
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,10-Phenanthroline ring-opening mediated by cis-{Re(CO)2} complexes†
Purificación
Cañadas
 a,
Julio
Pérez
ab,
Ramón
López
c and
Lucía
Riera
*b
a,
Julio
Pérez
ab,
Ramón
López
c and
Lucía
Riera
*b
aDepartamento de Química Orgánica e Inorgánica, Universidad de Oviedo, Julián Clavería, 8, 33006 Oviedo, Spain
bCentro de Investigación en Nanomateriales y Nanotecnología (CINN), Consejo Superior de Investigaciones Científicas (CSIC), Avda. de la Vega, 4-6, 33940 El Entrego, Spain. E-mail: l.riera@cinn.es
cDepartamento de Química Física y Analítica, Universidad de Oviedo, Julián Clavería, 8, 33006 Oviedo, Spain
First published on 30th November 2022
Abstract
Pyridine ring-opening of a metal-coordinated 1,10-phenanthroline has been demonstrated for the first time. The reaction of stable, well-defined cis,trans-[Re(CO)2(N–N)(N-RIm)(PMe3)]OTf [N–N = 2,2′-bipyridine (bipy), N-RIm = N-alkylimidazole] compounds with KN(SiMe3)2 followed by the addition of an excess of electrophile (MeOTf) afforded bipy ring-opening products easily at room temperature. The new, electron rich analogous 1,10-phenanthroline (phen) cis-{Re(CO)2} complexes allowed also, under mild conditions, the ring-opening of phen, which can be regarded as a model of coordinated quinoline, one of the impurities in fuels most difficult to eliminate by the hydrodenitrogenation (HDN) process. The phenanthroline ring-opening products are regiochemically different from those obtained with bipy, and the results of computational calculations suggest that the difference can be traced to the avoidance of a larger loss of aromaticity. The new ring-opening products have been spectroscopically characterized in solution and by means of X-ray diffraction in the solid state.
Introduction
Metal-mediated transformations of pyridine, quinoline and other N-heteroaromatics are of fundamental importance, in part due to the stability of the substrates, which makes them challenging reactions. On the other hand, they are expected to provide insight into the industrially very important, yet mechanistically poorly understood, hydrodenitrogenation (HDN) process, which is catalyzed by transition metal oxides.1 This may lead to the development of more efficient transformations under milder conditions.2 There are few examples of aromatic C–N bond cleavage processes mediated by molecular complexes, mainly due to the strength of the C–N bond, which often requires high temperatures and H2 pressures to cleave.3Pyridine ring-opening has been achieved with group 4 and 5 metal complexes, capable of binding pyridines in the very rare κ2(C,N) mode, in the presence of strongly reducing agents.4,5 Mindiola and co-workers developed a completely different approach, namely ring-opening metathesis of pyridine with a metal–carbon triple bond.6 Afterwards, using the same highly reactive Ti(II) alkylidine complex, these authors achieved the ring-opening of bicyclic N-heterocycles quinoline and isoquinoline.7 Remarkably, the transition-metal mediated C–N bond cleavage of a fused aromatic N-heterocycle had not been previously reported, despite quinolines being the most challenging substrates to remove by HDN and also the most abundant in some of the fuels to be treated. On the other hand, ring opening of substituted imidazoles by lanthanides and uranium alkyl complexes has been reported.8,9 More recently, Luo and Hou reported the extrusion of a nitrogen atom from pyridine or quinoline rings in trinuclear titanium heptahydride complexes under relatively mild conditions.10
In a completely different way, we demonstrated pyridine ring-opening of the usually very inert bipy ligand coordinated to the cationic fac-tricarbonyl rhenium(I) fragment.11 Bipy and phen are among the most extensively employed ligands in all fields of coordination chemistry;12 however, nucleophilic addition to the coordinated bipy or phen was limited, prior to our work, to a few examples of inner-sphere hydride and alkyl metal-to-pyridine migrations.13,14 We found that deprotonation of the central CH group of 1-methylimidazole coordinated to the cationic fac-{Re(bipy)(CO)3} fragment generated a strong nucleophile, which then attacks position 6 of bipy, leading to C–C coupling and dearomatization. The subsequent treatment with excess MeOTf resulted in the C–N bond cleavage and pyridine ring-opening.11 Extension of this study to 4,4′-disubstituted-2,2′-bipyridine ligands15 showed that increasing the electron density of the bipy ligand (i.e. 4,4′-dimethoxy or 4,4′-dimethylamino-2,2′-bipyridine vs. parent bipy) makes it more reactive towards electrophiles once the C–C coupling reaction has taken place, yielding ring-opening products more easily.16 However, attempts to extend these results to the ring opening of coordinated phen failed, reflecting the more inert character of quinolines.
Herein we report the results obtained with cis,trans-[Re(CO)2(PMe3)(N–N)(N-RIm)]OTf (N–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) bipy, phen; N-RIm = N-alkylimidazole) complexes, related to those previously studied by our research group with the formula fac-[Re(CO)3(N–N)(N-RIm)]OTf, but in which a carbonyl ligand has been replaced by a phosphane. Our working hypothesis is that this change makes the complexes substantially more electron rich, which should make them, once deprotonated, more reactive toward electrophiles and therefore better candidates for mediating ring-opening reactions. In fact, here we report the pyridine ring-opening of a coordinated phen, a quinoline model, a reaction, as far as we know, without precedents.
bipy, phen; N-RIm = N-alkylimidazole) complexes, related to those previously studied by our research group with the formula fac-[Re(CO)3(N–N)(N-RIm)]OTf, but in which a carbonyl ligand has been replaced by a phosphane. Our working hypothesis is that this change makes the complexes substantially more electron rich, which should make them, once deprotonated, more reactive toward electrophiles and therefore better candidates for mediating ring-opening reactions. In fact, here we report the pyridine ring-opening of a coordinated phen, a quinoline model, a reaction, as far as we know, without precedents.
Results and discussion
Cationic cis-dicarbonyl trimethylphosphane imidazole Re(I) complexes containing bipy or phen ligands were prepared using a two-step procedure starting from fac-tricarbonyl [Re(CO)3(N–N)(PMe3)]OTf derivatives.17 First, the CO in trans disposition to the phosphane is replaced by acetonitrile (employed as a solvent), a reaction mediated by trimethylamine N-oxide, and, in a subsequent step, the labile nitrile ligand was replaced with an N-methyl- or N-mesityl- (mesityl = 2,4,6-trimethylphenyl) imidazole ligand (N-MeIm or N-MesIm, respectively) to afford the new cis,trans-[Re(CO)2(N–N)(N-RIm)(PMe3)]OTf18 compounds, as depicted in Scheme 1. | ||
| Scheme 1 Synthesis of cis,trans-[Re(CO)2(N–N)(N-RIm)(PMe3)]OTf compounds. (a) Me3NO·2H2O, MeCN, Δ and (b) N-RIm, THF, Δ. | ||
To start exploring the reactivity of the new cis-dicarbonyl imidazole Re(I) complexes containing bipy or phen ligands, a slight excess of KN(SiMe3)2 (1.2 eq.) was added to a solution of cis,trans-[Re(bipy)(CO)2(N-MeIm)(PMe3)]OTf (1a) in THF at −78 °C resulting in a color change from orange to dark red. Monitoring the reaction course by IR in solution in the carbonyl region showed the formation of a neutral product, as evidenced by the shifts in the IR bands to lower wavenumber νCO values (from 1921, 1847 cm−1 to 1894, 1818 cm−1). Therefore, the increase in electron density at the metal center does not preclude imidazole deprotonation by the potassium amide, although it makes the resulting neutral species too unstable for isolation. Electrophilic interception was then performed by the addition of MeOTf in CH2Cl2 to the reaction mixture, which afforded a new pyridine ring-opening product, 3a (see Scheme 2), which could be purified by crystallization. The molecular structure of compound 3a, determined by single crystal X-ray diffraction, consists of a triflate salt of a cis-dicarbonyl rhenium cation which displays a tridentate N-donor ligand. The latter results from the C–C coupling between the deprotonated N-methylimidazole and the bipy ligand (C15–C11 bond, Fig. 1 left). As a consequence of the double methylation at N12, the C11–N12 pyridine C–N bond is broken, resulting in pyridine ring-opening and the formation of cyclopentadienyl and dimethylamino moieties. Accordingly, the Re1–N12 [2.289(7) Å] bond distance is longer than that of Re1–N1 [2.197(5) Å] as expected for amino vs. imino moieties, respectively. The N12–C7 and C7–C11 bond distances [1.512(10) Å and 1.518(10) Å, respectively] are clearly indicative of single bonds, whereas the lengths of the C8–C9 and C10–C11 bonds [1.314(10) Å and 1.364(9) Å, respectively] reflect the multiple C–C bond character. The 1H NMR spectrum of compound 3a in CD2Cl2 is consistent with its solid-state structure, showing the signals corresponding to one pyridyl group, the three one-hydrogen signals of the cyclopentadienyl unit, and the two imidazole C–H groups. More significantly, in the aliphatic region three signals of three hydrogens each are found, two of which, at 2.63 and 3.18 ppm, correspond to the new dimethylamino group formed and the other, at 3.82 ppm, to the N-MeIm ligand. In the 13C NMR spectrum, the loss of the Cs symmetry is indicated by the presence of one signal for each carbon atom of the molecule, showing, for example, two different signals for the two carbonyl ligands at 202.2 and 204.6 ppm. Additional 13C NMR signals include that of the C2 atom, at 88.6 ppm, clearly indicating the Csp3 character, and the two methyl groups, at 56.5 and 47.6 ppm, that correspond to the double methylation of the nitrogen atom.
 | ||
| Scheme 2 Deprotonation followed by methylation of cis,trans-[Re(bipy)(CO)2(N-RIm)(PMe3)]OTf (1a,b) compounds. | ||
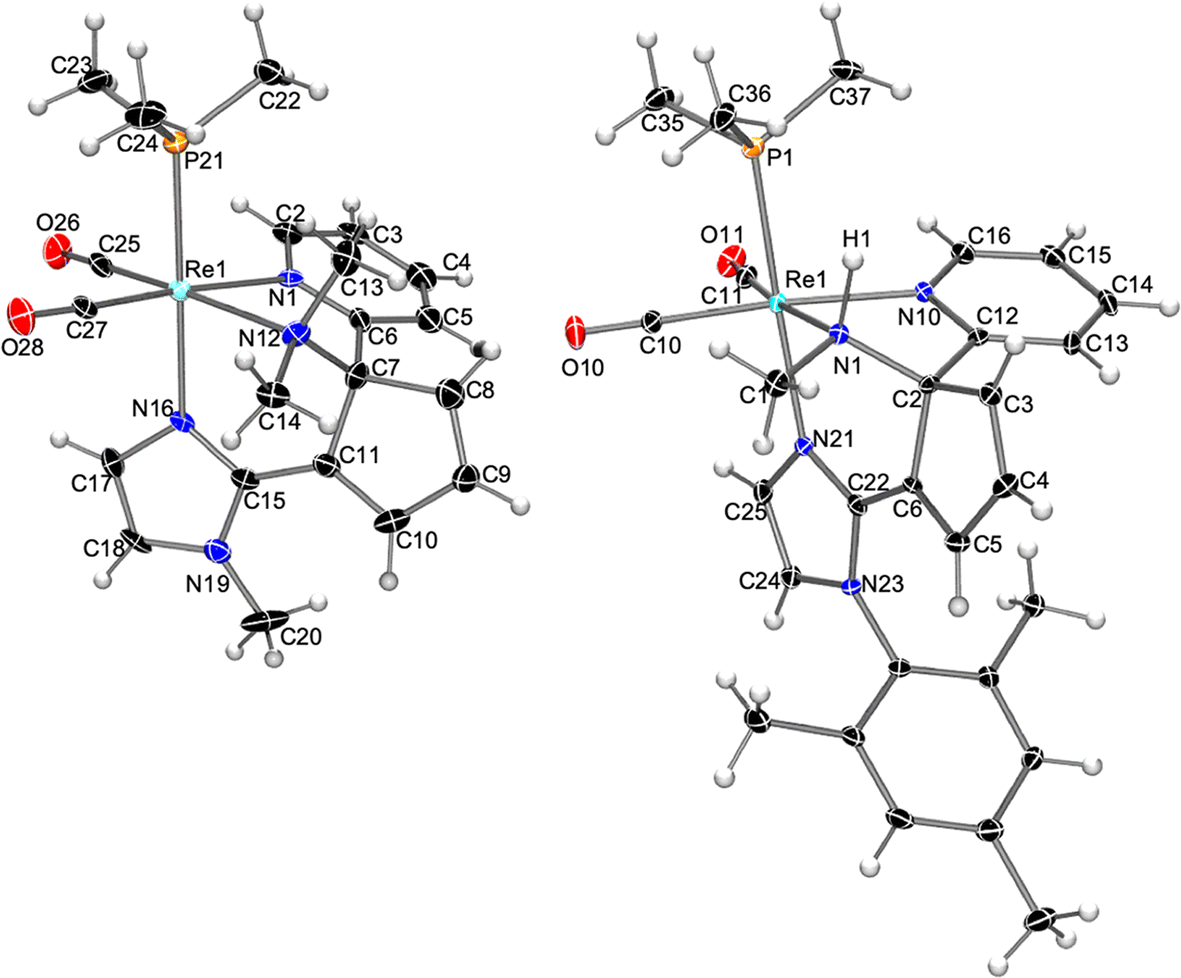 | ||
| Fig. 1 Molecular structure of the cation of compounds 3a (left) and 4b (right), showing thermal ellipsoids at the 30% probability level. | ||
This behavior is reminiscent of that previously found for compound fac-[Re(bipy)(CO)3(N-MeIm)]OTf,11 which shows that the greater electron richness of the new cis-dicarbonyl Re(I) complexes does not lead to a qualitative difference in their reactivity.
We extended our study to the N-MesIm derivative, a ligand that, with the tricarbonyl complexes, had not led to a pyridine ring-opening reaction, but stopped at the C–C coupling and dearomatization product (see below).11 The reaction of compound cis,trans-[Re(bipy)(CO)2(N-MesIm)(PMe3)]OTf (1b) with KN(SiMe3)2 (1.2 eq.) followed by MeOTf (2 eq.) led, as the NMR data in solution showed, to a mixture of two organometallic Re(I) compounds, one of which is a pyridine ring-opening product, 3b, analogous to the previously described with the N-MeIm ligand, 3a (Scheme 2). The molecular structure of the second product, 4b, determined by X-ray diffraction, was found to correspond also to a pyridine ring-opening species. As shown in Fig. 1 right, the coordination of the rhenium cation of compound 4b is analogous to that described above for 3a, showing a C–C coupling between the imidazole and bipy ligands, and the pyridine ring opening of the latter, affording a central cyclopentadienyl unit. The main difference with 3b is found in the nitrogen atom (N1), extruded from pyridine, which is now both methylated and protonated, instead of being doubly methylated. The Re1–N1 bond distance, of 2.236(4) Å, is consistent with an amino-type nitrogen as it is similar to that found in 3a [2.289(7) Å]. The N1–C1 and N1–C2 bond distances [1.484(7) and 1.506(6) Å, respectively] are typical for single C–N bonds. The formation of 3b or 4b seems to be a competitive transformation19 that can be due to the high reactivity towards electrophiles of the intermediates formed and to the inevitable presence of traces of protic impurities in the reaction media.
These results reveal that the substitution of one CO ligand by trimethylphosphane was sufficient to promote the ring-opening of a pyridine ring of bipy with the N-MesIm ligand, with which no ring-opening occurred for the tricarbonyl system.
Once confirmed that the new imidazole Re(I) complexes are good candidates to mediate, under mild conditions, pyridine ring-opening reactions of a bipy ligand, we extended our studies to the analogous yet more challenging 1,10-phenanthroline complexes, cis,trans-[Re(CO)2(N-RIm)(phen)(PMe3)]OTf (R = Me, 2a; Mes, 2b). As mentioned previously, well-defined metal complexes able to break the C–N bond of fused, bicyclic N-heterocycles are hardly known. We have chosen the phen ligand as a model of this class of substrates, like quinoline or isoquinoline, which are the most difficult impurities to activate in industrial HDN and, accordingly, the ring-opening of this type of derivatives has been found to be particularly challenging.
Thus, the reaction of the phen derivatives 2a,b with KN(SiMe3)2 (1.2 eq.) followed by MeOTf (2 eq.), was then performed to determine if it is able to promote the pyridine ring-opening reaction. In the case of the N-methylimidazole starting compound, 2a, reaction workup and slow diffusion of hexane into a concentrated CH2Cl2 solution at −20 °C afforded a crystalline material that was found to be a mixture of two Re(I) organometallic compounds, 5a (36%) and 6a (31%), which could be separated by fractional recrystallization (Scheme 3). The molecular structure of 5a, determined by single X-ray diffraction, showed it to be a triflate salt of a metallic cation resulting from double methylation of the nitrogen atom of a pyridine dearomatized ring (depicted in Fig. 2 left) that resembles the bipy ring-opening product 3a (see above). The central imidazole carbon atom (C2), deprotonated by the strong base, acts as a nucleophile and attacks an ortho phen CH group, affording a new C–C bond [C2–C6, 1.493(6) Å]. The double methylation of the nitrogen atom of the dearomatized pyridine ring (N1) caused the N1–C17 bond cleavage, and the formation of the C6–C17 bond [1.526(6) Å], with the concomitant remotion of N1 from the aromatic system. The resulting ligand is somewhat different from that obtained from the ring-opening of the bipy ligand, as in this case the carbon atom that has been attacked by the deprotonated imidazole (C6) is the same to which the dimethylamino group remains bonded. Therefore, the ligand can be described as a cyclopenta[h]quinoline bearing dimethylamino and imidazolyl groups at the 9 position (C6), which are both coordinated to the cis-{Re(CO)2(PMe3)} fragment. Probably this difference with the regiochemistry encountered with the bipy system is imposed by the rigidity of the fused central aromatic ring of the phen ligand and is crucial to the planarity of the resulting cyclopentaquinoline ligand. As far as we know, the ring opening reaction of a phen ligand has no precedents. NMR spectra of compound 5a in CD2Cl2 were consistent with the solid-state structure. The 1H NMR spectrum shows seven one-hydrogen signals, between 6.84 and 9.42 ppm, for the new conjugated cyclopentaquinoline unit, and three signals, in the aliphatic region, integrating three hydrogens each, that correspond to the N-MeIm and dimethylamino groups, along with the signal for the PMe3 ligand (a 9-hydrogen doublet at 1.52 ppm). In the 13C NMR, the asymmetry of the molecule is evidenced by the presence of one signal for each carbon atom. The signals at 55.4 and 45.7 ppm correspond to the two new methyl groups of the dimethylamino moiety, and the chemical shift of the signal of the C2 atom, at 81.2 ppm, clearly indicates that it is no longer an aromatic carbon atom.
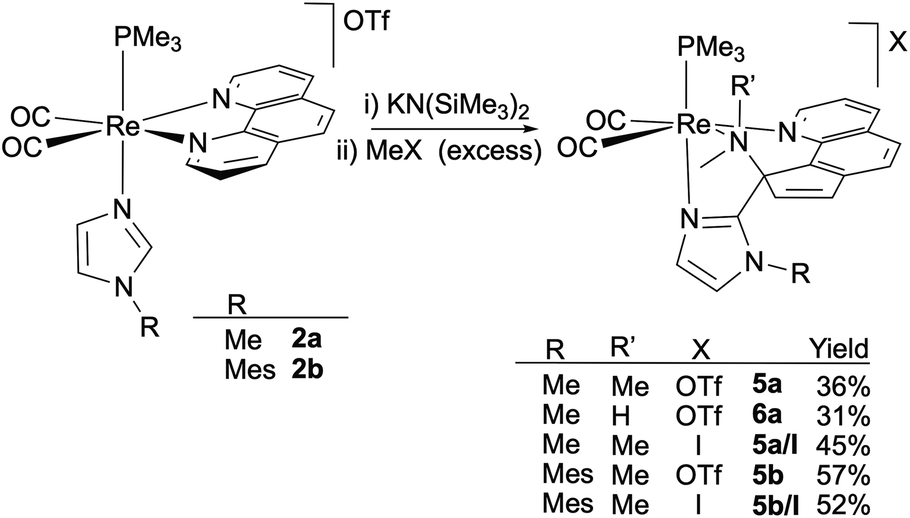 | ||
| Scheme 3 Deprotonation/methylation sequence of 1,10-phenanthroline compounds cis,trans-[Re(CO)2(N-RIm)(phen)(PMe3)]OTf (2a,b). | ||
 | ||
| Fig. 2 Molecular structure of the cation of compounds 5a (left) and 6a (right), showing thermal ellipsoids at the 30% probability level. | ||
Spectroscopic characterization of compound 6a showed similar features to those observed for 5a. The IR νCO bands in CH2Cl2, at 1917 and 1836 cm−1, indicate the persistence of the cis-{CO}2 unit and are at significantly higher wavenumbers than those in the neutral precursor (at 1895 and 1817 cm−1 in THF) in accordance with the reaction with an electrophile. The 1H NMR spectrum is, in contrast, quite surprising as it shows a set of seven signals, in the 6.71–9.68 ppm range, similar to that observed for the cyclopentaquinoline moiety in compound 5a, but only one extra signal for a methyl group is shown in the aliphatic region. Furthermore, this signal, which integrates for three hydrogen atoms at 2.51 ppm, is a doublet showing a small coupling constant (5.9 Hz), and a correlation in the 1H–1H COSY spectrum with a one hydrogen broad-singlet at 6.11 ppm, supporting the presence of an NH group. The 13C NMR is consistent, and only two signals, at 36.4 and 35.2 ppm, are observed for the two methyl groups (apart from the signal of the PMe3 ligand at 18.6 ppm), and the signal of the C2 atom is observed also in the aliphatic region, at 75.0 ppm, indicating its non-aromatic character. It seems, therefore, that, as for the bipy derivative 1b, a protonated-methylated ring-opened product is also formed (Scheme 2). The molecular structure of the rhenium cation contained in 6a is shown in Fig. 2 right (the triflate anion has been omitted for clarity). The solid-state structure of the Re(I) complex in 6a is consistent with the NMR data, showing a cyclopentaquinoline moiety, similar to that found in 5a, bonded, through the phen carbon atom C2, to nitrogen N1, which is also methylated and protonated. Another remarkable difference between the rhenium cation of 6a and that of the dimethylated analog found in 5a is the cis disposition of the PMe3 ligand to the N-MeIm moiety, whereas in 5a a trans disposition is observed.
Density functional theory calculations20 were performed in order to rationalize the difference found in the ring-opening products of the reactions of Re(I) complexes containing bipy or phen ligands (Schemes 2 and 3, respectively). To this end, we optimized the structures of the experimentally found products 3a and 5a (IIIa and Va′, respectively, in Fig. 3) and those of their isomers (IIIa′ and Va, respectively, in Fig. 3). The prime symbol refers to the isomer in which dimethylamino and imidazolyl groups are bonded to the same carbon atom of the cyclopentadienyl moiety (for the phen ligand, this is the experimentally obtained isomer). For bipy, in IIIa the diene moiety of the cyclopentadienyl unit is almost in the same plane of the imidazole ring, whereas in the IIIa′ isomer it is the pyridine ring the one it is coplanar with (see Fig. 3). As a result, in both isomers the diene fragment of the cyclopentadienyl ring formed in the reaction is conjugated with an aromatic system: the pyridyl ring in IIIa′ and the imidazolyl unit in IIIa. The nucleus-independent chemical shift (NICS) values were calculated 1 Å above the ring critical points (RCP) of the electron density located on the pyridyl, cyclopentadienyl, and imidazolyl rings (RCP and NICS(1): green and black dots in Fig. 3, respectively). Keeping in mind that typical NICS(1) values, in parts per million (ppm), are ca. −10.2 for benzene,21,22 or −10.1 for pyridine,23,24 our computations do not show significant differences in the NICS(1) values of both complexes. As expected, pyridine (NICS(1) = −9.4/−9.1 ppm, for IIIa/IIIa′ respectively) and imidazole (NICS(1) = −9.1/−9.2 ppm, for IIIa/IIIa′ respectively) show notably larger π-electronic delocalization than that of the cyclopentadienyl (NICS(1) = −3.3/−3.7 ppm, for IIIa/IIIa′ respectively) rings. For bipy, the IIIa isomer (the one experimentally obtained) is 5.0 kcal mol−1 in Gibbs energy in CH2Cl2 solution more stable than isomer IIIa′.25 In contrast, for phen the isomer Va′ is 24.5 kcal mol−1 more stable than Va, which is in agreement with the fact that 5a is the experimentally obtained product. This notable difference in stability can be, at least in part, ascribed to the loss of aromaticity of the central ring of the phen ligand in Va (see Fig. 3). This loss of aromaticity is clearly evidenced by the nucleus-independent chemical shift (NICS) values, as our computations reflect the strong aromatic character of the phen central ring in Va′ (NICS(1) = −10.2 ppm) but not in Va (NICS(1) = −0.2 ppm). The other rings exhibit similar NICS(1) values for both isomers Va and Va′. On the other hand, the formation of Va would imply a severe distortion of the geometry, with concomitant loss of planarity and conjugation of the cyclopentadienyl unit and the two six-membered rings from phen which undoubtedly would imply a destabilizing effect (see Fig. 3). Considering jointly all of this, it can be proposed that the transition state for the formation of the Va isomer would be significantly higher in energy than that corresponding to the formation of Va′, as a consequence of the loss of resonance energy.
 | ||
| Fig. 3 CPCM-B3LYP/6-31+G(d) optimized structures of the two possible ring-opening products found for both the bipy and phen ligands. The NICS(1) values in ppm are provided in parentheses. | ||
Interestingly, the addition of freshly purified (by filtering through an alumina plug) methyl iodide instead of MeOTf in CH2Cl2, once compound cis,trans-[Re(CO)2(N-MeIm)(phen)(PMe3)]OTf (2a) has been deprotonated, afforded, after eight hours at room temperature, selectively the ring-opening product 5a/I as the only rhenium complex in moderate yield. Compound 5a/I was fully characterized in solution by means of IR and NMR, showing practically identical features to those found for 5a.
Application of this deprotonation–methylation (MeI) sequence to the mesitylimidazole analog 2b yielded the doubly methylated ring-opened species as the main organometallic product 5b/I, in 52% yield (Scheme 3). It is remarkable that the ring-opening of a pyridine unit of a Re(I) coordinated phen ligand is exclusively found not only for the more electron rich complex 2a, bearing an N-MeIm ligand, but also for the mesitylimidazole analog 2b. Slow diffusion of hexane into a concentrated solution of 5b [obtained by reaction of 2b with KN(SiMe3)2 and MeOTf (2 eq.)] yielded orange crystals that allowed the molecular structure determination by X-ray diffraction. Fig. 4 shows the structure of the cation of 5b, confirming the structure proposed on the basis of the spectroscopic data in solution, i.e. the double methylation of the nitrogen N1, which originates from the phen ligand, and, which as a consequence was extruded from the aromatic ligand yielding a cyclopentaquinoline moiety. The latter is also coupled through position 9 (C2 in Fig. 4) to the central N-MesIm carbon atom (C22), so altogether there is a tridentate N-donor ligand facially coordinated to the Re atom through amino (N1), imino (N10) and imidazolyl (N21) nitrogen atoms.
 | ||
| Fig. 4 Molecular structure of the cation of compound 5b, showing thermal ellipsoids at the 30% probability level. | ||
Conclusions
The use of electron rich cis-{Re(CO)2} trimethylphosphane complexes allowed the pyridine ring-opening of a phen ligand for the first time. The use of the more electron rich cis,trans-[Re(CO)2(N–N)(N-RIm)(PMe3)]OTf (N–N bipy, phen; R = Me, Mes) compounds [compared to the previously studied fac-[Re(CO)3(N–N)(N-RIm)]OTf (N–N bipy, phen; R = Me, Mes)] does not preclude the deprotonation of the imidazole central CH group upon addition of the strong base KN(SiMe3)2. The resulting dearomatized C–C coupling products are highly reactive towards electrophiles to afford, under mild conditions, products resulting from the ring opening of a pyridyl unit of the bipy or phen ligands. Bipy and phen ring-opening products differ in the C–N bond which is broken in the reaction: for bipy, it is the N1–C6 bond, and for phen, it is the other C–N bond [N1–C11]. Computational calculations reveal that for the latter there is a large difference in the stability of both isomers, mainly due to the loss of aromaticity of the phen central ring if the isomer analogue to the bipy one was formed.
Data availability
Crystallographic data for compounds 3a, 4b, 5a, 5b and 6a have been deposited at the CCDC under CCDC 2192108–2192112† The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
LR and JP conceived the project. PC performed the investigations and analysed data with input from LR. RL performed and analysed the computational results. LR prepared the manuscript with the contribution of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Ministerio de Ciencia, Innovación y Universidades/FEDER (grant PGC2018-100013-B-I00). The authors thank Servicios Científico-Técnicos de la Universidad de Oviedo for technical support and the use of RIAIDT-USC analytical facilities.References
- S. S. Bello, C. Wang, M. Zhang, H. Gao, Z. Han, L. Shi, F. Su and G. Xu, A review on the reaction mechanism of hydrodesulfurization and hydrodenitrogenation in heavy oil upgrading, Energy Fuels, 2021, 35, 10998 CrossRef; E. Furimsky and F. E. Massoth, Hydrodenitrogenation of petroleum, Catal. Rev. Sci. Eng., 2005, 47, 297 CrossRef; R. Prins, Catalytic hydrodenitrogenation, Adv. Catal., 2001, 46, 399 Search PubMed; T. C. Ho, Hydrodenitrogenation Catalysis, Catal. Rev. Sci. Eng., 1988, 30, 117 CrossRef.
- M. Bachrach, T. J. Marks and J. M. Notestein, Understanding the hydrodenitrogenation of heteroaromatics on a molecular level, ACS Catal., 2016, 6, 1455 CrossRef; C. Bianchini, A. Meli and F. Vizza, Modelling the hydrodenitrogenation of aromatic N-heterocycles in the homogeneous phase, Eur. J. Inorg. Chem., 2001, 43 CrossRef.
- K. J. Weller, P. A. Fox, S. D. Gray and D. E. Wigley, Homogeneous models for hydrodenitrogenation catalysis, Polyhedron, 1997, 16, 3139 CrossRef; J. R. Katzer and R. Sivasubramanian, Process and catalyst needs for hydrodenitrogenation, Catal. Rev. Sci. Eng., 1979, 20, 155 CrossRef.
- K. J. Weller, I. Filippov, P. M. Briggs and D. E. Wigley, Pyridine degradation intermediates as models for hydrodenitrogenation catalysis: preparation and properties of a metallapyridine complex, Organometallics, 1998, 17, 322 CrossRef; K. J. Weller, I. Filippov, P. M. Briggs and D. E. Wigley, Isolation and characterization of a monomeric metallapyridine complex: models for pyridine hydrodenitrogenation intermediates, J. Organomet. Chem., 1997, 528, 225 CrossRef; K. D. Allen, M. A. Bruck, S. D. Gray, R. P. Kingsborough, D. P. Smith, K. J. Weller and D. E. Wigley, Quinoline binding mode as a function of oxidation state in aryloxide-supported tantalum complexes: models for hydrodenitrogenation catalysis, Polyhedron, 1995, 14, 3315 CrossRef; K. J. Weller, S. D. Gray, P. M. Briggs and D. E. Wigley, Mechanistic aspects of carbon-nitrogen bond cleavage in an η2(N,C)-pyridine complex: intimate details of metal to ligand aryl migrations and their relevance to hydrodenitrogenation catalysis, Organometallics, 1995, 14, 5588 CrossRef; S. D. Gray, K. J. Weller, M. A. Bruck, P. M. Briggs and D. E. Wigley, Carbon-nitrogen bond cleavage in an η2(N,C)-pyridine complex induced by intramolecular metal-to-ligand alkyl migration: models for hydrodenitrogenation catalysis, J. Am. Chem. Soc., 1995, 117, 10678 CrossRef; S. D. Gray, D. P. Smith, M. A. Bruck and D. E. Wigley, Regioselective carbon-nitrogen bond cleavage in an η2(N,C)-coordinated pyridine and an η1(N)→η2(N,C) bonding rearrangement in coordinated quinoline: models for hydrodenitrogenation catalysis, J. Am. Chem. Soc., 1992, 114, 5462 CrossRef.
- J. B. Bonanno, A. S. Veige, P. T. Wolczanski and E. B. Lobkovsky, Amide derivatives of tantalum and niobium-promoted ring opening of 3,5-lutidine, Inorg. Chim. Acta, 2003, 345, 173 CrossRef; T. S. Kleckley, J. L. Bennet, P. T. Wolczanski and E. B. Lobkovsky, Pyridine C=N bond cleavage mediated by (silox)3Nb (silox = tBu3SiO), J. Am. Chem. Soc., 1997, 119, 247 CrossRef; K. J. Covert, D. R. Neithamer, M. C. Zonnnevylle, R. E. LaPointe, C. P. Schaller and P. T. Wolczanski, Pyridine and related adducts, (silox)3ML (M = scandium, titanium, vanadium, tantalum): η1-pyridine-N vs η2-pyridine-N,C ligation, Inorg. Chem., 1991, 30, 2494 CrossRef; D. R. Neithamer, L. Parkanyi, J. F. Mitchell and P. T. Wolczanski, η2-(N,C)-Pyridine and μ-η2(1,2): η2(4,5)-benzene complexes of (silox)3Ta (silox=t-Bu3SiO−), J. Am. Chem. Soc., 1988, 110, 4421 CrossRef.
- A. R. Fout, B. C. Bailey, J. Tomaszewski and D. J. Mindiola, Cyclic denitrogenation of N-heterocycles applying a homogeneous titanium reagent, J. Am. Chem. Soc., 2007, 129, 12640 CrossRef; B. C. Bailey, H. Fan, J. C. Huffman, M. H. Baik and D. J. Mindiola, Room temperature ring-opening metathesis of pyridines by a transient Ti≡C linkage, J. Am. Chem. Soc., 2006, 128, 6798 CrossRef PubMed.
- S. Baek, T. Kurogi, D. Kang, M. Kamitani, S. Kwon, D. P. Solowey, C.-H. Chen, M. Pink, P. J. Carroll, D. J. Mindiola and M.-H. Baik, Room-temperature ring-opening of quinoline, isoquinoline, and pyridine with low-valent titanium, J. Am. Chem. Soc., 2017, 139, 12804 CrossRef.
- M. J. Monreal, S. Kjan and P. L. Diaconescu, Beyond C-H activation with uranium: a cascade of reactions mediated by a uranium dialkyl complex, Angew. Chem., Int. Ed., 2009, 48, 8352 CrossRef; C. T. Carver, D. Benitez, K. L. Miller, B. N. Williams, E. Tkatchouk, W. A. Goddard III and P. L. Diaconescu, Reactions of Group III biheterocyclic complexes, J. Am. Chem. Soc., 2009, 131, 10269 CrossRef PubMed; C. T. Carver and P. L. Diaconescu, Ring-opening reactions of aromatic N-heterocycles by scandium and yttrium alkyl complexes, J. Am. Chem. Soc., 2008, 130, 7558 CrossRef.
- W. Yi, J. Zhang, S. Huang, L. Weng and X. Zhou, Reactivity of TpMe2-supported yttrium alkyl complexes toward aromatic N-heterocycles: ring-opening or C=C bond formation directed by C-H activation, Chem. – Eur. J., 2014, 20, 867 CrossRef CAS PubMed; Y. Zhang, J. Zhang, J. Hong, F. Zhang, L. Weng and X. Zhou, Versatile reactivity of β-diketiminato-supported yttrium dialkyl complex toward aromatic N-heterocycles, Organometallics, 2014, 33, 7052 CrossRef.
- S. Hu, G. Luo, T. Shima, Y. Luo and Z. Hou, Hydrodenitrogenation of pyridines and quinolines at multinuclear titanium hydride framework, Nat. Commun., 2017, 8, 1866 CrossRef.
- M. A. Huertos, J. Pérez and L. Riera, Pyridine ring opening at room temperature at a rhenium tricarbonyl bipyridine complex, J. Am. Chem. Soc., 2008, 130, 5662 CrossRef CAS.
- A. P. Smith and C. L. Fraser, Bipyridine ligands, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier Pergamon, Oxford, 2004, vol. 1, pp. 1–23 Search PubMed.
- C. Weetman, M. S. Hill and M. F. Mahon, Magnesium-catalysed hydroboration of pyridines: kinetic analysis and poly-pyridine dearomatisation, Polyhedron, 2016, 103, 115 CrossRef; S. Leelasubcharoen, K.-C. Lam, T. E. Concolino, A. L. Rheingold and K. H. Theopold, Unusual transformations of a bipyridine ligand in attempts to trap a terminal chromium(III) alkylidine, Organometallics, 2001, 20, 182 CrossRef; L. M. Kobriger, A. K. McMullen, P. E. Fanwicck and I. P. Rothwell, Complexation and intramolecular alkylation of bipyridyl ligands by Zr(OAr)2R2 (R = CH3, CH2Ph) compounds, Polyhedron, 1989, 8, 77 CrossRef.
- An example of dearomatization of a 2,2′:6′,2′′-terpyridine coordinated to Lu(III) has been reported, see: K. C. Jantunen, B. L. Scott, P. J. Hay, J. C. Gordon and J. L. Kiplinger, Dearomatization and functionalization of terpyridine by lutetium(III) alkyl complexes, J. Am. Chem. Soc., 2006, 128, 6322 CrossRef PubMed.
- A computational study on the effect of different substituents onto the electronic properties of a 2,2′-bipyridine coordinated to Rh(III) has been reported: D. R. Pahls, J. T. Groves, T. B. Gunnoe and T. R. Cundari, Theoretical study of reductive functionalization of methyl ligands of group 9 complexes supported by two bipyridyl ligands: a key step in catalytic hydrocarbon functionalization, Organometallics, 2014, 33, 1936 CrossRef.
- M. Espinal-Viguri, S. Fombona, D. Álvarez, J. Díaz, M. I. Menéndez, R. López, J. Pérez and L. Riera, Regiochemistry control by bipyridine substituents in the deprotonation of ReI and MoIIN-alkylimidazole complexes, Chem. – Eur. J., 2019, 25, 92533 CrossRef PubMed.
- The method used herein is a variation of that previously reported in: R. Arévalo, R. López, L. Falvello, L. Riera and J. Pérez, Building C(sp3) molecular complexity on 2,2′-bipyridine and 1,10-phenanthroline in rhenium tricarbonyl complexes, Chem. – Eur. J., 2021, 27, 379 CrossRef PubMed; R. Arévalo, M. I. Menéndez, R. López, I. Merino, L. Riera and J. Pérez, Nucleophilic additions to coordinated 1,10-phenanthroline: intramolecular, intermolecular, reversible and irreversible, Chem. – Eur. J., 2016, 22, 17972 CrossRef PubMed . The main advantages are that it is carried out at room temperature, affords pure fac-[Re(CO)3(N–N)(PMe3)]OTf compounds, and avoids the use of potentially explosive perchlorate salts.
- See the ESI† for further experimental details.
- Based on the integration of the signals in the 31P NMR spectrum of the reaction mixture, the 3b
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4b ratio is 1.05:1.
:4b ratio is 1.05:1. - The geometry and energy of the optimized ring-opening products investigated were studied at the CPCM-B3LYP/6-31+G(d)-LANL2DZ level of theory (see the ESI† for more details).
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Nucleus-independent chemical shifts (NICS) as an aromaticity criterion, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- L. Andjelkovic, M. Peric, M. Zlatar, S. Grubisic and M. Gruden-Pavlovic, Magnetic criteria of aromaticity in a benzene cation and anion: how does the Jahn-Teller effect influence the aromaticity?, Tetrahedron Lett., 2012, 53, 794 CrossRef CAS.
- D. S. J. Shobe, Calculations on the Group 15 intraring substituted benzenes (pyridine to bismin series) and their datively bonded oxides and sulfides, Phys. Chem. A, 2005, 109, 9118 CrossRef CAS PubMed.
- I. Pausescu, M. Medeleanu, M. Stefanescu, F. Peter and R. Pop, A DFT study on the stability and aromaticity of heterobenzenes containing group 15 elements, Heteroat. Chem., 2015, 26, 206 CrossRef CAS.
- The possible formation of IIIa from IIIa′ has been analyzed given the small energy difference found between both isomers, but it has been discarded considering the high energy barrier found (see the ESI† for more details).
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and computational details, including preparative procedures, spectroscopic data, the computational matrix, and NMR spectra for the characterization of compounds. CCDC 2192108–2192112. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi01890j |
| This journal is © the Partner Organisations 2023 |