
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective semisynthesis of uzarigenin and allo-uzarigenin†
Sarah
Al Muthafer
a,
Christoph
Schissler
a,
Vanessa
Koch
 a,
Hannes
Kühner
a,
Martin
Nieger
b and
Stefan
Bräse
*ac
a,
Hannes
Kühner
a,
Martin
Nieger
b and
Stefan
Bräse
*ac
aInstitute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu; Fax: (+49)-721-608-48581; Tel: (+49)-721-608-42903
bDepartment of Chemistry, University of Helsinki, P. O. Box 55, 00014 University of Helsinki, Finland
cInstitute of Biological and Chemical Systems – Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 9th January 2023
Abstract
Herein, we report the concise semisynthesis of the natural cardenolide uzarigenin and its diastereoisomer allo-uzarigenin in nine and seven steps, respectively, starting from the broadly available epi-androsterone. For this purpose, the synthetic strategy for the stereoselective introduction of the β-hydroxy group at C-14 via Mukaiyama oxidation is discussed. Additionally, the installation of the butenolide ring at C-17 is performed using a Stille-cross-coupling reaction with subsequent stereoselective hydrogenation of the C-16/C-17 double bond to exclusively give allo-uzarigenin. By directing the hydrogenation via a protecting group strategy, the C-17β isomer can also be obtained stereoselectively.
Introduction
Uzarin (1) and its aglycone uzarigenin (2) belong to the natural product class of cardiac glycosides, and have been known for their medicinal use since centuries (Fig. 1).1 They can be obtained from the Uzara plant (Xysmalobium undulatum), which belongs to the milkweed family (Asclepiadoideae). In African folk medicine, the extracts isolated from the Uzara root were used for a long time to treat wounds, diarrhea, spasms, menstrual cramps, and headaches. Unlike other cardiac glycosides such as digitoxin (3), Uzara glycosides have a low cardiotonic effect, making intoxication less likely to occur.2–4 Uzarin (1) and its aglycone uzarigenin (2) exhibit several structural characteristics of the steroid class of cardiac glycosides, such as the β-orientated unsaturated lactone ring at C-17 and the β-hydroxy group at C-14, resulting in a cis-fusion of the C/D rings. Like many other cardiac glycosides derived from the plant family of Asclepiadoideae, they feature a typical trans A/B ring junction, whereas most other cardiac glycosides, such as the well-investigated digitoxin (3), have cis-fused A/B rings.5 Uzarigenin (2), therefore, can be assigned to the 5α-configured family of cardenolides, to which the better-known calotropin (4) also belongs.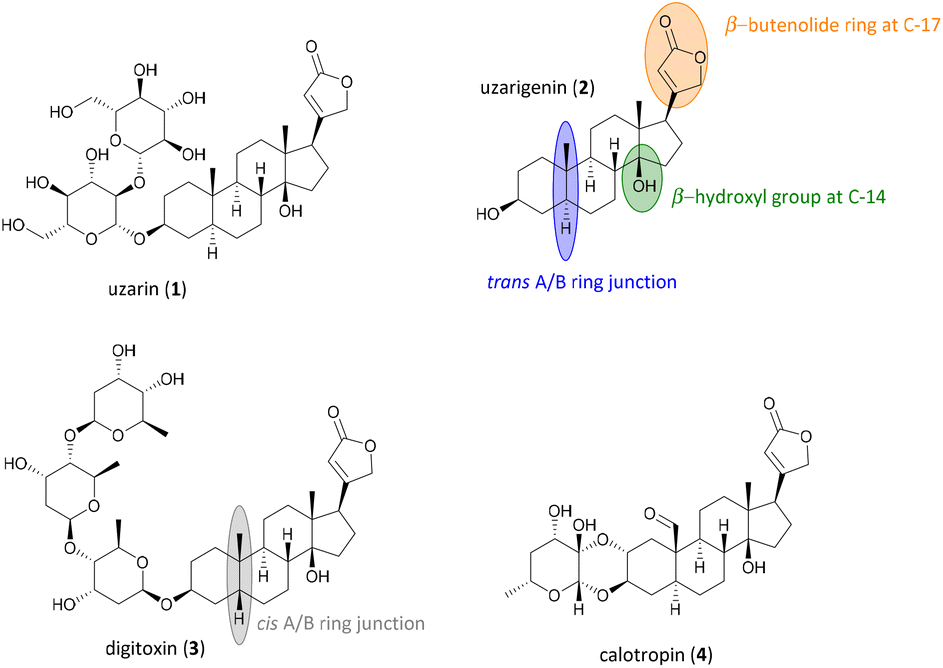 | ||
| Fig. 1 Molecular structures of the cardiac glycosides uzarin (1), its aglycone uzarigenin (2), digitoxin (3) and calotropin (4). | ||
The correspondence of concurrent reactions in the two configurationally and energetically different series (5α and 5β) is not self-evident. For example, a reaction sequence leading to digitoxigenin in the 5β-series is not generally transferable to the 5α-series.6 Nevertheless, Kurt Radscheit and co-workers succeeded in converting 15α-hydroxy-cortexon into 3-oxo-5α-carda-14,20(22)-dienolide and claimed to have successfully synthesized uzarigenin (2) via two different synthetic routes. However, the stereochemistry at C-14 was not further discussed.7 The same applies to Khristulas’ uzarigenin (2) semisynthesis, which started from 3β-acetoxy-5α-pregn-16-en-20-one and did not provide any direct proof of the configuration of the synthesized compounds.8 Emil Angliker managed to introduce a 14β-hydroxy group but struggled with the configuration at C-17 and, therefore, could only report the synthesis of allo-uzarigenin.9 Wicha et al. investigated the D ring chemistry in more detail and achieved the stereoselective synthesis of 3-OMe uzarigenin; however, the introduction of the butenolide ring at C-17 in the β-position involved 13 steps and the ether moiety at C-3 was not removed at the end of the sequence.10,11
So far, uzarigenin (2) with unambiguously correct stereochemistry can only be obtained with the support of nature: for example, Bauer and Gonzalez were able to extract uzarigenin (2) from Asclepias syriaca L. or leaves of Scrophulariaceae.12,13 Sauer et al. instead used Strophanthus kombé, a member of the Apocynaceae family, to convert 5α-pregnanolone into uzarigenin (2) via a foliar application method.14,15 Okada and Anjyo successfully converted the more widely occurring digitoxigenin to uzarigenin (2) via epimerization at the C-5 position.16
Besides these nature-supported methods, none of the pioneering syntheses described earlier give access to the natural version of uzarigenin (2) and the syntheses suffer furthermore from quite lavish procedures and a lack of stereocontrol. Therefore, we aimed to develop a straight and high-yielding synthesis route, which puts emphasis on well-characterized compounds by using 2D NMR spectra and X-ray analysis for the efficient semisynthesis of uzarigenin (2) and its C-17 epimer allo-uzarigenin (21).
Results and discussion
Inexpensive and readily available epi-androsterone (5) was chosen as the starting material for the semisynthesis of uzarigenin (2), requiring the introduction of the butenolide ring at C-17 and the inversion of the stereogenic center at C-14. To substitute the hydrogen atom attached in the α-position at C-14 with the aimed β-hydroxyl group, a three-step sequence was envisioned, with the first step comprising the synthesis of the Michael system 8. As presented in Scheme 1, epi-androsterone (5) was lithiated at −78 °C and subsequently the TMS enol ether 7 was formed by adding trimethylsilyl chloride.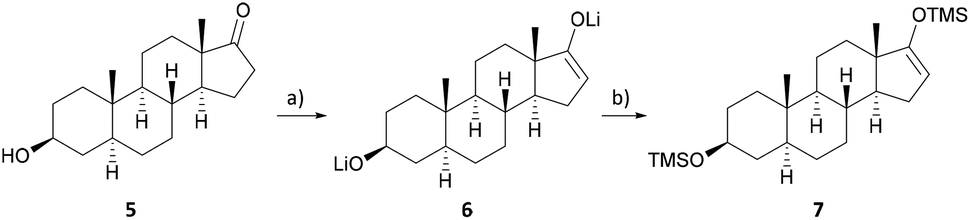 | ||
| Scheme 1 Synthesis of the precursor for the Saegusa–Ito oxidation. (a) LDA, THF, −78 °C, 1 h; (b) TMSCl, NEt3, −78 °C to r.t., 1.5 h. | ||
Without further purification, the enol ether 7 was subjected to the Saegusa–Ito oxidation, which is a well-known method for steroid compounds.17–23 However, most of the procedures use stoichiometric amounts of palladium, although many reoxidants have been described in the literature, including copper(II) salts such as copper(II) acetate or copper(II) chloride, 1,4-benzoquinone, oxygen, and Oxone®.24–28 With the aim of using palladium in catalytic amounts, different reaction conditions were screened, focusing on inexpensive oxygen and copper(II) acetate as reoxidants (see Table 1 and Table SI-1 in the ESI†).
|
|
|||
|---|---|---|---|
| Entry | Reagentsa | Reaction conditions | Isolated yield [%] |
| a 20 mol% Pd(OAc)2 and 40 mol% Cu(OAc)2 were used if not stated otherwise. b 1.00 equiv. of Pd(OAc)2 were used. c 10 mol% Pd(OAc)2 and 15 mol% Cu(OAc)2 were used. | |||
| 1 | Pd(OAc)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
CH2Cl2/MeCN (3:1), r.t., 16 h |
48 |
| 2 | Pd(OAc)2, Cu(OAc)2 | CH2Cl2/MeCN (2:3), r.t., 24 h |
43 |
| 3 | Pd(OAc)2, O2 | CH2Cl2/MeCN (2:3), r.t., 24 h |
39 |
| 4 | Pd(OAc)2, Cu(OAc)2 | MeCN, 60 °C, 24 h | 45 |
| 5 | Pd(OAc) 2 , Cu(OAc) 2 |
CH
2
Cl
2
/DMSO (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :3), r.t., 24 h :3), r.t., 24 h
|
66 |
| 6 | Pd(OAc) 2 , O 2 |
CH
2
Cl
2
/DMSO (2:3), r.t., 24 h
|
62 |
| 7 | Cu(OAc)2, O2 | CH2Cl2/DMSO (2:3), r.t., 24 h |
0 |
| 8 | Pd(OAc)2, Cu(OAc)2c |
CH2Cl2/DMSO (2:3), r.t., 24 h |
55 |
Using catalytic amounts of palladium and oxygen or copper(II) acetate as the reoxidant, comparable yields to that obtained when using stoichiometric amounts of palladium could be obtained (entries 1–3). Varying the solvent apparently influenced the yield, with a mixture of CH2Cl2/DMSO achieving the highest yields (entries 5 and 6). A further decrease in the catalyst amount resulted in only a minor drop in yield (compare entries 5 and 8). Furthermore, no reaction took place without palladium(II) acetate (entry 7), proving the latter to be the active metal in the reaction. The use of copper(II) chloride, Oxone® or 1,4-benzoquinone as the reoxidant did not give any further improvement in yields (see ESI Table SI-1†).
The rearrangement of the conjugated Δ15 to the isolated Δ14 double bond has already been effectively applied in many natural product syntheses17–21,29 and should once more serve as a starting point for the introduction of the C-14 hydroxy group in this work. Analogously to Johns et al.,17,30–32 isomerization was carried out with p-toluene sulfonic acid in refluxing toluene to give the desired product 9 in 61% yield after a reaction time of 15–20 minutes (Scheme 2).
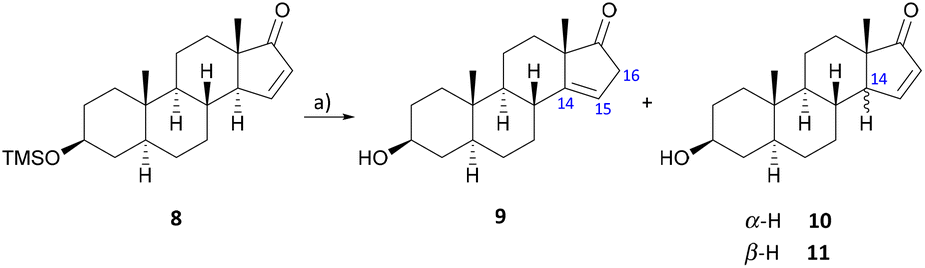 | ||
| Scheme 2 Rearrangement reaction at the steroidal D ring double bond. (a) p-TsOH, toluene, 130 °C, 15–20 min, 61% (9). | ||
In the course of this, deprotected starting material 10 and its 14β-epimer 11 could also be isolated by column chromatography on silica gel and reused for the rearrangement reaction. The molecular structure of 11 could be determined by X-ray crystallographic analysis (see the ESI†).
Starting from the isolated Δ14 double bond, the following step aimed to diastereoselectively attach the hydroxy group at C-14 in the β-position. Epoxidation with subsequent ring opening indicated the formation of many different products, and thus the Mukaiyama oxidation should accomplish the installation of the tertiary hydroxy group.33–36 To investigate whether diastereoselectivity can be observed for this reaction, a wide range of reaction conditions were tested by varying the catalyst, solvent, additives, reaction time and the addition rate and amount of the reductant (see Table 2 and Table SI-2 in the ESI†).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | PhSiH3 (equiv., a.r.) | Catalyst | Solvent | Additive | Reaction time | Yield [%], dr (β-OH:α-OH) |
| Reaction conditions: catalyst (30 mol%); yields refer to the isolated mixture; the diastereomeric ratio was determined by integration of the resonance of the angular methyl group C-18 in the 13C NMR spectra of the isomer mixture after prolonged relaxation delay (d1 = 10 s).a HPLC quality.b Absolute solvent.c 2.00 equiv. of PPh3 were used.d 10 mol% Co(acac)2 was used.e No full conversion. a.r. rate of addition. | |||||||
| 1 | H | 3.0, 1 h | Co(acac)2 | 1,4-Dioxanea | — | 3 h | 0 |
| 2 | H | 3.0, 1 h | Co(acac)2 | 1,4-Dioxaneb | — | 3 h | 44 |
| 3 | H | 4.5, 2 h | Co(acac)2 | 1,4-Dioxaneb | 4 Å MS | 20 h | 53 |
| 4 | TBDMS | 4.5, 2 h | Co(acac)2 | 1,4-Dioxaneb | 4 Å MS | 20 h | 64 |
| 5 | Bn | 4.5, 2 h | Co(acac)2 | 1,4-Dioxaneb | 4 Å MS | 20 h | 62 (2.0:1) |
| 6 | Bn | 4.5, 2 h | Co(dpm)2 | 1,4-Dioxaneb | 4 Å MS | 20 h | 49 (2.0:1) |
| 7 | Bn | 4.5, 2 h | Mn(acac)2 | 1,4-Dioxaneb | 4 Å MS, PPh3c |
20 h | 33 (2.3:1) |
| 8 | Bn | 4.5, 2 h | Co(acac)2 | 1,4-Dioxaneb | 4 Å MS, PPh3c |
20 h | 60 (2.1:1) |
| 9 | Bn | 4.5, 2 h | Co(acac)2 | MeCNb | 4 Å MS | 20 h | 71 (2.4:1) |
| 10 | Bn | 4.5, 2 h | Co(acac)2d |
MeCNb | 4 Å MS | 20 h | 59 (2.5:1) |
| 11 | Bn | 4.5, 2 h | Co(acac)2 | MeOHb | 3 Å MS | 20 h | 45e (4.7:1) |
| 12 | Bn | 4.5, 2 h | Co(acac) 2 | EtOH | 4 Å MS | 20 h |
73 (9.4:1)
|
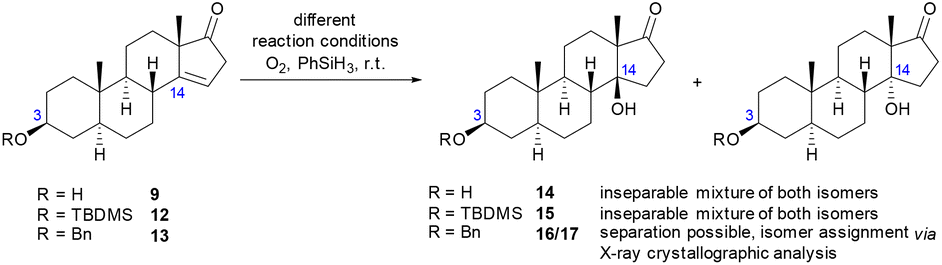
For the Mukaiyama oxidation of 9, no product formation could be observed in non-anhydrous 1,4-dioxane, making the use of an absolute solvent crucial (entries 1 and 2). Increasing the amount of reductant and adding molecular sieves to the reaction mixture resulted in a slight improvement in the yield from 44% to 53% (entry 3). To prevent the oxidation of the secondary hydroxyl group at C-3 as a possible side reaction, a TBDMS protecting group was introduced at C-3 prior to the oxidation, which led to a significant increase in the yield from 53% to 64% (entry 4). Unfortunately, an inseparable mixture of both hydroxy epimers was obtained in all experiments described above. The ratio of epimer A to epimer B was determined to be 2:1 by integration of the resonances of the angular methyl protons in the 1H NMR spectra. Considering the improved yield, a benzyl-protecting group was introduced at C-3, which enabled the separation of the two epimers (entry 5), whose absolute configuration was determined by X-ray crystallographic analysis (see Fig. 2), and the desired 14β-epimer 16 was identified as the main product. Having the two epimers separated and assigned, the reaction was further optimized in terms of yield and particularly the diastereomeric ratio. Varying the catalyst or adding PPh337 as an additive did neither improve the yield nor the diastereomeric ratio significantly (entries 6–8). Moreover, contrary to the literature,38 no reaction took place at all with Co(III) or Mn(III) species (Table SI-2 in the ESI†). Ultimately, varying the solvent not only improved the yield (entries 9 and 12), but the use of a polar protic solvent also shifted the diastereomeric ratio significantly from 2.0:1 to 4.7:1 for MeOH (entry 11) and eventually gave a dr of 9.4:1 in favour of the desired epimer 16 when ethanol was used as the solvent (entry 12).
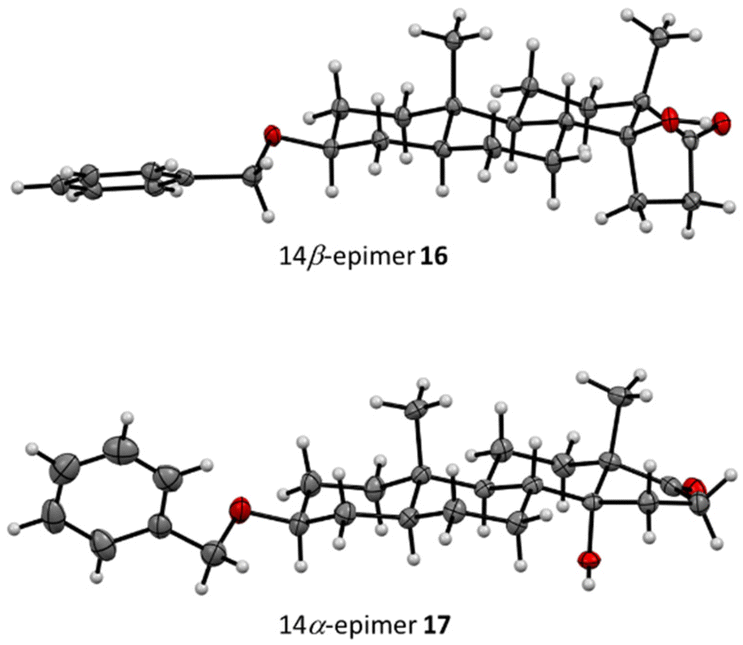 | ||
| Fig. 2 Crystal structures of 16 and 17 (displacement parameters are drawn at the 50% probability level). The impact of the β-position of the C-14 substituent can be observed clearly: the 14α-epimer is mostly flat due to its all-trans ring linkages, while the 14β-epimer exhibits a more bowed shape due to the cis-linkage of the C-/D-rings. | ||
After the successful diastereoselective introduction of the hydroxy group at C-14, the lactone ring has to be diastereoselectively attached to C-17. Thus ketone 16 was converted to the respective vinyl iodide 18 by following Barton's procedure.39 For this purpose, the carbonyl functionality was treated successively with hydrazine and iodine to yield vinyl iodide 18 in 51% yield (Scheme 3). The sp2-hybridized C-17 could now undergo a cross-coupling reaction following the procedure developed by Stille et al.,40 which has also been successfully applied by us previously,41 yielding cardadienolide 20 in 67% yield.
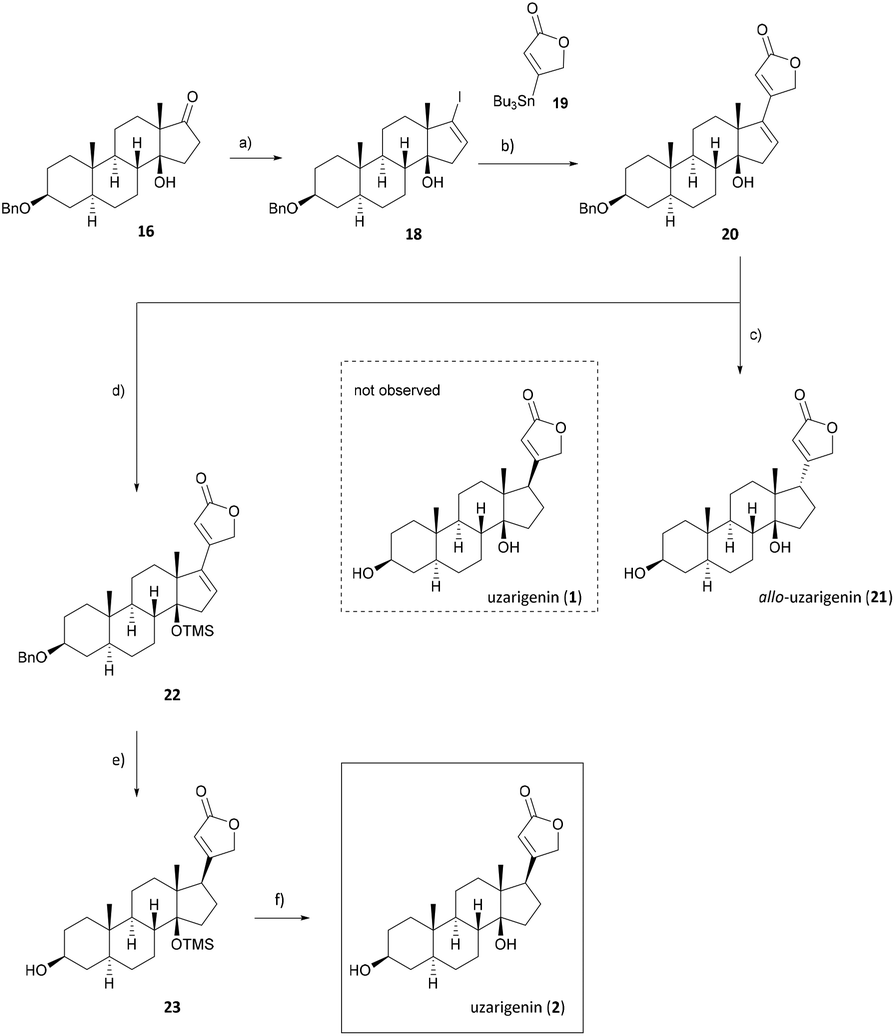 | ||
| Scheme 3 Synthesis of uzarigenin (1) and allo-uzarigenin (21). (a) N2H4, NEt3, EtOH, 50 °C, 16 h; I2, NEt3, THF, 0 °C to r.t., 3 d, 51%; (b) Pd(PPh3)4, LiCl, CuCl, DMF, 60 °C, 24 h, 67%; (c) H2, Pd/C (10 wt%), EtOAc, r.t., 30 min, 76%; (d) TMSCl, imidazole, DMF, r.t., 16 h, 94%; (e) H2, Pd/C (10 wt%), EtOAc, r.t., 30 min, 62%; (f) 3 M HCl, MeOH, r.t., 4 h, 79%. | ||
Cardadienolide 20 should then undergo a chemo- and diastereoselective reduction of the Δ16 double bond with simultaneous removal of the benzyl protecting group at C-3 using palladium on carbon as a catalyst. The reaction indeed proceeded diastereoselectively, as the formation of only one isomer was observed. Unfortunately, the reduction yielded the undesired isomer (allo-uzarigenin (21)) in 76% yield, which is consistent with the literature.42,43 In comparison, the exclusive formation of the desired C-17β isomer was observed upon hydrogenation of 3β-hydroxy-5α,14α-carda-(16,20)-dienolide (SI-06, see the ESI†), suggesting a directing effect of the hydroxy group in the present substrate 20. The stereochemistry at C-17 of 21 was determined by NOE correlation between the angular 18-CH3 and 17-CH as shown in Fig. 3. Precise tuning of the reaction time is mandatory for a chemoselective reaction since longer reaction times also result in a reduction of the double bond within the lactone ring, evident from the loss of the olefinic proton at δ = 5.77–5.83 ppm as shown for the C-14α-H system SI-07 in the ESI.†
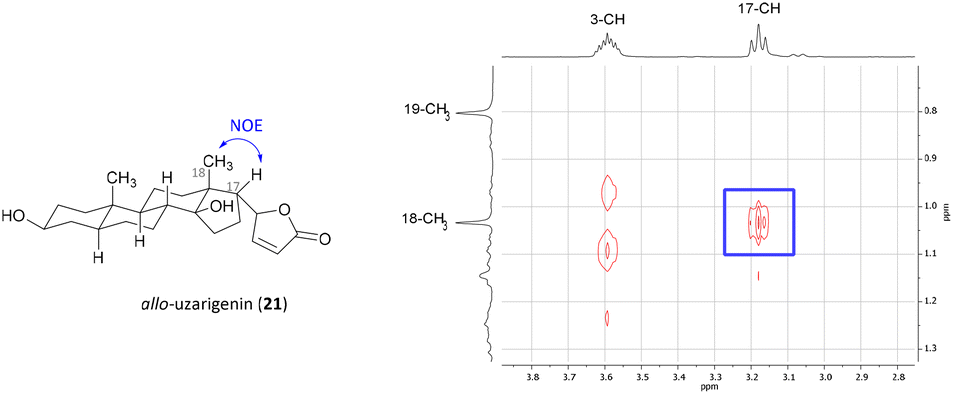 | ||
| Fig. 3 Excerpt from the NOESY spectrum of allo-uzarigenin (21). A strong NOE correlation between 18-CH3 and 17-CH indicates spatial proximity and thus hydrogenation from the β-face. | ||
Since the hydrogenation of Δ16-olefin 20 occurred from the convex β-face, affording exclusively the C-17α product (allo-uzarigenin (21)), TMS protection of the C-14 hydroxy group was conducted to circumvent a possible directing effect of the hydroxy group while introducing an even greater steric hindrance to the β-face at the same time (Scheme 3).42,44,45 A subsequent reduction of 22 gave exclusively the desired C-17β product, which yielded the natural product uzarigenin (2) after acidic deprotection with 3 M HCl. The stereochemistry at C-17 was determined by 2D NOESY spectroscopy as shown in Fig. 4. NOE correlation between 22-CH and 18-CH3 indicates spatial proximity of the lactone ring and the 18-methyl group and thus the successful hydrogenation from the α-face. Furthermore, no NOE correlation between 17-CH und 18-CH3 can be seen, as exemplarily shown for the NOESY spectrum of uzarigenin (2). Moreover, the analytical data of allo-uzarigenin (21) and uzarigenin (2) synthesized in this work were compared with those from the literature and are in agreement (Tables SI-3–SI-5 in the ESI†).
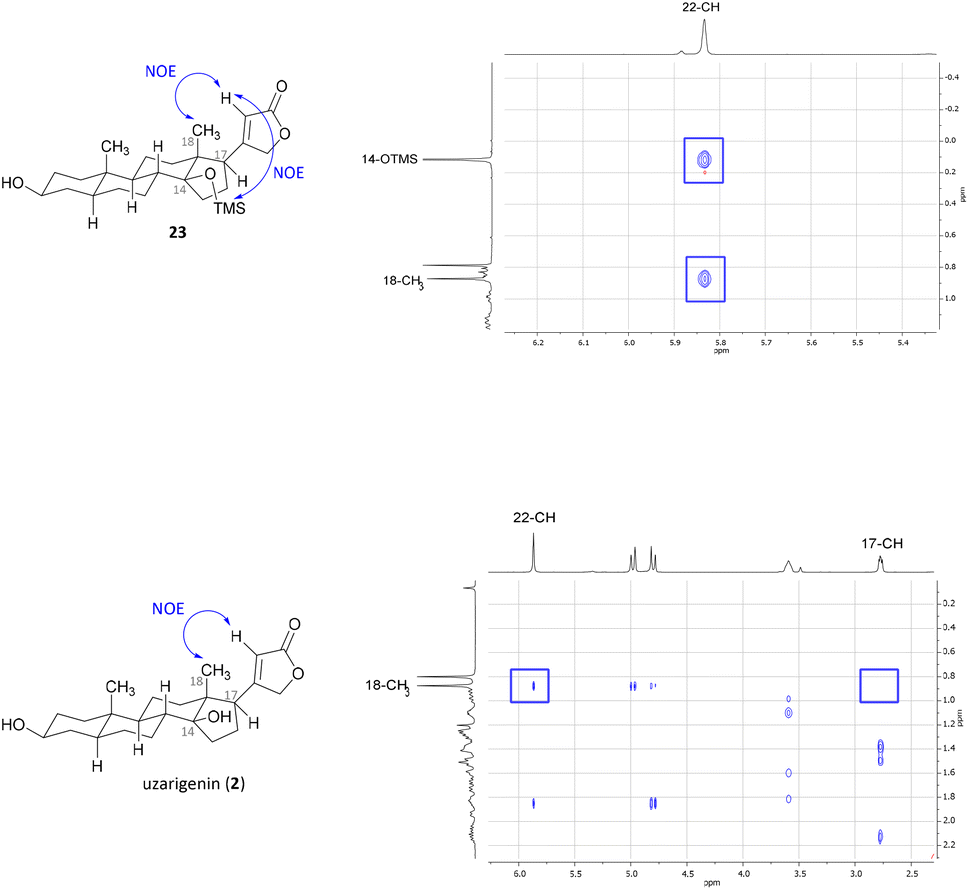 | ||
| Fig. 4 Excerpt of the NOESY spectra of TMS protected uzarigenin 23 (top) and uzarigenin (2) (bottom). In both cases, a clear NOE correlation between 22-CH and 18-CH3 indicates spatial proximity of the lactone ring and the 18-methyl group. Furthermore, a NOE cross peak between 22-CH and 14-OTMS can be seen, which further supports the successful hydrogenation from the α-face. | ||
Experimental section
The synthetic procedures for all synthesized compounds are available in the ESI.†Conclusions
In summary, we have reported the first stereoselective semisynthesis of uzarigenin (2) and allo-uzarigenin (21) starting from the widely available epi-androsterone (5) in nine and seven steps, respectively. This synthesis route offers efficient access to uzarigenin (2) by using moderate to high-yielding stereoselective reactions. Correct stereochemistry was confirmed by 2D NMR experiments and X-ray analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. M. and C. S. gratefully acknowledge the Fonds der Chemischen Industrie and the graduate program of the federal state of Baden-Württemberg (LGF) for financial support. V. K. gratefully acknowledges the Studienstiftung des Deutschen Volkes for not exclusively financial support. H. K. acknowledges support from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—2082/1—390761711 and the Collaborative Research Center TRR 88 “3MET”.References
- J. W. Estes and P. D. White, William Withering and the Purple Foxglove, Sci. Am., 1965, 212, 110–119 CrossRef CAS PubMed.
- I. Vermaak, G. M. Enslin, T. O. Idowu and A. M. Viljoen, Xysmalobium undulatum (uzara)–review of an antidiarrhoeal traditional medicine, J. Ethnopharmacol., 2014, 156, 135–146 CrossRef CAS PubMed.
- B.-E. Van Wyk, The potential of South African plants in the development of new medicinal products, S. Afr. J. Bot., 2011, 77, 812–829 CrossRef.
- M. Ghorbani, M. Kaloga, H. H. Frey, G. Mayer and E. Eich, Phytochemical reinvestigation of Xysmalobium undulatum roots (Uzara), Planta Med., 1997, 63, 343–346 CrossRef CAS PubMed.
- H. R. El-Seedi, S. A. M. Khalifa, E. A. Taher, M. A. Farag, A. Saeed, M. Gamal, M.-E. F. Hegazy, D. Youssef, S. G. Musharraf, M. M. Alajlani, J. Xiao and T. Efferth, Cardenolides: Insights from chemical structure and pharmacological utility, Pharmacol. Res., 2019, 141, 123–175 CrossRef CAS PubMed.
- F. Sondheimer, Syntheses in cardiac-active steroid field, Chem. Br., 1965, 1, 454–464 CAS.
- U. Stache, W. Fritsch, W. Haede, K. Radscheit and K. Fachinger, Herstellung von ungesättigten Lactonen der Steroid-Reihe, (IV1) Synthese von Uzarigenin, Liebigs Ann. Chem., 1969, 726, 136–144 CrossRef CAS.
- F. S. Khristulas, M. B. Gorovits and N. K. Abubakirov, Synthesis of uzarigenin, Chem. Nat. Compd., 1970, 6, 563–568 CrossRef.
- E. Angliker, Synthese von allo-Uzarigenin; Beitrag zur Konstitution der allo-Aglykone, Doctoral Thesis, ETH Zürich, 1948 Search PubMed.
- G. Groszek, A. Kurek-Tyrlik and J. Wicha, Synthesis of a cardenolide, 3-O-methyl uzarigenin, with 14β-hydroxyandrost-16-ene as the key intermediate, Tetrahedron, 1989, 45, 2223–2236 CrossRef CAS.
- V. Koch, M. Nieger and S. Bräse, Towards the synthesis of calotropin and related cardenolides from 3-epiandrosterone: A-ring related modifications, Org. Chem. Front., 2020, 7, 2670–2681 RSC.
- Š. Bauer, L. Masler, O. Bauerová and D. Šikl, Uzarigenin and desglucouzarin from Asclepias syriaca L, Experientia, 1961, 17, 15–15 CrossRef PubMed.
- A. González, J. Bretón, E. Navarro, J. Trujillo, J. Boada and R. Rodríguez, Phytochemical Study of Isoplexis chalcantha, Planta Med., 1985, 51, 9–11 CrossRef PubMed.
- H. H. Sauer, R. D. Bennett and E. Heftmann, Conversion of 5α-pregnanolone to uzarigenin by Strophanthus kombé, Phytochemistry, 1969, 8, 839–842 CrossRef CAS.
- H. H. Sauer, R. D. Bennett and E. Heftmann, Conversion of pregnenolone to digifologenin by Digitalis lanata, Naturwissenschaften, 1967, 54, 226–226 CrossRef CAS PubMed.
- M. Okada and T. Anjyo, Conversion of Digitoxigenin to Uzarigenin, Chem. Pharm. Bull., 1974, 22, 464–467 CrossRef CAS.
- V. Koch, Entwicklung neuer Synthesemethoden zur Darstellung von Calotropin und verwandten Cardenoliden als Leitstrukturen für potentielle Anti-Tumor-Wirkstoffe, Logos Verlag, 2018 Search PubMed.
- H. Renata, Q. Zhou, G. Dünstl, J. Felding, R. R. Merchant, C.-H. Yeh and P. S. Baran, Development of a Concise Synthesis of Ouabagenin and Hydroxylated Corticosteroid Analogues, J. Am. Chem. Soc., 2015, 137, 1330–1340 CrossRef CAS PubMed.
- H. Renata, Q. Zhou and P. S. Baran, Strategic Redox Relay Enables A Scalable Synthesis of Ouabagenin, A Bioactive Cardenolide, Science, 2013, 339, 59–63 CrossRef CAS PubMed.
- U. Egner, K.-H. Fritzemeier, W. Halfbrodt, N. Heinrich, J. Kuhnke, A. Müller-Fahrnow, G. Neef, K. Schöllkopf and W. Schwede, 7α,15α-Ethano bridged steroids. Synthesis and progesterone receptor interaction, Tetrahedron, 1999, 55, 11267–11274 CrossRef CAS.
- P. J. Hilton, W. McKinnon, E. C. Gravett, J.-M. R. Peron, C. M. Frampton, M. G. Nicholls and G. Lord, Selective inhibition of the cellular sodium pump by emicymarin and 14β anhydroxy bufadienolides, Steroids, 2010, 75, 1137–1145 CrossRef CAS PubMed.
- N. Isaka, M. Tamiya, A. Hasegawa and M. Ishiguro, A Concise Total Synthesis of the Non-peptide Bradykinin B1 Receptor Antagonist Velutinol A, Eur. J. Org. Chem., 2012, 665–668 CrossRef CAS.
- M. E. Krafft, O. A. Dasse and Z. Fu, Synthesis of the C/D/E and A/B Rings of Xestobergsterol-(A), J. Org. Chem., 1999, 64, 2475–2485 CrossRef CAS.
- Y. Ito, T. Hirao and T. Saegusa, Synthesis of α, β-unsaturated carbonyl compounds by palladium(II)-catalyzed dehydrosilylation of silyl enol ethers, J. Org. Chem., 1978, 43, 1011–1013 CrossRef CAS.
- T. Z. Wang and L. A. Paquette, Photochemical route to a representative 1, 8-annulated 7-methylene-1, 3, 5-cyclooctatriene, J. Org. Chem., 1986, 51, 5232–5234 CrossRef CAS.
- S. F. Martin and D. E. Guinn, Stereoselective synthesis of (+)-Prelog-Djerassi lactone from furanoid intermediates, J. Org. Chem., 1987, 52, 5588–5593 CrossRef CAS.
- R. C. Larock, T. R. Hightower, G. A. Kraus, P. Hahn and D. Zheng, A simple, effective, new, palladium-catalyzed conversion of enol silanes to enones and enals, Tetrahedron Lett., 1995, 36, 2423–2426 CrossRef CAS.
- Y. Lu, P. L. Nguyen, N. Levaray and H. Lebel, Palladium-catalyzed Saegusa–Ito oxidation: synthesis of α, β-unsaturated carbonyl compounds from trimethylsilyl enol ethers, J. Org. Chem., 2013, 78, 776–779 CrossRef CAS PubMed.
- A. Cleve, G. Neef, E. Ottow, S. Scholz and W. Schwede, Synthesis of 14β-H antiprogestins, Tetrahedron, 1995, 51, 5563–5572 CrossRef CAS.
- W. S. Johnson and W. F. Johns, 14-Isoestrone Methyl Ether and its Identity with Totally Synthetic Material, J. Am. Chem. Soc., 1957, 79, 2005–2009 CrossRef CAS.
- M. Sakakibara and A. Ogawa Uchida, Syntheses of (14β,17α,)-14-Hydroxy- and (14β,17α)-2,14-Dihydroxyestradiols and Their Activities, Biosci. Biotechnol. Biochem., 1996, 60, 411–414 CrossRef CAS PubMed.
- M. Sakakibara and A. Ogawa Uchida, Syntheses of (14β,15β,16β,17α)- and (14β,15α,16α,17α,)-1,3,5(10)-Estratriene-2,3,14,15, 16,17-hexaols, Possible Candidates for the Inagami—Tamura Endogenous Digitalis-like Factor, and Their Activity, Biosci. Biotechnol. Biochem., 1996, 60, 405–410 CrossRef CAS PubMed.
- M. Teruaki, I. Shigeru, I. Satoshi, K. Koji, Y. Tohru and T. Toshihiro, Oxidation-Reduction Hydration of Olefins with Molecular Oxygen and 2-Propanol Catalyzed by Bis(acetylacetonato)cobalt(II), Chem. Lett., 1989, 18, 449–452 CrossRef.
- K. Koji, Y. Tohru, T. Toshihiro, I. Satoshi and I. Shigeru, Catalytic Oxidation–Reduction Hydration of Olefin with Molecular Oxygen in the Presence of Bis(1,3-diketonato)cobalt(II) Complexes, Bull. Chem. Soc. Jpn., 1990, 63, 179–186 CrossRef.
- I. Satoshi, K. Koji, T. Toshihiro, I. Shigeru, Y. Tohru and M. Teruaki, Bis(trifluoroacetylacetonato)cobalt(II) Catalyzed Oxidation-Reduction Hydration of Olefins: Selective Formation of Alcohols from Olefins, Chem. Lett., 1989, 18, 515–518 CrossRef.
- I. Shigeru and M. Teruaki, A New Method for Preparation of Alcohols from Olefins with Molecular Oxygen and Phenylsilane by the Use of Bis(acetylacetonato)cobalt(II), Chem. Lett., 1989, 18, 1071–1074 CrossRef.
- Y. Y. See, A. T. Herrmann, Y. Aihara and P. S. Baran, Scalable C–H Oxidation with Copper: Synthesis of Polyoxypregnanes, J. Am. Chem. Soc., 2015, 137, 13776–13779 CrossRef CAS PubMed.
- S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed.
- D. H. R. Barton, R. E. O'Brien and S. Sternhell, A new reaction of hydrazones, J. Chem. Soc., 1962, 470–476 RSC.
- J. K. Stille, The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef.
- V. Koch, M. Nieger and S. Bräse, Stille and Suzuki Cross-Coupling Reactions as Versatile Tools for Modifications at C-17 of Steroidal Skeletons - A Comprehensive Study, Adv. Synth. Catal., 2017, 359, 832–840 CrossRef CAS.
- K. Mukai, D. Urabe, S. Kasuya, N. Aoki and M. Inoue, A Convergent Total Synthesis of 19-Hydroxysarmentogenin, Angew. Chem., Int. Ed., 2013, 52, 5300–5304 CrossRef CAS PubMed.
- H. Renata, Q. Zhou, G. Dünstl, J. Felding, R. R. Merchant, C.-H. Yeh and P. S. Baran, Development of a Concise Synthesis of Ouabagenin and Hydroxylated Corticosteroid Analogues, J. Am. Chem. Soc., 2015, 137, 1330–1340 CrossRef CAS PubMed.
- S. Watanabe, T. Nishikawa and A. Nakazaki, Total Synthesis of the Cardiotonic Steroid (+)-Cannogenol, J. Org. Chem., 2021, 86, 3605–3614 CrossRef CAS PubMed.
- B. Bhattarai and P. Nagorny, Enantioselective Total Synthesis of Cannogenol-3-O-α-l-rhamnoside via Sequential Cu(II)-Catalyzed Michael Addition/Intramolecular Aldol Cyclization Reactions, Org. Lett., 2018, 20, 154–157 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2207135 (11), 2207136 (13), 2207137 (16), 2207138 (17), and 2207139 (SI-06). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo01718k |
| This journal is © the Partner Organisations 2023 |