DOI:
10.1039/D3RA07035B
(Paper)
RSC Adv., 2023,
13, 35240-35250
Isatin-based benzyloxybenzene derivatives as monoamine oxidase inhibitors with neuroprotective effect targeting neurogenerative disease treatment†
Received
16th October 2023
, Accepted 26th November 2023
First published on 4th December 2023
Abstract
Eighteen isatin-based benzyloxybenzaldehyde derivatives from three subseries, ISB, ISFB, and ISBB, were synthesized and their ability to inhibit monoamine oxidase (MAO) was evaluated. The inhibitory activity of all synthesized compounds was found to be more profound against MAO-B than MAO-A. Compound ISB1 most potently inhibited MAO-B with an IC50 of 0.124 ± 0.007 μM, ensued by ISFB1 (IC50 = 0.135 ± 0.002 μM). Compound ISFB1 most potently inhibited MAO-A with an IC50 of 0.678 ± 0.006 μM, ensued by ISBB3 (IC50 = 0.731 ± 0.028 μM), and had the highest selectivity index (SI) value (55.03). The three sub-parental compounds, ISB1, ISFB1, and ISBB1, had higher MAO-B inhibition than the other derivatives, indicating that the substitutions of the 5-H in the A-ring of isatin diminished the inhibition of MAO-A and MAO-B. Among these, ISB1 (para-benzyloxy group in the B-ring) displayed more significant MAO-B inhibition when compared to ISBB1 (meta-benzyloxy group in the B-ring). ISB1 and ISFB1 were identified to be competitive and reversible MAO-B inhibitors, having Ki values of 0.055 ± 0.010, and 0.069 ± 0.025 μM, respectively. Furthermore, in the parallel artificial membrane penetration assay, ISB1 and ISFB1 traversed the blood–brain barrier in the in vitro condition. Additionally, the current study found that ISB1 decreased rotenone-induced cell death in SH-SY5Y neuroblastoma cells. In docking and simulation studies, the hydrogen bonding formed by the imino nitrogen in ISB1 and the pi–pi stacking interaction of the phenyl ring in isatin significantly aided in the protein–ligand complex's stability, effectively inhibiting MAO-B. According to these observations, the MAO-B inhibitors ISB1 and ISFB1 were potent, selective, and reversible, making them conceivable therapies for neurological diseases.
Introduction
Parkinson's disease (PD) is a progressive neurological condition associated with motor impairment, with approximately 2% of people over 65 years of age affected by this condition worldwide.1,2 Dopamine (DA) levels in the putamen and caudate nucleus are reduced in people with PD. Dopaminergic neurons are selectively reduced in the substantia nigra pars compacta and cytoplasm. The symptoms of this illness include Lewy bodies with aggregated α-synuclein.3–5 Although the exact trigger of PD is unknown, numerous studies reveal that, besides the DA depletion, additional factors such as neuroinflammation, protein aggregation, a lack of support from neurotrophic factors, oxidative stress, dysregulation of the autophagy-lysosomal pathway, and mitochondrial dysfunction contribute to the advancement of the disease. The 1960s marked the introduction of monoamine oxidase (MAO) inhibitors, but small biogenic molecules containing 3,4-dihydroxyphenylalanine (L-DOPA) have been used to treat PD symptoms since the end of the 1950s.6,7 MAO-B inhibitors have been widely employed for the symptomatic treatment of PD,8 because they reduce the rate of oxidative deamination catalyzed by MAO-B and subsequently decrease the formation of reactive oxygen species (ROS).9,10 MAO-B inhibitors have remarkable efficacy and safety when used in the early stages of PD and as adjunctive therapy for advanced illnesses.8 Dopamine is released into the synaptic cleft and absorbed by glial cells, where it is metabolized by MAO-B. An age-related increase in glial cells has been linked to elevated MAO-B activity in neurodegenerative disorders.11–13 MAO-B inhibitors suppress brain MAO-B activity, inhibit dopamine catabolism, improve dopamine signaling, and elevate synaptic dopamine levels.14,15 Numerous studies have suggested that irreversible MAO-B inhibitors, such as rasagiline and selegiline, can successfully fix the motor dysfunction and prevent dopamine degradation in PD patients. However, the target disruption, a longer half-life, de novo formation of MAO enzyme in the human brain, decreased sensitivity to ADME testing and immunogenicity of the MAO-B-inhibitor adduct include the pitfalls of irreversible MAO-B inhibitors.16–18 These characteristics aided in creating effective and reversible MAO-B inhibitors for treating PD. Recently, the European Union (EU) and the US Food and Drug Administration (USFDA) approved safinamide, which is a reversible MAO-B inhibitor to combat mid- to late-stage PD.19–21
A considerable number of reports have been published on the synthesis, biological assessment, and formulation of MAO inhibitors. Most drugs frequently contain specific pharmacophores with strong MAO-B inhibitory action and selectivity. Such pharmacophores include compounds that contain benzyloxy groups with strong MAO inhibitory effects.22–24 Benzyloxy substituents have been reported as selective MAO-B inhibitors in a series of oxadiazon derivatives.25 A series of compounds are reported to have improved molecular hydrophobicity as a result of the benzyloxy group attached to the fifth position of the indole ring, which is consistent with the environment of the active site and is crucial for MAO-B selectivity.26 Studies have confirmed that a benzyloxy group, which is substituted at the indole ring's fifth position, is found crucial to establish the selectivity of MAO-B inhibitory activity.27,28 Safinamide is the first anti-Parkinson's medication to be recognized in the last 10 years that also inhibits glutamate release and the reuptake of DA and serotonin. Early anti-Parkinson's medications include selegiline and rasagiline, of which selegiline has a reversible effect and good pharmacokinetic profile and a bioavailability29 of about 95%. Safinamide, on the other hand, has a distinctive structural makeup with an apparent benzyloxy pharmacophore on the phenyl ring and an aminoamide in the proper para position.30 It has been reported that safinamide31,32 and indolalkylamines33,34 contain benzyloxy groups and that halogenation of the benzyloxy pharmacophore of the chalcone framework could enhance MAO-B inhibition.35
Isatin is an indoledione, a pharmacologically active molecule that is the 2,3-diketo derivative of indole.36,37 Derivatives of isatin are widely known for their effective blood–brain barrier (BBB) penetration as well as their selectively reversible MAO-B inhibitory action.38 Human MAO-A and MAO-B are both reversibly inhibited by isatin, with Ki values of 15 μM and 3 μM, respectively.39,40 Isatin is an extremely reactive compound that has been employed as both an electrophile and a nucleophile in synthetic chemistry. Isatin derivatives possess several biological properties, including anticancer,41–43 antibiotic,44 antidepressant, anxiogenic, sedative, anticonvulsant,45,46 antibacterial, antidiabetic,47 carboxyesterase,48,49 anti-platelet, and anti-HIV50 effects. The most prevalent electrophilic reaction of isatin is a nucleophilic addition to the third-position keto group. There have been many published investigations on the synthesis of isatin spiro analogues at the C-3 position.28
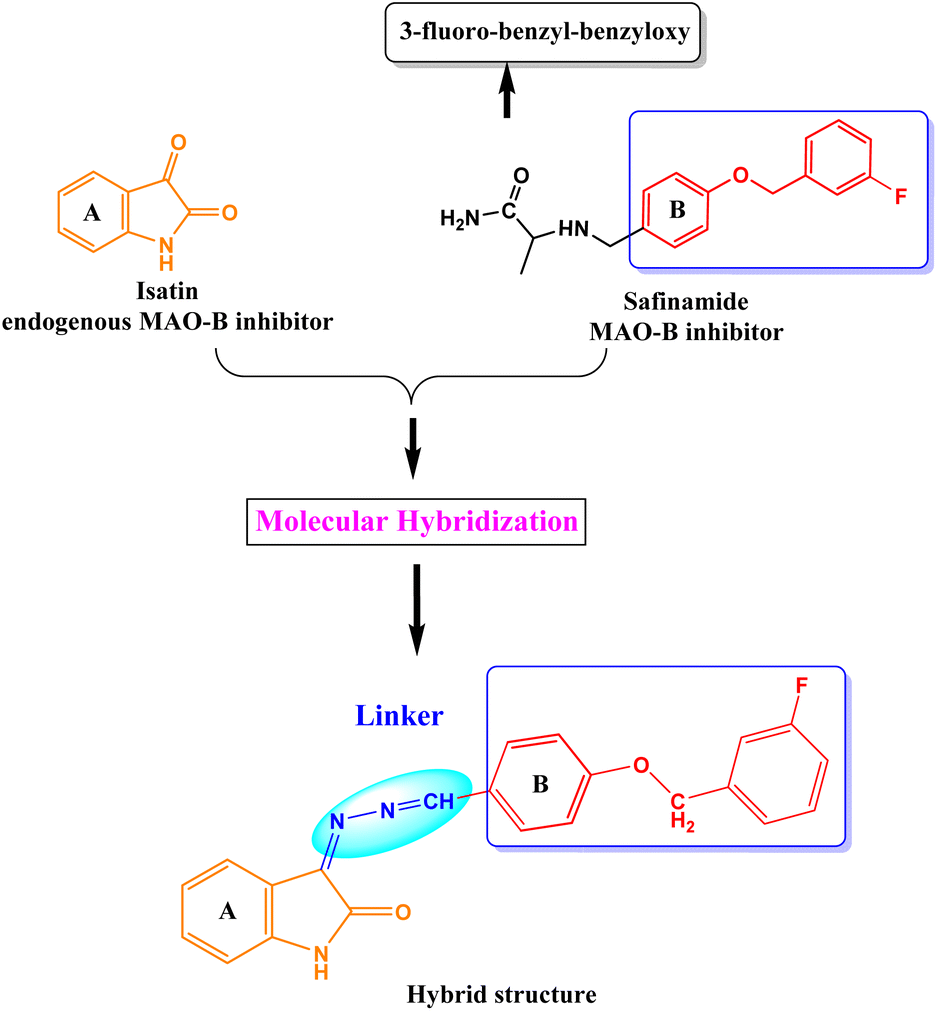
We developed a versatile chemical methodology (Fig. 1) that employs the molecular hybridization of isatin and safinamide to produce benzyloxy isatin hydrazones. Isatin is an endogenous MAO inhibitor, and substituting C-3 position with the lipophilic group markedly enhances the MAO-B inhibition.39,40 Main pharmacophore of the FDA-approved drug safinamide is 3-fluoro-(benzyl)-benzyloxy unit. Our design was focused on generating a hybrid class of compounds with these pharmacophores to produce a novel class of chemical entities in the category of MAO inhibitors. A hydrazone linker was used to link the isatin scaffold to a benzyloxy pharmacophore. This chemistry uses a microwave-mediated attachment of an isatin hydrazone linker to different benzyloxy benzaldehyde functionalities catalyzed by glacial acetic acid. Isatin hydrazone-based derivatives are selective, reversible, and effective inhibitors of MAO-B.51 According to earlier research, all isatin hydrazone-based inhibitors bind to both the substrate (Ile168, Tyr60, Tyr398, Cys172, Phe343, Tyr435, and Gln206) and entrance (Tyr326, Leu171, and Ile199) cavities, which is similar to the behavior of the reference MAO-B inhibitor, safinamide.28,51,52
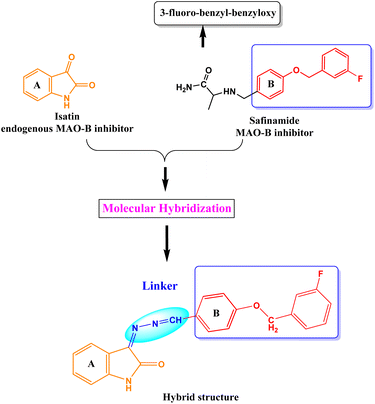 |
| | Fig. 1 Design strategy of benzyloxy isatin hydrazones. | |
Results and discussion
Chemistry
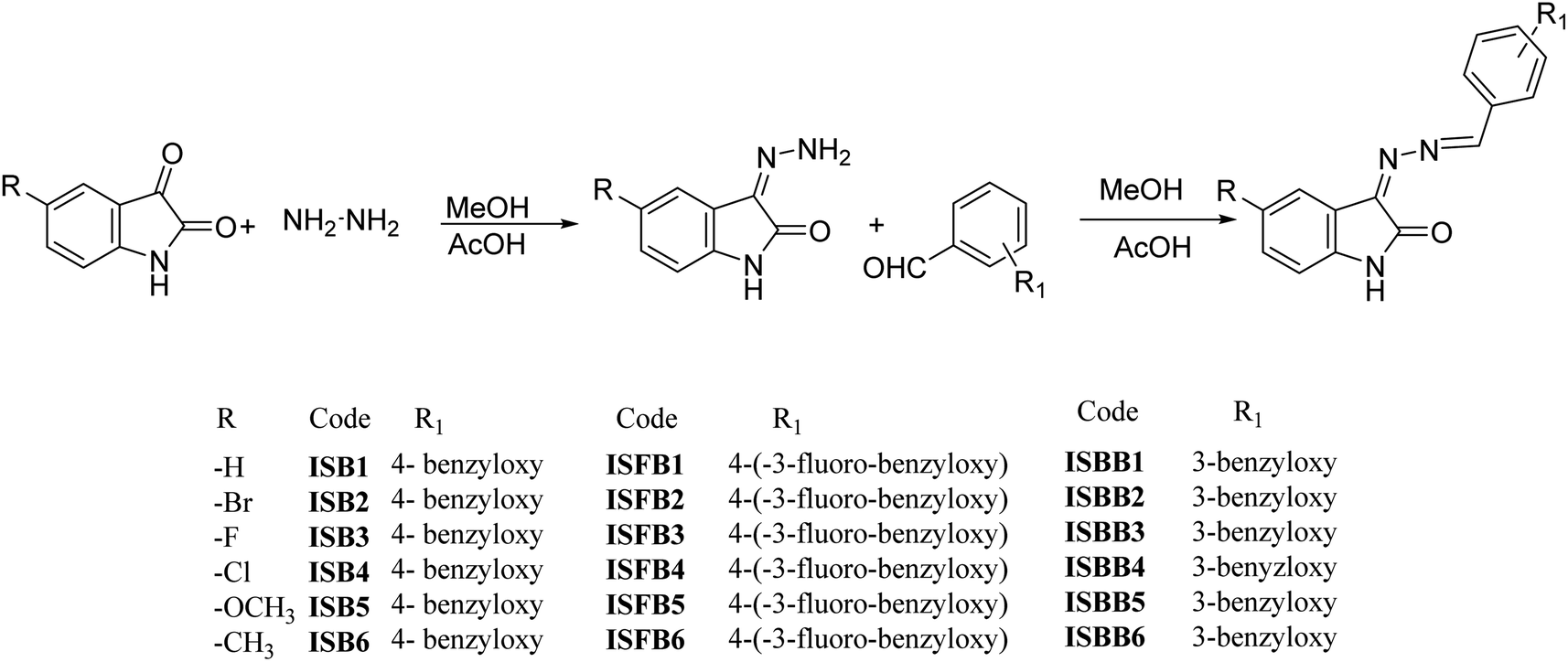
With various substituted isatin hydrazides and para-substituted benzyloxy benzaldehyde, the isatin-based benzyloxy-benzaldehyde derivatives were synthesized in a microwave reactor (Monowave 50, Anton Paar, Graz, Austria) by means of a nucleophilic addition reaction (Fig. 2). Using 1H NMR, 13C NMR (Bruker Advance Neo 500 MHz NMR spectrometer, Billerica, MA, USA), mass spectroscopy (Waters Xevo G2-XS QTOF, Milford, MA, USA), and HPLC (Shimadzu Prominence-i LC-2030C 3D Plus, Ramsey, MN, USA), structures of all final derivatives were investigated. Isatin with NH atom for proton occurs inside the 10.66–11.00 ppm di-shielded region. Benzyloxy–CH2 atom shielded proton in 5.23–5.33 ppm. The extensively di-shielded area 163–164.24 ppm included sp2 carbon of carbonyl carbon of isatin. In the di-shielded region of 159–163 ppm, C–F coupling also occurred (source data supporting).†
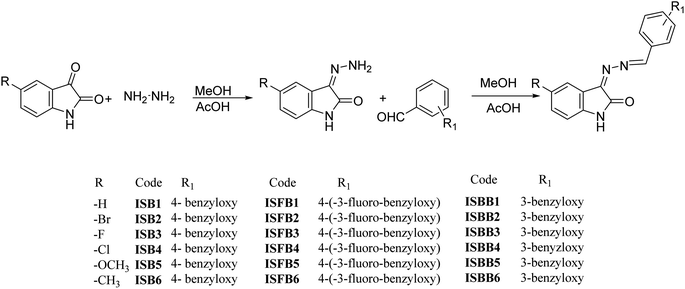 |
| | Fig. 2 Synthesis of ISB, ISFB, and ISBB compounds. | |
Inhibition studies of MAO-B and MAO-A
There were 18 compounds in this series, divided into three subseries and three sub-parental compounds: ISB1, ISFB1, and ISBB1. In this series, all of the compounds displayed more profound MAO-B inhibitory actions than MAO-A (Table 1). Compound ISB1 most potently inhibited MAO-B, with an IC50 = 0.124 μM, followed by ISFB1 (IC50 = 0.135 μM) (Table 1). These IC50 values were lower than methylthiosemicarbazone SM5 (IC50 = 5.48 μM) and piperazine-substituted chalcones PC10 and PC11 (IC50 = 0.65 and 0.71 μM, respectively)53 but higher than benzyloxy pharmacophore B10 (IC50 = 0.067 μM).54 Compound ISFB1 inhibited MAO-A with an IC50 = 0.678 μM, followed by ISBB3 (IC50 = 0.731 μM). The selectivity index (SI) for compounds ISB1 and ISFB1 were 55.03 and 5.02, respectively, for MAO-B compared to MAO-A, indicating that these compounds were selective MAO-B inhibitors (Table 1).
Table 1 Inhibition of MAO-A and MAO-B by the 18 compounds of ISB seriesa
| Compound |
Residual activity at 10 μM (%) |
IC50 (μM) |
SIb |
| MAO-A |
MAO-B |
MAO-A |
MAO-B |
| Results are the means ± standard errors of duplicate or triplicate experiments. Selectivity index (SI) values are expressed for MAO-B compared to MAO-A. |
| ISB1 |
38.78 ± 0.25 |
−8.77 ± 0.14 |
6.824 ± 0.049 |
0.124 ± 0.007 |
55.03 |
| ISB2 |
87.76 ± 1.11 |
64.91 ± 1.24 |
>40 |
14.182 ± 0.250 |
>2.82 |
| ISB3 |
74.49 ± 0.95 |
59.65 ± 1.11 |
>40 |
13.943 ± 0.204 |
>2.87 |
| ISB4 |
84.69 ± 1.55 |
54.39 ± 2.01 |
>40 |
9.764 ± 1.349 |
>4.10 |
| ISB5 |
85.71 ± 2.04 |
66.67 ± 1.35 |
>40 |
17.622 ± 0.202 |
>2.27 |
| ISB6 |
89.80 ± 1.12 |
40.35 ± 0.99 |
>40 |
6.563 ± 0.206 |
>6.09 |
| ISFB1 |
4.17 ± 0.32 |
−3.03 ± 0.24 |
0.678 ± 0.006 |
0.135 ± 0.002 |
5.02 |
| ISFB2 |
88.54 ± 1.38 |
57.58 ± 1.10 |
>40 |
16.043 ± 0.308 |
>2.49 |
| ISFB3 |
53.13 ± 0.44 |
24.50 ± 1.03 |
10.937 ± 0.220 |
1.189 ± 0.362 |
9.20 |
| ISFB4 |
84.38 ± 1.24 |
63.64 ± 0.15 |
>40 |
20.768 ± 0.027 |
>1.93 |
| ISFB5 |
87.50 ± 1.88 |
50.00 ± 0.38 |
>40 |
8.986 ± 0.079 |
>4.45 |
| ISFB6 |
84.38 ± 1.52 |
30.30 ± 0.77 |
>40 |
4.100 ± 0.311 |
>9.76 |
| ISBB1 |
4.84 ± 1.61 |
1.99 ± 0.86 |
1.570 ± 0.048 |
0.540 ± 0.029 |
2.91 |
| ISBB2 |
81.52 ± 1.64 |
46.88 ± 0.88 |
>40 |
8.127 ± 0.383 |
>4.92 |
| ISBB3 |
35.87 ± 1.02 |
29.69 ± 0.54 |
0.731 ± 0.028 |
0.607 ± 0.011 |
1.20 |
| ISBB4 |
73.91 ± 0.04 |
40.63 ± 0.63 |
>40 |
6.058 ± 0.318 |
>6.60 |
| ISBB5 |
58.70 ± 0.29 |
48.44 ± 0.44 |
11.054 ± 0.060 |
12.061 ± 3.567 |
0.92 |
| ISBB6 |
65.22 ± 0.87 |
39.06 ± 0.21 |
14.422 ± 0.670 |
15.157 ± 0.419 |
0.95 |
| Toloxatone |
|
|
1.080 ± 0.025 |
— |
|
| Lazabemide |
|
|
— |
0.110 ± 0.016 |
|
| Clorgyline |
|
|
0.007 ± 0.0007 |
— |
|
| Pargyline |
|
|
— |
0.140 ± 0.006 |
|
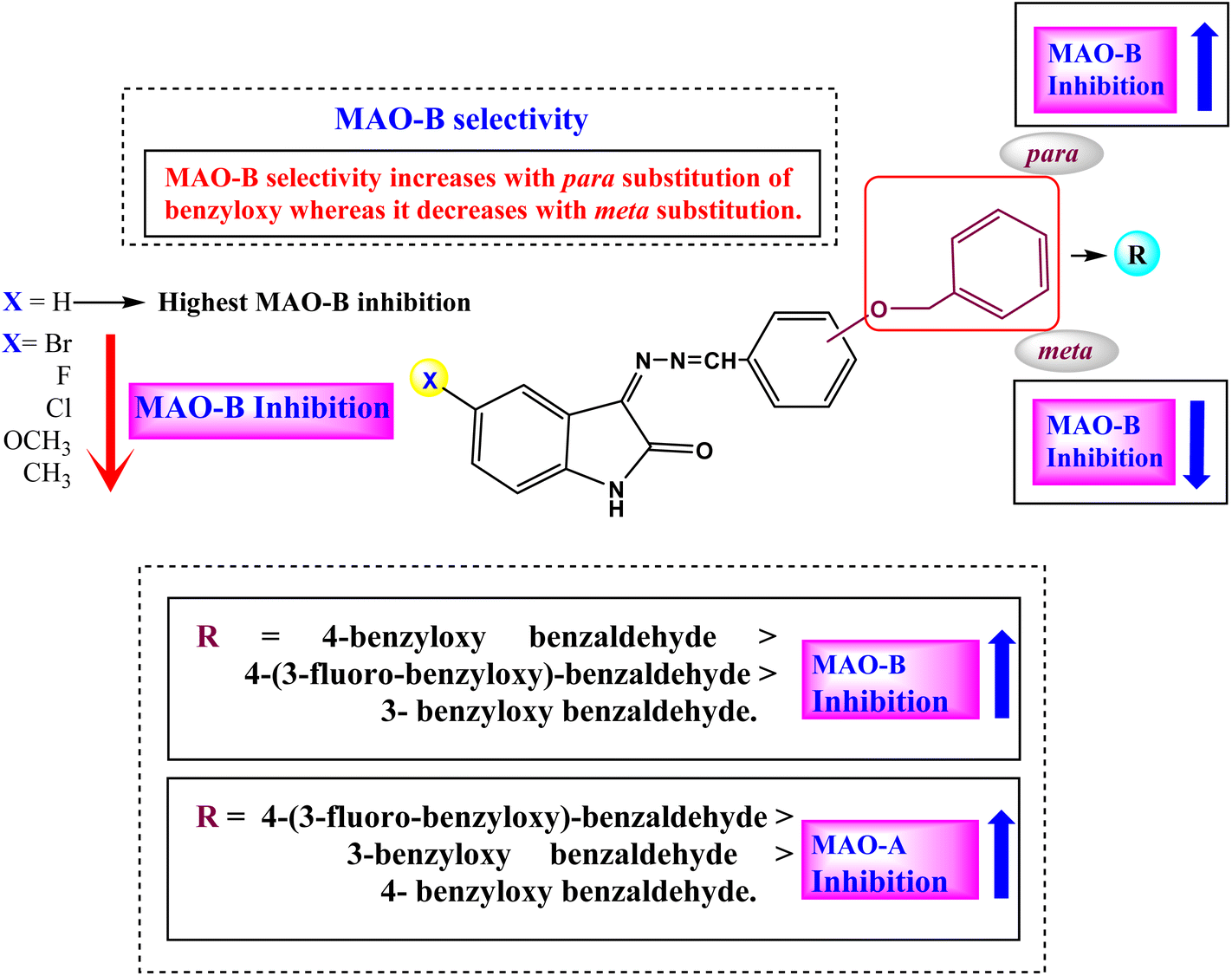
The structure–activity relationship (SAR) for the ISB derivatives showed that ISB1 (5-H in A-ring of isatin and para-benzyloxy group in B-ring) had 52.93- to 142.11-times higher MAO-B inhibition than other ISB derivatives with the substituents of –H > –H3C > –Cl > –F > –Br > –H3CO at the 5-position in the A-ring. In the ISFB derivatives, ISFB1 (5-H in A-ring of isatin and para-benzyloxy group containing meta-F in B-ring) had 8.81- to 153.84-times higher MAO-B inhibition than other ISFB derivatives with the substituents of –H > –F > –H3C > –H3CO > –Br > Cl at 5-position in the A-ring. Among the ISBB derivatives, ISBB1 (5-H in the A-ring and meta-benzyloxy group in the B-ring) showed 1.12- to 28.07-times higher MAO-B inhibition than the other ISBB derivatives with –H > –F > –Cl > –Br > –H3CO > –H3C at the 5-position in the A-ring. When we compared the B-ring, the B-ring at the para-position (ISB1, IC50 = 0.124 μM) had higher MAO-B inhibition than that in the meta-position (ISBB1, IC50 = 0.540 μM), and MAO-B inhibition increased in order by –H ISB1 > –F (ISFB1, IC50 = 0.135 μM). Overall, most compounds with –H substituents showed high MAO-B inhibition (Fig. 3 and Table 1). Collectively, the basic structures ISB1, ISFB1, and ISBB1 with 5-H in the A-ring of isatin showed higher MAO-B inhibition than the other derivatives, suggesting that the substitution of the –H group of the A-ring contributed to a decrease in MAO-B inhibition. Among these, ISB1 (para-benzyloxy group in the B-ring) showed higher MAO-B inhibition than ISBB1 (meta-benzyloxy group in the B-ring). However, three sub-parental compounds, ISB1, ISFB1, and ISBB1 (5-H substituent in the A-ring) displayed potent MAO-A inhibition, with IC50 values of 6.824, 0.678, and 1.570 μM, respectively. In addition, ISBB3 (5-F in the A-ring and meta-benzyloxy group in the B-ring) showed high inhibitory activity (IC50 = 0.731 μM). In each subseries, the substitution of 5-H in the A-ring with other groups generally decreased MAO-A inhibition (Fig. 3 and Table 1). According to these results, ISB1 and ISFB1 were strong and selective MAO-B inhibitors.
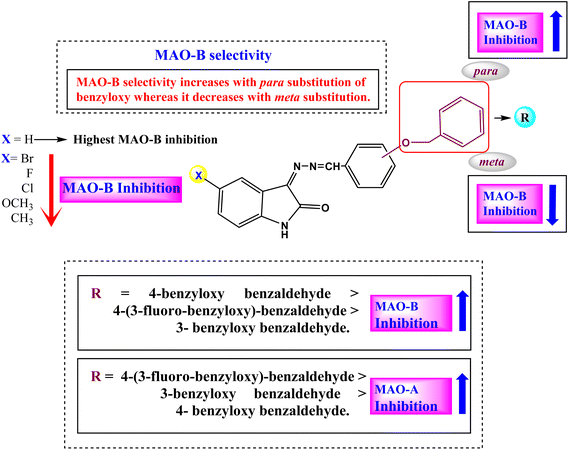 |
| | Fig. 3 Structure–activity relationships (SARs) of benzyloxybenzaldehyde derivatives. | |
Reversibility studies
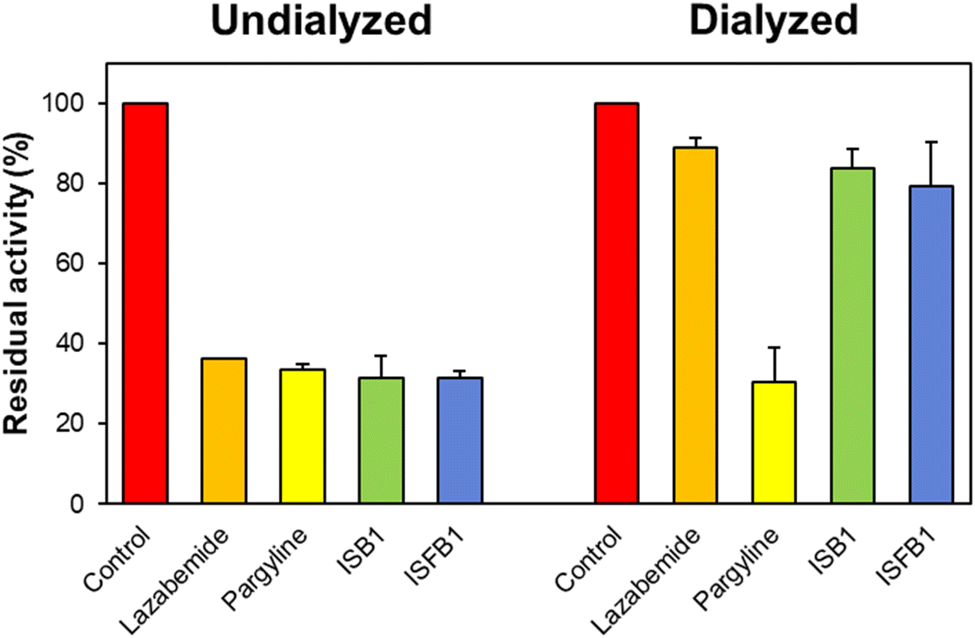
Using the dialysis approach, the reversibility of inhibition of the ISB series was evaluated using concentrations that were ∼2.0 times their respective IC50 values for ISB1 and ISFB1 (0.24 and 0.28 μM, respectively). Recovery patterns were determined by comparing the undialyzed (AU) and dialyzed (AD) relative activities. Compounds ISB1 and ISFB1 recovered from 31.61% to 83.77% and from 31.42% to 79.32%, respectively (Fig. 4). The compounds' recovery values were comparable to those of lazabemide (from 36.30% to 89.00%) and contrary to those of pargyline (from 33.55% to 30.46%). These findings demonstrated ISB1 and ISFB1 were reversible MAO-B inhibitors.
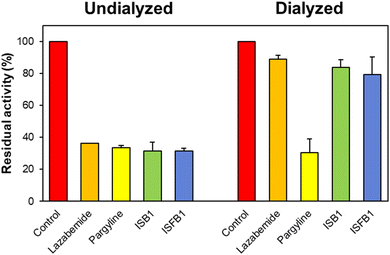 |
| | Fig. 4 Recovery of MAO-B inhibition by ISB1 and ISFB1 using dialysis experiments. The concentrations of ISB1 and ISFB1 were ∼2.0-times of their IC50 values (0.24 and 0.28 μM, respectively). After 30 min preincubation, the mixtures were dialyzed for 6 h with two buffer changes. | |
Enzyme kinetics
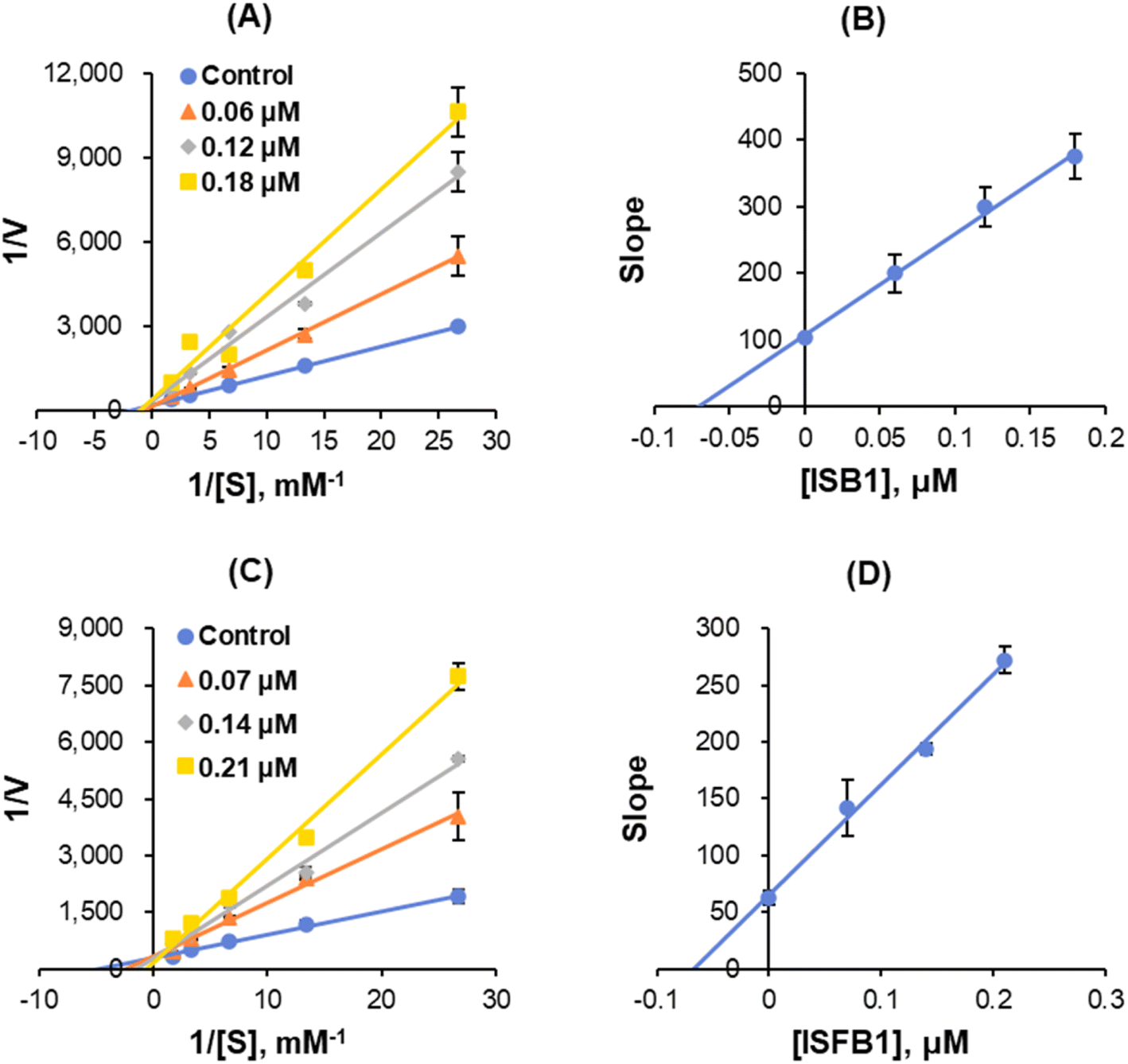
The enzyme kinetics of MAO-B and the inhibition of five concentrations of benzylamine as a substrate and three inhibitor concentrations were analyzed. Lineweaver–Burk plots illustrated that ISB1 and ISFB1 appeared to be competitive MAO-B inhibitors (Fig. 5A and C). Secondary plots revealed that the Ki values were 0.055 ± 0.010 and 0.069 ± 0.025 μM, respectively, for ISB1 and ISFB1 (Fig. 5B and D). These results suggested that ISB1 and ISFB1 are competitive inhibitors of MAO-B.
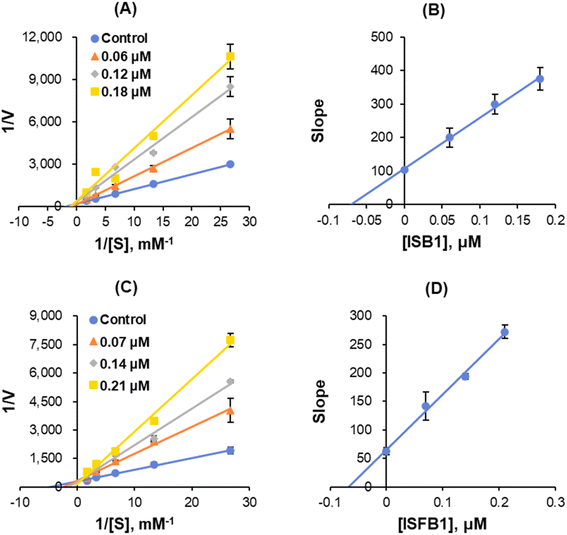 |
| | Fig. 5 Lineweaver–Burk plots for MAO-B inhibition (A and C, respectively), and respective secondary plots of the slopes vs. inhibitor concentrations (B and D, respectively) after ISB1 and ISFB1 treatment. The experiments were conducted using five concentrations of benzylamine as a substrate and at three different inhibitor concentrations. | |
PAMPA for BBB permeation study
PAMPA revealed that the central nervous system (CNS) bioavailability and permeability of the benzyloxy isatin hydrazones (ISB1 and ISFB1) were significant, with Pe values higher than 4.0 × 10−6 cm s−1 (Table 2). Brain penetration is an important requirement for the effective delivery of CNS medications. The PAMPA-BBB was implemented in the present study to evaluate the brain penetration of all derivatives. Using the equation and the compound's effective permeability, the rate of permeation was computed (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Pe). Molecules were categorized as potentially non-BBB permeable (CNS−), if their Pe value was less than 2.0 × 10−6 cm s−1 and as potentially permeable (CNS+), if their Pe value was greater than 4.0 × 10−6 cm s−1.
Pe). Molecules were categorized as potentially non-BBB permeable (CNS−), if their Pe value was less than 2.0 × 10−6 cm s−1 and as potentially permeable (CNS+), if their Pe value was greater than 4.0 × 10−6 cm s−1.
Table 2 BBB assay of key benzyloxy chalcone compounds using the PAMPA methoda
| Compound |
Experimental Pe (×10−6 cm s−1) |
Prediction |
| Pe (10−6 cm s−1) > 4.00: CNS+ (high permeation). |
| ISB1 |
4.93 ± 0.13 |
CNS+ |
| ISFB1 |
4.09 ± 0.24 |
CNS+ |
| Selegiline |
5.69 ± 0.04 |
CNS+ |
Effect of ISB1 on rotenone-induced cell viability
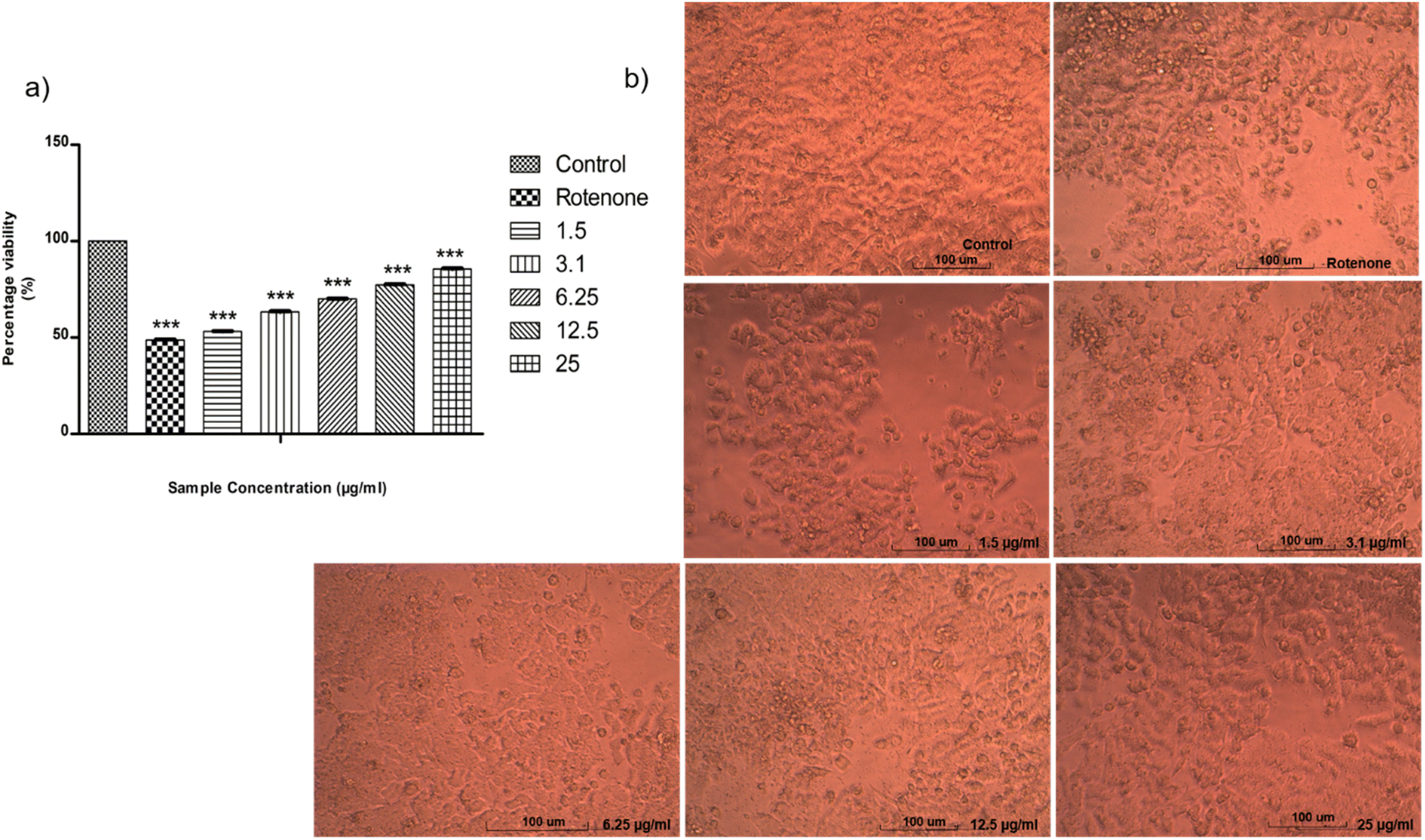
SH-SY5Y-human bone marrow neuroblastoma cell line was supplied from National Center for Cell Science (NCCS), Pune, India. MTT assay test was used to determine neuroprotective effect of ISB1 on rotenone-induced SH-SY5Y cells. ISB1 increased cell viability in a dose-dependent manner for rotenone-treated SH-SY5Y cells (Fig. 6a). The cell viability was increased from about 50% at 1.5 μg ml−1 to 85.59% at 25 μg ml−1 of ISB1. The fundamental morphological changes of the cells on each concentration were observed by direct microscopic method (Fig. 6b).
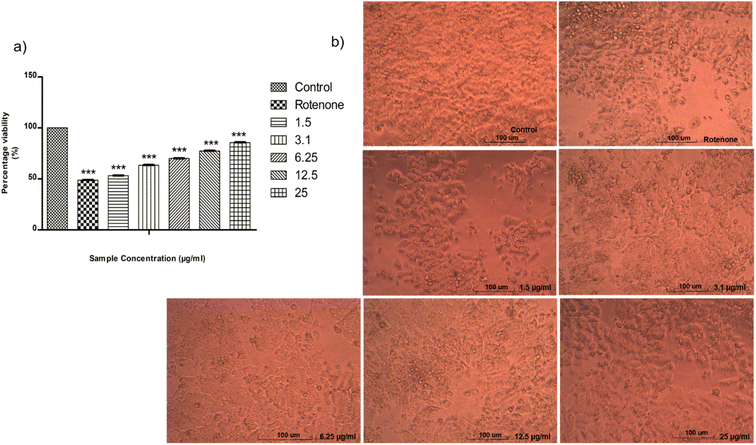 |
| | Fig. 6 (a) Graphical representation depicting the neuroprotective effect of ISB1 on SH-SY5Y cells by MTT assay. Co-treatment with ISB1 attenuated vulnerability of SH-SY5Y to rotenone. (b) The morphology changes of the cells. | |
Molecular docking
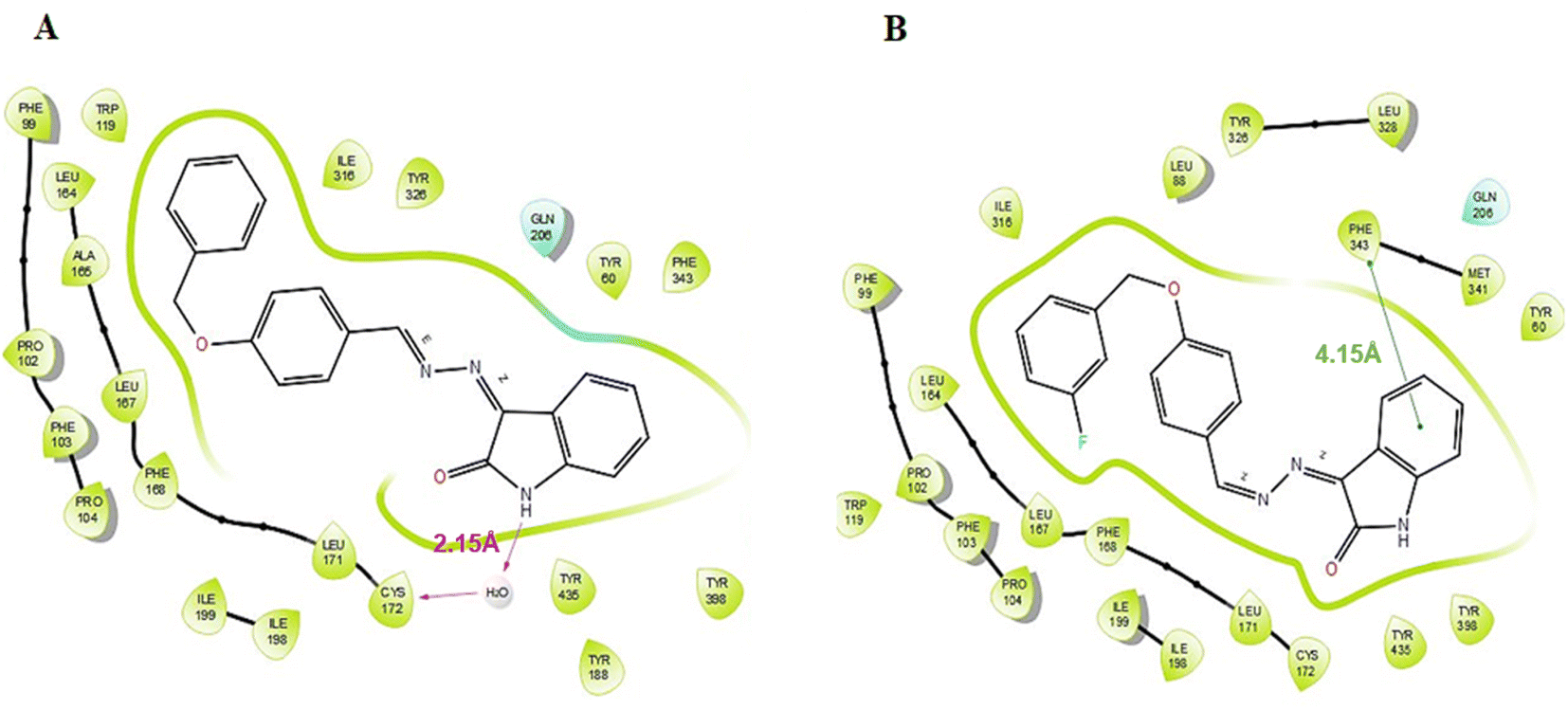
According to the results of enzyme assays, ISB1 and ISFB1 were determined to be the most effective and selective analogues of the series studied against human MAO-B enzymes. Thus, docking analyses were conducted to examine the binding mechanisms of compounds ISB1 and ISFB1 and to evaluate the effects of structural modifications on the MAO-B inhibitory activity of these compounds. The X-ray crystal structure of human MAO-B was found using the Protein Data Bank (PDB ID:2V5Z). ISB1 and ISFB1 had substantial docking scores for MAO-B, corroborating the findings of the enzyme activity assay and implying the selectivity for MAO-B (Table 3). ISB1 had a docking score of −10.413 (XP Glide Score) for MAO-B. Fig. 7A displays the binding interactions between compound ISB1 and MAO-B. The docking poses of hMAO-B displayed that the NH group of isatin was linked to Cys172 through a water molecule (2.15 Å) via hydrogen bonding. ISB1 makes polar contact with Gln206, while the Tyr398, Tyr435, Phe343, Tyr60, Tyr326, Ile316, Trp119, Leu164, Ala165, and Leu171 amino acid side chains interacted hydrophobically with ISB1. As neither of the other compounds displayed a comparable interaction, it was assumed that this interaction was crucial for ISB1 and would help to explain its stronger inhibitory effect. Thus, the presence of hydrogen bonding interactions between the –NH of isatin and Cys172 may significantly influence its biological activity. Compound ISFB1 had a docking score of 9.370 (XP Glide Score) for MAO-B. Fig. 7B displays the interactions between compound ISFB1 and MAO-B, which illustrates that the aromatic benzene ring of isatin in ISFB1 was extended to hydrophobic residues and engaged in π–π stacking with Phe343 (4.15 Å) and that ISFB1 made polar contact with Gln206. The binding modalities of the novel compounds in the 2V5Z complex were predicted using hydrophobic interactions established with the adjacent amino acids compared to the reversible monoamine oxidase inhibitor, safinamide.
Table 3 XP docking scores of promising compounds to the MAO-B (2V5Z) active sites
| Compound |
Docking score (kcal mol−1) |
| ISB1 |
−10.413 |
| ISFB1 |
−9.370 |
| Safinamide |
−11.029 |
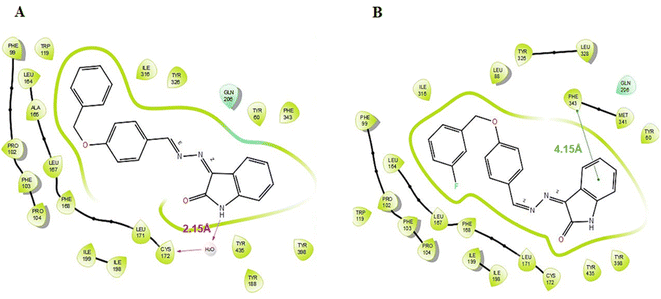 |
| | Fig. 7 Binding interactions of compounds ISB1 (A) and ISBF1 (B) to the active site of the MAO-B (PDB ID 2V5Z). | |
MD simulation
Since MD provides a single snapshot of the interaction between a protein and its ligands, it could possibly be used to describe all biological systems as dynamic networks of molecular interactions. To sample all of the conformations that this complex might acquire in a solvated state, we employed an explicit water model to simulate the dynamic behavior of the docked ISB1-MAO-B complex for a period of 100 ns. The main objective was to keep the ligand occupied in the substrate-binding pocket of the MAO-B enzyme and to conserve the major interactions found in the best docked posture throughout the MD simulation.
RMSD
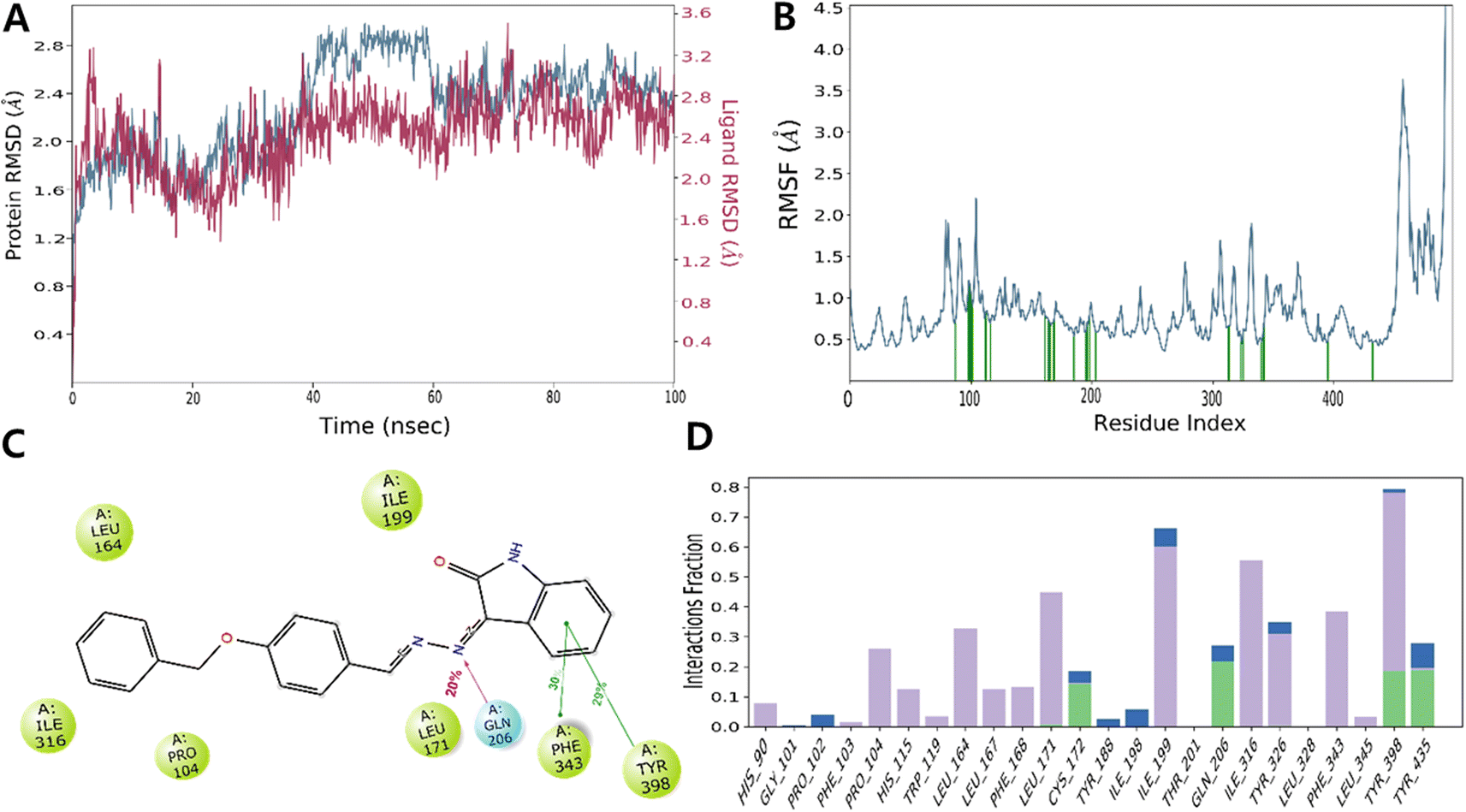
The RMSD value obtained from the MD simulation trajectory is one of the crucial metrics that demonstrates fluctuations in the structural conformation of the protein backbone over time during system equilibration; low and consistent RMSD values signify the stability of a protein structure. The RMSD of MAO-B with ISB1 ranged from 2.00 and 3.60 Å, with the complex's RMSD value reported at 2.28 Å on average. In the simulation trajectory, the ligand RMSD displayed little change and was estimated to be between 40 and 60 ns. Thereafter, the simulation's remaining frame marked gradual convergence. Analysis of the trajectory revealed that the phenyl ring of the free-moving benzyloxy moiety was the main cause of this fluctuation. Overall, there was little departure from the docked stance in the ISB1-hMAO-B docked complex (Fig. 8).
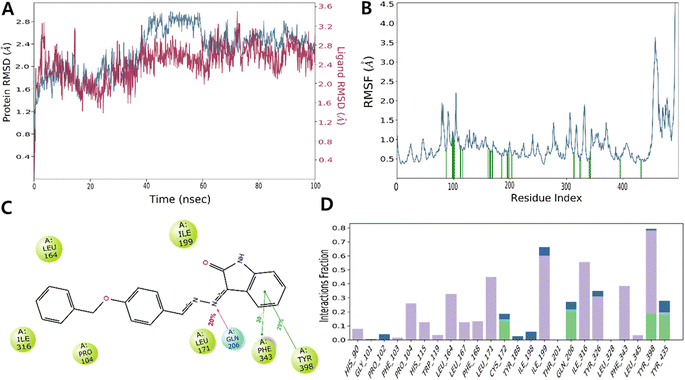 |
| | Fig. 8 Molecular docking (MD) simulation analysis of ISB1-MAO-B complex. Root-mean-square deviation (RMSD) (A) and root-mean-square fluctuation (RMSF) of various amino acids (B). A 2D interaction diagram of ISB1-MAO-B complex (C). Protein–ligand contact analysis of the MD trajectory (D). In panel (A), the RMSD of protein and ISB1 are depicted in blue and red, respectively. | |
In addition to RMSD, the RMSF value of a protein is widely used to assess ligand-induced variations in the protein's internal chains. The RMSF renders details on the flexibility and mobility of specific amino acids. A lower RMSF value indicates that the residue is less flexible and mobile. In ligand–protein interactions, the ligand establishes stronger binding with the protein when the RMSF value is low at the active site residues. Low values reflect the existence of secondary structures, including sheets and helices, which determine structural stability. The presence of twists, loops, terminal ends, and loose bonding, on the other hand, is represented by greater RMSF values (peaks), which implies structural flexibility.55–60 Thus, the protein RMSF plot analysis provided details of the flexible areas of the protein. Our analysis found that ISB1 made contact with 24 amino acids of the hMAO-B protein, which included His90 (0.715 Å), Gly101 (1.212 Å), Pro102 (1.16 Å), Phe103 (1.02 Å), Pro 104 (0.892 Å), His 115 (0.832 Å), Trp 119 (0.699 Å), Leu 164 (0.803 Å), Leu 167 (0.735 Å), Phe 168 (0.701 Å), Leu 171 (0.703 Å), Cys 172 (0.778 Å), Tyr 188 (0.542 Å), Ile 198 (0.588 Å), Ile 199 (0.716 Å), Thr 201 (0.832 Å), Gln 206 (0.592 Å), Ile 316 (0.666 Å), Tyr 326 (0.523 Å), Leu 328 (0.627 Å), Phe 343 (0.474 Å), Leu 345 (0.677 Å), Tyr398 (0.475 Å), and Tyr 435 (0.457 Å). The catalytic pocket residues displayed considerably less fluctuation than other residues, with the exception of the terminal 498–502 residues near the N-terminus, implying that the conformation of the residue remained reasonably stable during the simulation (Fig. 8).
Materials and methods
Chemical synthesis
Isatin hydrazone (1 equivalent) was synthesized using the chemical reaction of isatin substituted with hydrazine hydrate (2.5 equivalent) and glacial acetic acid under microwave radiation using methanol as a solvent. The mixture of isatin hydrazone (1 equivalent) and a substituted benzyloxy benzaldehyde (1 equivalent) was dissolved in 7.5 mL methanol using glacial acetic acid as a catalyst, and placed in the microwave reactor for 10 min at 120 °C. The precipitate obtained as a solid product was then filtered. The mixture was washed with methanol and the dried products were recrystallized (Fig. 2). Employing the solvent system hexane:ethyl acetate (2:1), thin layer chromatography (TLC) was performed on pre-coated TLC plates (silica gel 60-120#) to monitor all of the reactions.
Chemicals
For synthesis, isatin derivatives and hydrazine hydrate were purchased from Sigma-Aldrich (St. Louis, MO, USA) and TCI chemical. Substrates (benzylamine and kynuramine) and reference inhibitors (clorgyline, lazabemide, pargyline, and toloxatone) as well as recombinant human MAO-A and MAO-B were purchased from Sigma-Aldrich. For reversibility test was performed by using Dialyzer 6–8 kDa DiaEasy™ (BioVision, St. Grove, MA, USA).
Inhibition studies of MAO-B and MAO-A
The MAO-B and MAO-A activities were evaluated using and 0.3 mM benzylamine and 0.06 mM kynuramine, respectively.61 The previously reported continuous assay method (MAO-A at 316 nm and MAO-B at 250 nm) was used to determine the absorbance.62 The inhibition of the compounds was compared to that of the reference inhibitors of MAO-A (toloxatone and clorgyline) and MAO-B (lazabemide and pargyline). Utilizing version 5 of the GraphPad Prism software, the compounds' IC50 values were determined.63 The selectivity indices (SI) of the compounds were estimated by: (IC50 of MAO-A)/(IC50 of MAO-B).64
Enzyme kinetics
The inhibition types of the ISB1 and ISFB1 lead compounds for MAO-B were determined at five distinct substrate concentrations (0.0375–0.60 μM benzylamine for MAO-B),61,65 and at three different inhibitor concentrations that were 0.5, 1.0, and 1.5 times their IC50 values.63 By comparing Lineweaver–Burk plots and their secondary plots, the patterns of enzyme inhibition and the Ki values were determined.62
Reversibility studies
By comparing the residual activities of dialyzed and undialyzed samples, the reversibility for MAO-B inhibition was determined for lead compounds ISB1 and ISFB1 at a concentration of approximately 2.0-times the IC50 value after 30 min incubation, as previously described.61,65 The activities of the compounds were juxtaposed with those of the reference drugs lazabemide and pargyline, which are reversible and irreversible inhibitors of MAO-B, respectively. The reversibility patterns were determined by comparing the activities of the undialyzed (AU) and dialyzed (AD) compounds.61,65
BBB permeability study
The parallel artificial membrane permeation assay (PAMPA) method was first used in drug research to predict the passive transcellular penetration of pharmaceuticals over the BBB. Using a previous technique developed by our lab, the PAMPA test was carried out on 96-well microtiter and Millipore filter plates with 0.45 micron-sized pores and 125 microns thick (ipvh). The drug concentrations in the receiver, donor and reference wells were determined using UV spectroscopy. The penetration rate was calculated using the formula below.35,66
| logPe = −ln[1 − CA/equilibrium]/A × (1/VD + 1/VA) × t. |
Effect of ISB1 on rotenone-induced cell viability
The neuroprotective effect of ISB1 on rotenone-induced SH-SY5Y cells was analyzed by MTT assay. The cell line was cultured in two 25 cm-tissue culture flasks with DMEM (Sigma-Aldrich, D5648) supplemented with 10% FBS (Gibco, US Origin), L-glutamine (Sigma-Aldrich, G3126), sodium bicarbonate, and antibiotic solution. Cultured cell lines were kept at 37 °C in a humidified 5% CO2 incubator (NBS Eppendorf, Germany). In 96-well plates, SH-SY5Y cells (5 × 104 cells per well) were cultivated. Cells were pre-treated with rotenone (10 μM) (Sigma-Aldrich, R-8875) at different concentrations in the presence or absence of ISB1 (1.5, 3.1, 6.25, 12.5, 25 μg in 500 μl of 5% DMEM). After 24 h of incubation, the sample material in wells was removed and 30 μl of reconstituted MTT (Sigma-Aldrich, M-5655) solution was added. After 4 h of incubation at 37 °C in a humidified 5% CO2 incubator, the supernatant was removed and 100 μl of MTT solubilization solution (DMSO) was added to solubilize the formazan crystals. At a wavelength of 540 nm, the absorbance values were measured using a microplate reader (Erba Lisa Scan EM, Mannheim, Germany).67
Molecular docking
Using a in silico technique, the binding affinities of ISB1 and ISFB1 to the binding sites of human MAO-B with PDB ID:2V5Z were identified. The three-step Protein Preparation Wizard (PPW) was utilized to generate the protein crystal structures, including the program's pre-processing, optimization, and protein energy minimization steps.68,69 LigPrep was employed during the ligand preparation for docking studies.70 Receptor grid generation module to grid file was generated at the site of the co-crystallized ligand. Ligand docking was performed in Extra Precision mode (XP).
Molecular dynamic (MD) simulation
By docking poses, the lead molecule ISB1, in the hMAO-B enzyme with the least negative score (e.g., the top-docking poses), was the subject of molecular dynamics (MD) research utilizing Desmond in NVIDIA Quadro 6000 graphics processing unit.71–73 In order to construct trajectories of 1000 frames each to explore interactions between protein and ligand, the default parameter was employed for the systems under research. This included 100 ns MD generation with coordinates recorded at 100 ps. According to previous studies, additional data for MD investigations including box type, estimates of long- and short-range interactions, thermostat and barometer settings, were taken into consideration.74–78 Finally, stability analysis was performed by employing a tool for simulating interaction diagrams.
Conclusions
Novel classes of eighteen isatin-based benzyloxybenzaldehyde derivatives were synthesized, and their effectiveness in inhibiting MAOs was tested. The lead molecules (3Z)-3-((4-(benzyloxy)benzylidene)hydrazineylidene)indolin-2-one (ISB1) and (3Z)-3-((4-((3-fluorobenzyl)oxy)benzylidene)hydrazineylidene)indolin-2-one (ISFB1) exhibited potent MAO-B inhibitory activity with IC50 values of 0.124 ± 0.007 and 0.135 ± 0.002 μM, respectively. The leading compounds ISB1 and ISFB1 appeared to be competitive MAO-B inhibitors with the Ki values of 0.055 ± 0.010 and 0.069 ± 0.025 μM, respectively. The reversible mode of MAO-B inhibition and the BBB permeation nature of the lead molecules also claimed its CNS-drug likeness. Finally, the present study revealed that ISB1 significantly reduced rotenone-induced cell death in SH-SY5Y neuroblastoma cells. The docking and simulation studies indicated a pi–pi stacking interaction in the phenyl ring of isatin and hydrogen bonding formed by the imino nitrogen in ISB1 that maintained the stability of the protein–ligand complex. In short, our findings imply that ISB1 and ISFB1 are considered to be candidate agents for the treatment of PD based on the reversible inhibition of MAO-B.
Author contributions
Conceptualization, B. M. and H. K.; synthesis, F. B., S. K., M. M. G., and M. S. A. B.; biological tests, J. M. O. and M. A. A.; docking simulation, N. K. and J. J.; writing—original draft preparation, F. B., J. M. O., and S. K.; writing—review and editing, B. M. and H. K.; supervision, B. M. and H. K.; funding acquisition, H. K. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors are thankful to AlMaarefa University for their support. This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2022R1A2B5B01002536).
References
- L. M. de Lau and M. M. Breteler, Lancet Neurol., 2006, 5, 525–535 CrossRef.
- D. Robakis and S. Fahn, CNS Drugs, 2015, 29, 433–441 CrossRef CAS PubMed.
- P. Maiti, J. Manna and G. L. Dunbar, Transl. Neurodegener., 2017, 6, 28 CrossRef PubMed.
- N. Eriksen, A. K. Stark and B. Pakkenberg, in Birth, Life and Death of Dopaminergic Neurons in the Substantia Nigra, Springer Vienna, Vienna, 2009, pp. 203–213 Search PubMed.
- J. Zheng, X. Zhang and X. Zhen, ACS Chem. Neurosci., 2019, 10, 783–791 CrossRef CAS PubMed.
- G. Gao, Z. Wang, L. Lu, C. Duan, X. Wang and H. Yang, Biomed. Pharmacother., 2017, 96, 1380–1388 CrossRef CAS PubMed.
- N. Plotegher, G. Berti, E. Ferrari, I. Tessari, M. Zanetti, L. Lunelli, E. Greggio, M. Bisaglia, M. Veronesi, S. Girotto, M. Dalla Serra, C. Perego, L. Casella and L. Bubacco, Sci. Rep., 2017, 7, 40699 CrossRef CAS.
- Y.-Y. Tan, P. Jenner and S.-D. Chen, J. Parkinson's Dis., 2022, 12, 477–493 CAS.
- J. C. Shih, J. Neural Transm., 2018, 125, 1553–1566 CrossRef CAS.
- M. Bortolato, K. Chen and J. C. Shih, Adv. Drug Delivery Rev., 2008, 60, 1527–1533 CrossRef CAS PubMed.
- C. J. Fowler, A. Wiberg, L. Oreland, J. Marcusson and B. Winblad, J. Neural Transm., 1980, 49, 1–20 CrossRef CAS PubMed.
- S. Carradori, M. D'Ascenzio, P. Chimenti, D. Secci and A. Bolasco, Mol. Diversity, 2014, 18, 219–243 CrossRef CAS PubMed.
- J. Y. Heo, M.-H. Nam, H. H. Yoon, J. Kim, Y. J. Hwang, W. Won, D. H. Woo, J. A. Lee, H.-J. Park, S. Jo, M. J. Lee, S. Kim, J.-E. Shim, D.-P. Jang, K. I. Kim, S. H. Huh, J. Y. Jeong, N. W. Kowall, J. Lee, H. Im, J. H. Park, B. K. Jang, K. D. Park, H. J. Lee, H. Shin, I.-J. Cho, E. M. Hwang, Y. Kim, H. Y. Kim, S.-J. Oh, S. E. Lee, S. H. Paek, J. H. Yoon, B. K. Jin, G. R. Kweon, I. Shim, O. Hwang, H. Ryu, S. R. Jeon and C. J. Lee, Curr. Biol., 2020, 30, 276–291e9 CrossRef CAS PubMed.
- J. J. Chen and D. M. Swope, J. Clin. Psychopharmacol., 2005, 45, 878–894 CAS.
- H. Juárez Olguín, D. Calderón Guzmán, E. Hernández García and G. Barragán Mejía, Oxid. Med. Cell. Longevity, 2016, 2016, 1–13 CrossRef PubMed.
- H. H. Chan, M. K. Tse, S. Kumar and L. Zhuo, Eur. J. Pharmacol., 2018, 818, 254–262 CrossRef CAS.
- B. Mathew, J. M. Oh, M. A. Abdelgawad, A. Khames, M. M. Ghoneim, S. Kumar, L. R. Nath, S. T. Sudevan, D. G. T. Parambi, C. Agoni, M. E. S. Soliman and H. Kim, ACS Omega, 2022, 7, 8184–8197 CrossRef CAS.
- S. Carradori and R. Silvestri, J. Med. Chem., 2015, 58, 6717–6732 CrossRef CAS PubMed.
- S. Perez-Lloret and O. Rascol, Expert Rev. Neurother., 2016, 16, 245–258 CrossRef CAS PubMed.
- L. Dézsi and L. Vécsei, Expert Opin. Invest. Drugs, 2014, 23, 729–742 CrossRef.
- P. Sanchez Alonso, B. De La Casa-Fages, A. Alonso-Cánovas and J. C. Martínez-Castrillo, Brain Sci., 2023, 13, 276 CrossRef CAS.
- T. Müller, Neurodegener. Dis. Manage., 2020, 10, 195–204 CrossRef.
- S. T. Sudevan, T. M. Rangarajan, A. G. Al-Sehemi, A. S. Nair, V. P. Koyiparambath and B. Mathew, Arch. Pharm., 2022, 355, 2200084 CrossRef CAS.
- B. Mathew, S. Carradori, P. Guglielmi, M. S. Uddin and H. Kim, Curr. Med. Chem., 2020, 28, 266–283 CrossRef PubMed.
- F. Mazouz, S. Gueddari, C. Burstein, D. Mansuy and R. Milcent, J. Med. Chem., 1993, 36, 1157–1167 CrossRef CAS PubMed.
- M. Naoi and K. Yagi, Arch. Biochem. Biophys., 1980, 205, 18–26 CrossRef CAS.
- V. Pérez, J. L. Marco, E. Fernández-Álvarez and M. Unzeta, Br. J. Pharmacol., 1999, 127, 869–876 CrossRef.
- S. Kumar, A. S. Nair, M. A. Abdelgawad and B. Mathew, ACS Omega, 2022, 7, 16244–16259 CrossRef CAS.
- M. Fabbri, M. M. Rosa, D. Abreu and J. J. Ferreira, Neurodegener. Dis. Manage., 2015, 5, 481–496 CrossRef.
- M. Joy, B. Mathew and C. Sudarsanakumar, Chem. Data Collect., 2018, 17–18, 404–414 CrossRef.
- C. Binda, J. Wang, L. Pisani, C. Caccia, A. Carotti, P. Salvati, D. E. Edmondson and A. Mattevi, J. Med. Chem., 2007, 50, 5848–5852 CrossRef CAS.
- F. Leonetti, C. Capaldi, L. Pisani, O. Nicolotti, G. Muncipinto, A. Stefanachi, S. Cellamare, C. Caccia and A. Carotti, J. Med. Chem., 2007, 50, 4909–4916 CrossRef CAS.
- I. Bolea, J. Juárez-Jiménez, C. de los Ríos, M. Chioua, R. Pouplana, F. J. Luque, M. Unzeta, J. Marco-Contelles and A. Samadi, J. Med. Chem., 2011, 54, 8251–8270 CrossRef CAS.
- I. Bolea, M. A. Colivicchi, C. Ballini, J. Marco-Contelles, K. F. Tipton, M. Unzeta and L. Della Corte, CNS Neurosci. Ther., 2014, 20, 641–650 CrossRef CAS PubMed.
- S. T. Sudevan, J. M. Oh, M. A. Abdelgawad, M. A. S. Abourehab, T. M. Rangarajan, S. Kumar, I. Ahmad, H. Patel, H. Kim and B. Mathew, Sci. Rep., 2022, 12, 22404 CrossRef CAS.
- R. I. Al-Wabli, A. A. Almomen, M. S. Almutairi, A. B. Keeton, G. A. Piazza and M. I. Attia, Drug Des., Dev. Ther., 2020, 14, 483–495 CrossRef CAS.
- P. K. Shukla, M. P. Singh and R. Patel, J. Appl. Pharm. Sci. Res., 2018, 1, 16–22 CAS.
- D. Bonivento, E. M. Milczek, G. R. McDonald, C. Binda, A. Holt, D. E. Edmondson and A. Mattevi, J. Biol. Chem., 2010, 285, 36849–36856 CrossRef CAS.
- M. Tavari, S. F. Malan and J. Joubert, MedChemComm, 2016, 7, 1628–1639 RSC.
- F. Hubálek, C. Binda, A. Khalil, M. Li, A. Mattevi, N. Castagnoli and D. E. Edmondson, J. Biol. Chem., 2005, 280, 15761–15766 CrossRef.
- C. Liang, J. Xia, D. Lei, X. Li, Q. Yao and J. Gao, Eur. J. Med. Chem., 2014, 74, 742–750 CrossRef CAS PubMed.
- K. Kopka, A. Faust, P. Keul, S. Wagner, H.-J. Breyholz, C. Höltke, O. Schober, M. Schäfers and B. Levkau, J. Med. Chem., 2006, 49, 6704–6715 CrossRef CAS.
- Q. Zhang, Y. Fu, Y. Zhao, S. Cui, J. Wang, F. Liu, Y. Yuan, H. Galons, P. Yu and Y. Teng, RSC Adv., 2019, 9, 36690–36698 RSC.
- P. Pakravan, S. Kashanian, M. M. Khodaei and F. J. Harding, Pharmacol. Rep., 2013, 65, 313–335 CrossRef CAS.
- F. D. Popp, R. Parson and B. E. Donigan, J. Pharm. Sci., 1980, 69, 1235–1237 CrossRef CAS.
- S. N. Pandeya, S. Smitha, M. Jyoti and S. K. Sridhar, Acta Pharm., 2005, 55, 27–46 CAS.
- V. Varun, S. Sonam and R. Kakkar, MedChemComm, 2019, 10, 351–368 RSC.
- J. L. Hyatt, T. Moak, M. J. Hatfield, L. Tsurkan, C. C. Edwards, M. Wierdl, M. K. Danks, R. M. Wadkins and P. M. Potter, J. Med. Chem., 2007, 50, 1876–1885 CrossRef CAS PubMed.
- R. Nath, S. Pathania, G. Grover and M. J. Akhtar, J. Mol. Struct., 2020, 1222, 128900 CrossRef CAS.
- T. R. Bal, B. Anand, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2005, 15, 4451–4455 CrossRef CAS PubMed.
- M. S. Vishnu, V. Pavankumar, S. Kumar and A. S. Raja, ChemMedChem, 2019, 14, 1359–1376 CrossRef CAS.
- E. M. Milczek, C. Binda, S. Rovida, A. Mattevi and D. E. Edmondson, FEBS J., 2011, 278, 4860–4869 CrossRef CAS.
- G. E. Mathew, J. M. Oh, K. Mohan, A. Tengli, B. Mathew and H. Kim, J. Biomol. Struct. Dyn., 2021, 39, 4786–4794 CrossRef CAS PubMed.
- B. Mathew, J. M. Oh, R. S. Baty, G. E.-S. Batiha, D. G. T. Parambi, N. Gambacorta, O. Nicolotti and H. Kim, Environ. Sci. Pollut. Res., 2021, 28, 38855–38866 CrossRef CAS PubMed.
- Y. Boulaamane, I. Ahmad, H. Patel, N. Das, M. R. Britel and A. Maurady, J. Biomol. Struct. Dyn., 2023, 41, 2326–2340 CrossRef CAS.
- D. Osmaniye, Ş. Karaca, B. Kurban, M. Baysal, I. Ahmad, H. Patel, Y. Özkay and Z. Asım Kaplancıklı, Bioorg. Chem., 2022, 122, 105709 CrossRef CAS PubMed.
- S. Ghosh, S. Das, I. Ahmad and H. Patel, J. Indian Chem. Soc., 2021, 98, 100272 CrossRef CAS.
- U. Acar Çevik, I. Celik, A. Işık, I. Ahmad, H. Patel, Y. Özkay and Z. A. Kaplancıklı, J. Biomol. Struct. Dyn., 2023, 41, 1944–1958 CrossRef PubMed.
- R. Zrieq, I. Ahmad, M. Snoussi, E. Noumi, M. Iriti, F. D. Algahtani, H. Patel, M. Saeed, M. Tasleem, S. Sulaiman, K. Aouadi and A. Kadri, Int. J. Mol. Sci., 2021, 22, 10693 CrossRef CAS.
- Y. O. Ayipo, I. Ahmad, Y. S. Najib, S. K. Sheu, H. Patel and M. N. Mordi, J. Biomol. Struct. Dyn., 2023, 41, 1959–1977 CrossRef CAS.
- H. W. Lee, H. W. Ryu, M.-G. Kang, D. Park, S.-R. Oh and H. Kim, Bioorg. Med. Chem. Lett., 2016, 26, 4714–4719 CrossRef CAS.
- J. M. Oh, H.-J. Jang, W. J. Kim, M.-G. Kang, S. C. Baek, J. P. Lee, D. Park, S.-R. Oh and H. Kim, Int. J. Biol. Macromol., 2020, 151, 441–448 CrossRef CAS PubMed.
- J. M. Oh, Y. Kang, J. H. Hwang, J.-H. Park, W.-H. Shin, S.-K. Mun, J. U. Lee, S.-T. Yee and H. Kim, Int. J. Biol. Macromol., 2022, 217, 910–921 CrossRef CAS PubMed.
- S. C. Baek, M. H. Park, H. W. Ryu, J. P. Lee, M.-G. Kang, D. Park, C. M. Park, S.-R. Oh and H. Kim, Bioorg. Chem., 2019, 83, 317–325 CrossRef CAS.
- H. W. Lee, H. W. Ryu, M.-G. Kang, D. Park, H. Lee, H. M. Shin, S.-R. Oh and H. Kim, Int. J. Biol. Macromol., 2017, 97, 598–605 CrossRef CAS.
- L. Di, E. H. Kerns, K. Fan, O. J. McConnell and G. T. Carter, Eur. J. Med. Chem., 2003, 38, 223–232 CrossRef CAS.
- S. Pakrashi, J. Chakraborty and J. Bandyopadhyay, Neurochem. Res., 2020, 45, 1962–1973 CrossRef CAS PubMed.
- N. C. Desai, A. S. Maheta, A. M. Jethawa, U. P. Pandit, I. Ahmad and H. Patel, J. Heterocycl. Chem., 2022, 59, 879–889 CrossRef CAS.
- B. Chaudhari, H. Patel, S. Thakar, I. Ahmad and D. Bansode, In Silico Pharmacol., 2022, 10, 10 CrossRef PubMed.
- R. Girase, I. Ahmad, R. Pawara and H. Patel, SAR QSAR Environ. Res., 2022, 33, 215–235 CrossRef CAS.
- N. C. Desai, S. B. Joshi, A. G. Khasiya, D. J. Jadeja, H. K. Mehta, M. Pandya, I. Ahmad and H. Patel, J. Mol. Struct., 2022, 1270, 134000 CrossRef CAS.
- D. Osmaniye, Ş. Karaca, B. Kurban, M. Baysal, I. Ahmad, H. Patel, Y. Özkay and Z. Asım Kaplancıklı, Bioorg. Chem., 2022, 122, 105709 CrossRef CAS PubMed.
- I. Ahmad, R. Pawara and H. Patel, Mol. Simul., 2022, 48, 1639–1649 CrossRef CAS.
- H. A. Radwan, I. Ahmad, I. M. M. Othman, M. A. M. Gad-Elkareem, H. Patel, K. Aouadi, M. Snoussi and A. Kadri, J. Mol. Struct., 2022, 1264, 133312 CrossRef CAS.
- M. M. Farhan, M. A. Guma, M. A. Rabeea, I. Ahmad and H. Patel, J. Mol. Struct., 2022, 1269, 133781 CrossRef CAS.
- U. Acar Çevik, I. Celik, A. Işık, I. Ahmad, H. Patel, Y. Özkay and Z. A. Kaplancıklı, J. Biomol. Struct. Dyn., 2023, 41, 1944–1958 CrossRef.
- M. S. Tople, N. B. Patel, P. P. Patel, A. C. Purohit, I. Ahmad and H. Patel, J. Mol. Struct., 2023, 1271, 134016 CrossRef CAS.
- I. Ahmad, R. H. Pawara, R. T. Girase, A. Y. Pathan, V. R. Jagatap, N. Desai, Y. O. Ayipo, S. J. Surana and H. Patel, ACS Omega, 2022, 7, 21820–21844 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: TLC, HPLC, 1H-, 13C-NMR and MS spectra of all compounds. See DOI: https://doi.org/10.1039/d3ra07035b |
| ‡ Authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence g,
Hoon Kim
g,
Hoon Kim