
Ginseng-derived exosome-like nanovesicles extracted by sucrose gradient ultracentrifugation to inhibit osteoclast differentiation†
Kwansung
Seo‡
 a,
Ji Hye
Yoo‡
b,
Jisu
Kim
c,
Sung Jun
Min
a,
Dong Nyoung
Heo
d,
Il Keun
Kwon
*d and
Ho-Jin
Moon
*d
a,
Ji Hye
Yoo‡
b,
Jisu
Kim
c,
Sung Jun
Min
a,
Dong Nyoung
Heo
d,
Il Keun
Kwon
*d and
Ho-Jin
Moon
*d
aDepartment of Dentistry, Graduate School, Kyung Hee University, 26 Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Republic of Korea
bDepartment of Biomedical Science and Technology, Graduate School, Kyung Hee University, 26 Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Republic of Korea
cDepartment of Pharmaceutics, School of Pharmacy, Fudan University & Key Laboratory of Smart Drug Delivery, Ministry of Education, Shanghai 201203, China
dDepartment of Dental Materials, School of Dentistry, Kyung Hee University, 26 Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Republic of Korea. E-mail: kwoni@khu.ac.kr; 3216@khu.ac.kr; Tel: +82-2-961-0350 Tel: +82-2-961-0771
First published on 1st March 2023
Abstract
Plant-derived extracellular nanovesicles contain RNA and proteins with unique and diverse pharmacological mechanisms. The extracellular nanovesicles encapsulating plant extracts resemble exosomes as they have a round, lipid bilayer morphology. Ginseng is anti-inflammatory, anti-cancer, immunostimulant, and osteogenic/anti-osteoporotic. Here, we confirmed that ginseng-derived extracellular nanovesicles (GDNs) inhibit osteoclast differentiation and elucidated the associated molecular mechanisms. We isolated GDNs by centrifugation with a sucrose gradient. We measured their dynamic light scattering and zeta potentials and examined their morphology by transmission electron microscopy. We used bone marrow-derived macrophages (BMMs) to determine the potential cytotoxicity of GDNs and establish their ability to inhibit osteoclast differentiation. The GDNs treatment maintained high BMM viability and proliferation whilst impeding osteoclastogenesis. Tartrate-resistant acid phosphatase and F-actin staining revealed that GDNs at concentrations >1 μg mL−1 strongly hindered osteoclast differentiation. Moreover, they substantially suppressed the RANKL-induced IκBα, c-JUN n-terminal kinase, and extracellular signal-regulated kinase signaling pathways and the genes regulating osteoclast maturation. The GDNs contained elevated proportions of Rb1 and Rg1 ginsenosides and were more effective than either of them alone or in combination at inhibiting osteoclast differentiation. In vivo bone analysis via microcomputerized tomography, bone volume/total volume ratios, and bone mineral density and bone cavity measurements demonstrated the inhibitory effect of GDNs against osteoclast differentiation in lipopolysaccharide-induced bone resorption mouse models. The results of this work suggest that GDNs are anti-osteoporotic by inhibiting osteoclast differentiation and are, therefore, promising for use in the clinical prevention and treatment of bone loss diseases.
Introduction
Bone remodeling is a lifelong process and vital to healthy bone tissue function. It is systematically controlled via osteoclast and osteoblast metabolism, and it repairs microdamage in bone tissue, transforms old to new bone tissue, and mechanically alters bone to balance the anatomical structure of bone tissue and the forces applied to it.1,2When bone resorption by osteoclasts exceeds bone neogenesis by osteoblasts, osteoporotic low fracture resistance may ensue.3 Osteoporosis may occur in menopausal women and elderly men in response to hormonal changes. There are two therapeutic approaches for osteoporosis, namely (1) decreasing osteoclast activity and suppressing bone resorption, and (2) increasing osteoblast activity and promoting bone formation.
Bisphosphonates have been widely applied clinically to treat osteoporosis as they have high prophylactic efficacy, are cost-effective, and are skeletal-specific.4 However, they may also induce severe side effects such as gastroesophageal irritation and osteonecrosis of the jaw (BRONJ).5 Therefore, safe and efficacious new anti-osteoporotic agents are urgently needed.
Several recent studies have empirically demonstrated the potential of certain plants as alternative osteogenic agents that could inhibit bone loss. Chauhan et al. (2018) reported that Bombax ceiba had osteogenic activity in vitro and prevented estrogen deficiency-induced osteoporosis.6 Wang et al. (2021) reported that the antioxidant sesquiterpene from Curcuma zedoaria impeded binding between IPMK and TRAF6 and attenuated osteoclast activity in an OVX-induced bone loss model.7 Another study showed the efficacy of safflower seed extract at inhibiting osteoclast differentiation. Safflower seed extract suppressed reactive oxygen species generation, significantly downregulated the p38 and IκBα pathways, and inhibited NFATc1 expression in osteoclast precursors.8
A few techniques can effectively isolate the secondary metabolites in plants for use in experiments. (1) The active principles are extracted with water, ethanol, or methanol. (2) The active ingredients are extracted in the form of exosome-like extracellular nanovesicles.9 The traditional extraction methods isolate single molecules without any capsulation whereas the nanovesicle extraction methods encapsulate various plant natural products into lipid bilayer spheres. These nanovesicles also contain small RNAs, lipids, and proteins resembling those of mammalian-derived exosomes, and they have various beneficial effects on human cells. Unlike mammalian exosomes, however, plant-derived nanovesicles are not detected by the human immune system and are highly biocompatible. A few studies have used grapefruit-derived nanovesicles and lemon juice-derived vesicles. The former were absorbed, exhibited anti-inflammatory efficacy, and mediated cell-nanovesicle communication by upregulating haem oxygenase-1 (HO-1) levels and downregulating IL-1β and TNF-α levels in intestinal macrophages.10 The latter suppressed cancer cell proliferation by inducing TRAIL-mediated apoptosis in various tumor lines in vitro and in vivo.11
Ginseng is also promising for nanovesicle extraction as it contains ginseng polysaccharides (glucose, sucrose, and so on), ginseng polypeptides, volatile oils (sesquiterpenes, panasinsene, and β-elemene), vitamins, alkaloids, lignins, and ginsenosides or ginseng glycosides (Rb1, Rb2, Rc, Rd, Rh2, Rg1, Re, Rf, Rh1, and Ro).12 Ginsenosides are regarded as the main active principles in ginseng13 and have anti-inflammatory14–17 and anti-osteoclastogenic efficacies.18–21 Rh3 is anti-inflammatory by modulating the AMPK pathway and its downstream signaling16 whilst Rg3, Rb1, and Rg1 induce M2 polarization in microphages and microglia.17 Rb1 and Rh2 inhibit osteoclast differentiation by downregulating the NF-κB and/or MAPK signaling pathways.20,21
Ginseng nanovesicles have been recently investigated. They inhibited melanoma growth by controlling polarization in immune cells22 and exerted anti-senescence effects in human skin cells by downregulating melanogenesis-related proteins and ageing-associated molecules.23 To the best of our knowledge, however, no prior research has explored the impact of ginseng-derived nanovesicles (GDNs) on bone metabolism.
In the present study, we investigated the effects of GDNs on osteoclast differentiation. We extracted GDNs from ginseng by the sucrose gradient ultracentrifugation method and performed actin ring and TRAP staining and RT-PCR to confirm the effects of GDNs on the suppression of osteoclast differentiation. We then identified the signaling pathways modulated by GDNs and conducted an in vivo assay to assess their potential as an innovative osteoporosis treatment.
Materials and methods
Isolation and characterization of GDNs
GDNs were isolated from ginseng using a modified centrifugation method and a sucrose gradient cushion system, as previously reported.24 Ginseng was purchased from Geumsan (South Chungcheong, Korea) and sequentially washed with tap water, distilled water, and 1× phosphate-buffered saline (PBS). The material was then ground in a mixer for 2 min and subjected to serial low-velocity centrifugation to eliminate large impurities and debris (ESI Fig. S1†). Sucrose gradient cushions were loaded to preserve spherical GDNs morphology and prevent protein contamination. The sucrose solutions (68%/27%, 60%/45%/30%/8%) were gently loaded to maintain interfaces between layers, and the material was subjected to high-velocity ultracentrifugation. The GDNs were then collected between the 8% and 30% and between the 30% and 45% sucrose concentrations. Thereafter, the protein content of GDNs extracted per weight of ginseng was confirmed. After freeze-drying, approximately 1.875 g of GDNs were extracted per 100 g of ginseng; on dissolving 100 mg of GDNs after freeze-drying in 1 mL PBS, approximately 0.94 mg ml−1 of protein was quantified.GDNs size and zeta potential were analyzed using dynamic light scattering (DLS) with a Zetasizer (Nano-ZS90; Malvern Instruments, Malvern, UK). Ten milligrams of GDNs were dissolved in 1 mL of 1× PBS, and the samples were loaded into disposable cuvettes (46145; SPL Lifer Sciences, Gyeonggi-do, Korea) and disposable capillary cells (DTS1070; Malvern Instruments) for the zeta potential measurements. Each sample was measured in triplicate. To determine GDNs size and concentration, 10 mg of GDNs was dissolved in 1 mL of 1× PBS and gently pipetted. Each sample was diluted to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 with 1× PBS for nanoparticle tracking analysis (NTA). GDNs size and concentration were evaluated using NanoSight (NS300; Malvern Instruments). To confirm GDNs morphology, 10 μL of 10 μg GDNs suspension was deposited onto formvar carbon-coated copper grids (01800-F; Ted Pella Inc., Redding, CA, USA). Each sample was stained with 2% (w/v) phosphotungstic acid hydrate (PTA) at room temperature for 1 min and dried at room temperature overnight. GDNs morphology was then observed via transmission electron microscopy (TEM; JEM 2100F, JEOL Ltd., Tokyo, Japan) at 200 kV.
:500 with 1× PBS for nanoparticle tracking analysis (NTA). GDNs size and concentration were evaluated using NanoSight (NS300; Malvern Instruments). To confirm GDNs morphology, 10 μL of 10 μg GDNs suspension was deposited onto formvar carbon-coated copper grids (01800-F; Ted Pella Inc., Redding, CA, USA). Each sample was stained with 2% (w/v) phosphotungstic acid hydrate (PTA) at room temperature for 1 min and dried at room temperature overnight. GDNs morphology was then observed via transmission electron microscopy (TEM; JEM 2100F, JEOL Ltd., Tokyo, Japan) at 200 kV.
Cell culture for osteoclast differentiation
Osteoclast differentiation was achieved using primary mouse bone marrow-derived macrophage (BMM) cultures. To generate bone marrow-derived osteoclasts, monocytes were isolated from the femurs and tibiae of 6-wk-old ICR mice (Young Bio, Sungnam, Korea). Cells were seeded into 100 mm plates (2 × 106 per well) with 10 ng mL−1 macrophage colony-stimulating factor (M-CSF) for 24 h and cultured in the presence of 30 ng mL−1 M-CSF for 72 h. The cells were then cultured in minimum essential medium alpha (alpha-MEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Thermo Fisher Scientific) and 1% (w/v) antibiotic–antimycotic mixture under a humidified 5% CO2 atmosphere.Cell proliferation assay
BMMs were seeded into 48-well plates (2.5 × 104 per well) along with various GDNs protein concentrations and incubated for 1, 3, and 7 d. The specimens were then washed out, and the cells adhering to the substrates were rinsed with PBS. Cell counting kit (CCK-8) proliferation reagents (Dojindo Laboratories, Kumamoto, Japan) were added to each well plate. After 2 h of incubation at 37 °C, the reagents were then carefully transferred to 96-well plates. OD450 was then determined with a microplate reader (Thermo Scientific Multiskan GO, Thermo Fisher Scientific).Live/dead assay
Cell viability in the presence of GDNs was assessed by live/dead staining. BMMs were seeded into 48-well plates (5.0 × 104 per well) along with GDNs and tissue culture plate. After 24 and 48 h of incubation, the cells were rinsed with PBS and incubated with fluorescein diacetate (FDA)-propidium iodide (PI) live/dead staining at room temperature for 30 min. Live (viable) cells (green) and dead cells (red) were enumerated using fluorescence microscope (EVOS; Invitrogen, Carlsbad, CA, USA).Tartrate-resistant acid phosphatase (TRAP) staining
BMMs were seeded into 48-well plates (2.5 × 104 per well) with 100 ng mL−1 RANKL (R&D Systems, Minneapolis, MN, USA) and 30 ng mL−1 M-CSF (PeproTech, Cranbury, NJ, USA) along with GDNs or Rb1 and Rg1 (Sigma-Aldrich Corp., St. Louis, MO, USA). After RANKL and M-CSF addition, the TRAP-positive multinuclear cells were observed for 4 d. The cells were fixed by soaking in them in 3.7% (v/v) formaldehyde for 20 min, placed in 0.1% (v/v) Triton X-100 for 10 min, washed, and incubated in the dark at 37 °C for 30 min using the mixture of solutions included in a TRACP assay kit (TaKaRa Bio Inc., Shiga, Japan) following the manufacturer's instructions. TRAP-positive cells containing ≥3 nuclei were considered osteoclasts.Actin ring formation assay
Osteoclast actin rings were detected by staining the actin filaments with rhodamine phalloidin. BMMs were seeded into 48-well plates (2.5 × 104 per well) in the presence of 100 ng mL−1 RANKL and 30 ng mL−1 M-CSF and cultured 4 d for the actin ring formation assay. The cells were fixed in 3.7% (v/v) formaldehyde for 20 min and placed in 0.1% (v/v) Triton X-100 for 10 min. The fixed cells were then stained with rhodamine-conjugated phalloidin for 30 min to highlight the actin filaments and then with DAPI (4′,6-diamidino-2-phenylindole) to highlight the nuclei (Thermo Fisher Scientific). The osteoclast actin rings were stained with rhodamine-conjugated phalloidin. The actin rings were visualized and detected under an inverted fluorescence microscope (EVOS; Invitrogen).Reverse transcription-polymerase chain reaction (RT-qPCR)
To confirm the expression levels of the genes encoding osteoclast differentiation, RT-qPCR was performed at 4 d. Total RNA was purified with TRIzol® reagent (Invitrogen), and cDNA was synthesized from 1 μg of total RNA using an AccuPower® CycleScript RT PreMix (Bioneer, Daejeon, Korea). RbTaq™ qPCR PreMIX-SYBR Green (Enzynomics, Daejeon, Korea) was used to amplify the selected genes according to the manufacturer's protocol. Oligonucleotide primers were used to measure TRAP, OSCAR, NFATc1, and c-Fos expression (Table 1). PCR amplification was performed as follows: 10 s at 95 °C, 15 s at 60 °C, and 15 s at 72 °C for 40 cycles after initial denaturation for 3 min at 95 °C. Gene expression was quantified using the 2−ΔΔCt method. The expression levels of the targeted genes were normalized against the geometric average of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene. All results were confirmed by repeating the experiment in triplicate. Fold change (FC) deviation from the control was set at 1× and the ratio of the normalized FC was calculated.| Gene | Forward/sense 5′-3′ | Reverse/antisense 5′-3′ |
|---|---|---|
| TRAP | CTG GAG TGC ACG ATG CCA GCG ACA | TCC GTG CTC GGC GAT GGA CCA GA |
| OSCAR | CTG CTG GTA ACG GAT CAG CTC CCC AGA | CCA AGG AGC CAG AAC CTT CGA AAC T |
| NFATc1 | CTC GAA AGA CAG CAC TGG AGC AT | CGG CTG CCT TCC GTC TCA TAG |
| c-Fos | CTG GTG CAG CCC ACT CTG GTC | CTT TCA GCA GAT TGG CAA TCT C |
| GAPDH | ACT TTG TCA AGC TCA TTT CC | TGC AGC GAA CTT TAT TGA TG |
Western blotting
Cells were lysed with cold radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific) and protease and phosphatase inhibitors. The lysates were then incubated on ice for 30 min and centrifuged at 13000 rpm for 10 min. Thirty micrograms protein was subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane. EveryBlot blocking buffer (Bio-Rad Laboratories, Hercules, CA, USA) was added to the membrane for 10 min. The membrane was then probed with anti-phospho c-JUN N-terminal kinase (JNK), phospho extracellular signal-regulated kinase (ERK), phospho p38 MAP kinase (P38), phospho protein kinase B (AKT), and phospho IκBα and incubated with an appropriate secondary antibody conjugated to horseradish peroxidase (HRP). The membrane was stripped and reprobed with anti-JNK, anti-ERK, anti-p38, anti-AKT, anti-IκBα, and anti-β-actin. Signals were detected in a ChemiDoc XRS system (Bio-Rad Laboratories).
LC-MS analysis of ginsenosides in GDNs
The ginsenoside components of the GDNs were identified and quantitated through liquid chromatography-mass spectrometry (LC-MS). To isolate the ginsenosides, 1 mL GDNs and 500 μL of the upper clear phase in the liquid were mixed with 1 mL distilled water and butanol (2:1 (v/v)). The upper phase was transferred to a fresh tube and centrifuged at 5000g for 10 min. The solution was then evaporated in a rotary vacuum evaporator water bath at 80 °C for 10 min to remove the butanol. The sample was then dissolved with 100 μL methanol.
For LC-MS, the components were eluted through an Acquity UPLC BEH C18 column (1.7 μm; 2.1 × 100 mm; Waters Corporation, Milford, MA, USA) at 0.4 mL min−1 flow rate and separated with a mobile phase gradient. Mobile phase A was 0.1 mM ammonium acetate in distilled water, and mobile phase B was acetonitrile. The run time was 25 min, and the gradient was changed in mobile phase B to 20% at 0 min, 30% at 3 min, 33% at 5 min, 35% at 6 min, 42% at 8 min, 47% at 16 min, 52% at 17 min, 80% at 19 min, 95% at 20 min, 20% at 21 min, and 20% at 24 min and ended at 25 min. The ginsenosides were analyzed in the LCMS-8060 instrument (SHIMADZU Corp., Kyoto, Japan).
Lipopolysaccharide (LPS)-induced calvarial bone loss animal model
Healthy male 11-wk-old ICR mice weighing 39–42 g were purchased from Young Bio (Sungnam, Korea). They were divided into four experimental groups of five mice per group. These included the PBS treatment (control), the LPS treatment (8 mg kg−1), the GDNs (1 mg kg−1) with LPS treatment, and the GDNs (5 mg kg−1) with LPS treatment. The mice were subcutaneously injected with PBS, LPS, or GDNs in the tissue over the periosteum of the calvarial surface on days 1 and 7. They were then anaesthetized 7 d after the first injection. The calvarial bones were washed with PBS and fixed with 4% (v/v) paraformaldehyde (PFA). The calvarial bones were then examined by high-resolution microcomputed tomography (micro-CT; 1173_Skyscan v. 1.0; Skyscan, Kontich, Belgium) to obtain 3D images (Dataviewer_Skyscan v. 1.5.2.4; Skyscan). The relative percentage of bone resorption was quantitated by micro-CT imaging. The bone tissues were demineralized in 15% (w/v) ethylenediaminetetraacetic acid (EDTA) for 14 d, embedded in paraffin, and sectioned at 4 μm thickness. For the histological analyses, the sections were stained with hematoxylin and eosin (H&E) and TRAP. This study was conducted with the approval of the Animal Experimental Ethics Committee of Kyung Hee University (approval number: KHSASP-22-115) and performed under the Institutional Animal Care and Use Committee (IACUC) guidelines.Microcomputed tomography (micro-CT)
Micro-CT was performed at the Advanced Institutes of Convergence Technology (Genoss Co. Ltd, Gyeonggi-do, Korea). The amount of bone in the calvaria was determined by micro-CT analysis (Skyscan, 1173 micro-tomography system; Skyscan) at 100 kV and 80 μA. A total of 658 projections was collected at a resolution of 9.94 μm per pixel. Scanned images were reconstructed using the manufacturer's software (Bruker Belgium nv, Kontich, Belgium). The volume of interest (VOI) was set in the calvarial defect region and the ratio of the volume of regenerated bone to the VOI was calculated using CTAn software (Brucker Belgium nv).Histological analyses (hematoxylin and eosin [H&E] and TRAP staining)
Calvarial bones were isolated, fixed with 4% (v/v) PFA, and decalcified with 4% (w/v) EDTA. The extracted bones were washed, dehydrated with ethanol, embedded in paraffin, and cut to 4 μm thickness. The sectioned specimens were then stained with H&E and the bone was visualized. Other sections were stained with TRAP to visualize the osteoclasts. Microscopic images of the stained samples were acquired using a digital slide scanner (Panoramic 250 Flash III, 3DHISTECH Ltd., Budapest, Hungary).Statistical analysis
Each experiment was performed at least thrice. All quantitative data are expressed as the mean ± standard deviation. Statistical differences were identified using two-way ANOVA. *P < 0.05, **P < 0.01, and ***P < 0.001 indicated statistical significance.Results and discussion
Isolation and characterization of GDNs
Exosome-like nanovesicles are isolated from plants by the gold standard method of ultracentrifugation and gradient sucrose cushion.25,26 Here, the GDNs were sequentially extracted from ginseng by ultracentrifugation using various relative centrifugal forces (RCF; 2000–200000) and sucrose concentrations (8–68% w/v) (ESI Fig. S1†). The morphology, size, size dispersion, and zeta potential of the GDNs were measured by TEM, DLS, and NTA analysis to characterize them as particulate materials.
GDN morphology was observed by field effect (FE)-TEM and determined to be spherical with a lipid bilayer membrane (Fig. 1A). This structure is characteristic of mammalian exosomes. Hence, the GDNs were successfully extracted as exosome-like nanoparticles. Moreover, spherical nanomaterials with lipid layers can be transported intracellularly via the cell membrane.27 The GDNs had an average diameter of 71.42 nm (Fig. 1B) according to TEM and DLS analysis and their polydispersity indices (PDI) were low. The average zeta potential for the GDNs was −18.8 mV (Fig. 1C). The GDNs had uniform size dispersion (4.18 × 1011 particles/0.1 g lyophilized GDNs) (Fig. 1D). The foregoing results suggest that the GDNs were successfully extracted in the form of nanovesicles from the ginseng.
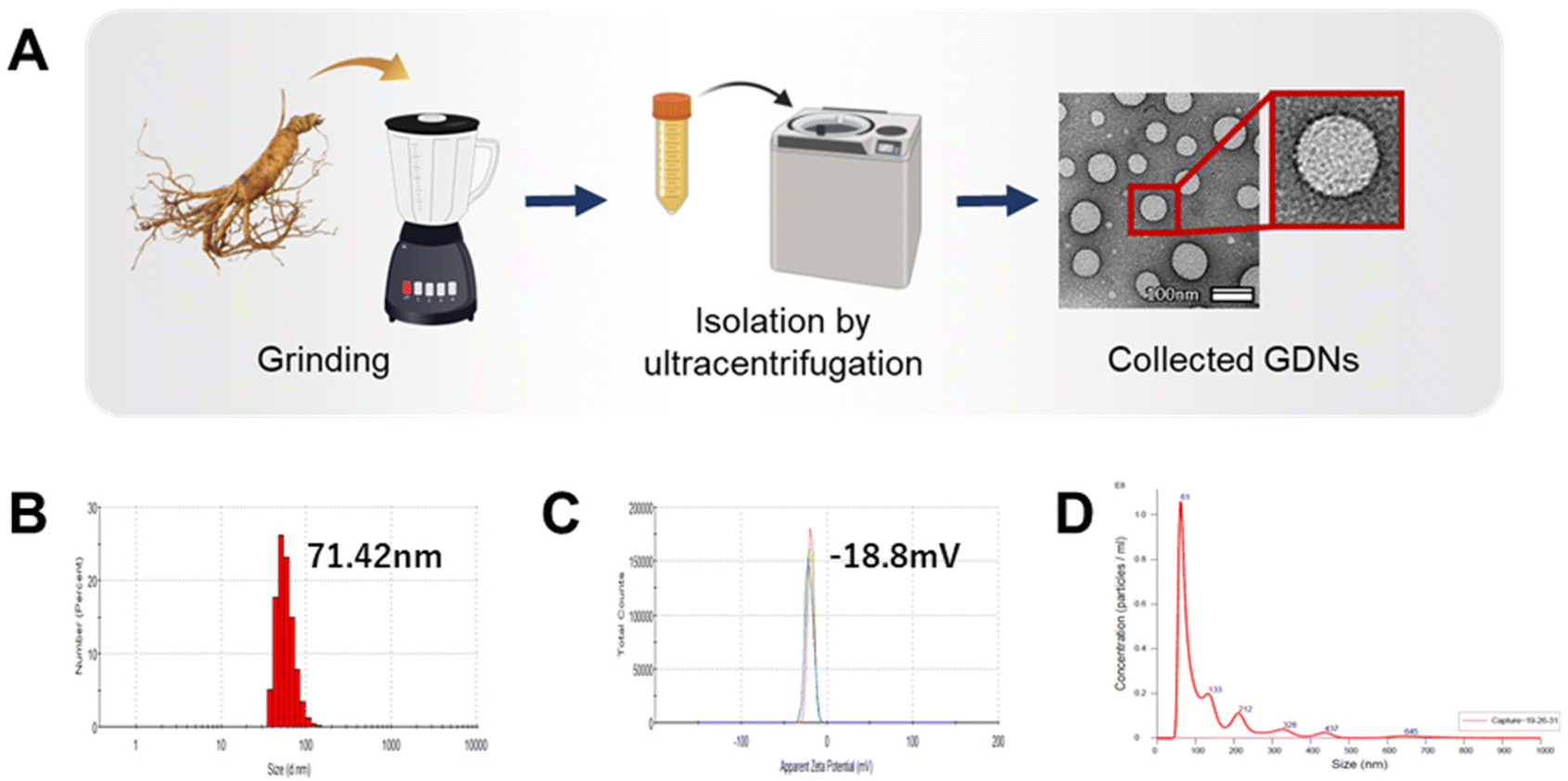 | ||
| Fig. 1 Characterisation of ginseng-derived nanovesicles. (A) Schematic showing extraction of GDNs from ginseng. (B and C) Size distribution and zeta potential of GDNs determined by DLS and (D) NTA analysis. | ||
Viability and proliferation of GDNs
Prior to determining the effects of the GDNs on osteoclast differentiation, we first investigated whether they had an impact on BMM viability. We subjected BMMs to α-MEM containing 30 ng mL−1 M-CSF and 0, 3, 5, 7, 10, 30, 50, or 100 μg mL−1 GDNs for 1, 3, or 7 d (Fig. 2A). The BMM proliferation rates had significantly increased with GDNs concentration by day 7. At 3, 5, 7, and 10 μg mL−1 GDNs, BMMs proliferation increased by 876%, 935%, 957%, and 1,074%, respectively, whereas the BMMs proliferation for the control had increased by only 663% at the same time compared to that for day 1 of the control.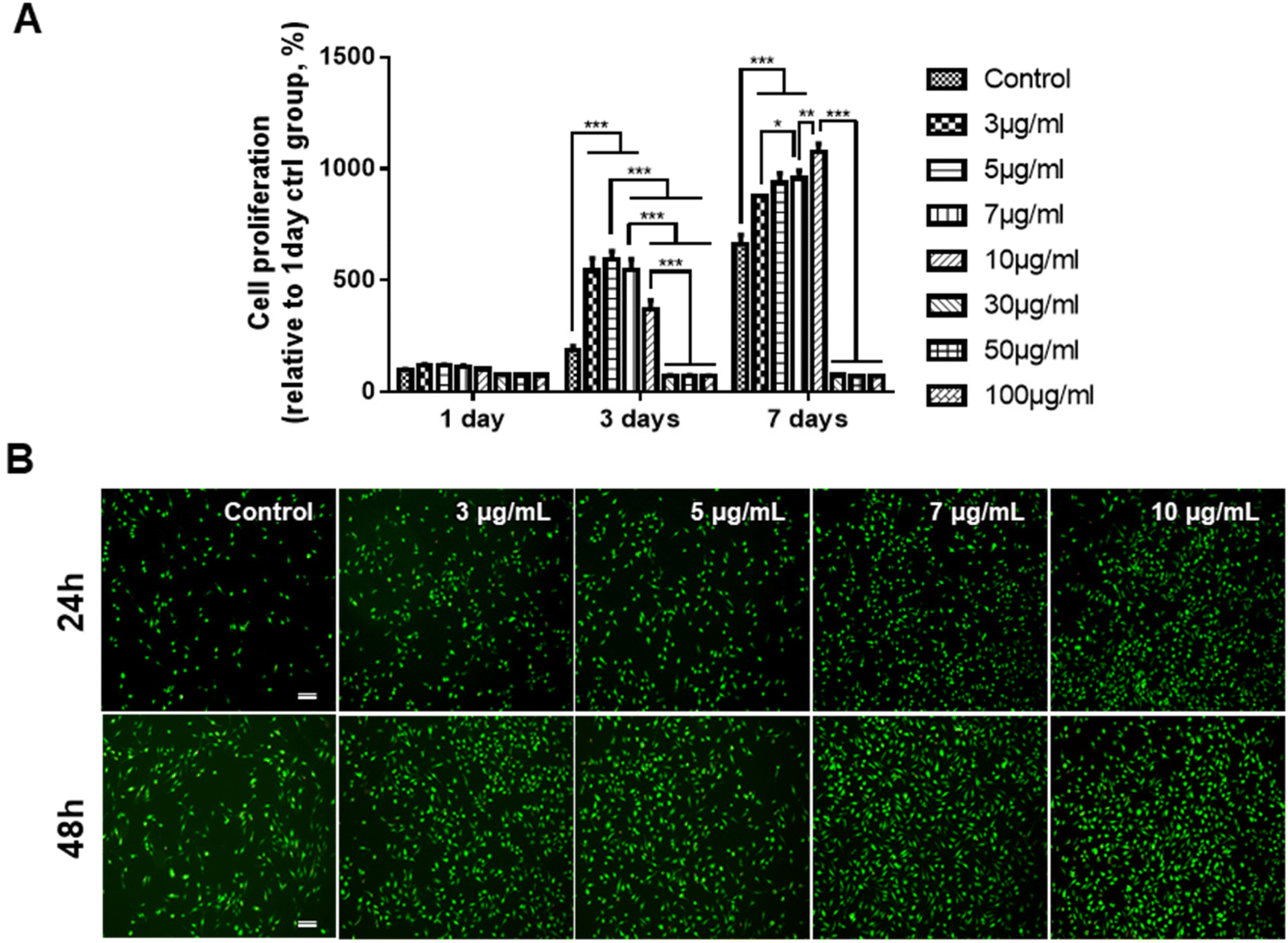 | ||
| Fig. 2 Proliferation assay of BMMs subjected to ginseng-derived nanovesicles. (A) CCK-8 assay (*P < 0.05, **P < 0.01, and ***P < 0.001 significantly different from control). (B) Live/Dead assay (scale bar: 200 μm). | ||
In contrast, the 30, 50, and 100 μg mL−1 GDN treatments presented with slow BMM growth rates (<80%) relative to the control at all time points. Hence, 30, 50, and 100 μg mL−1 GDNs had poor proliferative efficacy on BMMs whereas 3, 5, 7, and 10 μg mL−1 GDNs strongly promoted BMMs on day 7 and without requiring any supplementary M-CSF after the initial M-CSF treatment. In fact, the first M-CSF dosage sufficed to ensure macrophage survival and proliferation for ≤3 d. Thus, the GDNs induced rapid initial BMM growth and promoted BMM proliferation to a greater extent than the control.
Non-biocompatible materials generally cause cytotoxicity within ∼48 h. We conducted Live/Dead assays after 24 h and 48 h to assess the initial cytotoxicity of 3, 5, 7, and 10 μg mL−1 GDNs. The GDN-treated groups presented with more live cells than the control after 24 h and 48 h. Dead cells were not detected in any treatment group (Fig. 2B). These results were consistent with those of the proliferation test and underscored that 3, 5, 7, and 10 μg mL−1 GDNs were not cytotoxic. Rapid initial BMM growth in the presence of <10 μg mL−1 GDNs indicated that this dosage induced faster proliferation than the control. Other ginseng-derived materials also promoted cell proliferation possibly by regulating the ERK and AKT/mTOR pathways in HaCaT, HUVEC, and BJ cells.28 Niu et al. found that Rg1 and Rb1 from Panax ginseng affected colony-forming unit-granulocyte-macrophage (CFU-GM) proliferation and increased the colony formation rates in human marrow granulocyte-macrophage progenitor cells.29
In vitro characterization of the inhibitory effects of GDNs against RANKL-induced osteoclast differentiation
We investigated RANKL-induced osteoclast differentiation in the presence of M-CSF with GDNs and conducted TRAP and F-actin staining to determine the effects of GDNs on osteoclast differentiation. We exposed BMMs to various GDNs concentrations in the presence of RANKL (100 ng mL−1) and M-CSF (30 ng mL−1), and they differentiated into osteoclasts within 3–4 d. TRAP staining was conducted when a control group presented with abundant multinucleated cells. First, we conducted TRAP+ staining with experimental groups up to 10 μg mL−1, exhibiting excellent BMM proliferation and inhibiting osteoclast differentiation in all groups; no difference between groups could be confirmed (ESI Fig. S2†). Therefore, the concentration in experimental groups was adjusted to 100 ng mL−1, 500 ng mL−1, and 1 μg mL−1. As a results, the control group demonstrated large, TRAP-positive multinucleated cells with characteristic morphology. By contrast, the single cells were mostly round and lacked any thin or sharp branches. Conversely, the number and size of the TRAP-positive cells significantly decreased with increasing GDN concentration (Fig. 3A). The 100 and 500 ng mL−1 GDN groups exhibited fewer TRAP-positive cells than the control. GDN concentrations of 1 μg mL−1 suppressed BMM differentiation into osteoclasts compared to the other treatment groups (Fig. 3A). BMMs treated with 0, 100, 500, and 1 μg mL−1 GDNs had 95, 40, 35, and 18 TRAP+ cells, respectively (Fig. 3B). As there was no difference in the inhibitory effect on 1–10 μg mL−1 concentration, all subsequent experiments were conducted based on the 1 μg mL−1 concentration.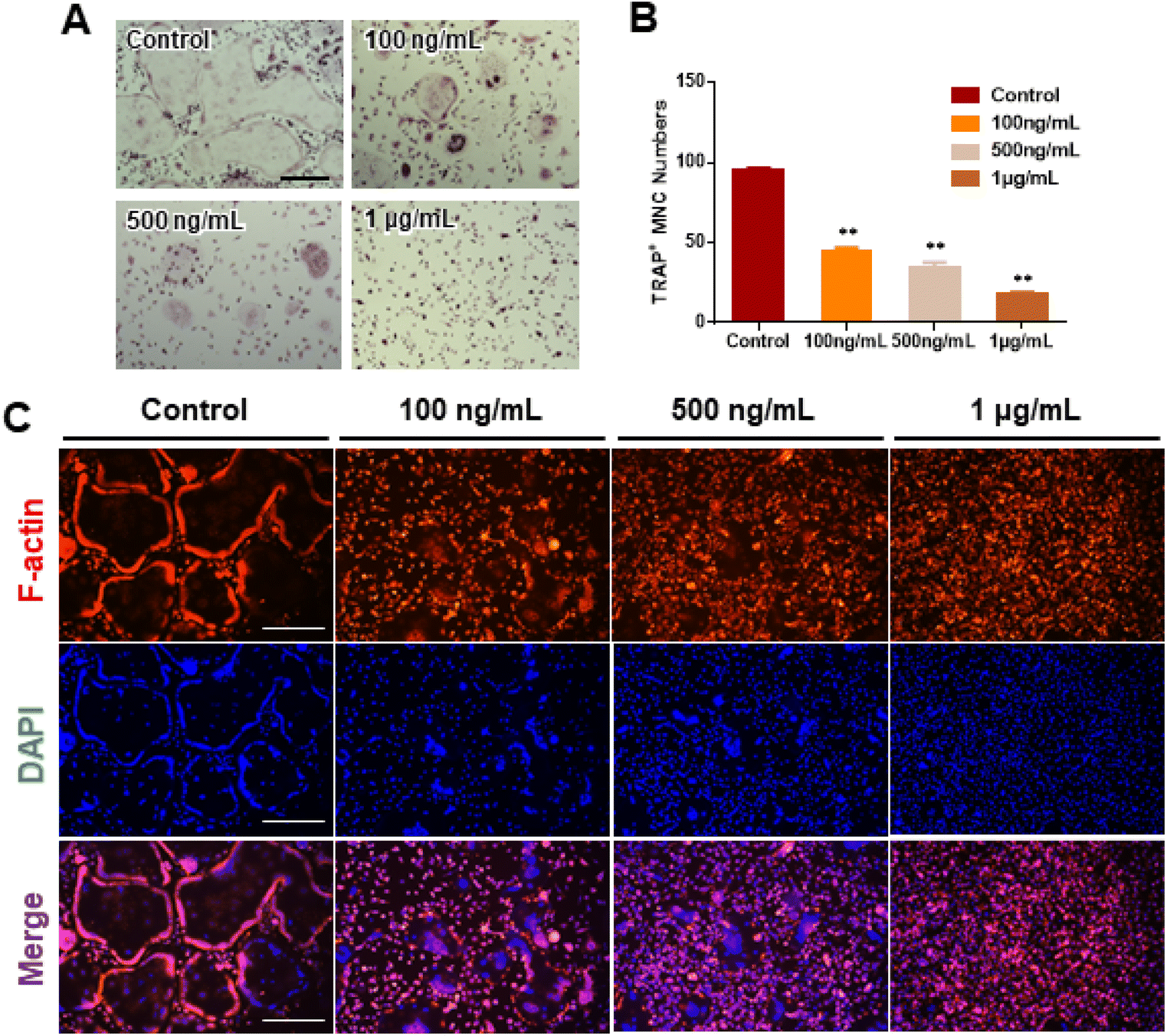 | ||
| Fig. 3 Inhibitory effect of osteoclast differentiation by ginseng-derived nanovesicles. (A) Assessment of multinucleated TRAP+ cell formation by TRAP staining (scale bar = 300 μm). (B) TRAP+ multinucleated cell counts (**P < 0.01, *P < 0.05 significantly different from control). (C) F-actin staining to confirm inhibitory effect of GDNs on osteoclast differentiation in BMMs (scale bar: 300 μm). | ||
The actin of osteoclasts differs from those of other normal cells. The actin ring or sealing zone is a resorption area wherein osteoclasts enter into contact with bone tissue, absorb the bone matrix, isolate the microenvironments between resorption areas, and surround the extracellular spaces on the skeletal sites.30 After osteoclast differentiation was induced in the BMMs through RANKL, M-CSF, and various GDN concentrations, the control presented with clear, thick actin rings and the interior parts of the latter had low F-actin and nuclear densities (Fig. 3C). By contrast, 100 and 500 ng mL−1 GDNs induced the formation of unclear actin rings in a dose-dependent manner. This finding was consistent with the results of the TRAP staining in that the multinucleated cell boundary was not obvious. However, we detected the convergence of small numbers of what appeared to be premature multinucleated cells. For the 1 μg mL−1 GDN group, the BMMs occurred as single cells, were evenly distributed, and did not coalesce. Virtually no actin ring formation was observed in the 1 μg mL−1 GDN group. Because the actin ring is characteristic of activated osteoclasts, GDNs might suppress differentiation into the bone resorption stage. Hence, actin ring staining demonstrated that GDNs effectively suppress osteoclast differentiation by inhibiting actin ring formation.
Signaling pathways associated with the suppression of RANKL-induced osteoclast differentiation by GDNs
We evaluated the mRNA expression levels of the genes encoding tartrate-resistant acid phosphatase (TRAP), osteoclast-associated receptor (OSCAR), nuclear factor of activated T cells c1 (NFATc1), and c-Fos to confirm the inhibitory effect of GDNs on osteoclast differentiation. TRAP and OSCAR are characteristic of mature osteoclast cells and representative factors that identify osteoclast differentiation. NFATc1 and c-Fos are generalized transcription factors (TFs). The GDNs treatments downregulated the foregoing genes in a dose-dependent manner compared to the control (Fig. 4A). For the 1 μg mL−1 GDN group, TRAP, OSCAR, NFATc1, and c-Fos were significantly downregulated by 0.14, 0.12, 0.03, and 0.13, respectively, relative to the control. The signaling pathways related to osteoclast differentiation were identified through western blotting to elucidate the molecular mechanisms by which GDNs inhibit osteoclast differentiation.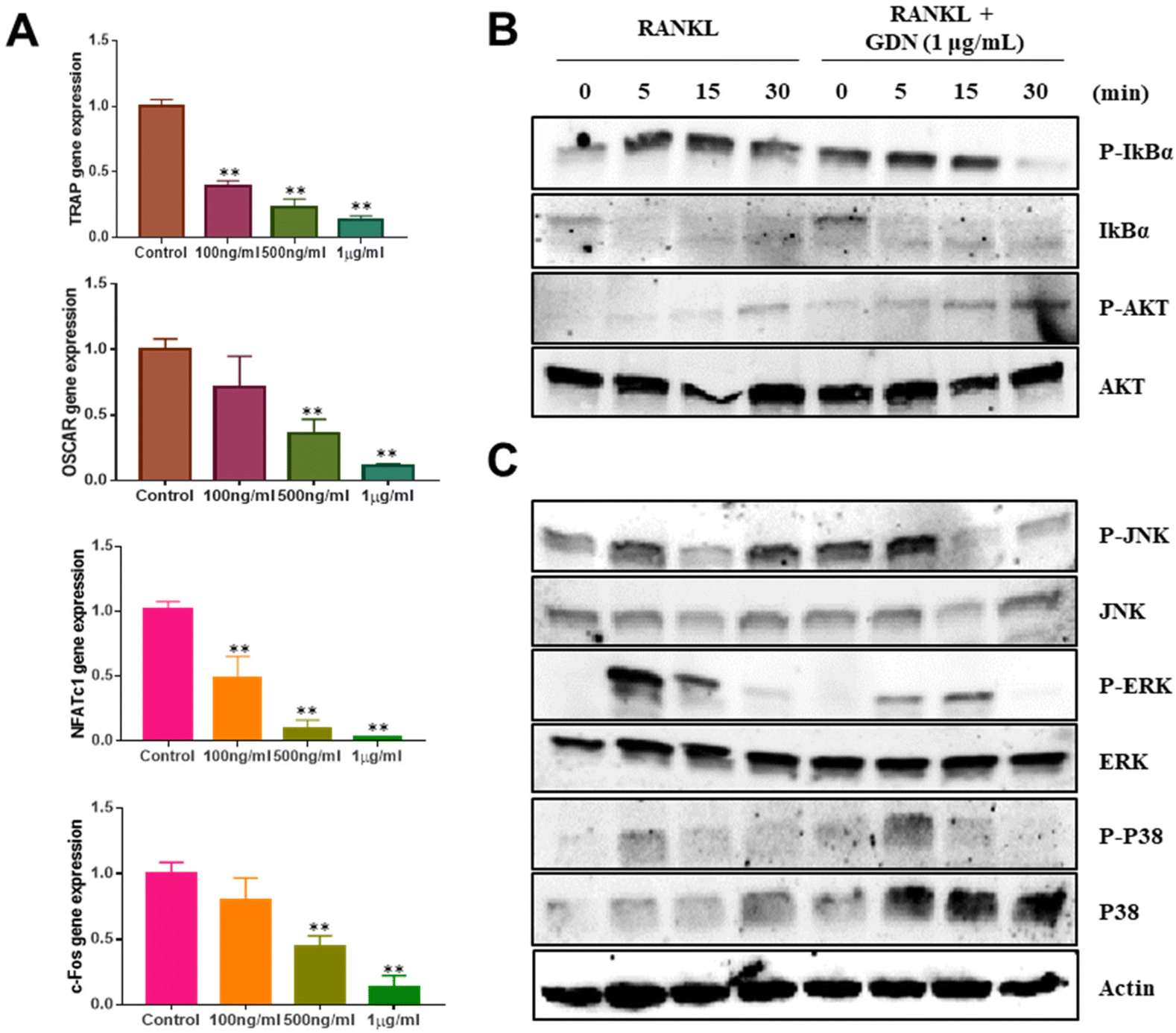 | ||
| Fig. 4 Effects of ginseng-derived nanovesicles on mRNA expression and osteoclast differentiation signaling pathways in bone-marrow macrophage cells. (A) TRAP, OSCAR, NFATc1, and c-FOS genes (**P < 0.01, *P < 0.05 significantly different from control). (B) IκBα and AKT signal proteins. (C) MAPK signal proteins (JNK, ERK, and p38). | ||
The IκBα, AKT, JNK, ERK, and p38 protein expression levels were assessed by western blotting to identify the signaling pathways that are inhibited by 1 μg mL−1 GDNs after BMM exposure to RANKL and M-CSF with or without GDNs. The p38 signaling pathway was not significantly suppressed. However, the GDNs substantially inhibited RANKL-induced IκBα, JNK, and ERK activation and, by extension, osteoclast formation (Fig. 4B and C). On the other hand, the GDNs slightly increased AKT activation (Fig. 4B). Thus, GDNs could increase BMM proliferation (Fig. 2A).
Ginsenoside composition of GDNs and comparison of single ginsenoside vs. GDNs
We compared the inhibitory effects of GDNs on osteoclast differentiation against those of single/mixed ginsenosides by TRAP staining to identify the osteoclastogenesis-suppressing capacity of GDNs as complex ginsenoside mixtures. Before comparing single/mixed ginsenosides against GDNs, we used LC-MS to measure the ratios of the internal components of GDNs with ginsenosides Rb1, Rg1, Rg3, Rg5, and R1, which are reputed to affect bone metabolism.18 Rb1 comprised 64.5% of GDNs, followed by Rg1 (31.1%), Rg3 (2.3%), R1 (2.0%), and Rg5 (0.1%) (Fig. 5A). It was previously empirically demonstrated that Rb1 and Rg1 suppress osteoclast differentiation. Rb1 inhibits RANKL-induced osteoclast differentiation by suppressing NF-κB upstream of numerous crucial TFs including c-Fos and NFATc1. Rb1 also inhibits the phosphorylation of mitogen-activated protein kinases (MAPKs) such as JNK and p38 but not ERK.20 Rg1 regulates the mRNA receptors of calcitonin and estrogen.31 Based on the preceding results, Rb1 and Rg1 were selected for use in subsequent comparison against GDNs and were assumed to be their major components.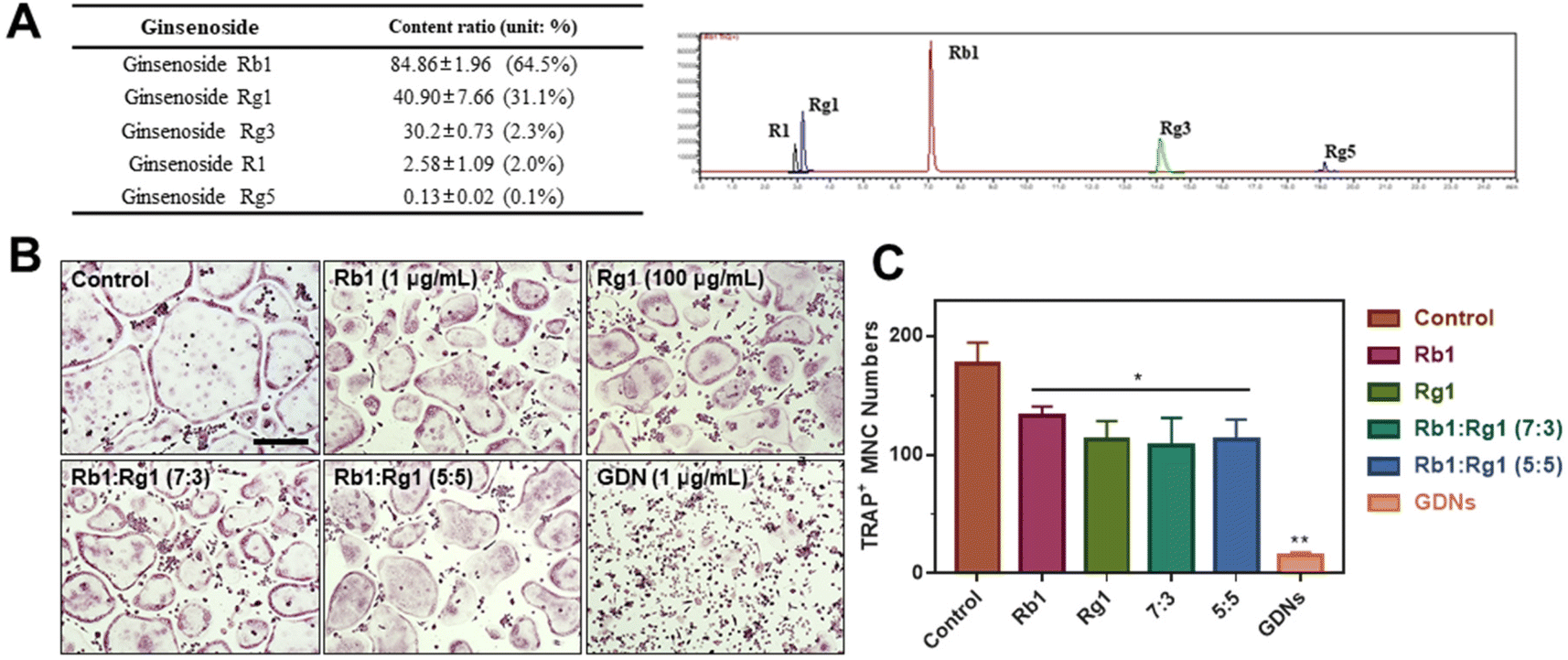 | ||
| Fig. 5 Ginsenoside compositions and their effects on inhibition of osteoclast differentiation compared with ginseng-derived nanovesicles. (A) LC-MS of GDNs. (B) TRAP staining. Scale bar: 300 μm (C) TRAP+ multinucleated cell counts (**P < 0.01, *P < 0.05 significantly different from control). | ||
The experimental groups included (1) control, (2) Rb1, (3) Rg1, and their mixtures at (4) 7:3 and (5) 5:5 ratios and (6) GDNs. The ratio of 7:3 was established based upon the LC-MS results. Because it is difficult to assume that the extracted GDNs always maintain a ratio of 7:3 on Rb1 and Rg1, another concentration groups with a 5:5 ratio was added. The ratios were established based upon the LC-MS results. To select the Rb1 and Rg1 concentrations, we compared Rb1 and Rg1 against GDNs via TRAP staining according to prior reports on the suppressive effects of Rb1 (100, 500, and 1 μg mL−1) and Rg1 (1, 10, and 100 μg mL−1).20,31 The highest concentrations of Rb1 and Rg1 (1 μg mL−1 and 100 μg mL−1, respectively) were suitable for comparison against GDNs (ESI Fig. S3†). The GDNs concentration used was set to 1 μg mL−1 as it previously demonstrated the strongest efficacy at inhibiting osteoclast differentiation. The TRAP staining assays on the GDNs showed that they suppressed osteoclastogenesis more effectively than Rb1, Rg1, or Rb1–Rg1 mixtures (Fig. 5B and C). Few osteoclasts were detected in the GDNs group. Therefore, GDNs composed of different ginsenosides have superior efficacy at inhibiting osteoclastogenesis than either single or mixed ginsenosides. One possibly explanation is that the spherical morphology of the GDNs is conducive to their being surrounded by lipid membranes. Anti-inflammatory drugs encapsulated in grape-derived exosome-like nanoparticles were less toxic to intestinal stem cells than the free drugs.32 Here, GDNs had significantly higher efficacy at suppressing osteoclast differentiation than either single or mixed ginsenosides.
In vivo characterization of bone destruction inhibition by GDNs in LPS-induced bone loss animal model
Lipopolysaccharides (LPS) are detected by TLR 4, activate NF-κB, and induce a proinflammatory cytokine signaling cascade. LPS is a potent monocyte and macrophage activator.33,34 The LPS-induced bone loss model has been used to evaluate osteoclast inhibition efficacy.35–37 Here, the LPS-induced mouse calvaria bone loss model was used to characterize the efficacy of GDNs at inhibiting osteoclast differentiation in vivo. Mice were injected with LPS alone, LPS plus GDNs L (1 mg kg−1 GDNs), or GDNs H (5 mg kg−1 GDNs). Over 7 d, the mice were injected with same solution every 3 d. Six days after the first injection, the mice were anaesthetized and examined by micro-CT, H&E, and TRAP staining (Fig. 6A).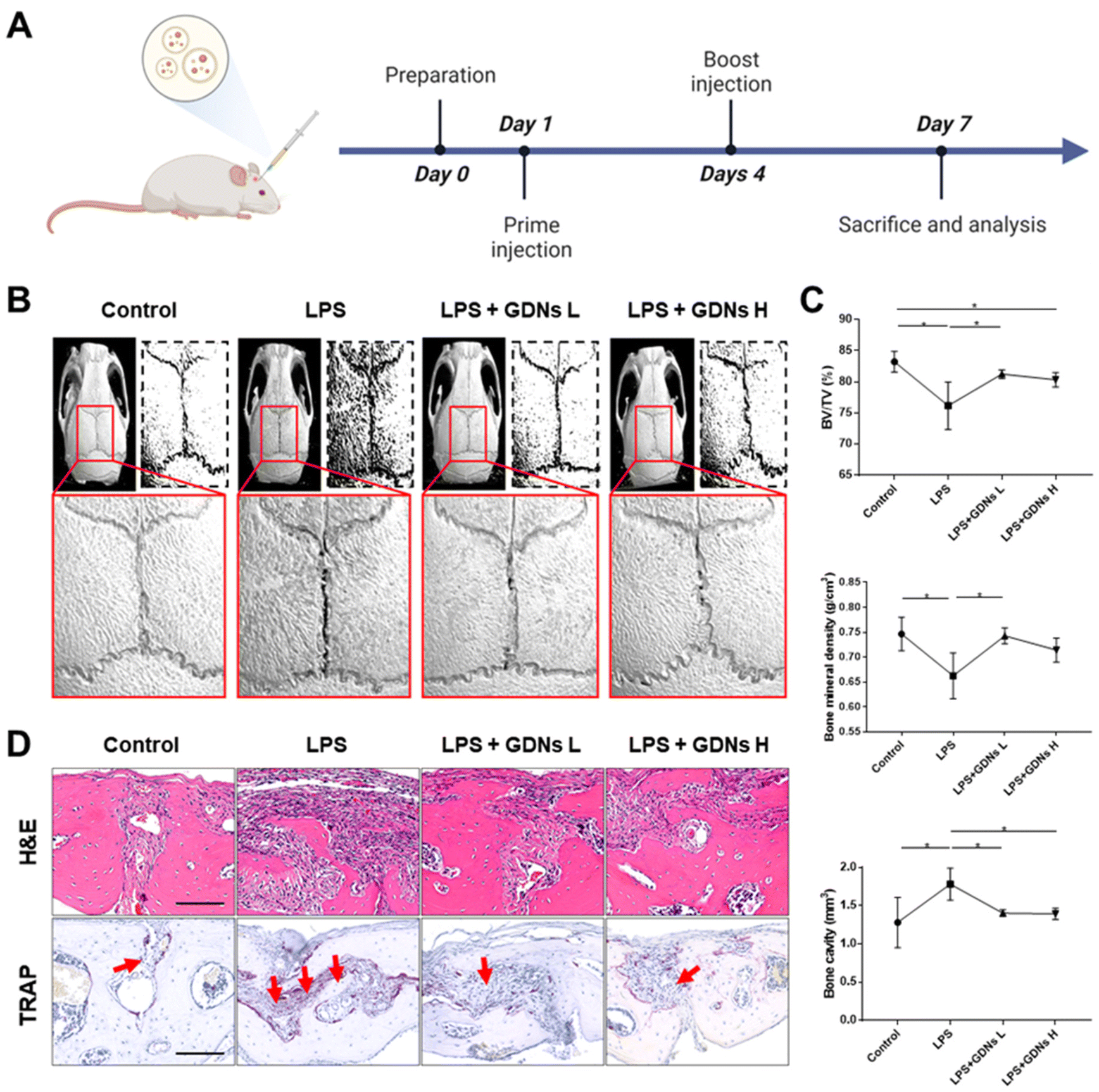 | ||
| Fig. 6 Inhibitory effects of GDNs on LPS-induced inflammation on in mouse calvaria. (A) Schematic timeline of the animal experiment for in vivo analysis. (B) Micro-CT images of mouse calvaria injected with LPS only/alone or with GDNs. (C) Bone volume (BV/TV), bone mineral density (BMD), and bone cavity values (*P < 0.05). (D) H&E and TRAP staining of calvaria cross section of calvaria. Red arrows indicate stained osteoclast cells (scale bar: 100 μm). GDNs L and GDNs H indicate 1 and 5 mg kg−1 of GDN-treated groups, respectively. | ||
In the LPS group, inflammation created a rough pore structure morphology on the calvarial bone surfaces and the suture lines. By contrast, the LPS + GDNs L and the LPS + GDNs H groups presented with significantly suppressed bone loss (Fig. 6B). We also accurately measured bone loss by measuring BV/TV (%), bone mineral density (BMD, g cm−3), and bone cavity (mm3). The results of these parameters statistically differed between the LPS and control groups. The former treatment induced an inflammatory reaction on the calvarial bone surface. However, the LPS + GDNs groups exhibited similar BV/TV, BMD, and bone cavity rates compared with the control (Fig. 6C). The preceding findings suggest that GDNs suppressed LPS-induced inflammation and bone loss by inhibiting osteoclast differentiation.
All treatment groups were subjected to H&E and TRAP staining to determine the histological mechanism by which GDNs inhibit osteoclastogenesis at the calvarial bone site induced by LPS. Abundant inflammatory sites and osteoclasts were observed in the LPS group whereas the LPS + GDNs groups showed relatively less inflammation and fewer osteoclasts (Fig. 6D). These discoveries indicated that GDN administration to bone tissue subjected to LPS can suppress LPS-induced inflammation and, by extension, bone loss. Overall, the results of this work revealed that GDNs effectively blocked bone loss both in vitro and in vivo by inhibiting osteoclast differentiation.
Conclusion
In the present study, GDNs were extracted from ginseng in the form exosome-like nanovesicles via sucrose gradient ultracentrifugation. They effectively inhibited BMM differentiation into osteoclasts and to a significantly greater extent than single or mixed Rb1 and Rg1 ginsenosides. They downregulated the IκBα, JNK, and ERK signaling pathways as well as the c-Fos, c-Jun, and NFATc1 genes. In this manner, the GDNs suppressed TRAP and OSCAR protein synthesis and inhibited RANKL-induced osteoclast differentiation in BMMs (Fig. 7). Future research should clarify the molecular mechanisms of GDNs and their osteogenic effects in the bone life cycle. As nano-sized carriers, GDNs could be injected along with scaffolds and/or hydrogels for tissue engineering applications. The findings of this work suggest that GDNs are promising as nano-sized prophylactic and/or therapeutic agents against osteoporosis.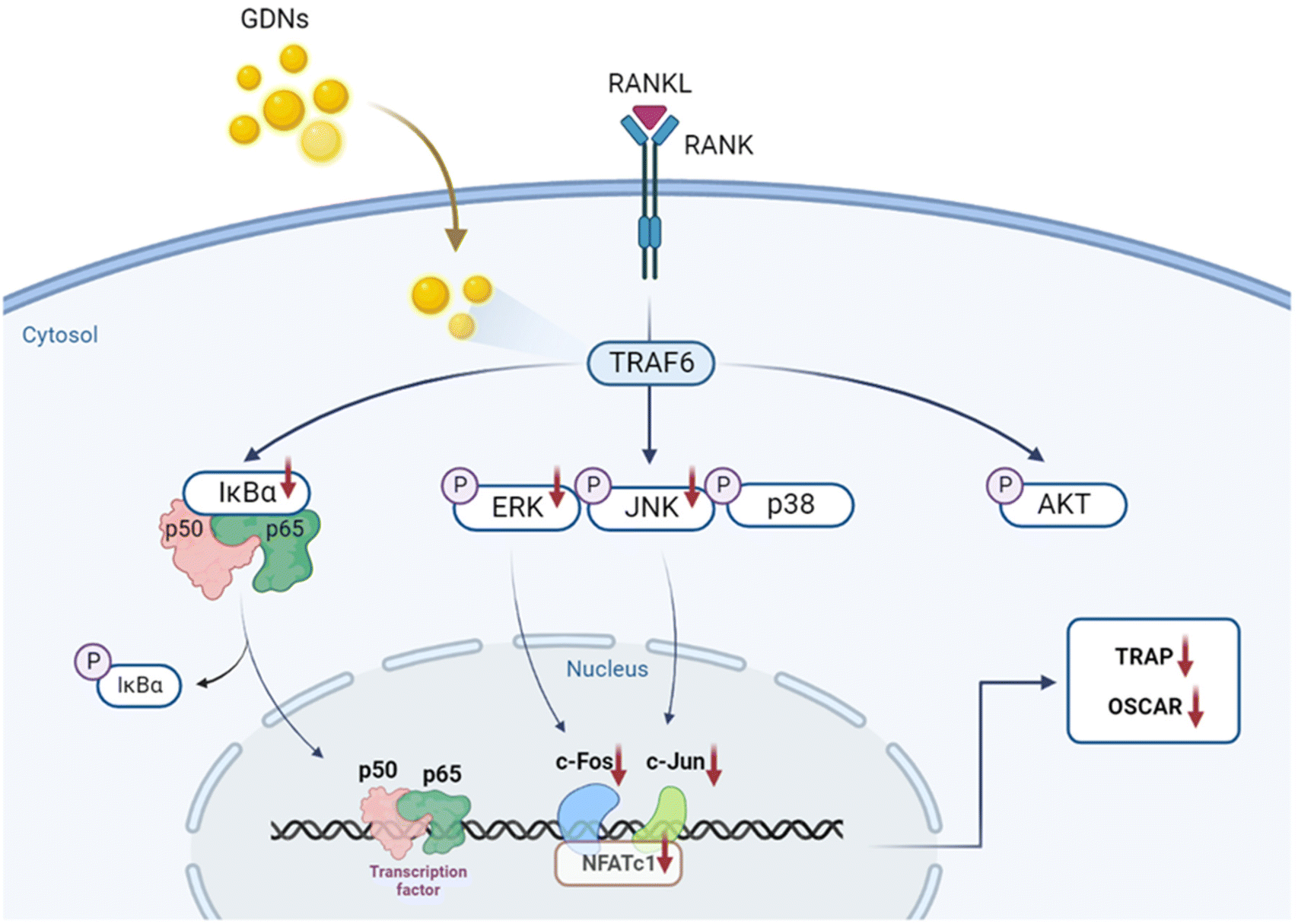 | ||
| Fig. 7 Schematic diagram of pathways by which GDNs suppress osteoclastogenesis in BMMs. | ||
Author contributions
K. Seo: methodology, data curation, formal analysis. writing – original draft. J. H. Yoo: methodology, investigation, validation, data curation, formal analysis, writing – original draft, visualization. J. Kim: methodology, investigation, validation. S. J. Min: methodology, validation, formal analysis. D. N. Heo: methodology, validation, visualization. I. K. Kwon: project supervision, funding acquisition, writing–review & editing. H. J. Moon: conceptualization, data curation, formal analysis, writing – review & editing, visualization, project supervision. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants from the Korean government (MSIT) (NRF-2020R1A4A1019456 and NRF-2020R1A2C2011937) and from the Ministry of Education (2021R1I1A1A01050431).References
- R. Zhang, Z. G. Liu, C. Li, S. J. Hu, L. Liu, J. P. Wang and Q. B. Mei, Bone, 2009, 45, 553–559 CrossRef CAS PubMed.
- W. J. Boyle, W. S. Simonet and D. L. Lacey, Nature, 2003, 423, 337–342 CrossRef CAS PubMed.
- G. A. Rodan and T. J. Martin, Science, 2000, 289, 1508–1514 CrossRef CAS PubMed.
- S. Khosla, J. P. Bilezikian, D. W. Dempster, E. M. Lewiecki, P. D. Miller, R. M. Neer, R. R. Recker, E. Shane, D. Shoback and J. T. Potts, J. Clin. Endocrinol. Metab., 2012, 97, 2272–2282 CrossRef CAS PubMed.
- K. A. Kennel and M. T. Drake, Mayo Clin. Proc., 2009, 84, 632–637 CrossRef CAS PubMed.
- S. Chauhan, A. Sharma, N. K. Upadhyay, G. Singh, U. R. Lal and R. Goyal, BMC Complementary Altern. Med., 2018, 18, 233 CrossRef PubMed.
- S. Wang, Q. Ma, Z. Xie, Y. Shen, B. Zheng, C. Jiang, P. Yuan, Q. An, S. Fan and Z. Jie, J. Bone Miner. Res., 2021, 36, 1850–1865 CrossRef CAS PubMed.
- H. J. Moon, E. K. Kim, Y. S. Nam, J. H. Kim, W. K. Ko, J. M. Lee, C. H. Lee, J. B. Jang, K. S. Lee and I. K. Kwon, Phytother. Res., 2012, 26, 1648–1655 CrossRef CAS PubMed.
- L. Yu, Z. Deng, L. Liu, W. Zhang and C. Wang, Front. Bioeng. Biotechnol., 2020, 8, 584391 CrossRef PubMed.
- B. Wang, X. Zhuang, Z. B. Deng, H. Jiang, J. Mu, Q. Wang, X. Xiang, H. Guo, L. Zhang, G. Dryden, J. Yan, D. Miller and H. G. Zhang, Mol. Ther., 2014, 22, 522–534 CrossRef CAS PubMed.
- S. Raimondo, F. Naselli, S. Fontana, F. Monteleone, A. Lo Dico, L. Saieva, G. Zito, A. Flugy, M. Manno, M. A. Di Bella, G. De Leo and R. Alessandro, Oncotarget, 2015, 6, 19514–19527 CrossRef PubMed.
- Y. Jin, R. Cui, L. Zhao, J. Fan and B. Li, Cell Proliferation, 2019, 52, e12696 Search PubMed.
- T. Ramesh, S. W. Kim, J. H. Sung, S. Y. Hwang, S. H. Sohn, S. K. Yoo and S. K. Kim, Exp. Gerontol., 2012, 47, 77–84 CrossRef CAS PubMed.
- K. S. Baek, Y. S. Yi, Y. J. Son, S. Yoo, N. Y. Sung, Y. Kim, S. Hong, A. Aravinthan, J. H. Kim and J. Y. Cho, J. Ginseng Res., 2016, 40, 437–444 CrossRef PubMed.
- S. Y. Han, J. Kim, E. Kim, S. H. Kim, D. B. Seo, J. H. Kim, S. S. Shin and J. Y. Cho, J. Ginseng Res., 2018, 42, 496–503 CrossRef PubMed.
- Y. Y. Lee, J. S. Park, E. J. Lee, S. Y. Lee, D. H. Kim, J. L. Kang and H. S. Kim, J. Agric. Food Chem., 2015, 63, 3472–3480 CrossRef CAS PubMed.
- D. S. Im, Biomolecules, 2020, 10, 444 CrossRef CAS PubMed.
- N. Yang, D. Liu, X. Zhang, J. Li, M. Wang, T. Xu and Z. Liu, Chin. Med., 2020, 15, 1–15 CrossRef PubMed.
- M. H. Siddiqi, M. Z. Siddiqi, S. Ahn, S. Kang, Y. J. Kim, N. Sathishkumar, D. U. Yang and D. C. Yang, J. Ginseng Res., 2013, 37, 261–268 CrossRef CAS PubMed.
- B. Cheng, J. Li, J. Du, X. Lv, L. Weng and C. Ling, Food Chem. Toxicol., 2012, 50, 1610–1615 CrossRef CAS PubMed.
- L. He, J. Lee, J. H. Jang, S. H. Lee, M. H. Nan, B. C. Oh, S. G. Lee, H. H. Kim, N. K. Soung, J. S. Ahn and B. Y. Kim, Bone, 2012, 50, 1207–1213 CrossRef CAS PubMed.
- C. C. Frohne, E. M. Llano, A. Perkovic, R. D. Cohen and J. J. Luke, J. Immunother. Cancer, 2019, 7, 1 CrossRef PubMed.
- E. G. Cho, S. Y. Choi, H. Kim, E. J. Choi, E. J. Lee, P. J. Park, J. Ko, K. P. Kim and H. S. Baek, Cells, 2021, 10, 486 CrossRef CAS PubMed.
- J. Kim, Y.-H. Lee, J. Wang, Y. K. Kim and I. K. Kwon, SN Appl. Sci., 2022, 4, 63 CrossRef CAS.
- M. Y. Konoshenko, E. A. Lekchnov, A. V. Vlassov and P. P. Laktionov, BioMed Res. Int., 2018, 2018, 8545347 Search PubMed.
- C. Stanly, I. Fiume, G. Capasso and G. Pocsfalvi, Unconventional Protein Secretion, Springer, 2016, pp. 259–269 Search PubMed.
- M. Yue, P. Huang, C. Wang, H. Fan, T. Tian, J. Wu, F. Luo, Z. Fu, X. Xia, P. Zhu, J. Li, Y. Han, Y. Zhang and W. Hou, Immunol. Invest., 2021, 50, 1–11 CrossRef CAS PubMed.
- S. Yang, S. Lu, L. Ren, S. Bian, D. Zhao, M. Liu and J. Wang, J. Ginseng Res., 2023, 47, 133–143 CrossRef PubMed.
- Y. P. Niu, J. M. Jin, R. L. Gao, G. L. Xie and X. H. Chen, Zhongguo Shiyan Xueyexue Zazhi, 2001, 9, 178–180 Search PubMed.
- P. P. Eleniste and A. Bruzzaniti, J. Signal Transduction, 2012, 2012, 296450 Search PubMed.
- H.-Y. Lee, S.-H. Park, S.-W. Chae, N.-K. Soung, M.-J. Oh, J. S. Kim, Y. O. Kim and H. J. Chae, J. Funct. Foods, 2015, 13, 192–203 CrossRef CAS.
- S. Ju, J. Mu, T. Dokland, X. Zhuang, Q. Wang, H. Jiang, X. Xiang, Z. B. Deng, B. Wang, L. Zhang, M. Roth, R. Welti, J. Mobley, Y. Jun, D. Miller and H. G. Zhang, Mol. Ther., 2013, 21, 1345–1357 CrossRef CAS PubMed.
- K. Suda, N. Udagawa, N. Sato, M. Takami, K. Itoh, J. T. Woo, N. Takahashi and K. Nagai, J. Immunol., 2004, 172, 2504–2510 CrossRef CAS PubMed.
- E. Hausmann, L. G. Raisz and W. A. Miller, Science, 1970, 168, 862–864 CrossRef CAS PubMed.
- K. R. Park, E. C. Kim, J. T. Hong and H. M. Yun, Theranostics, 2018, 8, 3087–3098 CrossRef CAS PubMed.
- Y. H. Cheon, J. Y. Kim, J. M. Baek, S. J. Ahn, H. Y. Jun, M. Erkhembaatar, M. S. Kim, M. S. Lee and J. Oh, J. Bone Miner. Res., 2016, 31, 403–415 CrossRef CAS PubMed.
- L. Wei, W. Chen, L. Huang, H. Wang, Y. Su, J. Liang, H. Lian, J. Xu, J. Zhao and Q. Liu, Pharmacol. Res., 2022, 184, 106400 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr07018a |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |