
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble C–H bond functionalization for C–C coupling at the β-position of thiophenes using palladium-catalyzed 1,4-migration associated with direct arylation†
Linhao
Liu
 ,
Marie
Cordier
,
Thierry
Roisnel
and
Henri
Doucet
*
,
Marie
Cordier
,
Thierry
Roisnel
and
Henri
Doucet
*
Univ. Rennes, 35042 Rennes, France. E-mail: henri.doucet@univ-rennes1.fr; Tel: +33 (0)2 23 23 63 84
First published on 31st January 2023
Abstract
The functionalization of C–H bonds at β-positions of 5-membered ring heteroarenes such as thiophene is generally more challenging than at α-positions. By using Pd-catalyzed 1,4-migration associated with direct arylation, under appropriate conditions, the functionalization of such thienyl β-positions of 2-arylthiophenes is possible. The oxidative addition of 2-(2-bromoaryl)thiophenes to palladium followed by Pd 1,4-migration activates these β-positions. Then, Pd-catalyzed direct coupling with heteroarenes provides β-heteroarylated 2-arylthiophene derivatives. In the course of this coupling reaction, a new C–C bond arises from the functionalization of two C–H bonds. Conversely, the Suzuki reaction using such 2-(2-bromoaryl)thiophenes provides 1,2-diheteroaryl-substituted benzene derivatives. These regiodivergent heteroarylations tolerate a range of substituents on the benzene ring and also several heteroarenes and provide a new route to π-extended polycyclic heteroaromatics. Moreover, these procedures employ easily available air-stable catalysts and inexpensive bases.
Introduction
When specific C–H bonds of organic molecules can be directly functionalized via catalytic reactions, it often provides simple synthetic methods.1 Metal-catalyzed C–H bond activation/functionalization of heteroarenes, reported by Ohta et al. 1990, now represents one of the most effective methods for the preparation of heteroarylated arenes.2 For such coupling reactions, initially in most cases, only the “most reactive” C–H bond of (hetero)arenes could be functionalized.3–7 Nevertheless, the C–H bond functionalization methodology will only be really synthetically useful when it will be possible to activate a specific C–H bond on the molecules. Therefore, the discovery of conditions allowing the regioselective functionalization of several bonds of the same molecule, also called regiodivergent functionalization, is a very important aspect of the current research on metal-catalyzed reactions.So far, the Pd-catalyzed reactions such as Suzuki coupling of heteroarylboronic acids with thiophenes bearing a 2-halobenzene substituent selectively lead to 1,2-diheteroarylated benzene derivatives (Scheme 1a).8 On the other hand, the C3-heteroarylation of 2-aryl-3-halothiophenes for the access to the corresponding heteroaryl-substituted thiophene derivatives via metal-catalyzed reactions has been rarely described (Scheme 1b).9,10 This might be due to the tedious multi-step route to many 3-halothiophene derivatives.11 To the best of our knowledge, the synthesis of 2-aryl-3-heteroarylthiophenes from 2-(2-bromoaryl)thiophenes has not been reported so far (Scheme 1c, bottom). We have recently reported that in the course of the heteroarylation of 1,2-dihalobenzenes by thiophenes, a partial Pd 1,4-migration occurred12,13 in some cases, leading to a mixture of 1,2-diheteroarylated benzenes and also to aryl-substituted biheteroarenes such as 2′-aryl-2,3′-bithiophenes.14 This Pd 1,4-migration on thiophene derivatives provides an appealing method for functionalizing the C–H bonds at the β-position of thiophenes which are often more difficult to activate than those at the α-position.15 In addition, so far, the preparation of 2′-aryl-2,3′-bithiophene derivatives requires multi-step synthesis and the substrate scope is very limited.16 Therefore, the potential of Pd-catalyzed 1,4-migration associated with direct arylation for the access to 2-aryl-3-heteroarylthiophenes via double C–H bond functionalization needed to be explored. Herein, we report (i) on the regioselectivity of the Pd-catalyzed direct arylations vs. Suzuki couplings of 2-(2-bromoaryl)thiophenes and on the scope of the Pd-catalyzed 1,4-migration associated with direct arylation for the formation of 2-aryl-3-heteroarylthiophenes via double C–H bond functionalization (Scheme 1c).
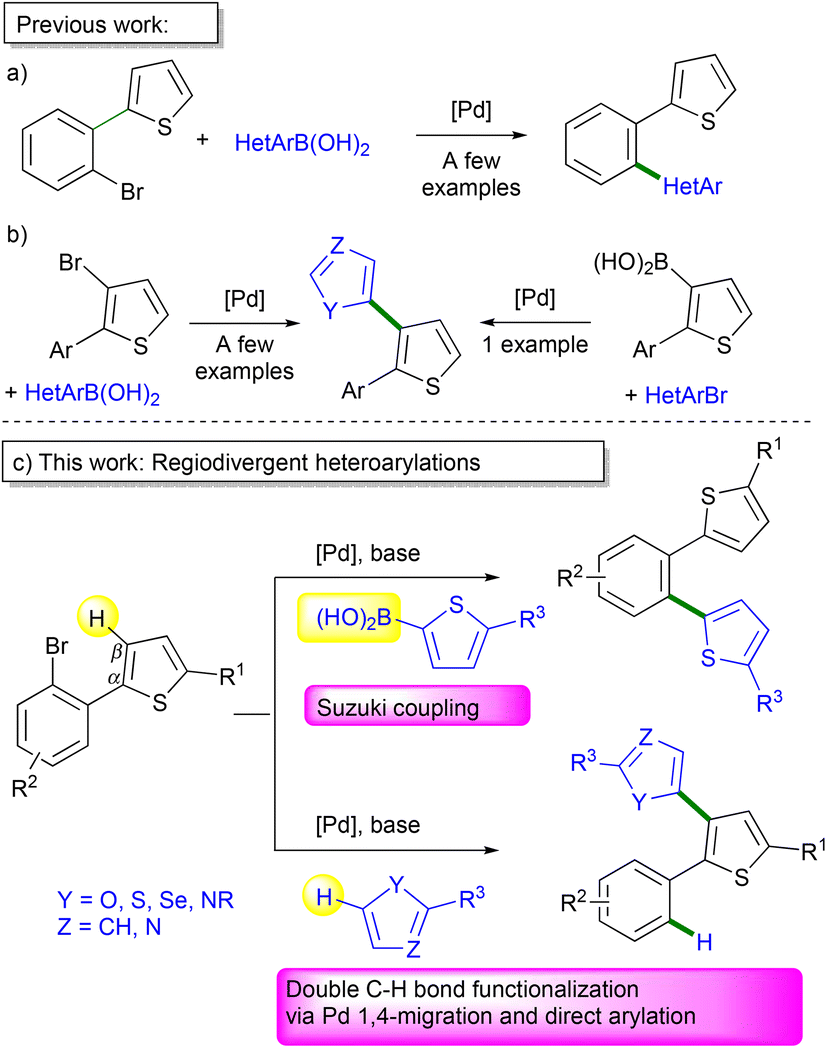 | ||
| Scheme 1 Pd-catalyzed direct arylation vs. Suzuki coupling of 2-(2-bromoaryl)thiophenes and related compounds. | ||
Results and discussion
First, we prepared a set of 2-(2-bromoaryl)thiophenes via Suzuki coupling or base deprotonation (Scheme 2). All the desired products 1a–1g were obtained in good yields.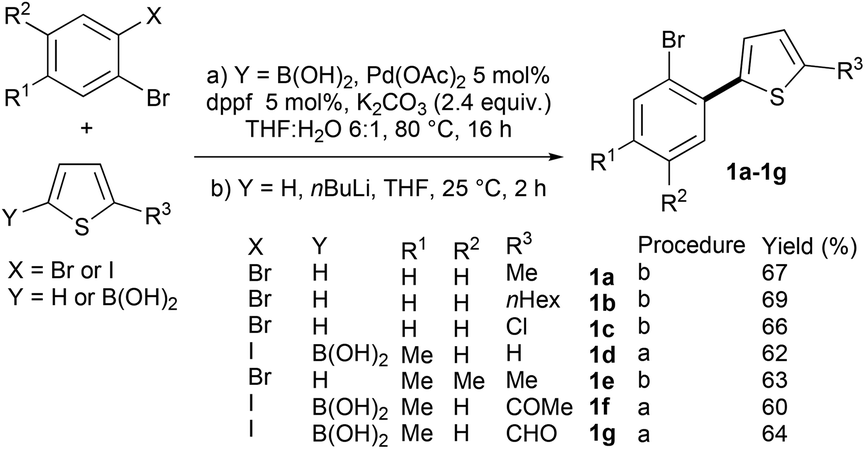 | ||
| Scheme 2 Preparation of 2-(2-bromoaryl)thiophenes 1a–1g. | ||
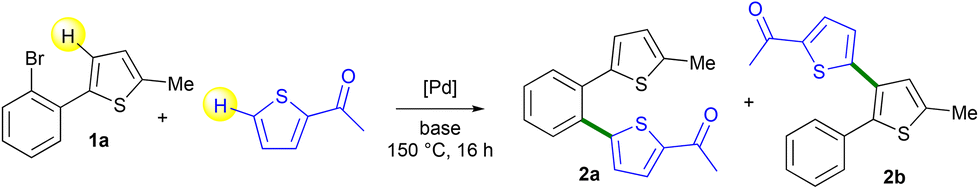
Then, we evaluated the selectivity for the Pd-catalyzed coupling of 2-(2-bromophenyl)-5-methylthiophene 1a with 2-acetylthiophene (Table 1). The best yield of the desired isomer 2b arising from the Pd-catalyzed 1,4-migration associated with direct arylation was obtained using 2 mol% Pd(OAc)2 catalyst and the KOAc base in DMA at 150 °C (Table 1, entry 1). Under these conditions, a mixture of 2a and 2b was obtained in a 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :81 ratio and 2b was isolated in 64% yield. The use of other bases such as Cs2CO3, K2CO3, NaOAc, CsOAc or KOPiv led to lower yields of 2b either due to lower conversions or poor selectivities (Table 1, entries 2–6). In the presence of the PdCl(C3H5)(dppb)17 catalyst, a quite similar 2a:2b selectivity and yield of 2b were obtained (Table 1, entry 7). The influence of a few solvents was also examined but both DMF and NMP led to lower selectivities in 2b, whereas xylene and diethyl carbonate (DEC) were ineffective (Table 1, entries 8–11). Finally, a lower reaction temperature of 130 °C instead of 150 °C gave 2b in only 53% yield due to partial conversion of 2-(2-bromophenyl)-5-methylthiophene 1a (Table 1, entry 12). It should be mentioned that this methodology is limited to the use of 2-(2-bromoaryl)thiophenes, as from 3-(2-bromoaryl)thiophenes, complex mixtures of products were obtained.18
:81 ratio and 2b was isolated in 64% yield. The use of other bases such as Cs2CO3, K2CO3, NaOAc, CsOAc or KOPiv led to lower yields of 2b either due to lower conversions or poor selectivities (Table 1, entries 2–6). In the presence of the PdCl(C3H5)(dppb)17 catalyst, a quite similar 2a:2b selectivity and yield of 2b were obtained (Table 1, entry 7). The influence of a few solvents was also examined but both DMF and NMP led to lower selectivities in 2b, whereas xylene and diethyl carbonate (DEC) were ineffective (Table 1, entries 8–11). Finally, a lower reaction temperature of 130 °C instead of 150 °C gave 2b in only 53% yield due to partial conversion of 2-(2-bromophenyl)-5-methylthiophene 1a (Table 1, entry 12). It should be mentioned that this methodology is limited to the use of 2-(2-bromoaryl)thiophenes, as from 3-(2-bromoaryl)thiophenes, complex mixtures of products were obtained.18
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Base | Conv. (%) | Ratio 2a:2b |
Yield in 2b (%) |
|
a [Pd] (0.02 equiv.), 2-acetylthiophene (2 equiv.), 2-(2-bromophenyl)-5-methylthiophene 1a (1 equiv.), base (3 equiv.), 150 °C, 16 h, 2a:2b ratios, determined by 1H NMR and GC/MS analyses of the crude mixtures, isolated yields.
b 130 °C.
|
||||||
| 1 | Pd(OAc)2 | DMA | KOAc | 100 | 19:81 |
64 |
| 2 | Pd(OAc)2 | DMA | Cs2CO3 | 5 | 56:44 |
nd |
| 3 | Pd(OAc)2 | DMA | K2CO3 | 100 | 46:54 |
nd |
| 4 | Pd(OAc)2 | DMA | NaOAc | 47 | 19:81 |
nd |
| 5 | Pd(OAc)2 | DMA | CsOAc | 84 | 40:60 |
nd |
| 6 | Pd(OAc)2 | DMA | KOPiv | 100 | 33:67 |
nd |
| 7 | PdCl(C3H5)(dppb) | DMA | KOAc | 100 | 24:76 |
61 |
| 8 | Pd(OAc)2 | DMF | KOAc | 100 | 26:74 |
50 |
| 9 | Pd(OAc)2 | NMP | KOAc | 100 | 24:76 |
52 |
| 10 | Pd(OAc)2 | Xylene | KOAc | 0 | — | 0 |
| 11 | Pd(OAc)2 | DEC | KOAc | 0 | — | 0 |
| 12 | Pd(OAc)2 | DMA | KOAc | 86 | 20:80 |
53b |
Next, the scope of this reaction using several heteroarenes and 2-(2-bromoaryl)thiophenes was investigated (Scheme 3). Using 2-isopropylthiazole and 1a, a similar a:b ratio was obtained and product 3b arising from the Pd 1,4-migration was isolated in 65% yield. 2-(2-Bromophenyl)-5-hexylthiophene 1b in the presence of 2-acetylthiophene or cyclopropyl-2-thienylketone afforded products 4b and 5b in 57% and 61% yields, respectively. No side-reaction on the cyclopropyl unit was detected in the course of this reaction. Then, the β-heteroarylation of the 2-chloro-substituted thiophene derivative 1c was studied. This reaction is particularly interesting as the access to 4-bromo-2-chlorothiophenes that could provide an alternative synthetic scheme for the preparation of similar products is very challenging.19 In the presence of 2-methylthiophene or 2-methyl-2-(thiophen-2-yl)-1,3-dioxolane, very high selectivities in favor of the formation of products b were observed with the formation of 6b and 7b in 69% and 71% yields, respectively. No cleavage of the thienyl C–Cl bond was observed allowing further transformations. In the presence of selenophene, 1c gave a lower yield of 52% of 8b due to the formation of unidentified side-products. 2-Isopropyl-4-methylthiazole, imidazo[1,2-a]pyrazine and 3,5-dimethylisoxazole in the presence of 1c afforded the target Pd 1,4-migration products 9b–11b in 35–65% yields. These three reactions were performed at a lower temperature of 130 °C due to a partial C–Cl bond cleavage at 150 °C. The reaction also tolerated the presence of two methyl substituents on the benzene ring. However, with this more electron-rich 2-arylthiophene derivative 1e, 5 mol% PdCl(C3H5)(dppb) catalyst had to be employed to reach a high conversion. With this catalyst, the target products 12b and 13b were obtained in 74% and 59% yields, respectively using 2-butylfuran and imidazo[1,2-a]pyrazine as the reaction partners. The reaction also tolerated a 2-acetyl substituent on the thienyl ring. From 2-acetyl-5-arylthiophene 1h and thiophene, furan and thiazole derivatives as reaction partners, very high selectivities (88–94%) of isomers b were observed and the target products 14b–16b were obtained in 72–81% yields. Finally, the reaction of the 2-formyl-5-arylthiophene derivative 1g and 2-methylthiophene or 2-isobutylthiazole gave products 17b and 18b with 95% and 87% selectivity and in 68% and 57% yields, respectively.
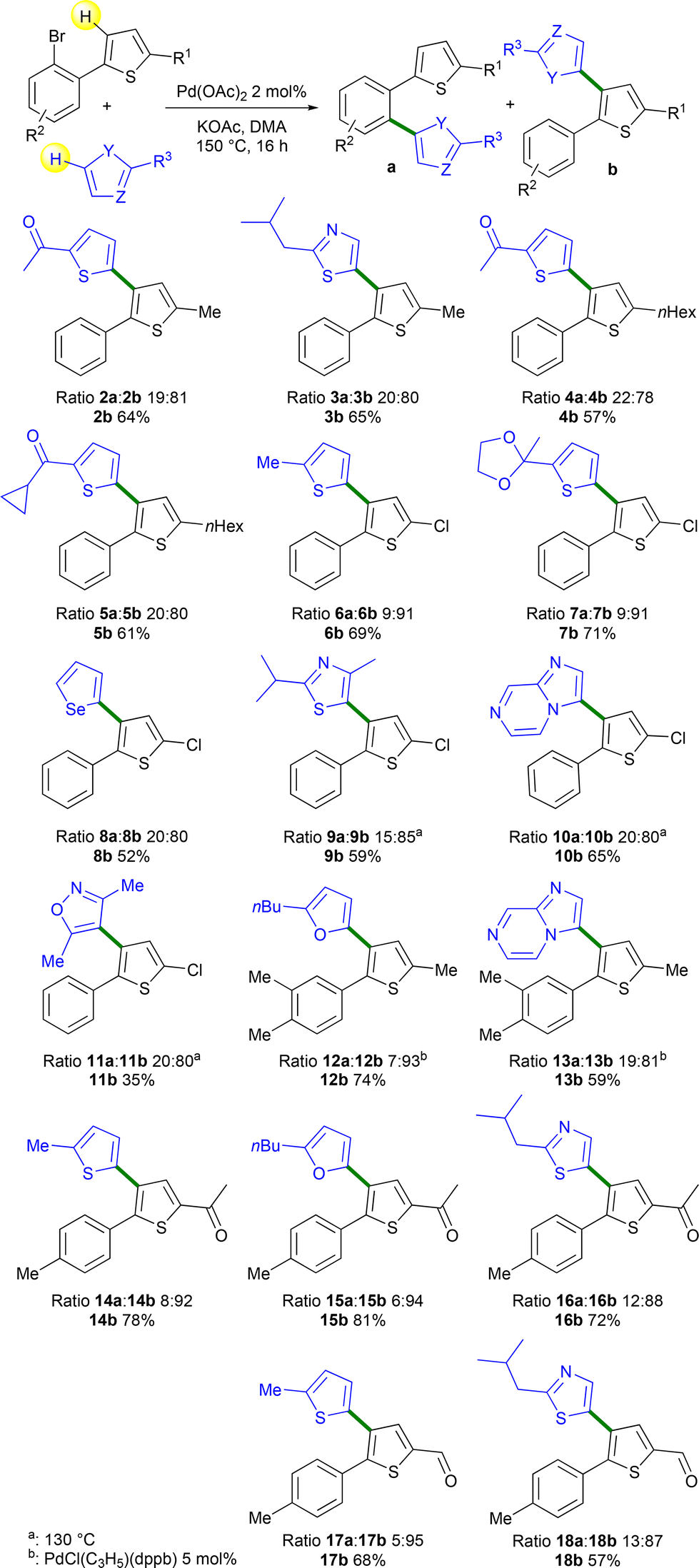 | ||
| Scheme 3 Heteroarylations of thienyl rings via Pd-catalyzed 1,4-migration associated with direct arylation. | ||
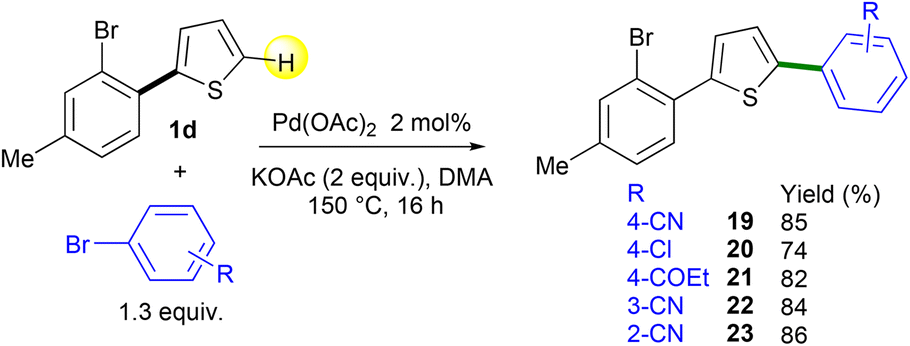
The use of Pd-catalyzed 1,4-migration associated with direct arylation should allow the introduction of an heteroarene regioselectively at one of the two β-positions of the thienyl ring of non-symmetrical 2,5-diarylthiophenes. The heteroarylation should occur at the β-C–H bond on the same side of the thienyl ring bearing the 2-bromoaryl substituent. The starting materials 19–23 were prepared in 74–86% yields by Pd-catalyzed direct C5-arylation from 2-(2-bromoaryl)thiophene 1d and a set of aryl bromides using the Pd(OAc)2 catalyst with the KOAc base in DMA (Scheme 4). Under these conditions – slight excess (1.3 equiv.) of an electron-poor aryl halide as the reaction partner – no cleavage of the C–Br bond of the bromotoluene unit of 1d was observed.
 | ||
| Scheme 4 Pd-catalyzed direct C5-arylation of the thienyl ring of 2-(2-bromoaryl)thiophene 1d. | ||
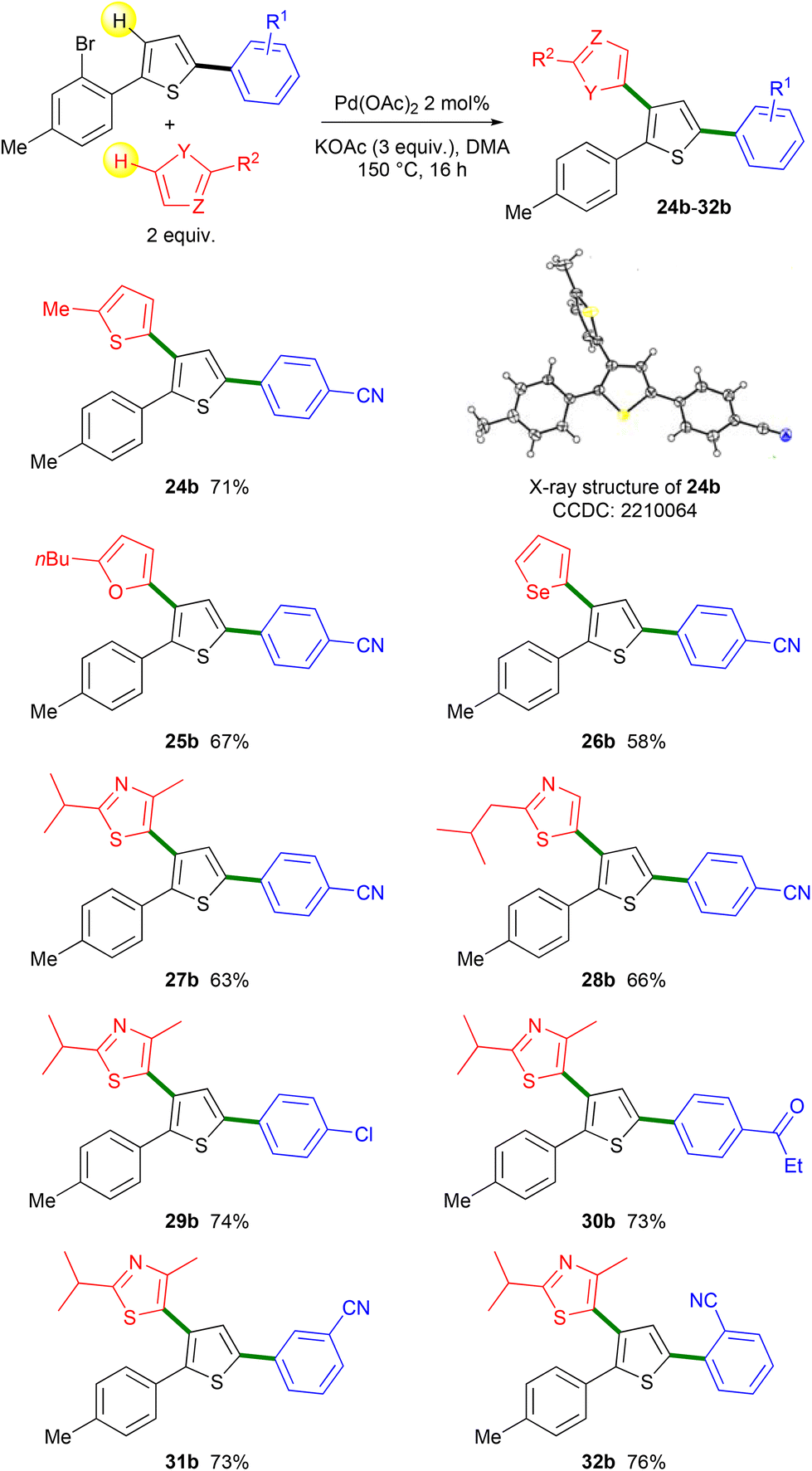
Then, the regioselectivity of the direct heteroarylation of 2,5-diarylthiophenes 19–23 was studied (Scheme 5). From 4-(5-(2-bromophenyl)thiophen-2-yl)benzonitrile 19 and 2-methylthiophene, the C4-heteroarylated product 24b was regioselectively obtained. The regioselectivity of the coupling reaction was determined by X-ray analysis, which confirms that there was no further Pd-migration after the initial Pd 1,4-migration. The reaction also tolerated furan, selenophene and thiazole derivatives as the reaction partners, affording products 25b–28b in 58–67% yields. From chloro- and propionyl-substituted 2,5-diarylthiophenes 20 and 21, the expected products 29b and 30b were also obtained in high yields. Again, under these conditions, no cleavage of the aryl C–Cl bond was observed allowing further transformations. No significant influence of the position of the nitrile substituent on the benzene ring was observed. Both meta- and ortho-nitrile-substituted 2,5-diarylthiophenes 22 and 23 led to the desired products 31b and 32b in similar yields. It should be mentioned that for all these heteroarylations of 2,5-diarylthiophenes 19–23, complete regioselectivities in favor of the formation of products b arising from Pd 1,4-migration were observed. This selectivity could be due to the conformation of 2,5-diarylthiophenes 19–23 which would favor the Pd 1,4-migration.
 | ||
| Scheme 5 Heteroarylations of thienyl rings via Pd-catalyzed 1,4-migration associated with direct arylation. | ||
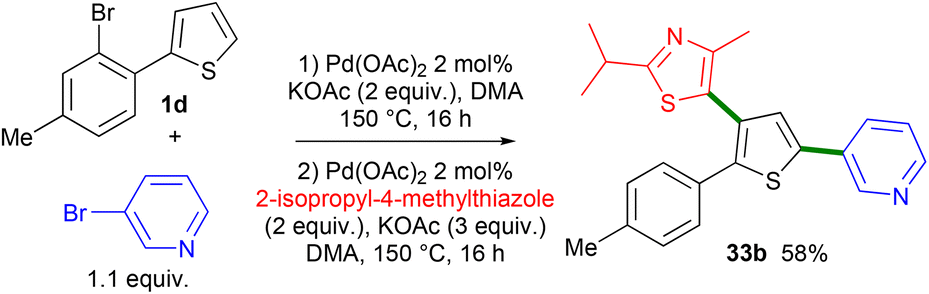
The synthesis of 2,5-diaryl-4-heteroarylthiophene without the isolation of the first arylation compound was also attempted (Scheme 6). From 2-(2-bromo-4-methylphenyl)thiophene 1d, 3-bromopyridine and 2 mol% Pd(OAc)2 catalyst, the 2,5-diarylated thiophene derivative was obtained in an impure form via filtration on a plug of silica. Then, 2-isopropyl-4-methylthiazole, 2 mol% Pd(OAc)2 and KOAc were added to the crude mixture and heated again at 150 °C for 16 h, affording the desired product 33b in 58% yield.
 | ||
| Scheme 6 Preparation of a tri(hetero)arylated thiophene derivative via successive direct heteroarylations. | ||
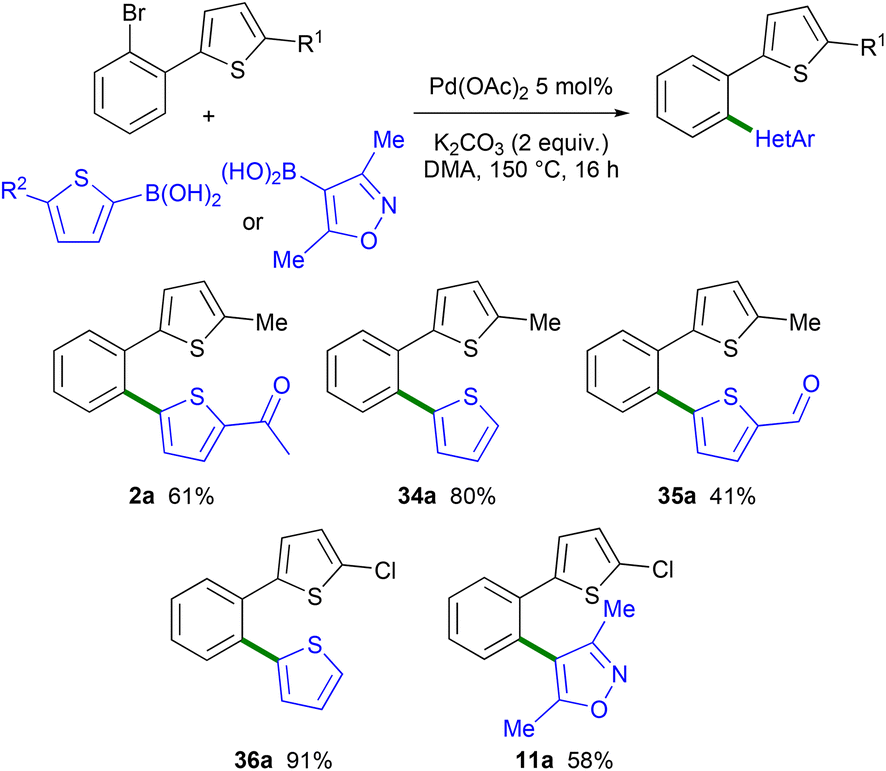
In order to compare the selectivity of the direct arylation reaction with Suzuki coupling, a few arylations of 2-(2-bromophenyl)-5-methylthiophene 1a and 2-(2-bromophenyl)-5-chlorothiophene 1c using arylboronic acids as the reaction partners were performed (Scheme 7). Again using 2 mol% Pd(OAc)2 catalyst and DMA as the solvent, but the K2CO3 base instead of a carboxylate base, the formation of the expected 1,2-diheteroaryl-substituted benzene derivatives 2a, 34a and 35a was observed with complete selectivity. It should be mentioned that in the presence of the KOPiv base instead of K2CO3, isomer 2a was obtained with 73% selectivity (27% of 2b) and in 52% yield indicating that the presence of a carboxylate base favors the Pd 1,4 migration. The reaction of 1c with 2-thienylboronic acid and the K2CO3 base also selectively led to the formation of the expected chloro-substituted 1,2-di(thiophen-2-yl)benzene 36a. Using 1c and (3,5-dimethylisoxazol-4-yl)boronic acid as the reaction partner, product 11a was also obtained with complete selectivity. These results demonstrate that the combination of Pd-catalyzed 1,4 migration reactions associated with direct arylation and Suzuki coupling can be employed as a tool to control regiodivergent heteroarylations.
 | ||
| Scheme 7 Heteroarylations of benzene rings via Suzuki coupling. | ||
π-Extended polycyclic heteroaromatics20 have found several applications in the preparation of optoelectronic devices. The topology of the ring fusion and the substituents at their periphery are essential to tune their electronic properties. Therefore, we also attempted to apply the Pd-catalyzed 1,4-migration associated with direct heteroarylation to the preparation of such products. First, we employed a 4-bromothiophene derivative as a reaction partner with 1a (Scheme 8a). The desired naphtho[1,2-b:3,4-b′]dithiophene 37 was obtained in a moderate yield using an extended reaction time (48 h). Intermediate 38 was also isolated in a low yield in an impure form. We also studied the reaction outcome of 2-(2-bromophenyl)-5-methylthiophene 1a in the absence of the heteroaryl coupling partner (Scheme 8b). The formation of the desired fused polyheteroaromatic naphtho[1,2-b:3,4-c′]dithiophene 39 in 65% yield was observed. The structure of 39 was confirmed by X-ray analysis. The formation of 39 likely proceeded via the initial formation of 2-(2-bromophenyl)-2′-phenyl-3,3′-bithiophene. Then, an intramolecular Pd-catalyzed direct arylation gives naphtho[1,2-b:3,4-c′]dithiophene 39. The use of 1e also provides the corresponding naphtho[1,2-b:3,4-c′]dithiophene 40 in a moderate yield. This one-pot multi-step synthesis of π-extended polycyclic heteroaromatics is very attractive, as so far very few methods allow the preparation of such fused polyheteroaromatics.21
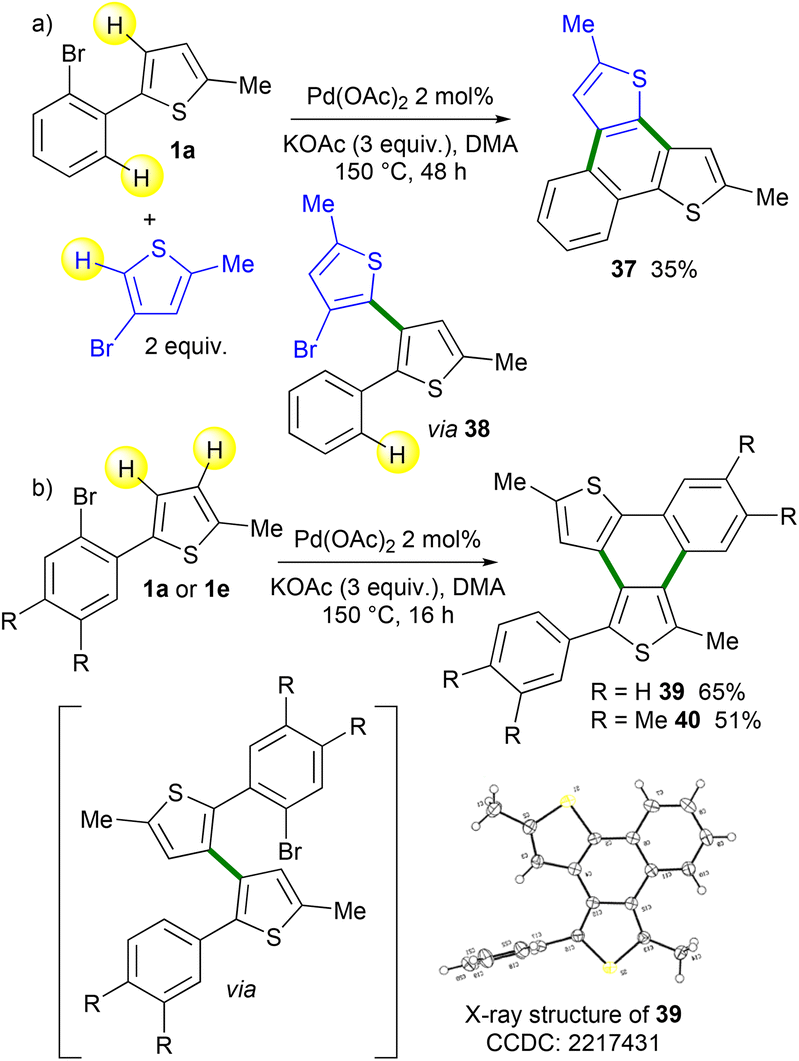 | ||
| Scheme 8 Application of the Pd-catalyzed 1,4-migration associated with direct arylation methodology for the preparation of π-extended polycyclic heteroaromatics. | ||
The access to 1,2-bis(heteroaryl)benzenes a proceeds certainly via a classical Suzuki coupling22 mechanism, whereas the access to 2′-aryl-2,3′-biheteroarenes b occurs likely via a Pd 1,4-migration followed by a direct arylation23 as described in Scheme 9. In both cases, the first step of the catalytic cycle involves the oxidative addition of 2-(2-bromoaryl)thiophene to give intermediate A. For Suzuki coupling with arylboronic acids, classical transmetalation followed by reductive elimination gives 1,2-bis(heteroaryl)benzenes a. By contrast, with simple heteroarenes using the KOAc base, a Pd 1,4-migration12 occurs to give intermediate B. Then, after a concerted metallation deprotonation (CMD)23 of the heteroarene coupling partner, intermediate C is obtained. Finally, reductive elimination provides 2′-aryl-2,3′-biheteroarenes b with the regeneration of the catalyst.
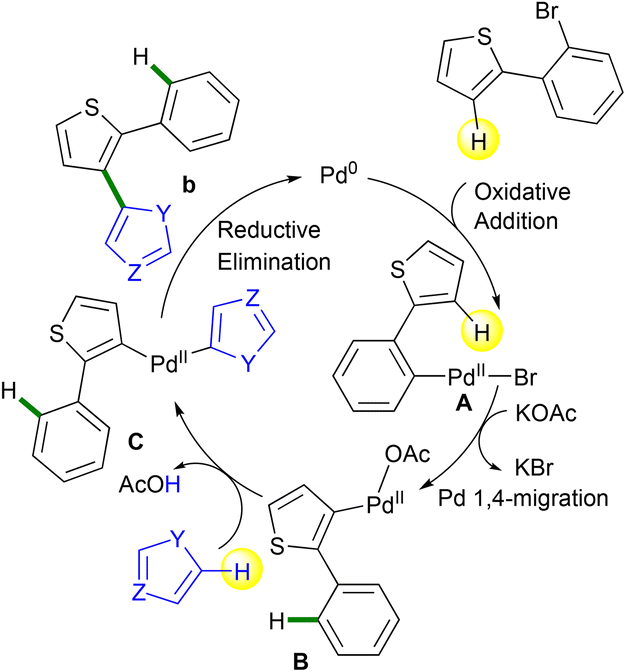 | ||
| Scheme 9 Proposed catalytic cycle for the access to b products. | ||
Conclusions
In summary, the use of either Pd-catalyzed 1,4-migration/direct arylation or Suzuki coupling allowed the regiodivergent heteroarylation of 2-(2-bromoaryl)thiophenes. Using Suzuki coupling, 1,2-diheteroarylbenzenes were obtained. Conversely, the use of C–H heteroarylation conditions led to 2-aryl-3-heteroarylthiophenes due to a Pd 1,4-migration between the aryl and β-position of the thienyl units before heteroarylation. In the course of this coupling reaction, a new C–C bond arises from the functionalization of two C–H bonds. In addition to the advantage of being able to synthesize two classes of products from the same molecular building block, heteroarylation at the β-position of thiophenes by this method is synthetically very attractive because very few β-bromothiophenes, which could allow access to the same products by a classical direct arylation, are commercially available at an affordable cost. The reaction tolerates a variety of heteroarenes and substituents at the C5-position on 2-(2-bromoaryl)thiophenes and provides a new route to π-extended polycyclic heteroaromatics. Moreover, these reactions employ a low loading of air stable palladium sources associated with inexpensive bases. Indeed, we believe that the development of this methodology using other coupling partners for regiodivergent couplings will lead to a powerful tool for organic synthesis.Experimental
General
DMA (99+%) extra pure and KOAc (99%) were purchased from ACROS and used without purification. All reagents were weighed and handled in air. All reactions were carried out under an inert atmosphere with standard Schlenk techniques. 1H, 19F and 13C NMR spectra were recorded on a Bruker Avance III 400 MHz spectrometer. High-resolution mass spectra were measured on a Bruker MaXis 4G spectrometer. Melting points were determined with a Kofler hot bench system. Chromatography purifications were performed using a CombiFlash NextGen 300 with Buchi FlashPure cartridges containing 40 μm irregular silica.Preparation of the PdCl(C3H5)(dppb) catalyst17
An oven-dried 40 mL Schlenk tube equipped with a magnetic stirring bar under an argon atmosphere was charged with [Pd(C3H5)Cl]2 (182 mg, 0.5 mmol) and dppb (426 mg, 1 mmol). 10 mL of anhydrous dichloromethane was added, and then the solution was stirred at room temperature for twenty minutes. The solvent was removed under vacuum. The yellow powder was used without purification. 31P NMR (162 MHz, CDCl3) δ = 19.3 (s).General procedures for the preparation of 2-(2-bromophenyl)thiophenes 1a–1g
Procedure a: As a typical experiment, a mixture of the 1,2-dihalobenzene derivative (3 mmol), the thiophen-2-ylboronic acid derivative (4.5 mmol), 2.0 M K2CO3 aq. (3.6 mL), Pd(OAc)2 (33.6 mg, 0.15 mmol) and dppf (83.1 mg, 0.15 mmol) in THF (18 mL) was stirred at 80 °C for 16 h. After being allowed to cool to room temperature, the resulting mixture was extracted with Et2O (3 × 10 mL). The combined organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel to give 2-(2-bromoaryl)thiophenes.Procedure b: As a typical experiment, to a solution of the thiophene derivative (12.0 mmol) in THF (25 mL), nBuLi was added dropwise (1.55 M in hexane, 7.70 mL, 12.0 mmol) at 0 °C. After being stirred for 1 h, the 1,2-dihalobenzene derivative (10.0 mmol) was added to the solution at the same temperature. Then, the mixture was warmed to room temperature and stirred for 2 h. The residue was purified by column chromatography on silica gel to afford 2-(2-bromoaryl)thiophenes.
1H NMR (400 MHz, CDCl3): δ 7.66 (dd, J = 8.0, 1.3 Hz, 1H), 7.46 (dd, J = 7.8, 1.7 Hz, 1H), 7.31 (td, J = 7.5, 1.3 Hz, 1H), 7.16 (td, J = 7.5, 1.7 Hz, 1H), 7.11 (d, J = 3.5 Hz, 1H), 6.77 (d, J = 3.5 Hz, 1H), 2.54 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 140.9, 139.4, 135.7, 133.8, 131.9, 128.8, 127.9, 127.5, 125.4, 122.8, 15.4.
LRMS calcd for [M]+ C11H9BrS 252, found: 252.
1H NMR (400 MHz, CDCl3): δ 7.68 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 7.21–7.12 (m, 2H), 6.81 (d, J = 3.5 Hz, 1H), 2.88 (t, J = 7.4 Hz, 2H), 1.84–1.69 (m, 2H), 1.52–1.29 (m, 6H), 0.95 (t, J = 7.4 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 147.1, 139.1, 135.8, 133.8, 131.8, 128.7, 127.6, 127.4, 124.1, 123.7, 31.7 (m), 30.3, 29.0, 22.7, 14.2.
LRMS calcd for [M]+ C16H19BrS 324, found: 324.
1H NMR (400 MHz, CDCl3): δ 7.67 (dd, J = 8.0, 1.3 Hz, 1H), 7.43 (dd, J = 7.8, 1.7 Hz, 1H), 7.33 (td, J = 7.6, 1.3 Hz, 1H), 7.19 (td, J = 7.7, 1.7 Hz, 1H), 7.06 (d, J = 3.8 Hz, 1H), 6.93 (d, J = 3.8 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ 140.4, 134.6, 133.9, 131.8, 130.7, 129.5, 127.7, 127.2, 126.2, 122.8.
LRMS calcd for [M]+ C10H6BrClS 274, found: 274.
1H NMR (400 MHz, CDCl3): δ 7.51 (s, 1H), 7.39–7.34 (m, 2H), 7.27 (dd, J = 5.6, 1.2 Hz, 1H), 7.13 (d, J = 7.7 Hz, 1H), 7.10 (dd, J = 5.2, 3.6 Hz, 1H), 2.36 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 142.0, 139.5, 134.2, 132.5, 131.8, 128.4, 127.6, 127.0, 125.9, 122.7, 20.9.
LRMS calcd for [M]+ C11H9BrS 252, found: 252.
1H NMR (400 MHz, CDCl3): δ 7.47 (s, 1H), 7.27 (s, 1H), 7.11 (d, J = 3.5 Hz, 1H), 6.79 (d, J = 3.5 Hz, 1H), 2.57 (s, 3H), 2.29 (s, 3H), 2.26 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 140.3, 139.6, 137.8, 136.1, 134.4, 132.8, 132.7, 127.4, 125.3, 119.2, 19.3, 19.2, 15.4.
LRMS calcd for [M]+ C13H13BrS 280, found: 280.
1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 3.5 Hz, 1H), 7.51 (s, 1H), 7.36 (d, J = 7.8 Hz, 1H), 7.27 (d, J = 3.5 Hz, 1H), 7.15 (d, J = 7.8 Hz, 1H), 2.57 (s, 3H), 2.36 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 190.8, 150.4, 144.2, 140.7, 134.5, 132.3, 131.5, 131.4, 128.7, 128.6, 122.3, 26.8, 20.9.
LRMS calcd for [M]+ C13H11BrOS 296, found: 296.
1H NMR (400 MHz, CDCl3): δ 9.91 (s, 1H), 7.73 (d, J = 3.5 Hz, 1H), 7.52 (s, 1H), 7.39–7.34 (m, 2H), 7.16 (d, J = 7.8 Hz, 1H), 2.36 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 183.0, 151.9, 143.6, 141.0, 136.2, 134.5, 131.5, 131.2, 128.8, 128.7, 122.3, 20.9.
LRMS calcd for [M]+ C12H9BrOS 280, found: 280.
General procedure for the preparation of 2′-aryl-2,3′-biheteroarenes 2b–18b
As a typical experiment, the reaction of the 2-(2-bromoaryl)thiophene derivatives 1a–1g (1 mmol), heteroarene (2 mmol) and KOAc (0.294 g, 3 mmol) at 150 °C for 16 h in DMA (4 mL) in the presence of Pd(OAc)2 (4.5 mg, 0.02 mmol) under argon afforded the coupling product after evaporation of the solvent and purification on silica gel. The a:b ratios were determined by 1H NMR and GC/MS analyses of the crude mixtures.
:81 ratio and 2b was isolated in 64% (0.191 g) yield as a white solid: mp 92–94 °C. Eluent pentane, Rf2a = 0.30, Rf2b = 0.33.
1H NMR (400 MHz, CDCl3): δ 7.46 (d, J = 3.9 Hz, 1H), 7.40–7.32 (m, 5H), 6.91 (s, 1H), 6.79 (d, J = 3.9 Hz, 1H), 2.51 (s, 3H), 2.49 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 190.6, 147.5, 142.6, 139.4, 138.7, 133.8, 133.0, 129.8, 129.7, 128.8, 128.4, 127.5, 126.5, 26.7, 15.3.
HRMS calcd for [M + H]+ C17H15OS2 299.0559, found: 299.0557.
:80 ratio and 3b was isolated in 65% (0.203 g) yield as a colorless oil. Eluent pentane, Rf3a = 0.20, Rf3b = 0.23.
1H NMR (400 MHz, CDCl3): δ 7.47 (s, 1H), 7.39–7.30 (m, 5H), 6.86 (s, 1H), 2.77 (d, J = 7.6 Hz, 2H), 2.50 (s, 3H), 2.09–1.99 (m, 1H), 0.95 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 169.5, 139.9, 139.1, 137.5, 133.9, 132.5, 129.9, 128.7, 128.2, 127.6, 127.5, 42.4, 29.8, 22.4, 15.3.
HRMS calcd for [M + H]+ C18H20NS2 314.1032, found: 314.1029.
:78 ratio and 4b was isolated in 57% (0.210 g) yield as a yellow oil. Eluent pentane, Rf4a = 0.27, Rf4b = 0.30.
1H NMR (400 MHz, CDCl3): δ 7.46 (d, J = 4.0 Hz, 1H), 7.41–7.32 (m, 5H), 6.92 (s, 1H), 6.80 (d, J = 4.0 Hz, 1H), 2.81 (t, J = 7.6 Hz, 2H), 2.49 (s, 3H), 1.72 (quint., J = 7.6 Hz, 2H), 1.49–1.39 (m, 2H), 1.39–1.30 (m, 4H), 0.91 (t, J = 7.6 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 190.6, 147.6, 145.6, 142.5, 138.4, 133.9, 133.0, 129.8, 129.5, 128.8, 128.3, 126.5, 126.3, 31.7, 31.5, 30.1, 28.9, 26.7, 22.7, 14.2.
HRMS calcd for [M + H]+ C22H25OS2 369.1341, found: 369.1341.
:80 ratio and 5b was isolated in 61% (0.240 g) yield as a yellow oil. Eluent pentane/EtOAc 80/20, Rf5a = 0.29, Rf5b = 0.33.
1H NMR (400 MHz, CDCl3): δ 7.59 (d, J = 4.0 Hz, 1H), 7.42–7.32 (m, 5H), 6.93 (s, 1H), 6.83 (d, J = 4.0 Hz, 1H), 2.81 (t, J = 7.6 Hz, 2H), 2.50–2.39 (m, 1H), 1.72 (quint., J = 7.6 Hz, 2H), 1.49–1.38 (m, 2H), 1.38–1.28 (m, 4H), 1.25–1.18 (m, 2H), 1.02–0.95 (m, 2H), 0.91 (t, J = 7.6 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 192.8, 147.0, 145.5, 143.0, 138.3, 134.0, 131.9, 129.8, 129.6, 128.8, 128.3, 126.6, 126.4, 31.7, 31.6, 30.1, 29.0, 22.7, 17.9, 14.2, 11.3.
HRMS calcd for [M + H]+ C24H27OS2 395.1498, found: 395.1503.
:91 ratio and 6b was isolated in 69% (0.201 g) yield as a colorless oil. Eluent pentane, Rf6a = 0.30, Rf6b = 0.33.
1H NMR (400 MHz, CDCl3): δ 7.42–7.31 (m, 5H), 7.01 (s, 1H), 6.66 (d, J = 3.5 Hz, 1H), 6.55 (d, J = 3.5 Hz, 1H), 2.41 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 139.9, 136.2, 135.0, 133.3, 130.8, 130.0, 128.8, 128.7, 128.5, 128.4, 126.1, 125.5, 15.4.
HRMS calcd for [M + H]+ C15H12ClS2 291.0064, found: 291.0064.
:91 ratio and 7b was isolated in 71% (0.257 g) yield as a yellow solid: mp 128–130 °C. Eluent pentane/EtOAc 80/20, Rf7a = 0.23, Rf7b = 0.27.
1H NMR (400 MHz, CDCl3): δ 7.40–7.31 (m, 5H), 7.02 (s, 1H), 6.82 (d, J = 3.7 Hz, 1H), 6.67 (d, J = 3.7 Hz, 1H), 4.05–3.98 (m, 2H), 3.98–3.91 (m, 2H), 1.74 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 147.0, 137.0, 136.9, 133.1, 130.3, 129.9, 129.0, 128.8, 128.6, 128.4, 125.9, 124.4, 107.2, 65.1, 27.5.
HRMS calcd for [M + H]+ C18H16ClO2S2 363.0275, found: 363.0277.
:80 ratio and 11b was isolated in 52% (0.168 g) yield as a colorless oil. Eluent pentane, Rf8a = 0.29, Rf8b = 0.33.
1H NMR (400 MHz, CDCl3): δ 7.85 (dd, J = 5.6, 1.2 Hz, 1H), 7.43–7.36 (m, 5H), 7.15 (dd, J = 5.6, 3.8 Hz, 1H), 7.11 (dd, J = 3.8, 1.2 Hz, 1H), 7.06 (s, 1H).
13C NMR (100 MHz, CDCl3): δ 142.7, 136.6, 133.0, 132.8, 131.0, 130.3, 129.6, 128.9, 128.8, 128.5, 128.3.
HRMS calcd for [M + H]+ C14H10ClSSe 324.9351, found: 324.9350.
:85 ratio and 9b was isolated in 59% (0.197 g) yield as a yellow solid: mp 91–93 °C. Eluent pentane/EtOAc 80/20, Rf9a = 0.26, Rf9b = 0.30.
1H NMR (400 MHz, CDCl3): δ 7.31–7.15 (m, 5H), 6.89 (s, 1H), 3.23 (sept., J = 7.6 Hz, 1H), 2.01 (s, 3H), 1.37 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 176.8, 149.0, 140.2, 133.2, 130.1, 128.9, 128.8, 128.5, 128.2, 127.3, 123.8, 33.5, 23.3, 15.8.
HRMS calcd for [M + H]+ C17H17ClNS2 334.0486, found: 334.0489.
:80 ratio and 10b was isolated in 65% (0.203 g) yield as a white solid: mp 140–142 °C. Eluent pentane/EtOAc 60/40, Rf10a = 0.21, Rf10b = 0.23.
1H NMR (400 MHz, CDCl3): δ 9.08 (d, J = 1.5 Hz, 1H), 7.78 (s, 1H), 7.64 (d, J = 4.6 Hz, 1H), 7.49 (dd, J = 4.6, 1.5 Hz, 1H), 7.27–7.17 (m, 3H), 7.09 (d, J = 8.2 Hz, 2H), 7.03 (s, 1H).
13C NMR (100 MHz, CDCl3): δ 144.2, 141.0, 140.9, 135.7, 132.6, 130.6, 129.7, 129.4, 129.0, 128.9, 127.5, 122.6, 121.2, 116.9.
HRMS calcd for [M + H]+ C16H11ClN3S 312.0357, found: 312.0354.
:80 ratio and 11b was isolated in 35% (0.101 g) yield as a white solid: mp 86–88 °C. Eluent pentane/EtOAc 80/20, Rf11a = 0.30, Rf11b = 0.33.
1H NMR (400 MHz, CDCl3): δ 7.33–7.27 (m, 3H), 7.23–7.19 (m, 2H), 6.79 (s, 1H), 2.06 (s, 3H), 1.98 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 166.2, 159.2, 140.1, 133.2, 129.2, 129.1, 129.0, 128.2, 128.0, 125.0, 111.2, 11.4, 10.5.
HRMS calcd for [M + H]+ C15H13ClNOS 290.0401, found: 290.0402.
:93 ratio and 12b was isolated in 74% (0.240 g) yield as a yellow oil. Eluent pentane, Rf12a = 0.14, Rf12b = 0.17.
1H NMR (400 MHz, CDCl3): δ 7.23 (s, 1H), 7.19 (d, J = 7.7 Hz, 1H), 7.11 (d, J = 7.7 Hz, 1H), 6.99 (q, J = 1.1 Hz, 1H), 6.01 (d, J = 3.2 Hz, 1H), 5.88 (d, J = 3.2 Hz, 1H), 2.57 (t, J = 7.6 Hz, 2H), 2.49 (d, J = 1.1 Hz, 3H), 2.29 (s, 3H), 2.27 (s, 3H), 1.57 (quint., J = 7.6 Hz, 2H), 1.35 (sext., J = 7.6 Hz, 2H), 0.91 (t, J = 7.6 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 155.0, 149.0, 138.3, 136.5, 136.2, 135.1, 132.3, 130.8, 129.7, 127.6, 127.1, 125.6, 107.0, 106.3, 30.4, 27.9, 22.4, 19.9, 19.7, 15.3, 14.0.
HRMS calcd for [M + H]+ C21H25OS 325.1621, found: 325.1625.
:81 ratio and 13b was isolated in 59% (0.188 g) yield as a colorless oil. Eluent pentane/EtOAc 60/40, Rf13a = 0.20, Rf13b = 0.23.
1H NMR (400 MHz, CDCl3): δ 9.08 (s, 1H), 7.78 (s, 1H), 7.63 (d, J = 4.6 Hz, 1H), 7.46–7.43 (m, 2H), 7.21 (s, 1H), 6.41–6.35 (m, 2H), 2.38 (s, 3H), 2.33 (s, 3H), 2.29 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 143.9, 140.9, 140.8, 139.1, 139.0, 136.7, 135.6, 133.6, 132.6, 131.4, 129.3, 126.5, 125.8, 125.5, 123.3, 116.9, 19.8, 19.5, 15.3.
HRMS calcd for [M + H]+ C19H18N3S 320.1216, found: 320.1214.
:92 ratio and 14b was isolated in 78% (0.243 g) yield as a yellow oil. Eluent pentane/EtOAc 80/20, Rf14a = 0.14, Rf14b = 0.17.
1H NMR (400 MHz, CDCl3): δ 7.71 (s, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 6.71 (d, J = 3.5 Hz, 1H), 6.59 (d, J = 3.5 Hz, 1H), 2.57 (s, 3H), 2.73 (s, 3H), 2.38 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 190.6, 146.8, 141.7, 140.1, 139.1, 134.8, 134.6, 132.2, 130.3, 129.5, 129.4, 126.3, 125.5, 26.6, 21.4, 15.3.
HRMS calcd for [M + H]+ C18H17OS2 313.0715, found: 313.0715.
:94 ratio and 15b was isolated in 81% (0.274 g) yield as a yellow oil. Eluent pentane/EtOAc 80/20, Rf15a = 0.33, Rf15b = 0.37.
1H NMR (400 MHz, CDCl3): δ 7.88 (s, 1H), 7.36 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 6.03 (d, J = 3.5 Hz, 1H), 5.91 (d, J = 3.5 Hz, 1H), 2.64–2.55 (m, 5H), 2.40 (s, 3H), 1.58 (quint., J = 7.6 Hz, 2H), 1.35 (sext., J = 7.6 Hz, 2H), 0.92 (t, J = 7.6 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 190.8, 155.9, 147.4, 145.4, 142.3, 139.1, 132.3, 130.7, 129.5, 129.4, 129.3, 108.2, 106.6, 30.3, 27.8, 26.7, 22.4, 21.5, 13.9.
LRMS calcd for [M]+ C21H22O2S 338, found: 338.
HRMS calcd for [M + H]+ C21H23O2S 339.1413, found: 339.1414.
:88 ratio and 16b was isolated in 58% (0.256 g) yield as a yellow solid: mp 87–89 °C. Eluent pentane/EtOAc 80/20, Rf16a = 0.20, Rf16b = 0.23.
1H NMR (400 MHz, CDCl3): δ 7.71 (s, 1H), 7.51 (s, 1H), 7.27 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 2.80 (d, J = 7.6 Hz, 2H), 2.57 (s, 3H), 2.38 (s, 3H), 2.10–2.02 (m, 1H), 0.96 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 190.5, 170.4, 148.4, 142.3, 140.6, 139.6, 134.2, 131.1, 129.7, 129.6, 129.4, 128.7, 42.4, 29.8, 26.7, 22.3, 21.5.
HRMS calcd for [M + H]+ C20H22NOS2 356.1137, found: 356.1136.
:95 ratio and 17b was isolated in 68% (0.203 g) yield as a yellow oil. Eluent pentane/EtOAc 80/20, Rf17ª = 0.23, Rf17b = 0.27.
1H NMR (400 MHz, CDCl3): δ 9.88 (s, 1H), 7.79 (s, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 6.72 (d, J = 3.5 Hz, 1H), 6.60 (d, J = 3.5 Hz, 1H), 2.44 (s, 3H), 2.40 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 182.9, 148.2, 141.2, 140.4, 139.5, 138.4, 134.4, 132.6, 130.2, 129.6, 129.4, 126.5, 125.6, 21.5, 15.4.
HRMS calcd for [M + H]+ C17H15OS2 299.0559, found: 299.0557.
:87 ratio and 18b was isolated in 57% (0.194 g) yield as a yellow solid: mp 90–92 °C. Eluent pentane/EtOAc 80/20, Rf18a = 0.10, Rf18b = 0.12.
1H NMR (400 MHz, CDCl3): δ 9.90 (s, 1H), 7.80 (s, 1H), 7.53 (s, 1H), 7.28 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 8.0 Hz, 2H), 2.80 (d, J = 7.6 Hz, 2H), 2.39 (s, 3H), 2.10–2.02 (m, 1H), 0.97 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 182.7, 170.6, 149.8, 141.6, 140.8, 140.0, 138.0, 130.8, 129.7, 129.5, 129.4, 129.1, 42.4, 29.8, 22.3, 21.5.
HRMS calcd for [M + H]+ C19H20NOS2 342.0981, found: 342.0981.
General procedure for the preparation of 2,5-diarylthiophenes 19–23
As a typical experiment, the reaction of 2-(2-bromo-4-methylphenyl)thiophene 1d (0.253 g, 1 mmol), aryl bromide (1.3 mmol) and KOAc (0.196 g, 2 mmol) at 150 °C for 16 h in DMA (4 mL) in the presence of Pd(OAc)2 (4.5 mg, 0.02 mmol) under argon affords the coupling product after evaporation of the solvent and purification on silica gel.1H NMR (400 MHz, CDCl3): δ 7.68 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 8.3 Hz, 2H), 7.52 (s, 1H), 7.40–7.36 (m, 2H), 7.28 (d, J = 3.7 Hz, 1H), 7.15 (d, J = 8.1 Hz, 1H), 2.36 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 143.6, 142.1, 140.0, 138.5, 134.4, 132.8, 131.6, 131.4, 129.0, 128.5, 125.9, 125.1, 122.3, 118.9, 110.6, 20.8.
LRMS calcd for [M]+ C18H12BrNS 353, found: 353.
1H NMR (400 MHz, CDCl3): δ 7.55 (d, J = 8.3 Hz, 2H), 7.53 (s, 1H), 7.40 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 8.3 Hz, 2H), 7.29–7.23 (m, 2H), 7.15 (d, J = 8.1 Hz, 1H), 2.38 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 143.4, 141.7, 139.6, 134.3, 133.4, 132.9, 132.1, 131.5, 129.2, 128.7, 128.5, 127.0, 123.4, 122.4, 20.8.
LRMS calcd for [M]+ C17H12BrClS 364, found: 364.
1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 8.3 Hz, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.52 (s, 1H), 7.43–7.38 (m, 2H), 7.28 (d, J = 3.7 Hz, 1H), 7.15 (d, J = 8.1 Hz, 1H), 3.00 (q, J = 7.6 Hz, 2H), 2.37 (s, 3H), 1.25 (t, J = 7.6 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 200.1, 143.3, 143.0, 139.9, 138.6, 135.7, 134.4, 132.0, 131.5, 129.0, 128.9, 128.5, 125.6, 124.5, 122.4, 31.9, 20.9, 8.4.
LRMS calcd for [M]+ C20H17BrOS 384, found: 384.
1H NMR (400 MHz, CDCl3): δ 7.89 (s, 1H), 7.82 (d, J = 8.3 Hz, 1H), 7.57–7.51 (m, 2H), 7.48 (t, J = 7.8 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.33 (d, J = 3.7 Hz, 1H), 7.27 (d, J = 3.7 Hz, 1H), 7.16 (d, J = 8.1 Hz, 1H), 2.37 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 143.0, 141.8, 140.0, 135.7, 134.4, 131.8, 131.5, 130.7, 129.9, 129.8, 129.1, 128.9, 128.5, 124.4, 122.4, 118.7, 113.3, 20.9.
LRMS calcd for [M]+ C18H12BrNS 353, found: 353.
1H NMR (400 MHz, CDCl3): δ 7.74 (d, J = 8.3 Hz, 1H), 7.68–7.63 (m, 2H), 7.59 (t, J = 7.8 Hz, 1H), 7.52 (s, 1H), 7.41 (d, J = 8.3 Hz, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.31 (d, J = 3.7 Hz, 1H), 7.15 (d, J = 8.1 Hz, 1H), 2.37 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 143.9, 139.9, 139.7, 137.4, 134.5, 134.4, 133.1, 131.7, 131.6, 129.5, 128.7, 128.5, 127.6, 127.5, 122.3, 119.0, 109.8, 20.8.
LRMS calcd for [M]+ C18H12BrNS 353, found: 353.
General procedure for the preparation of heteroarylated 2,3-diarylthiophenes 24b–32b
As a typical experiment, the reaction of the 2-(2-bromophenyl)-5-arylthiophene derivative 19–23 (1 mmol), heteroarene (2 mmol) and KOAc (0.294 g, 3 mmol) at 150 °C for 16 h in DMA (4 mL) in the presence of Pd(OAc)2 (4.5 mg, 0.02 mmol) under argon affords the coupling product after evaporation of the solvent and purification on silica gel.1H NMR (400 MHz, CDCl3): δ 7.69 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 7.45 (s, 1H), 7.35 (d, J = 8.1 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 6.74 (d, J = 3.5 Hz, 1H), 6.60 (d, J = 3.5 Hz, 1H), 2.45 (s, 3H), 2.40 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 139.9, 139.8, 139.7, 138.6, 138.4, 135.4, 132.9, 132.5, 130.6, 129.6, 129.5, 127.6, 126.1, 125.8, 125.6, 119.0, 110.8, 21.5, 15.4.
>HRMS calcd for [M + H]+ C23H18NS2 372.0875, found: 372.0874.
1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 7.63 (s, 1H), 7.40 (d, J = 8.1 Hz, 2H), 7.22 (d, J = 8.1 Hz, 2H), 6.08 (d, J = 3.2 Hz, 1H), 5.93 (d, J = 3.2 Hz, 1H), 2.62 (t, J = 7.6 Hz, 2H), 2.42 (s, 3H), 1.59 (quint., J = 7.6 Hz, 2H), 1.37 (sext., J = 7.6 Hz, 2H), 0.94 (t, J = 7.6 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 155.7, 148.0, 140.0, 138.7, 138.5, 138.4, 132.8, 131.0, 129.6, 129.4, 129.3, 125.8, 125.3, 119.0, 110.7, 107.9, 106.6, 30.3, 27.9, 22.4, 21.4, 13.9.
HRMS calcd for [M + H]+ C26H24NOS 398.1573, found: 398.1574.
1H NMR (400 MHz, CDCl3): δ 7.90 (dd, J = 4.8, 1.8 Hz, 1H), 7.71 (d, J = 8.3 Hz, 2H), 7.67 (d, J = 8.3 Hz, 2H), 7.50 (s, 1H), 7.35 (d, J = 8.1 Hz, 2H), 7.22–7.16 (m, 4H), 2.40 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 143.3, 140.3, 139.7, 138.9, 138.3, 134.5, 132.9, 130.9, 130.3, 129.9, 129.7, 129.6, 128.5, 127.6, 125.9, 118.9, 110.9, 21.5.
HRMS calcd for [M + H]+ C22H16NSSe 406.0163, found: 406.0164.
1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 7.35 (s, 1H), 7.22 (d, J = 8.1 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 3.26 (sept., J = 7.6 Hz, 1H), 2.35 (s, 3H), 2.04 (s, 3H), 1.40 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 176.7, 148.9, 143.4, 139.6, 138.4, 138.2, 132.9, 130.6, 129.7, 129.3, 128.9, 128.2, 125.8, 124.2, 118.9, 110.9, 33.5, 23.3, 21.4, 15.9.
HRMS calcd for [M + H]+ C25H23N2S2 415.1297, found: 415.1296.
1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 8.3 Hz, 2H), 7.67 (d, J = 8.3 Hz, 2H), 7.55 (s, 1H), 7.47 (s, 1H), 7.30 (d, J = 8.1 Hz, 2H), 7.19 (d, J = 8.1 Hz, 2H), 2.80 (d, J = 7.6 Hz, 2H), 2.40 (s, 3H), 2.11–2.03 (m, 1H), 0.97 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 170.2, 141.7, 140.4, 140.3, 139.1, 138.1, 133.0, 131.6, 130.0, 129.6, 129.5, 129.0, 127.3, 125.9, 118.9, 111.1, 42.5, 29.9, 22.4, 21.5.
HRMS calcd for [M + H]+ C25H23N2S2 415.1297, found: 415.1295.
1H NMR (400 MHz, CDCl3): δ 7.54 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H), 7.22 (s, 1H), 7.21 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.1 Hz, 2H), 3.26 (sept., J = 7.6 Hz, 1H), 2.34 (s, 3H), 2.04 (s, 3H), 1.40 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 176.5, 148.7, 141.5, 140.8, 138.0, 133.7, 132.5, 131.0, 129.6, 129.3, 128.4, 128.2, 127.6, 126.9, 124.7, 33.5, 23.3, 21.4, 15.9.
HRMS calcd for [M + H]+ C24H23ClNS2 424.0955, found: 424.0952.
1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 8.3 Hz, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.36 (s, 1H), 7.23 (d, J = 8.1 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 3.27 (sept., J = 7.6 Hz, 1H), 3.02 (q, J = 7.6 Hz, 2H), 2.35 (s, 3H), 2.05 (s, 3H), 1.40 (d, J = 7.6 Hz, 6H), 1.25 (t, J = 7.6 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 200.1, 176.6, 148.9, 142.7, 140.6, 138.2, 138.1, 135.9, 130.9, 129.6, 129.0, 128.7, 128.3, 125.5, 124.5, 33.6, 31.9, 23.3, 21.4, 15.9, 8.5.
HRMS calcd for [M + H]+ C27H28NOS2 446.1607, found: 446.1608.
1H NMR (400 MHz, CDCl3): δ 7.88 (s, 1H), 7.82 (d, J = 8.3 Hz, 1H), 7.57 (d, J = 8.3 Hz, 1H), 7.50 (t, J = 7.8 Hz, 1H), 7.30 (s, 1H), 7.21 (d, J = 8.1 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 3.27 (sept., J = 7.6 Hz, 1H), 2.35 (s, 3H), 2.04 (s, 3H), 1.40 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 176.7, 148.9, 142.7, 139.2, 138.3, 135.3, 130.9, 130.6, 130.0, 129.7, 129.6, 129.0, 128.7, 128.6, 128.3, 124.3, 118.6, 113.5, 33.5, 23.3, 21.4, 15.9.
HRMS calcd for [M + H]+ C25H23N2S2 415.1297, found: 415.1296.
1H NMR (400 MHz, CDCl3): δ 7.75 (d, J = 8.3 Hz, 1H), 7.66 (d, J = 8.3 Hz, 1H), 7.61 (t, J = 7.8 Hz, 1H), 7.39 (t, J = 7.8 Hz, 1H), 7.59 (s, 1H), 7.23 (d, J = 8.1 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 3.26 (sept., J = 7.6 Hz, 1H), 2.34 (s, 3H), 2.12 (s, 3H), 1.39 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 176.6, 149.0, 143.6, 138.3, 137.2, 137.1, 134.6, 133.2, 131.5, 130.5, 129.6, 129.4, 128.6, 128.5, 127.8, 124.2, 118.9, 109.9, 33.5, 23.3, 21.4, 15.9.
HRMS calcd for [M + H]+ C25H23N2S2 415.1297, found: 415.1302.
1H NMR (400 MHz, CDCl3): δ 8.90 (d, J = 2.4 Hz, 1H), 7.53 (dd, J = 4.8, 1.6 Hz, 1H), 7.88 (dt, J = 8.0, 1.5 Hz, 1H), 7.33 (ddd, J = 8.0, 4.8, 0.8 Hz, 1H), 7.30 (s, 1H), 7.22 (d, J = 8.1 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 3.26 (sept., J = 7.6 Hz, 1H), 2.35 (s, 3H), 2.05 (s, 3H), 1.40 (d, J = 7.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): δ 176.6, 148.8, 146.8, 142.4, 138.2, 138.1, 132.8, 130.8, 130.1, 129.6, 128.6, 128.4, 128.3, 124.5, 123.8, 33.5, 23.3, 21.4, 15.9.
HRMS calcd for [M + H]+ C23H23N2S2 391.1297, found: 391.1298.
General procedure for the preparation of 1,2-di(heteroaryl)benzenes 2a, 11a and 34a–36a
As a typical experiment, the reaction of the 2-(2-bromophenyl)thiophene derivative 1 (1 mmol), heteroarylboronic acid (2 mmol) and K2CO3 (0.276 g, 2 mmol) at 150 °C for 16 h in DMA (4 mL) in the presence of Pd(OAc)2 (11.3 mg, 0.05 mmol) under argon affords the coupling product after evaporation of the solvent and purification on silica gel.1H NMR (400 MHz, CDCl3): δ 7.54 (d, J = 3.9 Hz, 1H), 7.50–7.46 (m, 2H), 7.42–7.32 (m, 2H), 6.88 (d, J = 3.9 Hz, 1H), 6.64 (d, J = 3.5 Hz, 1H), 6.61 (d, J = 3.5 Hz, 1H), 2.53 (s, 3H), 2.45 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 190.8, 152.0, 144.2, 141.1, 139.6, 134.4, 132.8, 132.6, 131.4, 131.0, 129.0, 128.3, 127.9, 127.4, 125.6, 26.8, 15.5.
HRMS calcd for [M + H]+ C17H15OS2 299.0559, found: 299.0558.
1H NMR (400 MHz, CDCl3): δ 7.54–7.45 (m, 2H), 7.39–7.31 (m, 2H), 7.28 (dd, J = 5.1, 1.2 Hz, 1H), 6.98 (dd, J = 5.1, 3.5 Hz, 1H), 6.93 (dd, J = 3.5, 1.2 Hz, 1H), 6.67 (d, J = 3.8 Hz, 1H), 6.61 (d, J = 3.8 Hz, 1H), 2.46 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 143.0, 140.7, 140.4, 134.4, 133.7, 131.2, 131.0, 128.0, 127.7, 127.2, 127.1, 127.0, 126.0, 125.3, 15.4.
HRMS calcd for [M + H]+ C15H13S2 257.0453, found: 257.0453.
1H NMR (400 MHz, CDCl3): δ 9.86 (s, 1H), 7.62 (d, J = 3.9 Hz, 1H), 7.52–7.47 (m, 2H), 7.44–7.34 (m, 2H), 6.98 (d, J = 3.9 Hz, 1H), 6.64 (d, J = 3.5 Hz, 1H), 6.61 (d, J = 3.5 Hz, 1H), 2.45 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 183.1, 153.6, 143.7, 141.3, 139.4, 136.4, 134.5, 132.5, 131.5, 130.9, 129.3, 128.5, 128.0, 127.5, 125.6, 15.4.
HRMS calcd for [M + H]+ C16H13OS2 285.0402, found: 285.0400.
1H NMR (400 MHz, CDCl3): δ 7.53–7.43 (m, 2H), 7.40–7.33 (m, 2H), 7.31 (dd, J = 5.1, 1.2 Hz, 1H), 7.00 (dd, J = 5.1, 3.5 Hz, 1H), 6.93 (dd, J = 3.5, 1.2 Hz, 1H), 6.77 (d, J = 3.8 Hz, 1H), 6.68 (d, J = 3.8 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ 142.2, 141.5, 133.8, 133.2, 131.4, 130.9, 130.4, 128.4, 128.2, 127.5, 127.2, 126.4, 126.3, 126.2.
HRMS calcd for [M + H]+ C14H10ClS2 276.9907, found: 276.9909.
1H NMR (400 MHz, CDCl3): δ 7.56 (d, J = 7.8 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.20 (d, J = 7.8 Hz, 1H), 6.78 (d, J = 3.8 Hz, 1H), 6.69 (d, J = 3.8 Hz, 1H), 2.18 (s, 3H), 1.95 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 166.3, 159.5, 140.9, 134.3, 132.1, 130.8, 129.8, 129.0, 128.3, 128.0, 126.4, 125.5, 116.0, 11.4, 10.5.
HRMS calcd for [M + H]+ C15H13ClNOS 290.0401, found: 290.0402.
1H NMR (400 MHz, CDCl3): δ 8.30–8.25 (m, d1H), 8.10–8.04 (m, 1H), 7.65 (q, J = 1.2 Hz, 1H), 7.58–7.51 (m, 2H), 7.23 (q, J = 1.2 Hz, 1H), 2.73 (d, J = 1.2 Hz, 3H), 2.71 (d, J = 1.2 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 141.1, 138.4, 134.7, 133.1, 132.6, 132.0, 127.3, 126.8, 125.7, 125.5, 124.7, 124.2, 120.8, 120.7, 16.3, 16.2.
HRMS calcd for [M + H]+ C16H13S2 269.0453, found: 296.0456.
The intermediate 3-bromo-5,5′-dimethyl-2′-phenyl-2,3′-bithiophene 38 was also isolated in a low yield in an impure form as a colorless oil:
1H NMR (400 MHz, CDCl3): δ 7.33–7.21 (m, 5H), 6.82 (q, J = 1.2 Hz, 1H), 6.63 (q, J = 1.2 Hz, 1H), 2.52 (s, 3H), 2.41 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 140.2, 138.2, 134.4, 131.4, 129.3, 128.7, 128.6, 128.5, 128.2, 127.4, 108.9, 15.6, 15.5.
HRMS calcd for [M + H]+ C16H14BrS2 348.9715, found: 348.9717.
1H NMR (400 MHz, CDCl3): δ 8.41 (d, J = 7.2 Hz, 1H), 7.86 (d, J = 7.8, 2.0 Hz, 1H), 7.61–7.53 (m, 2H), 7.53–7.40 (m, 5H), 6.64 (q, J = 1.3 Hz, 1H), 3.06 (s, 3H), 2.42 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 138.7, 135.7, 135.6, 132.4, 132.3, 131.3, 130.7, 129.2, 128.8, 128.7, 128.6, 128.4, 126.4, 126.1, 125.5, 124.0, 123.3, 18.4, 16.2.
HRMS calcd for [M + H]+ C22H17S2 345.0766, found: 345.0767.
1H NMR (400 MHz, CDCl3): δ 8. 15 (s, 1H), 7.60 (s, 1H), 7.34 (s, 1H), 7.30 (d, J = 7.0 Hz, 1H), 7.22 (d, J = 7.0 Hz, 1H), 6.73 (q, J = 1.3 Hz, 1H), 3.04 (s, 3H), 2.45 (s, 3H), 2.43 (s, 3H), 2.42 (s, 3H), 2.38 (s, 3H), 2.33 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 137.6, 136.7, 136.6, 135.3, 135.0, 134.7, 132.9, 132.2, 131.8, 130.8, 130.5, 130.4, 129.8, 129.1, 128.5, 126.9, 126.8, 126.1, 124.5, 123.3, 20.7, 20.0, 19.9, 19.8, 18.2, 16.1.
HRMS calcd for [M + H]+ C26H25S2 401.1392, found: 401.1393.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the CSC for a grant to L. L. We thank the CNRS and “Rennes Metropole” for providing financial support.References
- For reviews on metal-catalyzed C–H bond functionalization: (a) L. Ackermann, Carboxylate-assisted transition-metal-catalyzed C–H bond functionalizations: mechanism and scope, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Ruthenium(II)-catalyzed C–H bond activation and functionalization, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (c) L. Theveau, C. Schneider, C. Fruit and C. Hoarau, Orthogonal palladium-catalyzed direct C–H bond arylation of heteroaromatics with aryl halides, ChemCatChem, 2016, 8, 3183–3194 CrossRef CAS; (d) S. Ruiz, P. Villuendas and E. P. Urriolabeitia, Ru-catalysed C–H functionalisations as a tool for selective organic synthesis, Tetrahedron Lett., 2016, 57, 3413–3432 CrossRef CAS; (e) S. Agasti, A. Dey and D. Maiti, Palladium-catalyzed benzofuran and indole synthesis by multiple C–H functionalizations, Chem. Commun., 2017, 53, 6544–6556 RSC; (f) T. Gensch, M. J. James, T. Dalton and F. Glorius, Increasing catalyst efficiency in C−H activation catalysis, Angew. Chem., Int. Ed., 2018, 57, 2296–2306 CrossRef CAS PubMed; (g) J. Kalepu, P. Gandeepan, L. Ackermann and L. T. Pilarski, C4–H indole functionalisation: precedent and prospects, Chem. Sci., 2018, 9, 4203–4216 RSC; (h) K. Hirano and M. Miura, A lesson for site-selective C–H functionalization on 2-pyridones: radical, organometallic, directing group and steric controls, Chem. Sci., 2018, 9, 22–32 RSC; (i) A. M. Prendergast and G. P. McGlacken, Transition metal mediated C–H activation of 2-pyrones, 2-pyridones, 2-coumarins and 2-quinolones, Eur. J. Org. Chem., 2018, 6068–6082 CrossRef CAS; (j) S. Mao, H. Li, X. Shi, J.-F. Soulé and H. Doucet, Environmentally benign arylations of 5-membered ring heteroarenes by Pd-catalyzed C−H bonds activations, ChemCatChem, 2019, 11, 269–286 CrossRef CAS; (k) S. Rej, Y. Ano and N. Chatani, Bidentate directing groups: An efficient tool in C–H bond functionalization chemistry for the expedient construction of C–C bond, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed; (l) A. Saha, M. Shankar, S. Sau and A. K. Sahoo, Multiple annulations of inert C(sp2)–H bonds with alkynes, Chem. Commun., 2022, 58, 4561–4587 RSC.
- (a) Y. Akita, A. Inoue, K. Yamamoto, A. Ohta, T. Kurihara and M. Shimizu, Palladium-catalyzed coupling reaction of chloropyrazines with indole, Heterocycles, 1985, 23, 2327–2333 CrossRef CAS; (b) A. Ohta, Y. Akita, T. Ohkuwa, M. Chiba, R. Fukunaga, A. Miyafuji, T. Nakata, N. Tani and Y. Aoyagi, Palladium-catalyzed arylation of furan, thiophene, benzo[b]furan and benzo[b]thiophene, Heterocycles, 1990, 31, 1951–1958 CrossRef CAS.
- (a) R. Rossi, F. Bellina, M. Lessi and C. Manzini, Cross-coupling of heteroarenes by C–H functionalization: recent progress towards direct arylation and heteroarylation reactions involving heteroarenes containing one heteroatom, Adv. Synth. Catal., 2014, 356, 17–117 CrossRef CAS; (b) H.-Y. Huang, A. Benzai, X. Shi and H. Doucet, Effective tools for the metal-catalyzed regiodivergent direct arylations of (hetero)arenes, Chem. Rec., 2021, 21, 343–356 CrossRef CAS PubMed.
- For selected examples of direct arylations of thiophenes: (a) K. Masui, A. Mori, K. Okano, K. Takamura, M. Kinoshita and T. Ikeda, Syntheses and properties of donor−acceptor-type 2,5-diarylthiophene and 2,5-diarylthiazole, Org. Lett., 2004, 6, 2011–2014 CrossRef CAS PubMed; (b) E. David, S. Pellet-Rostaing and M. Lemaire, Heck-like coupling and Pictet–Spengler reaction for the synthesis of benzothieno[3,2-c]quinolines, Tetrahedron, 2007, 63, 8999–9006 CrossRef CAS; (c) H. A. Chiong and O. Daugulis, Palladium-catalyzed arylation of electron-rich heterocycles with aryl chlorides, Org. Lett., 2007, 9, 1449–1451 CrossRef CAS PubMed; (d) J. Roger, F. Požgan and H. Doucet, Ligand-less palladium-catalyzed direct 5-arylation of thiophenes at low catalyst loadings, Green Chem., 2009, 11, 425–432 RSC; (e) B. Liégault, I. Petrov, S. I. Gorlesky and K. Fagnou, Modulating reactivity and diverting selectivity in palladium-catalyzed heteroaromatic direct arylation through the use of a chloride activating/blocking group, J. Org. Chem., 2010, 75, 1047–1060 CrossRef PubMed.
- For selected examples of direct arylations of furans: (a) M. Parisien, D. Valette and K. Fagnou, Direct arylation reactions catalyzed by Pd(OH)2/C: Evidence for a soluble palladium catalyst, J. Org. Chem., 2005, 70, 7578–7584 CrossRef CAS PubMed; (b) B. Liégaut, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, Establishment of broadly applicable reaction conditions for the palladium-catalyzed direct arylation of heteroatom-containing aromatic compounds, J. Org. Chem., 2009, 74, 1826–1834 CrossRef PubMed; (c) J. J. Dong, J. Roger, F. Požgan and H. Doucet, Low catalyst loading ligand-free palladium-catalyzed direct arylation of furans: an economically and environmentally attractive access to 5-arylfurans, Green Chem., 2009, 11, 1832–1846 RSC.
- For selected examples of direct arylations of pyrroles or indoles: (a) F. Bellina, S. Cauteruccio and R. Rossi, Palladium- and copper-mediated direct C-2 arylation of azoles—including free (NH)-imidazole, -benzimidazole and -indole—under base-free and ligandless conditions, Eur. J. Org. Chem., 2006, 1379–1382 CrossRef CAS; (b) X. Wang, D. V. Gribkov and D. Sames, Phosphine-free palladium-catalyzed C–H bond arylation of free (N–H)-indoles and pyrroles, J. Org. Chem., 2007, 72, 1476–1479 CrossRef CAS PubMed; (c) J. Roger and H. Doucet, Regioselective C-2 or C-5 direct arylation of pyrroles with aryl bromides using a ligand-free palladium catalyst, Adv. Synth. Catal., 2009, 351, 1977–1990 CrossRef CAS.
- For selected examples of direct arylations of thiazoles: (a) A. Mori, A. Sekiguchi, K. Masui, T. Shimada, M. Horie, K. Osakada, M. Kawamoto and T. Ikeda, Facile synthesis of 2,5-diarylthiazoles via palladium-catalyzed tandem C−H substitutions. Design of tunable light emission and liquid crystalline characteristics, J. Am. Chem. Soc., 2003, 125, 1700–1701 CrossRef CAS PubMed; (b) A. Yokooji, T. Okazawa, T. Satoh, M. Miura and M. Nomura, Palladium-catalyzed direct arylation of thiazoles with aryl bromides, Tetrahedron, 2003, 59, 5685–5689 CrossRef CAS; (c) G. L. Turner, J. A. Morris and M. F. Greaney, Direct arylation of thiazoles on water, Angew. Chem., Int. Ed., 2007, 46, 7996–8000 CrossRef CAS PubMed; (d) L.-C. Campeau, M. Bertrand-Laperle, J.-P. Leclerc, E. Villemure, S. Gorelsky and K. Fagnou, C2, C5, and C4 azole N-oxide direct arylation including room-temperature reactions, J. Am. Chem. Soc., 2008, 130, 3276–3277 CrossRef CAS PubMed; (e) J. J. Dong, J. Roger, C. Verrier, T. Martin, R. Le Goff, C. Hoarau and H. Doucet, Carbonates: eco-friendly solvents for palladium-catalysed direct arylation of heteroaromatics, Green Chem., 2010, 12, 2053–2063 RSC.
- For examples of Suzuki couplings with 2-(2-bromoaryl)thiophenes using heteroarylboronic acids: (a) M. Daniels, P. de Jong, T. Vandermeeren, L. Van Meervelt, M. Van der Auweraer and W. Dehaen, Bay-substituted thiaza[5]helicenes: Synthesis and implications on structural and spectroscopic properties, J. Org. Chem., 2019, 84, 13528–13539 CrossRef CAS PubMed; (b) C. Maeda, S. Nomoto, K. Akiyama, T. Tanaka and T. Ema, Facile synthesis of azahelicenes and diaza[8]circulenes through the intramolecular Scholl reaction, Chem. – Eur. J., 2021, 27, 15699–15705 CrossRef CAS PubMed; (c) F. Chen, J. Kim, Y. Matsuo, Y. Hong, D. Kim, T. Tanaka and A. Osuka, ortho-Phenylene-bridged hybrid nanorings of 2,5-pyrrolylenes and 2,5-thienylenes, Asian J. Org. Chem., 2019, 8, 994–1000 CrossRef CAS.
- For examples of C3-heteroarylations of thiophenes using Suzuki or Stille couplings with 2-aryl-3-halothiophenes: (a) R. Sanz, V. Guilarte, E. Hernando and A. M. Sanjuan, Synthesis of regioselectively functionalized benzo[b]thiophenes by combined ortho-lithiation–halocyclization strategies, J. Org. Chem., 2010, 75, 7443–7446 CrossRef CAS PubMed; (b) G. Balaji, T. S. Kale, A. Keerthi, A. M. Della Pelle, S. Thayumanavan and S. Valiyaveettil, Low band gap thiophene–perylene diimide systems with tunable charge transport properties, Org. Lett., 2011, 13, 18–21 CrossRef CAS PubMed; (c) D. Tian, Y. Zhou, Z. Li, S. Liu, J. Shao, X. Yang and J. Shao, Thieno[3,2-b]thiophene-based discotic liquid crystal mesogens: Rational synthesis, physical properties and self-assembly, ChemistrySelect, 2017, 2, 8137–8145 CrossRef CAS.
- For a rare example of C3-heteroarylation of thiophene via Suzuki couplings with 2-aryl-3-thiophenesboronic acid using a heteroaryl halide: R. Asato, C. J. Martin, J. P. Calupitan, R. Mizutsu, T. Nakashima, G. Okada, N. Kawaguchi, T. Yanagida and T. Kawai, Photosynergetic amplification of radiation input: from efficient UV induced cycloreversion to sensitive X-ray detection, Chem. Sci., 2020, 11, 2504–2510 RSC.
- For the preparation of 3-bromothiophenes: (a) P. Blanchard, P. Verlhac, L. Michaux, P. Frere and J. Roncali, Fine tuning of the electronic properties of linear π-conjugated oligomers by covalent bridging, Chem. – Eur. J., 2006, 12, 1244–1255 CrossRef CAS PubMed; (b) M. He and F. Zhang, Synthesis and structure of alkyl-substituted fused thiophenes containing up to seven rings, J. Org. Chem., 2007, 72, 442–451 CrossRef CAS PubMed; (c) J. I. Tietz, A. J. Seed and P. Sampson, Preparation of brominated 2-alkoxythiophenes via oxidation and etherification of 2-thienyltrifluoroborate salts, Org. Lett., 2012, 14, 5058–5061 CrossRef CAS PubMed.
- For reviews on 1,4-migration of Pd in catalytic organometallic reactions: (a) S. Ma and Z. Gu, 1,4-Migration of rhodium and palladium in catalytic organometallic reactions, Angew. Chem., Int. Ed., 2005, 44, 7512–7517 CrossRef CAS PubMed; (b) Y. Yasui, Intramolecular palladium migration, J. Synth. Org. Chem., Jpn., 2008, 66, 251–252 CrossRef CAS; (c) X. Dong, H. Wang, H. Liu and F. Wang, Recent advances in transition metal migration involving reactions, Org. Chem. Front., 2020, 7, 3530–3556 RSC; (d) M.-Y. Li, D. Wei, C.-G. Feng and G.-Q. Lin, Tandem Reactions involving 1,4-Palladium Migrations, Chem. – Asian J., 2022, 17, e202200456 CAS.
- For selected examples of Pd-catalyzed reactions involving Pd-migration: (a) R. C. Larock, Y. D. Lu, A. C. Bain and C. E. Russell, Palladium-catalyzed coupling of aryl iodides, nonconjugated dienes and carbon nucleophiles by palladium migration, J. Org. Chem., 1991, 56(15), 4589–4590 CrossRef CAS; (b) Q. Huang, A. Fazio, G. Dai, M. A. Campo and R. C. Larock, Pd-catalyzed alkyl to aryl migration and cyclization: An efficient synthesis of fused polycycles via multiple C−H activation, J. Am. Chem. Soc., 2004, 126, 7460–7461 CrossRef PubMed; (c) J. Zhao, D. Yue, M. A. Campo and R. C. Larock, An aryl to imidoyl palladium migration process involving intramolecular C−H activation, J. Am. Chem. Soc., 2007, 129, 5288–5295 CrossRef CAS PubMed; (d) M. A. Campo, H. Zhang, T. Yao, A. Ibdah, R. D. McCulla, Q. Huang, J. Zhao, W. S. Jenks and R. C. Larock, Aryl to aryl palladium migration in the Heck and Suzuki coupling of o-halobiaryls, J. Am. Chem. Soc., 2007, 129, 6298–6307 CrossRef CAS PubMed.
- (a) L. Liu and H. Doucet, One pot access to 2′-aryl-2,3′-bithiophenes via twofold palladium-catalyzed C−X/C−H coupling associated to a Pd-1,4-migration, Adv. Synth. Catal., 2022, 364, 2783–2795 CrossRef CAS; (b) L. Liu and H. Doucet, Scope and Limitations of the Palladium-Catalyzed Direct 1,2-Diheteroarylation of 1,2-Dihalobenzene Derivatives, Synthesis DOI:10.1055/a-1981-3213.
- For examples of Pd-catalyzed direct C3-arylations of thiophene derivatives: (a) T. Okazawa, T. Satoh, M. Miura and M. Nomura, Palladium-catalyzed multiple arylation of thiophenes, J. Am. Chem. Soc., 2002, 124, 5286–5287 CrossRef CAS PubMed; (b) J. J. Dong, J. Roger and H. Doucet, Palladium-catalysed direct 3- or 4-arylation of thiophene derivatives using aryl bromides, Tetrahedron Lett., 2009, 50, 2778–2781 CrossRef CAS; (c) K. Ueda, S. Yanagisawa, J. Yamaguchi and K. Itami, A general catalyst for the β-selective C–H bond arylation of thiophenes with iodoarenes, Angew. Chem., Int. Ed., 2010, 49, 8946–8949 CrossRef CAS PubMed; (d) D.-T. D. Tang, K. D. Collins, J. B. Ernst and F. Glorius, Pd/C as a catalyst for completely regioselective C–H functionalization of thiophenes under mild conditions, Angew. Chem., Int. Ed., 2014, 53, 1809–1813 CrossRef CAS; (e) K. Yuan and H. Doucet, Benzenesulfonyl chlorides: new reagents for access to alternative regioisomers in palladium-catalysed direct arylations of thiophenes, Chem. Sci., 2014, 5, 392–396 RSC.
- For rare examples of synthesis of 2′-aryl-2,3′-bithiophene derivatives: (a) S. Varello and S. T. Handy, Double couplings of dibromothiophenes using boronic acids and boronates, Synthesis, 2009, 138–142 CAS; (b) A. Acharya, G. Parameshwarappa, B. Saraiah and H. Ila, Sequential one-pot synthesis of tri- and tetrasubstituted thiophenes and fluorescent push–pull thiophene acrylates involving (het)aryl dithioesters as thiocarbonyl precursors, J. Org. Chem., 2015, 80, 414–427 CrossRef CAS PubMed; (c) Y. P. Zhong, J. M. Gao, P. Liu and W. J. Deng, Synthesis and electrochromic properties of star-shaped conjugated oligomers, Mater. Res. Innovations, 2015, 19, s51–s54 CrossRef CAS; (d) J.-H. Li, Q. Huang, W. Rao, S.-Y. Wang and S.-J. Ji, A trisulfur radical anion (S3(˙−) involved sulfur insertion reaction of 1,3-enynes: Sulfide sources control chemoselective synthesis of 2,3,5-trisubstituted thiophenes and 3-thienyl disulfides, Chem. Commun., 2019, 55, 7808–7811 RSC.
- T. Cantat, E. Génin, C. Giroud, G. Meyer and A. Jutand, Structural and kinetic effects of chloride ions in the palladium-catalyzed allylic substitutions, J. Organomet. Chem., 2003, 687, 365–376 CrossRef CAS.
- From a mixture of 4-(2-bromophenyl)-2-methylthiophene and 2-methylthiophene or 2-acetylthiophene, under the same conditions, complex mixtures of products were obtained.
- For rare examples of the synthesis of 4-bromo-2-chlorothiophenes: (a) S. Dapperheld, M. Feldhues, H. Litterer, F. Sistig and P. Wegener, Selektive debromierung von thiophen-derivaten durch elektrochemische reduction, Synthesis, 1990, 403–405 CrossRef CAS; (b) L. N. Lucas, J. Van Esch, R. M. Kellogg and B. L. Feringa, A new synthetic route to symmetrical photochromic diarylperfluorocyclopentenes, Tetrahedron Lett., 1999, 40, 1775–1778 CrossRef CAS; (c) C. Oberdorf, D. Schepmann, J. M. Vela, H. Buschmann, J. Holenz and B. Wunsch, Thiophene bioisosteres of spirocyclic σ receptor ligands: Relationships between substitution pattern and σ receptor affinity, J. Med. Chem., 2012, 55, 5350–5360 CrossRef CAS PubMed.
- W. Hagui, H. Doucet and J.-F. Soulé, Application of palladium-catalyzed C(sp2)–H bond arylation to the synthesis of polycyclic (hetero)aromatics, Chem, 2019, 5, 2006–2078 CAS.
- Y. Koga, T. Kaneda, Y. Saito, K. Murakami and K. Itami, Synthesis of partially and fully fused polyaromatics by annulative chlorophenylene dimerization, Science, 2018, 359, 435–439 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Palladium-catalyzed cross-coupling reactions of organoboron compounds, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- For a concerted metalation deprotonation mechanism: (a) D. L. Davies, S. M. A. Donald and S. A. Macgregor, Computational study of the mechanism of cyclometalation by palladium acetate, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed; (b) M. Lafrance and K. Fagnou, Palladium-catalyzed benzene arylation: Incorporation of catalytic pivalic acid as a proton shuttle and a key element in catalyst design, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed; (c) S. I. Gorelsky, D. Lapointe and K. Fagnou, Analysis of the concerted metalation-deprotonation mechanism in palladium-catalyzed direct arylation across a broad range of aromatic substrates, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed; (d) S. I. Gorelsky, Origins of regioselectivity of the palladium-catalyzed (aromatic)C–H bond metalation–deprotonation, Coord. Chem. Rev., 2013, 257, 153–164 CrossRef CAS; (e) D. L. Davies, S. A. Macgregor and C. L. McMullin, Computational studies of carboxylate-assisted C–H activation and functionalization at group 8–10 transition metal centers, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed; (f) R. A. Alharis, C. L. McMullin, D. L. Davies, K. Singh and S. A. Macgregor, The importance of kinetic and thermodynamic control when assessing mechanisms of carboxylate-assisted C–H activation, J. Am. Chem. Soc., 2019, 141, 8896–8906 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2210064 and 2217431. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo00018d |
| This journal is © the Partner Organisations 2023 |