
Nano vs. bulk: surfactant-controlled photophysical and morphological features of luminescent lanthanide MOFs†
Moritz
Maxeiner
 a,
Lea
Wittig
a,
Alexander E.
Sedykh
a,
Thomas
Kasper
a and
Klaus
Müller-Buschbaum
*ab
a,
Lea
Wittig
a,
Alexander E.
Sedykh
a,
Thomas
Kasper
a and
Klaus
Müller-Buschbaum
*ab
aInstitute of Inorganic and Analytical Chemistry, Justus-Liebig-University Giessen, Heinrich-Buff-Ring 17, 35392 Giessen, Germany. E-mail: kmbac@uni-giessen.de; Web: https://www.uni-giessen.de/de/fbz/fb08/Inst/iaac/mueller-buschbaum
bCenter for Materials Research (LAMA), Justus-Liebig-University Giessen, Heinrich-Buff-Ring 16, 35392 Giessen, Germany
First published on 9th October 2023
Abstract
A surfactant-assisted bottom-up synthesis route provides access to nanoscale metal–organic frameworks (nMOFs) that exploit size-dependent property advantages. This includes an increased surface-to-volume ratio, improved dispersibility and superior morphological properties by a narrow size distribution compared to the bulk analogues. Photophysical properties such as photoluminescence are also influenced by particle size, surfactant components and post-synthetic modification. The series of the related organic linkers (H2bdc, H2bpdc and H2bpydc) together with trivalent lanthanides (Ln3+ = Eu3+, Tb3+) in synthesis and post-synthetic modification together with surface-active agents (CTAB, PVP) were used to assemble luminescent nMOFs for three archetype MOFs: nLn3+-bdc, nDUT-5:Ln3+ and nMOF-253:Ln3+. Both, dispersibility and morphology benefit from the CTAB- and PVP-controlled bottom-up particle downsizing down to 35 nm. DLS confirms homogeneous, narrow particle size distributions down to ±5 nm, which is 21-times smaller than the bulk analogues. Moreover, excellent luminescence QYs of up to 78.1(3)% were determined for the Ln3+-containing nMOFs. Successful post-synthetic modification with trivalent lanthanide ions of nMOFs was accomplished showing an improved photoluminescence sensitization effect compared to the bulk MOFs and exhibiting increased Ln3+-to-linker emission intensity ratios. The amount and photophysical properties of surfactants encapsulating the nMOFs were further quantified by DTA/TG-MS and UV-Vis-DRS. Finally, this work aims to elaborate thoroughly on the previously mentioned properties of nMOFs by comparison with their bulk analogues. Since surfactants play a key role in this synthesis route, the pros and cons of this approach were also assessed concerning several nMOF features.
Introduction
Metal–organic frameworks (MOFs) combine a wide variety of chemical adaptability with intrinsic potentially occupiable interstices, leading to a vast range of applications such as gas storage,1–3 medical implementation,4–6 catalysis,7–9 sorption matrix,10–12 sensors13–15 or ionic conductors.16–18 The ability to play a role in such a variety of applications is given by the vast possibility of combinations of various inorganic building units (IBUs) and organic linkers. The properties of MOFs strongly depend on the structure, electronic states, functional groups, and synthesis parameters, making MOFs highly tuneable porous materials.19 Trivalent lanthanide (Ln3+)-containing MOFs are of a particular interest, as their unique photophysical properties render them highly suitable for the use as optical sensors.20–22 The light uptake by direct excitation of Ln3+ ions is low, due to the non-binding character of electronically isolated 4f-orbitals and the parity-prohibition of 4f–4f transitions according to Laporte's rule.23,24 However, the light uptake of Ln3+ can be significantly enhanced indirectly by a proper organic linker capable of a high light absorption followed by an energy transfer to Ln3+ ions, from which light is emitted. Thereby, this ‘antenna effect’ or ‘sensitization’ overall enhances the luminescence properties of the composite material.25 This includes lanthanide ions inside MOF pores.26 The range of emitted light covers both, the vis-region from 400 nm to 800 nm and also the NIR-region from 800 nm to 1700 nm is possible.27 This is an interesting endorsement of the photophysical capability of lanthanide MOFs and provides excess to applications such as luminescent MOF barcodes.28The interest in nanoparticles has grown enormously over the last two decades due to their unique size-dependent properties, which can be readily influenced by limiting their particle growth. As a result, they outperform their bulk analogues with higher surface-to-volume ratios, increased solvent affinity, enhanced particle penetration rates, and newly acquired photophysical properties.29 Nowadays, the importance of functionalized nanomaterials extends to a multitude of sectors, for instance, electronics30–32 and medical applications.33–35 However, the generation of novel nanomaterials is still challenging, as inevitable processes such as Ostwald ripening or agglomeration must be overcome. Therefore, well-designed synthesis routes with size limiting features are required. Several methods have been established using either chemical additives or physical concepts in bottom-up and top-down routes, respectively.36,37
The contradicting idea of reducing an extended three-dimensional network in size, which is defined by its long-range order, crystallinity, and an enormous number of pores as in bulk MOFs, consequently, takes these specific properties away but leads to nano-scaled MOF (nMOF) particles consisting of only a couple of unit cells. Furthermore, nMOFs offer the advantages stated above of bulk MOFs, plus those given by their nano-character.38 The bottom-up downsizing results in a higher specific surface area and less gravitational influence on dispersibility compared to bulk MOFs, as well as defined optical properties through the incorporation of luminophores.39 Therefore, distinct improvements can be achieved in the synthesis and application of composites containing nLn3+-MOFs compared to their bulk analogues. For instance, enhanced homogeneity and stability of a dispersion or ameliorated sorption efficiency.
In principle, nLn3+-MOFs can be synthesized by a bottom-up40 or top-down approach.41 In this work, the bottom-up approach implements a template-based method by using surface-active agents (surfactants).42Scheme 1 shows the behaviour of the cationic tenside N,N,N-trimethylhexadecan-1-ammonium bromide (CTAB) and polyvinylpyrrolidone (Mw = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, PVP40000) in solution prior to the synthesis of the nMOF, enclosing both the IBU (Ln3+ or Al3+) and the deprotonated linker in a confined synthesis space, which is the key to yield nMOFs and the method of choice in this work.
000 g mol−1, PVP40000) in solution prior to the synthesis of the nMOF, enclosing both the IBU (Ln3+ or Al3+) and the deprotonated linker in a confined synthesis space, which is the key to yield nMOFs and the method of choice in this work.
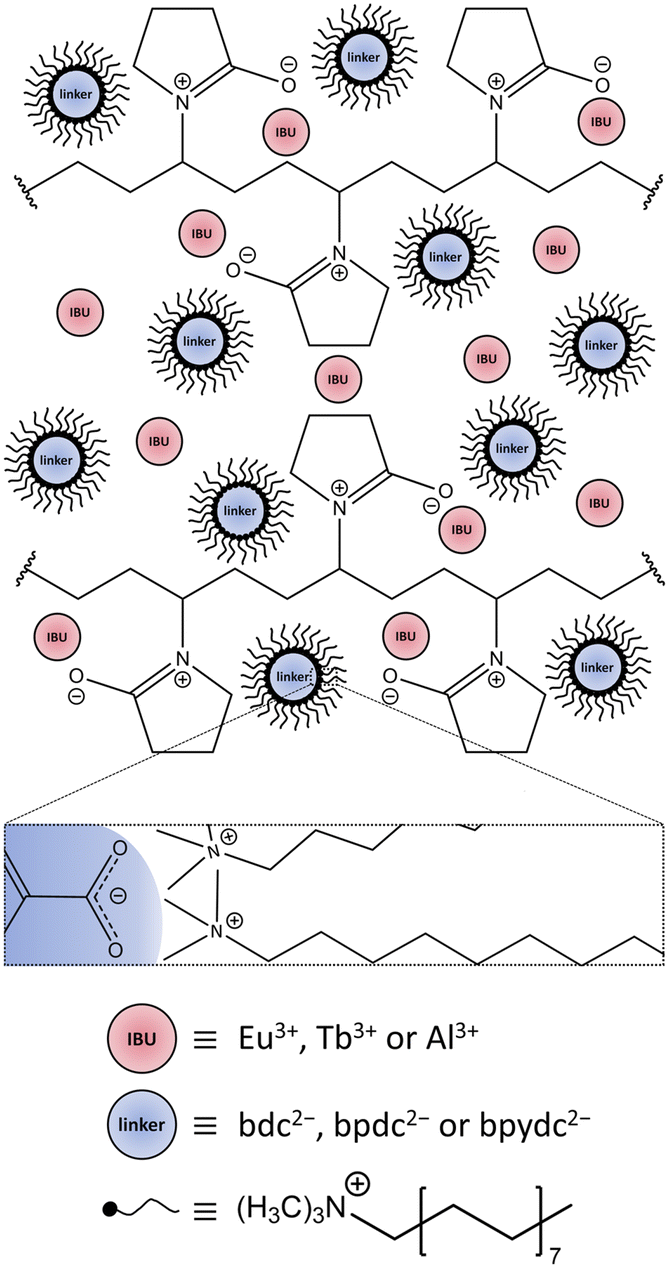 | ||
| Scheme 1 Depiction of the confined space made up by the surfactants CTAB and PVP40000, enclosing the IBU and linker ions prior to the synthesis. Red circles represent the IBU cations Eu3+ for Eu3+-bdc, Tb3+ for Tb3+-bdc, and Al3+ for DUT-5 and MOF-253, respectively. Blue circles represent the deprotonated linkers bdc2−, bpdc2− and bpydc2− for Ln3+-bdc, DUT-5 and MOF-253, respectively. The linkers are surrounded by black, tailed symbols representing reverse micelles, which are formed by CTAB. The enlarged rectangular box shows Coulomb-attraction between the negatively charged carboxylate group of the linker and the positively charged ammonium part of the CTAB in more detail. PVP40000 forms layers enclosing a limited amount of IBU and the linker-containing reverse micelles. Both, CTAB and PVP40000 set up the steric synthesis parameters for nMOFs. Solvent molecules, NO3− and Br− counterions have been omitted for clarity. The dimensions do not correspond to reality. | ||
Ln3+ were incorporated into MOFs either during the MOF synthesis or by post-synthetic modification of the synthesized MOF. The Ln3+-bdc (Ln3+ = Eu3+, Tb3+) has been prepared by mixing Eu3+- or Tb3+-salts as IBU with benzene-1,4-dicarboxylic acid (H2bdc) as linker. For the post-synthetic modification approach, MOFs have been synthesized with Al3+-salts as IBU with biphenyl-4,4′-dicarboxylic acid (H2bpdc) or 2,2′-bipyridine-5,5′-dicarboxylic acid (H2bpydc) as linkers. These MOFs are hereafter referred to as DUT-5:Ln3+ and MOF-253:Ln3+ (Ln3+ = Eu3+, Tb3+) according to their impregnation with Eu3+ ions or Tb3+ ions. An overview of the MOFs used in this work and the type of Ln3+-incorporation is given in Table 1.
A variety of analytical techniques were used to fully characterize the systems. Powder X-ray diffraction (PXRD), dynamic light scattering (DLS), scanning electron microscopy (SEM), microwave plasma – atomic emission spectroscopy (MP-AES), organic elemental analysis (OES) and simultaneous differential thermoanalysis/thermogravimetry combined with mass spectrometry (DTA/TG-MS) were carried out to entirely characterize the structure, composition, and particle morphology. In order to provide photophysical data, a spectrophotometer was used equipped with different setups for excitation and emission spectra (PL), luminescence decay (lifetime τ), quantum yield (QY) and linker triplet state determination. A UV-Vis diffuse reflectance spectrophotometer (UV-Vis-DRS) complemented the photophysical data acquisition.
For this work, three archetype MOFs were both synthesized as bulk (Ln3+-bdc, DUT-5, MOF-253, the latter two were post-synthetically modified with Ln3+: DUT-5:Ln3+, MOF-253:Ln3+) and nano (nLn3+-bdc, nDUT-5:Ln3+, nMOF-253:Ln3+) batch materials and discussed concerning their properties. A comparison of bulk MOFs and nMOFs on morphological and photophysical properties reveals the potential of nanomaterials.
Results and discussion
Crystal structures of Ln3+-bdc, DUT-5 and MOF-253
The successful bottom-up surfactant-assisted downsizing of Ln3+-bdc, DUT-5 and MOF-253 to nMOFs has been confirmed by PXRD in terms of crystallinity, purity, and potential side phases. Fig. 1 shows the PXRD results of nLn3+-bdc, nDUT-5 and nMOF-253 as well as the simulated diffractograms for the single-crystal data of Ln3+-bdc, DUT-5 and MOF-253, respectively. Moreover, the diffractograms of the corresponding bulk MOFs are shown in Fig. SI3–6.† All synthesized bulk MOFs and nMOFs correspond to the simulated powder patterns based on single crystal structure data. The crystal structures of the archetype MOFs Tb3+-bdc,43 DUT-544 and MOF-25345 are well known (for clarity, they are depicted in Fig. SI1 and 2†). PXRD reveals nEu3+-bdc and nTb3+-bdc to be isostructural and therefore called nLn3+-bdc (with Ln3+ = Eu3+, Tb3+). While the crystal structures of nDUT-5 and nMOF-253 are directly related. A broadening of the reflections is apparent for all nMOFs compared to the diffraction patterns of their bulk analogues. This can be explained by the reduction of crystallinity due to the decrease in particle size. The described phenomenon was already previously observed for other nMOFs.46 However, crystallite size determination using the Scherrer equation is not applicable due to broad, unseparated reflections. A slight overall shift of 2θ is noticeable in the diffractograms of MOF-253 and nMOF-253 compared to the simulated powder pattern of MOF-253, which was also previously observed by Deng et al. for the archetype MOF-253.47
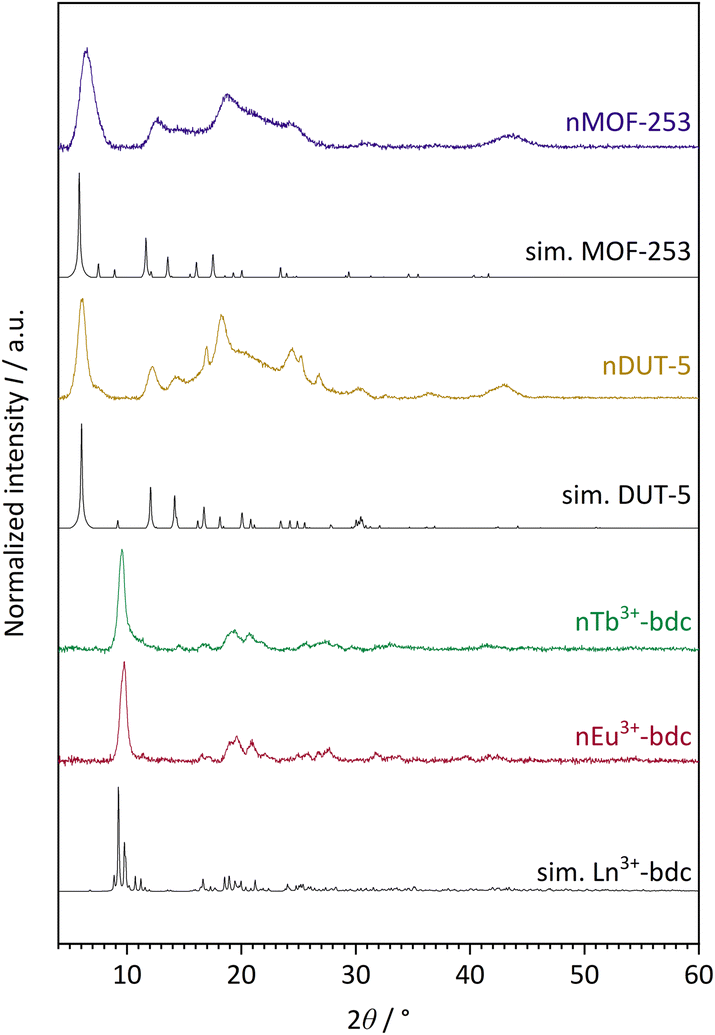 | ||
| Fig. 1 Experimental powder patterns of the investigated nMOFs: nEu3+-bdc, nTb3+-bdc, nDUT-5 and nMOF-253. Furthermore, simulated (sim.) powder patterns for single crystal data of Tb3+-bdc,43 DUT-5 (ref. 44) and MOF-253 (ref. 45) are shown. Since nEu3+-bdc and nTb3+-bdc are isostructural they are denoted as Ln3+-bdc (with Ln3+ = Eu3+, Tb3+). Both, experimental and single crystal data were acquired at room temperature and at ambient air atmosphere. | ||
Noteworthy is a broad reflection between 15–30° for nDUT-5 and nMOF-253, which belongs to the surface-located CTAB. This was also shown by Chhetri et al. for CTAB-functionalized MoS2. They described both CTAB attached and intercalated to the surface and in between MoS2 nanolayers, respectively.48 In our work, the cationic tenside CTAB forms reversible reverse micelles due to Coulomb-interactions with the unsaturated anionic carboxylate groups at the MOF surface. In contrast to micelles, the hydrophilic heads of reverse micelles face the centre, while at the outside, the tails form a hydrophobic interface with the solvent. A dynamic process of enclosing the MOF is essential to allow MOF synthesis. But the surfactant limits particle growth and also affects particle shape and dispersibility.49,50 Moreover, the presence of small amounts of residual amorphous polymer PVP40000 cannot be excluded by PXRD since this method is sensitive to the diffraction of crystalline materials only and therefore unable to detect thin layers of amorphous materials as PVP40000. In fact, PVP40000 forms non-linear layers in solution that resemble a confined space synthesis by encapsulating all the reactants. Particle growth becomes limited and unidirectional due to the limited amount of IBU and linker within this space.51
Besides, the results do not show reflections of the reagents, side phases or other non-assignable reflections, respectively.
Composition and morphology
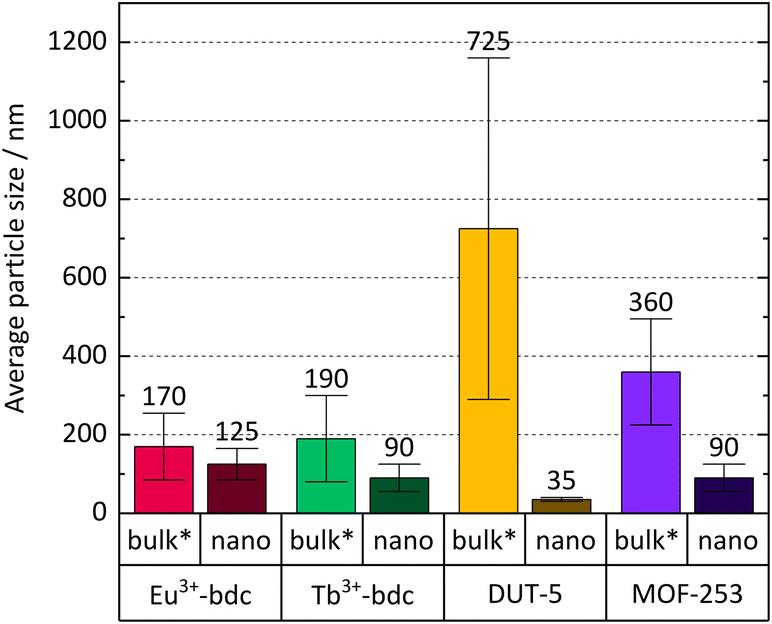 | ||
| Fig. 2 Comparison of average particle size and particle size distribution (error bars) of Ln3+-bdc, nLn3+-bdc (Ln3+ = Eu3+, Tb3+), DUT-5, nDUT-5, MOF-253 and nMOF-253. Bulk MOF sizes supposed to be larger than stated due to an unavoidable sedimentation of larger particles in the dispersion during the DLS measurement and are therefore marked with an asterisk. | ||
| Ln3+/wt% | Particle size/nm | Wavelength λmax/nm | QY/% | Lifetime τ/ms | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DLSa | SEM | PL ex | PL em | Ln3+ | Linker | τ 1 | τ 2 | |||
| MeCN | EtOH | |||||||||
| a Particle sizes of bulk MOFs are supposed to be even higher due to sedimentation. b Recording of linker-based emission spectrum. c No Ln3+ present in the sample. d Only linker-based emission observable, no sufficient energy transfer from linker to Tb3+. e No linker-based emission intensity observable in PL spectra. f An exponential fit of first order was used. g Measurements not carried out in MeCN. | ||||||||||
| Eu3+-bdc | 32 | n/ag | 170 ± 85 | >1000 | 288 | 614 | 33(2) | n/ae | 0.9055(6) | n/af |
| nEu3+-bdc | 32 | n/ag | 125 ± 40 | 20–60 | 296 | 614 | 23.7(3) | n/ae | 0.701(4) | 1.071(3) |
| Tb3+-bdc | 38 | n/ag | 190 ± 110 | >1000 | 300 | 544 | 94(2) | n/ae | 1.396(2) | n/af |
| nTb3+-bdc | 33 | n/ag | 90 ± 35 | 20–60 | 286 | 543 | 78.1(3) | n/ae | 1.04(4) | 1.612(3) |
| DUT-5 | n/ac | n/ag | 725 ± 430 | >400 | 307 | 376b | n/ac | 5.1(5) | 2.49(6) × 10−6 | n/af |
| nDUT-5 | n/ac | n/ag | 35 ± 5 | 10–20 | 326 | 378b | n/ac | 14.8(6) | 3.6(1) × 10−6 | n/af |
| DUT-5:Eu3+ | 2 | 265 ± 115 | 500 ± 90 | >400 | 307 | 616 | 2.1(2) | 10.4(3) | 0.288(6) | 0.596(6) |
| nDUT-5:Eu3+ | 2 | 175 ± 55 | 110 ± 10 | 20–50 | 310 | 616 | 3.6(1) | 4.5(3) | 0.306(8) | 0.70(1) |
| DUT-5:Tb3+ | 2 | 290 ± 110 | 385 ± 260 | >400 | 416 | 544 | n/ad | 11.5(3) | n/ad | n/ad |
| nDUT-5:Tb3+ | 1 | 160 ± 35 | 105 ± 10 | 20–50 | 427 | 544 | n/ad | 4.6(2) | n/ad | n/ad |
| MOF-253 | n/ac | n/ag | 360 ± 135 | >400 | 370 | 440b | n/ac | <1 | 1.19(6) × 10−6 | 6.4(1) × 10−6 |
| nMOF-253 | n/ac | n/ag | 90 ± 20 | 15–30 | 366 | 536b | n/ac | <1 | 0.559(7) × 10−6 | 2.13(5) × 10−6 |
| MOF-253:Eu3+ | 3 | 185 ± 180 | 150 ± 60 | >400 | 327 | 616 | 2.0(1) | n/ae | 0.204(3) | 0.432(3) |
| nMOF-253:Eu3+ | 4 | 135 ± 70 | 155 ± 80 | 20–50 | 310 | 616 | 1.79(3) | n/ae | 0.14(2) | 0.308(6) |
| MOF-253:Tb3+ | 7 | 155 ± 70 | 130 ± 45 | >400 | 374 | 544 | n/ad | <1 | n/ad | n/ad |
| nMOF-253:Tb3+ | 2 | 100 ± 10 | 155 ± 70 | 20–50 | 371 | 544 | n/ad | <1 | n/ad | n/ad |
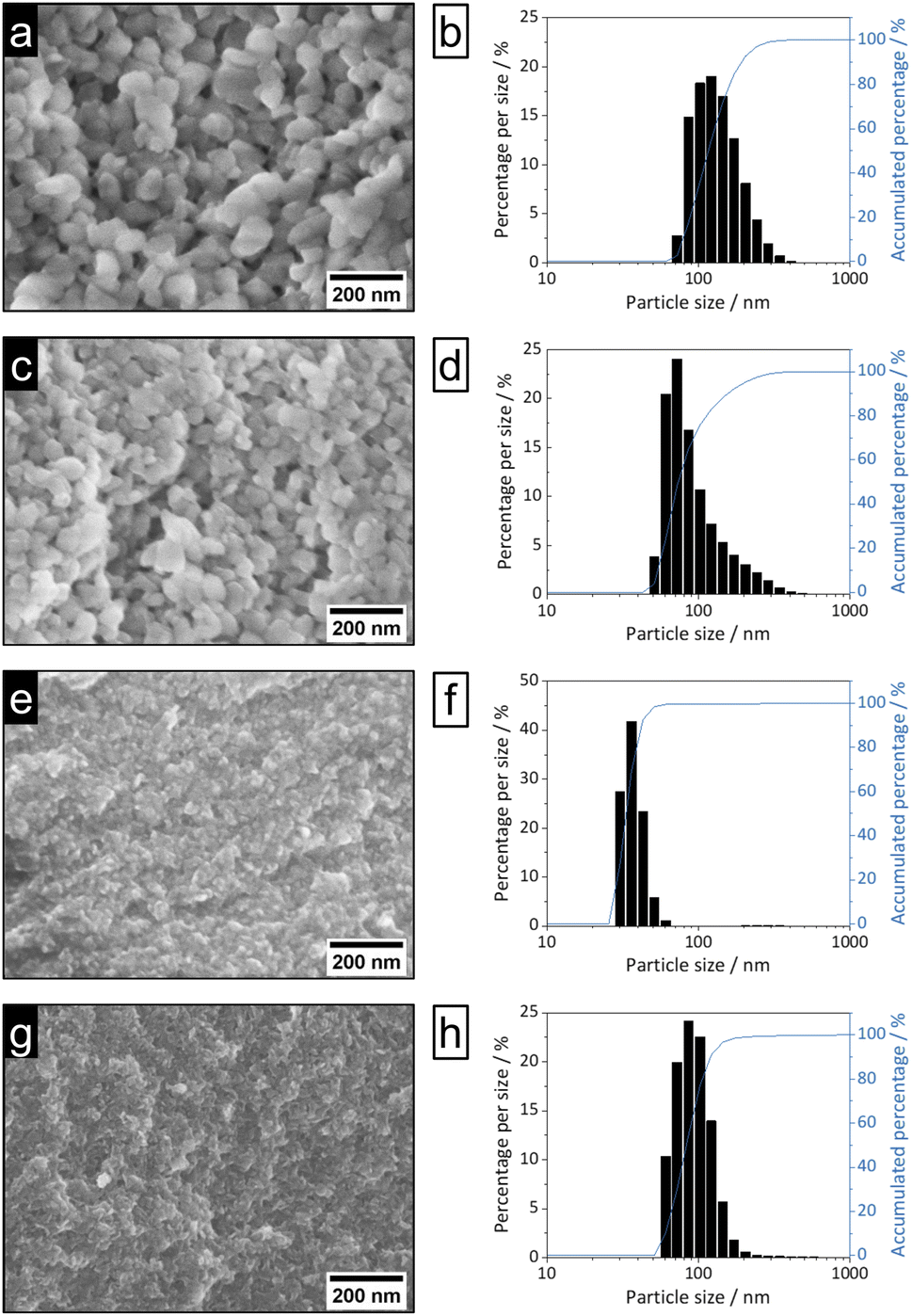 | ||
| Fig. 3 SEM images (100k magnification, left) and DLS particle size distributions (right) of nEu3+-bdc (a and b), nTb3+-bdc (c and d), nDUT-5 (e and f) and nMOF-253 (g and h). | ||
Bulk MOF synthesis results in clefted microparticles (>400 nm) for Ln3+-bdc, and spherical particles (>1000 nm) for DUT-5 and MOF-253, respectively, as SEM demonstrates. Furthermore, particle size determination based on DLS reveals particles sizes of 170 ± 85 nm for Eu3+-bdc, 190 ± 110 nm for Tb3+-bdc, 725 ± 430 nm for DUT-5 and 360 ± 135 nm for MOF-253. However, bulk MOF particle sizes again supposed to be even higher due to an unavoidable sedimentation process during the DLS data acquisition (Fig. SI10†).
As PXRD and SEM already indicated the presence of surfactants at the outside of the nMOFs, coverage with PVP40000 as well as the reverse micelle formation of CTAB lead to narrow particle size distributions and smaller particle sizes (Fig. 3b, d, f and h). In addition and beneficially, a dispersion with nMOFs is stabilized by residual surfactants and prevents sedimentation during DLS data acquisition. Scheme 2 illustrates how the surfactants are enclosing a nMOF particle which leads to the stabilization effect. It should be emphasized that nDUT-5 particles were significantly reduced in size by a factor of ≈21 and remarkably homogenized compared to DUT-5 (see Fig. 2).
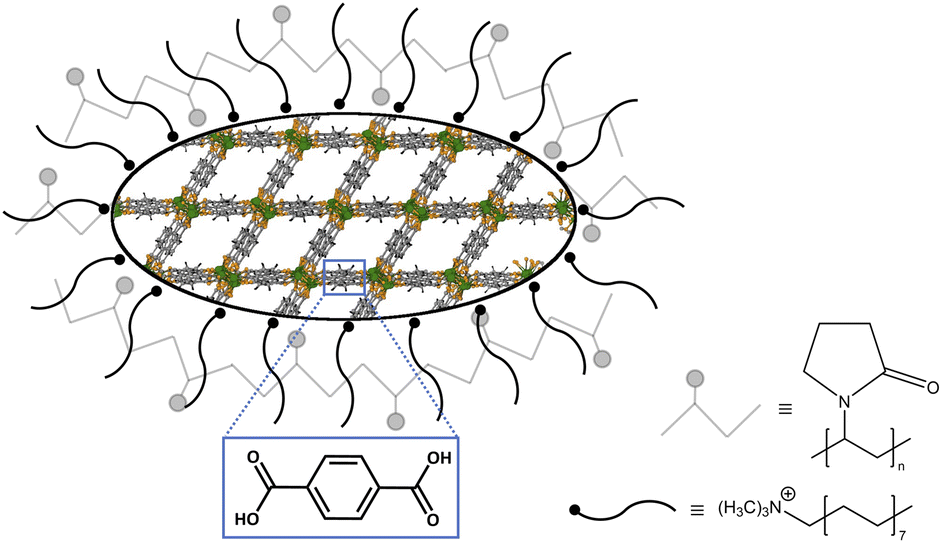 | ||
| Scheme 2 Depiction of a surfactant-stabilized nMOF particle. The Tb3+-bdc crystal structure is shown within the particle-shaped area and represents the MOFs used. Atom color code: green (Tb), yellow (O), grey (C), black (H). The nMOF is surrounded by black, tailed symbols representing reverse micelles, which are formed by CTAB. PVP40000 impacts the particle shape by enclosing the nMOF depicted as pale-grey chains. The blue box shows exemplarily the H2bdc in Tb3+-bdc as representative for the linkers used. | ||
Further information on the presence of surfactants on bulk MOFs and nMOFs was obtained by DTA/TG-MS using synthetic air as the working gas, which can oxidize the MOF instead of typical inert gases such as Ar/N2. DTA/TG-MS results are shown in Fig. 4 and as enlarged graphics in Fig. SI14–16 in the ESI.† DTA/TG confirm an exothermic mass loss of 15 wt% and 7 wt% for Tb3+-bdc (Fig. 4a) and nTb3+-bdc (Fig. 4b), respectively, at 230–270 °C. MS signals identify these mass losses as the solvent molecules H2O and DMF either located in the pores or at the surface of the MOFs. The mass loss of DUT-5 (Fig. 4c) with 40 wt% is higher than the mass loss of nDUT-5 (Fig. 4d) with 10 wt% at 100–250 °C. Consequently, both nTb3+-bdc and nDUT-5 contain less solvents than their bulk analogues, suggesting that surfactants partially replace solvents in the pores and on the surface of nMOFs.
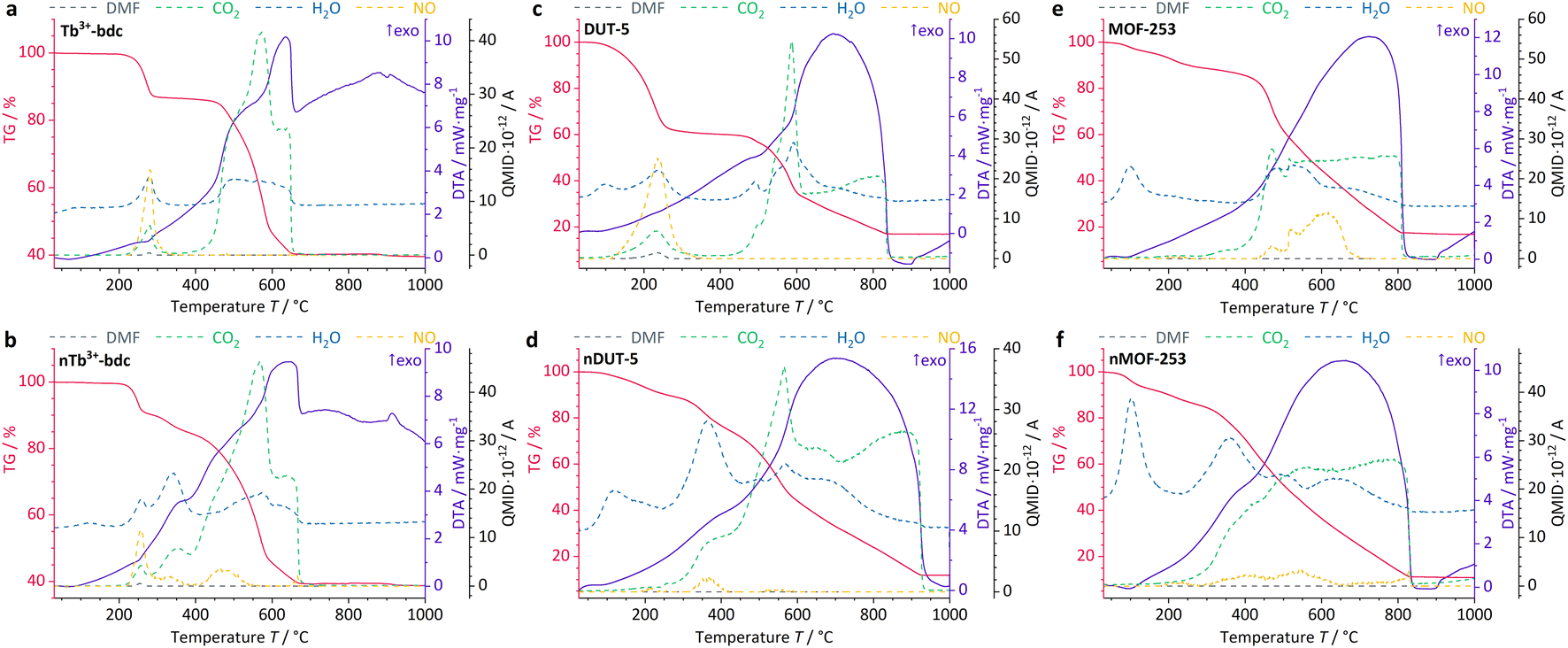 | ||
| Fig. 4 DTA/TG-MS results of investigated bulk MOFs and nMOFs at synthetic air atmosphere. Tb3+-bdc (a), nTb3+-bdc (b), DUT-5 (c), nDUT-5 (d), MOF-253 (e) and nMOF-253 (f). NO signal (yellow) is given in QMID × 10−13. A for a more detailed insight. | ||
In Fig. 4b, mass losses of 1 wt% at 305–345 °C and 13 wt% at 400–500 °C were observed for nTb3+-bdc, attributed to the oxidation of CTAB and PVP40000, respectively. Results from MS allow for the identification of CO2, NO and H2O as oxidation products, confirming the oxidation of surfactants, for the deprotonated linker bdc2− does not contain nitrogen. In the oxidation process, a shorter aliphatic chain as in CTAB requires less energy to be cracked than a longer polymeric chain as in PVP40000. In fact, this effect has been previously reported for CTAB52 and PVP40000 (ref. 53) for the above-mentioned temperature ranges. The powder pattern of nTb3+-bdc does not show any broad reflection at 12–30° (Fig. 1), therefore the amount of surfactant must be lower than for nDUT-5 and nMOF-253. In corroboration, both, nDUT-5 and nMOF-253 show a higher mass loss of 30 wt% at 300–470 °C (Fig. 4d and f). Moreover, NO signals at 310–420 °C and 300–470 °C for nDUT-5 and nMOF-253, respectively, are indicating surfactant oxidation and further confirm this assignment.
NO signals starting at 500 °C can be assigned to the oxidation of the N-containing linker bpydc2− (Fig. 4e and f). Additionally, the ongoing oxidation of organic linkers is also indicated by the CO2 signal starting at 400 °C for Tb3+-bdc and nTb3+-bdc while the oxidation of the linkers of DUT-5, nDUT-5, MOF-253 and nMOF-253 continues at 500 °C. The DTA/TG-MS results do not show any NO signals at 320–420 °C for the investigated bulk MOFs, thus the mass loss can be attributed to the amount of surfactants present in the investigated nMOFs by comparing bulk MOF and nMOF results.
At temperatures higher than 650 °C, Tb3+-bdc and nTb3+-bdc become fully oxidized accompanied by an altogether mass loss of 60 wt% with Tb4O7 being formed (Fig. 4a and b). Higher temperatures, starting at 850 °C are necessary to fully oxidize DUT-5 and nDUT-5 as well as MOF-253 and nMOF-253 to Al2O3 with an altogether mass loss of 85 wt% (Fig. 4c–f). Both, Tb4O7 and Al2O3 are confirmed as oxidation products by PXRD (Fig. SI9†).
The amount of Ln3+ was determined by MP-AES and the results are presented in Table 2. Ln3+-bdc and nLn3+-bdc contain 32–38 wt% Ln3+ depending on the lanthanide and MOF. In contrast, the post-synthetically modified DUT-5:Ln3+ and nDUT-5:Ln3+ contain only 1–2 wt% Ln3+, while MOF-253:Ln3+ and nMOF-253:Ln3+ contain 2–7 wt% Ln3+, again depending on lanthanide and MOF. The difference in the amount of Ln3+ between both synthesis approaches is explainable by the function of Ln3+ ions in the MOFs. As main connectivity centre, Ln3+ is much more present in Ln3+-bdc than by impregnation of DUT-5:Ln3+ and MOF-253:Ln3+. It can be seen that the surfactants do not have a distinct impact on the uptake of Ln3+ ions, as the amount of Ln3+ do not follow any comprehensible trend between bulk MOFs and nMOFs.
Powder patterns of DUT-5:Ln3+ and nDUT-5:Ln3+ (Fig. SI7†) as well as MOF-253:Ln3+ and nMOF-253:Ln3+ (Fig. SI8†) reveal stable crystal structures for MOF:Ln3+s impregnated with Eu3+ and Tb3+. As a side effect of the post-synthetic modification process, the broad reflection at 12–30° 2θ in the diffractograms of nDUT-5 and nMOF-253 is less intense than in the diffractograms of nDUT-5:Ln3+ and nMOF-253:Ln3+. Hence, SEM images of nDUT-5:Ln3+ and nMOF-253:Ln3+ modified in MeCN were recorded to check whether a reduced amount of surfactant can be observed. The partial purification from surfactants results in distinguishable particles for nDUT-5:Ln3+ and nMOF-253:Ln3+ (Fig. 5) meaning a lesser amount of surfactants are covering nMOF particles.
 | ||
| Fig. 5 SEM images (100k magnification) of (a) nDUT-5:Eu3+, (b) nDUT-5:Tb3+, (c) nMOF-253:Eu3+ and (d) nMOF-253:Tb3+. | ||
Complementary results on particle size and its distribution for the post-synthetically modified MOF:Ln3+ and nMOF:Ln3+ were obtained by DLS and are given in Table 2. DLS reveals the influence of the reduced amount of surfactants on the morphological properties and particle growth indicated by increasing particle sizes (130–140 nm for nDUT-5:Ln3+ and 10–50 nm for nMOF-253:Ln3+, respectively, see also Fig. SI11†). Processes such as crystal growth during post-synthetic modification due to a more accessible nMOF surface, which was previously covered with surfactants, or swelling of the crystal structure due to Ln3+ insertion are likely to be responsible for the determined increased particle sizes. In addition, penetration of the crystal structure by surfactants or formation of holes/defects by anions to compensate for the positive charge brought in by Ln3+ also contribute to the swelling effect.54 This increase in particle size is more prominent for nDUT-5:Ln3+ than for nMOF-253:Ln3+ due to the smaller particle size of nDUT-5. The smaller the particles, the higher the surface energy and the more distinct surface energy reducing mechanisms become, such as Ostwald-ripening, which consequently leads to crystal growth.55 In addition, bulk MOF:Ln3+ particle sizes are hardly comparable with non-modified bulk MOF particle sizes due to sedimentation processes described above during the DLS data acquisition, and therefore, do not follow any discernible trend (Fig. SI12 and 13†).
Altogether, these results point to the presence of residual surfactants after the post-synthetic modification process, that allows nDUT-5:Ln3+ and nMOF-253:Ln3+ to form stable and homogeneous dispersions with slightly increased particle sizes, due to a swelling of the crystal structure and less coverage by surfactants.
Photophysical properties
The evaluation of the photophysical properties of bulk MOFs and nMOFs was accomplished by comparing the obtained results of qualitative PL spectra, overall luminescence decay times τ, QYs and linker triplet state energy determinations, as well as UV-Vis-DRS.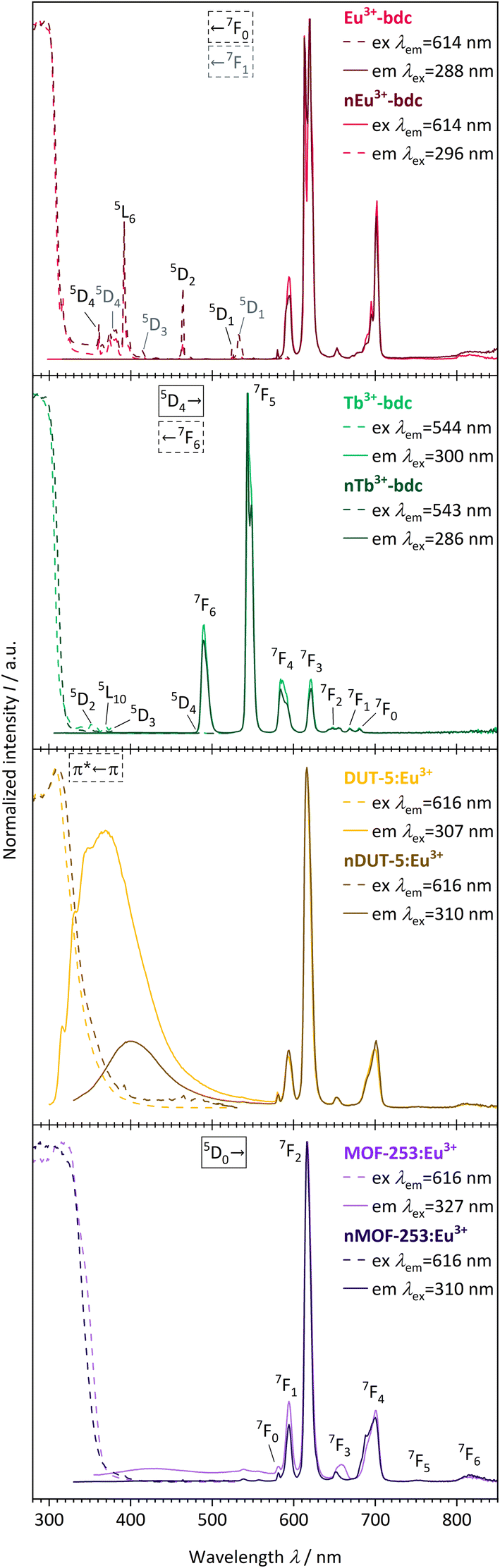 | ||
| Fig. 6 Qualitative PL excitation and emission spectra of Eu3+-bdc (pale red), nEu3+-bdc (dark red), Tb3+-bdc (pale green), nTb3+-bdc (dark green), DUT-5:Eu3+ (pale yellow), nDUT-5:Eu3+ (dark yellow), MOF-253:Eu3+ (pale violet) and nMOF-253:Eu3+ (dark violet). Electronic states are exemplarily denoted for clarity with the corresponding electronic transitions for the linker, Eu3+ and Tb3+, respectively.56,58 | ||
The emission spectra of the investigated Eu3+-containing MOFs and nMOFs show the typical Eu3+ transitions 5D0 → 7F0, 5D0 → 7F1, 5D0 → 7F2, 5D0 → 7F3, 5D0 → 7F4, 5D0 → 7F5 and 5D0 → 7F6, which correspond to the emission signals at 580 nm, 593 nm, 616 nm, 652 nm, 700 nm, 753 nm, and 810 nm, respectively. The emission bands differ in terms of relative intensity and shape. For instance, the coordinated Eu3+ in Eu3+-bdc and nEu3+-bdc causes a splitting of the 5D0 → 7F2 and 5D0 → 7F4 transitions, while Eu3+ inside the pores of DUT-5:Eu3+ and MOF-253:Eu3+ gives broader, single-maxima emission bands. This signal broadening is indicative for multiple, different local electric fields of Eu3+ in the pores or on the MOF surface, which differs from the distinct crystallographic Ln3+-positions in Ln3+-bdc and nLn3+-bdc. For the post-synthetic modification, the MOF pores allow for multiple locations inside the pores for Eu3+. In addition, the relative intensities of 5D0 → 7F2 and 5D0 → 7F4 vary between DUT-5:Eu3+ and MOF-253:Eu3+, depending on the specific Eu3+ position within the crystal structure. Both effects are well known in the literature56 and provide evidence for different electrostatic environments for Eu3+ inside the pores. Furthermore, the asymmetry ratio of Eu3+ provides valuable insights concerning its local environment and is defined as the ratio of the integrated intensities I(5D0 → 7F2) and I(5D0 → 7F1). As the 5D0 → 7F1 transition is predominantly unaffected by the local environment,57 a low asymmetry ratio indicates minimal distortions in the electric field, with 5D0 → 7F2 being comparatively weaker and Eu3+ is located at distinct positions. However, when the distortion in the electrostatic environment of Eu3+ increases, i.e. Eu3+ is located at positions with different local environments, the intensity of the 5D0 → 7F2 transition also increases, resulting in a higher asymmetry ratio. The calculated asymmetry ratios for bulk Eu3+-bdc and nEu3+-bdc are 5.07 and 6.57, respectively. This observation supports the assertion that surfactants remain present in the pores and/or at the surface of nEu3+-bdc after the washing procedure, leading to distortion of the local environment of Eu3+. The post-synthetically modified MOF-253:Eu3+ and nMOF-253:Eu3+ also following the described trend indicative by the asymmetry ratios 4.47 and 6.49, respectively. Additionally, in the case of the post-synthetically modified MOFs, the higher average diffusion pathway affords improved pore occupancy of Eu3+, resulting in even more different electrostatic environments than in Ln3+-bdc. Asymmetry ratios of 6.34 and 6.35 are observed for DUT-5:Eu3+ and nDUT-5:Eu3+, respectively. These ratios indicate a more distorted local environment for Eu3+ in DUT-5:Eu3+ compared to MOF-253:Eu3+ and Eu3+-bdc, while the environment of Eu3+ in nDUT-5:Eu3+ is similar to other nMOF-253:Eu3+ and Eu3+-bdc.
The emission spectra of Tb3+-bdc and nTb3+-bdc exhibit the typical emission pattern of Tb3+ with the transitions 5D4 → 7F6, 5D4 → 7F5, 5D4 → 7F4, 5D4 → 7F3,5D4 → 7F2, 5D4 → 7F1 and 5D4 → 7F0, which can be attributed to sharp bands at 489 nm, 543 nm, 584 nm, 621 nm, 647 nm, 669 nm, and 680 nm, respectively.58 Tb3+ does not show emission in post-synthetically modified DUT-5:Tb3+, nDUT-5:Tb3+, MOF-253:Tb3+ and nMOF-253:Tb3+ since the sensitization of Tb3+ by the energy transfer from the linkers to Tb3+ is not preferred (Fig. SI21 and 22†). In order to investigate the reasons for the neglectable energy transfer, the donor triplet state energies of the linkers were determined and subsequently compared with the emitting energy state level 5D4 of Tb3+ in terms of energy. Latva et al.59 state that donor–acceptor energy states should be optimally separated by 2000–4000 cm−1. The emitting state 5D4 of Tb3+ is located at 20470 cm−1,60 whereas the donor triplet states of Tb3+-bdc, DUT-5 and MOF-253 are located at 24691 cm−1, 21079 cm−1 and 20877 cm−1, respectively (Fig. SI23–25†). Since the energy states for DUT-5 and MOF-253 are energetically too close to the Tb3+ energy state 5D4, high energy back-transfer rates dominate the systems, and therefore no Tb3+-based emissions are observed. In contrast, Eu3+ has two energetic states 5D1,2 at 19026 cm−1 and 21499 cm−1, respectively, which get addressed to as acceptor states61 for the sensitization energy transfer from the linker to Eu3+ and is in accordance with Latva's rule.
Altogether, the comparison of Ln3+-containing MOFs and nMOFs show a benefit of nanoscale particles regarding the sensitizing efficiency as indicated by a strong decrease in the intensity of the linker-based emission bands.
The previously described emission processes were further characterised by performing luminescence overall decay time measurements to determine lifetimes τ (Fig. SI26–31†), with the results being given in Table 2. Lifetimes of the emitting states in millisecond range indicate quantum mechanically unfavoured phosphorescent emission processes caused by parity forbidden 4f–4f transitions of the Ln3+ ions. Typical lifetimes for phosphorescence processes are 0.9055(6) ms for Eu3+-bdc or 0.701(4) ms for nEu3+-bdc, for instance. However, for the non-Ln3+-containing MOFs, lifetimes in nanosecond range were recorded and are indicative for fluorescence processes of the linker-based emission with 2.49(6) ns for DUT-5 or 3.6(1) ns for nDUT-5, for instance.
In fact, an important aspect when considering the efficiency of the emission of the Ln3+-MOFs is its residual linker-based emission intensity. The presence and intensity of the emission band of the linker is a reciprocal indicator for the efficiency of the sensitizing effect. The better the energy transfer from the linker to Ln3+, the lower the emission band intensity of the linker. This energy transfer is strongly depending on the donor–acceptor distance i.e., linker and Ln3+. As nDUT-5:Ln3+ and nMOF-253:Ln3+ are smaller in size than their bulk analogues, the average Ln3+ diffusion pathway becomes shorter, and a better pore occupancy can be achieved. Accordingly, the linker-based emission intensity in the emission spectrum of nDUT-5:Eu3+ at 400 nm is lower than the bulk analogue. This is in corroboration with a decreased linker-based QY from 10.4(3)% to 4.5(3)% for DUT-5:Eu3+ and nDUT-5Eu3+, respectively. The linker-based emission band of nMOF-253:Eu3+ yet diminishes completely. Both reveal nicely the advantages of nanoparticles. Interestingly, although the MOF-253, nMOF-253, MOF-253:Tb3+ and nMOF-253:Tb3+ show linker-based emission bands in their PL spectra QYs are <1%, which, however, is consistent with the low emission intensities observed with the naked eye (Fig. 7). This is also evidence for energy release by rather non-radiative processes than light emission. Moreover, the undoped DUT-5 and nDUT-5 show linker-based QYs of 5.1(5)% and 14.8(6)%, respectively, which is in accordance with the observed higher intensity of nDUT-5 compared to its bulk analogue, accompanied by a bathochromic shift as explained later in more detail. On the other hand, Ln3+-bdc and nLn3+-bdc do not show any linker-based emission bands at all, which confirms a highly efficient energy transfer. This results in excellent Ln3+-based QYs as the ratio of emitted to absorbed photons of 94(2)% and 78.1(3)% for Tb3+-bdc and nTb3+-bdc, respectively (see Table 2). The respective Ln3+-based QYs of 33(2)% for Eu3+-bdc and 23.7(3)% for nEu3+-bdc are lower than the Ln3+-based QYs of their Tb3+-analogues indicating a better energetical match between the excited states of the sensitizer for Tb3+. Different to Ln3+-coordination sites of these MOFs, post-synthetically modified MOF:Ln3+s show lower Ln3+-QYs as the Ln3+-to-linker distances increase (Ln3+-based QY of 2.0(1)% for MOF-253:Eu3+, for instance). As discussed early, DUT-5:Tb3+ and nDUT-5:Tb3+ do not show Tb3+-based emission due to the mismatch of Tb3+-linker energy states while the linker-based QYs also decreasing from 11.5(3)% to 4.6(2)% from DUT-5:Tb3+ to nDUT-5:Tb3+. For nDUT-5:Eu3+, however, the Ln3+-based QY can exceed the bulk MOF (Ln3+-based QYs: 3.6(1)% compared to 2.1(2)%) corroborated by a longer lifetime τ (see Table 2). Accordingly, a decreased Ln3+-based QY may be concluded as a result of the surfactant influence, although there is no general trend.
 | ||
| Fig. 7 Photographs of powders of investigated bulk MOFs and nMOFs exposed to Vis-light (top) and UV-light (302 nm, bottom). A = Eu3+-bdc, B = nEu3+-bdc, C = Tb3+-bdc, D = nTb3+-bdc, E = DUT-5, F = nDUT-5, G = DUT-5:Eu3+, H = nDUT-5:Eu3+, I = DUT-5:Tb3+, J = nDUT-5:Tb3+, K = MOF-253, L = nMOF-253, M = MOF-253:Eu3+, N = nMOF-253:Eu3+, O = MOF-253:Tb3+, P = nMOF-253:Tb3+. | ||
In addition, light absorption was investigated by UV-Vis-DRS. In general, it reveals broad absorption from 200–320 nm for all investigated MOFs, which can be assigned to the π* ← π transition of the linkers and fits nicely the excitation spectra seen in Fig. 6. However, all absorption spectra of the investigated nMOFs show this broad absorption band starting at 200 nm but instead of tailing at 320 nm, the absorption band shows tailing up to 700 nm for nTb3+-bdc, for instance, which implies a contribution of further absorption processes along with the π* ← π transition. The range of this multiple-process absorption band varies with each investigated MOF and is displayed in Fig. SI17–20.† Since this broader absorption band appears exclusively in investigated nMOF absorption spectra, the absorption band is attributed to the surfactants CTAB and PVP40000. Therefore, the investigated nMOFs appear yellow at daylight compared to their colourless bulk analogues (Fig. 7 and SI33† as well as a video download). In addition, only Eu3+-bdc and nEu3+-bdc spectra show low-intensity, sharp absorption peaks at 395 nm, 466 nm and 535 nm which can again be assigned to the respective 4f–4f transitions 5L6 ← 7F0, 5D2 ← 7F1 and 5D1 ← 7F1 already observed in the excitation spectra of Eu3+-bdc and nEu3+-bdc.
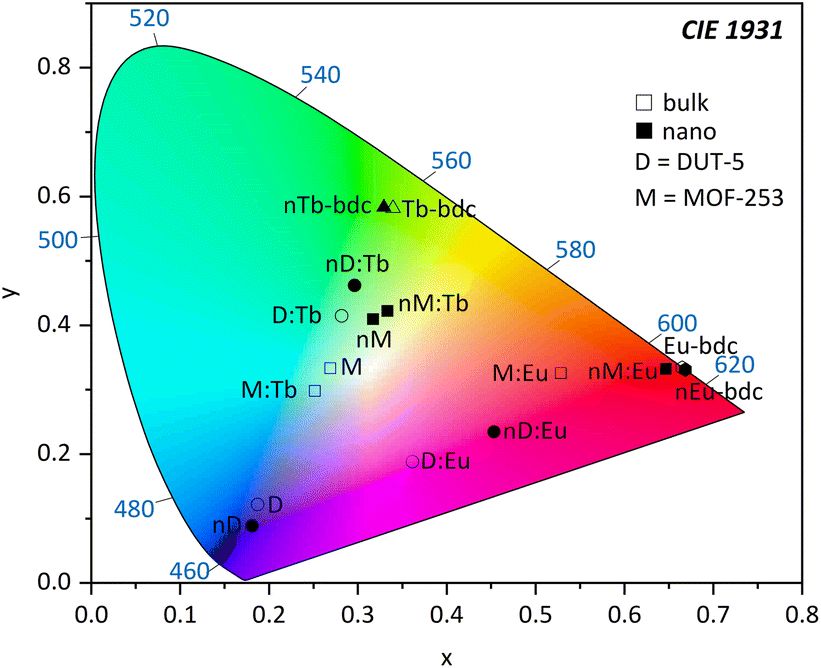 | ||
| Fig. 8 Chromaticity diagram according to CIE 1931 calculated from PL data of Ln3+-bdc, nLn3+-bdc, DUT-5, nDUT-5, MOF-253, nMOF-253, DUT-5:Ln3+, nDUT-5:Ln3+, MOF-253:Ln3+ and nMOF-253:Ln3+ (Ln3+ = Eu3+, Tb3+) investigated in this work. Hexagon = Eu3+-bdc; triangle = Tb3+-bdc; square = MOF-253 and MOF-253:Ln3+; circle = DUT-5 and DUT-5:Ln3+; D = DUT-5; and M = MOF-253. Empty symbols represent bulk MOFs, while filled symbols represent nMOFs. | ||
Conclusion
This work presents new aspects of the impact of nanoparticle formation on morphological and photophysical properties compared to the bulk archetype MOFs for Ln3+-MOFs and archetype MOFs doped post-synthetically modified by impregnation with lanthanide ions, shown for Ln3+-bdc, DUT-5:Ln3+ and MOF-253:Ln3+ (Ln3+ = Eu3+, Tb3+). Nanoscale variants of the three archetype MOFs have been successfully achieved by surfactant-assisted bottom-up synthesis as fully-characterised nMOFs.The nMOFs were synthesized with particle sizes down to 35 nm as nDUT-5 demonstrates (21-times smaller than DUT-5). Consequently, a particle consists of only a couple unit cells of the 3D framework. Furthermore, particle size distributions were tremendously narrowed down to ≈1% of their original bulk particle size distribution and stabilized in dispersion by PVP40000 and CTAB. After post-synthetic modification, the nMOFs:Ln3+ benefit from residual surfactants as good as non-post-synthetically modified nMOFs in terms of dispersibility and particle size. The shorter average free path of the diffusion of Ln3+ in nMOF:Ln3+ leads to a better pore occupancy and an improved energy transfer from the linker to the Ln3+. The importance of suitable energy differences of the excited states of the MOFs and incorporated Ln3+-ions becomes evident for the non-preferred sensitization of Tb3+ in nMOF:Tb3+ combinations. In contrast, MOFs and nMOFs with Ln3+ as connectivity centres show higher QYs up to 94% and 78% for Tb3+-bdc and nTb3+-bdc, as an energy transfer via the electronic system of the MOF is available. This results in chromaticity shifts between MOFs and nMOFs being either bathochromic or hypsochromic.
Altogether, this work provides insights into the photophysical and morphological properties of nano-sized lanthanide-containing MOFs, the pros and cons associated with the usage of surfactants as well as two different ways of functionalizing of these versatile three-dimensional compounds. We demonstrate, how the synthesis of nMOFs can be controlled by reagents such as surfactants, how the light emission of Ln3+-containing MOFs can be influenced either by changing the size and morphology of the particles or by incorporate the lanthanide at different stages of the synthesis process (early in the synthesis procedure or post-synthetic), as well as how the choice of IBU-linker-Ln3+ combination affects the energy transfer efficiency and luminescence efficiency.
Experimental
All chemicals were used as purchased without further purification. The rare-earth nitrates Ln(NO3)3·6H2O (Ln = Eu, Tb, Gd; 99.9% pur.); the organic linkers benzene-1,4-dicarboxylic acid (H2bdc, 98% pur.), biphenyl-4,4′-dicarboxylic acid (H2bpdc, 97% pur.) and 2,2′-bipyridine-5,5′-dicarboxylic acid (H2bpydc, 97% pur.); and the aluminium salt AlCl3·6H2O (99% pur.) have been supplied from abcr. In addition, the aluminium salts Al(NO3)3·9H2O (≥98% ACS gra.); the surfactants N,N,N-tri-methylhexadecan-1-ammonium bromide (CTAB, ≥96% pur.) and 1-ethenylpyrrolidin-2-one (PVP40000, avg. mol. wt. 40000, MQ: 200); and the reference material polytetrafluoroethylene (PTFE, 1 μm particle size) and magnesium oxide (MgO, MQ: 300) were purchased from Sigma-Aldrich (Merck). Moreover, the solvents N,N-dimethylformamide (DMF, 99% pur., purchased from Grüssing), ethanenitrile (MeCN, ≥99.8% pur., purchased from Chemsolute (Th. Geyer)), ethanol (EtOH, ≥99.8% abs., Fischer Scientific) and triethylamine (TEA, 99% pur., Fischer Scientific) did not undergo any additional drying process. Nitric acid (HNO3, tech. qual.) was purchased from STOCKMEIER Chemie. Furthermore, only demineralized water (H2O) was used for synthesis, analysis, and product purification processes.
Synthesis and post-synthetic modification procedure
565 g (rcf) for 10 min after each step.
Eu3+-bdc. Drying under vacuum gave 38 mg (0.026 mmol, 1472.72 g mol−1, 15%) of a colourless powder. Organic elemental analysis (3∞[Eu3(bdc)4.5]·3.3DMF·0.9H2O, found: C, 37.9; H, 3.0; N, 3.2. Calc. for Eu3C45.9H42.9N3.3O22.2: C, 37.4; H, 2.9; N, 3.1%).
Tb3+-bdc. Drying under vacuum gave 48 mg (0.032 mmol, 1510.96 g mol−1, 18%) of a colourless powder. Organic elemental analysis (3∞[Tb3(bdc)4.5]·3.7DMF·1.4H2O, found: C, 37.4; H, 3.0; N, 3.5. Calc. for Tb3C47.1H46.7N3.7O23.1: C, 37.4; H, 3.1; N, 3.4%).
000, separately, in 40 mL DMF under vigorous stirring. Subsequently, 190 mg (1.14 mmol) H2bdc and 3220 mg (8.78 mmol) CTAB were dissolved in 200 mL DMF inside a 500 mL three-necked round-bottomed flask equipped with a reflux condenser, under vigorous stirring at 55 °C for 60 min. Afterwards, the previously prepared solutions were then gradually added, followed by the addition of 200 μL of TEA. The reaction mixture became slightly opaque and was stirred vigorously for a further 60 min at 55 °C. Further heating at 90 °C for 6 h resulted in a thick opaque, pale yellowish dispersion, which was washed with 10 mL DMF (1×) and 10 mL EtOH (2×), consecutively, and centrifuged at 17855 g (rcf) for 10 min after each step.
nEu3+-bdc. Drying under vacuum gave 188 mg of a pale yellowish powder.
nTb3+-bdc. Drying under vacuum gave 121 mg of a pale yellowish powder.
000 and 417 mg (1.11 mmol) Al(NO3)3·9H2O were dissolved in 40 mL and 20 mL DMF, respectively, and gradually added to the reaction mixture. After 5 min of stirring, adding of 300 μL of TEA made the solution opaque. This was followed by a further 1 h of stirring at 55 °C. Afterwards, the reaction mixture was treated at 120 °C for 24 h in a reflux apparatus. First, the purification process was started by separating the precipitate by centrifugation at 17855 g (rcf) for 10 min. Second, washing with 15 mL DMF (4×) and 10 mL H2O (4×), followed by the above-mentioned centrifugation procedure after each step, yielded a pale brownish residue. Finally, the specimen was dried under vacuum to give 467 mg of a pale brownish powder.
565 g (rcf), 10 min) and subsequently reheated for 15 min in 20 mL DMF under reflux for purification. After a short cooling period, the product was centrifuged again (1690 g (rcf), 10 min) and the supernatant removed. Repeated purification with 20 mL DMF (1×) and 20 mL EtOH (2×), followed by vacuum drying yielded 324 mg (0.852 mmol, 380.29 g mol−1, 68%) of a colourless powder. Organic elemental analysis (3∞[Al(OH)bpydc]·1.3EtOH·1.9H2O, found: C, 46.1; H, 4.9; N, 7.4. Calc. for AlC14.6H18.6N2O8.2: C, 46.1; H, 5.0; N, 7.4%).
000 were dissolved in 16 mL DMF and gradually added to the reaction mixture. Additionally, 108 mg (0.447 mmol) AlCl3·6H2O were added as solid. After 5 min of stirring, adding of 120 μL of TEA made the solution opaque. This was followed by another 1 h of stirring at 55 °C. Afterwards, the reaction mixture was treated at 120 °C for 24 h in a reflux apparatus. First, the purification process was started by separating the precipitate by centrifugation at 17855 g (rcf) for 10 min. Second, washing with 10 mL DMF (4×) and 10 mL EtOH (4×), followed by the aforementioned centrifugation procedure after each step, yielded in a dark brownish residue. Finally, the specimen was dried under vacuum, yielding 136 mg of a dark brownish powder.
The first post-synthetic modification approach was carried out at room temperature (RT) in a snap-on cap glass vial. Previously, Ln(NO3)3·6H2O has been dissolved in EtOH. Then, 40 mg specimen were soaked with 1 mL of a 0.1 M Ln3+-solution. After 5 min of sonicating, the vial was closed and left untouched for one week. After rinsing the specimen out of the vial, it was centrifuged at 17855 g (rcf) for 10 min. Ultimately, the post-synthetically modified sample was washed with 2 mL of EtOH, centrifuged and dried under vacuum to obtain a powder.
The second post-synthetic modification approach was carried out at 65 °C in a closed pressure tube (Ace Glass) according to the procedure of Lu et al.,62 with some modifications. Previously, 45 mg Ln(NO3)3·6H2O were dissolved in 10 mL MeCN (0.01 M). After adding 40 mg MOF to the mixture, the mixture was again dispersed with 5 mL MeCN and sonicated for 5 min. This was followed by a heating step for 24 h and the final purification process, consisting of centrifugation (17855 g (rcf), 10 min), washing (10 mL MeCN) and sonication (5 min) three times consecutively. After drying under vacuum, the post-synthetically modified sample was obtained as powder.
Analytical methods
707 g (rcf), 15 min) was carried out to remove the linker (stem solution). Subsequently, the procedure was the same to all MOFs. Several dilutions were prepared from stem solution to theoretically match the concentration range of the external calibration. Intensity was recorded at characteristic atomic emission wavelengths (Al: 394.401 nm, 396.152 nm; Eu: 381.967 nm, 412.973 nm; Tb: 350.917 nm, 384.874 nm) were used to determine the concentration of the analytes. The instrument required 90 min for sample uptake, followed by 30 min for rinsing and 15 min for flow stabilization. This procedure was repeated after each dilution.
QY determination was performed using a Jobin Yvon Fluorolog 3 with the FluorEssence™ for Windows® (ver. 3.9.0.1) software equipped with a Quanta-φ Integrating Sphere F-3029 from HORIBA. For the measurement, the reference and sample, respectively, were filled into a micro cell cuvette 18-F/ST/C/Q/10 from Starna Scientific. The quantum yield determination was repeated three times for each specimen and related to the reference material MgO. An additional check using another standard material γ-[Tb4(OAc)12(ptpy)2] verified the calibration of the Quanta-φ Integrating Sphere (λex = 310 nm, λem = 450–700 nm; Φexp = 46(1)%, Φlit = 46(3)% (ref. 63)). Ln3+-based QYs were determined with λex = 310 nm and λem = 450–700 nm. Linker-based QYs were determined with λex = 310 nm, λem = 315–520 nm for DUT-5; λex = 310 nm, λem = 335–570 nm for nDUT-5; λex = 310 nm, λem = 330–540 nm for DUT-5:Tb3+ and nDUT-5:Tb3+; λex = 310 nm, λem = 330–550 nm for DUT-5:Eu3+ and nDUT-5:Eu3+ and λex = 360 nm, λem = 370–700 nm for MOF-253, nMOF-253, MOF-253:Tb3+ and nMOF-253:Tb3+.
Lifetime determinations were carried out by recording the overall luminescence decay times (τ) using a Jobin Yvon Fluorolog 3 with the Data Station (ver. 2.7.4) software for data acquisition and the Decay Analysis Software (ver. 6.8.16) for raw data fitting, respectively, from HORIBA. For this purpose, the instrument was upgraded with a TCSPC (time-correlated single-photon counting) and UV Xenon FX-1102 flashlamp from Excelitas Technologies. However, a pulsed Delta Diode™ 278 nm (HORIBA) was necessary for linker emission decay determination. Moreover, for this method, the sample was filled into a cylindrical synthetic-quartz glass cuvette (proQuarz) and placed in the focus of the beam. Subsequently after recording the overall emission decay, the lifetime has been calculated by exponentially fitting the data with I(t) = A + B1·e−(t−t0/τ1) + B2·e−(t−t0/τ2), where I(t) is the emission intensity depending on time, A is the baseline factor, B is the pre-exponential factor, t0 is the time offset, t is the time and τ is the lifetime.
Phosphorescence spectra were recorded using a pulsed UV Xenon FX-1102 flashlamp from Excelitas Technologies, each 41 ms a flash. Specimens were cooled down and measured at 77 K inside a specialized Dewar filled with liquid nitrogen. Parameters such as recording delay time, sample window time or counts per flash varied and are given next to the recorded spectra. Subsequently, triplet state determination carried out by using the software OriginPro 2023 (ver. 10.0.0.154) from OriginLab. The intersection of two linear fits marks the triplet state energy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is generously funded by the Deutsche Forschungs-gemeinschaft (DFG) within the project MU-1562/13-1 within the priority program SPP-1928 “COORNETs”.References
- L. Mu, B. Liu, H. Liu, Y. Yang, C. Sun and G. Chen, J. Mater. Chem., 2012, 22, 12246 RSC.
- D. Alezi, Y. Belmabkhout, M. Suyetin, P. M. Bhatt, L. J. Weseliński, V. Solovyeva, K. Adil, I. Spanopoulos, P. N. Trikalitis, A. H. Emwas and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137, 13308–13318 CrossRef CAS PubMed.
- S. Denning, A. A. Majid, J. M. Lucero, J. M. Crawford, M. A. Carreon and C. A. Koh, ACS Appl. Mater. Interfaces, 2020, 12, 53510–53518 CrossRef CAS PubMed.
- Z. He, Y. Dai, X. Li, D. Guo, Y. Liu, X. Huang, J. Jiang, S. Wang, G. Zhu, F. Zhang, L. Lin, J.-J. Zhu, G. Yu and X. Chen, Small, 2019, 15, 1804131 CrossRef PubMed.
- A. Wagner, Q. Liu, O. L. Rose, A. Eden, A. Vijay, Y. Rojanasakul and C. Z. Dinu, Int. J. Nanomed., 2019, 14, 7583–7591 CrossRef CAS PubMed.
- S. A. Ahmed, M. Nur Hasan, D. Bagchi, H. M. Altass, M. Morad, I. I. Althagafi, A. M. Hameed, A. Sayqal, A. E. R. S. Khder, B. H. Asghar, H. A. Katouah and S. K. Pal, R. Soc. Open Sci., 2020, 7, 200959 CrossRef CAS PubMed.
- R. Van Zeeland, X. Li, W. Huang and L. M. Stanley, RSC Adv., 2016, 6, 56330–56334 RSC.
- A. Herbst, A. Khutia and C. Janiak, Inorg. Chem., 2014, 53, 7319–7333 CrossRef CAS PubMed.
- S. Aguado, J. Canivet and D. Farrusseng, J. Mater. Chem., 2011, 21, 7582 RSC.
- A. D. Pournara, A. Margariti, G. D. Tarlas, A. Kourtelaris, V. Petkov, C. Kokkinos, A. Economou, G. S. Papaefstathiou and M. J. Manos, J. Mater. Chem. A, 2019, 7, 15432–15443 RSC.
- M. Wickenheisser, T. Paul and C. Janiak, Microporous Mesoporous Mater., 2016, 220, 258–269 CrossRef CAS.
- Z. Chen, P. Li, X. Zhang, P. Li, M. C. Wasson, T. Islamoglu, J. F. Stoddart and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2900–2905 CrossRef CAS PubMed.
- K. Müller-Buschbaum, F. Beuerle and C. Feldmann, Microporous Mesoporous Mater., 2015, 216, 171–199 CrossRef.
- V. Chernikova, O. Yassine, O. Shekhah, M. Eddaoudi and K. N. Salama, J. Mater. Chem. A, 2018, 6, 5550–5554 RSC.
- L. J. Small, S. E. Henkelis, D. X. Rademacher, M. E. Schindelholz, J. L. Krumhansl, D. J. Vogel and T. M. Nenoff, Adv. Funct. Mater., 2020, 30, 2006598 CrossRef CAS.
- H. Yang, B. Liu, J. Bright, S. Kasani, J. Yang, X. Zhang and N. Wu, ACS Appl. Energy Mater., 2020, 3, 4007–4013 CrossRef CAS.
- S.-S. Liu, Z. Han, J.-S. Yang, S.-Z. Huang, X.-Y. Dong and S.-Q. Zang, Inorg. Chem., 2020, 59, 396–402 CrossRef CAS PubMed.
- Z. Wei, R. Maile, L. M. Riegger, M. Rohnke, K. Müller-Buschbaum and J. Janek, Batteries Supercaps, 2022, 5(12), e202200318 CrossRef CAS.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- L. V. Meyer, F. Schönfeld, A. Zurawski, M. Mai, C. Feldmann and K. Müller-Buschbaum, Dalton Trans., 2015, 44, 4070–4079 RSC.
- K. Xu, F. Wang, S. Huang, Z. Yu, J. Zhang, J. Yu, H. Gao, Y. Fu, X. Li and Y. Zhao, RSC Adv., 2016, 6, 91741–91747 RSC.
- K. Mandel, T. Granath, T. Wehner, M. Rey, W. Stracke, N. Vogel, G. Sextl and K. Müller-Buschbaum, ACS Nano, 2017, 11, 779–787 CrossRef CAS PubMed.
- W. T. Carnall, in Non-metallic Compounds – I, 1979, vol. 3, pp. 171–208 Search PubMed.
- R. E. Whan and G. A. Crosby, J. Mol. Spectrosc., 1962, 8, 315–327 CrossRef CAS.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- F. Luo and S. R. Batten, Dalton Trans., 2010, 39, 4485 RSC.
- T. N. Nguyen, S. V. Eliseeva, A. Gładysiak, S. Petoud and K. C. Stylianou, J. Mater. Chem. A, 2020, 8, 10188–10192 RSC.
- K. A. White, D. A. Chengelis, K. A. Gogick, J. Stehman, N. L. Rosi and S. Petoud, J. Am. Chem. Soc., 2009, 131, 18069–18071 CrossRef CAS PubMed.
- M. Naito, T. Yokoyama, K. Hosokawa and K. Nogi, in Nanoparticle Technology Handbook, Elsevier Science, 2018, pp. 3–44 Search PubMed.
- F. Martinez-Julian, A. Guerrero, M. Haro, J. Bisquert, D. Bresser, E. Paillard, S. Passerini and G. Garcia-Belmonte, J. Phys. Chem. C, 2014, 118, 6069–6076 CrossRef CAS.
- S. S. El-Deen, A. M. Hashem, A. E. Abdel Ghany, S. Indris, H. Ehrenberg, A. Mauger and C. M. Julien, Ionics, 2018, 24, 2925–2934 CrossRef CAS.
- D.-H. Ha, M. A. Islam and R. D. Robinson, Nano Lett., 2012, 12, 5122–5130 CrossRef CAS PubMed.
- Y. L. F. Musico, C. M. Santos, M. L. P. Dalida and D. F. Rodrigues, ACS Sustain. Chem. Eng., 2014, 2, 1559–1565 CrossRef CAS.
- W. Wanas, S. A. Abd El-Kaream, S. Ebrahim, M. Soliman and M. Karim, Sci. Rep., 2023, 13, 27 CrossRef CAS PubMed.
- G. Prencipe, S. M. Tabakman, K. Welsher, Z. Liu, A. P. Goodwin, L. Zhang, J. Henry and H. Dai, J. Am. Chem. Soc., 2009, 131, 4783–4787 CrossRef CAS PubMed.
- S. L. Pal, U. Jana, P. K. Manna, G. P. Mohanta and R. Manavalan, J. Appl. Pharm. Sci., 2011, 1, 228–234 Search PubMed.
- M. Naito, T. Yokoyama, K. Hosokawa and K. Nogi, in Nanoparticle Technology Handbook, Elsevier Science, 2018, pp. 49–105 Search PubMed.
- J. Andreo, R. Ettlinger, O. Zaremba, Q. Peña, U. Lächelt, R. F. De Luis, R. Freund, S. Canossa, E. Ploetz, W. Zhu, C. S. Diercks, H. Gröger and S. Wuttke, J. Am. Chem. Soc., 2022, 144, 7531–7550 CrossRef CAS PubMed.
- C. D. Ma, C. Wang, C. Acevedo-Vélez, S. H. Gellman and N. L. Abbott, Nature, 2015, 517, 347–350 CrossRef CAS PubMed.
- X.-G. Wang, Q. Cheng, Y. Yu and X.-Z. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7836–7840 CrossRef CAS PubMed.
- T. Kundu, S. Mitra, P. Patra, A. Goswami, D. Díaz Díaz and R. Banerjee, Chem. – Eur. J., 2014, 20, 10514–10518 CrossRef CAS PubMed.
- R. Nagarajan and E. Ruckenstein, Langmuir, 1991, 7, 2934–2969 CrossRef CAS.
- X. Guo, G. Zhu, F. Sun, Z. Li, X. Zhao, X. Li, H. Wang and S. Qiu, Inorg. Chem., 2006, 45, 2581–2587 CrossRef CAS PubMed.
- I. Senkovska, F. Hoffmann, M. Fröba, J. Getzschmann, W. Böhlmann and S. Kaskel, Microporous Mesoporous Mater., 2009, 122, 93–98 CrossRef CAS.
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382–14384 CrossRef CAS PubMed.
- M. J. Neufeld, H. Winter, M. R. Landry, A. M. Goforth, S. Khan, G. Pratx and C. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 26943–26954 CrossRef CAS PubMed.
- X. Deng, J. Albero, L. Xu, H. García and Z. Li, Inorg. Chem., 2018, 57, 8276–8286 CrossRef CAS PubMed.
- S. Chhetri, N. Adak, P. Samanta, N. Murmu and T. Kuila, J. Compos. Sci., 2018, 2, 37 CrossRef.
- Y. Yuan, W. Wang, L. Qiu, F. Peng, X. Jiang, A. Xie, Y. Shen, X. Tian and L. Zhang, Mater. Chem. Phys., 2011, 131, 358–361 CrossRef CAS.
- C. Wang, J. Kim, J. Tang, M. Kim, H. Lim, V. Malgras, J. You, Q. Xu, J. Li and Y. Yamauchi, Chem, 2020, 6, 19–40 CAS.
- X. Xia, J. Zeng, L. K. Oetjen, Q. Li and Y. Xia, J. Am. Chem. Soc., 2012, 134, 1793–1801 CrossRef CAS PubMed.
- Z. Haghi and S. M. Masoudpanah, J. Sol-Gel Sci. Technol., 2019, 91, 335–341 CrossRef CAS.
- Y. K. Du, P. Yang, Z. G. Mou, N. P. Hua and L. Jiang, J. Appl. Polym. Sci., 2006, 99, 23–26 CrossRef CAS.
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Férey and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839–17847 CrossRef CAS PubMed.
- J. V Alemán, A. V Chadwick, J. He, M. Hess, K. Horie, R. G. Jones, P. Kratochvíl, I. Meisel, I. Mita, G. Moad, S. Penczek and R. F. T. Stepto, Pure Appl. Chem., 2007, 79, 1801–1829 CrossRef.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- P. A. Tanner, Chem. Soc. Rev., 2013, 42, 5090 RSC.
- D. Y. Medina-Velazquez, U. Caldiño, A. Morales-Ramirez, J. Reyes-Miranda, R. E. Lopez, R. Escudero, R. Ruiz-Guerrero and M. F. Morales Perez, Opt. Mater., 2019, 87, 3–10 CrossRef CAS.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4447–4449 CrossRef CAS.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4450–4455 CrossRef CAS.
- Y. Lu and B. Yan, J. Mater. Chem. C, 2014, 2, 7411–7416 RSC.
- A. E. Sedykh, M. Becker, M. T. Seuffert, D. Heuler, M. Maxeiner, D. G. Kurth, C. E. Housecroft, E. C. Constable and K. Müller-Buschbaum, ChemPhotoChem, 2023, 7, e202200244 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05219b |
| This journal is © The Royal Society of Chemistry 2023 |