
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Distinct insight into the use of difunctional double-decker silsesquioxanes as building blocks for alternating A–B type macromolecular frameworks†
Julia
Duszczak
ab,
Aleksandra
Mrzygłód
 ab,
Katarzyna
Mituła
ab,
Michał
Dutkiewicz
c,
Rafał
Januszewski
ab,
Monika
Rzonsowska
ab,
Beata
Dudziec
*ab,
Marek
Nowicki
bd and
Maciej
Kubicki
a
ab,
Katarzyna
Mituła
ab,
Michał
Dutkiewicz
c,
Rafał
Januszewski
ab,
Monika
Rzonsowska
ab,
Beata
Dudziec
*ab,
Marek
Nowicki
bd and
Maciej
Kubicki
a
aFaculty of Chemistry, Adam Mickiewicz University in Poznan, Uniwersytetu Poznanskiego 8, 61-614 Poznan, Poland. E-mail: beata.dudziec@gmail.com
bCentre for Advanced Technologies, Adam Mickiewicz University in Poznan, Uniwersytetu Poznanskiego 10, 61-614 Poznan, Poland
cAdam Mickiewicz University Foundation, Poznan Science and Technology Park, Rubiez 46, 61-612 Poznan, Poland
dInstitute of Physics, Poznan University of Technology, Piotrowo 3, 60-965 Poznan, Poland
First published on 12th December 2022
Abstract
Despite the rapid progress in the research on the synthesis of linear macromolecular systems with double-decker SQs included in the co-polymer chain, based on diverse catalytic processes, it still has limitations because of the formation of co-oligomers up to 20 units in a chain. Herein, we present a distinct look at known hydrosilylation reactions for forming hybrid copolymers. It is used as a synthetic protocol to synthesize DDSQ-based linear A–B alternating macromolecular systems and this is the first report on the formation of co-polymers with DDSQ with DPn over 1000. Additionally, this distinct insight concerns studies on the impact of Si–H and Si–Vi reactive group placement in DDSQ or in a respective co-reagent on the molecular weight distributions, degree of polymerization of the resulting copolymers as well as their selected physicochemical properties, i.e. thermal, mechanical, and hydrophobic properties. Understanding the basics of the catalytic processes leading to co-polymer SQ-based systems will pave the way for their use in the formation of hybrid materials of desired properties and respective applications.
Introduction
Linear block copolymers of a hybrid nature, in which an inorganic fragment is embedded within the main chain of the polymer, between organic units, are interesting systems that may not be formed by employing conventional hydrocarbon chemistry. The scientific significance of these structures is based on their enhanced thermal, mechanical, or optoelectronic properties, etc. which may exceed those of the organic analogues. From this perspective, the polyhedral oligomeric silsesquioxanes (SQs) offer beneficial properties as block co-monomers with inorganic Si–O–Si cores and organic coronae, which may be precisely modified. SQs belong to a large and diverse class of closed and open cage, three-dimensional organosilicon compounds of the general formula (RSiO3/2)n (where n = 6, 8, 10, 12).1,2 While their most important member is a cube-like T8 derivative (RSiO3/2)8, the double-decker (DDSQ) analogues (open M4T8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 or closed D2T84) have valuable utility.5–7 SQ applications are in line with their intrinsic unique properties and have found broad applications as (nano)materials (nanofillers and modifiers),8 in a variety of optic devices and sensors,9–11 in medicine12 or in catalysis.13
3 or closed D2T84) have valuable utility.5–7 SQ applications are in line with their intrinsic unique properties and have found broad applications as (nano)materials (nanofillers and modifiers),8 in a variety of optic devices and sensors,9–11 in medicine12 or in catalysis.13
The presence of reactive groups anchored onto the Si–O–Si core enables their precise modification and introduction of more complex functionalities. The reactions that govern this area of synthesis are based on catalytic transformations, chiefly catalyzed by Transition Metal (TM) complexes. The type of process used for these conversions depends on the nature of the reactive group at the SQ core and the most recognizable ones are Si–H, Si–HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, and Si–OH. They are involved in hydrosilylation (HS), cross-metathesis silylative or Heck coupling, dehydrocoupling condensation, O-silylation (metallation), and Friedel–Crafts reactions.14–21 These reactions lead to novel functionalized molecular and macromolecular silsesquioxane-based systems. While new catalytic methodologies have been developed, hydrosilylation is still one the most important synthetic methods of fundamental importance in organosilicon chemistry and also SQ functionalization, mediated by easily available Pt-based Karstedt or Speier catalysts.22,23 Even though this process is burdened with inconvenience, i.e. selectivity and formation of by-products (derived from side processes, e.g. dehydrogenative silylation, isomerization, or olefin hydrogenation), it is a convenient route to obtain compounds of desirable physical and chemical properties.24,25
CH2, and Si–OH. They are involved in hydrosilylation (HS), cross-metathesis silylative or Heck coupling, dehydrocoupling condensation, O-silylation (metallation), and Friedel–Crafts reactions.14–21 These reactions lead to novel functionalized molecular and macromolecular silsesquioxane-based systems. While new catalytic methodologies have been developed, hydrosilylation is still one the most important synthetic methods of fundamental importance in organosilicon chemistry and also SQ functionalization, mediated by easily available Pt-based Karstedt or Speier catalysts.22,23 Even though this process is burdened with inconvenience, i.e. selectivity and formation of by-products (derived from side processes, e.g. dehydrogenative silylation, isomerization, or olefin hydrogenation), it is a convenient route to obtain compounds of desirable physical and chemical properties.24,25
The concept of linear co-polymeric SQ-based systems, so-called Beads on a Chain (BoC) polymers, i.e. linear co-polymers with DDSQ fragments embedded in the polymer chain, is known.26,27 The methods of their formation included various reactions, i.e. cross-metathesis, silylative coupling, Piers–Rubinsztajn, or other types of condensation reactions.26–34 However, the number of reports on these copolymeric DDSQ-based systems is still limited, especially concerning hydrosilylation.28,31,35 Due to growing attention towards the interesting physical properties of these new materials, i.e. thermal (also insulating) and mechanical parameter enhancement, the transmittance of the prepared films, hydrophobic character, etc., an increase in the development of methodologies using varied comonomers to obtain new systems of tailored properties is observed.36–41
In this paper, we focus on the studies on Pt-catalysed hydrosilylation reactions using divinyl/dihydro-substituted double-decker silsesquioxanes to verify the impact of the placement of Functional Groups (FGs), i.e. Si–H and Si–HCCH2 on the reactivity of these systems. A novel, molecular DDSQ-based compound was disclosed, possessing an additional –OSi(Me2)– linker between the Si–O–Si core and Si–HCCH2. The versatility of the hydrosilylation procedure, elaborated for the synthesis of molecular compounds, was applied to obtain linear, macromolecular co-polymers with diorganosilicon fragments and the DDSQ-unit embedded in the main chain of the linear polymer. The possibility to introduce different diorganosilicon co-monomers and the presence of additional –OSi(Me2)– fragments were used to demonstrate and study their influence on the thermal, mechanical, and hydrophobic properties of the resulting DDSQ-based macromolecular systems.
Results and discussion
Our group has reported on the chemistry of double-decker silsesquioxanes with vinyl reactive groups that are susceptible to silylative coupling (SC) and cross-metathesis (CM) reactions leading to molecular and linear macromolecular compounds (depending on the number of vinyl moieties in the olefin reagent).19,30,42 Additionally, Si–H functionalized double-decker SQs are active in the hydrosilylation (HS) of arenes also resulting in analogous molecular and macromolecular DDSQ-based systems.28,31,35 Linear macromolecular compounds may be described as co-oligomers with DDSQ fragments embedded between the aryl moieties by an unsaturated ethenyl-bridge when SC/CM is used or a saturated ethyl-bridge when HS is applied for their syntheses. Encouraged by our previous research on the synthesis of double-decker SQ-based arene derivatives of molecular and macromolecular architecture via hydrosilylation, we decided to study this more comprehensively.31 Our goal was to verify the reactivity of Si–HCCH2 and Si–H difunctionalized DDSQ in hydrosilylation and apply them as reagents in the abovementioned reaction with dihydro- or divinyl-substituted organosilicon compounds, respectively.
The DDSQ-based reagents used in the studies, i.e. DDSQ-2SiH (1a) and DDSQ-2SiVi (1b) are well known, but we succeeded in obtaining the crystal structure of 1b for the first time (Fig. 1).19,31 For our studies, we also applied a novel DDSQ derivative possessing an additional –OSi(Me2)– linker between the Si–O–Si core and the –HCCH2 functionality (DDSQ-2OSiVi (1c)). It was obtained in a hydrolytic condensation reaction of 9,19-dihydroxy-octaphenylsilsesquioxane43 with chlorodimethyl-vinylsilane in a high yield (94%) and characterized by 1H, 13C, 29Si NMR, and FT-IR spectroscopy. Its structure was also confirmed by XRD for crystals obtained in MeCN during a slow (approx. two weeks) crystallization process (Fig. 1). Fig. 1 shows the perspective view of molecules 1b and 1c.
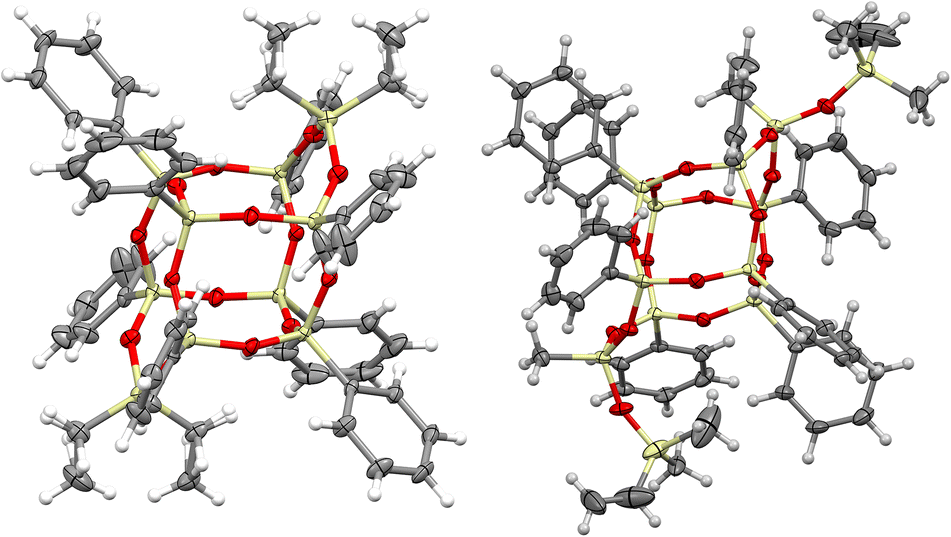 | ||
| Fig. 1 Perspective view of the compound 1b (left) and 1c (right); ellipsoids are drawn at the 50% probability level, hydrogen atoms are shown as spheres of arbitrary radii. For 1b only the higher-occupied part of the disordered fragment is shown. | ||
The crystal structures unambiguously confirm the proposed reaction products. The values of the geometrical parameters (bond lengths, angles) are very consistent and close to the typical bond lengths and angles in similar compounds. The structures are quite regular – the mean values of Si–O bonds are 1.617(1) Å in 1b and 1.616(1) Å in 1c, and the Si–O–Si angles are within a relatively (as compared with similar structures) narrow range, 144.10(9)°–163.31(9)° in 1b, and 139.3(2)°–162.5(2)° in 1c. Molecule 1b is Ci-symmetrical, and it lies across the inversion centre in the space group P21/c. In turn, the main molecule 1c skeleton (silsesquioxane cage) is only in good approximation Ci-symmetrical; the disposition of phenyl rings breaks the symmetry and lowers it to C1. The crystal architectures are determined mainly by weak van der Waals interactions.
Interestingly, the XRD analysis of 1c revealed the formation of only a cis-geometry (Fig. 2) which may suggest different susceptibility forms of cis- and trans-isomers, as both were obtained in the reaction (see 29Si NMR in the ESI, p. S-9†). It requires diverse procedures for their separation.44 This may also suggest that the presence of an –OSi(Me2)– bridge may affect the physical properties of this compound which in turn may alter the properties of its follow-up derivatives.
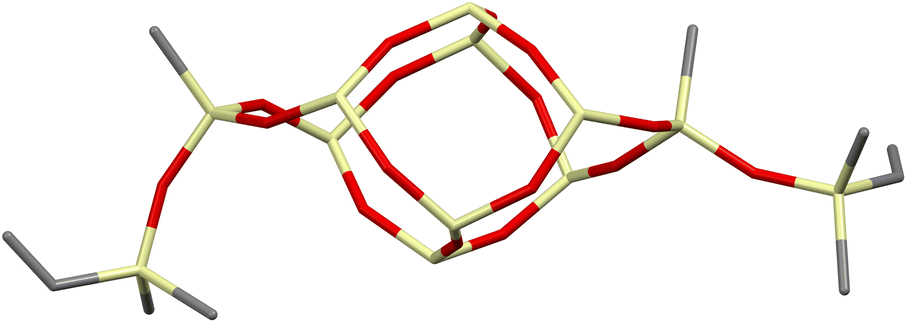 | ||
| Fig. 2 Perspective view of compound 1c; phenyl rings and hydrogen atoms are omitted to visualize the bare Si–O–Si DDSQ core. | ||
Initially, we studied hydrosilylation in two model reactions differing in the architecture of DDSQs, i.e. by varying the placement of the reactive Si–H moiety (1a or 2-H) vs. Si–HCCH2 (1b, 1c or 2-Vi) in hydrosilylation reagents as presented in Scheme 1.
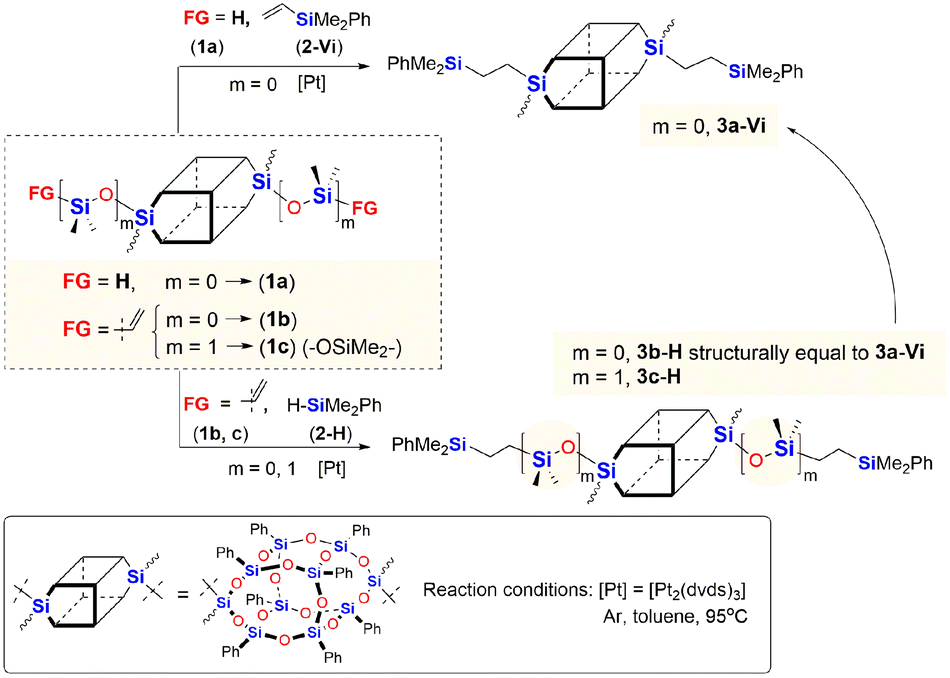 | ||
| Scheme 1 Model reactions for the hydrosilylation using organosilicon compounds with different locations of the Si–H functionality. | ||
The model reactions were carried out using real-time in situ FT-IR measurements (see the ESI†) to establish the time required for the complete conversion of Si–H bonds (based on changes in the surface area of the band at ῡ = 895 cm−1 characteristics for stretching vibrations of Si–H bonds) in the HS process to yield molecular DDSQ-based difunctionalized compounds. The catalyst used for this purpose is commercially available and it is still widely applied Pt-Karstedt's catalyst ([Pt2(dvds)3]) in 10−4 mol of Pt loading per mol of Si–H group. The reagent stoichiometry was maintained equivalent. We noted previously, that alkene excess may affect the reaction time but mainly its initial time and does not significantly reduce the total reaction time.16 However, the reaction conditions of these model reactions were to be applied in further copolymerization HS which is why the equimolar reagent stoichiometry was crucial. Hydrosilylation (besides possible side reactions) is generally selective towards the β-anti-Markovnikov addition product but the α-product may also occur. It should be emphasized that in our case, as previously noted, the process was regioselective towards the formation of β-hydrosilylation products (for details see the ESI†). As a result, two types of products differing in the presence of an additional –OSi(Me2)– linker between the Si–O–Si core and the SiMe2Ph substituent were formed (for m = 0, 3a-Vi, 3b-H; m = 1, 3c-H). The reaction profiles of the hydrosilylation progress for the model reaction are presented in Fig. 3.
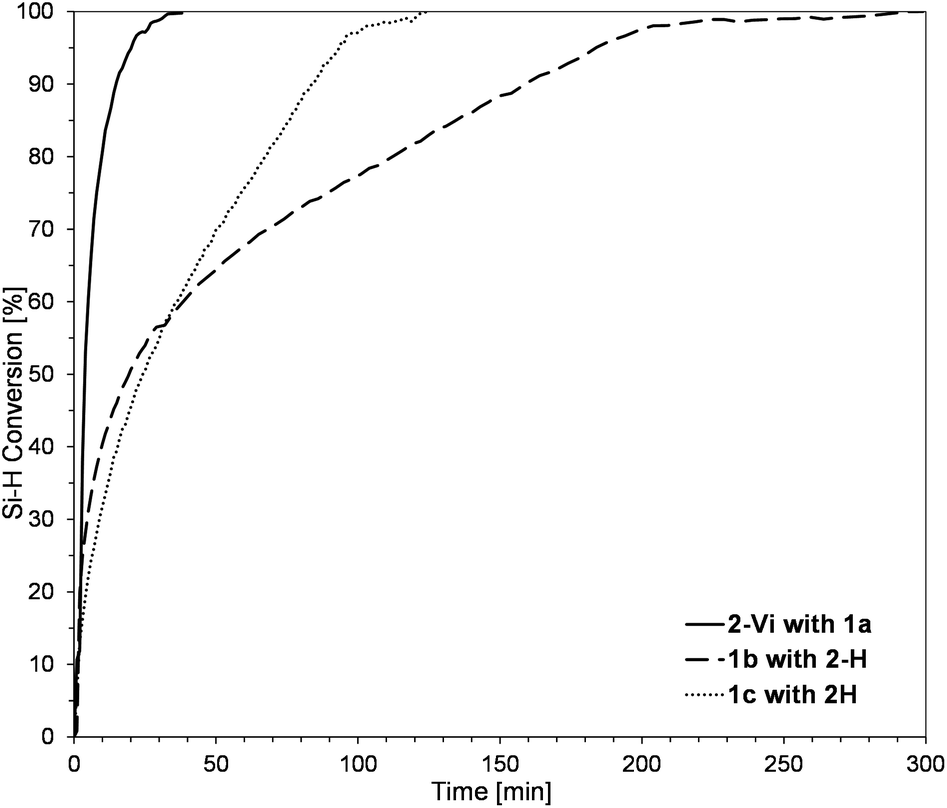 | ||
| Fig. 3 Reaction profile for the hydrosilylation of 2-Vi with 1a and 1b, 1c with 2-H determined by real-time in situ FT-IR spectroscopy. [Pt2(dvds)3] × 10−4 per mol of Si–H, equimolar stoichiometry of Si–H and Si–HCCH2 reagents, toluene (mSQ/Vtol = 33 mg mL−1), 95 °C. | ||
As shown in Fig. 3, rapid reagent conversion appeared within the first 10–40 min after [Pt2(dvds)3] addition (up to 50% of Si–H consumption). However, the real times of the reaction estimated by in situ FT-IR are as follows: 48 min for 1a, 290 min for 1b, and 120 min for 1c. The higher reactivity of 1a may be explained by the presence of D-type functional Si–H in its structure. The presence of vinyl moieties in 1b and 1c changes the electronic properties of these reagents and lowers the reactivity of 1b with the D-type functional Si–HCCH2 group may when compared with 1c – M-type of Si–HCCH2. It may be noted that the electron-withdrawing effect of the oxygen atom(s) present in the surrounding of the reactive Si–H (in 1a) promotes its reactivity but decreases the reactivity of Si–HCCH2 (in 1b and 1c). These observations are in accordance with the Chalk–Harrod HS mechanism.22 The conclusions derived from these model reactions were applied in further copolymerization HS and albeit the structures of the obtained products for m = 0 3a-Vi = 3b-H are structurally equal, the reagents built and reaction times varied along with different reaction profiles. We decided to conduct them as a studied model to observe if the electronic and steric effects of Si–H and Si–HCCH2 placement in DDSQ substrates affect the formation of copolymeric products.
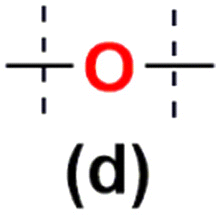
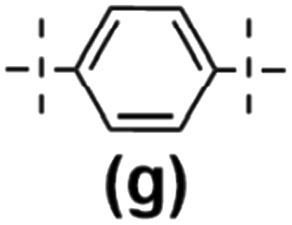
In the next stage of our studies, we transferred the information obtained from model reactions and performed copolymerization via hydrosilylation of 1b and 1c with a series of dihydrosubstituted (5d–h) organosilicon compounds and divinyl- (4d–g) organosilicon compounds with 1a derivative. In all tests, the reaction conditions were maintained the same, i.e. 10−3 mol of Pt-Karstedt's catalyst loading per mol of Si–H group, toluene (m1a/Vtol = 33 mg mL−1), and an equimolar stoichiometry of Si–H and Si–HCCH2 reagents (e.g. [1a]:[4d] = 1:1), 95 °C and 24 h (or 72 h when noted). The overview of this process that leads to the organic–inorganic co-oligomers with DDSQ in the main chain is presented in Scheme 2.
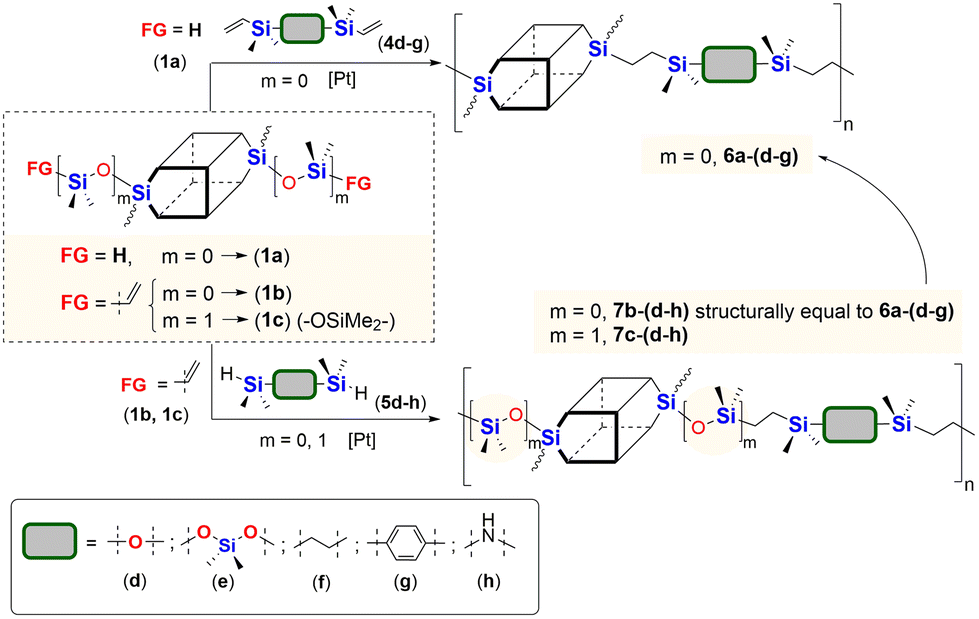 | ||
| Scheme 2 General synthetic procedure for alternating A–B type organic–inorganic co-oligomers with DDSQ and different organosilicon spacers via a hydrosilylation reaction. | ||
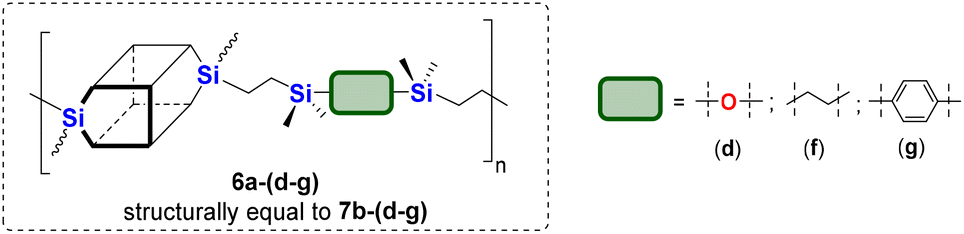
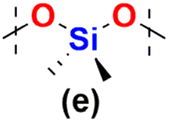
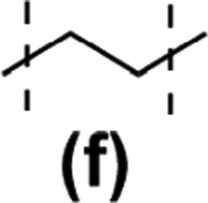
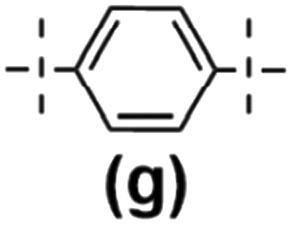
The model reactions enabled hydrosilylation oligomerization reactions under optimized reaction conditions. After several tests, a series of co-oligomeric products with DDSQ fragments with organosilicon spacers differing in the presence (7c(d–h))/absence (6a(d–g) = 7b(d–g)) of an additional –OSi(Me2)– linker between the DDSQ core and the organosilicon spacer was obtained. It is worth noting that hydrosilylation of 4d–g with 1a and 1b with 5d–g leads to products of analogous structure, i.e. of the identical architecture of mers and 6a(d–g) may be denoted as 7b(d–g) (Scheme 2). Nevertheless, they will be mentioned separately, to point out the differences in the degree of co-polymerization depending on the placement of the functional Si–H/Si–HCCH2 moiety in DDSQs (1a or 1b) and their reactivity in the hydrosilylation.
The obtained co-oligomers were purified by precipitation in methanol or n-hexane (with the eventual addition of water) and subsequent decantation (if possible), dried under reduced pressure to afford solids 6a(d–g) or viscous solids (waxes) 7c(d–h). Due to the possibility of solvent occlusion which was anticipated based on the product's chain structure and degree of viscosity, it should be advised to dry them under a vacuum with the use of liquid nitrogen. Isolated compounds were analysed by NMR and FT-IR spectroscopy to confirm their structures. The prepared co-oligomers were subjected to Gel Permeation Chromatography (GPC) to measure their molecular weights and polydispersity indexes. The measured Mn, Mw, and Đ values are presented in Tables 1 and 2.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Sample |
|
All fractions | |||
| M w (g mol−1) | M n (g mol−1) | Đ | DPn | |||
| Reaction conditions: [Pt2(dvds)3] and PtO2 – 10−3 per mol of Si–H, equimolar stoichiometry of Si–H and Si–HCCH2 reagents, e.g. [1a]:[4d] = 1:1, toluene (m1a/Vtol = 33 mg mL−1), 95 °C. Đ = dispersity index, DPn = degree of polymerization. |
||||||
| 1 | 6a–d-24h |
|
6500 | 4500 | 1.4 | 4 |
| 2 | 6a–d-72h | 7200 | 4900 | 1.5 | 4 | |
| 3 | 6a–d-24h PtO2 | 6100 | 4300 | 1.4 | 3 | |
| 4 | 6a–f-24h |

|
7700 | 5200 | 1.5 | 4 |
| 5 | 6a–f-72h | 12300 |
6900 | 1.8 | 5 | |
| 6 | 6a–f-24h PtO2 | 6500 | 4700 | 1.4 | 3 | |
| 7 | 6a–g-24h |
|
22800 |
9500 | 2.4 | 7 |
| 8 | 6a–g-72h | 26300 |
10000 |
2.6 | 7 | |
| 9 | 6a–g-24h PtO2 | 13800 |
7300 | 1.9 | 5 | |

|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Sample | DDSQ |
|
All fractions | |||
| M w | M n | Đ | DPn | ||||
| Reaction conditions: [Pt2(dvds)3] and PtO2 – 10−3 per mol of Si–H, equimolar stoichiometry of Si–H and Si–HCCH2 reagents, e.g. [1b/c]:[5d] = 1:1, toluene (m1b/c/Vtol = 33 mg mL−1), 95 °C, 24 h or 72 h. Đ = dispersity index, DPn = degree of polymerization. |
|||||||
| 1 | 7b–d-24h | 1b |
|
2300 | 9100 | 2.5 | 7 |
| 2 | 7b–d-72h | 42000 |
11700 |
3.6 | 9 | ||
| 3 | 7b–d-24h PtO2 | 8900 | 5500 | 1.6 | 4 | ||
| 4 | 7b–e-24h |
|
624300 |
23000 |
27.2 | 16 | |
| 5 | 7b–e-72h | 764600 |
21800 |
35.0 | 15 | ||
| 6 | 7b–f-24h |

|
4600 | 3700 | 1.2 | 3 | |
| 7 | 7b–f-72h | 7800 | 5200 | 1.5 | 4 | ||
| 8 | 7b–g-24h |
|
10600 |
5700 | 1.9 | 4 | |
| 9 | 7b–g-72h | 17700 |
9100 | 1.9 | 7 | ||
| 10 | 7b–g-24h PtO2 | 10300 |
5600 | 1.8 | 4 | ||
| 11 | 7b–h-24h |
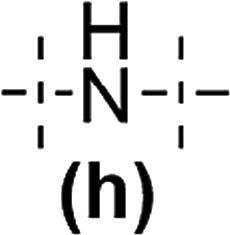
|
2400 | 1600 | 1.5 | 1 | |
| 12 | 7b–h-72h | 6800 | 2300 | 2.9 | 2 | ||
| 13 | 7b–h-24h PtO2 | 1400 | 1000 | 1.3 | 1 | ||
| 14 | 7c–d-24h | 1c |

|
18300 |
8400 | 2.2 | 6 |
| 15 | 7c–d-72h | 67000 |
15200 |
4.4 | 10 | ||
| 16 | 7c–e-24h |
|
519900 |
16100 |
32.4 | 10 | |
| 17 | 7c–e-72h | 950700 |
27400 |
34.7 | 18 | ||
| 18 | 7c–f-24h |
|
4900 | 4200 | 1.2 | 3 | |
| 19 | 7c–f-72h | 105200 |
20200 |
5.2 | 14 | ||
| 20 | 7c–g-24h |
|
11200 |
6100 | 1.8 | 4 | |
| 21 | 7c–g-72h | 21200 |
8500 | 2.5 | 6 | ||
| 22 | 7c–h-24h |

|
4500 | 3100 | 1.4 | 2 | |
| 23 | 7c–h-72h | 45500 |
11200 |
4.1 | 8 | ||
| 24 | 7c–h-24h PtO2 | 2000 | 1700 | 1.2 | 1 | ||
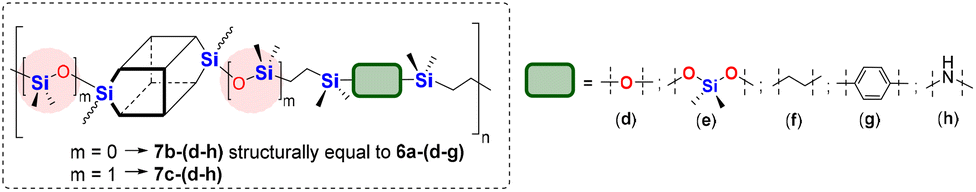

Tests verifying the reactivity of functional DDSQs by using 1a (with reactive Si–H moiety) in a hydrosilylation reaction using divinylsubstituted organosilicon derivatives (4d, 4f, and 4g) and their respective data are presented in Table 1.
The degree of polymerization (DPn) of 6a-(d–g) after 24 h was estimated as 5–7 units which seemed to be the expected result for these kinds of DDSQ-based linear systems.20,28,31,35,45 The additional reaction time (72 h) lead to a minor increase in molecular weights of the obtained copolymers and their Đ values. Based on the analysis of molecular weight distribution plots the low molecular fractions were converted to higher molecular weight products, e.g. as in the case of 6a–f – entries 4 and 5.30,31 This led to the question of what is the reason for such a slowdown in the course of the polymerization process? Is it caused by the Pt catalyst occluding the structure of the copolymer entangled chains resulting in its activity decrease, its deactivation, or steric hindrance and diffusion considerations? To answer this question, an independent experiment was carried out between 1a and 4d, performed under standard conditions. After 72 h a respective amount of [Pt2(dvds)3] (10−3 per mol of Si–H) was added and the reaction was carried out for an additional 24 h. GPC analysis showed only a minor change in the molecular weight of the resulting copolymer 6a–d (Mw = 7200, DPn = 4). This might suggest that the steric or diffusion reasons are to blame. An analogical test between 1a and 4d was performed and after 24 h a respective amount of [Pt2(dvds)3] (10−3 per mol of Si–H) was added along with the mixture of styrene and HSiMe2Ph (stoichiometry 1:1) and the reaction was continued for an additional 24 h. After reaction completion, the resulting 6a–d was precipitated in n-hexane and the decant was analysed via GC and GC-MS techniques. Styrene conversion was estimated as 92% after 24 h while the same reaction of styrene and HSiMe2Ph but without the presence of co-oligomer 6a–d enabled >99% styrene conversion in less than 8 h. In parallel, a similar test of styrene hydrosilylation by HSiMe2Ph in the post-reaction mixture of 6a–d was performed but without an additional amount of [Pt2(dvds)3] after 24 h. GC analysis revealed 68% conversion of styrene (for details see the ESI, p. S-6†). These studies may confirm that [Pt2(dvds)3] is partially occluded in the entangled chains of the copolymer 6a–d with the rigid DDSQ units. Again, it is consistent with the Chalk–Harrod HS mechanism.22 The possibility of using PtO2 as a catalyst known for its activity in hydrosilylation was verified.46,47 But the obtained results were noticeably worse compared to those for [Pt2(dvds)3] in terms of Mw and Mn of the obtained co-oligomers, DPn, and main polymer fraction content (Table 1, entries 3, 6 and 9). The GPC results considering the content of all fractions presented in Table 1 are consistent with the content of the main and highest volume fraction content, presented in Table S2 (in the ESI)† in the range of 63 to 100%.
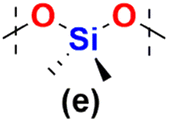
In the next step the reagents with changed Si–H and Si–Vi placement were studied in hydrosilylation, i.e. DDSQs with two –CHCH2 moieties attached directly to the DDSQ core (1b) or via the linker –OSi(Me2)– (1c) with the respective dihydrosubstituted organosilicon compounds (5d–h). The palette of dihydrosubstituted reagents was extended by 5e and 5h, i.e. trisiloxane (5e) and disilazane (5h) derivatives. The respective GPC data for the obtained copolymers – type 7b and 7c, are presented in Table 2 and Table S2 in the ESI† with additional information on the percentage content of the main and the highest Mw fractions.
In the cases of 1b and 1c, i.e. the DDSQ core with two ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–HCCH2 groups, hydrosilylation with dihydrosubstituted organosilicon derivatives (5d–h) resulted in the formation of the respective copolymers 7b and 7c type and was found to be dependent on the DDSQ core (1b and 1c) as well as the 5d–h co-monomer structure. The electronic impact (withdrawing or donating) of the spacer between Si–H moieties of 5d–h could be considered in these examples. For 7b type co-polymers, the DPn was estimated as 3–5 units for 5f – ethane bridged and 4–7 units for 5g – 1,4-phenylene-bridged (both electron-donating units), regardless of the reaction time (24 vs. 72 h) (Table 2, entries 6–9). This is similar to the data obtained for the aforementioned 6a–f and 6a–g systems, analogous in the structure, but obtained from 1a and 4f, 4g (reagents differing in Si–HCCH2 and Si–H placement) (Table 1, entries 4, 5 and 7, 8). Also, these results are respectively consistent with the content of the main and highest molecular weights fractions, see Table S2 (in the ESI),† entries 15–18 (in a range of 66–100%). In the case of 1c – DDSQ with a vinyl moiety attached to the core via linker –OSi(Me2)–, the respective copolymers 7c–f and 7c–g exhibited oligomeric structures with the DPn in the range of 3–4 after 24 h and their increase was noted after 72 h, i.e. DPn = 14 and DPn = 6, respectively (Table 2, entries 18–21), with a slightly higher Đ index in the range of 2.5–5.2. The values of the main and highest molecular weight fractions revealed the presence of ca. 82% of the content of higher molecular weights of 7c–f with DPn = 34 and ca. 39% of 7c–g with DPn = 20 measured for samples after 72 h (Table S2,† entries 27–30). This is a significant change in the molecular weights of DDSQ copolymers obtained via hydrosilylation which may be attributed not only to the steric availability of the Si–HCCH2 of 1c but also to the electronic impact of reagents.
Si–HCCH2 groups, hydrosilylation with dihydrosubstituted organosilicon derivatives (5d–h) resulted in the formation of the respective copolymers 7b and 7c type and was found to be dependent on the DDSQ core (1b and 1c) as well as the 5d–h co-monomer structure. The electronic impact (withdrawing or donating) of the spacer between Si–H moieties of 5d–h could be considered in these examples. For 7b type co-polymers, the DPn was estimated as 3–5 units for 5f – ethane bridged and 4–7 units for 5g – 1,4-phenylene-bridged (both electron-donating units), regardless of the reaction time (24 vs. 72 h) (Table 2, entries 6–9). This is similar to the data obtained for the aforementioned 6a–f and 6a–g systems, analogous in the structure, but obtained from 1a and 4f, 4g (reagents differing in Si–HCCH2 and Si–H placement) (Table 1, entries 4, 5 and 7, 8). Also, these results are respectively consistent with the content of the main and highest molecular weights fractions, see Table S2 (in the ESI),† entries 15–18 (in a range of 66–100%). In the case of 1c – DDSQ with a vinyl moiety attached to the core via linker –OSi(Me2)–, the respective copolymers 7c–f and 7c–g exhibited oligomeric structures with the DPn in the range of 3–4 after 24 h and their increase was noted after 72 h, i.e. DPn = 14 and DPn = 6, respectively (Table 2, entries 18–21), with a slightly higher Đ index in the range of 2.5–5.2. The values of the main and highest molecular weight fractions revealed the presence of ca. 82% of the content of higher molecular weights of 7c–f with DPn = 34 and ca. 39% of 7c–g with DPn = 20 measured for samples after 72 h (Table S2,† entries 27–30). This is a significant change in the molecular weights of DDSQ copolymers obtained via hydrosilylation which may be attributed not only to the steric availability of the Si–HCCH2 of 1c but also to the electronic impact of reagents.
Interestingly, the impact of the surrounding electron and steric properties of the reagents on the obtained results was visible in the case of dihydro-substituted organosilicon compounds with the presence of more electron-withdrawing moieties, i.e. slightly for 5d – disiloxane and especially for 5e – trisiloxane reacting with DDSQ-divinyl compounds, i.e.1b and 1c. For 7b–d and 7c and 7d the changes in the obtained molecular weights after 24 and 72 h were not that substantial with the DPn in the range of 7–10. Only the separation of the highest molecular weight fractions revealed the presence of ca. 46% content of the fraction with DPn = 21 after 24 h that changed after 72 h to ca. 25% content of the fraction with DPn = 68 (Table S2,† entries 10 and 11). In the cases of trisiloxane 5e and 1b, the obtained DPn values of 7b–e after 24 and 72 h were higher, i.e. in the range of 15–16 (Table 2, entries 4 and 5). But the Đ index showed a significantly wider molecular weight distribution. More adequate data were obtained for the main and highest molecular weight fraction identification (Table S2,† entries 13 and 14). It revealed 40% content of the main fraction with DPn = 7 after 24 h, and after 72 h there was a significant increase with DPn = 108 for ca. 39% of the content of 7b–e. Interestingly, the results presented for the highest molecular weight content were more impressive, with DPn = 1083 after 24 h and DPn = 1127 after 72 h for up to 23% sample content. What is more, the results obtained for 5e and 1c (with a –OSi(Me2)– linker) were even more extraordinary. Again, the DPn for all fractions of 7b and 7c equalled 10 (after 24 h) and 18 (after 72 h) with substantial Đ values (up to 35). But the detailed data on the main molecular weight fraction of ca. 40% content exhibited DPn = 10 (24 h) that notably increased until DPn = 79 after 72 h. Considering the highest molecular weight content fractions, they were estimated as DPn = 1412 after 24 h which changed to DPn = 1049 with 10% and ca. 23% fraction contents, respectively. It should be noted that the molecular weights of copolymers 7b–e and 7c–e are over 2837 × 103 g mol−1 and 3927 × 103 g mol−1 and are the first examples of copolymeric A–B alternating systems with a DDSQ moiety embedded in the linear structure of the polymer with such high values of Mw. In the end, the tests of dihydrodisilazane (5h) with 1b and 1c showed that the presence of an NH moiety significantly affected its catalytic reactivity when compared to the respective disiloxane 5d. The compounds 7b–h and 7c–h with DPn = 1–8 revealed their lower reactivity in the process which influenced the lower molecular weights of the respective copolymers (Table 2, entries 11–13 and 22–24). This may be attributed to the possible NH coordination to the metal centre, catalyst poisoning, or disilazane degradation, i.e. Si–H disproportionation.48–50 However, for 7c–h the molecular weights are ten times higher after 72 h (entries 22 and 23) when compared to 7b–h and a three-fold molecular weight increase is observed after 72 h (entries 11 and 12). This may be attributed to the steric and electronic structures of substrates 1bvs.1c (presence of a –SiO(Me2)– linker). It was also visible in the case of the aforementioned products 7c–evs. 7b–e. Finally, in the case of the PtO2 catalyst, the results were worse than those obtained for Karstedt's catalyst (Table 2, entries 3, 10, 13 and 24).
The presented results may suggest the electron-withdrawing impact of the siloxane unit activates Si–H and also the Si–HCCH2 moiety in the case of the DDSQ compound though only for its M type of functional Si atom, i.e. for 1c. Achieving efficient copolymerization seems to be important. The respective charts of molecular weight distributions exhibiting the electronic impact of dihydro- or divinylorganosilicon compounds (4gvs. 5g and 4dvs. 5d) are shown in Fig. 4 and 5. The impact of the DDSQ structure with or without the –Osi(Me2)– linker between the Si–HCCH2 moiety and the DDSQ core (1bvs. 1c) on the results of copolymerization is depicted in the chart in Fig. 6. In the case of the D units of the Si at the DDSQ core, the effect of the steric availability of the reactive Si–H and Si–HCCH2 units may seem to exceed the electronic influence. It should also be emphasized that other effects that may have a reasonable impact on the obtained results should also be considered, e.g. physical effects, i.e. diffusion within the entangled copolymer chains, the copolymer chain rigidity, or its susceptibility to rotation.
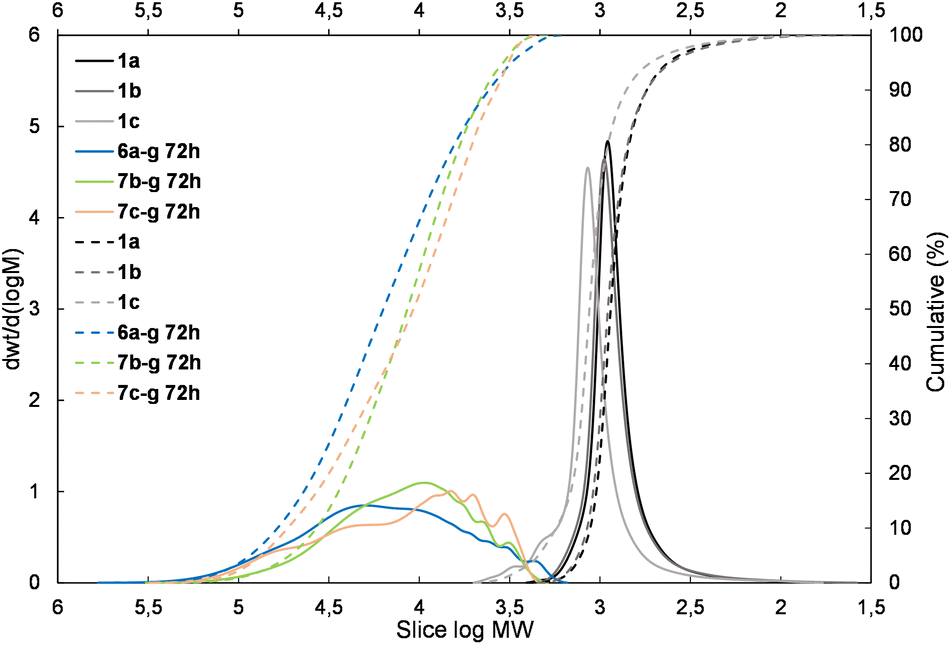 | ||
| Fig. 4 The molecular weight distribution chart for copolymers obtained with 1,4-bis(dimethylsilyl)phenylene (4g and 5g – electron donating moiety) – 6a–g, 7b–g, and 7c–g. | ||
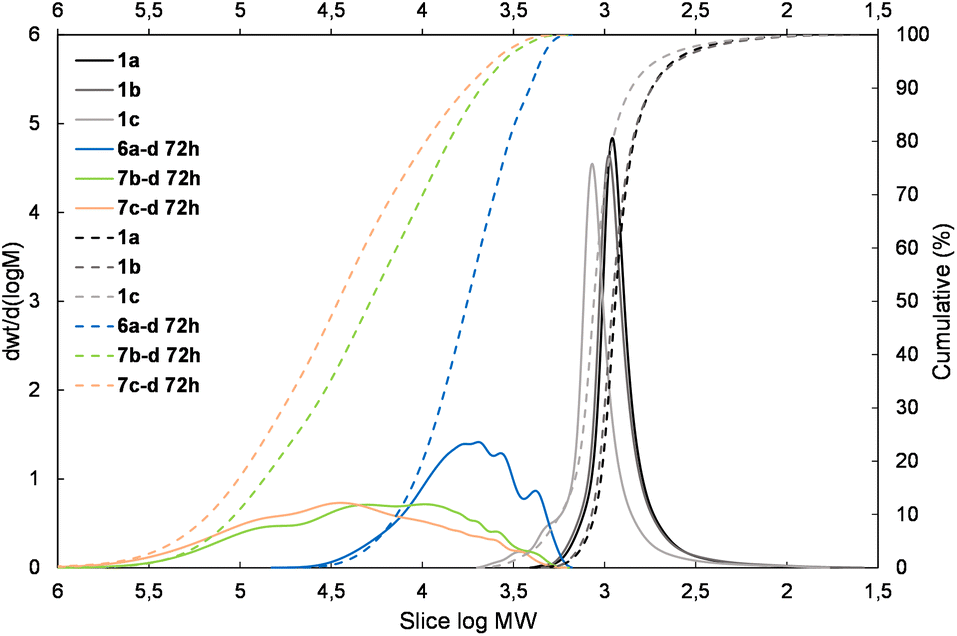 | ||
| Fig. 5 The molecular weight distribution chart for copolymers obtained with the 1,1,3,3-tetramethylsidiloxane unit (4d and 5d – electron-withdrawing moiety) – 6a–d, 7b–d, 7c–d. | ||
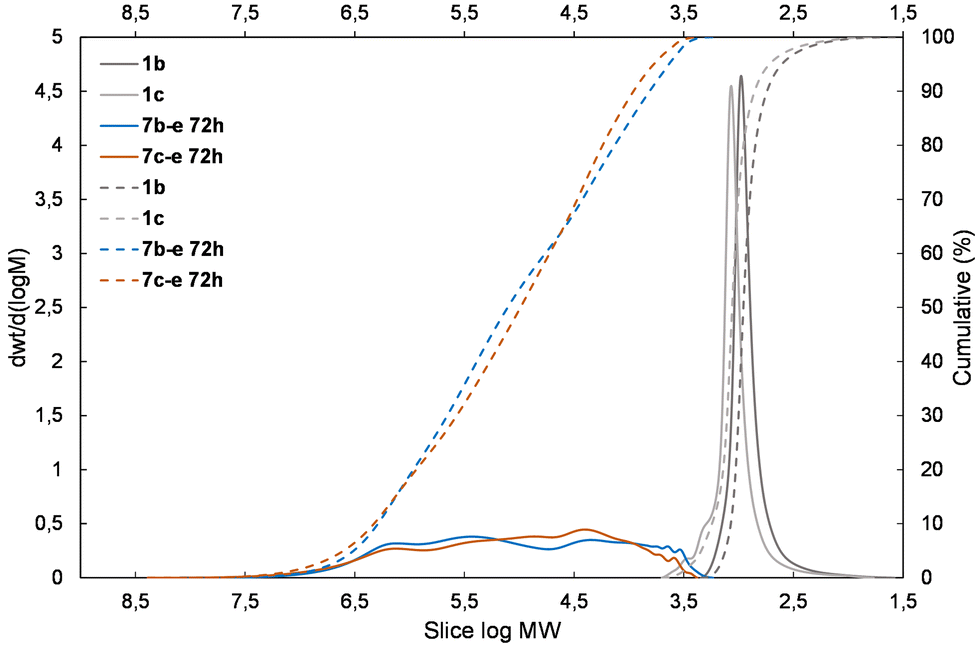 | ||
| Fig. 6 The molecular weight distribution chart for copolymers of 1bvs. 1c obtained with the 1,1,3,3,5,5-hexamethyltrisiloxane unit (5e) – 7b–e, 7c–e. | ||
The results of all spectroscopic analyses for the obtained copolymers along with the GPC data presented in the respective charts are available in the ESI.†
Selected samples 7b–g and 7c–g were verified in terms of their solubility, especially in terms of the presence of an additional –OSi(Me2)– linker between the DDSQ core and the organosilicon spacer. As expected and reported previously, these copolymers are similarly not soluble in methanol, n-hexane, and acetonitrile.51,52
However, in the case of dichloromethane (DCM), acetone, and tetrahydrofuran (THF) some discrepancies are observed. 7b–g exhibited two times better solubility in DCM and is over 10-times more soluble in acetone and THF than 7c–g. It seems that copolymers possessing –OSi(Me2)– linked to DDSQ and organosilicon fragments do not facilitate the solubility of the resulting macromolecular DDSQ-based systems (Table S4 in the ESI†). Probably the flexibility of the 7c–g linear chain influences its increased entanglement which in turn results in solubility lowering.
Thermogravimetric analysis
The prepared silsesquioxane-containing polymers were studied by thermogravimetric analysis (TGA) to verify the influence of their structure on thermal properties. The exemplary TGA curves recorded for polymers based on the 1c monomer are given in Fig. 7.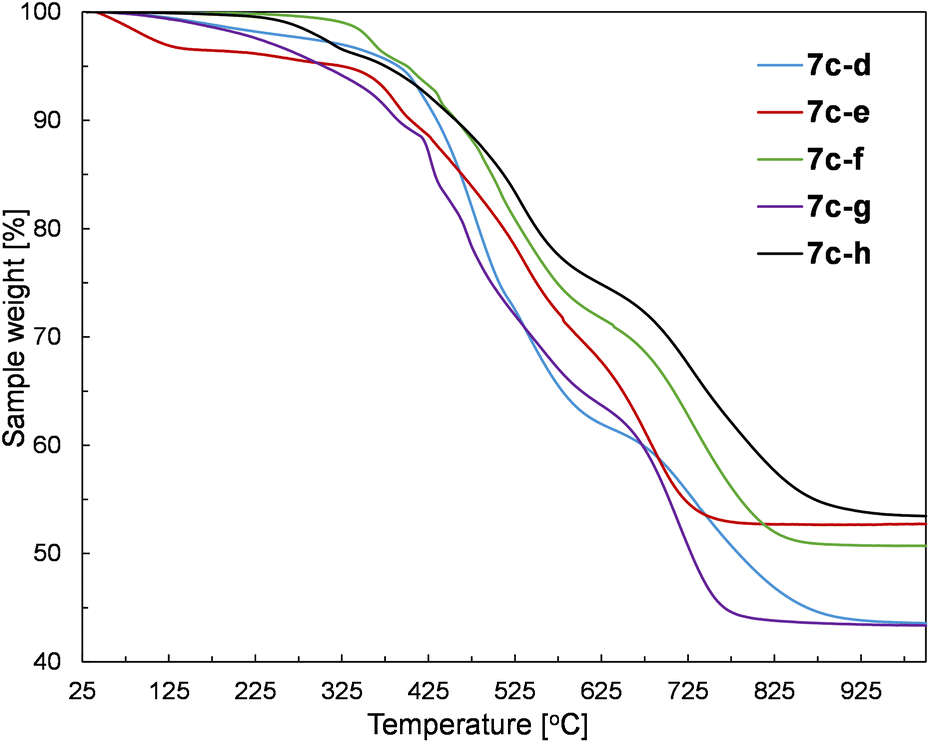 | ||
| Fig. 7 TGA analysis of 7c–(d–h) performed in air. | ||
The results of the TGA analysis containing initial degradation temperatures (T5%ds) and the residue values at 1000 °C are provided in Table S3 in the ESI.† The performed experiments in the air atmosphere revealed that there is no simple correlation between the structure and thermal stability. In our opinion, there are too many important factors that may influence the final thermal properties of the obtained copolymers. Namely, the combination of various SQs (1a–c) and divinyl- or dihydrosilyl comonomers (5d–h, 4d–g) have a significant impact on the polymerization process which affects the resulting polymer parameters such as molar mass, dispersity index, and polymerization degrees. Therefore, even the polymers based on the same SQ core contain completely different mentioned-above parameters, which impedes drawing appropriate conclusions. Nevertheless, the synthesized polymers have two common features, i.e. the main degradation of the samples occurs at temperatures higher than 400 °C, regardless of the type of SQ core and the structure of the comonomer. Moreover, all prepared polymers revealed high residues at 1000 °C (higher than 40%), which is typical for SQ-based compounds. Furthermore, the SQ-based alkenyl monomers (1b and 1c) showed to be more resistant to mass loss than the corresponding copolymers, which can be easily explained by the thermal activation of vinylsilyl-units and their subsequent polymerization/cross-linking. Therefore, the alkenyl moieties act as radical scavengers, which simply delays the degradation of the sample. This behaviour of alkenylsilsesquioxanes has already been reported.53 It should be also noticed that some of the samples showed the initiation of degradation at low temperatures (below 200 °C). It could be explained by the evaporation of the solvent traces and minor low molecular weight co-monomer residues that probably were occluded in the polymer chains.
Mechanical analysis
Due to the crystalline character of DDSQs30 and since the resulting co-polymeric products were mostly solids (or viscous solids), we decided to measure their mechanical parameters, i.e. Young's modulus and hardness. Selected co-polymer films were subjected to nanomechanical research using the nanoindentation technique (described in the ESI†). Thin films of selected specimens were fabricated through spin-coating. Glass plates were cleaned with acetone and isopropanol, and a 5 wt% solution of co-polymers in DCM was deposited on them at a spin speed of 100 rpm for 40 seconds. Measurements were taken on the surface of the resulting copolymer film by evaporating the solvent at RT. The film thickness was up to 100 μm. The values of Young's modulus and Hardness are presented in Fig. 8. Unfortunately, not all prepared samples of films were suitable for the measurements. A few photographs of the selected films are presented in Fig. 9. The coatings produced have viscoelastic properties. In some of them, viscous interactions are dominant and the material flows so fast that it is not possible to perform nanoindentation measurements (especially for the compounds with an additional –OSi(Me2)– linker, i.e.7c(d–h).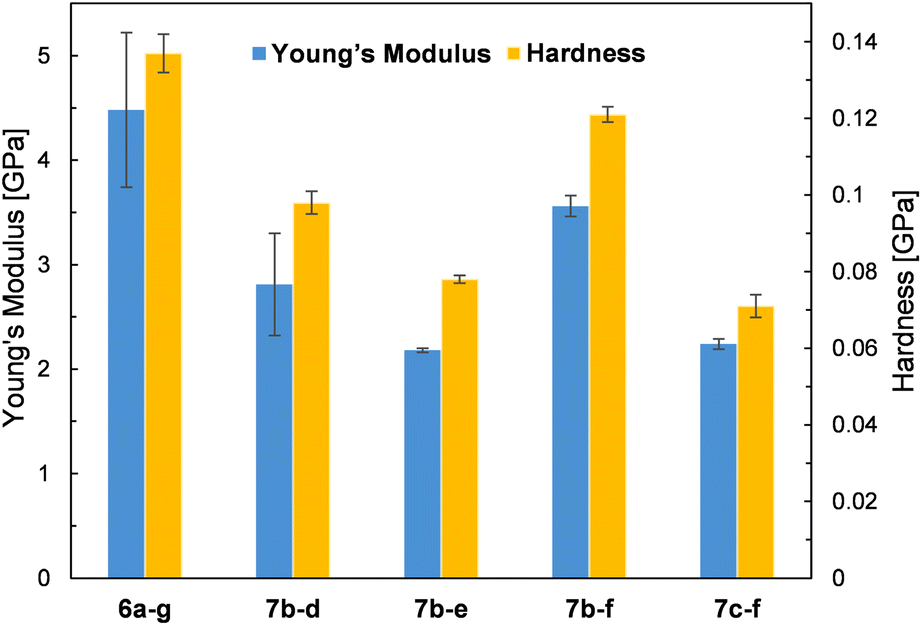 | ||
| Fig. 8 Graphical presentation of Young's modulus and hardness of the analyzed samples. | ||
 | ||
| Fig. 9 Photos of selected specimens of thin films on glass plates obtained for (a) 7b–e, (b) 7c–f, and (c) 7c–g. | ||
The measured values of Young's modulus varied between 2.18 and 4.48 GPa (the numerical values are presented in ESI Table S5†), and are in a few examples (entries 2 and 5, Table S5†) comparable with the results obtained for DDSQ-based linear co-oligomers with phenyl(s) spacers that we reported previously (1.81–2.67 GPa).30 For other specimens, the obtained values of Young's modulus are significantly higher.29 This may be attributed to the higher content of the rigid DDSQ core in the co-polymer chain in comparison with the presence of small spacers (type d–f). However, higher values than those for the respective polysiloxanes or polysilanes were observed.54 In comparison, for selected DDSQ-based composites, e.g. polyurethanes modified with the DDSQ derivative, the values of Young's modulus are incomparably smaller (however, increasing with the wt% of DDSQ used) than those of DDSQ-based co-oligomers with the A–B type alternating building block architecture, which is justified.55,56 One may find a trend in changes of values for Young's modulus, i.e. the results obtained for systems with Si–O–Si based fragments, e.g.7b–d and 7b–e, were lower than the values of the respective co-polymers with organic units, i.e.6a–g and 7b–f. This analogy may be also noted in the case of hardness data. Interestingly, the compounds with an additional –OSi(Me2)– linker, i.e.7c–f exhibited a reduction of the measured values of Young's modulus and hardness.
Moreover, it was found that the surface where the film was deposited and its adhesion to it may affect the values of Young's modulus (entries 3 and 4). It will be also a subject for further detailed studies. As noted, some of the film samples prepared for 7c type co-polymers were not suitable for the nanoindentation analysis due to their high viscosity preventing the measurement. In general, the composites and also co-polymers with the content of SQs exhibit enhanced mechanical parameters due to the intrinsic hardness of the rigid SQ segments.57,58
Surface properties – static water contact angle (WCA) measurements
The effects of DDSQs embedded in the main chain of the copolymers were evaluated also through the study of their water wettability by measurement of the static water contact angle of their thin films (prepared in the same manner as that for the nanoindentation measurements). The obtained samples in the form of colorless, transparent, or hazy films deposited on the surface of glass substrates were subjected to the WCA measurements. The selected samples are presented in Fig. 9. All of the measured samples for the 6 and 7 copolymer series exhibited contact angles in the range of 92 to 107° (see Table 3) which indicates their hydrophobic nature.| Sample | DDSQ |
|
Film appearance | WCA [°] |
|---|---|---|---|---|
| a Thin films in one layer prepared by a spin-coating technique using 5 wt% solutions of co-polymers in DCM that were deposited on the glass plates (spin speed of 100 rpm for 40 s.). b Surface structure of the coating with little cracks. | ||||
| 7b–d | 1b |
|
Hazyb | 105 |
| 7b–e | 1b |
|
Transparent | 95 |
| 6a–g | 1a |
|
Hazyb | 107 |
| 7b–f | 1b |
|
Transparentb | 98 |
| 7b–h | 1b |
|
Transparent | 97 |
| 7c–d | 1c |
|
Hazy | 102 |
| 7c–e | 1c |
|
Transparent | 92 |
| 7c–f | 1c |

|
Hazy | 103 |
| 7c–g | 1c |
|
Hazy | 103 |
| 7c–h | 1c |
|
Transparent | 97 |

The obtained parameters undoubtedly depend on the presence (and amount) of the inorganic–organic, rigid DDSQ core, but also on the type of fragments that are linking them which is confirmed by the literature results and is also reflected herein.29,30,51,59–62 Nevertheless, when analyzing the obtained results, it is easy to notice that the measured water contact angle values are not only correlated with the chemical structure of materials but the quality and form of the prepared films. Water contact angle values for hazy samples oscillated between 102 and 107°, while transparent samples were characterized by lower contact angles in the range of 92 to 97°. This phenomenon should be associated with the morphology and roughness of the surface of the obtained samples, which also affects both their appearance (transparency) and surface properties. Changes in the appearance of the obtained polymer films (see SEM images in Fig. 10(c), (e) and (f)) and their surface morphology may be the result of both differences in the chemical composition of the obtained copolymers and the resulting distinction in their physicochemical properties, e.g. solubility, which affect the speed and tendency to self-assemble, aggregate (Fig. 10(e) and (f)) and/or crystallize.
 | ||
| Fig. 10 Scanning electron microscopy images of samples: (a) 7b–e, (b) 7b–h, (c) 7c–d, (d) 7c–e, (e) 7c–f, and (f) 7c–g. | ||
Surface morphology – SEM
To assess the effect of the discussed copolymers’ chemical structures on the surface morphologies of their films selected specimens were also subjected to Scanning Electron Microscopy (SEM) imaging (Fig. 10). Samples were prepared on glass plates using 5% DCM solution using a spin coater as described before. The images of 5000× magnitude were selected as the most representative. The layers obtained from samples 7b–e, 7b–h, 7c–e, 7c–d, and 7c–f are quite homogeneous. They are smooth with small holes on the surface and they originated from solvent evaporation. The average distance between the holes is much larger on sample 7c–d and is about 5 micrometres. For 7c–f, it is about 1 micrometre. The hole density on sample 7c–f is much greater than that on 7c–d, but they are about 5× smaller in diameter. Sample 7c–g is more diverse. It has smooth areas with small holes similar to 7c–f, but at the same time, about 30% of the surface is occupied by complex structures of partially ordered lamellas protruding from its surface. Single lamellas are 1–5 micrometres long and nanometric wide, and the areas containing them do not exceed 10 micrometres at a time.Conclusions
In this work, a novel insight into the formation of linear BoC hybrid co-polymers bearing DDSQ fragments via hydrosilylation co-polymerization was presented. This synthetic approach was verified in terms of reactive Si–H and Si–HCCH2 reactive group placement in the reagents to reveal their electronic vs. electronic/steric impact on the degree of polymerization and molecular weight distribution of the obtained copolymers. Within conducted research, two unknown XRD structures of divinylfunctionalized DDSQ were resolved. As a result, a series of linear A–B alternating macromolecular hybrid systems were obtained and characterized spectroscopically (1H, 13C, 29Si NMR, FT-IR) via gel permeation chromatography and their thermal stability was analyzed. It is worth emphasizing that for dihydrosubstituted disiloxane and trisiloxane compounds reacting with divinylfunctionalized DDSQs (1b and 1c), the resulting copolymers were characterized by a 20% fraction content of Mw between 1500 and 2200 × 103 g mol−1. This is the first reported example of BoC DDSQ-based hybrid copolymers of DPn over 1000 units. Additionally, selected samples in the form of films obtained using a spin coater on glass plates were subjected to static water contact angle measurement as well as surface morphology measurement that revealed their hydrophobic properties. Also, the nanoindentation technique enabled the determination of mechanical parameters, i.e. Young's modulus and hardness. These studies exhibit a thorough synthetic approach leading to the complex verification of a series of aspects that should be taken into account while designing the formation of linear SQ-based hybrid systems. Overall, this work brings a new thought and approach to the construction of DDSQ-based BoC copolymeric hydrophobic coatings of high thermal stability for practical applications.63
Author contributions
B. D. conceived the experiments and designed the study. J. D., A. M., K. M., and M. R. participated in the planning of the study and carried out the synthesis and co-wrote the paper. M. D. performed GPC measurements. R. J. performed TG analysis. M. N. was responsible for nanoindentation and SEM techniques. M. K. performed the X-ray analysis of the obtained crystals. All authors discussed the results and contributed to the interpretation of data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Science Centre (Poland) Project OPUS UMO 2016/23/B/ST5/00201 and grant no. POWR.03.02.00-00-I026/16 co-financed by the European Union through the European Social Fund under the Operational Program Knowledge Education Development. The authors are grateful to Mr Jan Jarożek for the project of graphical abstract.References
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Recent developments in the chemistry of cubic polyhedral oligosilsesquioxanes, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- M. Laird, N. Herrmann, N. Ramsahye, C. Totée, C. Carcel, M. Unno, J. R. Bartlett and M. Wong Chi Man, Large Polyhedral Oligomeric Silsesquioxane Cages: The Isolation of Functionalized POSS with an Unprecedented Si18O27 Core, Angew. Chem., Int. Ed., 2021, 60, 3022–3027 CrossRef CAS.
- K. Yoshizawa, Y. Morimoto, K. Watanabe and N. Ootake, Silsesquioxane Derivative And Process For Producing The Same, US7319129B2, 2008.
- Y. Morimoto, K. Watanabe, N. Ootake, J. J. Inagaki, K. Yoshida and K. Ohguma, Silsesquioxane Derivative And Production Process For The Same, US7449539B2, 2008.
- Y. Morimoto, K. Watanabe, N. Ootake, J. Inagaki, K. Yoshida and K. Ohguma, Silsesquioxane Derivatives And Process For Production Thereof, US2004/0249103A1, 2004.
- Applications of Polyhedral Oligomeric Silsesquioxanes, ed. C. Hartmann-Thompson, Springer, London-New York, 2011 Search PubMed.
- B. Dudziec and B. Marciniec, Double-decker Silsesquioxanes : Current Chemistry and Applications, Curr. Org. Chem., 2017, 21, 2794–2813 Search PubMed.
- Q. Ye, H. Zhou and J. Xu, Cubic polyhedral oligomeric silsesquioxane based functional materials: Synthesis, assembly, and applications, Chem. – Asian J., 2016, 11, 1322–1337 CrossRef PubMed.
- Y. Liu, X. Wu, Y. Sun and W. Xie, POSS dental nanocomposite resin: Synthesis, shrinkage, double bond conversion, hardness, and resistance properties, Polymers, 2018, 10, 369–379 CrossRef PubMed.
- R. Kunthom, P. Piyanuch, N. Wanichacheva and V. Ervithayasuporn, Cage-like silsesequioxanes bearing rhodamines as fluorescence Hg2+ sensors, J. Photochem. Photobiol., A, 2018, 356, 248–255 CrossRef.
- R. M. Laine, Unconventional conjugation in macromonomers and polymers, Chem. Commun., 2022, 58, 10596–10618 RSC.
- H. Ghanbari, B. G. Cousins and A. M. Seifalian, A nanocage for nanomedicine: Polyhedral oligomeric silsesquioxane (POSS), Macromol. Rapid Commun., 2011, 32, 1032–1046 CrossRef CAS PubMed.
- C. Calabrese, C. Aprile, M. Gruttadauria and F. Giacalone, POSS nanostructures in catalysis, Catal. Sci. Technol., 2020, 10, 7415–7447 RSC.
- M. Walczak, R. Januszewski, M. Dutkiewicz, A. Franczyk and B. Marciniec, A facile approach for the synthesis of novel silsesquioxanes with mixed functional groups, New J. Chem., 2019, 43, 18141–18145 RSC.
- P. Żak, B. Marciniec, M. Majchrzak and C. Pietraszuk, Highly effective synthesis of vinylfunctionalised cubic silsesquioxanes, J. Organomet. Chem., 2011, 696, 887–891 CrossRef.
- J. Duszczak, K. Mituła, R. Januszewski, P. Żak, B. Dudziec and B. Marciniec, Highly efficient route for the synthesis of a novel generation of tetraorganofunctional double–decker type of silsesquioxanes, ChemCatChem, 2019, 11, 1086–1091 CAS.
- K. Stefanowska, A. Franczyk, J. Szyling and J. Walkowiak, Synthesis of functional 3−buten–1−ynes and 1,3−butadienes with silsesquioxane moiety via hydrosilylation of 1,3−diynes, ChemCatChem, 2019, 18, 4848–4853 CrossRef.
- J. Kaźmierczak, K. Kuciński and G. Hreczycho, Highly Efficient Catalytic Route for the Synthesis of Functionalized Silsesquioxanes, Inorg. Chem., 2017, 56, 9337–9342 CrossRef.
- P. Żak, B. Dudziec, M. Kubicki and B. Marciniec, Silylative Coupling versus Metathesis-Efficient Methods for the Synthesis of Difunctionalized Double-Decker Silsesquioxane Derivatives, Chem. – Eur. J., 2014, 20, 9387–9393 CrossRef.
- J. Guan, Z. Sun, R. Ansari, Y. Liu, M. Unno, A. Ouali, S. Mahbub, J. C. Furgal, N. Yodsin, S. Jungsuttiwong, D. Hashemi, J. Kieffer and R. M. Laine, Conjugated copolymers that shouldn't be, Angew. Chem., Int. Ed., 2021, 60, 11115–11119 CrossRef CAS PubMed.
- Y. Du and H. Liu, Cage-like Silsesquioxanes-based Hybrid Materials, Dalton Trans., 2020, 49, 5396–5405 RSC.
- B. Marciniec, C. Pietraszuk, P. Pawluć and H. Maciejewski, Inorganometallics (Transition Metal-Metalloid Complexes) and Catalysis, Chem. Rev., 2022, 122, 3996–4090 CrossRef CAS PubMed.
- D. Troegel and J. Stohrer, Recent advances and actual challenges in late transition metal catalyzed hydrosilylation of olefins from an industrial point of view, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef.
- X. Du and Z. Huang, Advances in Base-Metal-Catalyzed Alkene Hydrosilylation, ACS Catal., 2017, 7, 1227–1243 CrossRef.
- M. Zaranek and P. Pawluc, Markovnikov Hydrosilylation of Alkenes: How an Oddity Becomes the Goal, ACS Catal., 2018, 8, 9865–9876 CrossRef.
- J. H. Jung and R. M. Laine, Beads on a Chain (BOC) Polymers Formed from the Reaction of [NH2PhSiO1.5]x[PhSiO1.5]10−x and [NH2PhSiO1.5]x[PhSiO1.5]12−x Mixtures (x=2–4) with the Diglycidyl Ether of Bisphenol A, Macromolecules, 2011, 44, 7263–7272 CrossRef.
- J. H. Jung, J. C. Furgal, S. Clark, M. Schwartz, K. Chou and R. M. Laine, Beads on a Chain (BoC) Polymers with Model Dendronized Beads. Copolymerization of [(4-NH2C6H4SiO1.5)6(IPhSiO1.5)2] and [(4-CH3OC6H4SiO1.5)6(IPhSiO1.5)2] with 1,4-Diethynylbenzene (DEB) Gives Through-Chain, Extended 3-D Conjugation in the Excited State That Is an Average of the Corresponding Homopolymers, Macromolecules, 2013, 46, 7580–7590 CrossRef.
- M. Seino, T. Hayakawa, Y. Ishida, M. Kakimoto, K. Watanabe and H. Oikawa, Hydrosilylation Polymerization of Double-Decker-Shaped Silsesquioxane Having Hydrosilane with Diynes, Macromolecules, 2006, 39, 3473–3475 CrossRef CAS.
- S. Wu, T. Hayakawa, R. Kikuchi, S. J. Grunzinger, M. Kakimoto and H. Oikawa, Synthesis and Characterization of Semiaromatic Polyimides Containing POSS in Main Chain Derived from Double-Decker-Shaped Silsesquioxane, Macromolecules, 2007, 40, 5698–5705 CrossRef CAS.
- P. Żak, B. Dudziec, M. Dutkiewicz, M. Ludwiczak, B. Marciniec and M. Nowicki, A new class of stereoregular vinylene-arylene copolymers with double-decker silsesquioxane in the main chain, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1044–1055 CrossRef.
- M. Walczak, R. Januszewski, M. Majchrzak, M. Kubicki, B. Dudziec and B. Marciniec, Unusual cis- and trans- architecture of dihydrofunctional double-decker shaped silsesquioxane – design and construction of its ethyl bridged π-conjugated arene derivatives, New J. Chem., 2017, 41, 3290–3296 RSC.
- R. Sodkhomkhum and V. Ervithayasuporn, Synthesis of poly(siloxane/double-decker silsesquioxane) via dehydrocarbonative condensation reaction and its functionalization, Polymer, 2016, 86, 113–119 CrossRef CAS.
- M. G. Mohamed and S. W. Kuo, Functional Silica and Carbon Nanocomposites Based on Polybenzoxazines, Macromol. Chem. Phys., 2019, 220, 1800306 CrossRef.
- K. Tian, T. Y. Luh, X. Wang, C. Hao, X. Yang, Z. Li and G. Lai, Caterpillar-shaped polysilsesquioxanes, Chem. Commun., 2019, 55, 2613–2615 RSC.
- M. Miyasaka, Y. Fujiwara, H. Kudo and T. Nishikubo, Synthesis and characterization of hyperbranched polymer consisting of silsesquioxane derivatives, Polym. J., 2010, 42, 799–803 CrossRef.
- M. Soldatov and H. Liu, Hybrid porous polymers based on cage-like organosiloxanes: synthesis, properties and applications, Prog. Polym. Sci., 2021, 119, 101419 CrossRef.
- Q. Ge and H. Liu, Rational design and preparation of superhydrophobic photo-cured hybrid epoxy coating modified by fluorocarbon substituted silsesquioxane-based nanoparticles, Prog. Org. Coat., 2022, 172, 107089 CrossRef.
- F. Chen, F. Lin, Q. Zhang, R. Cai, Y. Wu and X. Ma, Polyhedral Oligomeric Silsesquioxane Hybrid Polymers: Well-Defined Architectural Design and Potential Functional Applications, Macromol. Rapid Commun., 2019, 40, 1900101 CrossRef.
- T. Hamada, Y. Nakanishi, K. Okada, S. Tsukada, A. Uedono and J. Ohshita, Thermal Insulating Property of Silsesquioxane Hybrid Film Induced by Intramolecular Void Spaces, ACS Appl. Polym. Mater., 2021, 3, 3383–3391 CrossRef CAS.
- S. Tsukada, Y. Nakanishi, T. Hamada, K. Okada, S. Mineoi and J. Ohshita, Ethylene-bridged polysilsesquioxane/hollow silica particle hybrid film for thermal insulation material, RSC Adv., 2021, 11, 24968–24975 RSC.
- H. Shi, J. Yang, M. You, Z. Li and C. He, Polyhedral Oligomeric Silsesquioxanes (POSS)-Based Hybrid Soft Gels: Molecular Design, Material Advantages, and Emerging Applications, ACS Mater. Lett., 2020, 2, 296–316 CrossRef CAS.
- P. Żak, M. Majchrzak, G. Wilkowski, B. Dudziec, M. Dutkiewicz, B. Marciniec, P. Zak, M. Majchrzak, G. Wilkowski, B. Dudziec, M. Dutkiewicz and B. Marciniec, Synthesis and characterization of functionalized molecular and macromolecular double-decker silsesquioxane systems, RSC Adv., 2016, 6, 10054–10063 RSC.
- D. Brząkalski, M. Walczak, J. Duszczak, B. Dudziec and B. Marciniec, Chlorine-Free Catalytic Formation of Silsesquioxanes with Si-OH and Si-OR Functional Groups, Eur. J. Inorg. Chem., 2018, 45, 4905–4910 CrossRef.
- A. Władyczyn, A. Gągor, K. Ślepokura and Ł. John, Hydroxyalkyl-substituted double-decker silsesquioxanes: effective separation of cis and trans isomers, Inorg. Chem. Front., 2022, 9, 3999–4008 RSC.
- J. Guan, Z. Zhang and R. M. Laine, Synthesis and Characterization of Rigid-Rod Polymers with Silsesquioxanes in the Main Chain, Macromolecules, 2022, 55, 5403–5411 CrossRef CAS.
- K. Stefanowska, A. Franczyk, J. Szyling, M. Pyziak, P. Pawluć and J. Walkowiak, Selective hydrosilylation of alkynes with octaspherosilicate (HSiMe2O)8Si8O12, Chem. – Asian J., 2018, 13, 2101–2108 CrossRef.
- N. Sabourault, G. Mignani, A. Wagner and C. Mioskowski, Platinum oxide (PtO2): A potent hydrosilylation catalyst, Org. Lett., 2002, 4, 2117–2119 CrossRef.
- J. Olejarka, A. Łącz, Z. Olejniczak and M. Hasik, Non-porous and porous materials prepared by cross-linking of polyhydromethylsiloxane with silazane compounds, Eur. Polym. J., 2018, 99, 150–164 CrossRef.
- M. Walczak, A. Franczyk and B. Marciniec, Synthesis of Monofunctionalized Silsesquioxanes (RSiMe2O)(iBu)7Si8O12 via Alkene Hydrosilylation, Chem. – Asian J., 2018, 13, 181–186 CrossRef PubMed.
- C.-W. Chen, H. Yu, M.-Y. Huang and Y.-Y. Jiang, Hydrogenation of m-Xylene Catalyzed by a Silica-supported Polysilazane-Platinum Complex, Polym. Adv. Technol., 1996, 7, 79–83 CrossRef.
- J. Xu, W. Zhang, Q. Jiang, J. Mu and Z. Jiang, Synthesis and properties of poly(aryl ether sulfone)s incorporating cage and linear organosiloxane in the backbones, Polymer, 2015, 62, 77–85 CrossRef CAS.
- J. Hao, Y. Wei, X. Li and J. Mu, Poly(arylene ether ketone)s with low dielectric constants derived from polyhedral oligomeric silsesquioxane and difluorinated aromatic ketones, J. Appl. Polym. Sci., 2018, 135(15), 46084 CrossRef.
- J. Duszczak, K. Mituła, A. Santiago-Portillo, L. Soumoy, M. Rzonsowska, R. Januszewski, L. Fusaro, C. Aprile and B. Dudziec, Double-Decker Silsesquioxanes Self-Assembled in One-Dimensional Coordination Polymeric Nano fibers with Emission Properties, ACS Appl. Mater. Interfaces, 2021, 13, 22806–22818 CrossRef CAS PubMed.
- E. Yilgör and I. Yilgör, Silicone containing copolymers: Synthesis, properties and applications, Prog. Polym. Sci., 2014, 39, 1165–1195 CrossRef.
- B. Zhao, K. Wei, L. Wang and S. Zheng, Poly(hydroxyl urethane)s with Double Decker Silsesquioxanes in the Main Chains: Synthesis, Shape Recovery, and Reprocessing Properties, Macromolecules, 2020, 53, 434–444 CrossRef CAS.
- B. Zhao, H. Ding, S. Xu and S. Zheng, Organic–Inorganic Linear Segmented Polyurethanes Simultaneously Having Shape Recovery and Self-Healing Properties, ACS Appl. Polym. Mater., 2019, 1, 3174–3184 CrossRef CAS.
- A. M. Díez-Pascual, M. A. Gómez-Fatou, F. Ania and A. Flores, Nanoindentation in polymer nanocomposites, Prog. Mater. Sci., 2015, 67, 1–94 CrossRef.
- M. Wang, H. Chi, K. S. Joshy and F. Wang, Progress in the Synthesis of Bifunctionalized Polyhedral Oligomeric Silsesquioxane, Polymers, 2019, 11, 2098–2118 CrossRef CAS PubMed.
- S. Wu, T. Hayakawa, M. A. Kakimoto and H. Oikawa, Synthesis and characterization of organosoluble aromatic polyimides containing POSS in main chain derived from double-decker-shaped silsesquioxane, Macromolecules, 2008, 40, 5698–5705 CrossRef.
- K. Wei, L. Wang and S. Zheng, Organic-inorganic copolymers with double-decker silsesquioxane in the main chains by polymerization via click chemistry, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4221–4232 CrossRef CAS.
- W. Zhang, J. Xu, X. Li, G. Song and J. Mu, Preparation, characterization, and properties of poly(aryl ether sulfone) systems with double-decker silsesquioxane in the main chains by reactive blending, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 780–788 CrossRef CAS.
- L. Wang, C. Zhang and S. Zheng, Organic-inorganic poly(hydroxyether of bisphenol A) copolymers with double-decker silsesquioxane in the main chains, J. Mater. Chem., 2011, 21, 19344–19352 RSC.
- Z. Zhang, J. Guan, R. Ansari, J. Kieffer, N. Yodsin, S. Jungsuttiwong and R. M. Laine, Further Proof of Unconventional Conjugation via Disiloxane Bonds: Double Decker Sesquioxane [vinylMeSi(O0.5)2(PhSiO1.5)8(O0.5)2SiMevinyl] Derived Alternating Terpolymers Give Excited-State Conjugation Averaging That of the Corresponding Copolymers, Macromolecules, 2022, 55, 8106–8116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Measurements, analytical data, along with GPC and TGA analysis of isolated compounds. CCDC 1972875 and 1891035. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi02161g |
| This journal is © the Partner Organisations 2023 |