DOI:
10.1039/D2RA06870B
(Paper)
RSC Adv., 2023,
13, 5324-5336
In vitro anti-inflammatory, in silico molecular docking and molecular dynamics simulation of oleanane-type triterpenes from aerial parts of Mussaenda recurvata†
Received
31st October 2022
, Accepted 30th January 2023
First published on 13th February 2023
Abstract
Bioactive-guided investigation of the aerial parts of Mussaenda recurvata Naiki, Tagane, and Yahara (Rubiaceae) led to the isolation of four triterpenes, including two new triterpenes recurvatanes A and B (1 and 2), along with two known compounds 3β,6β,23-trihydroxyolean-12-en-28-oic acid (3) and 3β,6β,19α,23-tetrahydroxyolean-12-en-28-oic acid (4). The chemical structures of the compounds were identified from spectroscopic data and by comparison with the literature. A comprehensive review of NMR data of the oleanane-type triterpenes bearing 3-hydroxy and 4-hydroxymethylene groups indicated the characteristic spectroscopic features in this series. Compounds 1–4 were evaluated for the inhibitory NO production in LPS-stimulated RAW264.7 cells. Compounds 2 and 3 showed a moderate reduction of nitrite accumulation with IC50 values of 55.63 ± 2.52 and 60.08 ± 3.17 μM, respectively. Molecular docking model dedicated to compound 3 or pose 420, which is the best candidate among docking poses of compounds 1–4 interacted well with the crystal structure of enzyme 4WCU: PDB. The best ligand molecule, pose 420 in terms of binding energy obtained from docking studies on molecular dynamics (MD) simulations for 100 ns exhibited non-bonding interactions with the protein and remained stable inside the active site.
1. Introduction
The molecular docking model is one method that allows the evaluation of biochemical activities between one ligand and one receptor target protein or enzyme and this model is based on the scoring function that estimates the free energy change upon binding.1,2 Most biochemical activities performed in silico such as anticancer,3–5 enzyme α-glucosidase inhibition,6 antibacterial activity, and anti-inflammatory are based on the inhibition mechanism explanations of inhibition of the biosynthesis of bacterial and fungal cell walls.7 The molecular docking model must assess the validation via the value of RMSD between one reference pose that is available in an enzyme or protein and a ranked pose or ligand.8–10 Enzyme 4WCU is a target enzyme for anti-inflammatory activity. It is a phosphodiesterase-4 enzyme that performed the degradation of the second messenger, cAMP; thus, the 4WCU enzyme contributed to the cell signal. This target developed a clinical drug, which is detailed for anti-inflammatory activity and others.11 A ligand is considered that it is interacting well with the enzyme when the three parts of the ligand, including the capping group, linker, and functional group bond well to the active center of the enzyme.12 MD generates a more accurate biophysical simulation of the protein–ligand complex binding to explain the model complex's stability.13 Molecular dynamics (MD) simulations have been documented to determine the accuracy of the predicted binding pose obtained from the molecular docking study.
The genus Mussaenda (Rubiaceae) is native to tropical and subtropical regions. It includes approximately 160 species.14 Some Mussaenda plants are believed to have valuable applications in Chinese traditional medicines.15 They are well-known as natural sources of saponins, terpenoids, iridoids, and flavonoids.16–27 These compounds also have various pharmaceutical properties as antioxidants, anticancer, anti-inflammatory, anticonvulsant, and thrombolytic agents.15,28–31 Some members of this genus (M. pubescens, M. glabrata, M. parviflora, M. shikokiana, and M. pilosissima) have been thoroughly investigated. However, little is known about the chemical and biological data of Mussaenda recurvata Naiki, Tagane, and Yahara. A previous study on the Vietnamese native plant indicated the presence of five saponins and two ursane-type triterpenes.32 These compounds exhibited potent anti-inflammatory activity.32 Bioactive-guided isolation of the Vietnamese plant M. recurvata indicated that the less polar fraction of this plant was the most active fraction and was selected for further isolation. Two new triterpenes recurvatanes A and B (1 and 2) along with two known compounds 3β,6β,23-trihydroxyolean-12-en-28-oic acid (3), 3β,6β,19α, 23-tetrahydroxyolean-12-en-28-oic acid (4) were isolated and structurally elucidated. Isolated compounds were evaluated for their inhibitory effects on LPS-induced NO production in RAW264.7 cells. A molecular docking study was conducted to determine the mechanism of the biological activity.
2. Results and discussion
2.1. Phytochemical identification of compounds 1–4
Compound 1, a white amorphous powder, exhibited a deprotonated molecular ion peak at [M − H]− 471.3488 on the HRESI mass spectrum, indicating its molecular formula as C30H47O4. Subsequent 1H NMR and HSQC analysis revealed one olefin proton (δH 5.16), one oxymethine (δH 3.44), one oxymethylene (δH 3.32 and 3.07), six tertiary methyls (δH 0.53, 0.72, 0.87 × 3, 1.10), three methines (δH 2.74, 1.50, and 1.11) and nine methylenes in the range 1.13–1.90 ppm. The 13C-NMR data, in accordance with HRESI mass data, showed 30 carbon signals, including one carboxylic acid carbon (δC 178.6), two olefinic carbons (δC 143.9 and 121.6), one oxymethine carbon (δC 70.3), one oxymethylene carbon (δC 64.5), six methyls (δC 32.8, 25.6, 23.4, 16.9, 15.5, and 12.6), three methines (δC 47.1, 46.4, and 40.8), and nine methylenes. These spectroscopic data indicated that compound 1 was an oleanane-type triterpenoid. NMR data of compound 1 were highly similar to those of oleanolic acid. The differences between them are in the structure of the A-ring. The first point is the oxygenation at C-23 in compound 1. HMBC correlations from the oxymethylene at δH 3.32 and 3.08 (H2-23), the methyl at δH 0.53 (H3-24), and the hydroxy group at δH 4.13 to carbon at δC 70.3 (C-3) (Fig. S1†) and from the methyl H3-24 to C-23 (δC 64.5) support the presence of the hydroxy group at C-23. The second difference is the change in the C-3 configuration as compared to that of oleanolic acid. The hydroxy group at C-3 was defined as being located at the α-position, based on the NOESY correlation of H-3 (δH 3.44) and H3-26 (δH 0.87). The relative configuration of compound 1 was further determined from the NOESY spectrum and comparison with the literature. NOESY correlations H2-23/H-5, H-5/H-9, H2-23/H-9, H-9/H3-27, and H-5/H-9 indicated the α orientations of H-5, H-9, H2-23, and H3-27. The chemical shift of H-3 of compound 1 was downfield, and the coupling pattern of this proton was changed compared to those of compounds 3 and 4, which were recorded in the same deuterated solvent (i.e., DMSO-d6, Table S1†). These points support the α-position of the 3-OH group in compound 1.
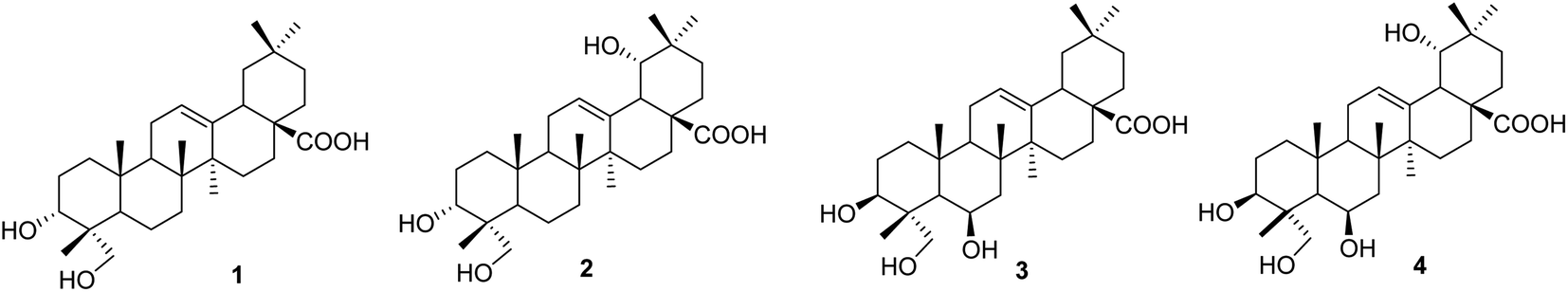
The literature review supports the relative configuration of compound 1. In particular, the NMR data of the series of oleanolic-acid derivatives bearing a 4-hydroxymethylene group are shown in Table S2.† In this series, two chiral centers, C-3 and C-4, provided four possible stereoisomers of 1 (Fig. S3†), namely wilforol C (1A),33,34 3α,24-dihydroxyolean-12-en-28-oic acid (1B),35 scutellaric acid (1C),36 and hederagenin (1D).37 NMR data of compound 1 were obviously different from those of 1A–1D. Notably, the chemical structure of compound 1 was identical to that described for wilforol C.32,33 However, the 13C chemical shifts of carbons C-23 and C-24 of wilforol C were more significantly shifted to downfield than those of the related compounds. As can be seen in Table S2,† the oxymethylene carbon was at δC 71.3 in wilforol C (1A) compared to other compounds (1B–1D, 2A, 3, 3A, 3B, and 4: δC 64–67), whereas the methyl carbon was downfield shifted (δC 18.3 in wilforol C vs. 13–15 ppm of other compounds) in different deuterated solvents. More specifically, the compounds coded 1C, 1D, 3, 3A, 3B, and 4 [recorded in the same deuterated pyridine-d5 as wilforol C (1A)] demonstrated different chemical shifts of C-23 and C-24 than those of wilforol C. These findings indicate that the structural elucidation of wilforol C can be regarded as dubious. Altogether, compound 1 was elucidated, as shown in Fig. 1, namely, recurvatane A.
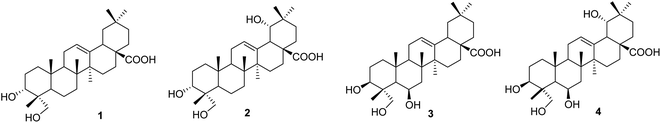 |
| | Fig. 1 Chemical structures of 1–4. | |
Compound 2 was isolated as a white amorphous powder. The HRESI mass spectrum exhibited a deprotonated molecular ion peak at [M − H]− 487.3441, indicating its molecular formula as C30H47O5. NMR and mass data of compound 2 indicated that 2 had one more hydroxy group than 1. The only difference between them was the presence of an additional hydroxy group at C-19 in 2. This was supported by HMBC correlations of H3-29 (δH 0.88) and H3-30 (δH 0.84) to C-19 (δH 80.1), and COSY correlation of H-18 (δH 2.93) and H-19 (δH 3.13). The 19-OH group was determined to be at α-orientation due to the small coupling constant of H-19. With two chiral centers, C-3 and C-4, four possible isomers of compound 2 (namely 2A, 2B, 2C, and 3B) existed (Fig. S3†). Among them, 2C and 3B were reported previously as spathodic acid and ilexosapogenin A, respectively. A careful comparison of NMR data of compounds 1 and 2 indicated that they shared the same configurations as C-3 and C-4. Altogether, compound 2 was elucidated as shown, namely recurvatane B.
2.2. The NO inhibition of compounds 1–4
Compounds 1–4 were evaluated for their inhibitory NO production in LPS-stimulated RAW264.7 cells (Table S3†). Compounds 2 and 3 showed a moderate reduction of nitrite accumulation in LPS-stimulated RAW 264.7 cells, with IC50 values of 55.63 ± 2.52 and 60.08 ± 3.17 μM, respectively, while compounds 1 and 4 were inactive compared to the positive control, L-NMMA (IC50 value of 29.35 ± 0.30 μM). The biological activities of compounds 1 and 4 are weaker than those of compounds 2 and 3, indicating the important role of the hydroxy substituents in this scaffold. Molecular docking studies of compounds 1–4 were conducted to clarify the inhibitory mechanism.
2.3. In silico molecular docking model
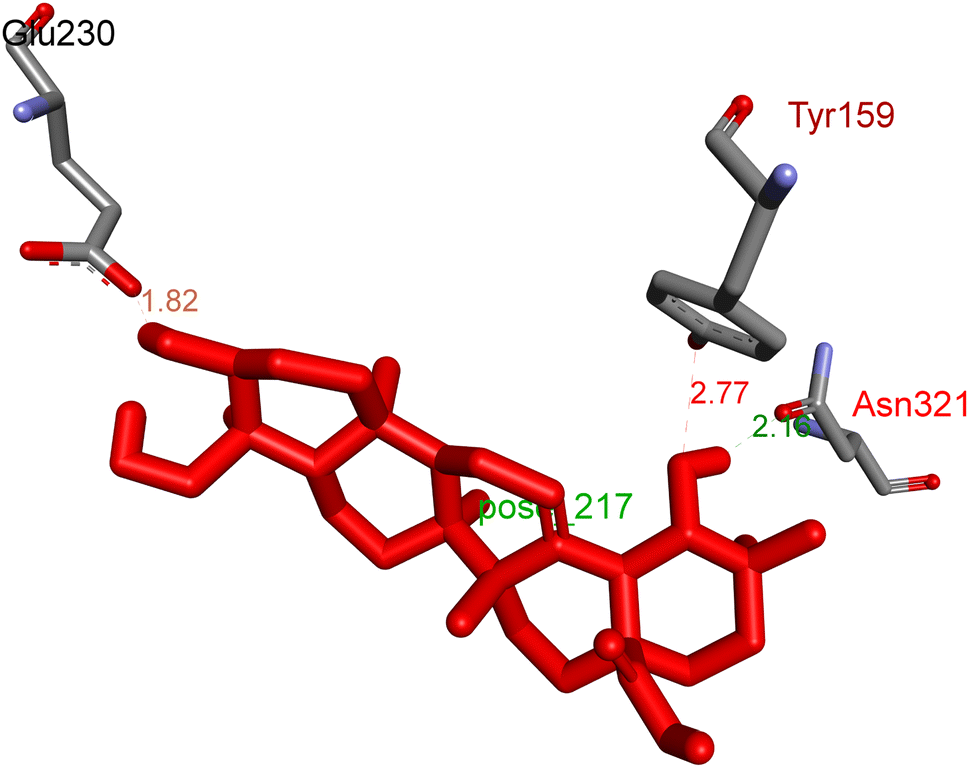
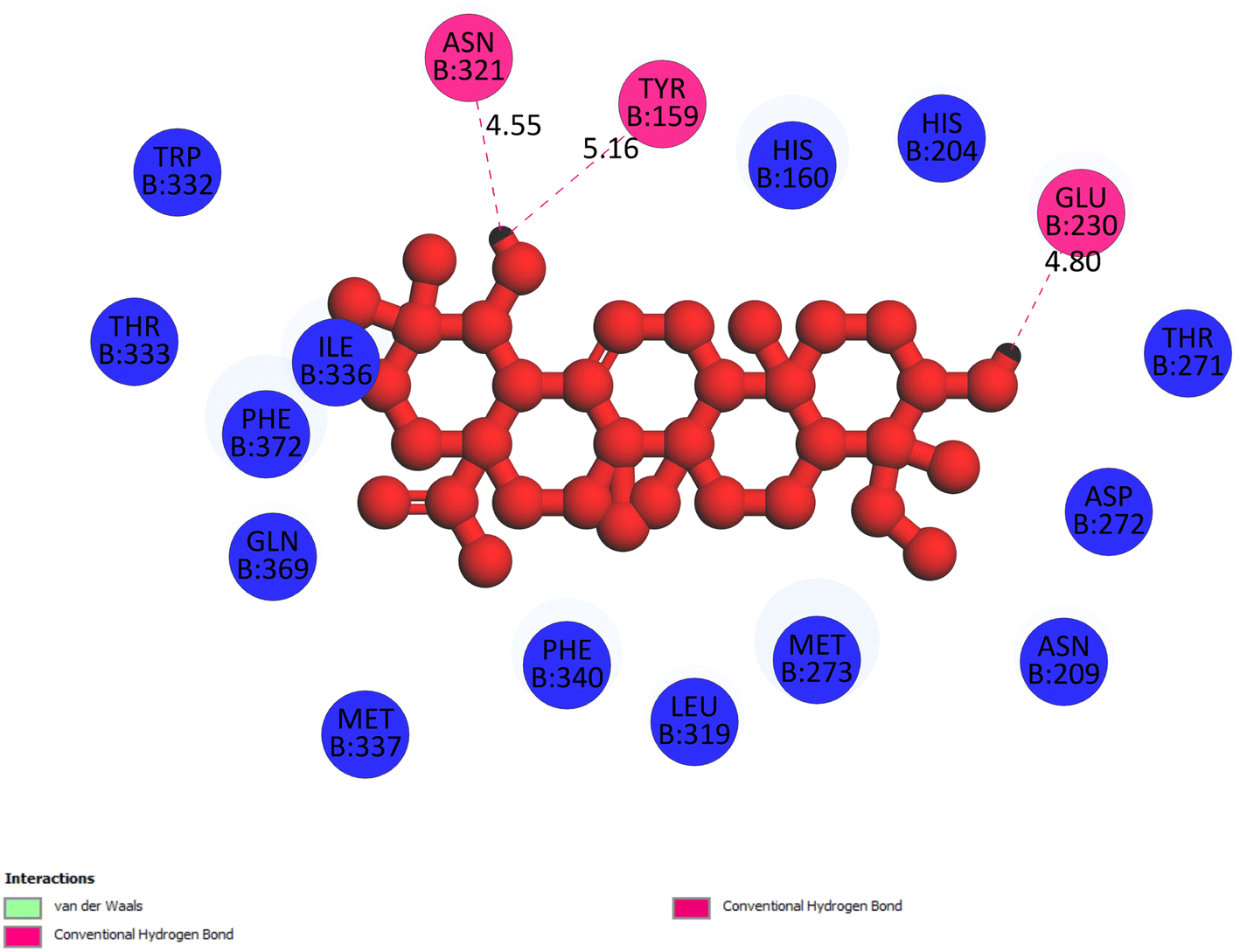
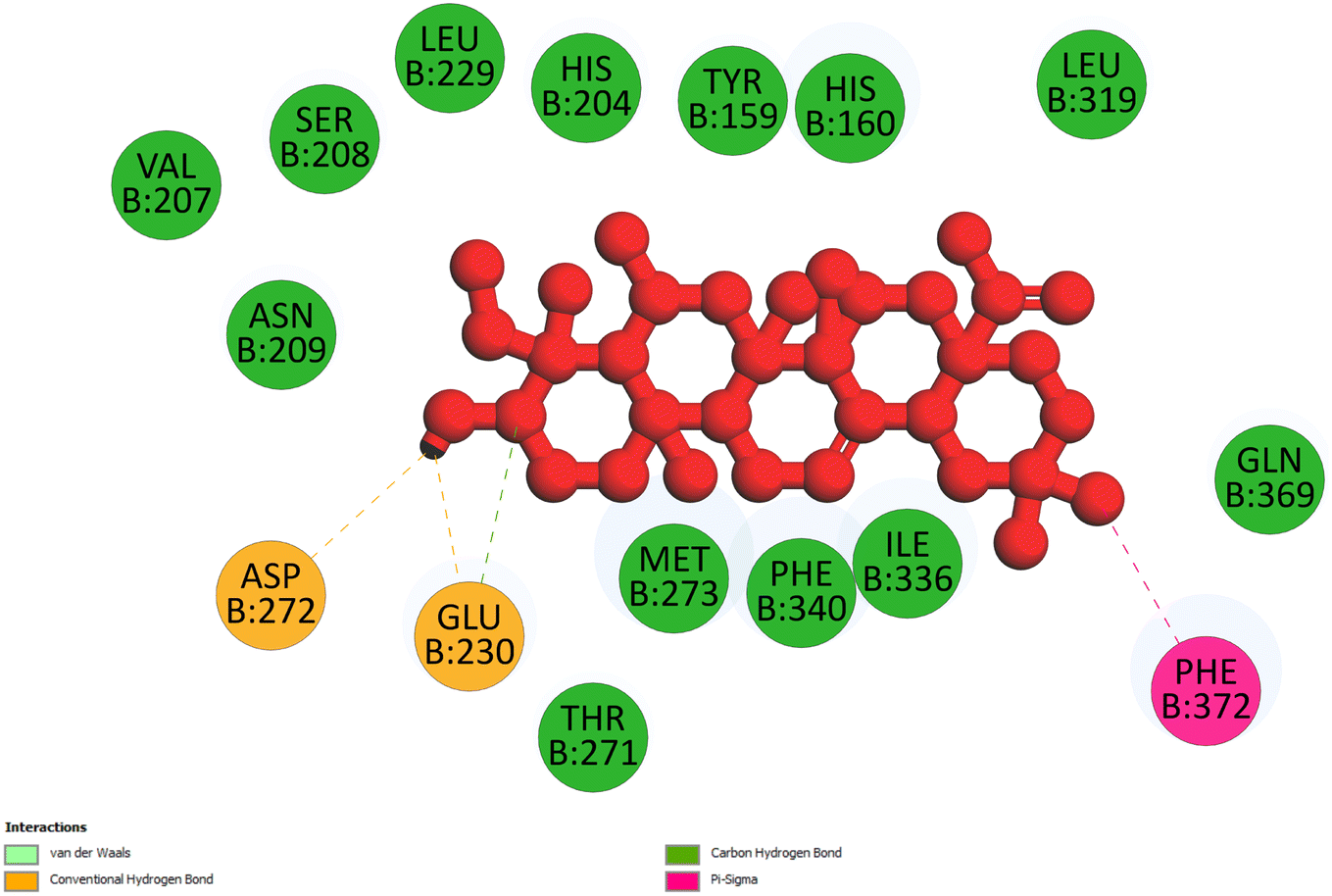
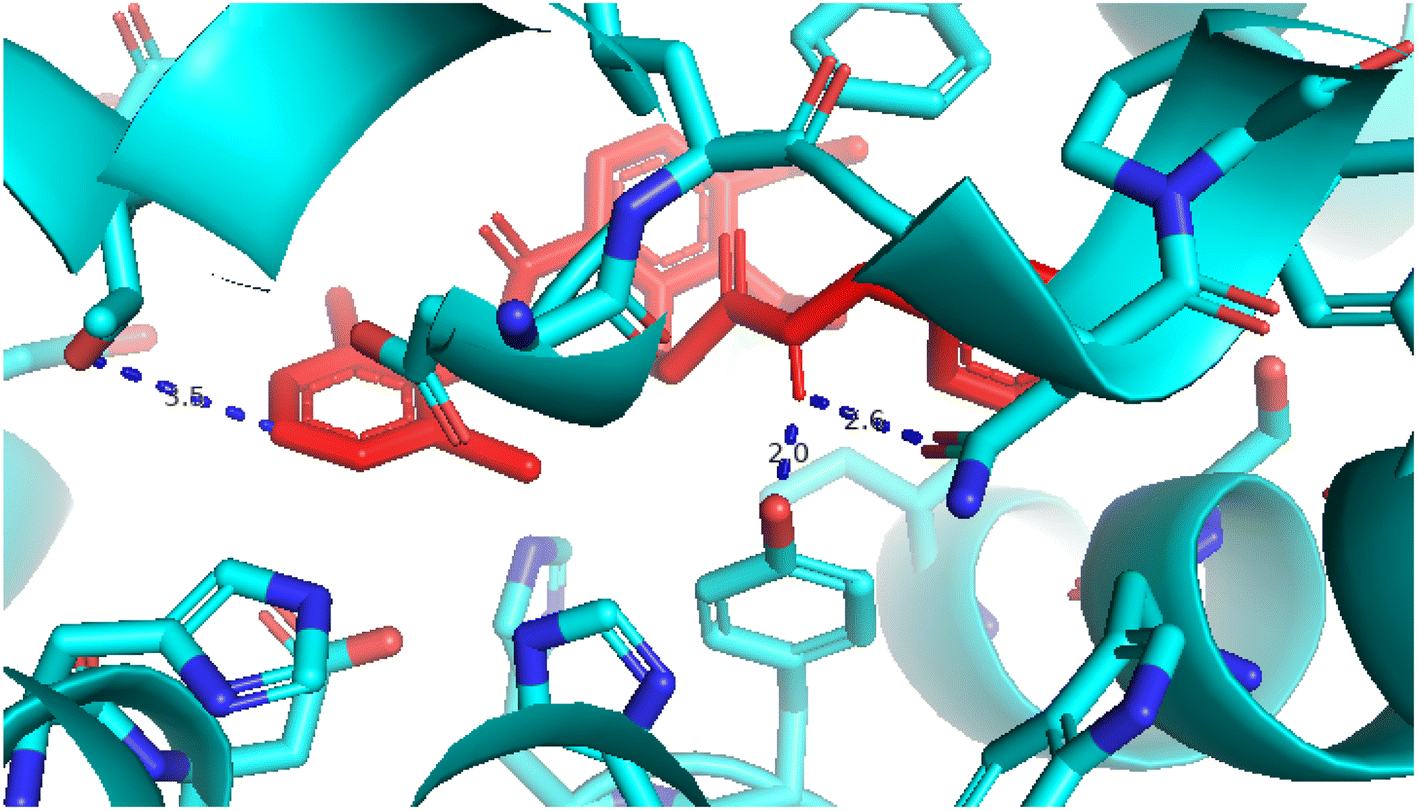
2.3.1 Pose 217. One ranked pose, the most stable conformation ligand of compound 2 interacted well with the 4WCU enzyme target with the values of affinity energy and inhibition constant of −7.21 kcal mol−1 and inhibition constant of 5.20 μM, respectively, as shown in Table 1. As shown in Fig. 2, three hydrogen bondings formed from the residual amino acids on one pocket enzyme, 4WCU to active atoms on the ligand or pose 217. Among the hydrogen bondings formed between pose 217 and one pocket enzyme or active center of enzyme 4WCU, the hydrogen bonding, hydrogen bonding linked from Glu230 to atom hydrogen on pose 217 is the strongest bonding because of the shortest bond length, 1.82 Å, as seen in Table 1. It is also the strongest hydrogen bonding among hydrogen bondings that was formed from the ranked pose and active pocket on the 4WCU enzyme due to the shortest bond length among hydrogen bondings, as indicated in Table 1. As shown in Fig. 3, one 2D diagram indicated the most significant ligand interactions formed between pose 217 and pocket enzyme of the crystal structure of enzyme 4WCU that included the capping group of pose (identification protein – interactions with aromatic ring or heterocyclic), connecting unit, CU or linker of pose (identification aliphatic chain or hydrophobic group) and functional group or ZBG (hydrogen bonding).38 For pose 217/compound 2, there are three hydrogen bonds linked from residual amino acids, Asn 321, Tyr 159, and Glu 230 to hydrogen atoms of alcohol hydroxyl of pose 217. The connecting unit and capping unit have no ligand interactions, so we consider that the protein and linker of this pose will not be detected. Pose 217, the best docking pose, and the most stable conformation of the ligand compound 2 do not interact well in the ligand model although the value of the affinity energy and the inhibition constant are low.
Table 1 The significant ligand interactions between the best docking pose and receptor, 4WCU (B chain)
| Entry |
Active posea |
Affinity energyb |
Inhibition constantc, Ki |
Hydrogen bond |
The property and bond lengthd |
| Active pose-based AutoDockTools-1.5.6rc3 package and the results indicated in dock.dlg file. Calculation based on AutoDockTools-1.5.6rc3 package and the results indicated in dock.dlg file, affinity energy in the unit of kcal mol−1. Calculation based on AutoDockTools-1.5.6rc3. Calculation based on AutoDockTools-1.5.6rc3, Ki in the unit of μM. |
| Compound 1 |
Pose 260/500 |
−7.98 |
1.42 |
2 |
Pose 260: H – B: Glu 230: O (2.02 Å); pose 260: H – B: Asp 201: O (2.09 Å) |
| Compound 2 |
Pose 217/500 |
−7.21 |
5.20 |
3 |
B: Tyr 159: O – pose 217: O (2.77 Å); pose 217: H – B: Glu 230: O (1.82 Å); pose 217: H – B: Asn 321: O (2.17 Å) |
| Compound 3 |
Pose 420/500 |
−5.36 |
118.09 |
2 |
B: Asp 272: O – pose 420: H (3.83); B: Glu: 230: N – pose 420 (5.23) |
| Compound 4 |
Pose 455/500 |
−5.87 |
49.93 |
1 |
Pose 455: H – B: Asn 321: O (4.19 Å) |
| Small ligand in B chain |
Pose 96/500 |
−8.30 |
0.00083 |
1 |
Small ligand: H – B: Tyr159: O (2.00 Å) |
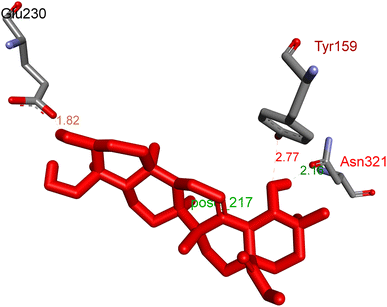 |
| | Fig. 2 The hydrogen bonding between pose 217, ranked pose of compound 2, and target enzyme, 4WCU. This pose formed 3 hydrogen bondings with residual amino acids, Tyr 159, Glu 230, and Asn 321. | |
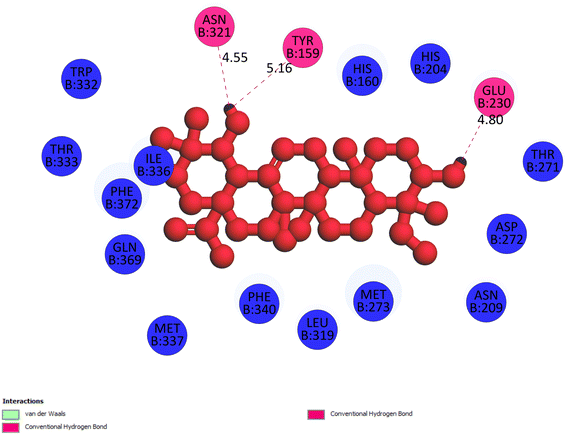 |
| | Fig. 3 The 2D diagram indicated the ligand interactions between pose 217/compound 2 and B chain of 4WCU. Three hydrogen bonding formed, and this pose did not interact well with the 4WCU enzyme. | |
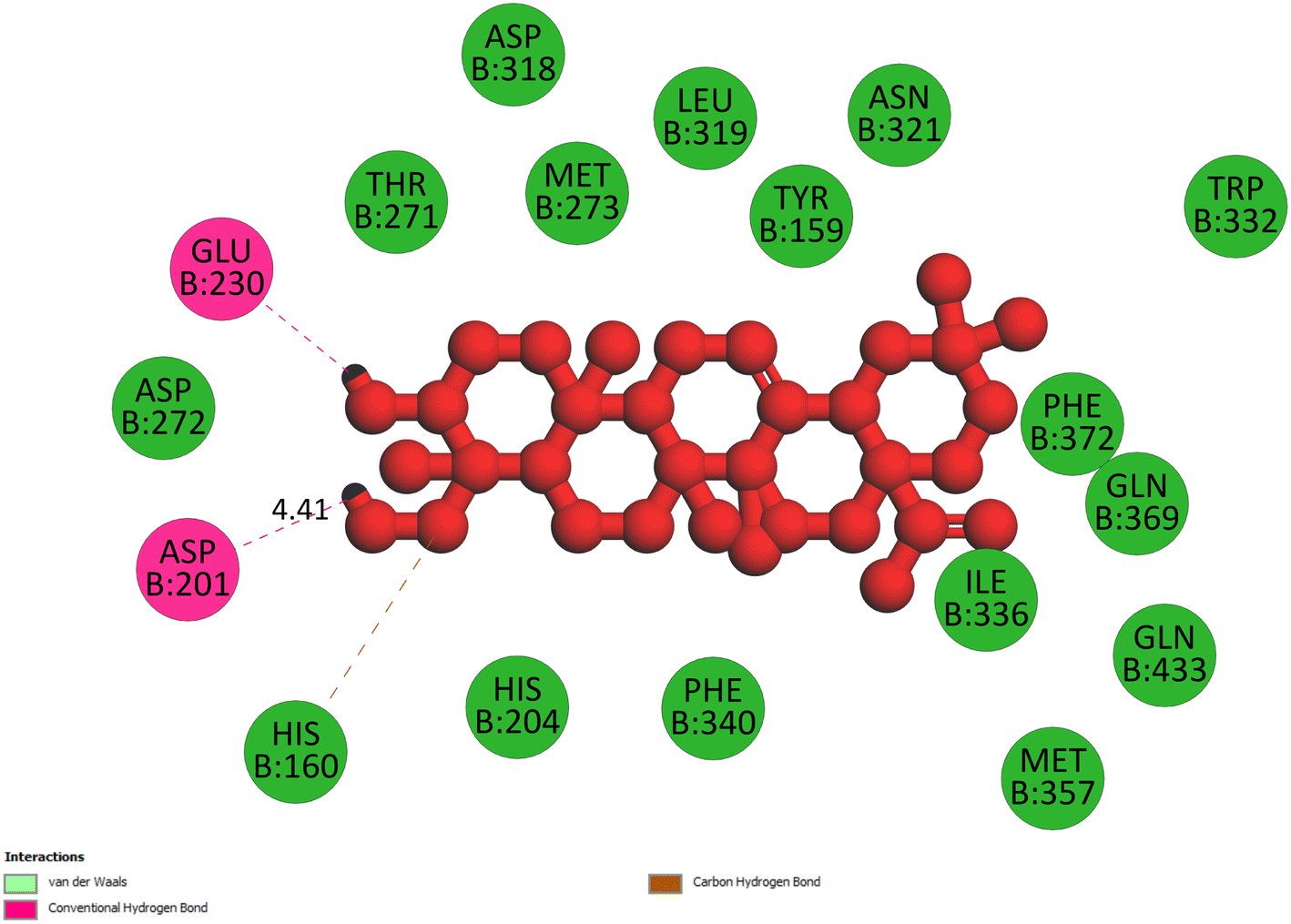
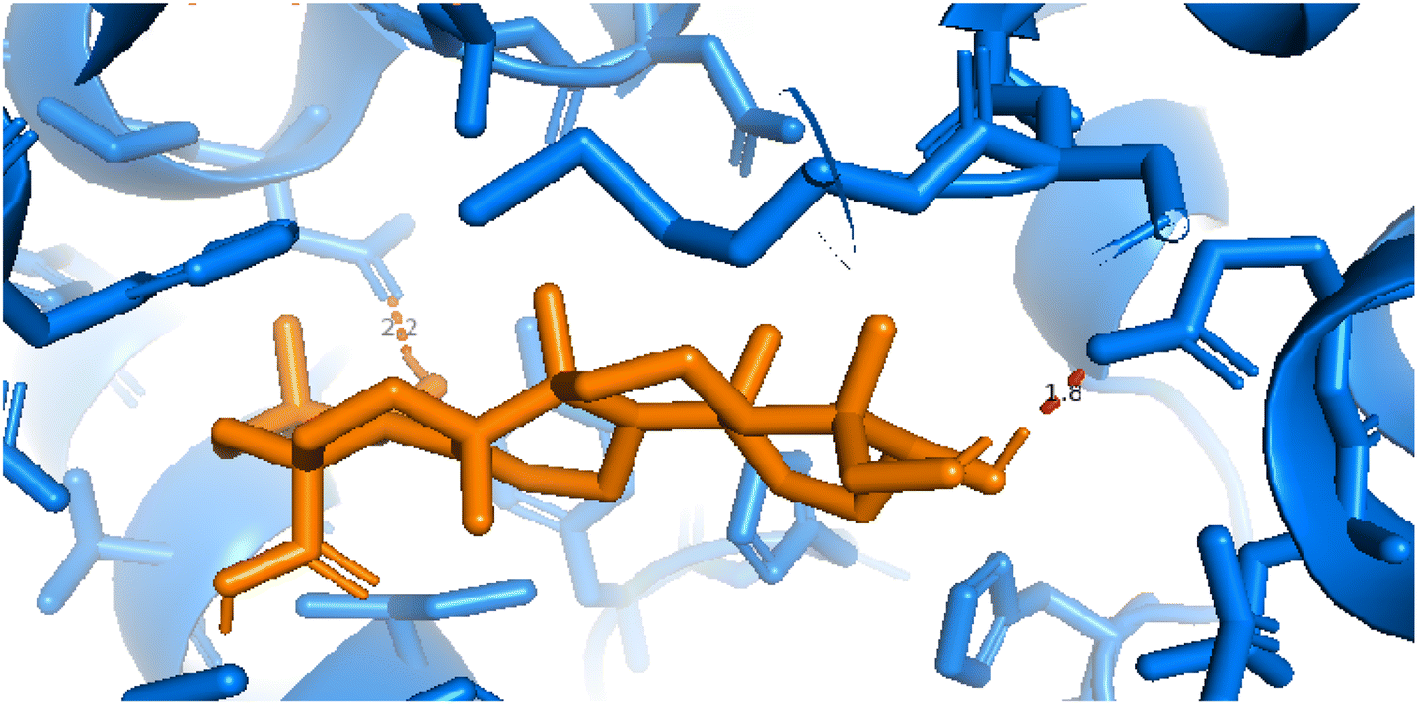
2.3.2 Pose 260. One ranked pose among 500 poses interacted with the range of the active enzyme pocket with the values of affinity energy and inhibition constant of −7.98 kcal mol−1 and 1.42 μM, respectively, as seen in Table 1. Pose 260 linked 2 hydrogen bondings from hydrogen atoms of alcohol hydroxyl on pose to Glu 230 and Asp 201 of the B chain, as shown in Table 1. For the ligand interaction model, the linker of pose 260 formed one carbon–hydrogen interaction – hydrophobic interaction from His 160 to atom carbon on this pose. The functional group of this pose detected 2 hydrogen bondings from Asp 201 and Glu 230 to hydrogen atoms of alcohol hydroxyl atoms on pose 260. The capping group of this pose has no interactions with the active enzyme pocket so the capping group of this pose is not identified. Pose 260 is not an active pose in the ligand interaction model, although the value of the affinity energy of pose 260 with the enzyme is very low (Fig. 4).
 |
| | Fig. 4 The 2D diagram indicated the ligand interactions between pose 260/compound 1 and B chain of 4WCU. Two hydrogen bondings from Glu 230 and Asp 201 to atoms on the pose and one carbon–hydrogen bond from His 160 to this pose. This pose does not have good interactions with the enzyme. | |
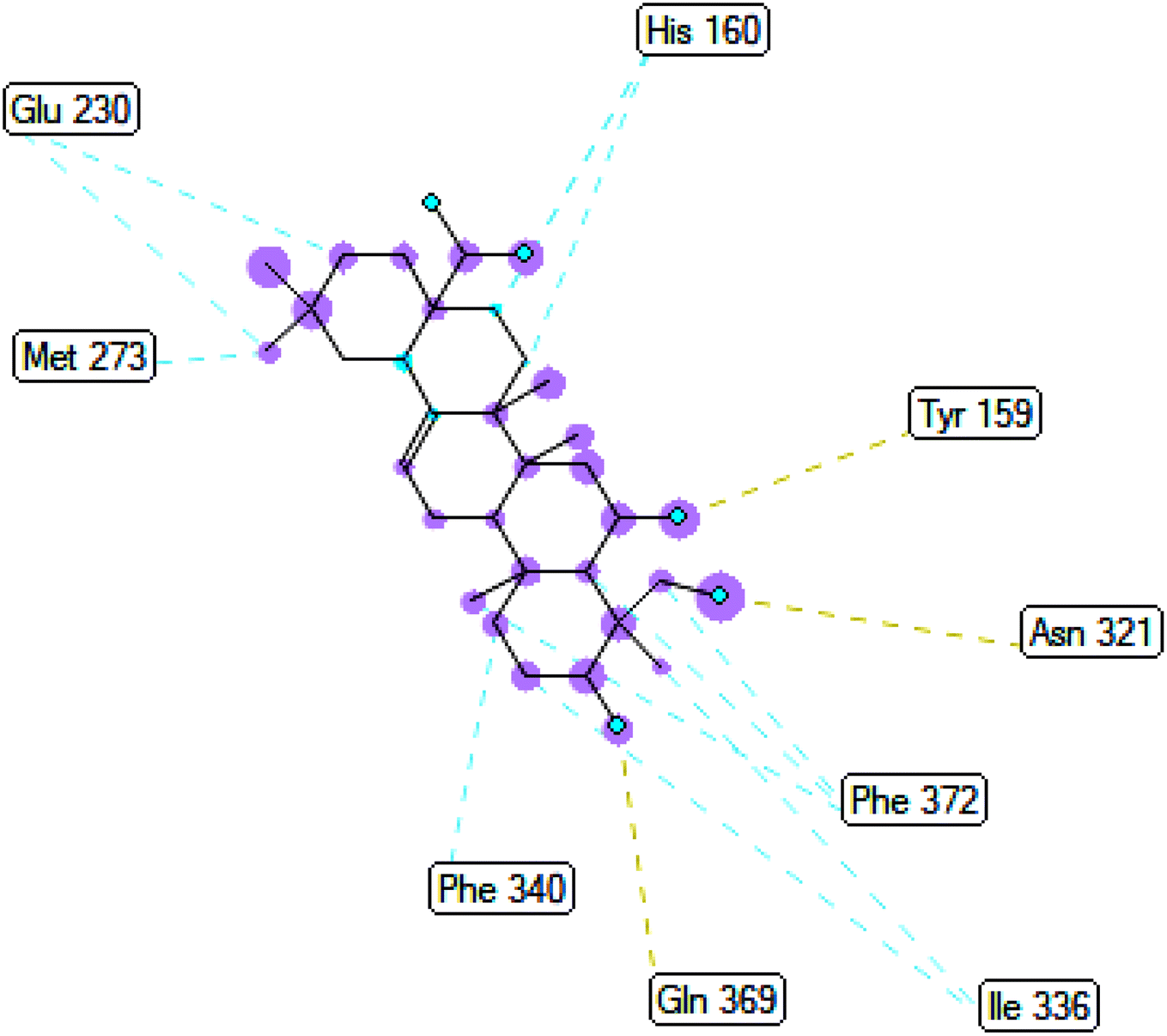
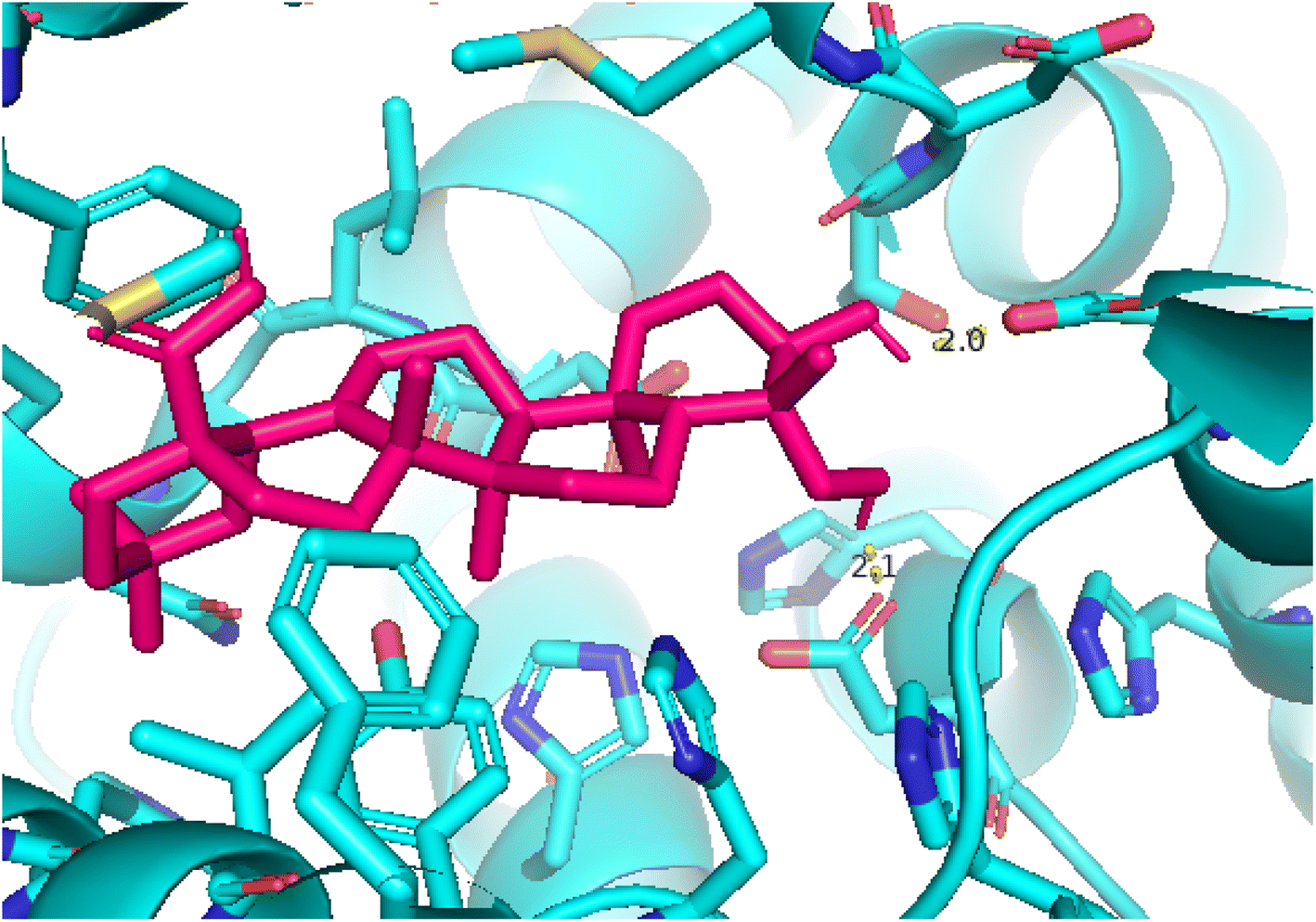
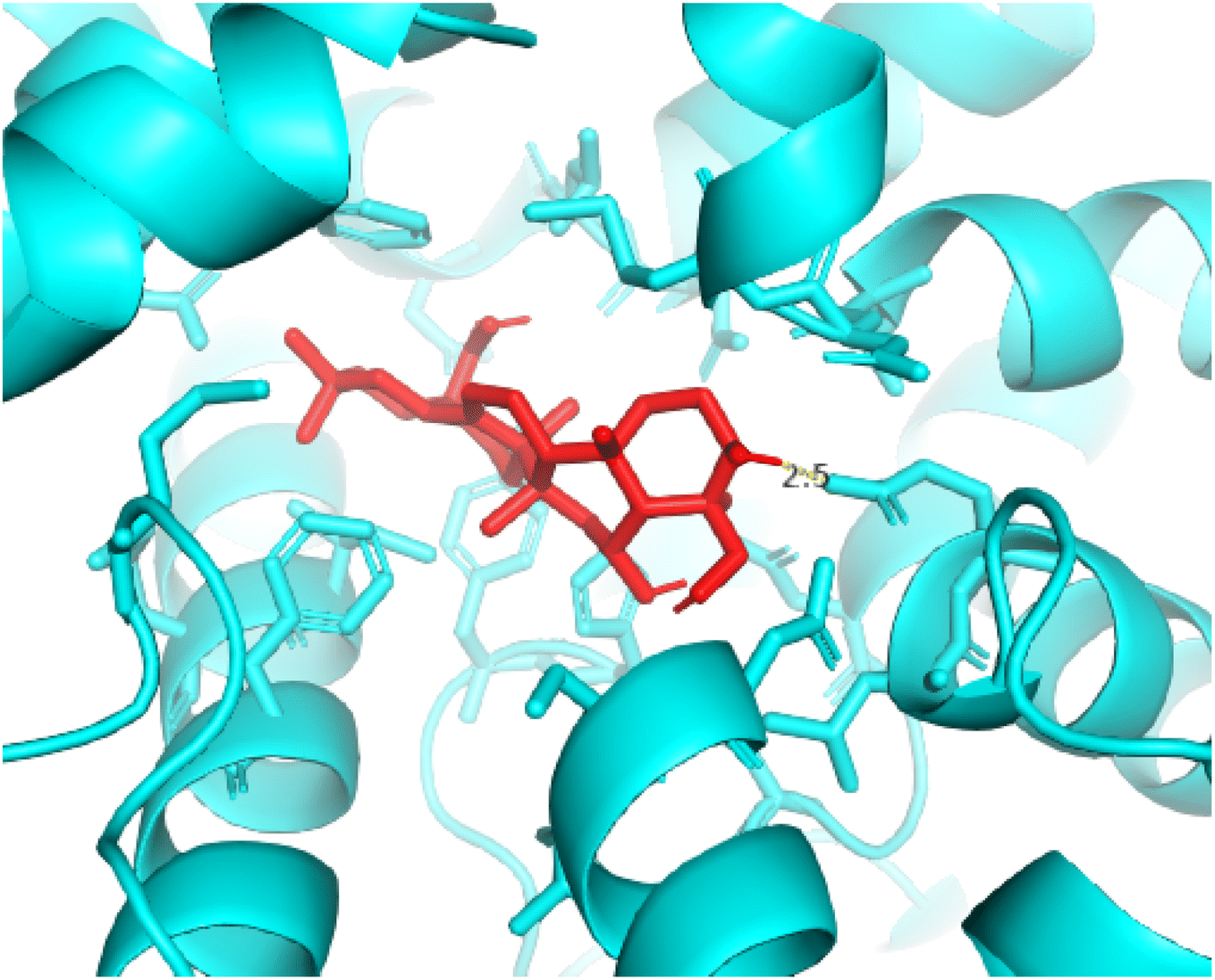
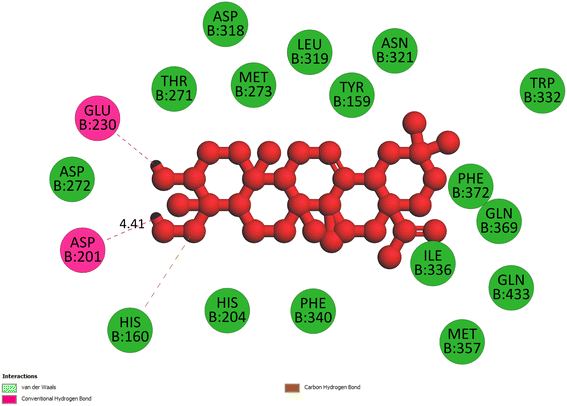
2.3.3 Pose 420. One ranked pose among 500 conformation ligands of compound 3 in molecular docking model docked to the range of 4WCU: PDB active pocket enzyme with the values of affinity energy and inhibition constant of −5.36 kcal mol−1 and 118.09 μM, respectively, as shown in Table 1. As shown in Fig. 5, the most significant ligand interactions indicated in one 2D diagram that included the capping group of poses were identified by carbon hydrogen interaction, hydrophobic interaction, or non-polar interaction from Glu 230 of the B chain to the carbon atom of the cyclohexyl group of this pose, the connecting unit or linker of this pose identified via pi–sigma, hydrophobic interaction from Phe 372 to one methyl group on this pose, and 2 hydrogen bondings, hydrophilic or polar interaction from Asp 272 and Glu 230 to one hydrogen atom of the alcohol hydroxyl of the cyclohexyl ring. They are characteristics, such as B: Asp 272: O – pose 420: H (3.83 Å) and B: Glu: 230: N – pose 420 (5.23 Å). Pose 420 was identified for full ligand interaction because 3 parts of pose 420 have ligand interactions with the active range of the pocket enzyme 4WCU. The second interaction between this pose and the enzyme is indicated on one ligand map, as indicated in the one ligand map in Fig. 6. As seen in Fig. 5, the ligand map indicated the hydrogen bondings, brown color lines from Tyr 159, Asn 321, and Glu 369 to the active center of this pose, the steric interactions, green lines being relative to Tyr 159, His 160, Glu 230, Me 273, Asn 321, Ile 336, Gln 369, and Phe 372. More steric interactions proved that pose 420 interacted strongly with the active range of the whole enzyme conformation processing. The overlap interactions, violet circles on atoms of the pose, and bigger size of violet circles indicate that the overlap interactions between the pose and enzyme are stronger.4
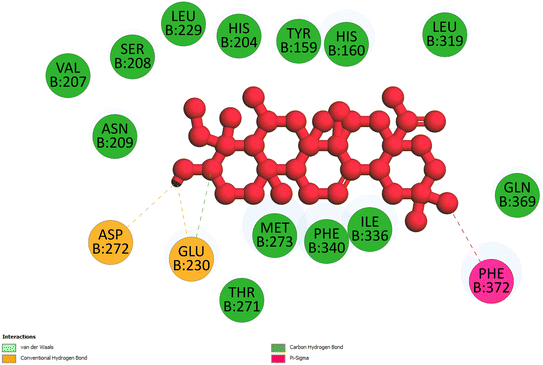 |
| | Fig. 5 The 2D diagram indicated the ligand interactions between pose 420, the best one stable conformation of compound 3 and the B chain of 4WCU. Two hydrogen bondings from Asp 272, Glu 230, one carbon–hydrogen, Thr 271, and one pi–sigma from Phe 372 to atoms on this pose. This pose interacted well with the 4WCU enzyme. | |
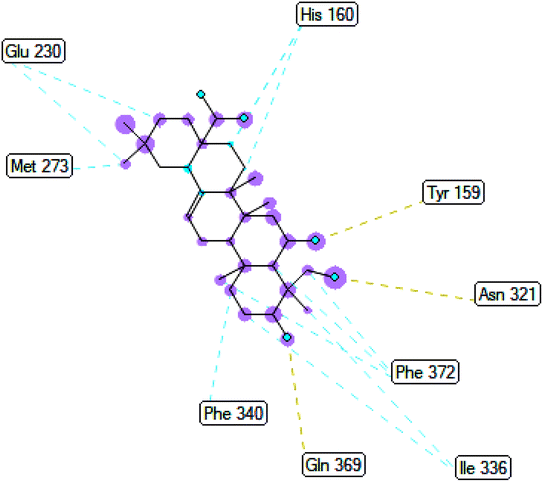 |
| | Fig. 6 The ligand map indicated the secondary interaction between pose 420/compound 3 and the crystal structure of the 4WCU enzyme. There are many interactions as steric interactions and hydrogen bondings made around pose 420. This proved that this pose interacted strongly with the 4WCU enzyme. | |
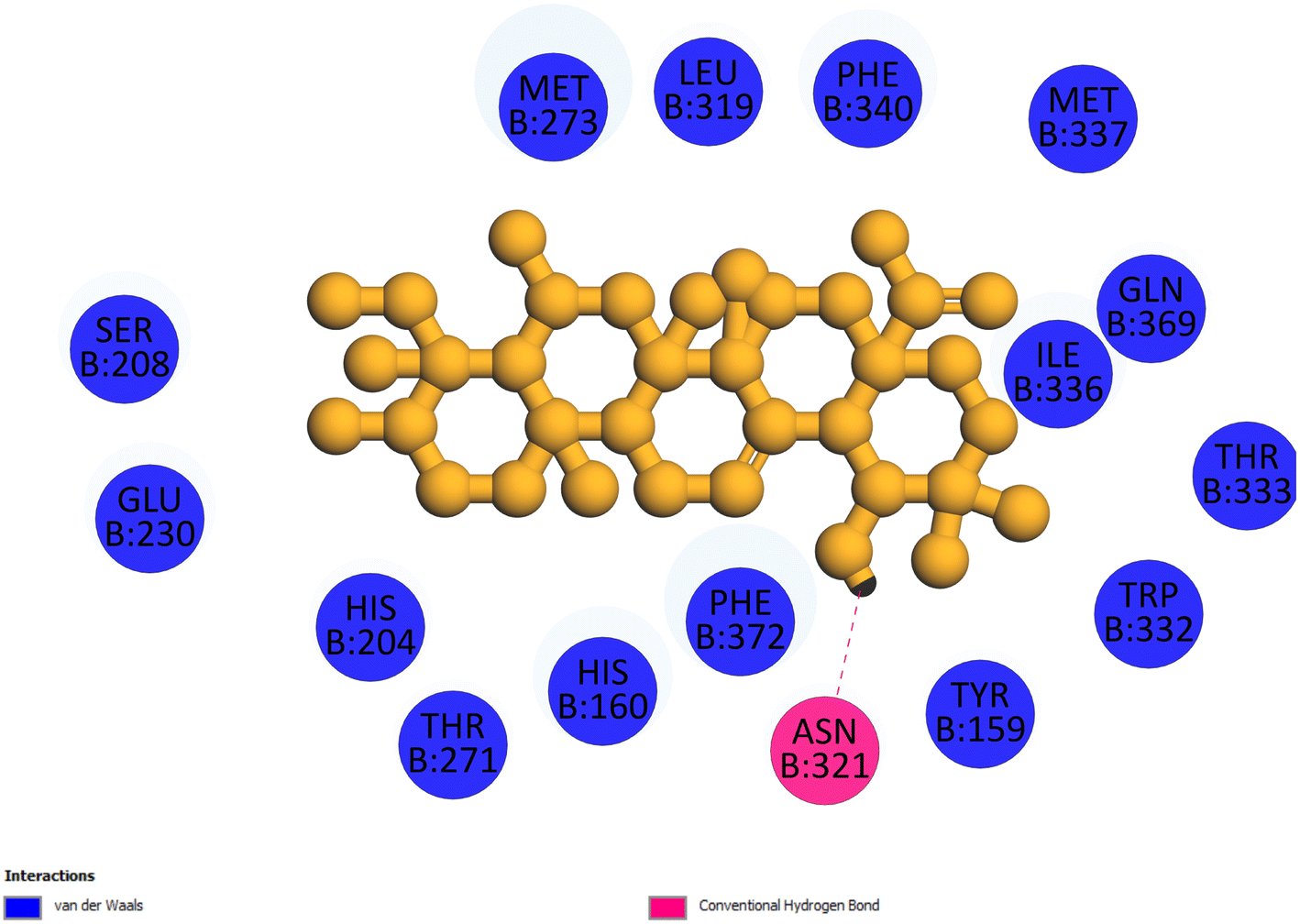
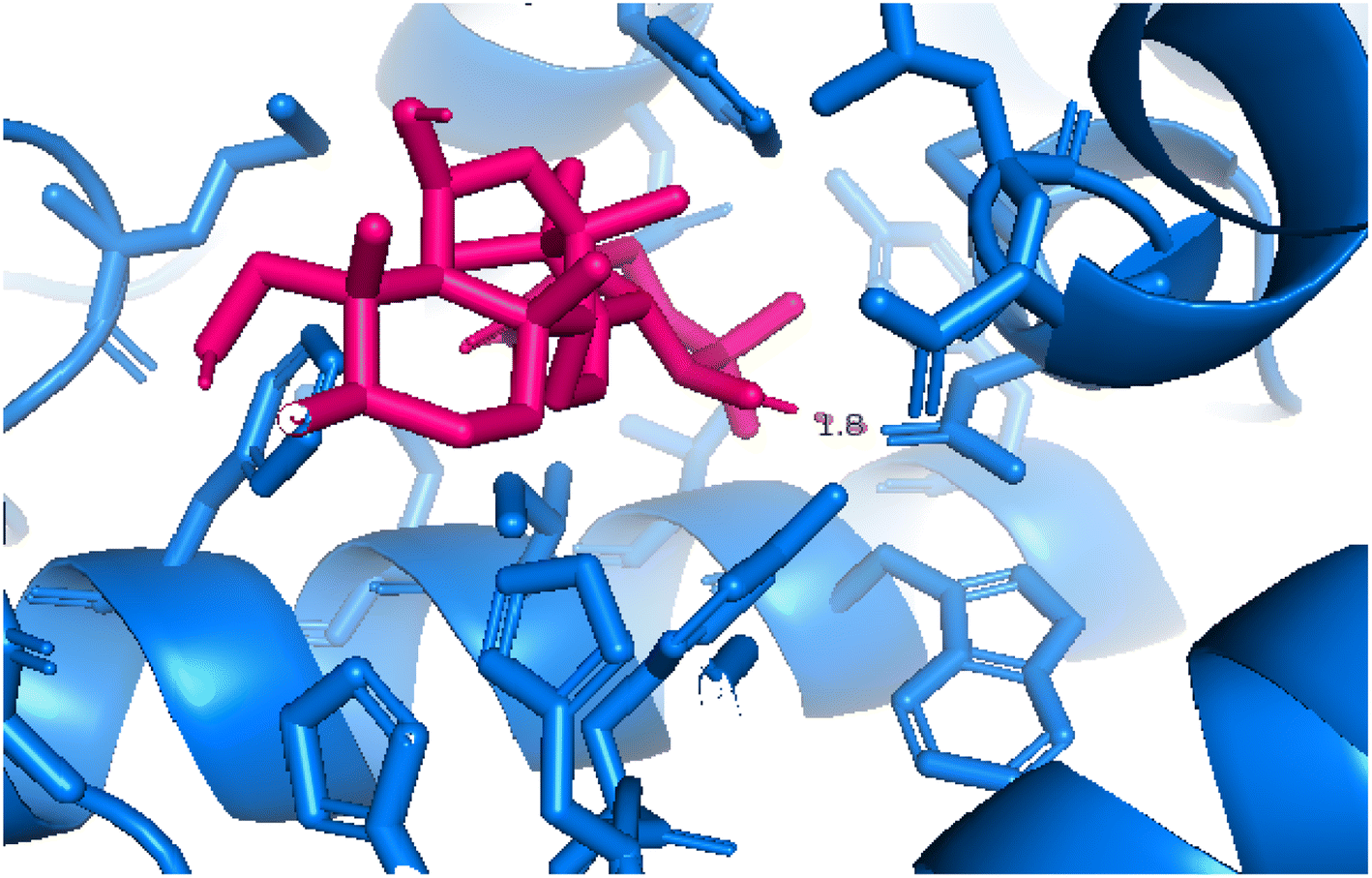
2.3.4 Pose 455. Ranked pose for the best conformation ligand of compound anchored to enzyme 4WCU with the values of affinity and inhibition constant of −5.87 kcal mol−1 and 49.93 μM, respectively, as shown in Table 1. This pose linked one hydrogen bonding from Asn 321 to a hydrogen atom on this pose at a distance of 4.19 Å, pose 455: H – B: Asn 321: O (4.19 Å), as shown in Table 1. This pose detected the functional group (ZBG) via one hydrogen bonding from Asn 321 to the hydrogen atom of alcohol hydroxyl of the cyclohexyl group on this pose, as indicated in Fig. 7. This pose had not identified the capping group and linker group, so it does not interact well with the 4WCU enzyme.
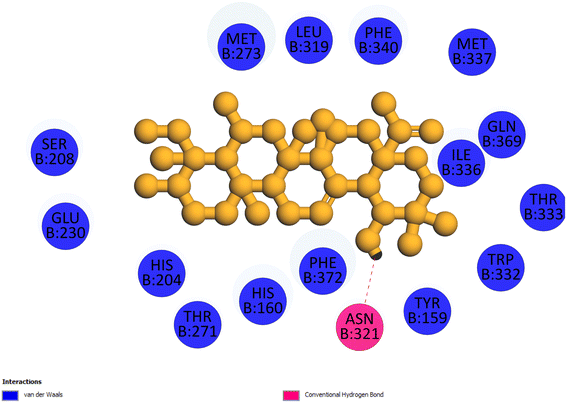 |
| | Fig. 7 The 2D diagram indicated the ligand interactions between pose 455, the best one stable conformation of compound 4 and the B chain of 4WCU. There is only one ligand interaction, hydrogen bonding and this pose did not interact well with the 4WCU enzyme. | |
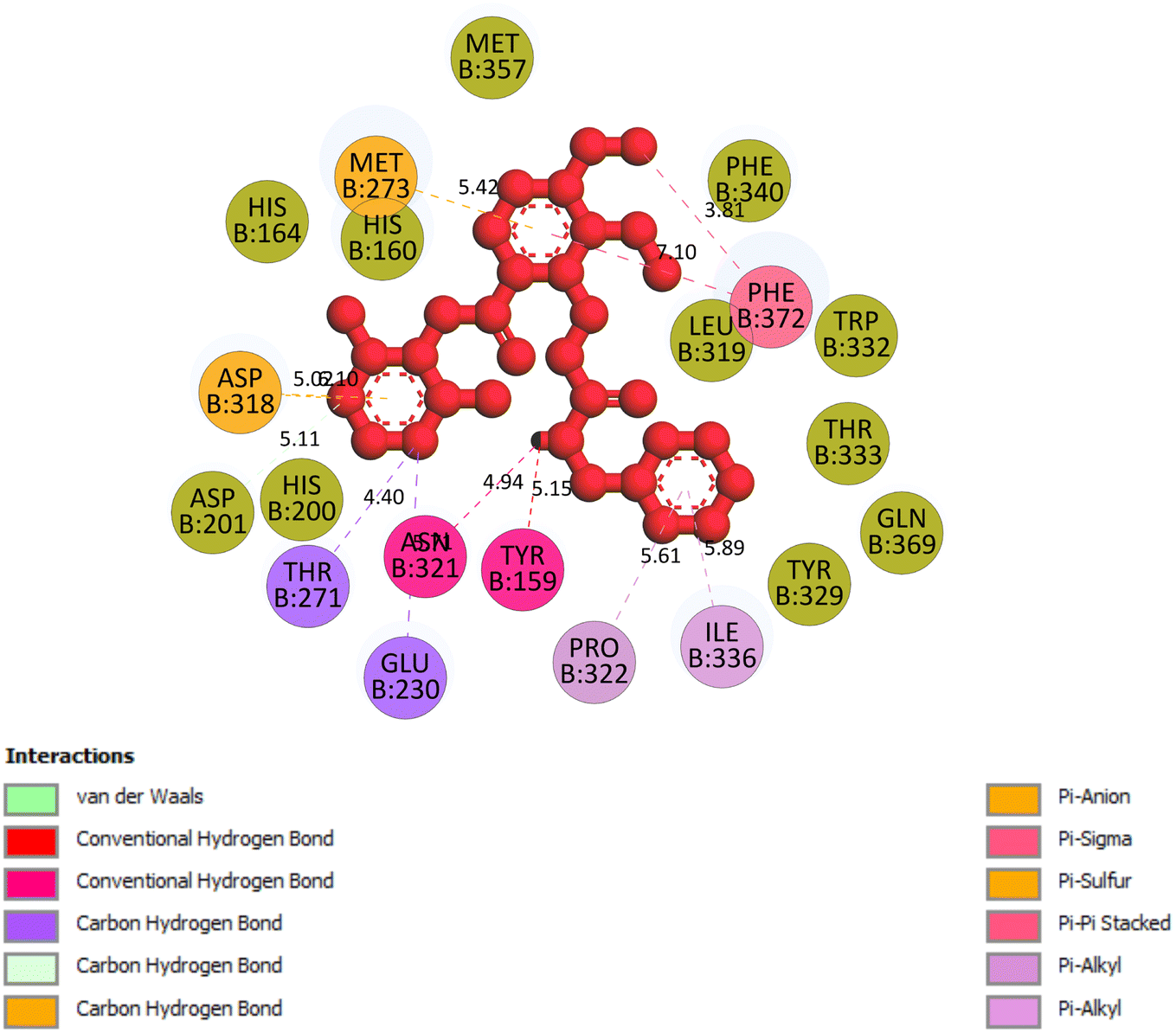
2.3.5 Pose 96. One ranked pose of a small ligand that is available in the crystal structure of 4WCU was redocked to assess the validation of the docking model. This pose docked to the receptor of the 4WCU enzyme with the values of affinity and inhibition constant of −8.30 kcal mol−1 and 0.83 nM, respectively, as seen in Table 1. At the thermodynamic site, this pose docked at the best crystal structure of 4WCU enzyme among those poses. As shown in Fig. 8, one 2D diagram indicated the most ligand interactions that identified parts of this pose at interaction sites. The capping unit was identified as an aromatic ring of pose via pi–alkyl (hydrophobic interaction) from Pro 322 and Ile 336 to the benzyl group, one pi–anion (electrostatic interaction) from Met 373 to phenyl group, one pi–pi stacked (hydrophobic interaction) from Phe 372 to a phenyl group, one pi–sulfur from Asp 318 to nitrogen heterocyclic, and 5 carbon–hydrogen interactions (hydrophobic interaction) from Asp 318, His 200, Thr 271, and Asn 321 to nitrogen heterocyclic. The linker of the pose was detected via one pi–sigma interaction (hydrophobic interaction) from Phe 372 to methyl of the methoxy group of the phenyl group. The functional group of poses or ZBG was identified via hydrogen bondings from Tyr 159 and Asn 321 to the hydrogen atom of the amide group on the pose. Pose 96 interacted well with the active enzyme pocket of 4WCU because 3 parts of pose 96 have full ligand interactions and pose 96 was considered as good enzyme inhibition via the ligand interaction model for both thermodynamic and interaction sites. The ability of the poses was evaluated as pose 96 (small ligand) > pose 420 (compound 3) > pose 217 (compound 2) > pose 260 (compound 1) > pose 455 (compound 4) (Fig. 9 and Table 2).
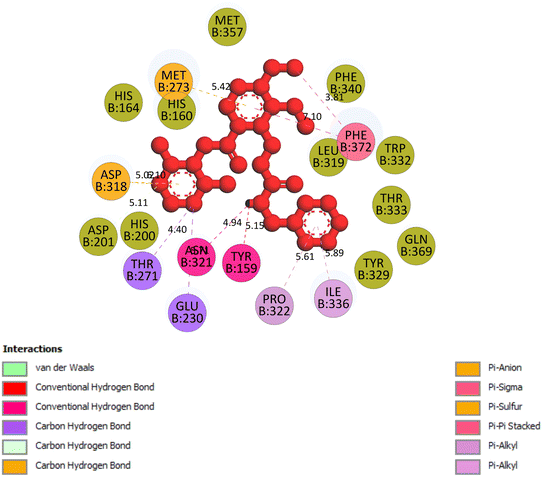 |
| | Fig. 8 The 2D diagram indicated the ligand interactions between the best stable conformation of small ligands, pose 96, and the B chain of 4WCU. This pose interacts well with the 4WCU enzyme. One hydrogen bonding from Tyr 159, a conventional hydrogen bond from Asn 321, two carbon–hydrogen (violet) from Thr 271, Glu 230, two pi–alkyl from Pro 322, Ile 336 (light violet), one pi–cation from Asp 318, one pi–anion from Met 273, and two pi–pi stacked from Phe 372 to atoms on the ligand. This pose interacted well with the 4WCU enzyme. | |
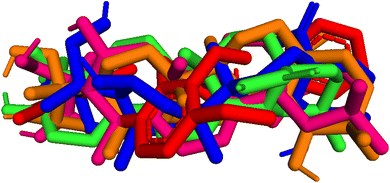 |
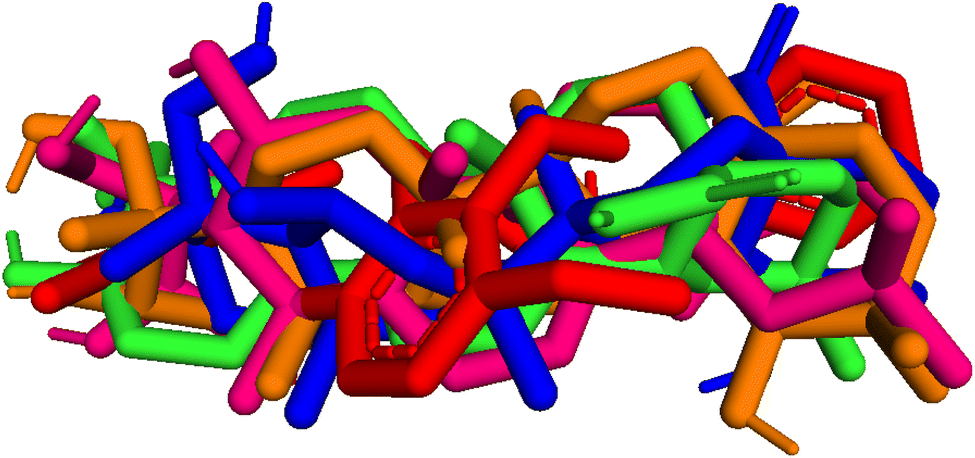
| | Fig. 9 Pose 455-blue color, pose 217-orange, pose 260-green color, and pose 420-magenta color aligned to pose 96-red color. Pose 217, pose 260, pose 420, and pose 455 aligned to pose 96, one the best docking that is available in enzyme and called reference pose. The values of RMSD between pair poses were calculated by PYmol software and presented in Table 2. | |
Table 2 The values of RMSDs between poses and reference pose, pose 96
| RMSD, Å |
Pose 96, red |
Pose 175, blue |
Pose 217, orange |
Pose 260, green |
Pose 420, magenta |
| Pose 96, reference pose |
0 |
4.009 |
3.413 |
3.392 |
3.600 |
2.4. Analysis of MD simulations
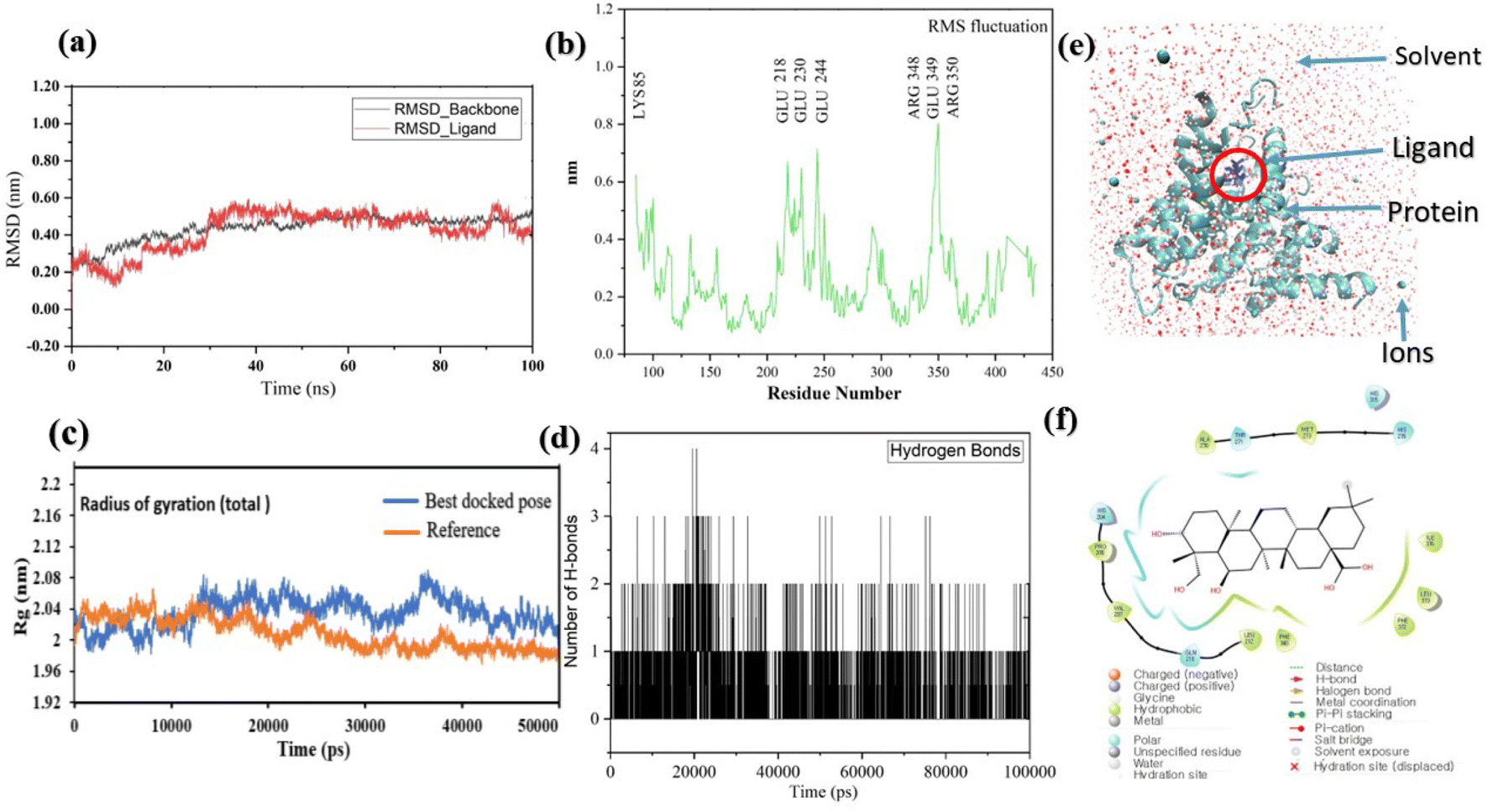
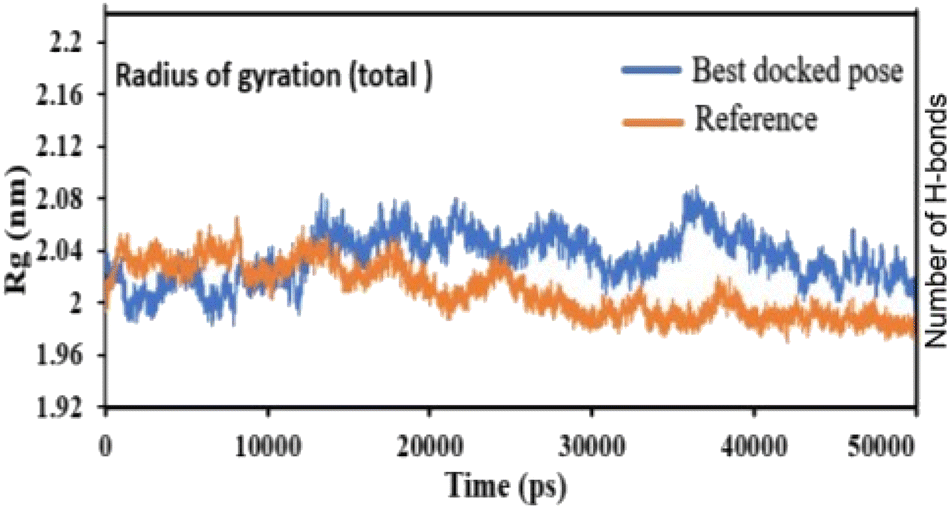
Pose 420, ranked the pose of compound 3 among the poses of compounds 1–4 and was selected to analyze the strength in the real environment with residual enzyme 4WCU. The ligand's ability to bind to the protein over time at the atomic level was tested using MD simulation. The radius of gyration, RMSD, RMSF, hydrogen bonding, and other factors, among others, all provide valuable information about the bond pattern in the protein–ligand complex. As shown in Fig. 10, a 100 ns MD simulation study was performed to evaluate the conformational stability of the ligand with the 3D structure of the receptor (PDB: 4WCU) to assess the overall stability and binding efficiency of the protein–ligand complex. To determine the conformational stability of a system, RMSD parametric analysis provides detailed structural information. Based on the simulation results obtained after 30 ns, the protein–ligand complex appears to be highly equilibrated and stable (Fig. 10a). As shown by the close interaction of the ligand complex, the RMSD value for the ligand to dock in the active sites of protein decreased to 0.11 nm during the first 9 ns (time), and then increased to 0.59 nm at 36 ns (time), to respect its original position, but it is within its limit of the RMSD of the trajectory (Fig. 10a). Based on the RMSD of the receptor analysis, it was determined that the protein remained stable throughout the MD simulations of the protein–ligand complex. As shown in Fig. 10b, root means square fluctuation (RMSF) measures the flexibility of the protein residues in the presence of a ligand. The fluctuation pattern of the protein–ligand complex is nearly identical, confirming the restricted movements over 100 ns of the simulation. Although some residues were not directly involved, such as Lys 85, Glu 218, Glu 244, Arg 348, Glu 349, and Arg 350, which had some mobility with a 0.6 to 0.8 nm fluctuation from their initial position (Fig. 10b, e and f), indicating non-significance, another Glu 230 was found to participate in carbon–hydrogen bond formation as well as van der Waals forces, leading to Glu 230 peak (Fig. 10b) elongation due to the interaction of the ligand with the protein during MD simulation process. These residue fluctuations were small in comparison to other residue fluctuations, as shown in Fig. 10b. The small fluctuations show that the ligand and protein are developing stable interactions. In addition, the radius of gyration (Rg) of a protein with ligand in active sites was estimated during a 100 ns MD simulation, as shown in Fig. S21.† Rg values of protein–ligand complexes are shown in nanometers in Fig. S21† due to the dynamic behavior of the protein–ligand complex with ions and solvent molecules during the MD simulation process. In the MD simulation, the Rg values for the best-docked model started at 2.02 nm, and the structure gradually grew and shrank within the limit. Rg remained almost the same throughout the process. Although the Rg value increased from 2.02 to 2.05 nm, then decreased to 1.98 nm during the 20–47 ns MD simulation process, and then increased again to return the system to its near original position, demonstrating that the receptor-ligand was stable and tightly packed. For comparison, an MD simulation of up to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ps was also performed with the best-docked model and the protein-bound reference molecule. The radius of gyration (Rg) in the presence of the best-docked pose and the crystallized protein molecule [N-benzyl-2-{6-[(3,5-dichloropyridin-4-yl)acetyl]-2,3-dimethoxyphenoxy}acetamide] as a reference for comparison, attached to the active sites (PDB: 4WCU), were also used to determine the level of protein compactness using MD simulation, as shown in Fig. 10c. The MD simulation results revealed that the best docking pose with protein (PDB: 4WCU) was a static representation with only minor fluctuations. Beginning at 2.02 nm (Rg), it decreased to 1.98 nm until 9000 ps, showing slightly more compactness. Following that, it increased slightly to 2.08 ns until 36000 ps and then was maintained at a constant Rg value of 2.02 nm until the MD simulation was complete. On the other hand, N-benzyl-2-{6-[(3,5-dichloropyridin-4-yl)acetyl]-2,3-dimethoxyphenoxy}acetamide, taken as a reference, exhibited a great deal of static depiction. Firstly, Rg values fluctuated slightly between 2.01 and 2.05 nm till the MD simulation time of 0 to 25000 ps, suggesting slightly reduced protein compactness with the reference molecule. Following that, the Rg value of the system remained constant at 1.98 nm throughout the simulation time of 25000–50000 ps, as depicted in Fig. 10c. As seen from the difference between the starting and final Rg values (2.01 − 1.98 = 0.02 nm), the protein bound to the reference molecule was determined to be more compact. Based on the radius of gyration (Rg) values derived from MD simulations for the compactness of the protein, the best docking pose corresponded most closely to the reference pattern. After binding with the receptor, the molecule was efficiently accepted into the active cavity of the protein, resulting in improved compactness, as was the case with the reference molecule (Fig. 10c, or more specifically, Fig. 16). The number of hydrogen bonds observed between a protein and ligand complex during the MD simulation was counted using the Gromacs package. Fig. 10d depicts the number of H-bonds formed between protein (4WCU: PDB) and active compound using 100 ns MD simulation trajectories. The majority of the hydrogen bonds in protein–ligand complexes in the MD simulation process are 1 > 2 > 3, with the fewest being 4 hydrogen bonds at roughly 20 ns. This equal number of hydrogen bonds produced during the process without abrupt changes in the hydrogen bonds (most of which are Hb; 1 to 3) that aids the stability of the ligand with no significant change in the RMSD value during the simulation period. By maintaining a steady state throughout the simulation, these results contributed to the stability of the protein–ligand complex (Fig. 11–16).
000 ps was also performed with the best-docked model and the protein-bound reference molecule. The radius of gyration (Rg) in the presence of the best-docked pose and the crystallized protein molecule [N-benzyl-2-{6-[(3,5-dichloropyridin-4-yl)acetyl]-2,3-dimethoxyphenoxy}acetamide] as a reference for comparison, attached to the active sites (PDB: 4WCU), were also used to determine the level of protein compactness using MD simulation, as shown in Fig. 10c. The MD simulation results revealed that the best docking pose with protein (PDB: 4WCU) was a static representation with only minor fluctuations. Beginning at 2.02 nm (Rg), it decreased to 1.98 nm until 9000 ps, showing slightly more compactness. Following that, it increased slightly to 2.08 ns until 36000 ps and then was maintained at a constant Rg value of 2.02 nm until the MD simulation was complete. On the other hand, N-benzyl-2-{6-[(3,5-dichloropyridin-4-yl)acetyl]-2,3-dimethoxyphenoxy}acetamide, taken as a reference, exhibited a great deal of static depiction. Firstly, Rg values fluctuated slightly between 2.01 and 2.05 nm till the MD simulation time of 0 to 25000 ps, suggesting slightly reduced protein compactness with the reference molecule. Following that, the Rg value of the system remained constant at 1.98 nm throughout the simulation time of 25000–50000 ps, as depicted in Fig. 10c. As seen from the difference between the starting and final Rg values (2.01 − 1.98 = 0.02 nm), the protein bound to the reference molecule was determined to be more compact. Based on the radius of gyration (Rg) values derived from MD simulations for the compactness of the protein, the best docking pose corresponded most closely to the reference pattern. After binding with the receptor, the molecule was efficiently accepted into the active cavity of the protein, resulting in improved compactness, as was the case with the reference molecule (Fig. 10c, or more specifically, Fig. 16). The number of hydrogen bonds observed between a protein and ligand complex during the MD simulation was counted using the Gromacs package. Fig. 10d depicts the number of H-bonds formed between protein (4WCU: PDB) and active compound using 100 ns MD simulation trajectories. The majority of the hydrogen bonds in protein–ligand complexes in the MD simulation process are 1 > 2 > 3, with the fewest being 4 hydrogen bonds at roughly 20 ns. This equal number of hydrogen bonds produced during the process without abrupt changes in the hydrogen bonds (most of which are Hb; 1 to 3) that aids the stability of the ligand with no significant change in the RMSD value during the simulation period. By maintaining a steady state throughout the simulation, these results contributed to the stability of the protein–ligand complex (Fig. 11–16).
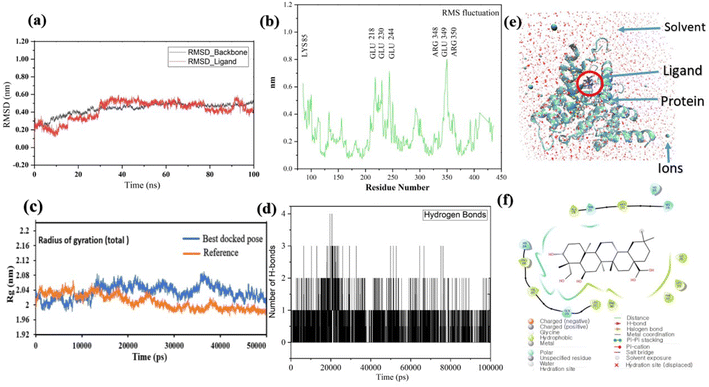 |
| | Fig. 10 (a) The graph of RMSD studies for the docked ligand to protein (PDB: 4WCU); (b) the root mean square fluctuation (RMSF); (c) the radius of gyration (Rg) of a protein in the presence of a ligand and compared with the presence of a reference up to 50000 ps; (d) the number of H bonds formed between the ligand and the crystal structure of protein determined using 100 ns simulation trajectories; (e) the protein–ligand complex in three-point transferable intermolecular potential solvent box and (f) some amino acids participate in the formation of non-bonding interactions. | |
 |
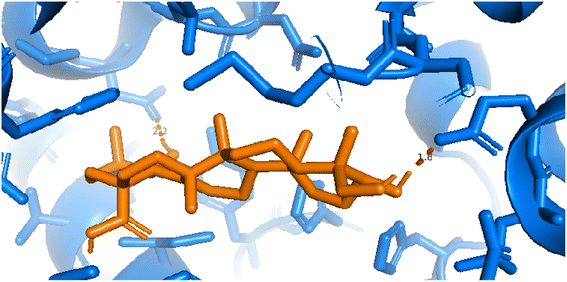
| | Fig. 11 Pose 260, the best pose docking was interacted with the active center of enzyme 4WCU as shown by PYmol. | |
 |
| | Fig. 12 Pose 217 was interacted with the active center of enzyme 4WCU, which is shown by PYmol software. | |
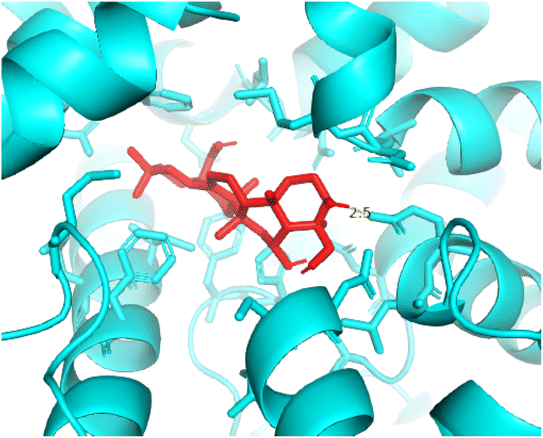 |
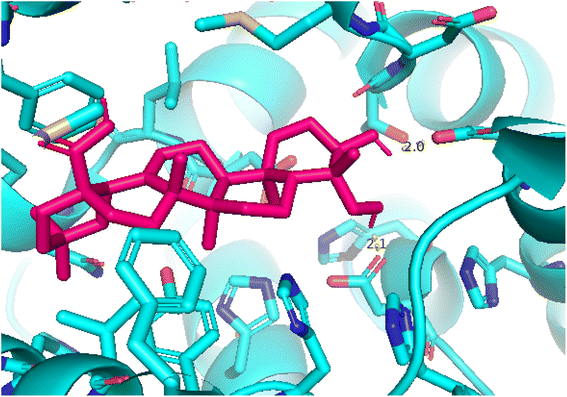
| | Fig. 13 Pose 420, the best pose docking of compound 3 bonds to the active site on 4WCU as presented by PYmol software. | |
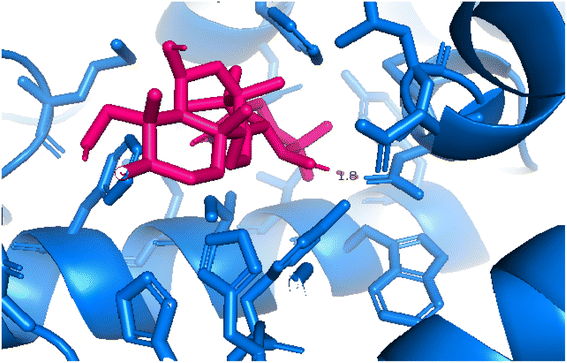 |
| | Fig. 14 Pose 455, the best pose docking of compound 4 interacted with the active site of 4WCU and was exposed by PyMol software. | |
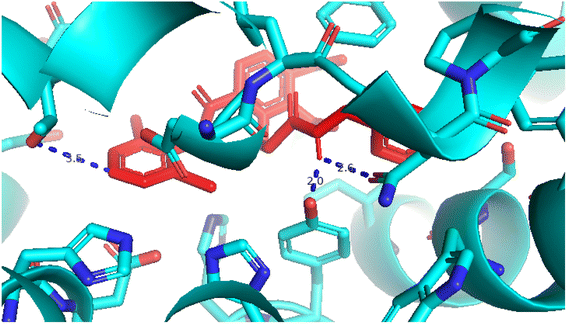 |
| | Fig. 15 Pose 96, one small ligand was available in 4WCU that was shown by PYmol software. | |
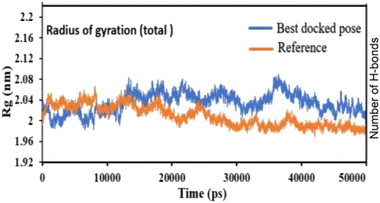 |
| | Fig. 16 The radius of gyration of protein-reference (reference pose), and protein-best pose (pose 420) complexes with the best-docked position as calculated using 50 ns MD simulations. | |
3. Experimental
3.1. General experimental procedures
NMR spectra (1D and 2D) were recorded on a Bruker Avance at 500 MHz for 1H NMR and 125 MHz for 13C NMR. HRESIMS were recorded on a Bruker MicrOTOF-Q II mass spectrometer. Thin layer chromatography was carried out on precoated Kieselgel 60 F254 or silica gel 60 RP-18 F254S (Merck). Spots were visualized by spraying with 20% H2SO4 solution, followed by heating.
3.2. Plant material
Aerial parts of Mussaenda recurvata were collected in the Hon Ba Nature Reserve (12°07′34.82′′N, 109°01′22.56′′E), Khanh Hoa province, Vietnam (October to November 2019). The scientific name of the plant was authenticated by the botanist Van-Son Dang, Institute of Tropical Biology, Vietnam Academy of Science and Technology. A voucher specimen (VNM_Dang356) was deposited in the VNM Herbarium, Institute of Tropical Biology, Vietnam Academy of Science and Technology.
3.3. Extraction and isolation
The dried Mussaenda recurvata aerial parts (6.0 kg) were powdered to produce a powder with 30 mesh (0.59 mm). This powder was exhaustively extracted with methanol at room temperature to produce the crude extract (420 g) after evaporation of the solvent. This extract was suspended in H2O and then successively partitioned with n-hexane, CH2Cl2, and EtOAc to yield n-hexane (95.0 g), CH2Cl2 (130.0 g), and EtOAc extracts (80.0 g), respectively. The CH2Cl2 (130.0 g) extract was subjected to silica gel column chromatography (CC) and was eluted with the gradient system of n-hexane–EtOAc (10:1–0:10, v/v) to produce ten fractions (C1–C10). Fraction C4 (28 g) was further chromatographed on silica gel CC and eluted with the solvent system of n-hexane–EtOAc (20:1, 10:1, 5:1, 1:1, v/v) to afford four subfractions (C4.1–C4.4). The subfraction C4.2 (3.1 g) was separated by silica gel CC using the mobile phase CHCl3–EtOAc (30:1, 20:1, 10:1, 5:1, 1:1 v/v) to yield three subfractions (C4.2.1–C4.2.13). Subfraction C4.2.2 (120 mg) was repeatedly chromatographed on a silica gel column using CHCl3–EtOAc (10:1 v/v) as an eluent to afford compounds 1 (7.2 mg) and 2 (8.1 mg). Fraction C5 (22 g) was applied to silica gel CC, eluted with n-hexane–EtOAc (15:1–0:1 v/v) to give five fractions (C5.1–C5.5). Fraction C5.3 (4.5 g) was subjected to silica gel CC using the solvent system of CHCl3–EtOAc (20:1, 10:1, 5:1, 1:1, 0:1 v/v) as a mobile phase to give five subfractions (C5.3.1–C5.3.5). Subfraction C5.3.2 (183 mg) was rechromatographed by silica gel CC eluting with CH2Cl2–EtOAc (5:1 v/v) to give two subfractions (S1 and S2). Subfraction S1 (49 mg) was further purified by C-18 reverse-phase silica gel CC eluted with MeOH–H2O (1:3, v/v) to provide compounds 3 (15 mg) and 4 (20 mg).
3.3.1 Recurvatane A (1). White amorphous powder; [α]25D +158 (c 0.1, MeOH). HRESIMS m/z [M − H]− 471.3488 (calcd for C30H47O4−, 471.3474); 1H-NMR (500 MHz, DMSO-d6) and 13C-NMR (125 MHz, DMSO-d6) See Table S1.†
3.3.2 Recurvatane B (2). White amorphous powder; [α]25D +311 (c 0.1, MeOH). HRESIMS m/z [M − H]− 487.3441 (calcd for C30H47O4−, 487.3423); 1H-NMR (500 MHz, DMSO-d6) and 13C-NMR (125 MHz, DMSO-d6) See Tables S1 and S2.†
3.4. Measurement of the NO production assay
Determination of NO production and cell viability assay were performed using the same method described in our previous report.30 Briefly, RAW 264.7 cells were cultured in the Dulbecco's Modified Eagle's Medium (DMEM) supplemented with L-glutamine (2 mM), HEPES (10 mM), sodium pyruvate (1 mM), and 10% fetal bovine serum (FBS). The cells were seeded in 96-well culture plates with 2 × 105 cells per well and incubated for 3 days at 37 °C in a humidified atmosphere containing 5% CO2. The amount of NO in the cultured medium was measured using the Griess reagent. The standard curve was created using known concentrations of sodium nitrite, and the absorbance was measured at 540 nm. L-NMMA was used as a positive control.
3.5. In silico molecular docking model
In silico molecular docking model of compounds 1–4 was calculated based on AutoDockTools-1.5.6rc3 package and built model as Discovery Studio Client 2021. The calculation docking model was conducted briefly in Scheme S1.† The receptor from PDB was used 4WCU.7 The grid parameters in dock.gpf file set up by one active center on enzyme 4WCU with coordinators of (29.286–50.843–26.674), spacing of 0.5 Å, and the number of elements on x, y, and z-axis of 50, 50, and 50, respectively. The docking parameters were determined in the output and input of genetic and algorithm and Lamarckian GA, respectively. The number of the installed models was 500 models. In the processing docking, enzyme 4WCU included 4 chains A, B, C, and D. The docking of compound/ligand to enzyme proved that only the B chain interacted with ligand/compound, so the B chain is used to calculate the molecular docking model. The validation of the model was based on the values of RMSD bet ranked pose and reference pose, one small ligand that is available in the target enzyme, 4WCU.39,40
3.6. Molecular dynamics simulation
A top-docked pose with high binding energy was used in an MD simulation employing the GROMACS software program41 with the GROMOS9643a1 force field to ensure conformational stability. Briefly, the MD simulation was run for 100 ns, beginning with the predicted docking pose of 4WCU and the compound. In the Periodic Boundary Conditions (PBC), the complex was solvated with TIP3 water molecules with a margin of 15 Å. To balance the system's overall charge, Na and Cl were added at constant temperature (300 K) and pressure (1.0 bar). The coordinates of the molecules were provided to the PRODRG2/SwissParam server, which changed them into topologies appropriate for GROMACS. For energy minimization, the steepest descent approach of the protein–ligand complex was applied. The compound and receptor were then subjected to 100 ns MD simulation. The crystal structure of PDB: 4WCU with the ligand complex was in the periodic boundary condition, and each simulation used 5000 frames. Hydrogen bond counting, the radius of gyration (Rg), root mean square deviation (RMSD), and root mean square fluctuation (RMSF), were analyzed using Xmgrace software (https://plasma-gate.weizmann.ac.il/Grace) and other tools such as OriginLab and VMD software.42 In addition to the above, the compactness of the protein with the best docking model using the value of the radius of gyration (Rg) was compared with that of a reference molecule co-crystallized with a receptor (PDB: 4WCU) up to 50000 ps of MD simulation.
4. Conclusions
From the most bioactive fraction of Mussaenda recurvata, four compounds 1–4 were isolated and structurally elucidated. Recurvatanes A and B (1 and 2) were determined to be new compounds. Compounds 2 and 3 showed moderate anti-inflammatory activity with IC50 values of 55.63 ± 2.52 and 60.08 ± 3.17 μM, respectively. The molecular docking model indicated that compound 3 is the best candidate to interact well with the crystal structure of enzyme 4WCU: PDB. Molecular dynamics simulations for 100 ns exhibit non-bonding interactions with the protein and remained stable inside the active site.
Conflicts of interest
The authors have no potential conflicts of interest to report.
Acknowledgements
This research is funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.01-2018.353.
References
- D. W. Ingersoll, P. M. Bronstein and J. Bonventre, Chemical modulation of agonistic display in Betta splendens, J. Comp. Physiol. Psychol., 1976, 90(2), 198 CAS.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Molecular docking: A powerful approach for structure-based drug discovery, Curr. Comput.-Aided Drug Des., 2011, 7(2), 146 CrossRef CAS PubMed.
- B. L. Staker, M. D. Feese, M. Cushman, Y. Pommier, D. Zembower, L. Stewart and A. B. Burgin, Structures of three classes of anticancer agents bound to the human Topoisomerase I−DNA covalent complex, J. Med. Chem., 2005, 48(7), 2336 CrossRef CAS PubMed.
- T. N. M. An, N. V. Cuong, N. M. Quang, T. V. Thanh and M. Alam, Green synthesis using PEG-400 catalyst, antimicrobial activities, cytotoxicity and in silico molecular docking of new carbazole based on α-aminophosphonate, ChemistrySelect, 2020, 5(21), 6339 CrossRef CAS.
- T. N. M. An, T. P. Pham, N. M. Quang, V. S. Nguyen, N. V. Cuong, V. T. Le, D. T. Mai, M. Alam and V. T. Pham, Synthesis, docking study, cytotoxicity, antioxidant, and anti-microbial activities of novel 2,4-disubstituted thiazoles based on phenothiazine, Curr. Org. Synth., 2019, 17(2), 151 CrossRef.
- T.-H. Duong, A. P. Devi, T. N. M. An, H.-V.-T. Phan, N.-V. Huynh, J. Sichaem, H.-D. Tran, M. Alam, T.-P. Nguyen, H.-H. Nguyen, W. Chavasiri and T.-C. Nguyen, Synthesis, α-glucosidase inhibition, and molecular docking studies of novel N-substituted hydrazide derivatives of atranorin as antidiabetic agents, Bioorg. Med. Chem. Lett., 2020, 30(17), 127359 CrossRef CAS PubMed.
- F. U. Eze, U. C. Okoro, D. I. Ugwu and S. N. Okafor, Biological activity evaluation of some new benzenesulphonamide derivatives, Front. Chem., 2019, 7, 634 CrossRef CAS PubMed.
- N. Uwabagira, B. K. Sarojini, M. K. Shankar and R. S. Gani, Potent pharmacophoric aminothiazole derivatives as FabH inhibitors for antibacterial activity: in vitro and in silico approach, SN Appl. Sci., 2019, 1(11), 1375 CrossRef CAS.
- E. W. Bell and Y. Zhang, DockRMSD: an open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism, J. Cheminf., 2019, 11(1), 40 Search PubMed.
- K. Sargsyan, C. Grauffel and C. Lim, How molecular size impacts RMSD applications in molecular dynamics simulations, J. Chem. Theory Comput., 2017, 13(4), 1518 CrossRef CAS.
- F. U. Eze, U. C. Okoro, P. O. Ukoha and D. I. Ugwu, New antioxidant agents bearing carboxamide moiety: Synthesis, molecular docking and in vitro studies of new benzenesulfonamide derivatives, Iran. J. Chem. Chem. Eng., 2021, 40(3), 853 CAS.
- G. Giannini, W. Cabri, C. Fattorusso and M. Rodriquez, Histone deacetylase inhibitors in the treatment of cancer: Overview and perspectives, Future Med. Chem., 2012, 4(11), 1439 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2019, 1–2, 19 Search PubMed.
- P. Chantaranothai, A synopsis of Mussaenda L. (Rubiaceae) in Thailand, Thai For. Bull. (Bot.), 2015, 43, 51 Search PubMed.
- M. Chowdury, M. Alam, S. Chowdhury, M. Biozid, M. Faruk, M. Mazumdar and A. Chowdhury, Evaluation of ex-vivo anti-arthritic, anti-inflammatory, anti-cancerous and thrombolytic activities of Mussaenda roxburghii leaf, Eur. J. Med. Plants, 2015, 10, 1 CrossRef.
- Y. Takeda, H. Nishimura and H. Inouye, Two new iridoid glucosides from Mussaenda parviflora and Mussaenda shikokiana, Phytochemistry, 1997, 16, 1401 CrossRef.
- W. Zhao, J. Xu, G. Qin and R. Xu, Saponins from Mussaenda pubescens, Phytochemistry, 1995, 39, 191 CrossRef CAS.
- W. Zhao, P. Wang, R. Xu, G. Qin, S. Jiang and H. Wu, Saponins from Mussaenda pubescens, Phytochemistry, 1996, 42, 827 CrossRef CAS PubMed.
- W. Zhao, R. Xu, G. Qin, T. Vaisar and M. S. Lee, Saponins from Mussaenda pubescens, Phytochemistry, 1996, 42, 1131 CrossRef CAS PubMed.
- W. Zhao, J.-L. Wolfender, K. Hostettmann, K. Cheng, R. Xu and G. Qin, Triterpenes and triterpenoid saponins from Mussaenda pubescens, Phytochemistry, 1998, 45, 1073 CrossRef PubMed.
- W. Zhao, J. Xu, G. Qin, R. Xu, H. Wu and G. Weng, New triterpenoid saponins from Mussaenda pubescens, J. Nat. Prod., 1994, 57, 1613 CrossRef CAS PubMed.
- W. Zhao, G. Yang, R. Xu and G. Qin, Three monoterpenes from Mussaenda pubescens, Phytochemistry, 1996, 41, 1553 CrossRef CAS.
- Y. Li, W. Qiao and K. Yuan, Isolation and structural elucidation of chemical constituents of Mussaenda hainanensis Merr, J. Med. Plants Res., 2011, 5, 1789 Search PubMed.
- B. Dinda, S. Debnath, S. Majumder, S. Arima, N. Sato and Y. Harigaya, Chemical Constituents of Mussaenda incana, Indian J. Chem., 2006, 44B, 2362 Search PubMed.
- B. Dinda, S. Majumder, S. Arima, N. Sato and Y. Harigaya, Iridoid glucoside and sterol galactoside from Mussaenda macrophylla, J. Nat. Med., 2008, 62, 447 CrossRef CAS PubMed.
- V. K. Thu, N. X. Bach, L. T. Anh, D. T. Trang, N. X. Nhiem, B. H. Tai, P. Van Kiem, C. Van Minh, S. Park, Y. Seo, W. Namkung and S. H. Kim, Discovery of cycloartane-type triterpene saponins from Mussaenda glabra, Phytochem. Lett., 2019, 33, 39 CrossRef CAS.
- V. K. Thu, N. X. Bach, C. T. Dung, N. T. Cuong, T. H. Quang, Y.-C. Kim, H. Oh and P. Van Kiem, Iridoids and cycloartane saponins from Mussaenda pilosissima valeton and their inhibitory NO production in BV2 cells, Nat. Prod. Res., 2021, 35, 4126 CrossRef CAS PubMed.
- N.-C. Kim, A. E. Desjardins, C. D. Wu and A. D. Kinghorn, Activity of triterpenoid glycosides from the root bark of Mussaenda macrophylla against two oral pathogens, J. Nat. Prod., 1999, 62, 1379 CrossRef CAS PubMed.
- D. B. Menon and J. M. Sasikumar, Antioxidant and anti-inflammatory activities of the root of Mussaenda glabrata, J. Pharm. Res., 2011, 4, 3320 CAS.
- L. D. M. Sahadevan and D. B. Menon, Mussaenin A isolated from Mussaenda glabrata induces apoptosis in the liver cancer cells via mitochondrial pathway, Int. J. Pharmacogn. Phytochem. Res., 2017, 9, 1266 Search PubMed.
- M. A. A. Sikder, R. B. Rashid, F. Islam, A. K. M. N. Hossian, A. B. Siddique, S. Kabir, M. R. Haque, M. S. Rahman and M. A. Rashid, Screening of ten medicinal plants of Bangladesh for analgesic activity on Swiss-albino mice, Orient. Pharm. Exp. Med., 2013, 13, 327 CrossRef.
- M. D. Tri, T. T. M. Tram, L. H. Ngoc, T. N. M. An, N. T. Phat, P. N. Minh, N. V. Kieu, D. Van Son, T.-P. Nguyen, T. T. N. Mai and T.-H. Duong, Recurvataside, a new saponin from aerial parts of Mussaenda recurvata, Nat. Prod. Res., 2022 DOI:10.1080/14786419.2022.2039137 , article in press..
- T. Morota, Triterpenes from Tripterigium wilfordii, Phytochemistry, 1995, 39(5), 1153 CrossRef CAS.
- J. Liu, Q. Wu, J. Shu, R. Zhang and L. Liu, A novel spermidine macrocyclic alkaloid from the roots of Tripterygium wilfordii, Chem. Nat. Compd., 2020, 56(3), 496 CrossRef CAS.
- M. Deepak and S. S. Handa, 3α,24-Dihydroxy-urs-12-en-28-oic acid from Verbena officinalis, Phytochemistry, 1998, 49(1), 269 CrossRef CAS.
- Y. H. Kuo, Y. L. Lin and S. M. Lee, Scutellaric Acid, a new triterpene from Scutellaria rivularis, Chem. Pharm. Bull., 1988, 36(9), 3619 CrossRef CAS.
- B. S. Joshi, K. L. Singh and R. Roy, Complete assignments of 1H and 13C NMR spectra of the pentacyclic triterpene hederagenin from Nigella sativa Linn, Magn. Reson. Chem., 1999, 37(4), 295 CrossRef CAS.
- G. Giannini, W. Cabri, C. Fattorusso and M. Rodriquez, Histone deacetylase inhibitors in the treatment of cancer: Overview and perspectives, Future Med. Chem., 2012, 4(11), 1439 CrossRef CAS.
- H. T. Nguyen HT, T.-T. Nguyen, T.-H. Duong, T. N. M. An, C. H. Nguyen, T.-H.-A. Nguyen and J. Sichaem, α-Glucosidase inhibitory and antimicrobial benzoylphloroglucinols from Garcinia schomburgakiana Fruits: In Vitro and In Silico studies, Molecules, 2022, 27(8), 2574 CrossRef PubMed.
- S. Hidalgo-Figueroa, A. Rodríguez-Luévano, J. C. Almanza-Pérez, A. Giacoman-Martínez, R. Ortiz-Andrade, I. León-Rivera and G. Navarrete-Vázquez, Synthesis, molecular docking, dynamic simulation and pharmacological characterization of potent multifunctional agent (dual GPR40-PPARγ agonist) for the treatment of experimental type 2 diabetes, Eur. J. Pharmacol., 2021, 907, 174244 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, flexible, and free, J. Comput. Chem., 2005, 26(16), 1701 CrossRef CAS.
- Y. Yamada, S. Gohda, K. Abe, T. Togo, N. Shimano, T. Sasaki, H. Tanaka, H. Ono, T. Ohba, S. Kubo, O. Takahiro and S. Satoshi, Carbon materials with controlled edge structures, Carbon, 2017, 122, 694 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Phan Nhat Minhab,
Mai Thanh Chiab,
Bui Xuan Haoc,
Tran Nguyen Minh An
ab,
Phan Nhat Minhab,
Mai Thanh Chiab,
Bui Xuan Haoc,
Tran Nguyen Minh An