
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The lignin challenge in catalytic conversion of biomass solids to chemicals and fuels
Mahdi M.
Abu-Omar
* and
Peter C.
Ford
 *
*
Department of Chemistry and Biochemistry, University of California, Santa Barbara, Santa Barbara, CA, 93106-9510 USA. E-mail: mabuomar@ucsb.edu; ford@chem.ucsb.edu
First published on 17th July 2023
Abstract
Presented here is an overview of progress in one approach to reducing greenhouse gas emission, namely substituting photosynthesis generated, woody biomass for fossil carbon as feedstock in the production of chemicals and liquid fuels. Particular emphasis is given to one of the most difficult aspects of biomass conversion, namely dealing with lignin, which is the second largest component (after cellulose) of woody materials. Lignin is largely composed of aromatic monomers linked (and cross-linked) in various bonding modes. As such it presents particular challenges for disassembling. However, it also represents an important opportunity for producing both liquid fuels for transportation and monomers with valuable properties as carbon-neutral precursors for various chemical applications including the synthesis of new polymers. Many examples of the approaches to these challenges are drawn from the authors' own research. While we have attempted to present a broader viewpoint, this Perspective is not intended as a comprehensive review of the extensive research from other laboratories worldwide.
Sustainability spotlightGlobal climate change is an existential challenge to human health correlated with the excessive generation of greenhouse gases by modern society. Multiple strategies to addressing this threat are essential. In this perspective, we address issues and opportunities arising from one such approach, the conversion of lignocellulose to liquid fuels and to higher value chemicals such as polymers. |
Introduction
This perspective discusses a key challenge of sustainability chemistry, the utilization of non-food biomass for the production of organic chemicals and liquid fuels. Successful, efficient and economic methods for converting renewable biomass solids to chemicals and liquid fuels would reduce society's dependence on non-renewable fossil carbon resources while easing the atmospheric carbon dioxide burden. The topic is obviously not new; humankind has been utilizing biomass as a fuel for heating, cooking and lighting since the discovery of fire. However, conversion of lignocellulose, which is the most abundant form of renewable biomass carbon, to the liquid fuels and chemicals that are linchpins of modern economies is a complex technical problem.When the Abu-Omar1 and Ford2 research groups independently initiated studies in biomass conversion about 15 years ago, we and many others were very aware of the growing threat of the atmospheric greenhouse gas (GHG) concentrations that inexorably continue to increase. For example, carbon dioxide, the most important GHG, has increased from an average atmospheric concentration of ∼383 ppm in 2007 to 419 ppm in late 2022, a 9% increase in just 15 years.3 It has been estimated that such CO2 increases since the pre-industrial revolution era (est. 280 ppm) leads to a trapping of ∼3.2 extra Watts per m2 of solar energy (average) over the surface of the Earth.4 Correspondingly, seven of the hottest years in history have been recorded in the last decade with 2021 being the 7th consecutive year (2015–2021) having a global temperature more than 1 °C above pre-industrial levels, according to the World Meteorological Organization.5 Concerns have been raised that further temperature rises will push the climate past a critical tipping point.6 Moreover, a number of extreme weather episodes in recent years has been attributed to global warming. With growing awareness of accelerating climate change and the accompanying social disruptions, sustainability gains a new sense of urgency. While no one action, or even one type of action, will ameliorate this existential crisis, multiple collective responses by many can make a difference.
Energy production via the combustion of fossilized carbon (coal, natural gas and petroleum) for transportation, electricity production, industrial activities and residences is the origin of the largest fraction of anthropologic GHG emissions. Solar, wind and other, less conventional, carbon-neutral methods have begun to decrease the GHG impact of electricity generation in the United States over the past decade,7 and it is obvious that, as the carbon-neutral electricity sector grows, other end-use applications will benefit.
The transportation sector directly contributes more than a quarter of the CO2 emissions in the United States7 and likely contributes a comparable percentage to anthropologic GHG emissions elsewhere. Currently, there are ∼1.4 billion 4-wheeled vehicles in use worldwide powered by internal combustion engines (ICEs) using liquid hydrocarbon fuels.8 Governmental programs in a number of countries encourage and provide incentives for purchasing of electric vehicles (EVs), and 13% of worldwide new car sales in 2022 were EVs and plug-in hybrid electric vehicles (PHEVs).9 To cite an example more local for the authors, the State of California in 2022 adopted rules banning the sale of new strictly gasoline and diesel powered automobiles and light trucks as of 2035.10 This follows the earlier passages of similar bans by New York State and by the United Kingdom and Germany in 2021. Because of such developments, and assuming the politicians have the courage to maintain these programs, there will be a major increase in the role of EVs in the marketplace. Nonetheless, there will be a continuing major need for liquid hydrocarbon fuels for many decades simply to fuel the ICE powered vehicles now in use and those ICEs and PHEVs that will undoubtedly be added to the world's fleet during the next generation.
Other major contributors to GHG emissions are aviation and international shipping. Current estimates place commercial, private and military aviation as contributing >2% of the worldwide CO2 emissions.11 Presently, there are no viable alternatives to liquid hydrocarbon aviation fuels given the inherent energy storage density needed to power the fleet of airplanes currently flying. Aviation is a growing industry that is difficult to decarbonize, so lignocellulose conversion to jet fuel can have a major impact on this particularly CO2 polluting industry. Thus, there is a huge, continuing need to develop renewable, carbon-neutral alternatives to supply this and other transportation sectors.
A very important question is whether liquid hydrocarbon fuels can be produced by sustainable methods that minimize further conversion of fossil-carbon to carbon dioxide. One potential approach would be to obtain such fuels from lignocellulosic biomass, which has stored chemical potential derived from solar energy passively collected over a distributed area and converted to reduced carbon from atmospheric CO2 by photosynthesis. Bioethanol and bio-diesel are currently used in transportation; however, both of these are relatively minor contributors and are largely derived from food crops (ethanol from corn in the USA and from sugarcane in Brazil; biodiesel from soy beans). Second generation bioethanol from lignocellulose has received significant financial investment but has been plagued by a number of challenges that left it economically not viable without subsidies and legislative mandates. Electrochemical CO2 reduction for production of liquid fuels and other chemicals is another approach gaining considerable attention. However, such processes are energy intensive and the electrochemical potential might be better used for direct applications such as residential environmental control (cooling and heating) and industrial processes like green ammonia production. Of course, there would be little environmental value unless the sources of that electrochemical energy were renewable or at least carbon-neutral.
In this perspective, we offer an overview of research in our laboratories and some related studies focused on using non-food biomass feedstocks for liquid transportation fuels as well as the chemicals and materials necessary for modern society. We are not attempting a comprehensive review but note that growing awareness of fossil fuel related climate change has led to rapidly expanding research into utilizing biomass in this manner.12–14
Conversion of lignocellulose
The major non-food component of biomass is lignocellulose, a matrix of the carbohydrates cellulose (40–60%, depending upon the source) and hemicellulose/xylan (10–30%) plus the irregularly-coupled, aromatic polymer lignin (20–35%) (Fig. 1).15 These are the three most plentiful biopolymers in nature and are products of photosynthetic carbon dioxide reduction. Cellulose is the crystalline polymer of glucose, while hemicellulose is a heterogeneous polymer of C5 and C6 sugars such as xylose and mannose. Lignin is a heterogeneous macromolecule largely formed from the three aromatic monomers coniferyl alcohol (G), sinapyl alcohol (S) and coumaryl alcohol (H) linked by various phenyl ether bonds as well as some much more recalcitrant carbon–carbon cross-linkages (Fig. 2).17,18 Major sources of lignocellulose are woody solids and grasses, and very substantial quantities are available as agricultural and forestry wastes.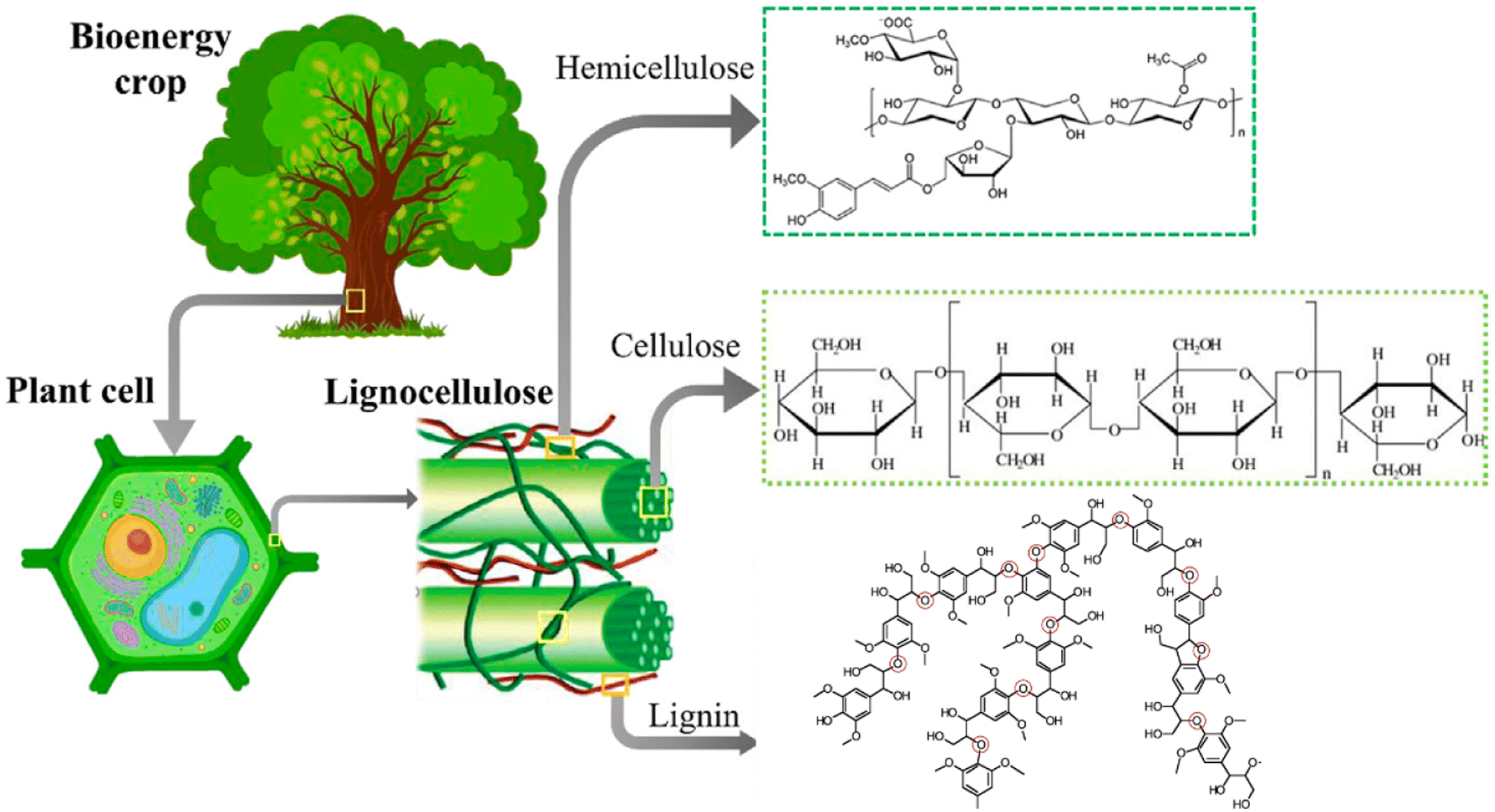 | ||
| Fig. 1 Lignocellulose components. This illustration was adapted from ref. 16. | ||
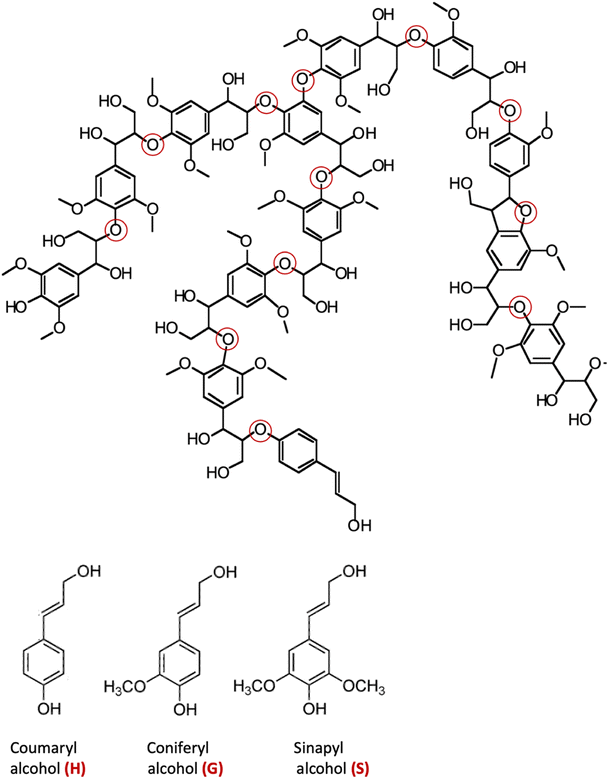 | ||
| Fig. 2 Top: A representative structure of lignin, the red circles indicating various ether linkages between aromatic monomer units. (Figure adapted from ref. 18) Bottom: Structures of the monomers coumaryl alcohol (H), coniferyl alcohol (G) and sinapyl alcohol (S). | ||
Lignocellulose is the most abundant source of renewable carbon on the planet, thus, is an obvious target for catalytic conversion to liquid transportation fuels and/or platform industrial chemicals. However, it is important to remember that the composition of lignocellulose is variable and is a function of the source from which it is derived.19
Current biorefineries largely utilize the polysaccharide components of lignocellulose feedstocks. Ethanol remains by far the most prominent product from such refineries owing to the built-in market defined by USA federal law dictating ethanol content in automobile gasoline. However, there has been considerable progress20–23 toward developing effective procedures for converting carbohydrates to platform chemicals such as furfural and 5-hydroxymethylfurfural and their derivatives (e.g., Fig. 3). These products can be transformed further to useful chemicals and fuels. For example, Bohre et al. have described the catalytic technology for upgrading furfurals into longer chain hydrocarbons for uses as drop-in fuels.24 However, substantial amounts of lignin are generated as by-products of the cellulose and hemi-cellulose utilization, and in many biorefineries, this component is burned. Clearly, greater biorefinery economic efficiency would be one major benefit of developing methodologies for the valorization of lignin to higher value products.25,26
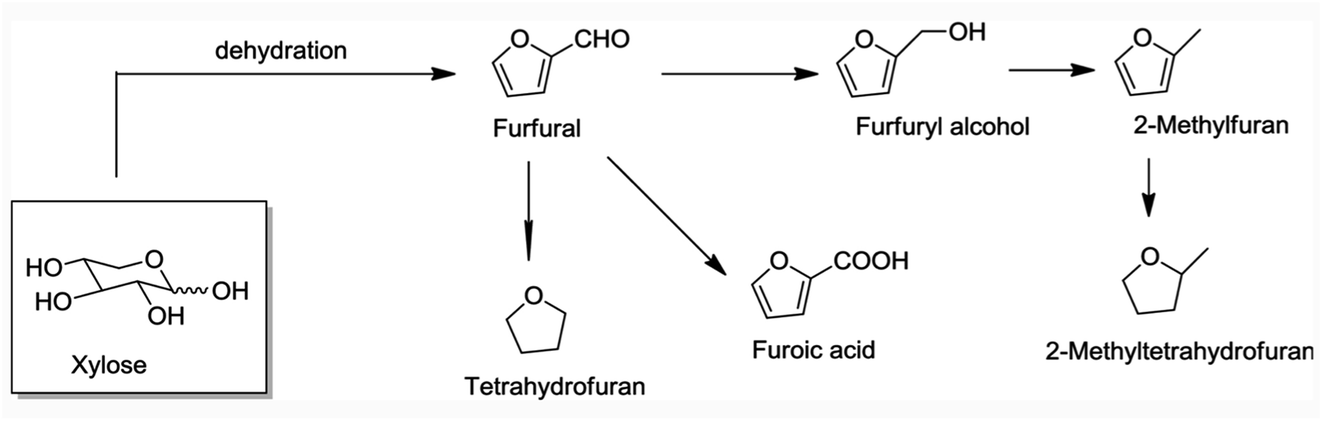 | ||
| Fig. 3 Steps leading to furfural and derivatives from a hemi-cellulose component of lignocellulose.23 | ||
Of interest would be procedures for the disassembly of lignin to aromatic monomers that may serve as drop-in feedstocks for current industrial processes. However, lignin is also a potential starting point for the synthesis of products not easily accessed from petroleum. For example, one can easily envision the synthesis of new polymeric materials from lignin-derived monomers, a topic that will be addressed below. Key issues that need addressing are how to separate lignin from the lignocellulose matrix without modifying it into a less tractable material, how to disassemble this biopolymer into monomeric units and, lastly, how to channel those monomers into products that enhance the economic viability of biorefineries.27
Radically different approaches are needed to effect facile conversion of lignocellulose solids to liquid fuels and to other chemical precursors without forming intractable and valueless side products such as tars and chars. Petroleum refineries are remarkably efficient at utilizing all components of the oil feedstock. Biorefineries need to develop comparable specificity and energetic efficiency to give economically sustainable product streams.
Petroleum feedstocks have high thermal stability and low functionality and can be depolymerized and fractionated by high temperature procedures. Petrochemical syntheses often involve adding functionality to saturated hydrocarbon structures to increase value. Lignocellulose, in contrast, is highly functionalized with nearly every framework carbon atom oxidized in some manner. Its major components have low thermal stability and little volatility. In general, functionality needs to be removed to give liquid fuels, and many, if not most, biomass derived chemicals. Different catalysts and mechanistic insights are necessary for lower temperature depolymerization of biopolymers and selective deoxygenation of the resulting fragments.3–6Fig. 4 illustrates the conceptually different approaches needed for a chemical economy drawing upon lignocellulose rather than petroleum.28 Technology that better exploits these renewable feedstocks promises to spawn entire new industries.
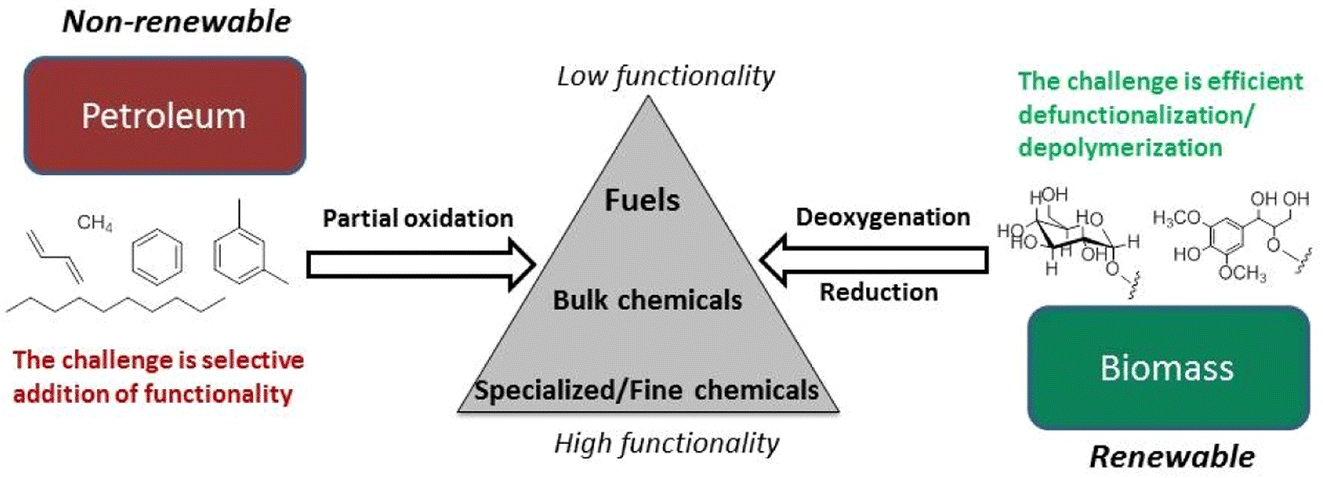 | ||
| Fig. 4 Fundamental differences in the conversion of petroleum- and biomass-based feedstocks. (Figure from ref. 28, Copyright 2014 American Chemical Society). | ||
The differences between lignocellulose and petroleum feedstocks as chemical precursors, however, should not be simply viewed as problematic. Instead, the extensive functionality of biomass components also offers new opportunities and potential access to small molecule synthons that otherwise may be difficult to prepare via traditional petrochemistry. Once the technology has been established and the economic incentives are sufficient, industry will adapt to the need to focus on renewable resources. History has taught us that the failure to respond to societal shifts will doom the recalcitrant.
Catalytic lignin disassembly
The Ford group's initial interest in lignocellulose conversion largely focused on the lignin component. The cellulose component has long been utilized in paper manufacture, and as noted above, methods were being developed for using the carbohydrate components as renewable feedstocks for the synthesis of new platform chemicals and for 2nd-generation ethanol production from lignocellulose. However, the lignin component is often devalued by simply burning as a low-grade heat source. Given that it is the second-most abundant biopolymer on Earth and by far the largest natural source of aromatic compounds, burning lignin represents a wasted opportunity. Although lignins can be converted by pyrolysis to bio-oils,29 these are low-quality fuels, since their high oxygen content makes them immiscible with petroleum-derived fuels, although new techniques for upgrading such bio-oils are under study.30 More efficient extraction of value from this renewable resource is essential for economically successful utilization of lignocellulose.In these contexts, Macala et al. initiated a study of porous metal oxides (PMOs) prepared by calcining copper doped Mg2+/Al3+ (3/1) hydrotalcites as catalysts for the disassembly of lignins.31 Similar transition metal doped PMOs had proved to be effective solid base catalysts for the transesterification of triglycerides with methanol to form fatty acid methyl esters as biodiesel fuels32 and for sequestering CO2.33 Thus, it was hypothesized that, in super-critical methanol (sc-MeOH), these solid bases would catalyze solvolysis of the lignin aromatic ether linkages. These early studies were done in mini-autoclaves such as illustrated in Fig. 5 owing to the high pressures from heating methanol to the super-critical state. The catalysts were Cu10-PMO or Cu20-PMO (prepared by replacing 10% or 20% of the Mg2+, respectively, with Cu2+ in the hydrotalcite before calcining). This one-pot system was used to investigate the reactions of a model substrate, 2,3-dihydrobenzofuran (DHBF),31 then of organosolv poplar lignin (OPL).18 Similar Cu doped PMO's have since found applications in other catalytic processes34,35 including the Guerbet condensation of ethanol to higher alcohols.36
 | ||
| Fig. 5 Photograph of 10 mL mini-reactors built from Swagelok junctions. (Figure from ref. 27, Copyright 2015 American Chemical Society). | ||
The Cu-doped PMOs proved to be very effective catalysts, but not in the solvolysis context originally envisioned. Macala et al.31 showed that with DHBF, the principal pathway in sc-MeOH at 300 °C involved hydrogenolysis (not solvolysis) of the alkyl carbon–oxygen bond of the cyclic ether to give (initially) 2-ethyl phenol (Scheme 1). However, under these conditions, this initial product proved quite susceptible to subsequent ring hydrogenation and methylation to give the cyclohexanol products. Some hydrodeoxygenation (HDO) to give hydrocarbons was also observed, but this pathway was relatively minor.
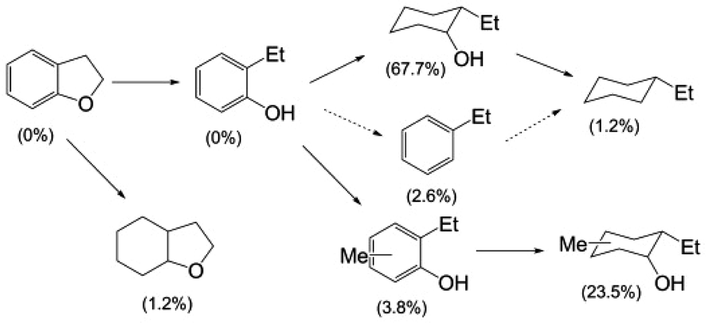 | ||
| Scheme 1 Products from the Cu20-PMO-catalysed reaction of DHBF in sc-MeOH at 300 °C. The yields after 13 h are indicated in parentheses.31 | ||
Mg–Al mixed oxides and those doped with Cu are catalysts for alcohol reforming37,38 and for the water gas shift reaction39 (eqn (1) and (2)), so these reactions are the likely sources of the hydrogen equivalents needed for the transformations illustrated in Scheme 1. This conclusion gained further support from gas chromatographic analysis of gases formed during catalytic runs. The principal component proved to be H2, with lesser amounts of CO and CO2 plus traces of methane.31 No gas was formed in the absence of the catalyst. Thus, the methanol can be effectively viewed as a “liquid syngas” under these reaction conditions, and reforming gives the hydrogen necessary for the desired hydrogenolysis as well as for the less desired hydrogenations.
| CH3OH ⇄ 2H2 + CO | (1) |
| CO + H2O ⇄ H2 + CO2 | (2) |
Barta et al.18 extended this system by demonstrating that OPL, which had been extracted from poplar wood shavings using hot, acidic methanol, is very effectively disassembled into monomeric and dimeric fragments without the formation of chars. However, a key complication was the multitude of products that resulted, in part due to the side reactions also observed above with the DHBF model substrate. Thus, while solvolysis of the aromatic ether linkages undoubtedly plays a role, hydrogenolysis of these bonds is apparently the key pathway in depolymerizing OPL, itself undoubtedly already a mixture of different species.40 Subsequent reactions under these conditions, ring hydrogenation and methylation as well as HDO, contributed to the complexity of the product mixtures. Notably, cleaner mixtures were later obtained by Barta et al.41 for lignin extracted from candlenut nutshell and disassembled using a similar reaction scheme, but at a lower, non-critical, reaction temperature. Subsequently, Hensen and coworkers42 studied soda lignin valorization with a similar CuMgAlOx catalyst in super critical ethanol (sc-EtOH), which was the source of reducing equivalents but which also served as an alkylating agent to cap reactive hydroxyl groups.
The same catalysts in sc-MeOH were also shown by Matson et al.43 to convert various samples of wood sawdust cleanly into organic liquids, again with the formation of very little or no char or other solid residues. With intact lignocellulose as the substrate, the organic liquids obtained could be separated into two fractions. The lighter products were C2 to C6 alkyl alcohols and ethers derived largely from the carbohydrate component while the heavier components were largely C9–C12 substituted cyclohexyl alcohols and ethers originating from the lignin. Reaction of cellulose alone under analogous conditions gave a mixture of C2 to C6 products like those seen from intact lignocellulose solids.43 Analysis of the gas phase again indicated a mixture of H2, CO, CO2 and traces of methane. Thus, the Cu-doped PMO catalyst combined with sc-MeOH proved to be capable of breaking down complex biomass derived substrates to markedly deoxygenated monomeric units with increased hydrogen content.
An important question is: How can a solid such as Cu20-PMO catalyze reactions with a solid substrate (lignocellulose) and do so effectively? It is here that the sc-MeOH likely plays a major role in partially solubilizing lignin and carbohydrate fragments for further reaction with the catalyst surface. Matson et al. speculated that unsaturated intermediates, formed by dehydration under the hydrothermal conditions,43 undergo catalytic hydrogenation more rapidly than do the competing condensation and re-polymerization reactions that would lead to tars and chars. Control reactions with intact lignocellulose solids, with cellulose solids or with OPL in the same medium, but without the catalyst, led to substantial char formation in each case.
Although the Cu-PMO/sc-MeOH system performs the relatively clean disassembly of OPL and of lignocellulose in one-pot processes, the multiplicity of products decreases the net value, although such a mixture may serve as a liquid fuel. In order to improve/optimize selectivity, subsequent studies of the Cu-PMO/sc-MeOH system involved exploring different reaction conditions (temperature, solvent, other additives, different substrates, etc.). In these investigations, various small molecule model compounds and OPL were utilized as the substrates.38,44–46 For example, much better selectivity toward aromatic products resulted with this catalytic system when dimethyl carbonate (DMC) was added to the MeOH as a co-solvent/co-reactant.44,45 These observations were rationalized on the premise that the phenolic fragments formed by hydrogenolysis of the phenyl ethers are a major culprit in the proliferation of products owing to subsequent reactions catalyzed by the Cu-doped PMOs. The DMC alkylates these phenolic intermediates in situ to give analogous anisole derivatives (Scheme 2) that were shown to be much less susceptible to hydrogenation or ring methylation than the analogous phenols.
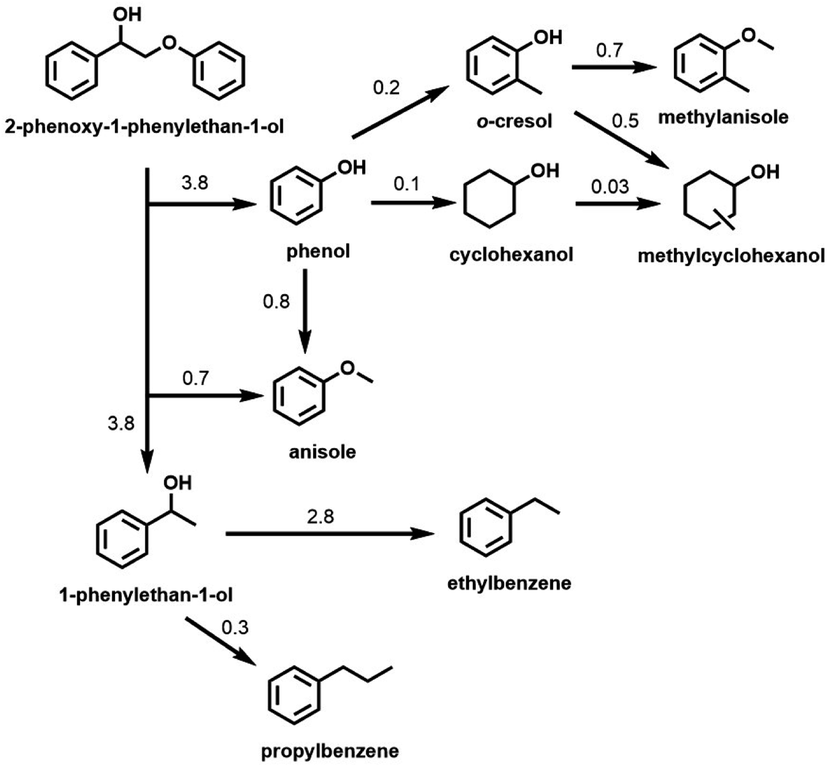 | ||
| Scheme 2 Hydrogenolysis of 2-phenoxy-1-phenylethan-1-ol and subsequent reactions of major products catalyzed by Cu20PMO in MeOH/DMC co-solvent at 300 °C. (Numbers represent the approximate rate constants for various steps in h−1). (Figure from ref. 45, Copyright 2016 American Chemical Society.) | ||
Scheme 2 illustrates the reactivity of 2-phenoxy-1-phenylethan-1-ol (PPE), which is a model for the reactivity of a β-O-4 phenyl ether bond, the most common inter-unit linkage in lignin. The scheme summarizes the rates of different steps based on the temporal evolution of PPE and of the hydrogenolysis products as catalyzed by Cu20PMO in the MeOH/DMC co-solvent at 300 °C. The β-O-4 bond was cleaved with 98% conversion within 1 h. Although some of the expected 1-phenylethanol was observed in the early stage, this intermediate underwent rapid HDO to ethylbenzene (76–92% yield). The lesser product propylbenzene (6–10%) was attributed to β-carbon methylation of 1-phenylethanol followed by HDO.45 Anisole (74% yield after 2 h) was formed as the result of o-methylation of the initial phenol product. After 3 h, aromatic products represented ∼91% of the theoretical yield, much higher than would have been seen in sc-MeOH alone. The aliphatic products formed were mostly cyclohexanols; the remainder were cyclohexanes. Material balance for each time point was ∼100%.
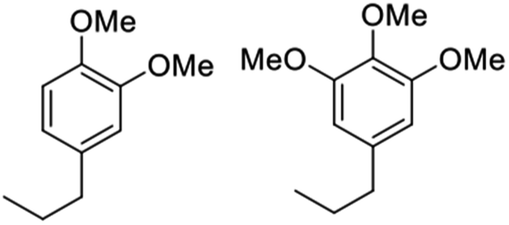
Notably, when OPL was used as the substrate under the same conditions in the DMC/MeOH cosolvent system, the range of products was much narrower than found when the reaction was run in sc-MeOH alone.45 The two major monomeric products were dimethoxy-propyl benzene (DMPB) and trimethoxy-propyl-benzene (TMPB), although quantitative yields were not determined (Fig. 6).
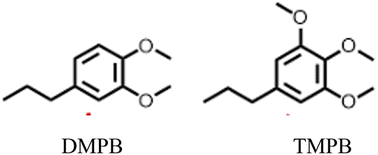 | ||
| Fig. 6 Principal products from the reductive disassembly of OPL in DMC/MeOH at 300 °C.45 | ||
The lignin-first strategy
Extracting the lignin as OPL duplicates to some degree the separation of the (currently) higher value carbohydrates from lignocellulose feedstocks in current industrial processes. It is now well recognized that conventional delignification methods like the soda, Kraft and steam explosion processes give lignins with additional interunit carbon–carbon bonds that are challenging to disassemble to monomers.47,48 The milder organosolv extraction using alcohol with dilute acid18,49,50 gives lignin closer to the native material but still may lead to some structural modifications.51 The OPL obtained thus by extraction from poplar wood can be further valorized by hydrogenation to monomeric lignols. However, the organosolv extraction steps do require the expenditure of solvents, time and energy, although using formaldehyde or diols as extractants has been demonstrated to improve lignin yields significantly.52–56An alternative lignin-first strategy involves the use of heterogeneous transition metal catalysts in methanol or ethanol to enable the selective conversion of lignin to phenolic monomers starting from the intact lignocellulosic biomass.57–60 This procedure has also been termed reductive catalytic fractionation (RCF). Typically, RCF reactions are run with noble metals such as Pd, Pt, Rh, or Ru supported on activated carbon in the presence or absence of H2 (3–4 MPa) at 200–250 °C with alcohol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :biomass ratio (by mass) of at least 20:1 for a period of hours. After reaction, the alcohol solvent contains methoxy phenols, some dimers and trimers originating from the native lignin in the biomass substrate as well as some small solubilized components of xylan. The remaining solid is composed of catalyst and the delignified carbohydrate fraction, cellulose and hemicellulose/xylan as illustrated in Fig. 7.58 The monomeric phenols composition of the liquid phase accounts in many cases for approximately 50% of the carbon of the starting lignin content in the biomass substrate (see below). Lignin is freed from the lignocellulose matrix by partial solvolysis followed by HDO to monomers,62 thus avoiding the cross-linking and recondensation pathways of lignin fragments that plague conventional extraction methods. Notably, an analogy can be drawn to the mechanism proposed for the total reductive dissolution of woody solids by the CuPMOs in sc-MeOH discussed above.43 In the latter case, char formation was avoided by the reductive capture of reactive intermediates, but the difference is that the carbohydrates were also solubilized giving a less desirable mixture of products.
:biomass ratio (by mass) of at least 20:1 for a period of hours. After reaction, the alcohol solvent contains methoxy phenols, some dimers and trimers originating from the native lignin in the biomass substrate as well as some small solubilized components of xylan. The remaining solid is composed of catalyst and the delignified carbohydrate fraction, cellulose and hemicellulose/xylan as illustrated in Fig. 7.58 The monomeric phenols composition of the liquid phase accounts in many cases for approximately 50% of the carbon of the starting lignin content in the biomass substrate (see below). Lignin is freed from the lignocellulose matrix by partial solvolysis followed by HDO to monomers,62 thus avoiding the cross-linking and recondensation pathways of lignin fragments that plague conventional extraction methods. Notably, an analogy can be drawn to the mechanism proposed for the total reductive dissolution of woody solids by the CuPMOs in sc-MeOH discussed above.43 In the latter case, char formation was avoided by the reductive capture of reactive intermediates, but the difference is that the carbohydrates were also solubilized giving a less desirable mixture of products.
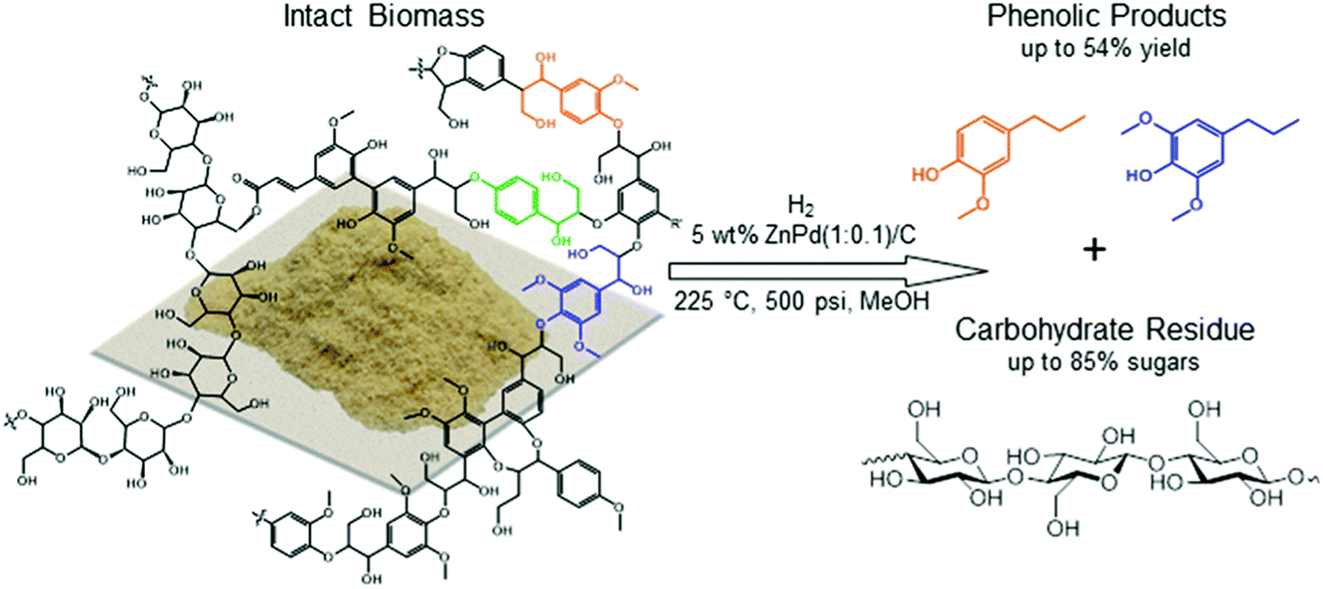 | ||
| Fig. 7 Selective depolymerization and HDO (hydrodeoxygenation) of lignin first from wood biomass to give a lignin-derived phenolic platform and a delignified carbohydrate solid residue, a process subsequently referred to in the literature as reductive catalytic fractionation (RCF). (Figure from ref. 58.) | ||
Development of the RCF approach points to viable bio-refinery strategies where greater valorization of the lignin components would be achieved.63 One advantage is the resulting preservation of the aromaticity since the resulting monolignols are phenols. The cellulose is delignified and remains intact as a solid, which allows its separation and subsequent utilization as a feedstock for production of ethanol, other liquid fuels and/or chemicals. Drawbacks of the lignin-first strategy include the required large solvent to biomass ratio, the need to separate catalyst from the solid cellulose residue (more on this below), the use of hydrogen or the solvent alcohol to generate hydrogen equivalents through reforming (similar to that described above for the CuPMO catalyst), and the high pressures (5–7 MPa) at the operating temperature.
Fig. 8 compares the efficiency of a lignin-first approach to ones where other extraction methods were first used to separate the lignins from the high-S poplar lignocellulose prior to hydrogenolysis of the extractant to give monomers.50 In dry, high-S poplar wood, lignin is a 22% (by wt) component, so a 10 g sample of that material contains 2.2 g of intact lignin. The extraction pathways examined included: refluxing with acidic methanol for 14 days (OPL), reacting with acidic formaldehyde in dioxane at 95 °C for 3.5 h (FPL), and extracting by heating in a formic acid/acetic acid mixture (FA/AA) or aqueous hydrochloric or sulfuric acid. The FPL, OPL and FA/AA methods gave the larger yields of extracted lignin, and these extracts were then subjected to RCF in MeOH under H2 (36 bar) using Pd–Zn/C catalyst at 225 °C. The effective transformation to the monomeric lignols would be the extraction efficiency times the monomer yield from the RCF. Of these procedures, the FPL/RCF pathway gave nearly 3 times the yield found for the OPL/RCF pathway, owing to the more efficient FPL extraction. However, direct RCF of high-S poplar sawdust itself under analogous catalytic conditions gave a significantly higher overall yield of monomers (61% of the initial 22% lignin).50 Furthermore, direct lignin-first procedure requires shorter overall processing times and less solvent and other reagents, thus is a greener process.
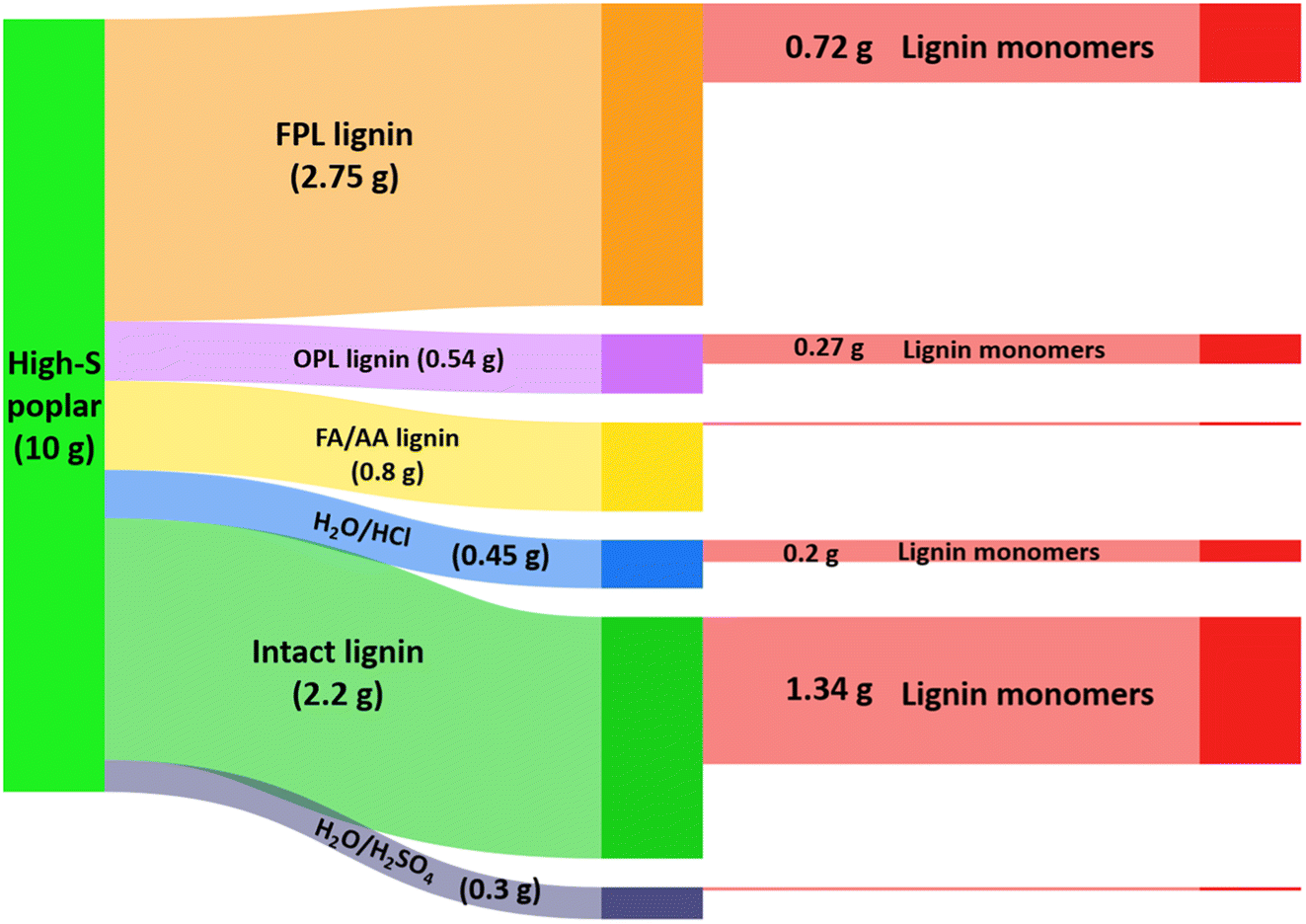 | ||
| Fig. 8 Illustration of organosolv lignin made from 10 g high-S poplar biomass and the further conversion of different types of lignin into monomers. (Figure from ref. 50, Copyright 2022 American Chemical Society). | ||
The ratio of mono versus dimethoxy phenol products depends on the starting lignocellulose substrate, since these reflect the S and G lignin composition (Fig. 2). For example, poplar (a hardwood) gives a dimethoxy:monomethoxy ratio of ∼2:1 while pine (a soft wood) gives exclusively monomethoxy phenol products. As illustrated by Fig. 9,57,58,64–67 the functionalities remaining on the phenolic products depends on the catalyst and reaction conditions. For example, catalysis by Pd/C gives the propyl alcohol side chain while Ru/C and Pd/Zn/C give the propyl group.64 Reactions under limiting hydrogen availability afford an alkenyl chain in the product such as isoeugenol. As will be demonstrated, such diversity enables the construction of different polymers. In addition, certain phenolic products from lignin are high value flavoring and fragrance compounds such as isoeugenol and dihydroeugenol, while exhaustive HDO upon further processing can afford C9 hydrocarbon fuels such as propylbenzene and propylcyclohexane (see below).68
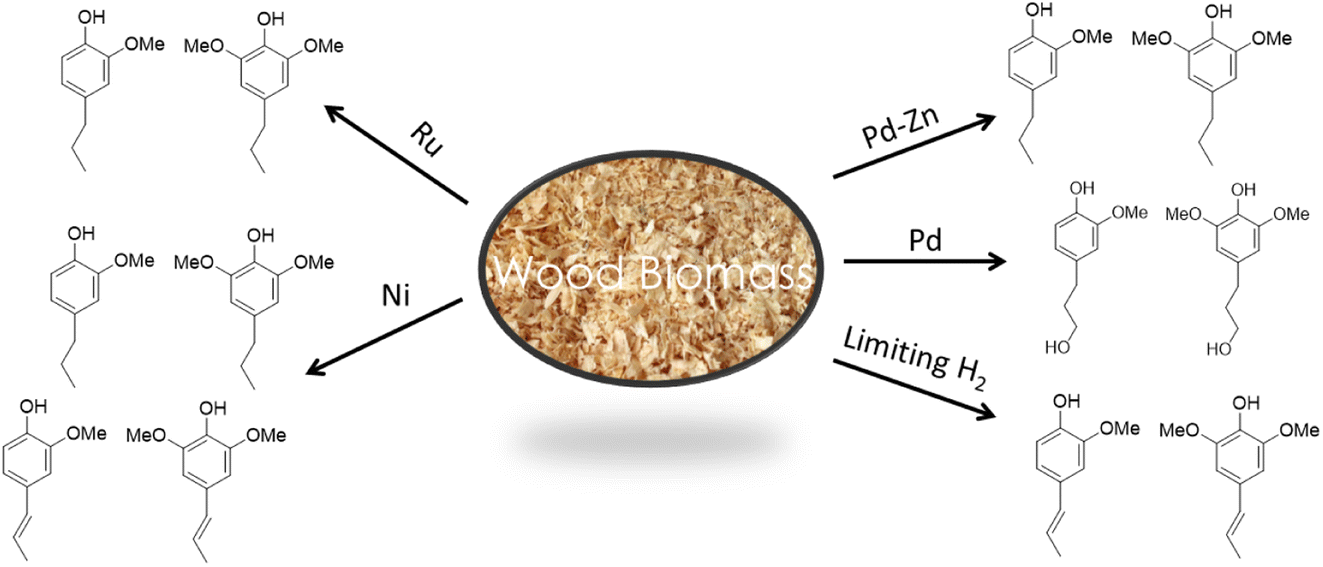 | ||
| Fig. 9 Changing the catalyst or conditions affects the functionality and oxygen content in the product phenols (figure based on results from ref. 57, 58 and 64–67). | ||
Two drawbacks mentioned above, catalyst separation from the cellulose co-product and solvent usage, have been addressed. In batch reactions, the catalyst has been placed in a cage which allows it to remain separate from the solid biomass reactant and the solid cellulose co-product.61 Obviously, as discussed previously for the sc-MeOH CuPMO system, a solid catalyst does not react with a solid biomass substrate. In RCF the alcohol solvent dissolves the lignin but not the cellulose, and the solubilized lignin substrate (polymeric and oligomeric) reacts with the catalyst. Therefore, as long as the solvent passes through the catalyst cage, the reaction ensues with comparable efficiency to the mixed slurry of lignocelluslose and catalyst. Another approach, which helps also with the large solvent demand and provides safer operating condition, is to run RCF as a plug flow through serial reactors (Fig. 10).69–71 The hot solvent passes first through the packed biomass to extract lignin; subsequently the lignin solution passes through a packed catalyst bed to depolymerize lignin to phenols. A chemistry technology startup Spero Renewables LLC (formerly Spero Energy Inc.) has developed SPERLU (Selective Process for Efficient Removal of Lignin and Upgrading), a process in which the solvent can be recycled and the catalyst regenerated.72 Nevertheless, process intensification continues to be a challenge because of the low density of biomass and the requirement of excess solvent (8–10× the mass of lignocellulose) even with solvent recycling regimes. Furthermore, slow reactions necessitate long residence times complicating processing of plug flow reactors.
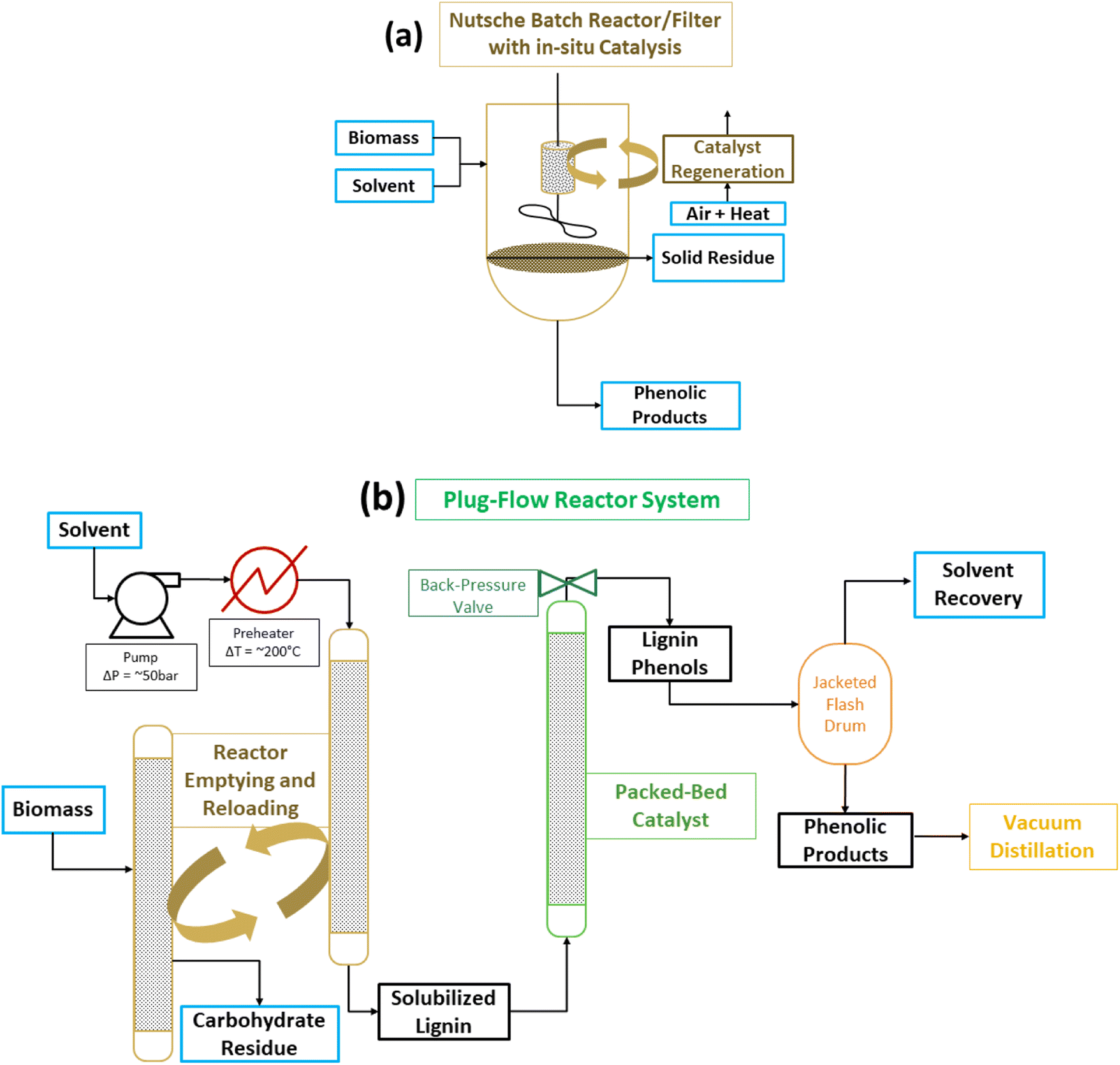 | ||
| Fig. 10 Process and reactor configuration for lignin first biorefining. (a) Batch reactor with the use of a catalyst cage to keep biomass and its solid products separated from the catalyst. (b) Plug-flow reactor system with solvent recovery and recycling as in the SPERLU process. (Figure is provided by Mr Sean Prager of Spero Renewables LLC.) | ||
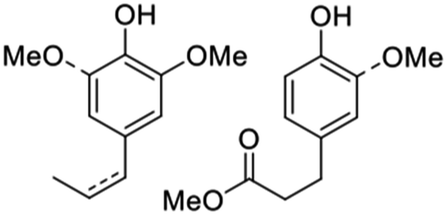
Nickel, which is much more Earth-abundant than the platinum metals, has also been demonstrated as a catalyst for depolymerization of lignins in methanol.57,66 In one example from Abu-Omar group, Ni on activated carbon converted lignin in miscanthus into aromatic phenols with a maximum yield of 68% based on the starting lignin.66 The recovered carbohydrates of miscanthus were treated with iron chloride to produce furfural and levulinic acid, useful platform chemicals. Thus, all three major biomass components (lignin, cellulose and hemicellulose) were effectively valorized into high value chemicals with 98% mass balance (Fig. 11).
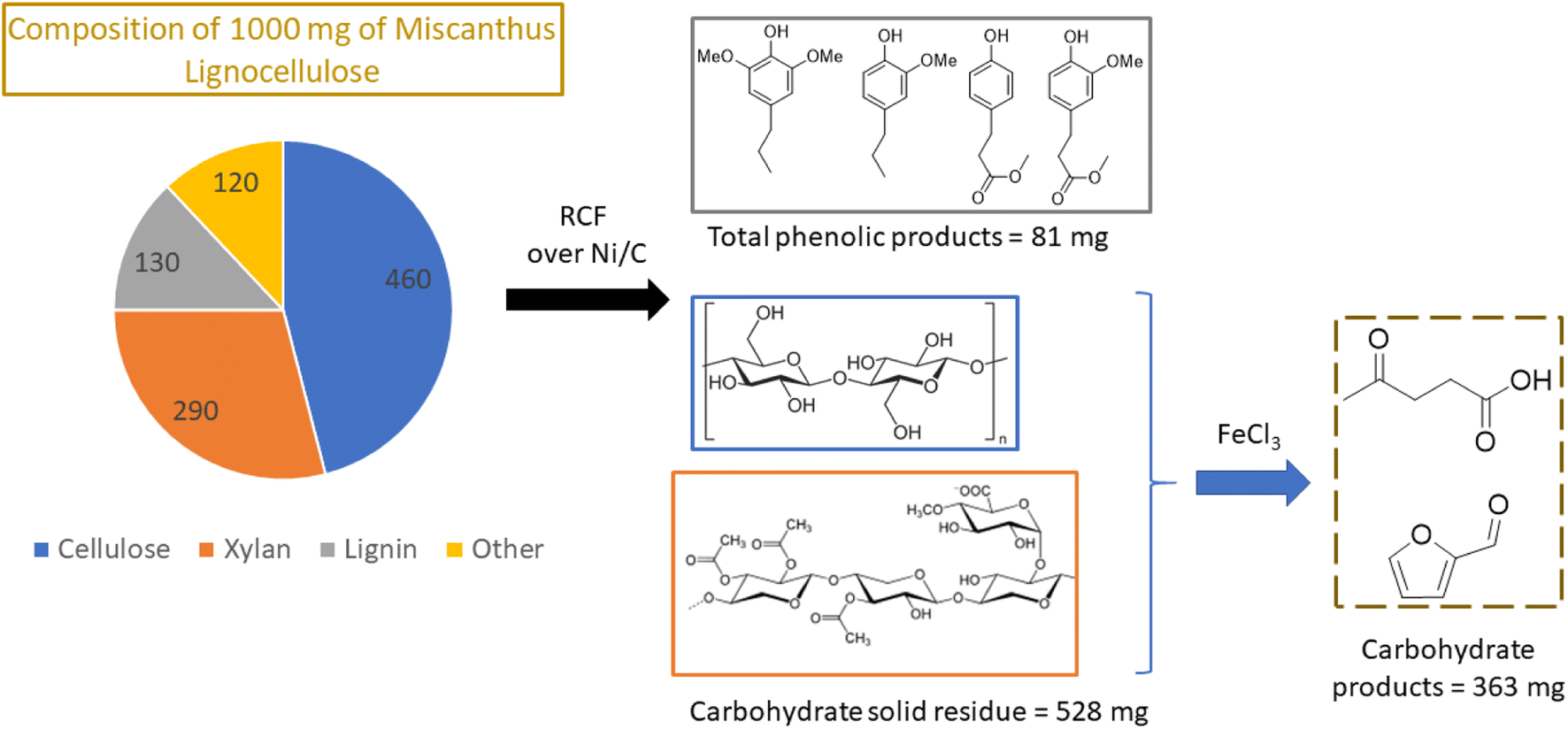 | ||
| Fig. 11 Two step transformation of whole biomass via lignin first RCF and Lewis acid catalyzed dehydration of carbohydrates to yield platform chemicals. (Figure based on results from ref. 66.) | ||
Table 1 presents a summary of the products obtained from various feedstocks.
| Bio-based feedstock | Conversion process | Products | % Yielda | Application |
|---|---|---|---|---|
| a Based on mass of feedstock. b Porous metal oxides (PMO) in supercritical MeOH or EtOH. c RCF = reductive catalytic fractionation. In MeOH under H2. d Formaldehyde protected organosolv lignin. | ||||
| Lignocellulose (poplar, stover, etc.) | Acid extraction | Organosolv lignin | 10–20% | Feedstock |
| Cellulose | 50% | Feedstock | ||
| Lignocellulose (wood shavings) | Catalytic Cu-PMOb | C2–C6 alkyl alcohols | ∼50% | Fuels |
| C9–C12 cyclohexyl alcohols | ∼20% | Chemicals and fuels | ||
| Organosolv poplar lignin (OPL) | Catalytic Cu-PMO | C9–C12 cyclohexyl alcohols | ∼90% | Chemicals and fuels |
| OPL | Cu-PMO in DMC/MeOH |
|
∼90% | Aromatic synthons, specialty chemicals |
| Lignocellulose (poplar, birch, etc.) | RCF | Aromatic phenols 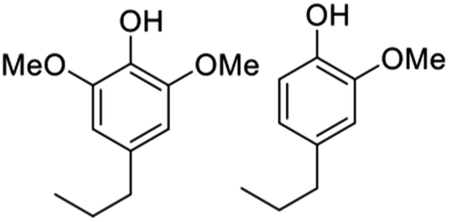 |
∼12% | Specialty chemicals (flavors and fragrance molecules) |
| Zn–Pd/C or Ru/Cc | Cellulose/xylan | ∼60% | Feedstock | |
| FPLd | RCF | Phenol monomers | ∼30% | Chemicals, monomers |
| Zn–Pd/C | ||||
| OPL | RCF | Phenol monomers | ∼50% | Chemicals, monomers |
| Zn–Pd/C | ||||
| Miscanthus lignocellulose | RCF |
|
∼10% | Specialty chemicals |
| Ni/C | ||||
| Cellulose/xylan (from RCF) | Catalytic FeCl3 | Levulinic acid and furfural | 70% | Chemicals |
| Phenol monomers (from RCF) | Catalytic | C9 hydrocarbons 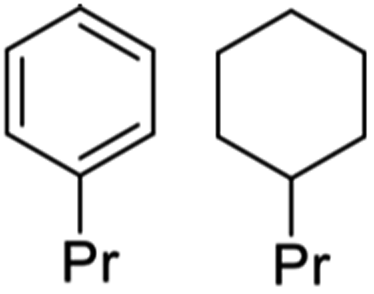 |
>90% | Fuels |
| Nb2O5 and Ru/C | ||||
Mechanistic aspects of the RCF reaction
The solvent in RCF plays two important roles. It solubilizes native lignin from the biomass without degrading the carbohydrates. As a result, the solubilized lignin can come in contact with the heterogeneous catalyst and undergo C–O bond cleavage and HDO. The second role of the alcohol solvent is preventing condensation of native lignin to form recalcitrant C–C linkages. One of the eminent reactions that lignin molecules undergo is formation of benzylic carbonium ion under acidic conditions, which give rise to condensation reactions with aromatic moieties in lignin and C–C bond formation. A solvent such as MeOH can scavenge carbonium ion and act as a protecting group reducing condensation reactions and as a result increase the yields of monomeric phenols from lignin under RCF. The methoxy protecting groups have been detected by 2D NMR spectroscopy and they are akin to protecting group strategy in producing organosolv lignin that is amenable to further upgrading by catalysis. The roles of the solvent in RCF are illustrated in Fig. 12.72,73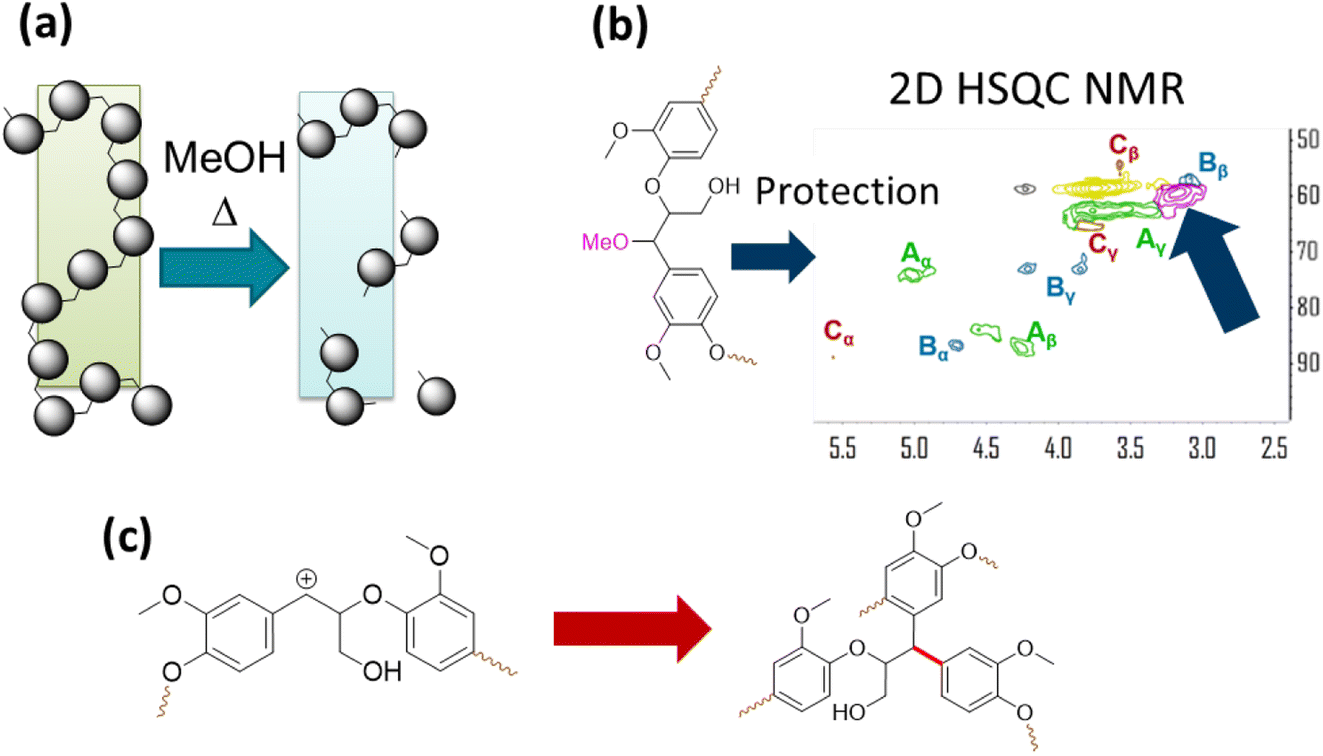 | ||
| Fig. 12 The role of the alcohol solvent in RCF. (a) Dissolution of lignin. (b) Protection group chemistry as observed by 2D HSQC NMR. (c) Self condensation reaction of lignin resulting from benzylic carbonium ion formation under acidic conditions. (Figure based on results reported in ref. 72 and 73.) | ||
Mechanistic features of the synergy between Pd and Zn in the Pd/Zn/C catalyst was investigated by the Abu-Omar group through studying lignin model compounds.74 Spectroscopic data coupled with reactivity studies on lignin model compounds suggested the crucial role Zn ions play in removal of the alcohol group on the γ-carbon of β-O-4-ether linkages. The proposed mechanism is shown in Fig. 13. In this same study several Lewis acids were investigated in combination with Pd for lignin fractionation and conversion from wild type as well as high S poplar wood. Zinc salts were the most successful and did not show dependence on the choice of anion used to introduce the zinc into the catalyst system.
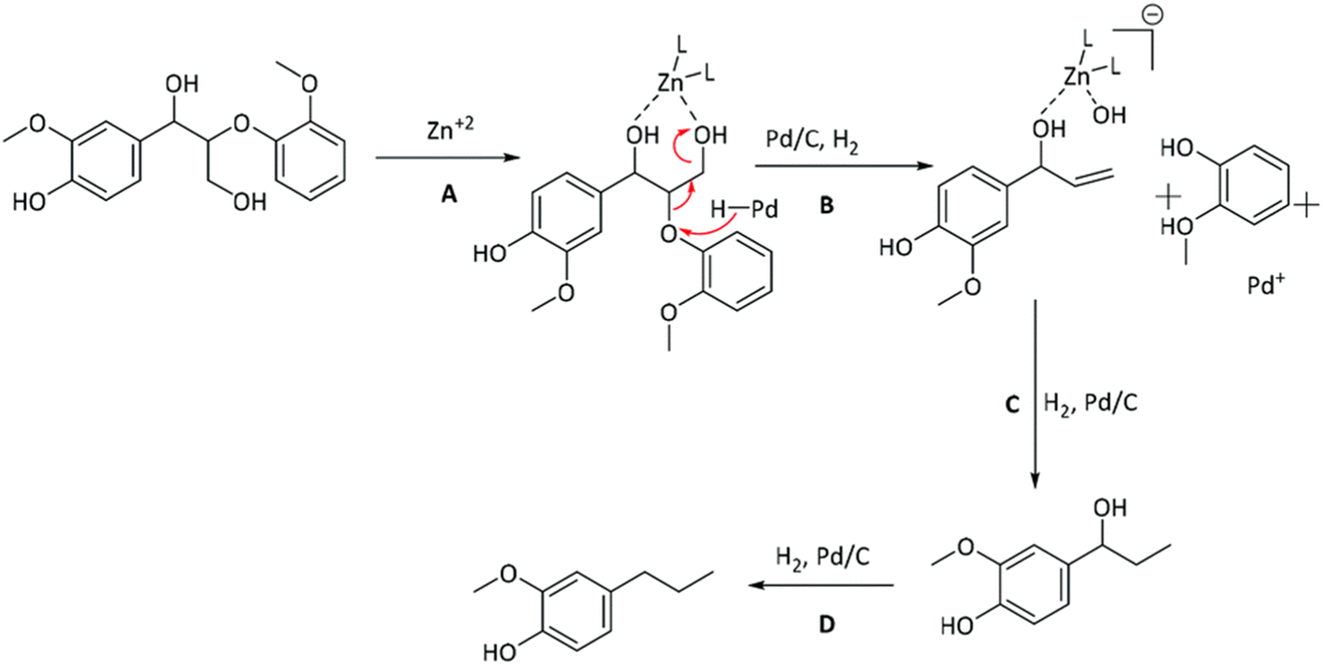 | ||
| Fig. 13 Proposed mechanism for the role of Zn and its synergy with Pd in lignin depolymerization and HDO. (Figure from ref. 74.) | ||
Reactions of the lignin monomers
A key issue is how to utilize such monomers once they have been isolated. One approach is hydrodeoxygenation (HDO) to a narrowly defined hydrocarbon stream useful as a feedstock for the synthesis of aliphatic and aromatic chemicals or as drop-in biofuels compatible with current ICEs. Most such HDO procedures require high temperatures and pressures (H2),75,76 so more subtle methodologies are desirable. An example is the report by Li et al.68 describing the hydroprocessing of dihydroeugenol (DHE) using a physical mixture of two catalysts, ruthenium on carbon (Ru/C) and calcined niobium pentoxide (Nb2O5). As a model, DHE includes the methoxy, hydroxy, and a propyl groups characteristic of the lignin monomers present in bio-oils. These two catalysts worked synergistically under relatively mild conditions (6 bar H2, T = 250 °C in aqueous media with small quantities of added MeOH) to convert DHE in high yields to propyl cyclohexane (e.g., eqn (3)), which has potential applications as a drop-in liquid fuel. Both propyl benzene and 4-propylcyclohexyl-1-ol were seen as intermediates in this transformation. The synergism of the dual catalyst system is evident from the observations that, with Nb2O5 catalyst alone, the product was principally 4-propylcatechol while with the Ru/C catalyst alone the product was largely 4-propylcyclohexyl-1-ol. Notably, the Nb2O5 calcination temperature had a major influence on that catalyst's activity, owing to the decrease in acidic sites when calcined at T > 450 °C.68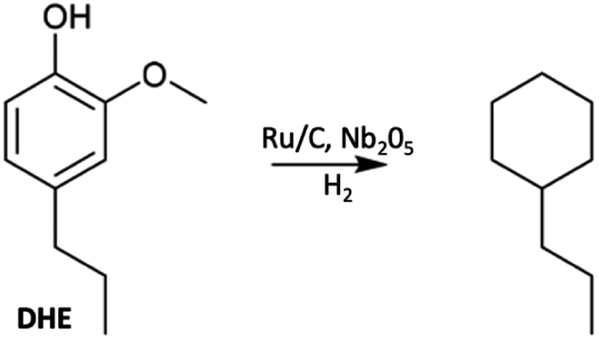 | (3) |
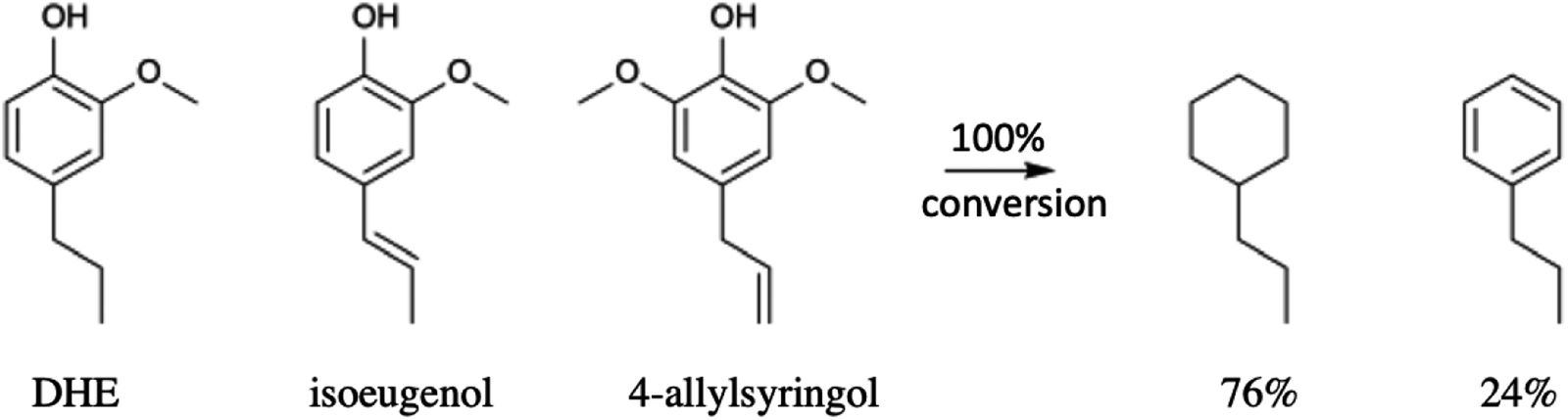
Li et al.68 also subjected a simulated bio-oil consisting of a roughly equal mixture of the common monolignols DHE, isoeugenol and 4-allylsyringol to HDO using the same dual catalyst system. The products after 100% conversion (eqn (4)) were the C9 hydrocarbons propyl-cyclohexane (76%) and propyl-benzene (24%). A 100% yield of the former could be achieved with an extended reaction time. Thus, this HDO catalysis system offers a viable strategy for funneling multiple monolignols from lignin disassembly into a much simpler mixture of C9 alkanes more compatible with fuel applications. Furthermore, given that cyclohexane dehydrogenation to benzene is known technology,77 this mixture could also provide an entry into BTX-type chemistry based on renewable feedstocks.
 | (4) |
The lignin monomers will be even more valuable if one can take advantage of their oxygenated forms, for example, by forming functional polymers based on monolignols or other lignin fragments.50,78–85 In this context, Zhou et al.78 synthesized epoxy nanocomposites based on DHE by following the sequence outlined in eqn (5) to prepare the epoxide precursors. The DHE was first converted to propylcatechol (DHEO) by HBr hydrolysis of the methoxy group. It should be noted that methoxy group deprotection on the phenol ring can be accomplished now under greener conditions with Nb2O5 catalyst in water (vide infra). Reaction of the DHEO with epichlorohydrin gave a mixture of the di-epoxy monomer and the benzodioxane byproduct, the latter accounting for ∼5% of the total. Reaction of this mixture with an amine hardener such as diethylenetriammine (DETA) gave a thermoset polymer, which when mixed with a nanoclay gave a high-performance epoxy nanocomposite.
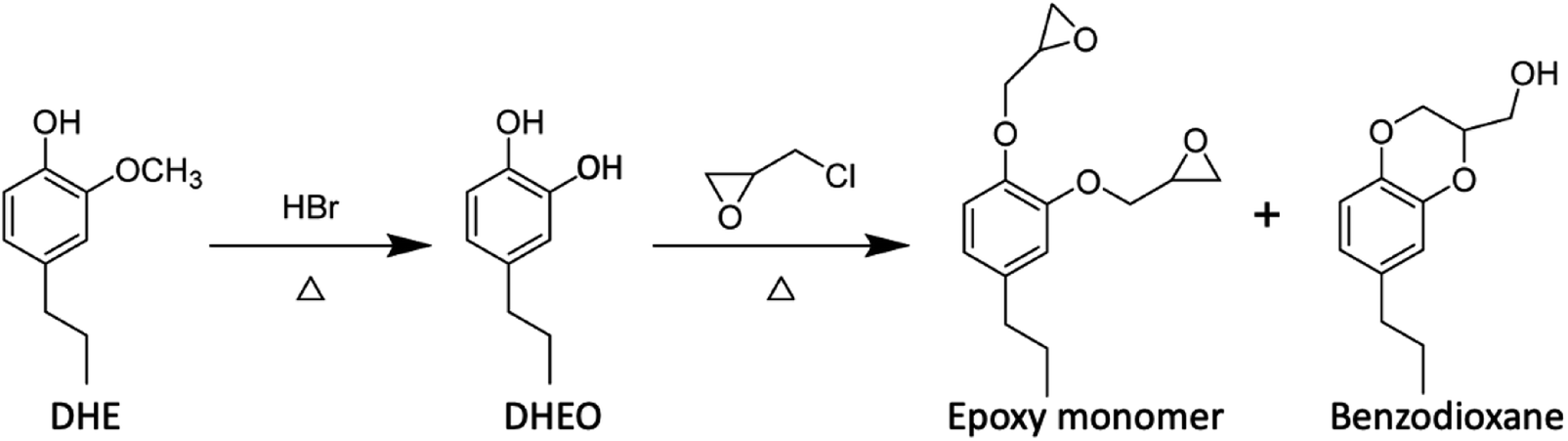 | (5) |
In an extension of the above study, Liu et al.50 extracted 4-propyl-2,6-dimethoxyphenol (DMPP) from poplar biomass that had been genetically modified to afford high-sinapyl (high-S) lignin. The DMPP was then converted to propyl-pyrogallol (DMPPO) in a reaction catalyzed by calcined Nb2O5 in water. Under optimized conditions, this reaction was nearly quantitative (96% yield), the other product being the monomethoxy species. Since the Nb2O5 catalyst could be reused several times before significant deactivation, then reactivated by calcining again, methoxy group hydrolysis/deprotection in this manner is greener than using HBr as noted in eqn (5). The resulting DMPPO mixture underwent quantitative reaction with epichlorohydrin (Scheme 3) to give largely the novel tri-epoxide of DMPPO plus the benzodioxane monoepoxide analogue. This epoxy mixture in turn, underwent reaction with DETA (1:1 stoichiometric epoxy:NH ratio) to give, after curing, a thermoset polymer.
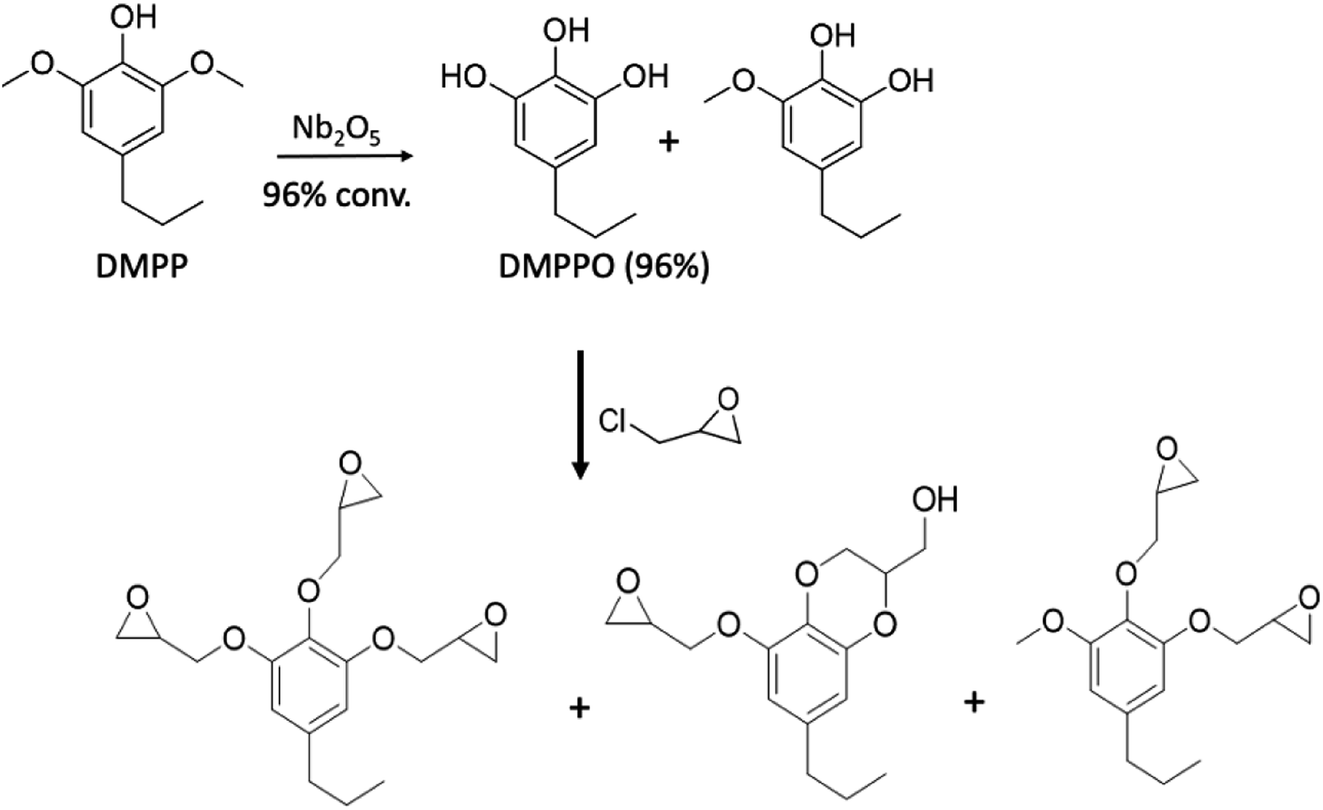 | ||
| Scheme 3 Formation of thermoset polymer precursors from biomass derived DMPP. (Figure from ref. 50). | ||
The promise of using lignin derived phenols as monomers in preparation of thermoset polymers is to have a renewable substitute to bisphenol A (BPA). However, the formation of benzodioxane as a result of the epoxide functional groups being in close proximity (ortho-) to each other leads to materials with lower Tg (glass transitions temperature) and storage modulus than thermoset materials made with BPA. Zhao et al. investigated the effect of cross-linking density on bio-based epoxy network by modifying DHE through o-demethylation and phenol–formaldehyde reactions.84 The cured thermoset polymers were evaluated using rubber elasticity theory from dynamic mechanical analysis (DMA) and thermogravimetric analysis (TGA). The monomer with oligomeric DHE and all oxygens on the aromatic rings are hydroxyl/phenolic groups –OH exhibited the most significant improvements with cross-linking density of 9.77 mol dm−3 and Tg = 139 °C (Fig. 14).
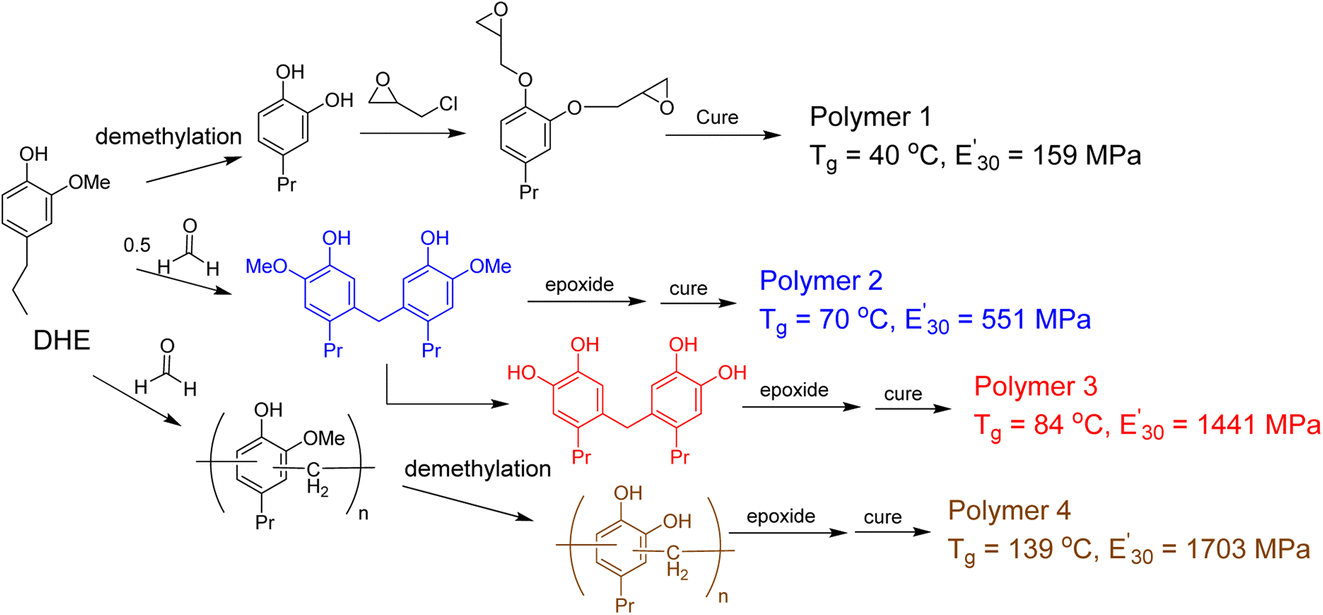 | ||
| Fig. 14 Modification of lignin monomeric phenol to improve the thermal and mechanical properties of the resulting thermoset polymers. (Figure based on results reported in ref. 84). | ||
In a similar vein, polyphenols were generated by combining lignin and carbohydrate derived monomers. For example, lignin based 4-methylcatechol is reacted with furfural (produced from xylose) or 5-hydroxymethylfurfural (HMF, produced from glucose) to prepare a novel bisphenol-furan polyphenol (Fig. 15).81 The structures of the resulting polyphenol are ideal for generating reactive epoxide precursors that are cured to an epoxy polymer networks. These thermoset polymers display excellent mechanical and thermal properties with Tg = 110 °C and glassy modulus of 10 GPa. This study illustrates the viability of making highly bio-based epoxy thermoset, moldable polymers, from wood.
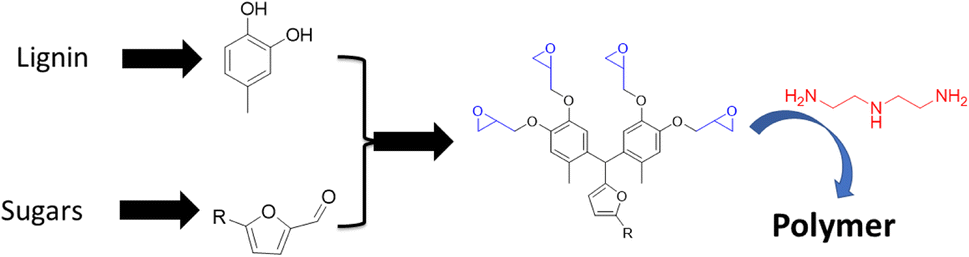 | ||
| Fig. 15 Thermoset polymers based on biobased polyphenols made from the reaction of lignin-based phenols with carbohydrate derived furans. | ||
More recently the alkenyl functionality on the propyl chain of lignin products such as isoeugenol has been utilized through the olefin metathesis reaction to create bisphenol akin to BPA, where the phenol groups are on opposite sides of the molecule.86 These bisphenols before and after hydrogenation of the linking double bond were used with different organic diacids such as succinic acid, a bio-based monomer made through fermentation of glucose, to synthesize bio-polyesters. The glass transition temperature can be tuned to match that of petroleum-based polyesters such as PET (Tg = 80 °C) by using mixtures of lignin-based bisphenols to obtain a Tg = 83 °C. In addition to being thermally tunable, these renewable polyesters can be 100% bio-based, for example, when succinic acid is used. With the newly emerging interest in circularity and upcycling of polymers, these bio polyesters can be chemically upcycled through cross-linking with lignin-based epoxide in the presence of an organic base to afford a bio-based thermoset polymer (Fig. 16).87 The mechanical properties showed storage moduli in the range of 5–19 MPa and tensile stress of up to 68 MPa. The use of polyester as a crosslinker for epoxy thermoset polymers is an interesting mechanism that remains largely under utilized, and it has the potential to expand the library of bio-based thermoset materials.
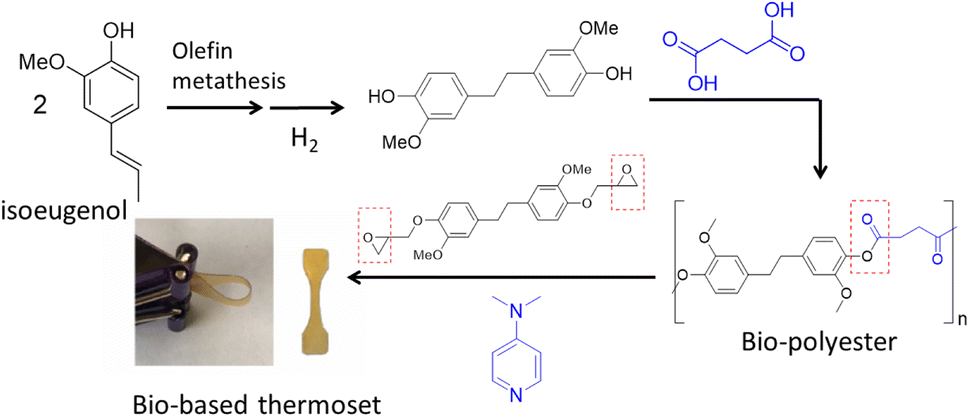 | ||
| Fig. 16 Synthesis of bio-polyester from isoeugenol via olefin metathesis and chemical upcycling of the polyester with lignin-based bis-phenol epoxide to thermoset polymer. (Figure based on results from ref. 86 and 87). | ||
These studies and a number of others point to the feasibility of using such lignin-based fragments as precursors of various biomass-based polymers. However, true sustainability will depend on demonstrating that production of such materials will be economically viable and has a carbon footprint significantly less than that for other polymer sources.88 Another key factor that would be demonstrating that any new biomass-derived polymers have superior physical or chemical properties, such as recyclability or bio-degradability. The resulting added value would provide an economic incentive for the necessary capital investment by society to make the transition to such materials.
Future directions
While lignin-first may prove an effective strategy for utilization of virgin lignocellulose, it is not applicable to the lignin residues from paper manufacturing where processes are optimized to maximize cellulose yield. The different approaches used industrially to separate lignin from the cellulose lead to considerable structural modification. For example, the commonly used Kraft processing generates over 50 million tons of lignin world-wide by reacting wood chips with sodium hydroxide and sodium sulfide at high temperature.89 The resulting technical lignin has a high sulfur content and markedly increased crosslinking via C–C bonds. Dealing with such crosslinking certainly adds to the challenge of utilizing this feedstock, and while there have been advances addressing this challenge,48,90 sulfur removal would also appear necessary before successful chemical valorization.Biomass is widely distributed, and transportation costs will limit the range over which forest or agricultural lignocellulose feedstock can be economically collected and transported to a centralized location. Therefore, processing of such geographically dispersed feedstocks would best be accomplished at moderate-sized, integrated biorefineries located in the regions where lignocellulose wastes or crops are generated. The impact would be a redistribution of industrial activity that could have major socio-economic consequences, including redistribution of wealth to rural communities, assuming that this is coupled to sound environmental management.
Ongoing studies are focused on characterizing and optimizing new catalytic processes that can be based on biomass feedstocks for the production of fuels and to chemical precursors for industrial products. For example, several recent reports have described studies to convert biomass to aviation fuels.91,92 There is also increasing emphasis on elucidating key mechanistic issues and on evaluating how the reaction variables control the yields and selectivity in product streams. Additionally, new tools such as machine learning and artificial intelligence will no doubt play a larger role in developing new catalysts and perhaps by discerning pathways to give higher values products.93
Successful development of economically viable processes has the potential for reducing the atmospheric CO2 burden without compromising food supplies. Implementing these developments to prepare chemicals and fuels present serious challenges, but also some real opportunities. One should be able to build new materials from biomass derived substrates having key functional advantages, including biodegradability. However, the task of scaling up from interesting, perhaps even precedent-shattering discoveries in the laboratory to economically-viable industrial operations is a daunting one94,95 that will require considerable engineering skill and perseverance.
These and related issues need to be the subjects of detailed techno-economic analysis to assess the viability of the new processes and products that are resulting from the already high, and increasing, research activity in biomass conversion. Important parameters that must eventually be included in such evaluations are environmental and societal damages that will result from continuing along our current path dependent on the energy and chemicals obtained from fossilized carbon.
This perspective has attempted to provide an overview of some current studies and progress in the conversion of biomass to chemicals and fuels that may alleviate some of modern societies dependence on petroleum and other fossil carbons. We have provided examples from our own laboratories and have in no way attempted to provide a comprehensive review of the very extensive research activities in other laboratories around the world. One should not view these activities in a competitive light, but in terms of the growing recognition that the problems of climate change and related environmental damage is truly existential and threatens modern civilization.
We conclude with the comment that problems that sometimes appear insurmountable often present incredible opportunities. However, some words of caution are appropriate. The authors firmly believe that biomass conversion, specifically of lignocellulose biomass, can make a substantial contribution to reducing GHG emissions as part of the transition to a more sustainable world. However, it is clear that this is only one solution. Furthermore, we must be committed to an environmentally just society. The production of fuels and chemicals from biomass must be balanced against the nutritional needs of the populace and the need for open natural space where other species are not challenged with extinction. The 21st century is the century of renewable energy, but let's bring about this inevitable transition with grace and justice and with respect for all of nature, including ourselves.
Conflicts of interest
MMAO is founder and part owner of Spero Renewables, LLC, a technology company making biomass-based renewable alternative to petrochemicals.Acknowledgements
Research in the Ford laboratory on biomass conversion was supported by the National Science Foundation Center for Sustainable Use of Renewable Feedstocks (CenSURF) (NSF CHE-1240194) and the UCSB PIRE-ECCI program (NSF Grant OISE-05302680). Preliminary studies received support from the Petroleum Research Fund of the American Chemical Society. Research in the Abu-Omar laboratory on biomass conversion was supported by Basic Energy Sciences, US Department of Energy through EFRC (Energy Frontier Research Center) C3Bio (Center for Catalytic Conversion of Biomass to Biofuels) under award no. DE-SC000097 and Catalysis Science Program under award no. DE-SC0019161; the Mellichamp Sustainability Initiative at UCSB; and the NSF (CHE-2154333).References
- J. E. Ziegler, M. J. Zdilla, A. J. Evans and M. M Abu-Omar, H-2-Driven Deoxygenation of Epoxides and Diols to Alkenes Catalyzed by Methyltrioxorhenium, Inorg. Chem., 2009, 48, 9998–10000 CrossRef CAS PubMed.
- G. S. Macala, A. W. Robertson, C. L. Johnson, Z. B. Day, R. S. Lewis, M. G. White, A. V. Iretskii and P. C. Ford, Transesterification catalysts from iron doped hydrotalcite-like precursors. Solid Bases for Biodiesel Production, Catal. Lett., 2008, 122, 205–209 CrossRef CAS.
- National Aeronautics and Space Administration (USA) news release July 2022, https://climate.nasa.gov/vital-signs/carbon-dioxide/.
- National Oceanographic and Atmospheric Administration (USA) Research News 23 May 2022, https://research.noaa.gov/article/ArtMID/587/ArticleID/2877/Greenhouse-gas-pollution-trapped-49-more-heat-in-2021-than-in-1990-NOAA-finds.
- Source: World Meteorological Organization: published 19 January 2022, https://public.wmo.int/en/media/press-release/2021-one-of-seven-warmest-years-record-wmo-consolidated-data-shows hottest years in history.
- D. I. A. McKay, A. Staal, J. F. Abrams, R. Winkelmann, B. Sakschewski, S. Loriani, I. Fetzer, S. E. Cornell, J. Rockstromand and T. M. Lenton, Exceeding 1.5°C global warming could trigger multiple climate tipping points, Science, 2022, 377, DOI:10.1126/science.abn7950.
- Environmental Protection Agency (USA) report 14 April 2022: “Inventory of U.S. Greenhouse Gas Emissions and Sinks 1990-2020”, https://www.epa.gov/ghgemissions/sources-greenhouse-gas-emissions#electricity Search PubMed.
- Hedges & Company blog, https://hedgescompany.com/blog/2021/06/how-many-cars-are-there-in-the-world/.
- EV Volumes.com (The electric vehicle world sales database), https://www.ev-volumes.com.
- California to ban gasoline-only cars, Science, ed. J. Brainard, 2022, vol. 377, p. 1024.
- (a) International Energy Agency report, https://www.iea.org/reports/aviation Search PubMed; (b) Our World in Data Report, https://ourworldindata.org/co2-emissions-from-aviation Search PubMed.
- Catalysis in Biomass Conversion, Advances in Inorganic Chemistry, ed. P. C. Ford and R. van Eldik, Academic Press, Oxford, UK, 2021, vol. 77 Search PubMed.
- R. Mori, Replacing all petroleum-based chemical products with natural biomass-based chemical products: a tutorial review, RSC Sustainability, 2023, 1, 179 RSC.
- F. Wang, D. Ouyang, Z. Zhou, S. J. Page, D. Liu and X. Zhao, Lignocellulosic biomass as sustainable feedstock and materials for power generation and energy storage, J. Energy Chem., 2021, 57, 247 CrossRef CAS.
- M. Carrier, A. Loppinet-Serani, D. Denux, J.-M. Lasnier, F. Ham-Pichavant, F. Cansell and C. Aymonier, Thermogravimetric analysis as a new method to determine the lignocellulosic composition of biomass, Biomass Bioenergy, 2011, 35, 298–307 CrossRef CAS.
- R. Sankaran, K. Markandan, K. S. Khoo, C. K. Cheng, V. Ashokkumar, B. Deepanraj and P. L. Show, The Expansion of Lignocellulose Biomass Conversion into Bioenergy via Nanobiotechnology, Front. Nanotechnol., 2021, 3, 793528 CrossRef.
- G. Brunow, Methods to Reveal the Structure of Lignin, in Lignin, Humic Substances and Coal, ed. M. Hofrichter and A. Steinbuchel, Wiley-VCH, Weinheim, Germany, 2001, vol. 1, pp. 89−116 Search PubMed.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Catalytic disassembly of an organosolv lignin via hydrogen transfer from supercritical methanol, Green Chem., 2010, 12, 1640–1647 RSC.
- X. Zhao, L. Zhang and D. H. Liu, Biomass recalcitrance. Part I: the chemical compositions and physical structures affecting the enzymatic hydrolysis of lignocellulose, Biofuels, Bioprod. Bioref., 2012, 6, 465–482 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Catalytic conversion of biomass to biofuels, Green Chem., 2010, 12, 1493–1513 RSC.
- S. Dutta, D. Sudipta, M. I. Alam, M. M. Abu-Omar and B. Saha, Direct conversion of cellulose and lignocellulosic biomass into chemicals and biofuel with metal chloride catalysts, J. Catal., 2012, 288, 8–15 CrossRef CAS.
- B. Saha and M. M. Abu-Omar, Advances in 5-hydroxymethylfurfural production from biomass in biphasic solvents, Green Chem., 2014, 16, 24–38 RSC.
- S. Takkellapati, T. Li and M. A. Gonzalez, An overview of biorefinery-derived platform chemicals from a cellulose and hemicellulose biorefinery, Clean Technol. Environ. Policy, 2018, 20, 1615–1630 CrossRef CAS PubMed.
- A. Bohre, S. Dutta, B. Saha and M. M. Abu-Omar, Upgrading Furfurals to Drop-in Biofuels: An Overview, ACS Sustainable Chem. Eng., 2015, 3, 1263–1277 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, T. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. Wyman, Lignin valorization: improving lignin processing in the biorefinery, Science, 2014, 44, 1246843 CrossRef PubMed.
- W. Arts, D. Ruijten, K. van Aelst, L. Trullemans and B. Sels, The RCF biorefinery: building on a chemical platform from lignin, Adv. Inorg. Chem., 2021, 77, 241–298 CrossRef CAS.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Valorization of Biomass: Deriving More Value from Waste, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- K. Barta and P. C. Ford, Catalytic Conversion of Non-food Woody Biomass Solids to Organic Liquids, Acc. Chem. Res., 2014, 47, 1503–1512 CrossRef CAS.
- D. Mohan, C. U. Pittman and P. H. Steele, Pyrolysis of Wood/Biomass for Bio-oil: A Critical Review, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- M. B. Figueirêdo, I. Hita, P. J. Deuss, R. H. Venderbosch and H. J. Heeres, Pyrolytic lignin: a promising biorefinery feedstock for the production of fuels and valuable chemicals, Green Chem., 2022, 24, 4680 RSC.
- G. S. Macala, T. D. Matson, C. L. Johnson, R. S. Lewis, A. V. Iretskii and P. C. Ford, Hydrogen Transfer from Supercritical Methanol over a Solid Base Catalyst: A Model for Lignin Depolymerization, ChemSusChem, 2009, 2, 215–217 CrossRef CAS PubMed.
- G. S. Macala, A. W. Robertson, C. L. Johnson, Z. B. Day, R. S. Lewis, M. G. White, A. V. Iretskii and P. C. Ford, Transesterification Catalysts from Iron Doped Hydrotalcite-like Precursors: Solid Bases for Biodiesel Production, Catal. Lett., 2008, 122, 205–209 CrossRef CAS.
- C. T. Yavuz, B. D. Shinall, A. V. Iretskii, M. G. White, T. Golden, M. Atilhan, P. C. Ford and G. D. Stucky, Markedly Improved CO2 Capture Efficiency and Stability of Gallium Substituted Hydrotalcites at Elevated Temperatures, Chem. Mater., 2009, 21, 3473–3475 CrossRef CAS.
- M. De bruyn, Z. Sun and K. Barta, The thousand faces of Cu-doped porous mixed oxides (Cu-PMO) in the conversion of renewable resources and beyond, Adv. Inorg. Chem., 2021, 77, 59–98 CrossRef CAS.
- J. A. Barrett, Enhancing Product Selectivity in Biomass and Bioalcohol Reactions over Cu-doped Porous Metal Oxides, Adv. Inorg. Chem., 2021, 77, 299–342 CrossRef CAS.
- J. A. Barrett, Z. R. Jones, C. Stickelmaier, N. Schopp and P. C. Ford, Pinch of Salt Improves n-Butanol Selectivity in the Guerbet Condensation of Ethanol over Cu-doped Mg/Al Oxides, ACS Sustainable Chem. Eng., 2018, 6, 15119–15126 CrossRef CAS.
- J. I. Di Cosimo, V. K. Diez, M. Xu, E. Iglesia and C. R. Apesteguia, Structure and Surface and Catalytic Properties of Mg-Al Basic Oxides, J. Catal., 1998, 178, 499–510 CrossRef CAS.
- C. M. Bernt, H. Manesewan, M. Chui, M. Boscolo and P. C. Ford, Temperature Tuning the Catalytic Reactivity of Cu-Doped Porous Metal Oxides with Lignin Models, ACS Sustain. Chem. Eng., 2018, 6, 2510–2516 CrossRef CAS.
- P. C. Ford, Water Gas Shift Reaction, Encyclopedia of Catalysis, J. Wiley and Sons, Hoboken, NJ, 2nd edn, 2010 Search PubMed.
- A. Ponnudurai, P. Schulze, A. Seidel-Morgenstern and H. Lorenz, Fractionation and Absolute Molecular Weight Determination of Organosolv Lignin and Its Fractions: Analysis by a Novel Acetone-Based SEC_MALS Method, ACS Sustain. Chem. Eng., 2023, 11, 766–776 CrossRef CAS.
- K. Barta, G. Warner, E. S. Beach and P. T. Anastas, Depolymerization of organosolv lignin to aromatic compounds over Cu-doped porous metal oxides, Green Chem., 2014, 16, 191–196 RSC.
- X. Huang, T. I. Koranyi, M. D. Boot and E. M. J. Hensen, Catalytic Depolymerization of Lignin in Supercritical Ethanol, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, One-pot catalytic conversion of cellulose and of woody biomass solids to liquid fuels, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- C. M. Bernt, G. Bottari, J. A. Barrett, S. L. Scott, K. Barta and P. C. Ford, Mapping reactivities of aromatic models with a lignin disassembly catalyst. Steps toward controlling product selectivity, Catal. Sci. Technol., 2016, 6, 2984–2994 RSC.
- J. A. Barrett, Y. Gao, C. M. Bernt, M. Chui, A. T. Tran, M. B. Foston and P. C. Ford, Enhancing Aromatic Production from Reductive Lignin Disassembly: in situ O-methylation of Phenolic Intermediates, ACS Sustain. Chem. Eng., 2016, 4, 6877–6886 CrossRef CAS.
- M. Chui, G. Metzker, C. M. Bernt, A. T. Tran, A. C. B. Burtoloso and P. C. Ford, Probing the lignin disassembly pathways with modified catalysts based on Cu-doped porous metal oxides, ACS Sustain. Chem. Eng., 2017, 5, 3158–3169 CrossRef CAS.
- B. Liu and M. M. Abu-Omar, Lignin Extraction and Valorization using Heterogeneous Transition Metal Catalysts, Adv. Inorg. Chem., 2021, 77, 137–174 CrossRef.
- L. Dong, L. Lin, X. Han, X. Si, X. Liu, Y. Guo, F. Lu, S. Rudi, S. F. Parker, S. Yang and Y. Wang, Breaking the Limit of Lignin Monomer Production via Cleavage of Interunit Carbon–Carbon Linkages, Chem, 2019, 5, 1521–1536 CAS.
- C. Cheng, J. Truong, J. A. Barrett, D. Shen, M. M. Abu-Omar and P. C. Ford, Hydrogenolysis of Organosolv Lignin in Ethanol/Isopropanol Media without Added Transition-Metal Catalyst, ACS Sustainable Chem. Eng., 2020, 8, 1023–1030 CrossRef CAS.
- B. Liu, M. Sanchez, J. Truong, P. C. Ford and M. M. Abu-Omar, Catalytic conversion of high S-lignin to a sustainable tri-epoxide polymer precursor, Green Chem., 2022, 24, 4958–4968 RSC.
- J. R. Meyer, H. Li, J. Zhang and M. B. Foston, Kinetics of Secondary Reactions Affecting the Organosolv Lignin Structure, ChemSusChem, 2020, 13, 4557–4566 CrossRef CAS PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Heroguel, Y. D. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- M. T. Amiri, G. R. Dick, Y. M. Questell-Santiago and J. S. Luterbacher, Fractionation of lignocellulosic biomass to produce uncondensed aldehyde-stabilized lignin, Nat. Protoc., 2019, 14, 921–954 CrossRef PubMed.
- D. S. Zijlstra, C. Lahive, C. A. Analbers, M. B. Figueiredo, Z. W. Wang, C. Lancefield and P. J. Deuss, Mild Organosolv Lignin Extraction with Alcohols: The Importance of Benzylic Alkoxylation, ACS Sustainable Chem. Eng., 2020, 8, 5119–5131 CrossRef CAS.
- C. Nitsos, U. Rova and P. Christakopoulos, Organosolv Fractionation of Softwood Biomass for Biofuel and Biorefinery Applications, Energies, 2018, 11, 50 CrossRef.
- A. De Santi, M. V. Galkin, C. W. Lahive, P. K. Deuss and K. Barta, Lignin-First Fractionation of Softwood Lignocellulose Using a Mild Dimethyl Carbonate and Ethylene Glycol Organosolv Process, ChemSusChem, 2020, 13, 4468–4477 CrossRef CAS PubMed.
- P. Ferrini and R. Rinaldi, Catalytic Biorefining of Plant Biomass to Non-Pyrolytic Lignin Bio-Oil and Carbohydrates through Hydrogen Transfer Reactions, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, A synergistic biorefinery based on catalytic conversion of lignin prior to cellulose starting from lignocellulosic biomass, Green Chem., 2015, 17, 1492–1499 RSC.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps, Energy Environ. Sci., 2015, 8, 1748–2176 RSC.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Reductive catalytic fractionation: state of the art of the lignin-first biorefinery, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. Luterbacher, J. Ralph, R. Rinaldi, Y. Roman-Leshkov, J. Samec, B. Sels and F. Wang, Guidelines for performing lignin-first biorefining, Energy Environ. Sci., 2020, 14, 262–292 RSC.
- T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttaemaa, F. Ribeiro, J. T. Miller and M. M. Abu-Omar, Cleavage and hydrodeoxygenation (HDO) of C-O bonds relevant to lignin conversion using Pd/Zn synergistic catalysis, Chem. Sci., 2013, 4, 806–813 RSC.
- W. Arts, D. Ruijten, K. Van Aelst, L. Trullemans and B. Sels, The RCF biorefinery: building on a chemical platform from lignin, Adv. Inorg. Chem., 2021, 77, 241–297 CrossRef CAS.
- H. Luo, I. M. Klein, Y. Jiang, H. Zhu, B. Y. Liu, H. I. Kenttaemaa and M. M. Abu-Omar, Total Utilization of Miscanthus Biomass, Lignin and Carbohydrates, Using Earth Abundant Nickel Catalyst, ACS Sustainable Chem. Eng., 2016, 4, 2316–2322 CrossRef CAS.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yua and J. Xu, Lignin depolymerization (LDP) in alcohol over nickel based catalysts via a fragmentation–hydrogenolysis process, Energy Environ. Sci., 2013, 6, 994 RSC.
- I. Klein, B. Sahaa and M. M. Abu-Omar, Lignin depolymerization over Ni/C catalyst in methanol, a continuation: effect of substrate and catalyst loading, Catal. Sci. Technol., 2015, 5, 3242 RSC.
- H. Li and G. Song, Ru-Catalyzed Hydrogenolysis of Lignin: Base-Dependent Tunability of Monomeric Phenols and Mechanistic Study, ACS Catal., 2019, 9, 4054–4064 CrossRef CAS.
- S. Li, B. Liu, J. Truong, J. Luo, P. C. Ford and M. M. Abu-Omar, One-pot hydrodeoxygenation (HDO) of lignin monomers to C9 hydrocarbons co-catalysed by Ru/C and Nb2O5, Green Chem., 2020, 22, 7406–7416 RSC.
- I. M. Klein and D. H. Coller, Lignin Conversion to Phenolic Molecules Using Transition Metal Catalysts, WO2019108959A1, 2018.
- E. M. Anderson, M. L. Stone, M. J. Hulsey, G. T. Beckham and Y. Roman-Leshkov, Kinetic Studies of Lignin Solvolysis and Reduction by Reductive Catalytic Fractionation Decoupled in Flow-Through Reactors, ACS Sustainable Chem. Eng., 2018, 6, 7951–7959 CrossRef CAS.
- F. Brandi, B. Pandalone and M. Al-Naji, Flow-through reductive catalytic fractionation of beech wood sawdust, RSC Sustainability, 2023, 1, 459 RSC.
- H. Luo and M. M. Abu-Omar, Lignin extraction and catalytic upgrading from genetically modified poplar, Green Chem., 2018, 20, 745–753 RSC.
- H. Yang, X. Zhang, H. Luo, B. Liu, T. M. Shig, X. Li, J. I. Kim, P. Rubinelli, J. C. Overton, V. Subramanyam, B. R. Cooper, H. Mo, M. M. Abu-Omar, C. Chapple, B. S. Donohoe, L. Makowski, N. S. Mosier, M. C. McCann, N. C. Carpita and R. Meilan, Overcoming cellulose recalcitrance in woody biomass for the lignin-first biorefinery, Biotechnol. Biofuels, 2019, 12, 171 CrossRef CAS.
- I. Klein, C. Marcum, H. Kenttamaaa and M. M. Abu-Omar, Mechanistic investigation of the Zn/Pd/C catalyzed cleavage and hydrodeoxygenation of lignin, Green Chem., 2016, 18, 2399–2405 RSC.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation, Energy Environ. Sci., 2014, 7, 103 RSC.
- Y. Wang, A. Akbarzadeh, L. Chong, J. Du, N. Tahir and M. K. Awasthi, Catalytic pyrolysis of lignocellulosic biomass for bio-oil production: a review, Chemosphere, 2022, 297, 134181 CrossRef CAS PubMed.
- L. Chen, P. Verma, K. Hou, Z. Qi, S. Zhang, Y.-S. Liu, J. Guo, V. Stavila, M. D. Allendorf, L. Zheng, M. Salmeron, D. Prendergast, G. A. Somorjai and J. Su, Reversible dehydrogenation and rehydrogenation of cyclohexane and methylcyclohexane by single-site platinum catalyst, Nat. Commun., 2022, 13, 1092 CrossRef CAS PubMed.
- S. Zhao and M. M. Abu-Omar, Biobased Epoxy Nanocomposites Derived from Lignin-Based Monomers, Biomacromolecules, 2015, 16, 2025–2031 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, From Lignin-derived Aromatic Compounds to Novel Biobased Polymers, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed.
- S. Zhao and M. M. Abu-Omar, Synthesis of Renewable Thermoset Polymers through Successive Lignin Modification Using Lignin-Derived Phenols, ACS Sustain. Chem. Eng., 2017, 5, 5059–5066 CrossRef CAS.
- Y. Jiang, D. Ding, S. Zhao, H. Zhu, H. T. Kenttamaaa and M. M. Abu-Omar, Renewable thermoset polymers based on lignin and carbohydrate derived monomers, Green Chem., 2018, 20, 1131–1138 RSC.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. Xu, Global lignin supply overview and kraft lignin potential as an alternative for petroleum-based polymers, Renew. Sustain. Energy Rev., 2020, 123, 109768 CrossRef CAS.
- X. Wu, M. De bruyn, G. Trimmel, K. Zangger and K. Barta, High-Performance Thermoplastics from a Unique Bicyclic Lignin-Derived Diol, ACS Sustainable Chem. Eng., 2023, 11, 2819–2829 CrossRef CAS PubMed.
- S. Zhao and M. M. Abu-Omar, Renewable Epoxy Networks Derived from Lignin-Based Monomers: Effect of Cross-Linking Density, ACS Sustain. Chem. Eng, 2016, 4, 6082–6089 CrossRef CAS.
- W. Li, H. Sun, G. Wang, W. Sui, L. Dai and C. Si, Lignin as a green and multifunctional alternative to phenol for resin synthesis, Green Chem., 2023, 25, 2241–2261 RSC.
- K. G. Hanson, C.-H. Lin and M. M. Abu-Omar, Preparation and properties of renewable polyesters based on lignin-derived bisphenol, Polymer, 2021, 233, 124202 CrossRef CAS.
- K. G. Hanson, C. H. Lin and M. M. Abu-Omar, Crosslinking of renewable polyesters with epoxides to form bio-based epoxy thermosets, Polymer, 2022, 238, 124363 CrossRef CAS.
- S. Fadlallah, P. S. Roy, G. Garnier, K. Saito and F. Allais, Are lignin-derived monomers and polymers truly sustainable? An in-depth green metrics calculations approach, Green Chem., 2021, 23, 149 RSC.
- I. F. Demuner, J. L. Colodette, A. J. Demuner and C. M. Jardim, Biorefinery review: wide-reaching products through kraft lignin, BioRes., 2019, 14, 7543–7581 Search PubMed.
- S. Guadix-Montero and M. Sankar, Review on Catalytic Cleavage of C–C Inter-unit Linkages in Lignin Model Compounds: Towards Lignin Depolymerisation, Top. Catal., 2018, 61, 183–198 CrossRef CAS.
- S. Chen, W. Wang, X. Li, P. Yan, W. Han, T. Sheng, T. Deng, W. Zhu and H. Wang, Regulating the nanoscale intimacy of metal and acidic sites in Ru/γ-Al2O3for the selective conversions of lignin-derived phenols to jet fuels, J. Energy Chem., 2022, 66, 576 CrossRef CAS.
- S. Yang, C. Shi, Z. Shen, L. Pan, Z. Huang, X. Zhang and J.-J. Zou, Conversion of lignin oil and hemicellulose derivative into high-density jet fuel, J. Energy Chem., 2023, 77, 452 CrossRef CAS.
- Z. Li, S. Wang and H. Xin, Toward artificial intelligence in catalysis, Nat. Catal., 2018, 1, 641–642 CrossRef.
- P. D. Kouris, X. Huang, M. D. Boot and E. J. M. Hensen, Scaling-Up Catalytic Depolymerisation of Lignin: Performance Criteria for Industrial Operation, Top. Catal., 2018, 61, 1901–1911 CrossRef CAS.
- E. Cooreman, T. Nicola, W. Arts, K. Van Aelst, T. Vangeel, S. Van den Bosch, J. Van Aelst, B. Lagrain, K. Thiele, J. Thevelein and B. F. Sels, The Future Biorefinery: The Impact of Upscaling the Reductive Catalytic Fractionation of Lignocellulose Biomass on the Quality of the Lignin Oil, Carbohydrate Products, and Pulp, ACS Sustainable Chem. Eng., 2023, 11, 5440–5450 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |