation of the protein@HKUST-1 synthetic procedure. XRPD patterns of the (B) protein@H0, (C) protein@H1, (D) protein@HK0 and (E) protein@HK1 biocomposites. | ||
The impact of protein encapsulation within dense (H0, H1) and their corresponding transformed porous phases (HK0, HK1) was evaluated by analyzing the activity of encapsulated CytC (MW 12.23 kDa, hydrodynamic diameter of 3.4 nm) a small protein involved in electron transfer in aerobic and anaerobic respiration.21 The redox ability was measured employing H2O2 and 3,3′,5,5′-tetramethylbenzidine (TMB) as substrates. H1 and related HK1 phases were preferred for further investigations, as CytC@H0 composite shows poor stability upon long term storage. H1 and HK1 control materials exhibit little peroxidase activity, attributed to the constitutive Cu2+ present in both materials (Fig. 5A).22–24 In the case of the biocomposites, CytC@H1 exhibits a similar activity than H1, while CytC@HK1 reveals a sharp activity enhancement, which is attributed to its characteristic permanent porosity that enables the transfer of substrates. Control analysis over the physical mixtures reveal that combination of CytC and H1 perform as free CytC (Fig. S12a, ESI†), whereas mixing of CytC and HK1 has a comparable activity than the related biocomposite, CytC@HK1. This trend is also observed in CytC@HK0 measured 24 h after synthesis (Fig. 5B).
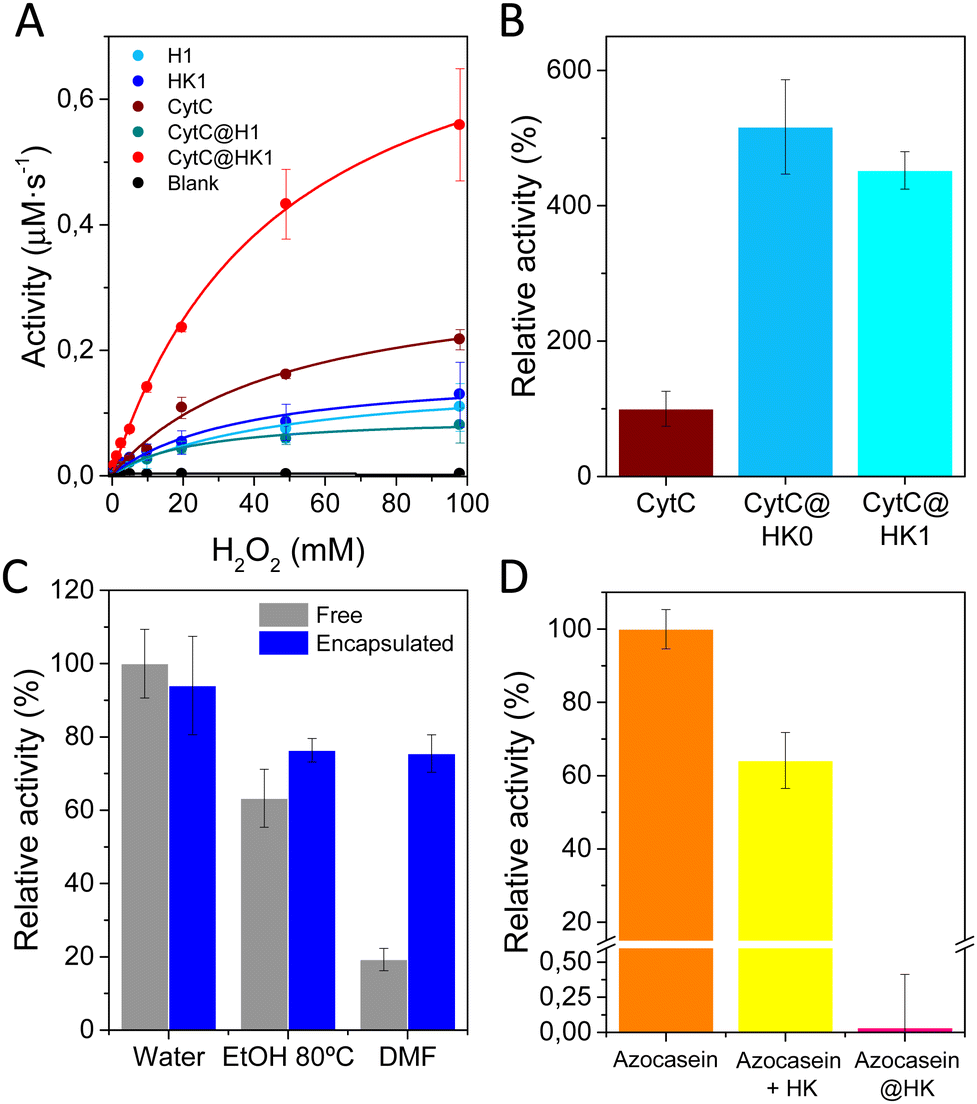 | ||
| Fig. 5 (A) Kinetic analysis of the free CytC, encapsulated CytC and CytC + MOFs physical mixtures. (B) Relative activity of CytC@HK0 and CytC@HK1 biocomposites (H2O2 3.27 mM). (C) Lysolytic activity retained by encapsulated Ly after exposure to different conditions. (D) Proteolysis of free azocasein, azocasein and HKUST-1 physical mixture, and encapsulated azocasein after treatment with protease. | ||
In view of the activity increase in both the CytC@HK1 and its related physical mixture (Table S5, ESI†), an interfacial activation is proposed.25,26 This mechanism was evidenced with a control experiment using the mixture of CytC and Cu(OAc)2, revealing a similar activity than the sum of CytC and Cu(OAc)2 and therefore dismissing an activator role for Cu2+ (Fig. S12b, ESI†). Thus, the synergistic effect observed on CytC@HK1 is attributed to the interaction of CytC with the HKUST-1. Similar peroxidase increase for CytC has been observed in Cu-based systems,27 as well as in MOFs28 and HOFs.29
In addition, it was found that Cu2+ exerts a negative impact in the activity of encapsulated SubC and Try proteases (Fig. S13 and S14a, ESI†), an inhibition that can be reversed to some extent when the protein is incubated with optimal concentrations of a chelating agent, EDTA (Fig. S14b and c, ESI†). Analogous enzymatic inhibition was observed for different enzymes in Cu-terephthalate systems,30 as many of them have reported Cu2+ inhibition,31,32 which indicates that HKUST-1 support may not be appropriate for certain proteins.
The effect of encapsulation was also evaluated with Ly, which retained a ∼90% of lysolytic activity as compared with the free form (Fig. 5C). In addition, the scaffold provides some degree of protection to external agents, as temperature or organic solvents (Fig. 5C), but most importantly, acts as a shell against proteases, as proved by the protection of the dyed derivative of casein, azocasein (Fig. 5D), thus validating the preferred encapsulation vs physical adsorption.
In summary, an alternative synthetic procedure to obtain protein@HKUST-1 composites using protein@Cu-BTC dense composites as precursors are presented. Essentially, a pH dependent in situ formation of H0 (Cu3(BTC)2·12H2O) or H1 (Cu(HBTC)·3H2O) and their direct transformations into HKUST-1 are reported. A systematic study with six different proteins reveals that encapsulation efficiency is directly affected by the pI of the protein, but also relies on the sequential or simultaneous addition of the MOF precursors. Characterization of the biocomposites demonstrate that the solid matrix confers protection against protein denaturation by organic solvents and proteolytic agents. The dense-to-porous reversible transformation enables fine-tuning of encapsulated CytC activity (4-fold increase) as a result of the change in mass transport across the support. We believe this alternative route will disclose new possible pathways for biomolecule encapsulation to overcome the elusive mineralization process of most MOFs.
This work has been supported by TED2021-132729A-I00 and PID2020-118564GA-I00 grants, funded by MCIN/AEI/10.13039/501100011033 and NextGenerationEU/PRTR, Generalitat Valenciana (PROMETEO CIPROM/2022/48), and 2022 Leonardo Grant, BBVA Foundation. M. G.-M. thank MICINN for a Ramón y Cajal (RYC2019-027902-I) fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- V. Lykourinou, Y. Chen, X. Wang, L. Meng, T. Hoang, L. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382–10385 CrossRef CAS PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 2017, 50, 1423–1432 CrossRef CAS PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 4–11 Search PubMed.
- S. Kumari, R. N. Ehrman and J. J. Gassensmith, Matter, 2023, 6, 2570–2573 CrossRef CAS.
- D. Jordahl, Z. Armstrong, Q. Li, R. Gao, W. Liu, K. Johnson, W. Brown, A. Scheiwiller, L. Feng, A. Ugrinov, H. Mao, B. Chen, M. Quadir, H. Li, Y. Pan and Z. Yang, ACS Appl. Mater. Interfaces, 2022, 14(46), 51619–51629 CrossRef CAS.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- B. Zhang, J. Zhang, C. Liu, X. Sang, L. Peng, X. Ma, T. Wu, B. Han and G. Yang, RSC Adv., 2015, 5, 37691–37696 RSC.
- Y. Mao, J. Li, W. Cao, Y. Ying, P. Hu, Y. Liu, L. Sun, H. Wang, C. Jin and X. Peng, Nat. Commun., 2014, 5, 5532 CrossRef.
- G. Majano, O. Martin, M. Hammes, S. Smeets, C. Baerlocher and J. Pérez-Ramírez, Adv. Funct. Mater., 2014, 24, 3855–3865 CrossRef CAS.
- Q.-W. Zhang and G.-X. Wang, Z. Kristallogr. - New Cryst. Struct., 2006, 221, 101–102 CAS.
- R. Pech and J. Pickardt, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 992–994 CrossRef.
- O. M. Yaghi, H. Li and T. L. Groy, J. Am. Chem. Soc., 1996, 118, 9096–9101 CrossRef CAS.
- A. Nowacka, P. Briantais, C. Prestipino, F. X. Llabrés and I. Xamena, Cryst. Growth Des., 2019, 19, 4981–4989 CrossRef CAS.
- Y.-H. Ma, P.-Z. Ma, T. Yao and J.-T. Hao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m538 CrossRef CAS.
- N. K. Maddigan, A. Tarzia, D. M. Huang, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Sci., 2018, 9, 4217–4223 RSC.
- G. Chen, X. Kou, S. Huang, L. Tong, Y. Shen, W. Zhu, F. Zhu and G. Ouyang, Angew. Chem., Int. Ed., 2020, 59, 2867–2874 CrossRef CAS PubMed.
- J. Cases Díaz, B. Lozano-Torres and M. Giménez-Marqués, Chem. Mater., 2022, 34, 7817–7827 CrossRef.
- L. R. Wetter and H. F. Deutsch, J. Biol. Chem., 1951, 192, 237–242 CrossRef CAS.
- J. B. DeCoste, G. W. Peterson, B. J. Schindler, K. L. Killops, M. A. Browe and J. J. Mahle, J. Mater. Chem. A, 2013, 1, 11922–11932 RSC.
- N. Mirkin, J. Jaconcic, V. Stojanoff and A. Moreno, Proteins: Struct., Funct., Bioinf., 2008, 70, 83–92 CrossRef CAS PubMed.
- M. Hesari, R. Jia and M. V. Mirkin, ChemElectroChem, 2022, 9, 5–9 CrossRef.
- L. Chen, C. Lin, X. Shi, L. Zhang and D. Sun, ACS Appl. Nano Mater., 2023, 6, 3835–3847 CrossRef CAS.
- X. Li, L. Gao and Z. Chen, Spectrochim. Acta, Part A, 2019, 213, 37–41 CrossRef CAS PubMed.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- T. Zisis, P. L. Freddolino, P. Turunen, M. C. F. Van Teeseling, A. E. Rowan and K. G. Blank, Biochemistry, 2015, 54, 5969–5979 CrossRef CAS.
- Z. Li, Y. Ding, S. Li, Y. Jiang, Z. Liu and J. Ge, Nanoscale, 2016, 8, 17440–17445 RSC.
- Y. Chen, F. Jimenez-Ángeles, B. Qiao, M. D. Krzyaniak, F. Sha, S. Kato, X. Gong, C. T. Buru, Z. Chen, X. Zhang, N. C. Gianneschi, M. R. Wasielewski, M. O. De La Cruz and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 18576–18582 CrossRef CAS PubMed.
- G. Chen, L. Tong, S. Huang, S. Huang, F. Zhu and G. Ouyang, Nat. Commun., 2022, 13, 4816 CrossRef CAS.
- Z. Li, H. Xia, S. Li, J. Pang, W. Zhu and Y. Jiang, Nanoscale, 2017, 9, 15298–15302 RSC.
- Y. Wu, L. Chu, W. Liu, L. Jiang, X. Chen, Y. Wang and Y. Zhao, RSC Adv., 2017, 7, 47309–47315 RSC.
- Y. Xianyu, K. Zhu, W. Chen, X. Wang, H. Zhao, J. Sun, Z. Wang and X. Jiang, Anal. Chem., 2013, 85, 7029–7032 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc04320g |
| This journal is © The Royal Society of Chemistry 2024 |