
From screening to the hectogram scale: sustainable electrochemical synthesis of mefenpyr-diethyl†
Martin
Linden‡
a,
Silja
Hofmann‡
 a,
Felix N.
Weber
a,
Robin M.
Bär
b,
Sherif J.
Kaldas
c,
Mark J.
Ford
b and
Siegfried R.
Waldvogel
*ad
a,
Felix N.
Weber
a,
Robin M.
Bär
b,
Sherif J.
Kaldas
c,
Mark J.
Ford
b and
Siegfried R.
Waldvogel
*ad
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, Mainz, 55128, Germany
bResearch & Development, Crop Science, Bayer AG, Alfred-Nobel-Str. 50, Monheim am Rhein, 40789, Germany
cChemical Process Development, Crop Science, Bayer AG, 41538, Dormagen
dInstitute of Biological and Chemical Systems – Functional Molecular Systems (IBCS FMS), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: waldvogel@uni-mainz.de; Web: https://www.aksw.uni-mainz.de/prof-dr-s-r-waldvogel/
First published on 1st August 2023
Abstract
To demonstrate the technical application potential of electrochemical reactions, we developed a synthetic method to access the commercially available herbicide safener mefenpyr-diethyl. In a simple undivided electrolysis cell with non-hazardous aqueous sodium iodide as supporting electrolyte and mediator, the target compound was synthesized via the (E)- or (Z)-hydrazone. Environmentally benign solvents or solvent-free conditions ensure a sustainable process and excellent recycling of the excess reagents. Finally, scale-up to the hectogram scale was performed with good to excellent yield.
Introduction
The global climate crisis necessitates action and has caused a shift towards a more sustainable economy. For the chemical industry, this will mean a renunciation of long-practiced processes. Nowadays, highly efficient synthesis with regards to atom economy and energy efficiency is necessary not only from an economical but also an ecological point of view. This has led to a need for a sustainable, circular economy.1 Furthermore, minimizing risks by circumventing the use of hazardous chemicals and establishing inherently safe processes is of utmost importance for modern synthetic methodologies.2,3Electrochemistry is linked to the principles of green chemistry and can contribute to a more sustainable chemical industry.3,4 Hazardous and often expensive redox reagents can be substituted with electric current.5 Electricity contributes not only to the sustainability of a process, but also to its safety.6 Control over the reaction is simply given by controlling the current density, as the initial electron transfer is confined to the electrode surface and runaway reactions can be avoided.7 Furthermore, organic electrochemistry facilitates resource efficiency, as unique reactivities can give access to synthetic shortcuts and additives, e.g., supporting electrolytes, can be easily recycled.8 Thus, a reaction can be considered practically waste- and pollutant-free, if the applied electricity originates from renewable resources.9
The structural motifs of pyrazoles and pyrazolines are widely found among biologically active substances. Their potential pharmaceutical applications include antimicrobial,10 antipsychotic,11 anti-inflammatory,12 anti-nociceptive,13 anti-cancer14 and anti-obesity drugs.15 Furthermore, a variety of agrochemicals featuring a pyrazole or pyrazoline moiety have been developed, such as the herbicide benzofenap (3)16 and the herbicide safener mefenpyr-diethyl (1)17 (Chart 1). The latter is commonly used for fenoxaprop-P-ethyl (2) detoxification in wheat and barley.18 Conventionally, pyrazolines and pyrazoles are synthesized by condensation of hydrazones with 1,3-diketones,19,20 2-ynones,21 or 2-enones.22 Yet, these strategies suffer from an often-unsatisfying regioselectivity and difficult-to-obtain starting materials. Other strategies rely on oxidative synthesis of hydrazonoyl halides; subsequent pyrazoline or pyrazole formation is achieved via base-promoted liberation of the corresponding nitrile imine and [3 + 2] cycloaddition with a dipolarophile.19,26,27 The yield for the latter route commonly varies as much as 38–95% over the two steps.27 However, often hazardous and expensive catalysts and oxidizers, e.g., N-chlorosuccinimide27 (NCS), are employed. Literature has several further examples on the electrochemistry of pyrazolines and pyrazoles. These include intramolecular synthesis,28 4-halogenation,29 dimerization,30 and N-arylation of pyrazoles.31 Yet, pyrazol(in)e syntheses are rather poorly explored. Reported examples additionally suffer from elaborate starting materials which themselves are borne out of complex synthetic routes.32 Recently, the electro-chemical generation of stable diazo compounds33 and a simple and scalable electrosynthesis of pyrazolines and pyrazoles34 starting from easily accessible hydrazones were published. The latter gave access to a broad variety of pyrazolines in up to excellent yields by using aqueous sodium iodide as mediator and supporting electrolyte in a biphasic system using ethyl acetate as solvent.
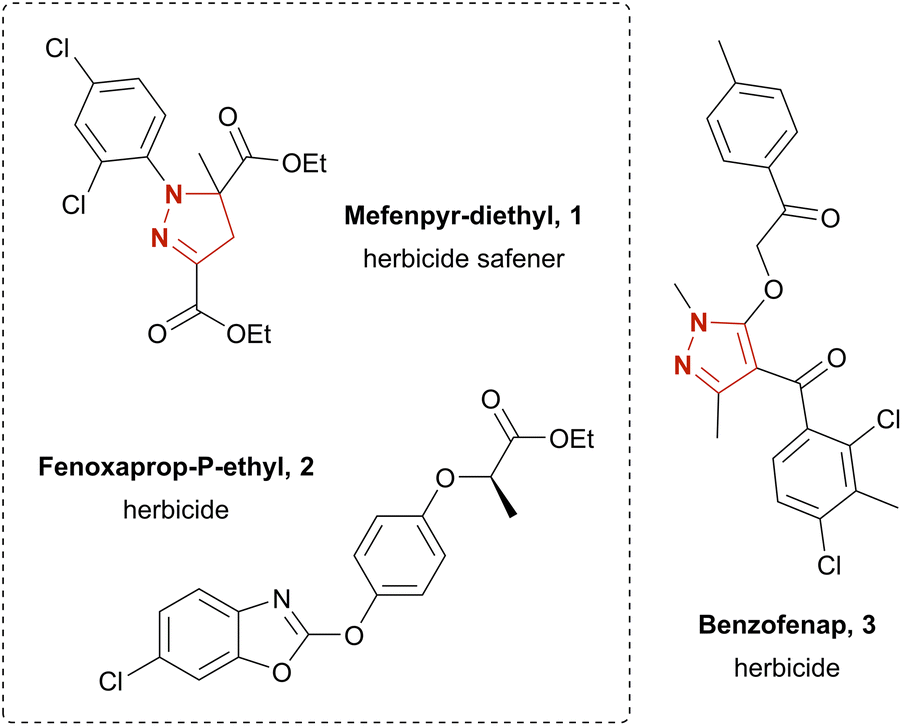 | ||
| Chart 1 Examples of commercial herbicides and herbicide safeners. | ||
The conventional synthetic route to mefenpyr-diethyl (1) follows a highly complex pathway.25,35 First, ethyl acetoacetate (4) is chlorinated in position 2. The key intermediate 6 is yielded via nucleophilic attack of ethyl 2-chloroacetoacetate (5) at the corresponding aryldiazonium species and deacetylation of the formed adduct. The hydrazonoyl chloride 6 is finally converted to mefenpyr-diethyl (1) by base-promoted cycloaddition as seen below (Scheme 1). To demonstrate the applicability of electrochemical techniques in modern industrially relevant processes,36 we developed an electrochemical approach for an environmentally benign synthesis of herbicide safener mefenpyr-diethyl (1). Aqueous sodium iodide is used in a dual role as mediator and supporting electrolyte. The use of inexpensive isostatic graphite (Cgr) as electrode material in environmentally benign solvents or under solvent-free conditions ensures a sustainable process and excellent recycling potential of the excess reagents. The simple setup of a galvanostatic beaker-type cell allows for easy scale-up to hectogram scale.
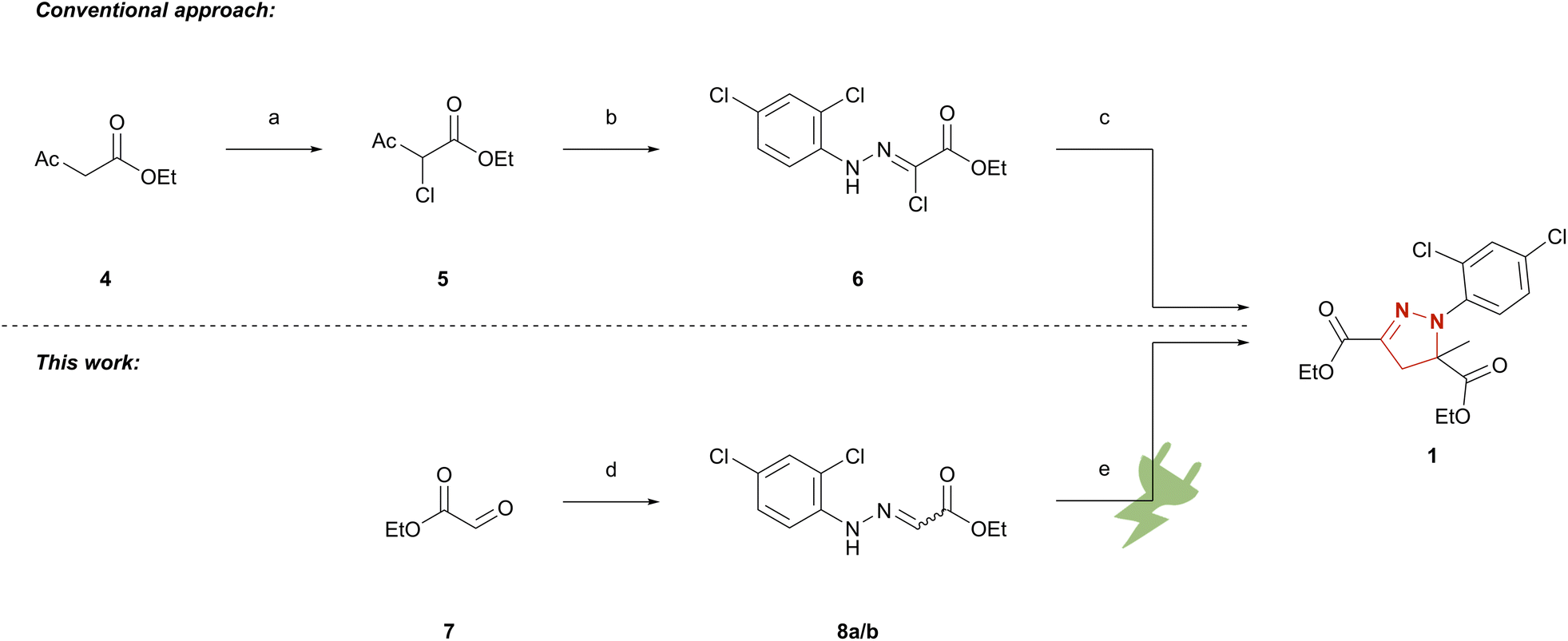 | ||
Scheme 1 Synthetic approaches to mefenpyr-diethyl (1). (a) DMSO, NCS, room temperature;23 (b) 2,4-dichloroaniline, NaNO2, aq. HCl, 0 °C, then 5, NaOAc, EtOH, room temperature;24 (c) NEt3/KHCO3, ethyl methacrylate, 60–65 °C;25 (d) 2,4-dichlorophenylhydrazine hydrochloride, THF, NEt3; EtOH 0 °C → room temperature or 2,4-dichlorophenylhydrazine hydrochloride, EtOH, AcOH, 80 °C; (e) Cgr||Cgr, 1 M aq. NaI, ethyl methacrylate, 27.9 mA cm−2, 5.4 F, 33 °C or Cgr||Cgr EtOH/MeCN 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v), ethyl methacrylate, NaI, 4.0 mA cm−2, 4.03 F, 25 °C. :1 (v/v), ethyl methacrylate, NaI, 4.0 mA cm−2, 4.03 F, 25 °C. | ||
Results and discussion
We chose the synthesis of mefenpyr-diethyl (1) as an example reaction to highlight the development of a technically relevant electrochemical reaction from screening to scale-up. The previously employed conditions for starting material synthesis led to selective formation of (Z)-hydrazone 8a in an excellent yield of 90% on 15 mmol scale.34 However, scale-up resulted in a massive decrease in yield of 8a, as significant amounts of the corresponding (E)-hydrazone 8b were formed according to 1H NMR. However, modification of this method allowed for selective synthesis of 8a even on larger scale. Therefore, treatment of the hydrazine hydrochloride with triethylamine in THF and filtration prior to addition to the corresponding glyoxylate 7 proved beneficial. Furthermore, an excess of aldehyde 7 was required to supress hydrazinolysis of the ethyl ester, yielding (Z)-hydrazone 8a in a satisfactory yield of 80% on 47 mmol scale. Further scale-up to hectogram scale (0.9 mol) again resulted in a decreased yield of 63%, but maintained the selectivity towards the (Z)-hydrazone 8a. Nevertheless, on this scale parameters such as efficient mixing to overcome concentration gradients, changes in temperature due to alteration in heat transfer, and the relative concentration as well as stability of products and starting materials need to be considered. Thus, transferring this synthesis to hectogram scale or beyond does require re-evaluation of the reaction conditions.37 Another common route to access hydrazones involves refluxing the corresponding hydrazine and aldehyde in methanol or ethanol with catalytic amounts of acid.38 This method allowed for selective synthesis of the (E)- hydrazone 8b in a very good yield of 89% on hectogram scale (0.75 mol). Thus, selective synthesis of both isomers in satisfactory yields was achieved by controlling solvent, temperature, and additives.The electrochemical synthesis of mefenpyr-diethyl was initially investigated with the (Z)-hydrazone 8a to re-evaluate the solvent system. Ethyl acetate, tert-butyl methyl ether, dichloromethane, and chlorobenzene were tested as organic solvents (phase ratio org. solvent/1 M aq. NaI of 1:4). Unfortunately, the chlorinated solvents were observed to give the highest yields (Table S7, ESI†). Thus, further reaction optimization aiming for a sustainable system was necessary. A statistically driven optimization via Design of Experiments (DoE)39 was pursued to ensure a comprehensive investigation of the system. In a two-step optimization via fractional factorial designs (both 23–1, resolution III with central point and rotatable axial points, each data point acquired thrice) regarding the applied amount of charge (Q), the current density (j), and the amount of acrylate, the following conditions were found: graphite electrodes, 27.9 mA cm−2, 5.4 F, 3.21 eq. ethyl methacrylate (Fig. 1). Through further linear optimization, solvent-free electrolytic conditions were achieved, employing 250 mg (0.96 mmol) hydrazone 8a per 1 mL aqueous phase at 33 °C. This enabled full conversion of 1 g (3.8 mmol) 8a in a 5 mL screening cell, accessing mefenpyr-diethyl (1) in 93% GC-yield. Hence, the reaction was transferred to a 50 mL jacketed beaker-type cell and run on 5 g-scale (19.1 mmol). This allowed for more efficient mixing of the reaction mixture due to more powerful cross-shaped stirring bars and the target compound 1 was obtained in a very good isolated yield of 86%.
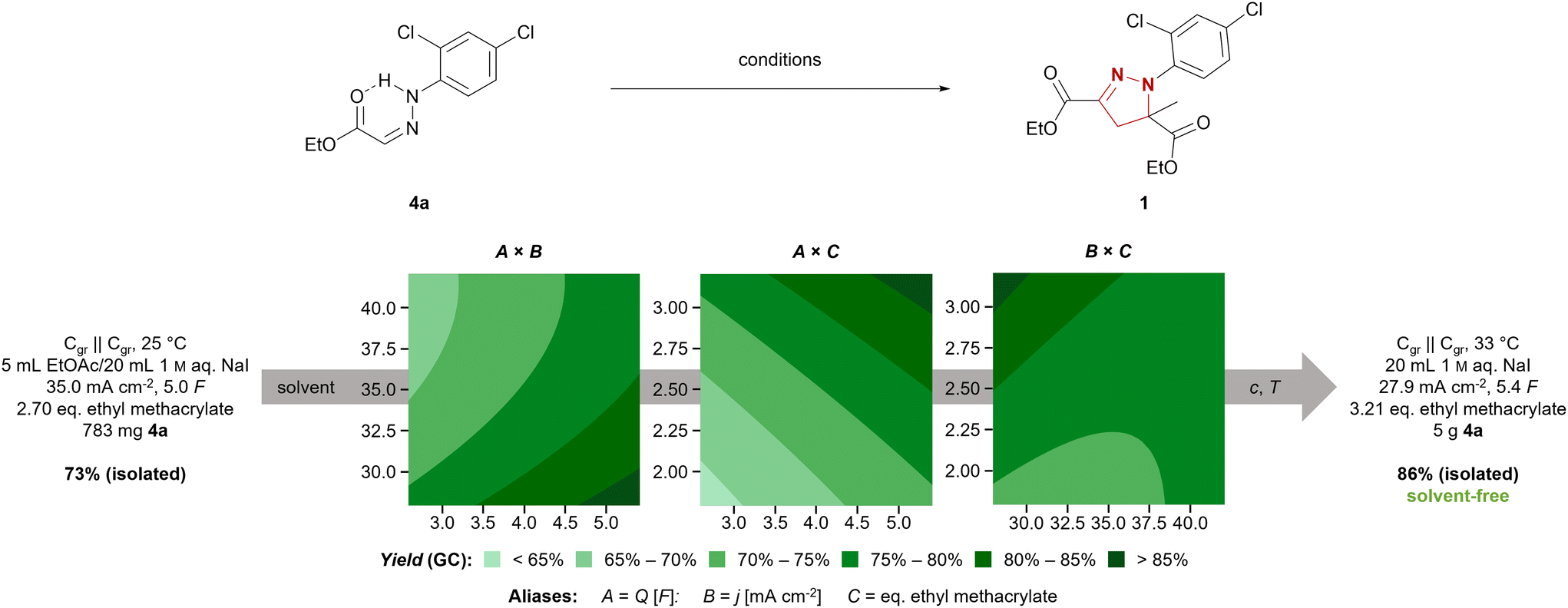 | ||
| Fig. 1 Contour plots of the yield (GC, 1,3,5-trimethoxybenzene as internal standard) for the optimization process via DoE for the synthesis of 1 form (Z)-hydrazone 8a. The solvent, substrate concentration c and reaction temperature T were optimized in a linear fashion. Synthesis conditions before and after optimization are shown on the left and right, respectively. Holding values: A = 5.41 F; B = 27.9 mA cm−2; C = 3.21 eq. GC yields determined with 1,3,5-trimethoxybenzene as internal standard after external calibration. | ||
Furthermore, the electrochemical conversion of the (E)-hydrazone 8b under previously optimized conditions was tested. Contrary to hydrazone 8a, application of hydrazone 8b in the reaction resulted in a rather poor yield of mefenpyr-diethyl (1) as well as incomplete conversion. According to LC-MS analysis, both isomers exhibit a pronounced difference in polarity (Fig. S22 and S23, ESI†). Whilst (Z)-hydrazone 8a is more polar than the formed product 1, the initial oxidation of the hydrazone can occur at the organic/aqueous phase boundary. Thus, even under solvent-free conditions the reaction is thought to follow the initially hypothesized mechanism.34 In contrast, (E)-hydrazone 8b shows increased lipophilicity compared to 8a and is even more apolar than mefenpyr-diethyl. This seems to strongly disable efficient oxidation of the hydrazone at the interface, explaining the insufficient conversions and poor yields of 1. Addition of tert-butyl methyl ether as solvent improved the results, but still gave un-satisfactory yields (GC). To circumvent the inhibited oxidation of the lipophilic (E)-hydrazone 8b at the phase boundary, application of a homogeneous system was investigated. Thus, sodium iodide was employed in a dual role as mediator and supporting electrolyte in various organic solvents (acetonitrile, methanol, ethanol, isopropanol). Generally, the homogeneous system proved suitable for conversion of the (E)-hydrazone 8b.
The best results were obtained using a mixture of acetonitrile/ethanol (1:1 v/v) as solvent system and further optimization was performed via DoE. A fractional factorial design (25–2, resolution III with central point and rotatable axial points, each data point acquired once, central point thrice) led to the following optimum conditions: graphite electrodes, acetonitrile/EtOH (1:1 v/v), 2.79 eq. NaI, 3.79 eq. ethyl methacrylate, 9.0 mA cm−2, 4.03 F, rt. An additional linear optimization of the current density revealed an optimum at 4.0 mA cm−2. With the adjusted conditions, full conversion of hydrazone 8b was achieved and mefenpyr-diethyl (1) was obtained in a satisfactory isolated yield of 71% on 2.85 mmol scale. Hence, the electrosynthesis of herbicide safener 1 seems to be strongly solvent dependent with regards to the substrate configuration.
After suitable reaction conditions for both hydrazones 8a and 8b were established, the reactions were investigated regarding scalability. The results are summarized in Table 1.
| Product | Hydrazone |
|
|
|
|---|---|---|---|---|
|
a Solvent-free, c = 0.96 M8a in 1 M NaI (aq.), 3.21 eq. ethyl methacrylate, graphite electrodes, 33 °C, 27.9 mA cm−2, 5.4 F.
b
c = 0.11 M8b in EtOH/MeCN 1:1 (v/v), 3.79 eq. ethyl methacrylate, 2.79 eq. NaI, 4.0 mA cm−2, 4.03 F.
|
||||
|
8a | 86% | 88% | 66% |
| 16.4 mmol | 88.2 mmol | 0.528 mol | ||
| 6.13 g | 32.9 g | 197 g | ||
| 8b | 71% | 66% | 63% | |
| 2.03 mmol | 14.9 mmol | 101 mmol | ||
| 757 mg | 5.59 g | 37.8 g | ||



To our delight, both established synthetic routes for conversion of hydrazones 8a and 8b to mefenpyr-diethyl (1) showed robustness in scale-up. Synthesis of 1 starting from (E)-hydrazone 8b was performed in a satisfactory yield of 66% on multigram-scale. Even on multi-decagram scale in a bipolar cell set-up,40 the homogeneous reaction proved to be stable, and mefenpyr-diethyl was obtained in a comparable yield of 63%. Similarly, (Z)-hydrazone 8a underwent the desired conversion smoothly under solvent-free conditions at different scales. Even when going from gram-scale to decagram-scale, no loss in yield was observed and target compound 1 was obtained in an excellent yield of 88%. Thus, we decided to transfer this reaction to even larger scale, employing 209 g (0.8 mol) of (Z)-hydrazone 8a. A stacked electrode setup was used,40 and mefenpyr-diethyl was obtained in 66% (197 g) on hectogram scale. On these scales, the counter electrode reaction represents an important process which must be taken into consideration. The counter electrode reaction for the represented example is the evolution of hydrogen. This is a well-known and extensively studied reaction, which can be easily controlled.41
To compare both approaches to herbicide safener 1, the yield over two steps including hydrazone formation in the first and electrochemical conversion in the second step must be considered. Overall, mefenpyr-diethyl (1) was obtained in a total yield of up to 70% over two steps via (Z)-hydrazone 8a. The competing sequence via (E)-hydrazone 8b yielded the desired product 1 in up to 63% (Scheme 2).
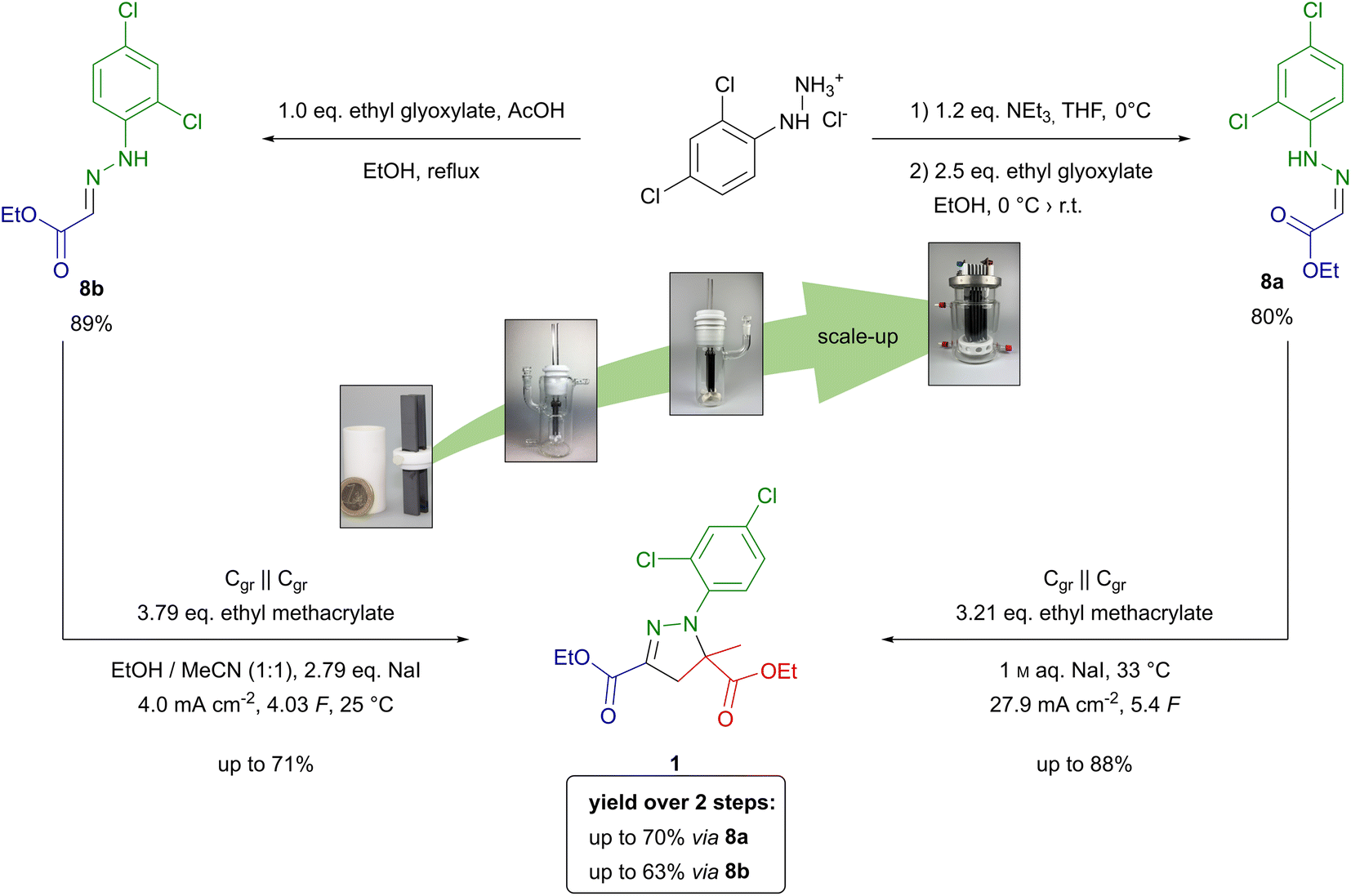 | ||
| Scheme 2 Synthesis from screening to scale-up of mefenpyr-diethyl (1) from 2,4-dichlorophenylhydrazone hydrochloride via hydrazones 8a and 8b. | ||
Overall, the synthesis of mefenpyr-diethyl (1) via (Z)-hydrazone 8a is slightly more favourable compared to the method using (E)-hydrazone 8b. However, both synthetic strategies demonstrate excellent performance regarding their scalability. Thus, these electro-organic procedures show potential for application on technically relevant scale.
Conclusions
A sustainable two-step protocol for the preparation of the industrially relevant herbicide safener mefenpyr-diethyl was established.The selective synthesis of (E)- or (Z)-hydrazone as substrates for the electrochemical conversion was achieved by solvent-control in very good yields. A substantial difference in the polarity of the hydrazones was found to require vastly different reaction conditions for a smooth synthesis of the title compound. Whereas the (E)-hydrazone required a homogeneous system for successful conversion, the (Z)-hydrazone was converted in a biphasic system under solvent-free conditions. A series of scale-up experiments revealed a good to excellent stability for both processes. Thus, the (E)-hydrazone yielded up to 71% mefenpyr-diethyl and a 50-fold scale-up to multi-decagram scale was performed successfully. In parallel, the conversion of the (Z)-hydrazone yielded 86–88% in up to multi-decagram scale and even a scale-up to hectogram scale was demonstrated.
In conclusion, the general application potential of this exemplary electrochemical conversion on technically relevant scale was proven. Both reaction sequences captivate with the simplicity of their electrochemical key steps: an undivided beaker-type cell equipped with inexpensive graphite electrodes and excess reagents can be recycled easily. By employing environmentally benign solvents or solvent-free conditions with abundant and non-hazardous sodium iodide in a dual role as supporting electrolyte and mediator, the established protocol offers a sustainable and feasible alternative to conventional synthetic methods.
Author contributions
M. L. and S. H. contributed equally to this work. M. L. and S. H. established the reaction, analysed experimental data, and performed the scale-up reactions. M. L., S. H., and F. N. W synthesized starting materials. M. L., S. H., and F. N. W. conducted the experiments for initial reaction optimization and DoE. M. L., S. H., R. M. B., S. J. K., M. J. F., and S. R. W. wrote the manuscript. All authors discussed the results and agreed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr R. Hamill for her assistance in editing the manuscript. Financial support by the Deutsche Forschungsgemeinschaft (WA 1276/17-2) is highly appreciated. Endorsement by SusInnoScience in frame of the Forschungsinitiative Rheinland-Pfalz was very helpful.References
-
(a) I. T. Horváth, Chem. Rev., 2018, 118, 369 CrossRef PubMed
; (b) N. Jain, Nat. Rev. Methods Primers, 2022, 2, 61 CrossRef
- P. T. Anastas, Green Chem., 2003, 5, G29 RSC
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC
-
(a) J. Seidler, J. Strugatchi, T. Gärtner and S. R. Waldvogel, MRS Energy Sustain., 2020, 7, E42 CrossRef
-
J. L. Röckl, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed, 2020, 59, 315 Search PubMed
-
(a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed
- S. B. Beil, D. Pollok and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 14750 CrossRef CAS PubMed
-
(a) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018 CrossRef PubMed
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386 RSC
- R. Verma, S. K. Verma, K. P. Rakesh, Y. R. Girish, M. Ashrafizadeh, K. S. Sharath Kumar and K. S. Rangappa, Eur. J. Med. Chem., 2021, 212, 113134 CrossRef CAS PubMed
- G. Li, Y. Cheng, C. Han, C. Song, N. Huang and Y. Du, RSC Med. Chem., 2022, 13, 1300 RSC
- S. Basu, D. A. Barawkar, V. Ramdas, M. Patel, Y. Waman, A. Panmand, S. Kumar, S. Thorat, M. Naykodi, A. Goswami, B. S. Reddy, V. Prasad, S. Chaturvedi, A. Quraishi, S. Menon, S. Paliwal, A. Kulkarni, V. Karande, I. Ghosh, S. Mustafa, S. De, V. Jain, E. R. Banerjee, S. R. Rouduri, V. P. Palle, A. Chugh and K. A. Mookhtiar, Eur. J. Med. Chem., 2017, 134, 218 CrossRef CAS PubMed
- I. F. Florentino, D. P. B. Silva, C. S. Cardoso, R. Menegatti, F. S. de Carvalho, L. M. Lião, P. M. Pinto, S. Peigneur, E. A. Costa and J. Tytgat, Biomed. Pharmacother., 2019, 115, 108915 CrossRef CAS PubMed
-
(a) H. Aziz, A. F. Zahoor and S. Ahmad, J. Chil. Chem. Soc., 2020, 65, 4746 CrossRef CAS
- A. Samat, B. Tomlinson, S. Taheri and G. N. Thomas, Recent Pat. Cardiovasc. Drug Discovery, 2008, 3, 187 CrossRef CAS PubMed
- W. C. Quayle, D. P. Oliver, S. Zrna and A. Fattore, J. Agric. Food Chem., 2007, 55, 5199 CrossRef CAS PubMed
-
(a) L. Bianchi, S. M. Perissato, V. M. Anunciato, R. C. Dias, D. M. Gomes, C. A. Carbonari and E. D. Velini, J. Environ. Sci. Health, Part B, 2021, 56, 163 CrossRef CAS PubMed
-
(a) L. Yuan, G. Ma, Y. Geng, X. Liu, H. Wang, J. Li, S. Song, W. Pan and Z. Hun, PLoS One, 2021, 2, e0256884 CrossRef PubMed
- S. Fustero, A. Simón-Fuentes and J. F. Sanz-Cervera, Org. Prep. Proced. Int., 2009, 41, 253 CrossRef CAS
-
(a) S. Fustero, R. Román, J. F. Sanz-Cervera, A. Simón-Fuentes, A. C. Cuñat, S. Villanova and M. Murguía, J. Org. Chem., 2008, 73, 3523 CrossRef CAS PubMed
- D. B. Grotjahn, S. Van, D. Combs, D. A. Lev, C. Schneider, M. Rideout, C. Meyer, G. Hernandez and L. Mejorado, J. Org. Chem., 2002, 67, 9200 CrossRef CAS PubMed
- A. E. Sarhan, A. A. Sediek, N. M. Khalifa and E. E. Hasan, Heterocycles, 2022, 104, 447 CrossRef CAS
- B. Sreedhar, P. Surendra Reddy and M. Madhavi, Synth. Commun., 2007, 37, 4149 CrossRef CAS
- S. Altomonte, G. L. Baillie, R. A. Ross and M. Zanda, RSC Adv., 2015, 5, 13692 RSC
-
G. Schlegel, US5908938A, 1999
- D. Li, S. Qiu, Y. Chen and L. Wu, ChemistrySelect, 2020, 5, 12034 CrossRef CAS
- L. Song, Y. Lai, H. Li, J. Ding, H. Yao, Q. Su, B. Huang, M.-A. Ouyang and R. Tong, J. Org. Chem., 2022, 87, 10550 CrossRef CAS PubMed
- S. A. Paveliev, O. O. Segida, O. V. Bityukov, H.-T. Tang, Y.-M. Pan, G. I. Nikishin and A. O. Terent'ev, Adv. Synth. Catal., 2022, 364, 3910 CrossRef CAS
-
(a) B. V. Lyalin and V. A. Petrosyan, Russ. Chem. Bull., 2014, 63, 360 CrossRef CAS
- F. Pragst and C. Böck, J. Electroanal. Chem. Interfacial Electrochem., 1975, 61, 47 CrossRef CAS
- S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS PubMed
- X. Wei and B. Speiser, Electrochim. Acta, 1997, 42, 73 CrossRef CAS
- N. Tanbouza, A. Petti, M. C. Leech, L. Caron, J. M. Walsh, K. Lam and T. Ollevier, Org. Lett., 2022, 24, 4665 CrossRef CAS PubMed
- M. Linden, S. Hofmann, A. Herman, N. Ehler, R. M. Bär and S. R. Waldvogel, Angew. Chem., Int. Ed., 2023, e202214820 CAS
-
G. Shi, H. Hu and H. Meng, CN111592493A, 2020
-
(a) D. Pollok, B. Gleede, A. Stenglein and S. R. Waldvogel, Aldrichimica Acta, 2021, 54, 3 CAS
- D. C. Hendershot and A. Sarafinas, Chem. Health Saf., 2005, 12, 29 CrossRef CAS
-
(a) N. Thamban Chandrika, E. K. Dennis, S. K. Shrestha, H. X. Ngo, K. D. Green, S. Kwiatkowski, A. G. Deaciuc, L. P. Dwoskin, D. S. Watt and S. Garneau-Tsodikova, Eur. J. Med. Chem., 2019, 164, 273 CrossRef CAS PubMed
-
(a) E. Babaoglu and G. Hilt, Chem. – Eur. J., 2020, 26, 8879 CrossRef CAS PubMed
- G. H. M. de Kruijff, T. Goschler, N. Beiser, A. Stenglein, O. M. Türk and S. R. Waldvogel, Green Chem., 2019, 21, 4815 RSC
- M. Klein and S. R. Waldvogel, Angew. Chem., Int. Ed., 2022, 61, e202204140 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02118a |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |