
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClarifying the local microenvironment of metal–organic frameworks and their derivatives for electrochemical CO2 reduction: advances and perspectives
Muhammad Kashif
Aslam
,
Kang
Yang
,
Sheng
Chen
 *,
Qiang
Li
* and
Jingjing
Duan
*
*,
Qiang
Li
* and
Jingjing
Duan
*
MIIT Key Laboratory of Thermal Control of Electronic Equipment, School of Energy and Power Engineering, School of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing, 210094, China. E-mail: jingjing.duan@njust.edu.cn
First published on 27th February 2023
Abstract
The conversion of carbon dioxide (CO2) into fuels, value-added products and electricity is one of the most effective ways to reduce the atmospheric CO2 concentration, and thus mitigate the greenhouse effect, simultaneously resolving the environmental and energy crises. In this case, metal–organic frameworks (MOFs) show good application prospect in the catalytic CO2 reduction reaction because of their well-defined porous structure, rich active sites and feasible functionalization. Herein, we summarize the latest research progress of MOFs and their derivatives for catalytic CO2 electroreduction and discuss their mechanism, kinetics, thermodynamics and catalytic performance. Moreover, the effect of first-, secondary and out-sphere coordination on the metal active centers and the local microenvironment of MOFs, which can be manipulated by adjusting their metal nodes, organic linkers, and solvents, are addressed in-depth to clarify the key to their great electrocatalytic performance. Based on this, the main challenges and future outlook of MOF catalysts are summarized and perspectives presented.
Broader contextRenewable energy technologies are trying to reshape the energy pathway of modern society, which will decrease the usage of fossil fuels, and thus reduce the risk of future environmental degradation. The efficient utilization of renewable energy is the key, and thus there is a great demand for appropriate energy conversion technologies. The electrochemical reaction is the foundation and key of various energy conversion technologies. The electroreduction of CO2 reaction (eCO2RR) promoted by renewable power for the conversion of CO2 into valuable industrial materials stores excess renewable energy as chemical energy in fuels. Therefore, eCO2RR is important technology to realize carbon recycling and renewable energy storage for a zero/negative carbon and energy cycle. In this case, the design of highly efficient electrocatalysts is crucial but challenging, which is significant for the actual use of conversion reactions. This work summarizes the key aspects and latest progress of MOFs and their derivatives for eCO2RR in recent years. In combination with the research trend, the metal nodes, coordination types and configuration and local microenvironment are discussed in-depth. Finally, the problems and future development strategies of MOFs and MOF-derived confined materials in CO2 reduction have been analyzed and prospected. |
1. Introduction
Presently, the rapid industrialisation and excessive use of fossil fuels as an energy source have resulted in a significant increase in the concentration of carbon dioxide in the atmosphere, reaching a high value never seen in the last tens of millions of years. As a typical greenhouse gas, the increasing CO2 concentration has increased the global temperature gradually,1 leading to extreme weather, rising sea levels, species extinction and other serious consequences. Accordingly, renewable energy technology is reshaping the energy layout of modern society, which will decrease the use of fossil fuels and reduce the risk of future environmental degradation.2–7 Here, the efficient utilization of renewable energy is the key, and thus there is a great demand for appropriate energy conversion technologies.8–10 Electrochemical reactions are the foundation and key of various energy conversion technologies. For example, the electrochemical oxygen reduction reaction (ORR) is a vital cathode reaction in proton exchange membrane fuel cells (PEMFCs) to realize effective energy conversion.11–13 Furthermore, techniques such as biological,14 thermochemical, photochemical, and electrochemical reactions are being widely studied for CO2 reduction. Thermal CO2 reduction can achieve the use of energy in the form of heat, where a pressurized reactant gas (CO2) and high-performance catalyst to reduce the amount of energy consumed during CO2 decomposition are required to utilize waste heat. In thermal CO2 reduction, the catalyst facilitates the breakage of the strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, which have a dissociation energy of 750 kJ mol−1 (∼9 × 104 K). The reduction of CO2 to CO was achieved at 1100 K using CeO2, ZrO2, LaFeO3, and lanthanum-based perovskites as catalysts.15–18 Alternatively, the electrochemical conversion of CO2 has several advantages over thermal reduction, including mild reaction conditions, controllable reaction rates and product selectivity through the applied potential, and wide scalability due to modular electrolyzer design. However, the electroreduction of CO2 reaction (CO2RR) uses renewable power for the conversion of CO2 into valuable industrial materials and stores excess renewable electric energy as chemical energy in fuels.19 Therefore, CO2RR is important technology to realize carbon recycling and renewable energy storage. Consequently, the design of high-efficiency electrocatalysts is crucial but challenging, which inhibit the actual use of conversion reactions.3,20–24 Nevertheless, in these electrochemical conversion applications, the slow electrode reaction and low gas diffusion have become increasingly prominent. Noble metals (such as Ag and Au) and their derivatives are considered to be the key catalysts for the electroreduction of CO2.25–29 However, their high cost and low abundance hinder the commercial applications of noble metal catalysts. Consequently, researchers have devoted their efforts to finding alternatives to noble metal catalysts for electrocatalysis. In this case, non-noble metal–carbon catalysts with high activity are known as the superior choice and receiving increasing attention.30Scheme 1 schematically illustrates the first CO2RR catalyst and some representative work.
O bonds, which have a dissociation energy of 750 kJ mol−1 (∼9 × 104 K). The reduction of CO2 to CO was achieved at 1100 K using CeO2, ZrO2, LaFeO3, and lanthanum-based perovskites as catalysts.15–18 Alternatively, the electrochemical conversion of CO2 has several advantages over thermal reduction, including mild reaction conditions, controllable reaction rates and product selectivity through the applied potential, and wide scalability due to modular electrolyzer design. However, the electroreduction of CO2 reaction (CO2RR) uses renewable power for the conversion of CO2 into valuable industrial materials and stores excess renewable electric energy as chemical energy in fuels.19 Therefore, CO2RR is important technology to realize carbon recycling and renewable energy storage. Consequently, the design of high-efficiency electrocatalysts is crucial but challenging, which inhibit the actual use of conversion reactions.3,20–24 Nevertheless, in these electrochemical conversion applications, the slow electrode reaction and low gas diffusion have become increasingly prominent. Noble metals (such as Ag and Au) and their derivatives are considered to be the key catalysts for the electroreduction of CO2.25–29 However, their high cost and low abundance hinder the commercial applications of noble metal catalysts. Consequently, researchers have devoted their efforts to finding alternatives to noble metal catalysts for electrocatalysis. In this case, non-noble metal–carbon catalysts with high activity are known as the superior choice and receiving increasing attention.30Scheme 1 schematically illustrates the first CO2RR catalyst and some representative work.
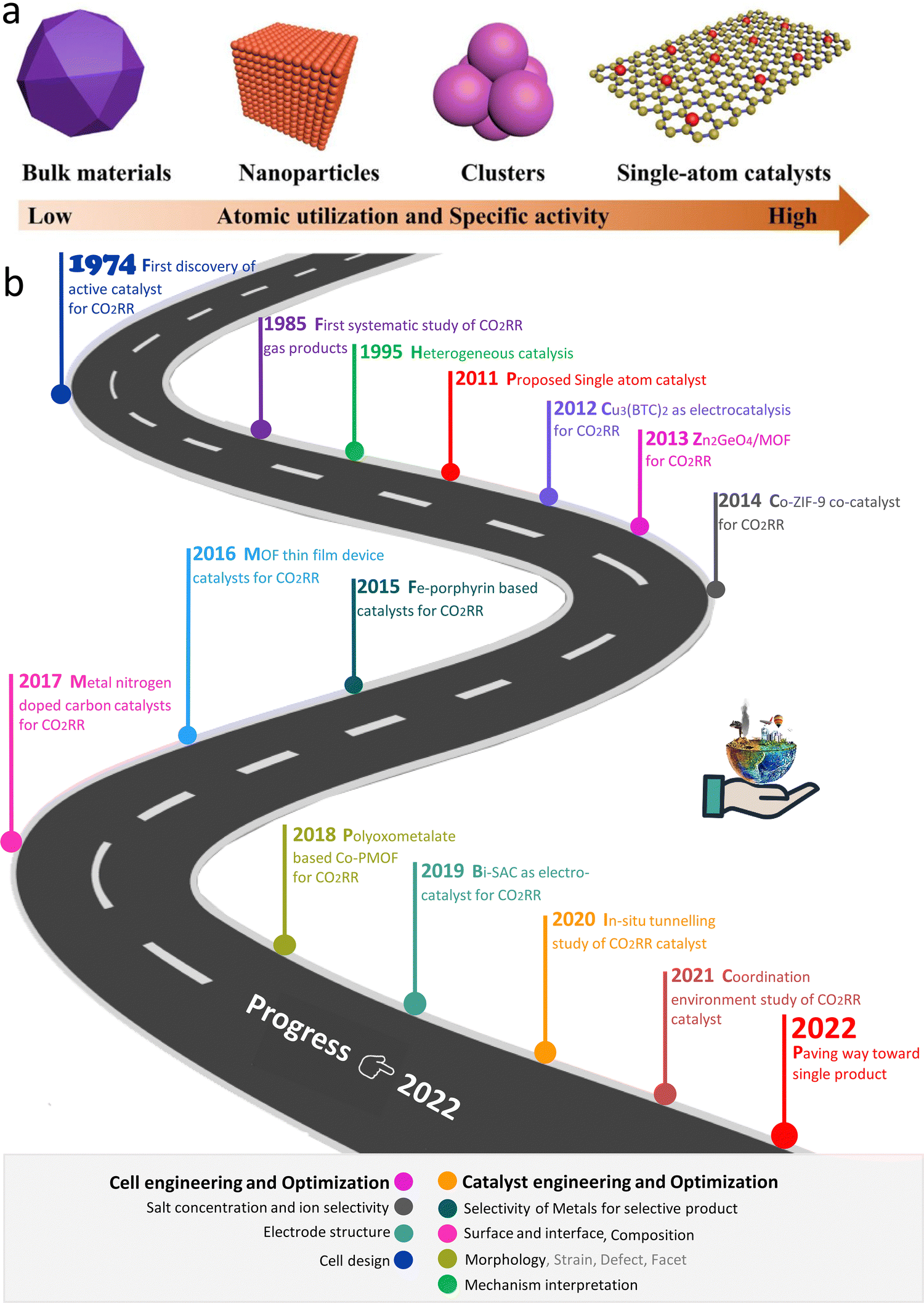 | ||
| Scheme 1 (a) Illustration of size-dependent catalytic activity of materials. (b) Systematic depiction of some historic progress and works on the CO2RR. | ||
Carbon materials with significant porosity have become the main catalysts to replace precious metals due to their outstanding physiochemical stability, high porosity with variable pore size, high surface area, and feasible synthesis method.31–34 Metal–organic frameworks (MOFs) are porous materials with well-defined crystallinity constructed of organic ligands and metal centers.35 MOF-confined carbon materials exhibit several advantages, as follows: (1) significantly porous, large surface area and variable pore size, which are beneficial for the enrichment and diffusion of gas molecules and conducive to mass transport.36 (2) The components (metal nodes and linkers) of MOFs can catalyse important reactions by creating charge transfer interactions with active sites through π bond coordination, thus chemically improving the microenvironment.37–40 (3) Compared with ordinary carbon catalysts, MOF-derived carbon shells can efficiently encapsulate the active center and prevent the leaching or agglomeration of particles, thus exhibiting excellent electrochemical stability during catalysis.41,42 (4) MOF materials possessing a transition metal are regularly used as active centers for electrocatalysis. The active centers in MOFs and MOF-derived catalysts are uniformly dispersed due their homogeneously arranged metal nodes and linkers.43 Therefore, MOF-derived carbon-containing materials are extensively used as electrocatalysts.44–46 The porous multifunctional structure of MOFs can be tuned via heteroatom-doping of carbon electrocatalysts obtained through calcination, such as N-, Fe-, Co-, and Ni-doping, whereas some studies have suggested the use of the as-prepared MOFs for CO2RR without further treatment. In addition to the above-mentioned valuable properties, the synthesis of MOF materials is cost-effective, and MOFs can be converted into single-atom catalysts, metal nanoparticle catalysts, metal oxides and carbon-based catalysts via single-step heat treatment.
Due to the diversity of MOF materials, many carbon materials having heteroatom doping, dispersed pore size and morphology can be obtained using MOF precursors. Usually, carbon materials with a large surface area can be obtained by proper heat treatment, and subsequently metal removal. Among the MOFs, Zn metal-based MOFs are the most frequently used materials because Zn can be reduced to metal Zn by the supporting carbon material at high temperatures. Also, the boiling point (908 °C) of metal Zn is low, and consequently Zn evaporates at about 900 °C via the simple calcination of Zn-based MOFs, resulting in the formation of carbon materials with high porosity and large surface area. In addition, MOFs and other metal ions can be used to prepare metallic single-atoms/clusters/nanoparticles with the confinement of porous carbon. The dispersion of metal species in the carbon matrix will be uniform and more active sites will be exposed for the electroactive metal species. The MOF-derived porous carbon base not only provides a conductive path for electron transfer, but also avoids the agglomeration of the metal species. Generally, the type of metal species (single atoms/clusters/nanoparticles) has an impact on the electronic structure of the catalytic material. Firstly, the type of material to be synthesized can be adjusted by regulating the proportion of metal source in the raw material.47 Secondly, the type of metal states finally synthesized can also be tuned by controlling the temperature during carbonization.48 However, at present, it is challenging to regulate the synthesis of specific types of materials, and thus the precise synthesis of metal types will be a fundamental strategy in the future. Recent research showed that MOF-derived carbon-based electrocatalysts under confinement engineering exhibit surprising results when used as electrocatalysts, as presented below. Firstly, the uniform distribution of metal centers in the carrier can be achieved because of the regular cavities/channels of MOFs, and also aggregation does not occur easily.
Specifically, spatial confinement can be used to stabilize and disperse individual atoms or nanoparticles to avoid agglomeration and advance the performance of the catalyst.22,49–51 Secondly, by coordinating with the organic ligand in the MOF, the metal center can be better stabilized on the support, thus stabilizing the long-term cyclic performance of the catalyst. In addition, the confinement engineering in materials can subtly change the microelectronic properties of the active sites and nearby microenvironment, enhancing the performance of the catalyst. To date, a relevant summary and review on MOF-derived and confinement-engineering-based electrocatalysis and the molecular-level interaction between the confinement engineering and reaction intermediates are limited. Because of the fast improvement in research in this direction and the continuous breakthroughs in catalyst performance (Scheme 2), herein, we summarize the key aspects and latest progress of new functional materials based on MOFs for CO2RR in the last three years. In combination with the research trend, the scope of the discussion is expanded, and three types of materials, i.e., active MOFs, MOF composites and MOF derivatives, are discussed, focusing on the analysis of their physiochemical properties. It can be seen from the comparison of these materials that their metal site properties, ligand environment, morphology and size of the particles are common concerns in the design of catalysts. Finally, the problems and future development strategies of MOF and MOF-derived confined materials in CO2 reduction are analyzed and prospected.
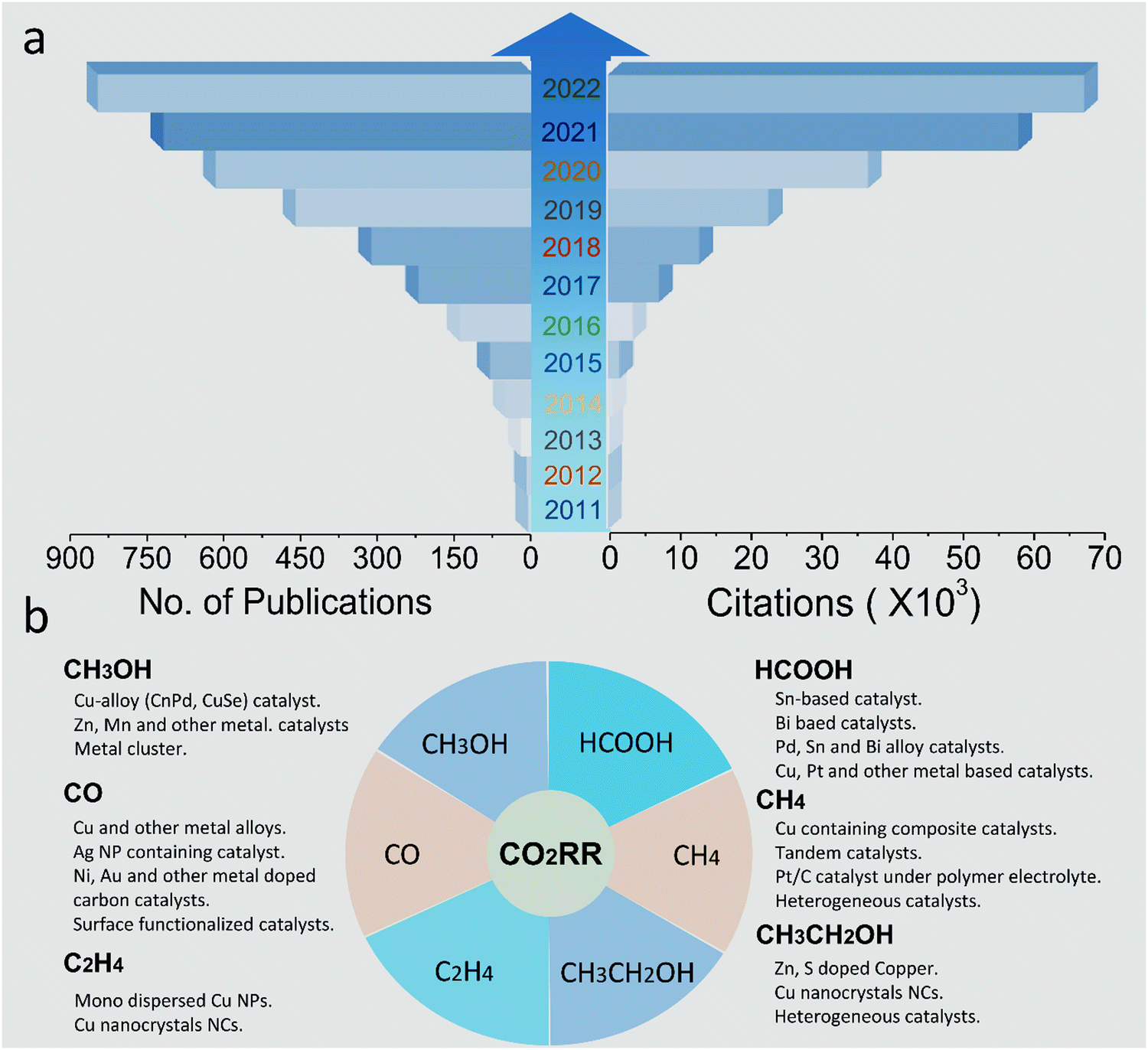 | ||
| Scheme 2 (a) Trend of publications and citations of electroreduction of carbon dioxide, with data collected from Web of Science. (b) Systematic illustration of metal-based CO2RR catalysts and their corresponding products. | ||
2. Structural engineering to regulate catalytic performance
Carbon-based materials derived from MOFs exhibit a significant confinement effect when applied as electrocatalysts because of their ordered pore size, huge surface area, high porosity, and strong chemical composition adjustability, as follows: (1) different metal atoms and linkers have an important synergistic effect with restricted active centers, modifying the microelectronic structure of the active centers, and thus improving the catalytic performance. (2) The spatial distribution and specific crystal structure of the metal atoms and linkers offer an ideal scenario for elucidating the possible catalytic mechanism. (3) The active centers (including single atoms, clusters and nanoparticles) are confined in the cavity/channel of MOFs to prevent agglomeration, which helps to improve the catalytic stability. (4) The porous structure of MOFs has significant potential to regulate the catalytic stability by adjusting the reaction rate. To regulate the electronic structure and maintain the stability of the active sites in carbon compounds derived from MOFs, the confinement effect is a key factor. To increase the rate of adsorption and desorption of the reaction intermediates, to lower the energy barrier in the catalysis process, speed up the reaction kinetics and enhance the catalytic activity, it is useful to regulate the microelectronic structure and stability of the active center.52–54 Generally, the improvement in the catalytic performance by altering the electronic environment by coordinating the metal active center with a metal node or ligand is called the chemical coordination confinement effect. Given that coordination structures mostly exist in monatomic catalysts, the confinement effect mostly applies to monatomic catalysts. The effect of improving stability is called the spatial confinement effect, which mostly applies to metal-nanoparticle catalysts. Here, we explore both spatial confinement and chemical coordination compared to the confinement effect of carbon-based materials derived from MOFs in electrocatalytic applications.2.1 Effect of chemical coordination confinement
The MOF precursor contains metal atoms and linkers, which are advantageous for the confinement of single-metal atoms in catalysts. To realize the dispersion of metal atoms, the desired metal atom can to some extent replace the original metal nodes in MOFs. The MOF precursor can also be used to stabilize the metal atoms. A certain degree of interactions is present between the confined metal atoms and the ligands in the MOF precursor, which stabilize the metal active centers.55 Furthermore, besides the confinement of metal atoms in metal nodes, other coordination structures can be assembled to immobilize metal atoms. In summary, the use of MOFs to confine a single-metal atoms is a simple and effective method.Due to the presence of defects in the coordination sites in linkers or MOFs, MOFs can be used to design single-metal atoms for many reactions. Chen et al.56 prepared a catalyst based on the nitrogen-rich tricyclic quinazoline (TQ), multi-catechol ligands and transition metal ions, which coordinated to form a two-dimensional (2D) porous sheet similar to graphene and denoted as M3(HHTQ)2. The M3(HHTQ)2 catalyst can be considered an SA catalyst, in which the Ni or Cu sites are regularly distributed in the lattice (Fig. 1a–h). This study revealed the strong dependence of the catalytic performance on the metal sites and N-rich linkers, indicating that the chemical confinement effect in the MOF coordinates with the organic ligands, having a noteworthy effect on the catalyst performance. Because the porphyrin unit has a clear planar symmetric anchor site, it can successfully anchor various noble metal atoms. For example, Li et al.57 effectively designed a Zr porphyrin-based MOF using a viable coordination approach, displaying SA catalytic sites and exceptionally high catalytic performance (Fig. 1i and j).
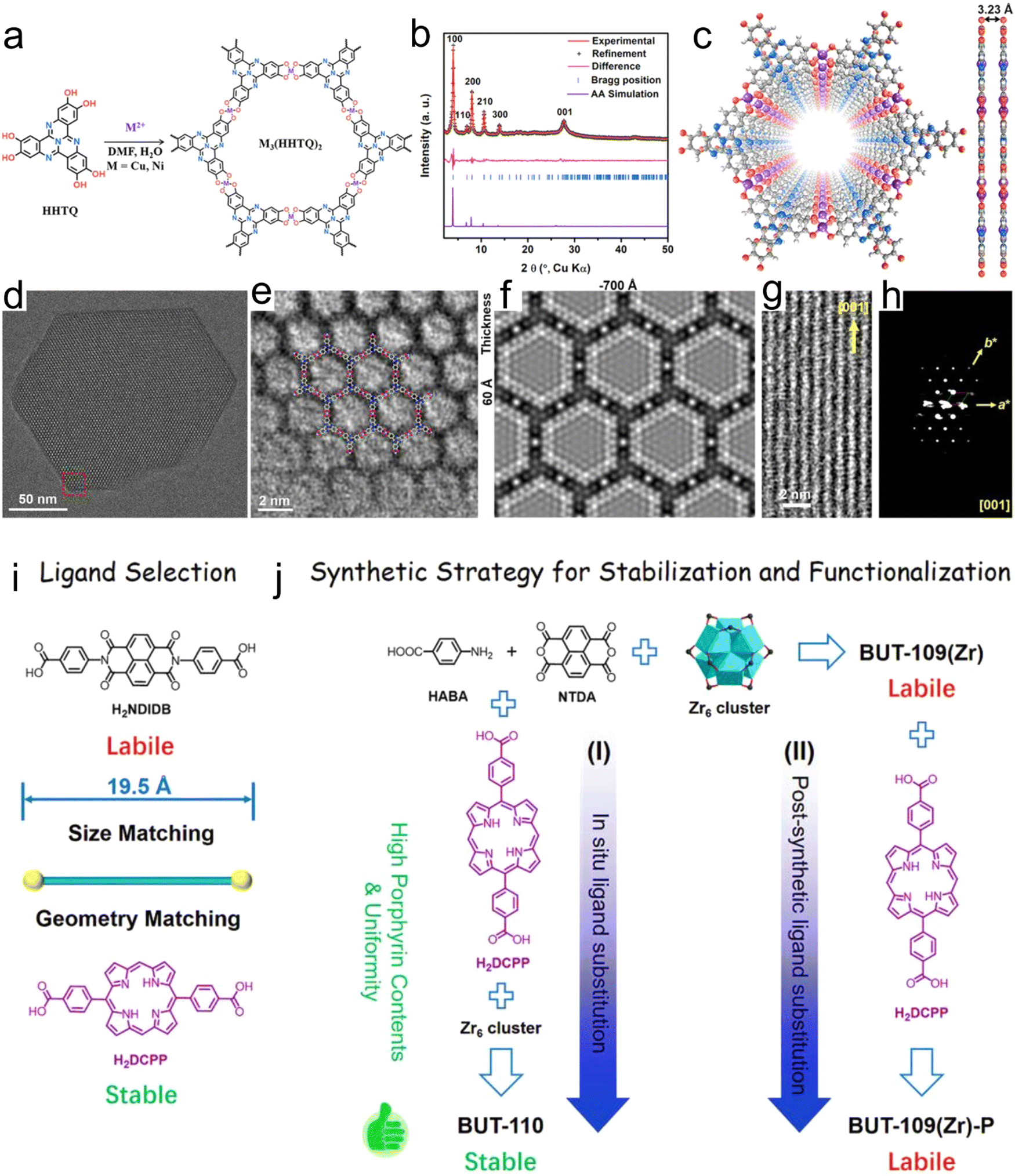 | ||
| Fig. 1 (a) Synthesis of M3(HHTQ)2 (M = Cu and Ni). (b) Experimental and Pawley-refined PXRD patterns of Cu3(HHTQ)2. (c) Structural model of Cu3(HHTQ)2. (d) TEM image of Cu3(HHTQ)2 taken along the c axis. (e) Magnified view of HRTEM image for Cu3(HHTQ)2 taken along the c axis, showing a hexagonal pore and ligand termination, overlaid with the structure model. (f) Simulated TEM image along the [001] direction. (g) HRTEM image along the [120] direction. (h) Projection view of 3D EDT data of Cu3(HHTQ)2 along the [001] direction. Reproduced with permission from ref. 56 Copyright 2021, Wiley. (i) Ligand selection and (j) synthetic strategies of BUT-110 through (I) in situ ligand substitution and (II) post-synthetic ligand substitution. Reproduced with permission from ref. 57 Copyright 2021, Wiley. | ||
In brief, the components of MOFs, i.e., their metal atoms and linkers, can confine single-metal atom catalysts and enhance the stability of the active centers. Consequently, the materials obtained by confinement in MOFs can be used as catalysts. Qin et al.58 reported a broad approach for the large-scale preparation of CoFe2O4/C nanocubes from MOFs through a combined exchange coordination and pyrolysis method (Fig. 2a). The strong restriction by the metal and organic MOF realizes the reconstruction of the crystal phase and composition, but not by simply introducing the metal oxide supported by Co2+. Compared with Co3O4Fe2O3/C, the MOF-derived CoFe2O4/C nanocubes possessed a higher surface area and better surface chemical properties, showing excellent catalytic activity and competitive water resistance.
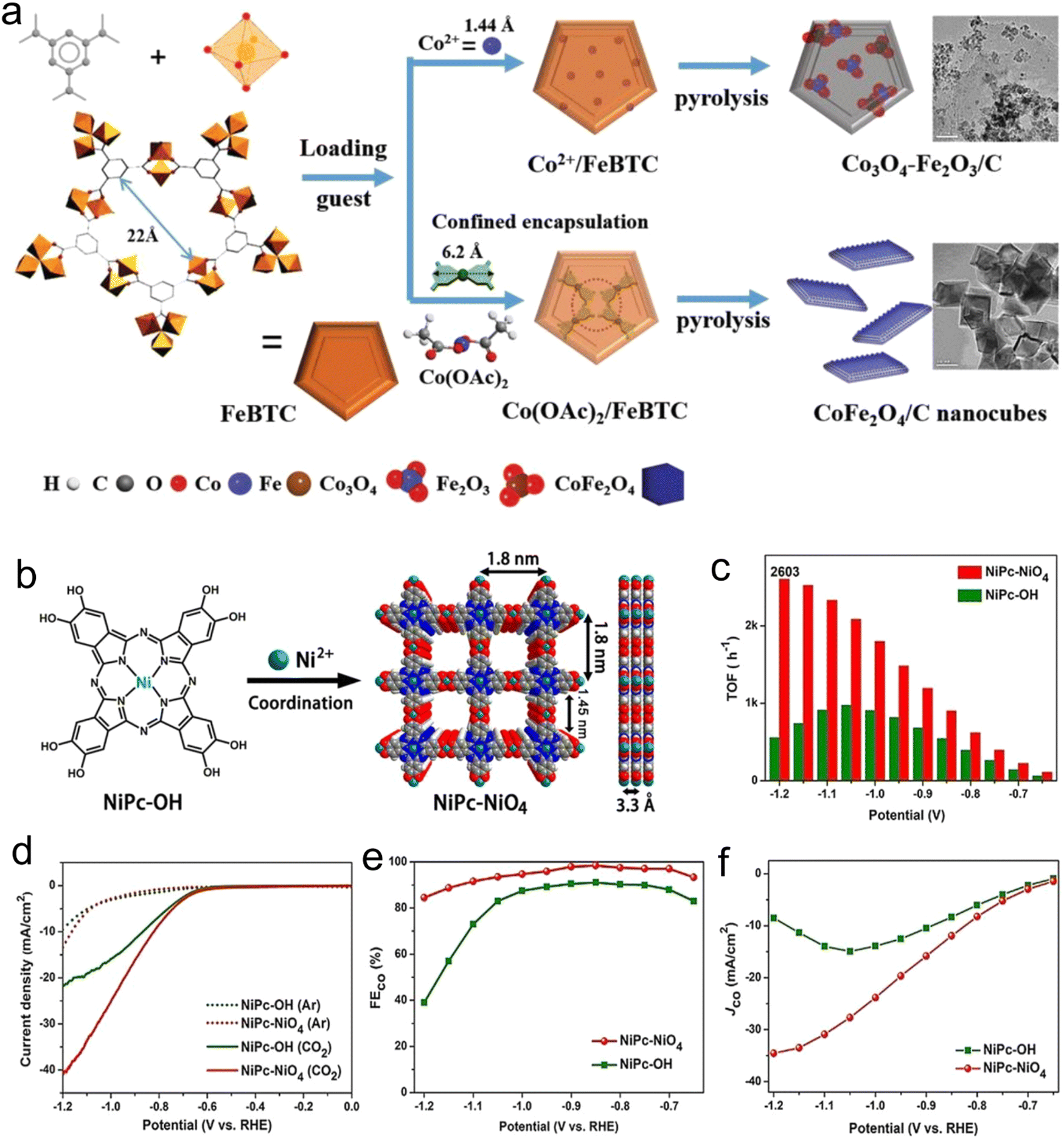 | ||
| Fig. 2 (a) Illustration of assembly MOF-derived bimetallic spinel oxide CoFe2O4 nanocubes through the combination of exchange–coordination and pyrolysis. Reproduced with permission from ref. 58 Copyright 2020, Wiley. (b) Synthesis. (c) Turnover frequency. (d) LSV curves in CO2. (e) FEs for CO. (f) CO partial current density of NiPc–NiO4. Reproduced with permission from ref. 59 Copyright 2021, Wiley-VCH. | ||
Also, MOF-derived catalysts are commonly prepared via the domain-confining effect. Cao et al.59 designed conductive 2D-phthalocyanine-based (NiPc–NiO4) MOF nanosheets, which were used as an electrocatalyst for CO2RR, efficiency producing CO because of its planar full π-d conjugation. The prepared NiPc–NiO4 nanosheets exhibited high selectivity (98.4%) and a CO partial current density of about 34.5 mA cm−2 (Fig. 2b–f). Single-metal atoms can be stabilized and dispersed through confinement via chemical coordination, and accordingly they can increase the stability and activity of the catalysts. However, to date, scarce research has been reported in the use of coordination confinement to stabilize nanoparticles. Consequently, designing new strategies for the synthesis and confining extra active centers is one of the main challenges in the development of efficient MOF-derived catalysts in the future.
2.2 Spatial confinement effect
MOFs are a new class of porous materials, which are appealing in many research fields because of their high specific surface area, uniform pore size, and exclusive tailorability and design. The presence of uniform porous structures allows MOFs to spatially disperse and confine metal active centers via appropriate confinement approaches. Xu et al.60 reported the use of covalent organic framework materials (COFs) as shells and MOF as the yolk shell structure of the inner yolk to construct yolk–shell COF@MOF (YS-COF@MOF) via a template-free solvothermal method. The comparative experiments showed that the formation of the cavity between the core and shell under the catalysis of pyrrolidine is due to the transformation from amorphous to crystalline and the simultaneous contraction of the shell. YS-COF@MOF combined the inherent structure adjustability and function of the COF and MOF and exhibited the space-limiting effect of MOFs on other materials (Fig. 3a–e). Wang et al.61 synthesized MIL-101 via the hydrothermal method, which possesed excellent thermal stability and highly ordered pore structure, and subsequently Au particles were immobilized and the content of Au was adjusted within a certain range. Based on the confining effect of MIL-101, the size of the Au nanoparticles could be controlled to less than 3 nm. The Au particles with this small size could provide good catalytic activity. Meanwhile, the pore confinement effect of MIF-101 ensured the uniform size and distribution of the nanoparticles.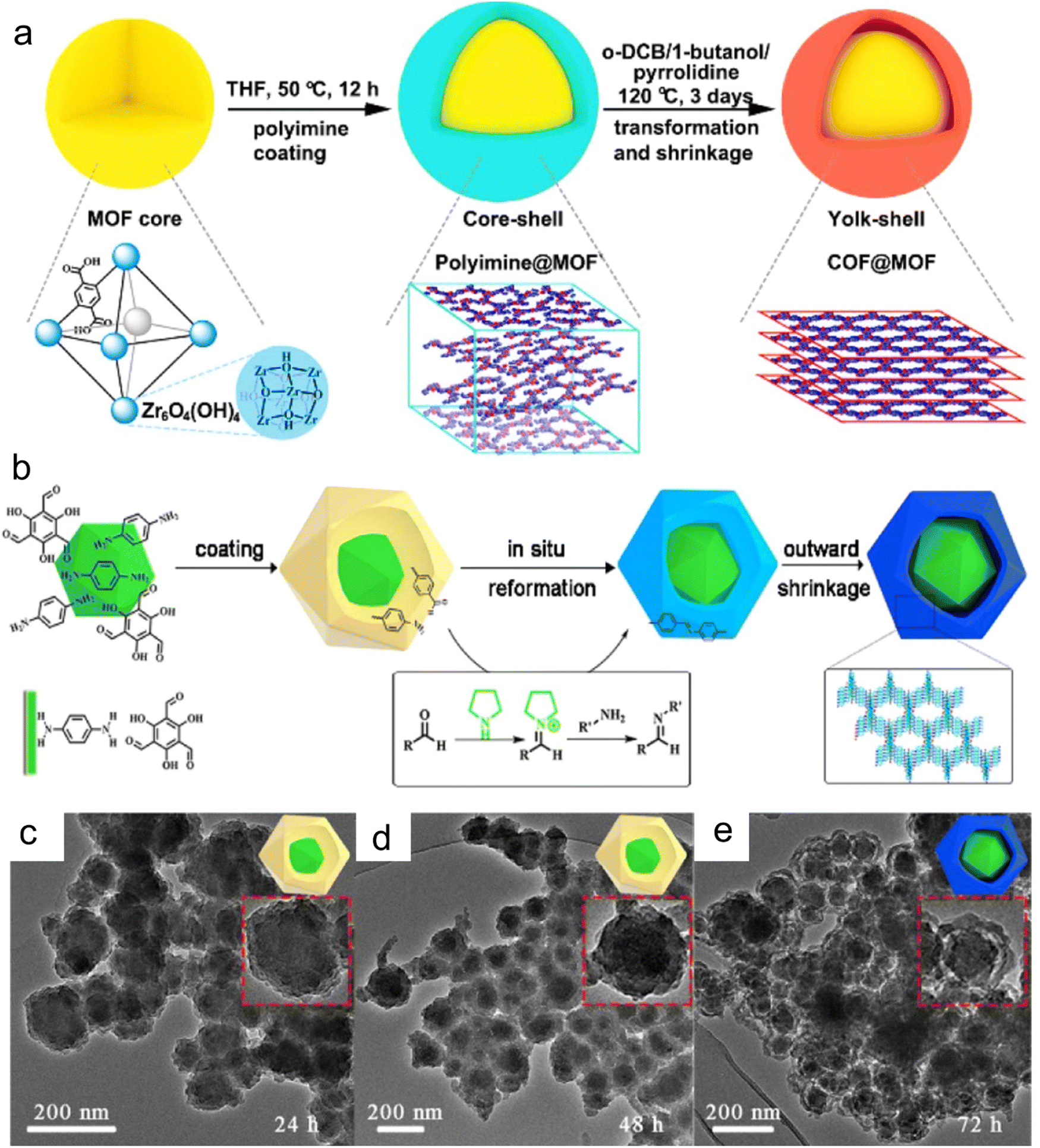 | ||
| Fig. 3 (a) Synthesis of yolk shell COF@MOF nanocomposites and the in situ transformation of the COF shell. (b) Proposed mechanism of the formation of yolk–shell-structured COF@MOF. (c–e) Time-dependent experiments of YS-TpPa@UiO-66-(COOH)2. Reproduced with permission from ref. 60 Copyright 2021, ACS. | ||
Kim et al.62 synthesized a bimetallic nanoparticle catalyst encapsulated in a conductive MOF (cMOF, Fig. 4a–e), in which the localization was guided by the site-specific nucleation induced by dipole interactions. The heterogeneous metal precursor was bound in the pores of the 2D cMOF through dipole interaction, and its subsequent reduction produced ultra-small and well-isolated PtRu-NPs with a size of ≈1.54 nm encapsulated in pores. Due to the bimetallic synergism of PtRu-NPs and the highly porous and large surface area of cMOFs, the PtRu-NPs-modified cMOFs exhibited a significantly enhanced NO2 sensing performance. Usman et al.63 demonstrated the confinement effect of a porous metal organic framework (HKUST-1) on the surface and wall of nanochannels by orbital etching in polyethylene terephthalate (np-PET) films using liquid phase epitaxy (LPE) (Fig. 4f). The composite film (HKUST-1/np-PET) showed the defect-free growth of the MOF, strong adhesion of the MOF to the carrier and high flexibility. The MOF has a preferred growth direction, which can capture CO2 in natural gas and olefins. Experimentally and theoretically (DFT), it was confirmed that the confined diffusion of the gas only occurs on np-PET substrates confined by nanoporous MOFs. These findings provide a new perception for the growth of other porous MOFs for artificially prepared nanochannels and prove that the MOF structure has a limiting effect on the gas reactants in energy conversion.
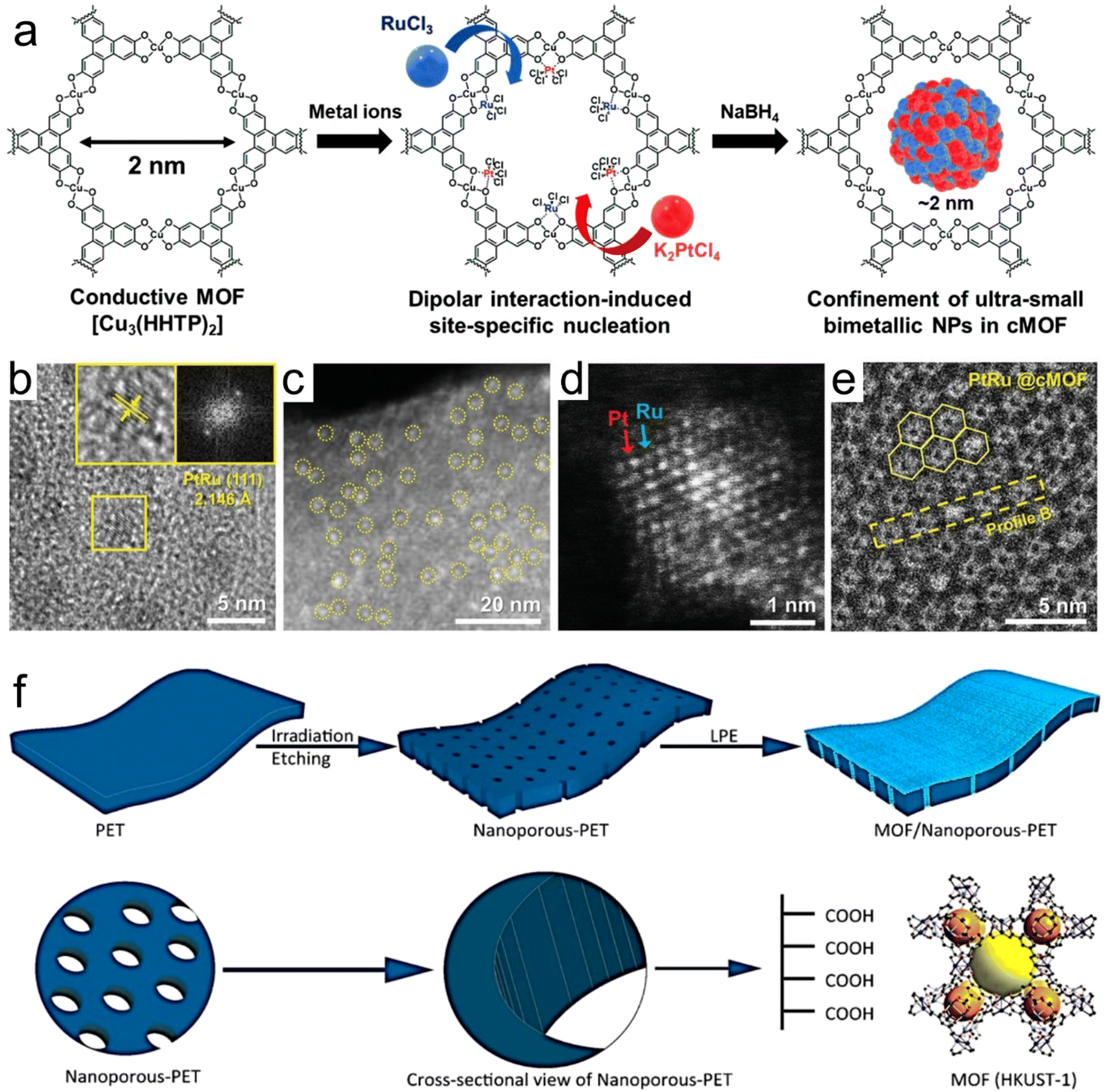 | ||
| Fig. 4 (a) Synthesis of PtRu@cMOF. (b) HR-TEM image of PtRu@cMOF. The inset shows magnified images, exhibiting the inter planar spacing of the PtRu(111) plane and corresponding fast Fourier transformation pattern. (c) HAADF-STEM image of PtRu@cMOF. (d) Z-contrast image. (e) Cryo-EM images of PtRu@cMOF. Reproduced with permission from ref. 62 Copyright 2021, Wiley-VCH. (f) Formation of nanochannels in the polymer membrane and subsequent confinement of the porous HKUST-1 metal–organic framework. Reproduced with permission from ref. 63 Copyright 2020, ACS. | ||
3. Electrocatalysis carbon dioxide reduction (CO2RR)
It is of great significance to effectively convert carbon dioxide into liquid fuels and high-value-added chemicals to alleviate global warming and the energy crisis at present, where the resource utilization technologies of CO2 include catalytic hydrogenation, photochemical reduction, photocatalytic reduction and electrochemical reduction.64 Among them, electrochemical technology has attracted wide attention because of its mild reaction conditions, which can effectively use the electric energy generated by renewable energy, for example, wind energy, solar energy and tidal energy, to convert CO2 into a variety of carbon-containing compounds, and its high catalytic efficiency. According to the number of electrons transferred, the CO2RR is generally divided into 2e−, 4e−, 6e−, 8e− or 12e− electron reactions, and the thermodynamic potential energy of several reactions that may occur is different, resulting in the formation of mixed products of several carbonaceous compounds (Table 1 and Fig. 5).65 At present, there are many challenges to realize the greatly effective CO2RR, including the competing HER, which leads to a reduction in the product selectivity; the stable molecular structure of CO2, and the high overpotential of CO2 generated by electrons. In recent years, researchers have developed new catalyst structures to reduce the overpotential and inhibit the HER side reaction to improve the reactivity and selectivity for the CO2RR.66–68 In this case, various nanocatalysts such as gold (Au), silver (Ag) and copper (Cu) show excellent catalytic performances.69 However, these traditional electrocatalysts exhibit several disadvantages, as follows: (1) they exhibit high overpotentials and slow kinetics for the multielectron CO2RR. (2) It is still difficult to control the reaction path to generate carbon-based products with high selectivity. (3) The CO2RR rate is mainly determined by the concentration of reactants, which limits the use of catalysts in a low-concentration CO2 environment.70 Therefore, the research on the application of MOF-based materials in the CO2RR has developed rapidly, which can be divided into three directions, including the design of active MOF materials, the formation of composites of MOFs and other catalytic materials, and the development of MOF-based derivatives.| Reduction reaction | Product | R. potential (V vs. SHE) |
|---|---|---|
| 2H+(g) + 2e− → H2(g) | Hydrogen | 0 |
| CO2(g) + e− → CO2˙− | −1.900 | |
| CO2(g) + 2H+(aq) + 2e− → CO(g) + H2O(l) | Carbon monoxide | −0.531 |
| CO2(g) + H2O(l) + 2e− → CO(g) + 2OH−(aq) | Carbon monoxide | −1.347 |
| CO2(g) + 2H+(aq) + 2e− → HCOOH(l) | Formic acid | −0.610 |
| CO2(g) + H2O(l) + 2e− → HCOO−(aq) + 2OH−(aq) | Formate | −1.491 |
| CO2(g) + 4H+(aq) + 2e− → HCHO(l) + H2O(l) | Formaldehyde | −0.480 |
| CO2(g) + 3H2O(l) + 4e− → HCHO(l) + 4OH−(aq) | Formaldehyde | −1.311 |
| CO2(g) + 6H+(aq) + 6e− → CH3OH(l) + H2O(l) | Methanol | −0.380 |
| CO2(g) + 5H2O (l) + 6e− → CH3OH(l) + 6OH−(aq) | Methanol | −1.225 |
| CO2(g) + 8H+(aq) + 8e− → CH4(g) + 2H2O(l) | Methane | −0.240 |
| CO2(g) + 6H2O(l) + 8e− → CH4 (g) + 8OH−(aq) | Methane | −1.072 |
| 2CO2(g) + 12H+(aq) + 12e− → C2H4(g) + 4H2O(l) | Ethene | −0.349 |
| 2CO2(g) + 8H2O(l) + 12e− → C2H4(g) + 12OH−(aq) | Ethene | −1.177 |
| 2CO2(g) + 12H+(aq) + 12e− → C2H5OH(l) + 3H2O(l) | Ethanol | −0.329 |
| 2CO2(g) + 9H2O(l) + 12e− → C2H5OH(l) + 12OH−(aq) | Ethanol | −1.157 |
| 3CO2(g) + 18H+(aq) + 18e− → C3H7OH(l) + 5H2O(l) | Propanol | −0.320 |
| 2CO2(g) + 2H+(aq) + 2e− → H2C2O4(l) | Oxalic acid | −0.913 |
| 2CO2(g) + 2e− → C2O42−(aq) | Oxalate | −1.003 |
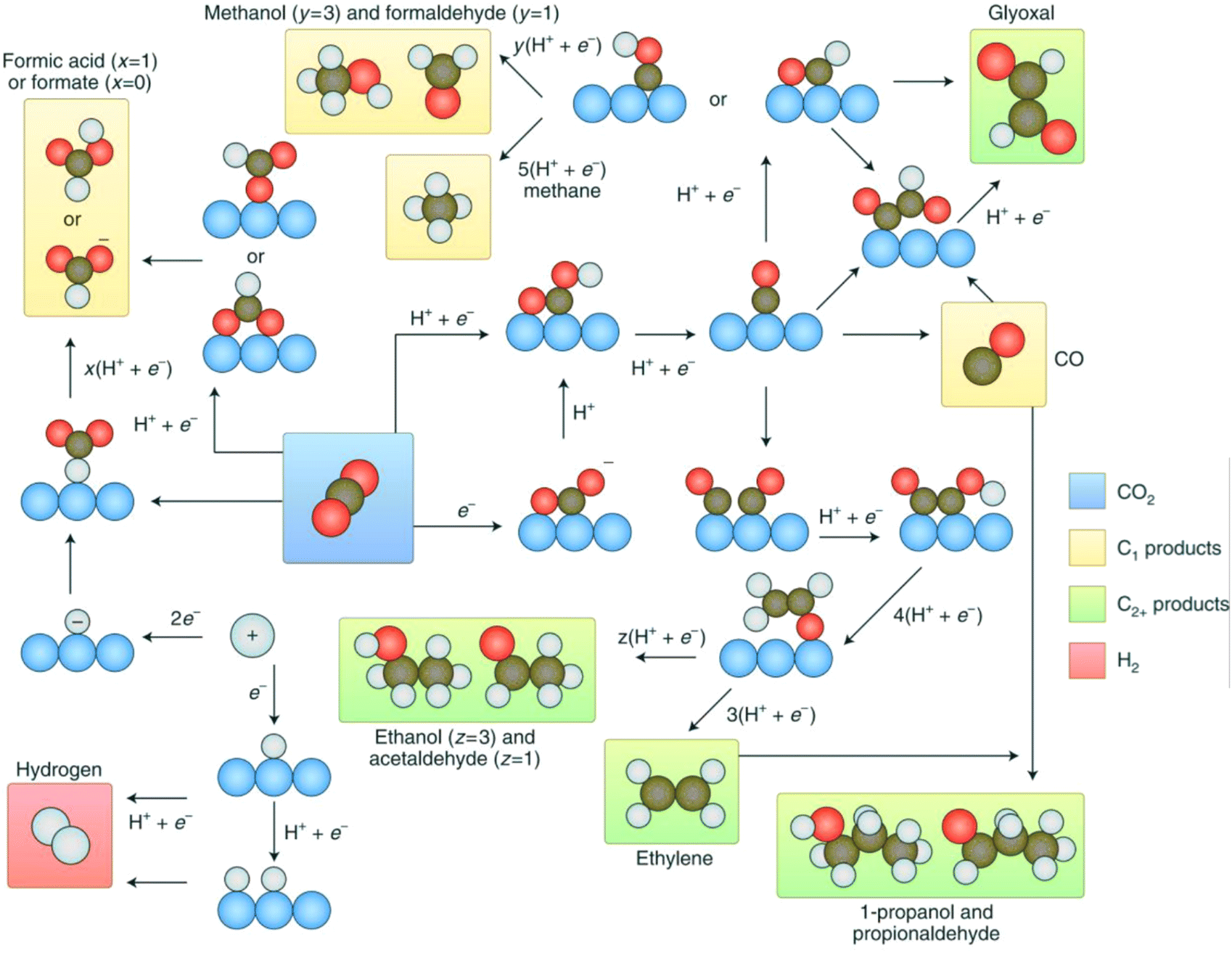 | ||
| Fig. 5 Reaction pathways of CO2 to different products. Reproduced with permission from ref. 65 Copyright 2019, Springer Nature. | ||
4. Mechanism, kinetics and thermodynamics of CO2RR
4.1 Mechanism and kinetics
The electrochemical conversion pathway of CO2 is thermodynamically favourable, where the energy barrier/overpotential decides the practical selectivity for the CO2RR products. The selectivity, current density and long-time stable working hours are the critical parameters for commercial viability.72 The mechanistic study of the CO2RR includes two steps including the formation of intermediate products and formation of the final products. The mechanism of the different products (C1, C2 and C2+) is different. Generally, the first step in the formation of mono carbon products (HCOOH and CO) is critical, where carbon dioxide can be activated by transferring of one electron from the catalyst surface (*CO2˙−).73–76 For the formation of the *COOH intermediate, proton-coupled electron transfer (PCET) occurs with *CO2˙−. Also, after the formation of the *COOH intermediate, PCET occurs again to convert this intermediate to CO. The *OCHO intermediate is generated by the interaction between *CO2˙− and the O atom, and then converted to formate via the electron-transfer protonation of the O atom.77 In the suggested mechanism for the production of methanol, methane, and formaldehyde by the CO2RR, theoretical and experimental studies illustrate that the first intermediate formed is *CO.73,78 Subsequently, the *HCO, *H2CO and * H3CO intermediates are generated by the hydrogenation of *CO and transformed into HCHO, CH4 and CH3OH, respectively. However, there are two different views supporting the transformation of the *CO intermediate to CH4, as follows: (1) it is suggested that *CO is reduced to *C through the *COH intermediate, and furthermore this reduction is expanded to *CH, *CH2 and *CH3, and lastly CH4 is generated.79,80 (2) The most favourable route according to experiments and theoretical calculations is *CO → *CHO → *CH → *CH2 → *CH3 → *+CH4.81In addition, the production of multi-carbon products generally follows the *CO intermediate route and the final product decided after the reduction of *CO depends on the experimental conditions.82,83 For example, after *CH is protonated to *CH3, ethane is generated by the PCET process via the *CH intermediate. The *CH3 intermediate produces C2H6 as the final product after dimerization. The addition of CO to *CH2 generates CH3COO−. Also, *CO–CO generates the *CHCH2O intermediate through successive proton and electron transfer, which is the rate-determining step (RDS) to produce ethylene and ethanol.83,84
In some of CO2RR experiments, bulk metals have been used for catalysis. According to the results, there is a theoretical trend,85–91 and bulk metal catalysts are divided into the following categories: (1) formic acid production, for example, Hg, Tl, Cd, Sn, In, Pb, and Bi. (2) Production of CO (Au, Ag, Ga, Zn and Pd). (3) H2-Producing metals, i.e., Fe, Ni, Ti and Pt. (4) Copper metal-based electrodes, which are known to produce multi-carbon products.92–94 According to studies, any electronic change or influence on the active centers can lead to changes in the selectivity and FE of the products. For example, Zn and Cu have the same electronic structures but their catalytic properties are quite different. Specifically, the Zn electrode displays 2e− reduction and produces CO upon CO2 reduction, while Cu generates multiple hydrocarbons.95,96 In some other reports, Zn-doped (Zn/Cu) electrodes generated different products, i.e., the production of alcohols.97,98 For example, a Cu4Zn catalyst produced ethanol with 29.1% FE.97 Similarly, a mixture of ZnO and CuO (with 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 wt%) produced CH3OH with an FE of 25.2%.99 However, Sn-doped Cu did not exhibit a good performance, which may be ascribed to the strong interactions of the intermediate (*CO) with the Cu surface.100,101 Consequently, the absorbance time of the intermediates on the catalyst surface is also an important factor to regulate the product selectivity in the CO2RR process, and it is suggested that the long-term interactions between the designed catalysts and intermediates may possibly generate multi-carbon (C2+) products.102–107 For example, recently, Song et al. revealed the transition state of the adsorbed intermediate of CO2 to realize selectivity for the formation of formic acid on the surface of an S-doped Cu catalyst during the CO2RR.107 They confirmed that S can stabilize the reduced Cu center.
:1 wt%) produced CH3OH with an FE of 25.2%.99 However, Sn-doped Cu did not exhibit a good performance, which may be ascribed to the strong interactions of the intermediate (*CO) with the Cu surface.100,101 Consequently, the absorbance time of the intermediates on the catalyst surface is also an important factor to regulate the product selectivity in the CO2RR process, and it is suggested that the long-term interactions between the designed catalysts and intermediates may possibly generate multi-carbon (C2+) products.102–107 For example, recently, Song et al. revealed the transition state of the adsorbed intermediate of CO2 to realize selectivity for the formation of formic acid on the surface of an S-doped Cu catalyst during the CO2RR.107 They confirmed that S can stabilize the reduced Cu center.
In recent years, it has also been suggested that the structural properties of catalytic materials can change the reaction kinetics, products, and selectivity. Accordingly, porous materials have received special consideration because of their special characteristics, for example, enlarged surface area ratio to active centers, adjustable pore size for reactant/product mass transfer, and controllable local pH value.104,108 He et al. applied the self-assemble strategy to synthesize gradient macro and mesoporous alloys (Cu/Zn). The catalysis outcomes displayed that the Cu5Zn8 alloy exhibited a superior performance in reducing carbon dioxide to ethanol with an FE of about 46% and excellent stability for 11 h.89 SnO2 and Au show almost the same catalytic performance for the CO2RR because they possess the same morphology.106,109 Nam et al. proposed the reaction kinetics for the CO2RR to multi-carbon products under different morphologies.102 They applied various copper-based catalytic materials with defined morphologies and revealed that the morphology also plays an active role in accelerating the coupling (C–C) and the stability can be controlled by the flow rate and pH.102,103 MOF materials generally have highly-tuneable large porosity and huge surface area, which are very important for CO2RR, and thus they have been extensively studied in recent years. For instance, a 2D Bi-MOF displayed an excellent performance for the CO2RR (FE for formic acid was 92%) compared with the reversible hydrogen electrode (RHE), with stability for 30 h. The specific mass ratio of formic acid was as high as 41.0 mA MgBi−1 and better than that of the commercially available Bi sheets and Bi2O3.110
4.2 Thermodynamics
CO2 bond cleavage requires an active catalyst for the conversion of CO2 to fuels and chemicals.113 The CO2 molecular orbital (MO) diagram, showing a fully interconnected and formed orbital, demonstrates that CO2 is a stable substance (Fig. 6a).111 In the first step to activate CO2 molecules, the transfer of electrons occurs (i.e., the reduction process), which have LUMO σ* to counter binding. Also, the HOMO takes the high electron density, which is localized as the lone pair on the oxygen plane, favouring the interaction with the electrophilic molecule. Accordingly, the bond length of C–O is enhanced, which is displayed as the first tilt in the linear molecule. The electrophilic and nucleophilic molecules can easily attack to the curved molecule through their charged local orbital boundaries. Nonetheless, the electronic reduction of CO2 to CO2− is an intricate pathway and requires high overpotentials (Fig. 6b and c).112 Because of the big barrier between the bent anion and linear-shape molecule, the subsequent step, specifically, PCET, is thermodynamically more favourable than the first step (Fig. 6d).65,114,115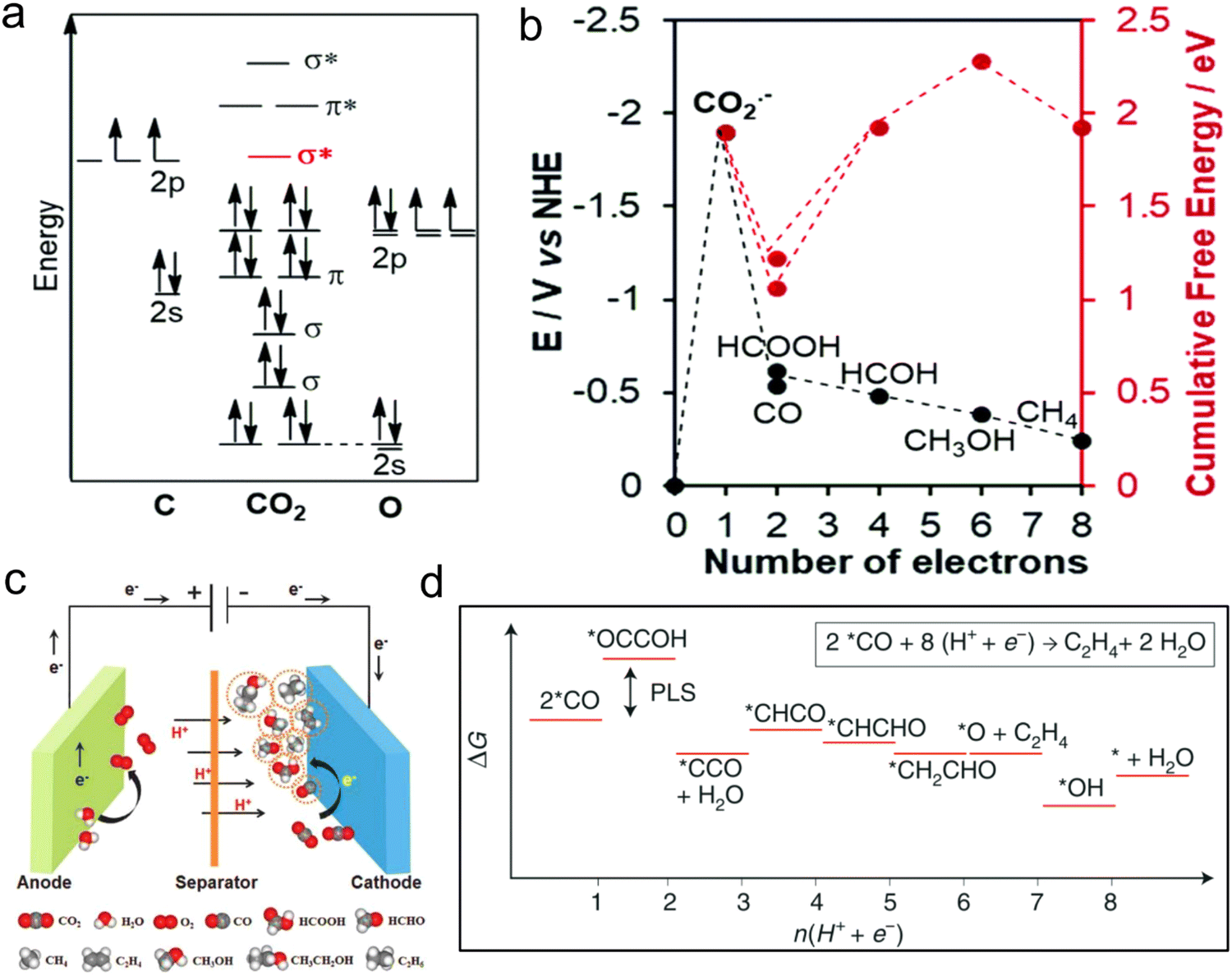 | ||
| Fig. 6 (a) CO2 molecular orbital (MO) diagram. (b) Thermodynamic of required activation energy to final product. Reproduced with permission from ref. 111 Copyright 2020, RSC. (c) Design of the cell for the conversion of CO2. Reproduced with permission from ref. 112 Copyright 2018, Wiley. (d) Free-energy diagram with favourable reaction path for CO2 to C2H4 on Cu surface. Reproduced with permission from ref. 65 Copyright 2019, Springer Nature. | ||
As designated in Fig. 6b and d, the transfer of 2, 4, 6, 8 or 12 electrons and protons during the CO2RR process leads to the formation of various products. The generation of different as well as specific products during the CO2RR process, for example, mono and multi-carbon products, mainly depends on the reaction conditions (electrolyte, overpotential, pH and design of the device) and nature of the catalyst (metal type and distribution of active centers). Many types of products are generated in the catalytic process, but one will be produced that is thermodynamically competitive with the others. Thus, the selectivity for the production of the target product is one of the important standards to evaluate the CO2RR performance.
5. MOFs for the CO2RR
The apparent performance of electrocatalysts is greatly affected by the electrode preparation process. However, the traditional electrode preparation methods such as drop coating usually result in low apparent activity. Alternatively, the in situ synthesis of MOFs on the electrode is expected to give full play to the intrinsic activity of the material. Zhu et al.116 reported the strategy of in situ electrosynthesis of a hollow copper-MOF and reduction to multi-stage Cu dendrite catalyst during the reaction. The preparation of thew Cu-MOF film only took 5 min. The Cu dendrite catalyst obtained by this strategy possessed a large surface area and open active sites, which are conducive to the CO2RR to HCOO− in ion–liquid electrolyte. The current density of the above-mentioned system was 102.1 mA cm−2 with an FE of 98.2%. Kang et al.117 used copper foam as the substrate and ligands with large steric hindrance to control the growth kinetics of an MOF, producing Cu-MOF with a large number of defects, i.e., (Cu2(L)) (H4l = 4,4′,4′′,4′′′-(1,4-phenylene)bis(pyridine-4,2,6-triacyl)) tetrabenzoic acid, which rapidly grew on the copper foam in acetonitrile ionic–liquid electrolyte to produce formate with an FE of 90%. The Cu2+ defect has strong binding ability to CO2 and mainly coordinates with the oxygen in CO2 in the form of O–Cu–O because the formation of Cu-MOF*OOCH is easier than Cu-MOF*COOH, leading to higher selectivity for formic acid. The high-density Cu2+ defects also generated formic acid at lower overpotentials, thus weakening other competitive reactions such as Cu2+ reduction and hydrogen generation.Kang et al.118 successfully deposited MFM-300 (In) on In foil within 10 min using an acetate ionic liquid as the electrolyte. The MOF prepared using this method had strong binding force with the electrode metal substrate, and the particles were evenly dispersed, which effectively enhanced the charge transfer efficiency in the reaction process. Compared with other MOFs and oxides, MFM-300 (In) is more stable and can maintain its structural integrity at a negative potential without being reduced to In. Besides, the FE of formate produced by the MFM-300 (In)/In electrode was as high as 99.1% due to the presence of defect sites and strong interactions with CO2. Compared with the MFM-300 (In)/carbon paper electrode prepared by the traditional thermal synthesis method, it could generate more ˙COOH radicals, thus promoting the efficient synthesis of formic acid.
The type and states of ligands also have a significant influence on the electrocatalytic activity of MO-based catalysts. Dou et al.119 reported an approach to improve the catalytic activity of MOFs for CO2 reduction by ligand doping. The electron donor 1,10-phenanthroline was doped in activated ZIF-8, promoting the charge transfer and endowing the sp2 hybrid atoms in the adjacent imidazole ligands with more electrons, making it easier to generate *COOH, and thus improving the catalytic activity and FE for CO production.
Conductive materials are beneficial for charge transfer and will show great advantages in electrocatalysis. Therefore, the development and application of cMOFs have become a research hotspot. Yi et al.59 prepared conductive phthalocyanine-based 2D nanosheets denoted as MOF-NiPc@NiO4 using nickel pyrocatechol (Fig. 2b), which efficiently converted CO2 to CO with an FE of 98.4% under a CO partial current density of 34.5 mA cm−2 (Fig. 2d). Compared with the NiO4 node, the CO2RR is more likely to occur on the nickel phthalocyanine center (NiPc) because the adsorption energy of CO2 on NiPc (0.23 eV) is higher than that of the NiO4 node (0.02 eV), which makes it easier to bind with NiPc, and the electron content of nickel in NiPc is greater than that in NiO4. When the CO2 molecule moves from NiPc to the NiO4 node, the LUMO shifts from −4.22 eV to −4.62 eV, which indicates that NiPc has better reducibility, and thus enables the catalyst to show higher selectivity and activity.
Majidi et al.122 constructed a 2D copper-based cMOF, i.e., copper tetrahydroxyquinone (Cu-THQ) nanosheet. In a mixed electrolyte (1 M (CH3)3N(Cl)CH2CH2OH and 1 M KOH), the CO2RR could be accomplished at a low overpotential of 16 mV, showing excellent catalytic activity. At an applied potential of −0.45 V, the current density was about 173 mA cm−2 and FECO was about 91%. In addition, the MOF can provide a good fixing platform for organic molecules with intrinsic electrocatalytic activity, and thus a large number of molecular catalysts can be fully contacted with the electrode, resulting in various advantages for heterogeneous catalysis. Hod et al.123 used electrophoresis technology to immobilize an Fe porphyrin molecular catalyst on the electrode surface on the Fe-MOF-525 platform. Simultaneously, the Fe porphyrin molecule was not the only catalytically active material, but also the ligand of the MOF acted as a bridge to connect the electrode and the outer catalytically active sites. Based on the porous platform provided by the MOF, the amount of effective electrochemically active centers on the catalyst surface increased to about 1015 cm2, which is nearly 3 times higher than the active site concentration obtained by direct monolayer-loaded Fe porphyrin molecules and nearly 1-fold higher than the reported maximum concentration of loaded molecular catalyst. In this system, the product of CO2RR is an equal mixture of CO and H2 with an FE of close to 100%. This work reflects the innovative application of MOFs due to their regular and ordered pore structures in the immobilization of molecular electrocatalysts. Furthermore, Table 2 presents a comparison of the state-of-the-art results for MOFs in the CO2RR.
| Material | Main product | Electrolyte | Faradaic efficiency [%] | Potential | Current density [mA cm−2] | Ref. |
|---|---|---|---|---|---|---|
| Bi MOF | HCOOH | 0.5 M KHCO3 | 95.5 | −0.86 V vs. RHE | −11.2 | 124 |
| ZIF-8 | CO | 0.5 M NaCl | 65.5 | −1.8 V vs. SCE | NA | 125 |
| ZIF-8 | CO | 0.25 M K2SO4 | 81 | −1.1 V vs. RHE | 6.9 | 126 |
| ZIF-108 | NA | −1.3 V vs. RHE | 12.8 | |||
| ZIF-A-LD | CO | 0.1 M KHCO3 | 90.57 | −1.1 V vs. RHE | NA | 127 |
| HKUST-1(Cu, Ru) | C2H5OH CH3OH | 0.5 M KHCO3 | 47.2 | NA | 20 | 128 |
| Cu@MOF | HCOOH | 0.1 M NaClO4 | 28 | −0.82 V vs. RHE | −1.2 | 129 |
| Cu2O@Cu-MOF | CH4 | 0.1 M KHCO3 | 63.2 | −1.71 V vs. RHE | 8.4 | 130 |
| Ag2O/layered ZIF | CO | 0.25 M K2SO4 | 80.6 | −1.2 V vs. RHE | 26.2 | 131 |
| RE-ndc-fcu-MOF @AuNMEs | CH4 | 0.1 M KHCO3 | 0.5 ± 0.1 | −0.5 V vs. RHE | −2.5 | 132 |
| Cu(bdc)·xH2O @AuNMEs | 1 ± 0.4 | −1.7 | ||||
| ZIF-8@AuNMEs | 0.9 ± 0.2 | −0.1 | ||||
| Al2(OH)2 TCPP@AuNMEs | C2H4 | 1.1 ± 0.4 | −3.1 | |||
| Co porphyrin MOF | CO | 0.5 M K2CO3 | 76 | −0.7 V vs. RHE | NA | 133 |
| Fe-MOF-525/FTO | CO + H2 | 1 M TBAPF6 | 100 | −1.3 V vs. NHE | 5.9 | 123 |
| Zn-BTC MOF/CP | CH4 | BmimBF4 | 80.1 ± 6.6 | −2.2 V vs. Ag/Ag+ | 3.1 ± 0.5 | 134 |
| BmimOTf | 85.4 ± 3.2 | 2.1 ± 0.3 | ||||
| BmimPF6 | 87.7 ± 5.1 | 1.6 ± 0.3 | ||||
| BmimClO4 | 88.3 ± 3.8 | 0.5 ± 0.2 | ||||
| Re MOF/FTO | CO | 0.1 M TBAH in CH3CN + 5% trifluoroethanol | 93 ± 5 | −1.6 V vs. NHE | 2.5 | 135 |
5.1 Role of central metal atoms
Usually, for the CO2RR to occur, it is assumed that a cathode potential is required, and most likely, the central metal atom acts as the active site for the CO2RR. Thus far, most of the used electrocatalysts in the CO2RR work as pre-catalytic converters. In the catalytic process, the central metal atom is oxidized or reduced, which is why it is difficult to distinguish the actual active site before the catalytic process.120 For example, in the case of transition metals, CuO-driven Cu has been extensively studied for the CO2RR because of its high selectivity. Correspondingly, in Cu-based MOFs, Kumar et al.136 detected different Cu(I)/Cu(II) and Cu(0)/Cu(I) as well as reversible redox reactions during cyclic voltammetry in 0.1 M KCl using a glassy carbon electrode coated with a Cu–BTC thin film. Chang et al.120 elaborated the transition phase of Cu-based electrodes using in situ XAS and explored their oxidation state together with local structure. The synthesis of Cu-based materials with Cu(0) is advantageous to produce multi-carbon products of the CO2RR rather than Cu(I), whereas the competitive oxidation reaction of Cu(0) decides the products and chemically active sites of CO2RR. Furthermore, in situ Raman was performed, confirming that the re-oxidation reaction determines the chemical state of Cu, as shown in Fig. 7a and b. In addition, Sargent et al.121 used in situ XAS to inspect the percentage of copper at different potentials (Fig. 7c and d). Additionally, they proposed a noteworthy relationship between the product distribution and the surface state of copper and noted the existence of copper-stabilized ethylene intermediates. Furthermore, Yaghi et al.133 explored the active oxidation states of other transition metals, e.g., they investigated the oxidation states of Co in a Co-MOF during the CO2RR using in situ techniques. This study displayed that most of the metal centers are available for redox reactions, where Co(II) is reduced to (I), and revealed that Co reduction is the first step in the reduction process. During the CO2RR process, the reduction of Co(II) to Co(0) is due to the adsorption of CO2 on the surface of the active center. However, this type of transition does not occur on MOF-derived metal active centers. Recently, Hu et al.137 reported the exceptional FE and low overpotential of an iron-based single-atomic catalyst (SAC) for CO. In addition, in situ XAS analysis indicated that the active Fe3+ species coordinated with the pyrrolic-N atom, which can maintain its +3 oxidation state during the electrocatalysis process. Furthermore, on MOF-74 impregnated with cobalt, nickel and zinc, the role of the metal center was also experimentally determined, where at −0.91 V vs. RHE, CO2 was reduced to 78.3% of H2 and 15.4% CO, which suggests that this heterogeneous catalyst can produce syngas efficiently.138 In addition, inorganic modification was performed to determine its effects on the selectivity and activity of Zn-MOF catalysts for the CO2RR. Wang et al.139 designed many zinc (ZIF-8)-based materials using different sources for Zn as reactants and studied the CO2RR process. The electrochemical findings showed that the ZIF-8 prepared using a ZnSO4 source exhibited a better catalytic performance for the CO2RR with a high FE of 65% and current density of 3 mA cm−2, giving new insight into the relationship between activity for the CO2RR and the zinc source, and it is appealing that the vital active centers are discrete zinc nodes. The boost in the activity and selectivity of Zn-MOF in the presence of SO42− ions is mainly ascribed to the interaction between the Zn node and SO42− ions, which can contribute to the exchange of the anion for charge balance on the oxidation of the Zn nodes. However, some reported works showed that the Zn metal sites of Zn MOFs are not the active sites for the catalysis process because of their fully filled of d-orbital.140–142 Bao et al.140 explored and confirmed the imidazole coordination with Zn in ZIF and that it serves as the catalytic center for the CO2RR using DFT calculations and in situ XAS. Therefore, the definite active center in MOFs is quite controversial, and thus further in-depth research is needed to answer this question. In this case, operando or in situ studies play an important role in the dynamic process of the electrocatalytic process, which can recognize the definite active centres and support the understanding of the reaction mechanism. However, with the help of powerful in situ technology, it is still necessary to develop next-generation catalysts.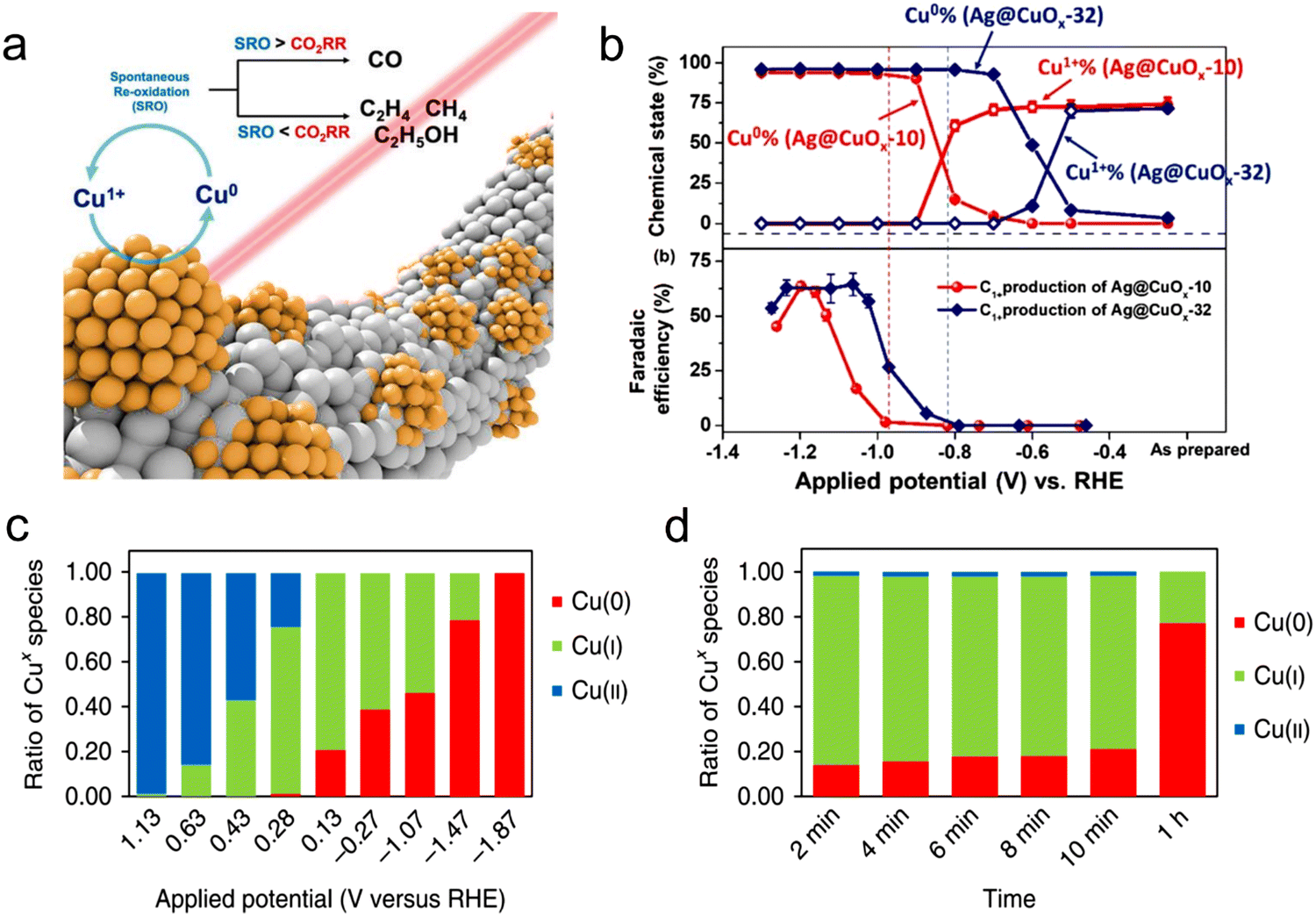 | ||
| Fig. 7 (a) Schematic process of Cu during the CO2RR. (b) FE and chemical state vs. applied potential. Reproduced with permission from ref. 120 Copyright 2019, ACS. (c) Oxidation state ratio of Cu vs. applied potential. (d) Oxidation state ratio vs. time. Reproduced with permission from ref. 121 Copyright 2018, Springer Nature. | ||
5.2 Role of organic linkers
The presence of ligands can change the catalytic performance of MOFs. For example, ligands with electron-donating ability can increase the catalytic activity of MOFs by inducing electrons to move from the active site to the anti-bonding orbital of CO2, thereby generating *COOH through proton coupling.119 Based on this, numerous works have been performed to check the influence of the surrounding ligands on the CO2RR process. Here, in this part, we reflect the microscopic and macroscopic influence of ligands on the catalytic performance of metal complexes and MOFs. The effect of ligands on the CO2RR product was demonstrated in the ground-breaking work of catalysis. It is worth noting that different Cu-MOFs generate different CO2RR products in the reduction process, which is due to the presence of different ligands connected to the same metal center.136,143 Kumar et al.136 used a ligand with an O donor atom, and consequently the CO2RR product was oxalate. Hinogami et al.143 designed an MOF containing S and N donor atoms and the CO2RR product was formic acid. Actually, the ligands and their donor atoms change the electronic configuration of the central active metal atom, resulting in different CO2RR products.To confirm this influence, four different copper-MOFs were synthesized, as follows: (1) copper AdeAce, (2) HKUST-1, (3) metal organic aerogel of mesoporous Cu-DTA, and (4) CuZn-DTA, and sprayed on GDE.144 The measured cumulative (methanol and ethanol) FE for the CO2RR was 15.9% using the HKUST-1 electrode at a current density of 10 mA cm−2. It was observed that the MOFs having unsaturated metal sites can contribute to CO2 reduction to alcohol. Furthermore, HKUST-1 displayed a stable output load for 17 h. Therefore, MOF catalysts are very flexible catalysts due to their tuneable features especially for CO2RR catalysis because of the excellent adsorption property for CO2 gas, which enhances the interaction between CO2 and the active sites of the catalysts. Jiang et al.140 synthesized the ZIF-8 MOF, which is a typical MOF material, and confirmed that the coordination between the imidazole ligand and Zn(II) acts as the active center for the CO2RR, which was confirmed by XAS and DFT calculations. In addition, they studied the topology structure of other ZIFs but with different organic linkers, for example, SIM-1 and ZIF-7, -8, and -108 for the CO2RR. Remarkably, among the catalysts, ZIF-8 showed a progressive FECO of 81%. Porphyrin-based Fe catalysts are high-efficiency homogeneous catalysts, which were first synthesized by Savéant et al.,145 and subsequently used for the CO2RR.146–148 Considering the constant use of Fe porphyrins, porphyrin-based constituents are introduced in MOF structures for two scientific purposes, as follows: (1) MOFs have potential electrocatalytic use and electron transfer capabilities, which are crucial for the CO2RR and (2) the catalytic structure is modified by introducing active molecules having characteristics including adjustable porosity, surface controllability, chemical stability, and access to active sites.149 For instance, Hod et al.123 combined an Fe-porphyrin with MOF-525 and applied it as a CO2RR catalyst, and confirm its structure by XRD (Fig. 8a and b). During the CO2RR process, a mixture of H2 and CO (FECO of 50%) was produced. These results indicate that Fe–porphyrins bound to the surface of MOFs can be used for the CO2RR (Fig. 8c and d). In addition, it has been observed that the porphyrin ring-coordinated Co atom is also a catalytically active site for the CO2RR. For example, Yaghi et al. designed a Co-based MOF with porphyrin as the organic linker, which was named as Al2(OH)2TCPP-Co, for the CO2RR.133 Consequently, an improved current density and catalytic performance of FECO 76% were achieved, and it was demonstrated that Co is the active center. Furthermore, they stated that the metal center of Co(II) is not actually the active center, instead Co(I) is the active center for the reduction of CO2.133 These preliminary investigations show that the electrocatalytic performance of MOFs for the CO2RR can be adjusted by changing their ligand.
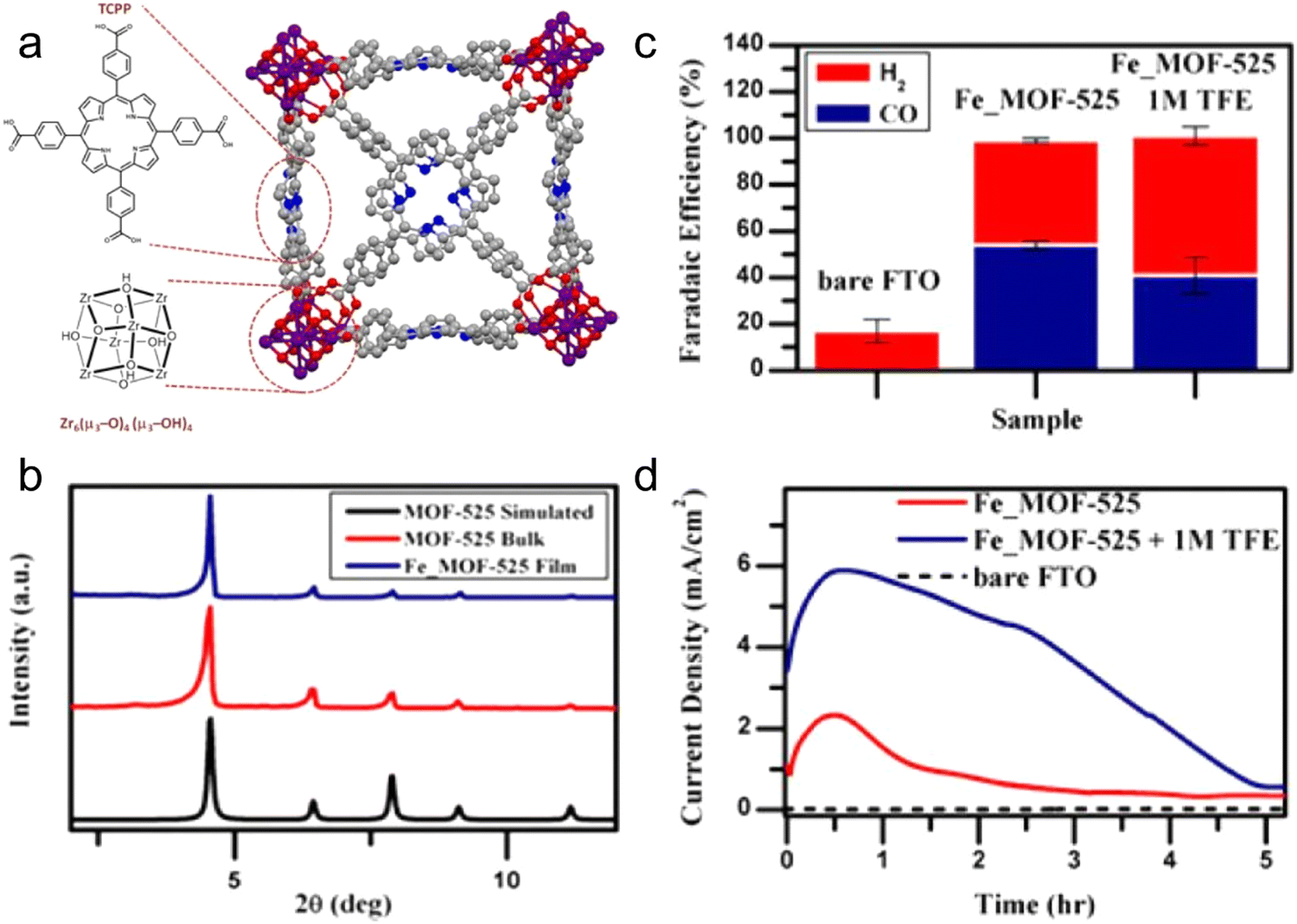 | ||
| Fig. 8 (a) Crystal structure of MOF-525 including the chemical structure of Zr-based node. (b) Comparison of PXRD of Fe-MOF-525 and other MOFs. (c) FE. (d) Current density vs. time. Reproduced with permission from ref. 123 Copyright 2015, ACS. | ||
5.3 Role of metal–ligand bonding
In addition, MOFs have the ability to resist the chemical environment for a long time. Actually, their chemical stability is very important for electrochemical reactions, which depends on the metal-linker bonding strength of the MOFs. Generally, the thermal and mechanical stability of MOFs are related to their capability to maintain their structural integrity when exposed to vacuum, pressure or heat.150 Because MOF-based materials are widely applied in the electrocatalytic process of different pH value electrolytes, their stability in a wide pH range is very important. For instance, it was reported that the highly stable Bi-MOF is an effective catalyst for the CO2RR process, which can yield formate with an FE of 92.2% and stability for 30 h. Moreover, a current density of 14 mA cm−2 was achieved under the low overpotential of 0.61 V. Further, in situ and ex situ XAFS were employed to study the structure-related features of Bi-MOF to reserve Bi3+, representing the chemical stability of Bi-based MOFs.110 Subsequently, Zhang et al. synthesized and studied a Bi-BTC MOF for the CO2RR via the simple hydrothermal process. They reported that Bi-BTC is an active catalyst for the production of HCOO− with an FE of 92 ± 3%. Interestingly, the FE was observed to be 90% after a long stability test of 12 h, which can be ascribed to the electrochemical stability of the MOF material.124 Yang et al.133 reported that Co–porphyrin has a better initial performance for the CO2RR with an FE of 76% at 0.7 V, but they reported the stability performance only of 7 h. Similarly, a zirconium-based MOF (MOF-525) containing Fe–porphyrin was used as an electrocatalyst for the CO2RR, where the stability of the MOF declined within 4 h, indicating its unsatisfactory stability.123 Although numerous studies synthesized novel MOFs with good stability using different synthesis methods, further expanded studies are needed to improve the stability of MOFs and systematically elucidate the degradation of the catalyst. In this case, more operando characterizations are needed to examine the electronic structural composition and morphological features.6. MOF-based composites
As is known, pristine MOFs possess low conductivity, and thus MOF-based composites with other materials (for example, metal, graphene, metal oxides and conductive carbon) are commonly used for the CO2RR. Yi et al.151 developed in situ uniformly distributed Cu2O(111) quantum dots on a porous conductive copper-based MOF. Because the catalyst has rich hydroxyl groups and uniform active sites (Fig. 9), it could efficiently reduce CO2 to methane. During this process, the hydroxyl group on the MOF can form a hydrogen bond with intermediate products such as *COOH, *CHO, and *CO, while its uniform active sites can reduce the occurrence of side reactions, thus effectively improving the selectivity for methane. The in situ-formed Cu2O@Cu-MOF having abundant active centers showed strong CO2 adsorption capacity. In the case of MOF composites, the MOF provides high stability and porosity, and transfers the scaffold in a limited space to achieve size-selective catalysis.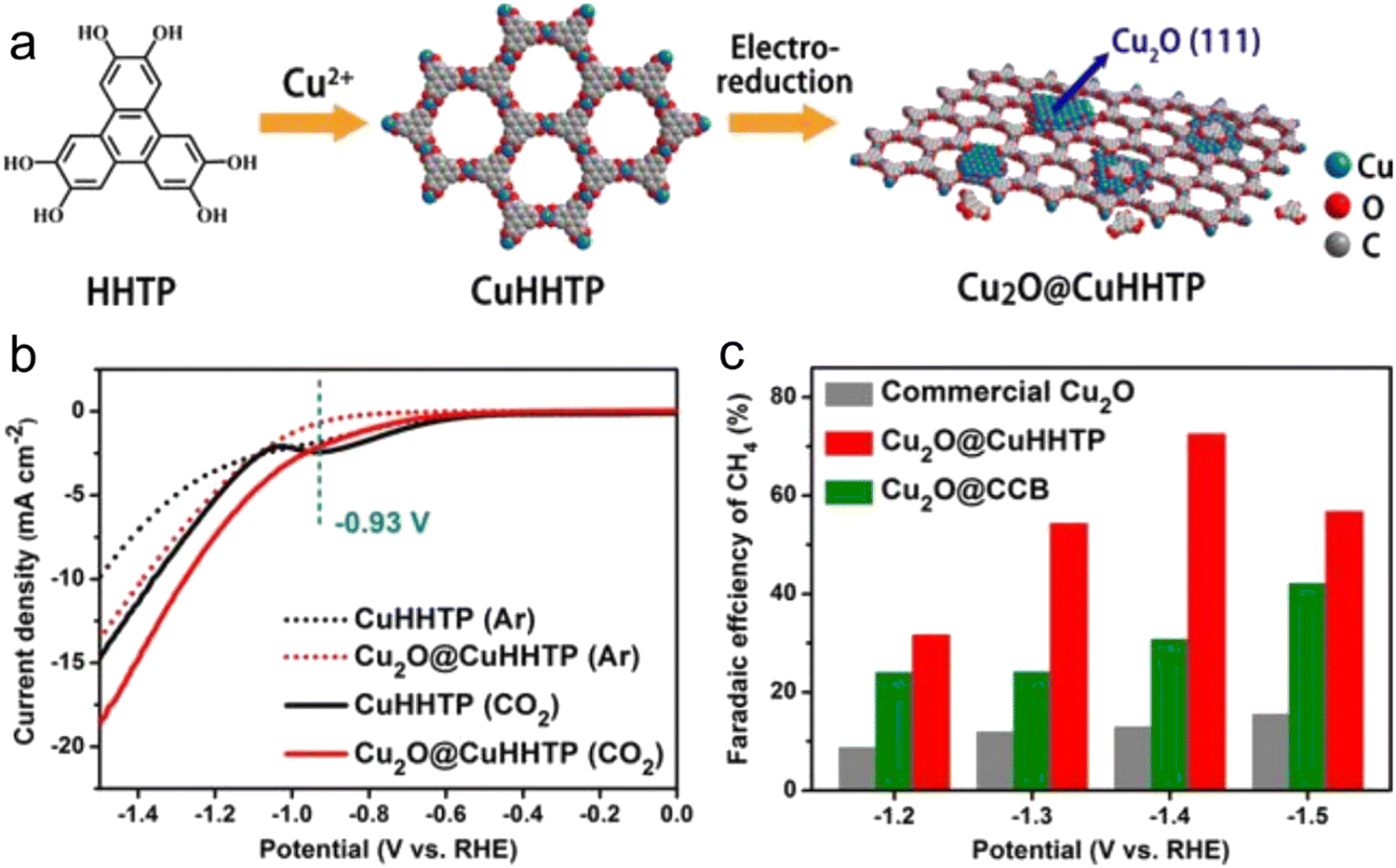 | ||
| Fig. 9 (a) Design of Cu2O@CuHHTP through electrochemical treatment. (b) LSV curves. (c) FE comparison. Reproduced with permission from ref. 151 Copyright 2020, Wiley-VCH. | ||
6.1 Polyoxometalate@MOF
In polyoxometalates (POMs), polyhedrons of metal oxides, MOx (x = 5, 6), are the basic unit and composed of polyanions of polyoxometalate and metal cations. The transition metal cations present in POMs usually have a high valence state, such as MoVI, WVI, TaV, NbV, and VV. As a typical class of cluster compounds, POMs have been extensively studied in catalytic applications.152–154 Exploiting the excellent characteristics of POMs, including redox ability, clear active center, adjustable light absorption, and reversible electron acceptor and donor, POM materials can be applied as photocatalysts, photosensitizers, co-catalysts and multi-electron sources for the CO2 reduction process.155–161 By integrating POMs in the MOF channels, they can promote electron transport, and ultimately improve the catalytic performance of the active centers. For instance, Lan et al.162 designed POM@PCN-222(Co) by linking a rich-electron POM structure with an MOF with a large specific surface area (Fig. 10). Surprisingly, the composite (H-POM@PCN-222(Co)) displayed significantly improved electron transfer ability and showed an excellent CO2RR performance with FECO of 96.2% at −0.8 V vs. RHE, and stability for up to 10 h. Fig. 11 shows the structure, synthesis, and reduction activity of CO2 of (PW12, Cp*Rh)@UiO-67, in which a simple strategy was used to fix the POM in the voids of the MOF, and after the encapsulation of the Cp*Rh(bpydc)Cl2 complex was done through the linker exchange post-synthesis.163 The catalytic activity of the composite was evaluated in the reduction of CO2 to formate. Compared with Cp*Rh@UiO-67 without the POM, the TON increased to high extent of 175 and the yield also doubled. The reference POM is considered as an electronic transfer material, but it does not impact on the electronic structure of the complex. In addition, [BW12O40]5−, [SiW12O40]4−, [PMo12O40]3− and [PTi2W10O40]7− with different redox potentials and acidities are also appealing for further studies.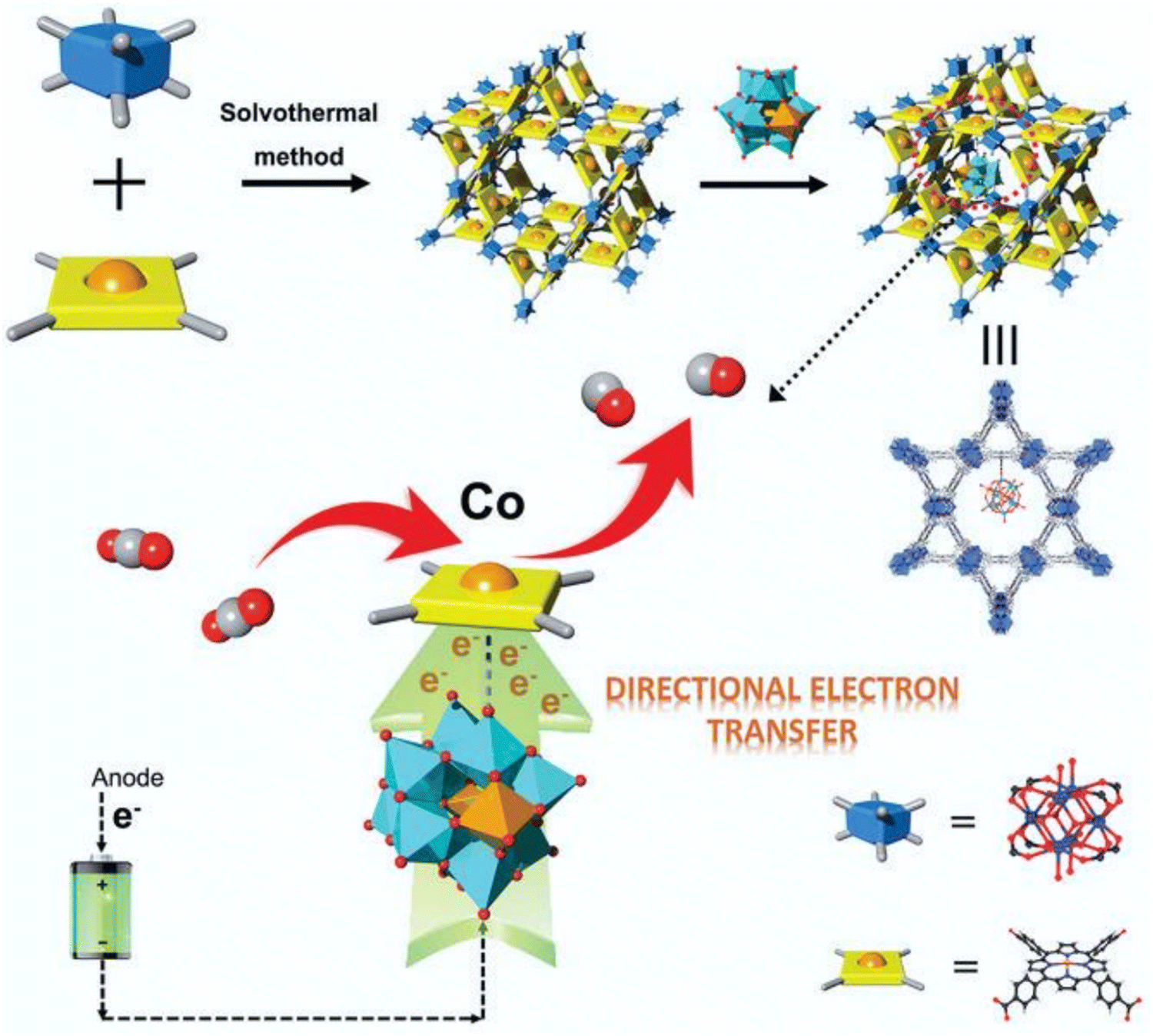 | ||
| Fig. 10 Top: Schematic of the synthetic process and detailed structure of the POM@PCN-222(Co) composite. Bottom: Proposed electron transfer scheme on the active single-metal site Co of H-POM@PCN-222(Co) for the CO2RR (Keggin-type POM builds an electron-transfer channel to the Co center from the electrode and accelerates this multi-electron-transfer process to enrich the electron density of the active Co center). Blue box: Zr-based second building unit, yellow square: TCPP linker, and yellow ball: Co metal center. Reproduced with permission from ref. 162 Copyright 2021, Wiley. | ||
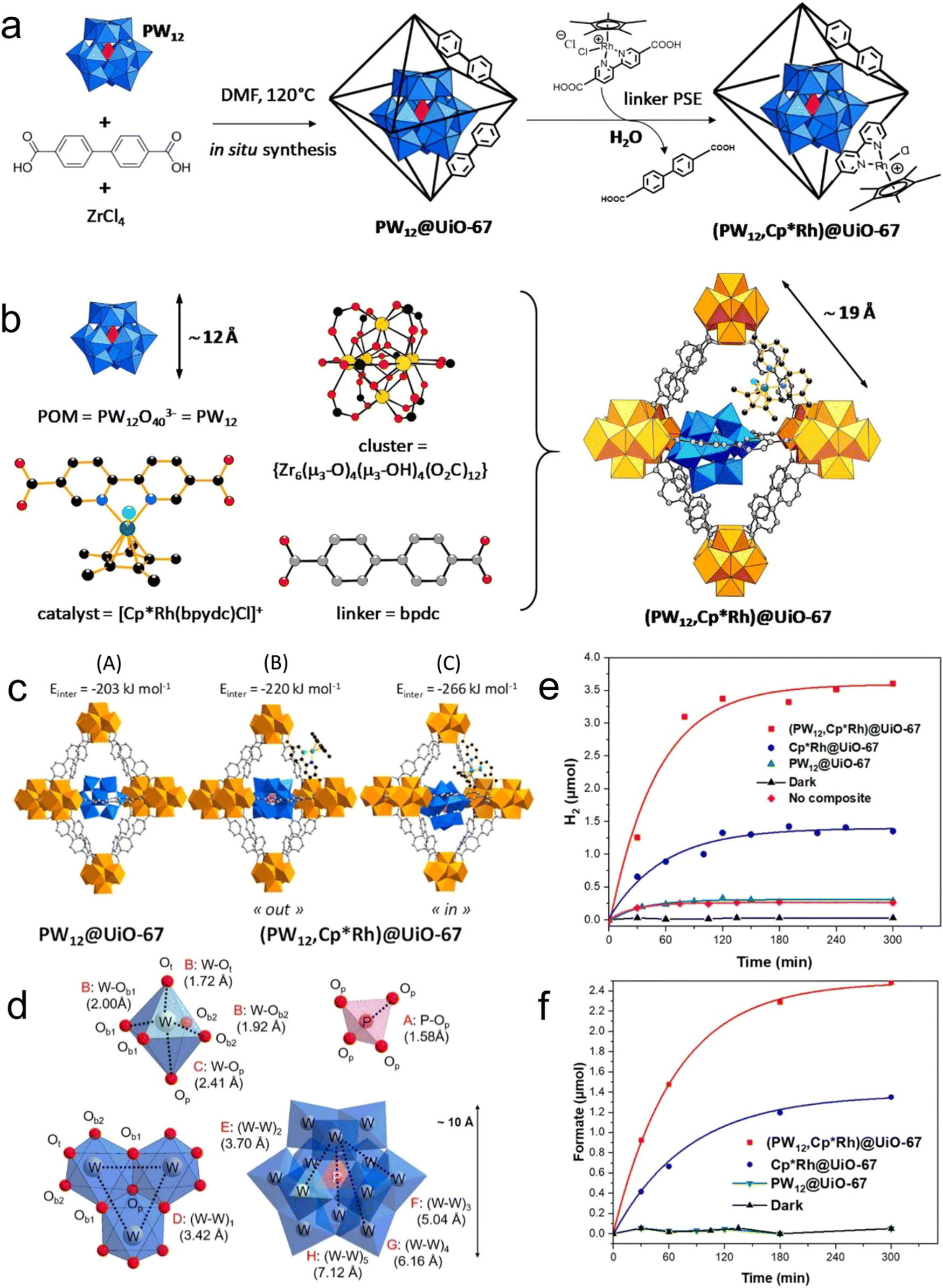 | ||
| Fig. 11 (a) Schematic of in situ synthesis. (b) Components of (PW12, Cp*Rh)@UiO-67. (c) Models of PW12@UiO-67 and (PW12,Cp*Rh)@UiO-67. (d) Distances between the components of POM. Production rate of (e) H2 and (f) HCOO−. Reproduced with permission from ref. 166 Copyright 2020, ACS. | ||
6.2 Metal oxide@MOF
It is very attractive to integrate copper oxide nanoparticles with the conductive network of MOFs, and the resulting electrocatalyst can display good catalytic activity. Accordingly, a Cu-based MOF (CuHHTP) with 2D conductive network was designed as the precursor material for the construction of quantum dots of Cu2O(111) on the porous surface of CuHHTP via an in situ strategy (Fig. 9). Operando IR spectroscopy and DFT calculations confirmed that Cu2O(111) can generate and stabilize *CH2O and *OCH3, which are the key intermediates of the CH4 reaction path by Cu2O, and hydrogen bonds. The FECH4 was high as 73%. The CuHHTP carrier with high conductivity could promote the electron transfer, resulting in a high current density.1516.3 Conductive support@MOF
The inherent low conductivity of MOFs greatly limit their electrochemical activities and applications. Accordingly, the decoration of MOFs on a conductive support improves their overall conductivity, and thus electrochemical activity toward the CO2RR. For instance, an MOF-55 Al–porphyrin containing cobalt porphyrin as the active center for the CO2RR to CO was reported. The conductive alumina film was formed via the atomic layer deposition (ALD) method, which can also work as a metal precursor, and then the MOF was generated by reacting the coated alumina with a linker (Fig. 12a).133 The thickness of the precursor could be simply controlled by the number of deposition cycles, which could adjust the thickness of the catalyst coating. The catalytic activity of the synthesized MOF was mainly enhanced by exploiting the thickness of the catalyst. The optimum catalyst with the thickness of 5 nm showed an FECO of 76% during the 7 h durability test (Fig. 12b and c). In addition, Wu et al.164 introduced a new strategy using FTO as a conductive support for the preparation of Cu2F(CuTCPP) MOF nanosheets, and the resulting catalyst displayed an improved performance for the CO2RR, generating formate with 68.4% FE at 1.55 V vs. Ag/AgCl (Fig. 12d). Sun et al.165 prepared highly oriented MOF films on FTO via liquid-phase epitaxy. The zinc acetate solution in ethanol and Re(CO)3C1 solution in ethanol were successively sprayed on the surface of FTO with a spray gun containing nitrogen, and a layer of film was prepared. Spraying was repeated 30 times to obtain 30 layers of Re-MOF film (Fig. 13a–d).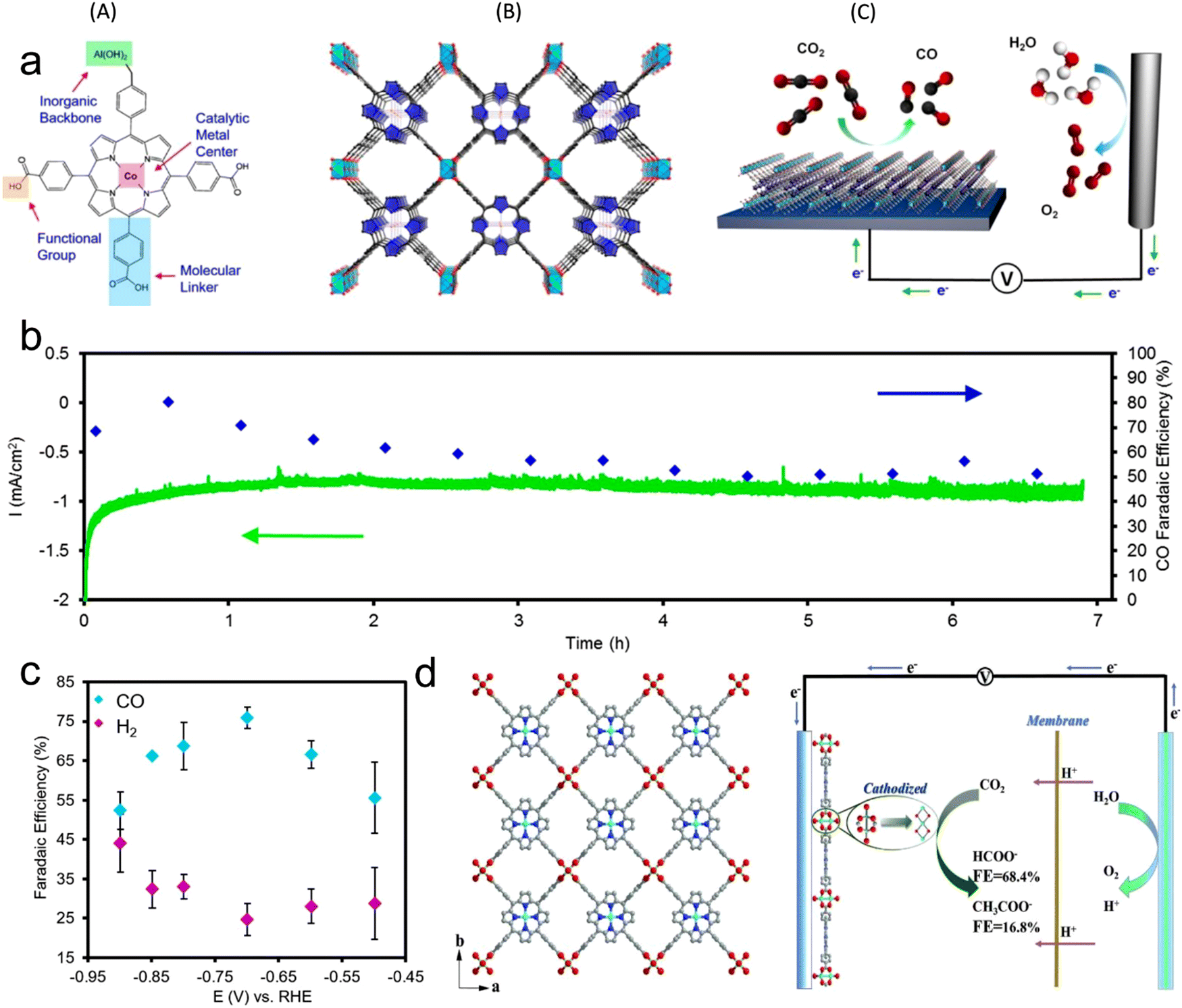 | ||
| Fig. 12 (a) Illustration of metal center, linkers, and functional groups and building unit in electrochemical system for CO2 reduction. (b) Stability of the MOF catalyst evaluated through chronoamperometric measurements in combination with faradaic efficiency measurements. (c) FEs. Reproduced with permission from ref. 133 Copyright 2015, ACS. (d) Crystalline structure of Cu2(CuTCPP) nanosheets in CO2 electrochemical reduction system. Reproduced with permission from ref. 164 Copyright 2019, RSC. | ||
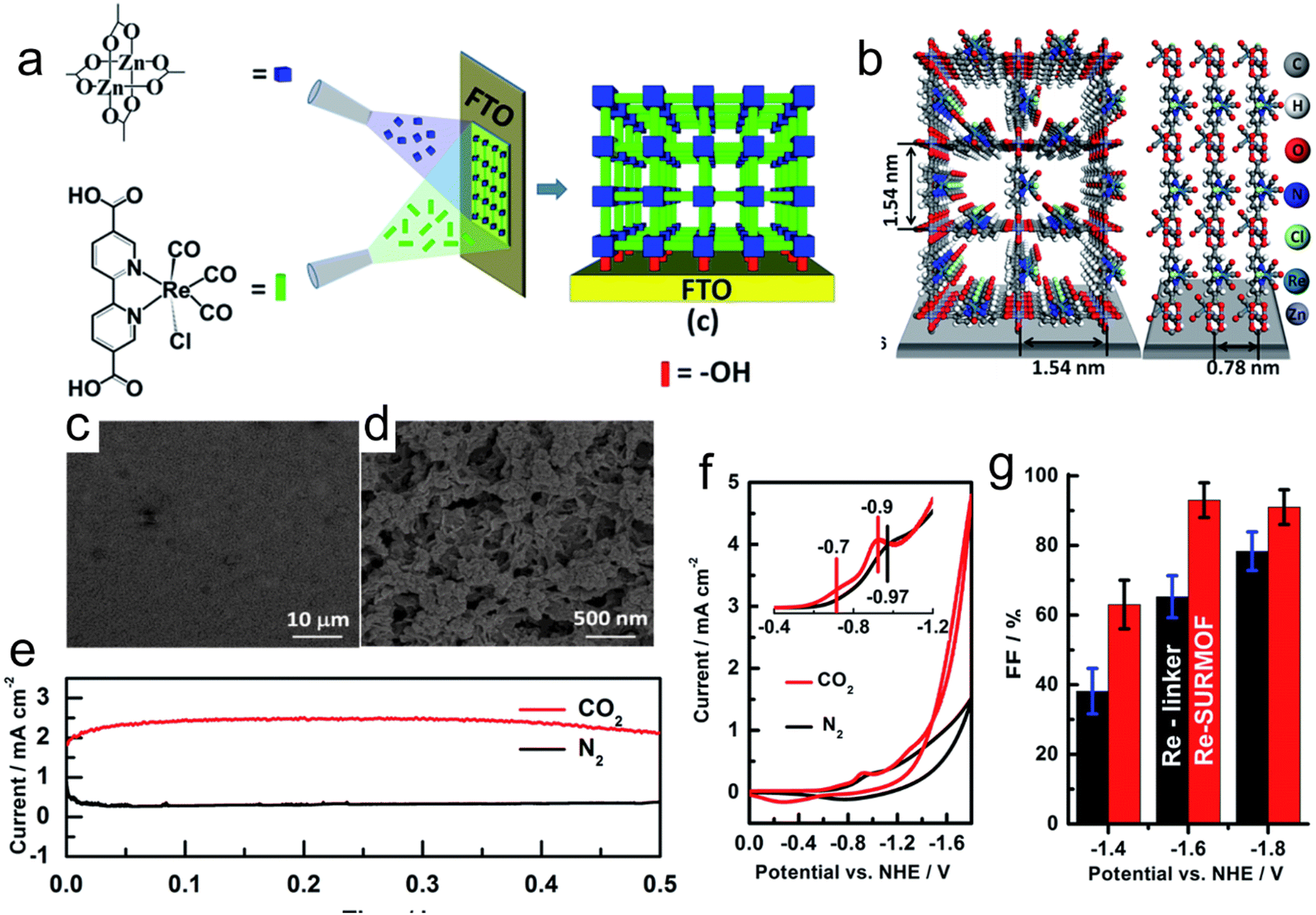 | ||
| Fig. 13 (a) Schematic synthesis of Re-SURMOF. (b) Crystal structure of Re-SURMOF. (c and d) SEM images of Re-SURMOF. (e) Current density during catalysis of CO2RR in CO2- and N2-saturated electrolyte. (f) Cyclic voltammograms of Re-SURMOF. (g) FE of Re-SURMOF. Reproduced with permission from ref. 165 Copyright 2016, RSC. | ||
In the CO2RR process, porous structures are conducive to the entry of CO2 dissolved in the organic solvent and the reaction with the inner catalyst, improving the effective active area of the Re-MOF catalyst. Simultaneously, they are also conducive to the escape of CO and H2 generated during the catalytic process, thus preventing the generated gas from gathering on the surface of the film to hinder the further reaction of the catalyst. This conductive supported MOF showed an FECO of about 96%, which can be attributed to the improved charge transfer by the conductive support (Fig. 13e–g). It is remarkable that the durability of the catalyst was low because of its degradation (Re(CO)3C1), and thus it is necessary to develop novel strategies to enhance the durability of the catalyst.
6.4 Molecular catalyst@MOF
It is well known that MOFs exhibit a large surface area, which is advantageous for the distribution of the active centers and can enhance the CO2 transportation and charge, resulting in improved CO2RR activity. Buonsanti et al.167 proposed that CO2 can be actively converted to CO using a mixed catalyst platform, for example, an MOF (Al-PMOF) was combined with colloidal crystals (Ag@PMOF). This study claimed that the hybrid material has greater ability to inhibit the HER than the single silver nanocrystal, and also contributes to the CO2RR. In addition, the electronic properties were also enhanced by the synergistic effect of the silver colloidal crystals and Al-PMOF and by partially removing the MOF ligands. Furthermore, Lan et al.168 also reported that the catalytic performance of a cobalt porphyrin increased to 99% FECO by the synergistic combination effect with POM in KHCO3 (0.5 M) electrolyte (Fig. 14a and b). Through theoretical calculations (DFT), it was confirmed that the free energy of adsorption for *COOH and *CO (intermediate products) is small, further confirming the superior activity of Co-PMOF for the CO2RR (Fig. 14c and d). Based on the previous perception, these findings verify that the cobalt atom in Co–porphyrins is the main active center, and POMs with electron-rich aggregates are helpful to boost the electron transport (Fig. 14e and f).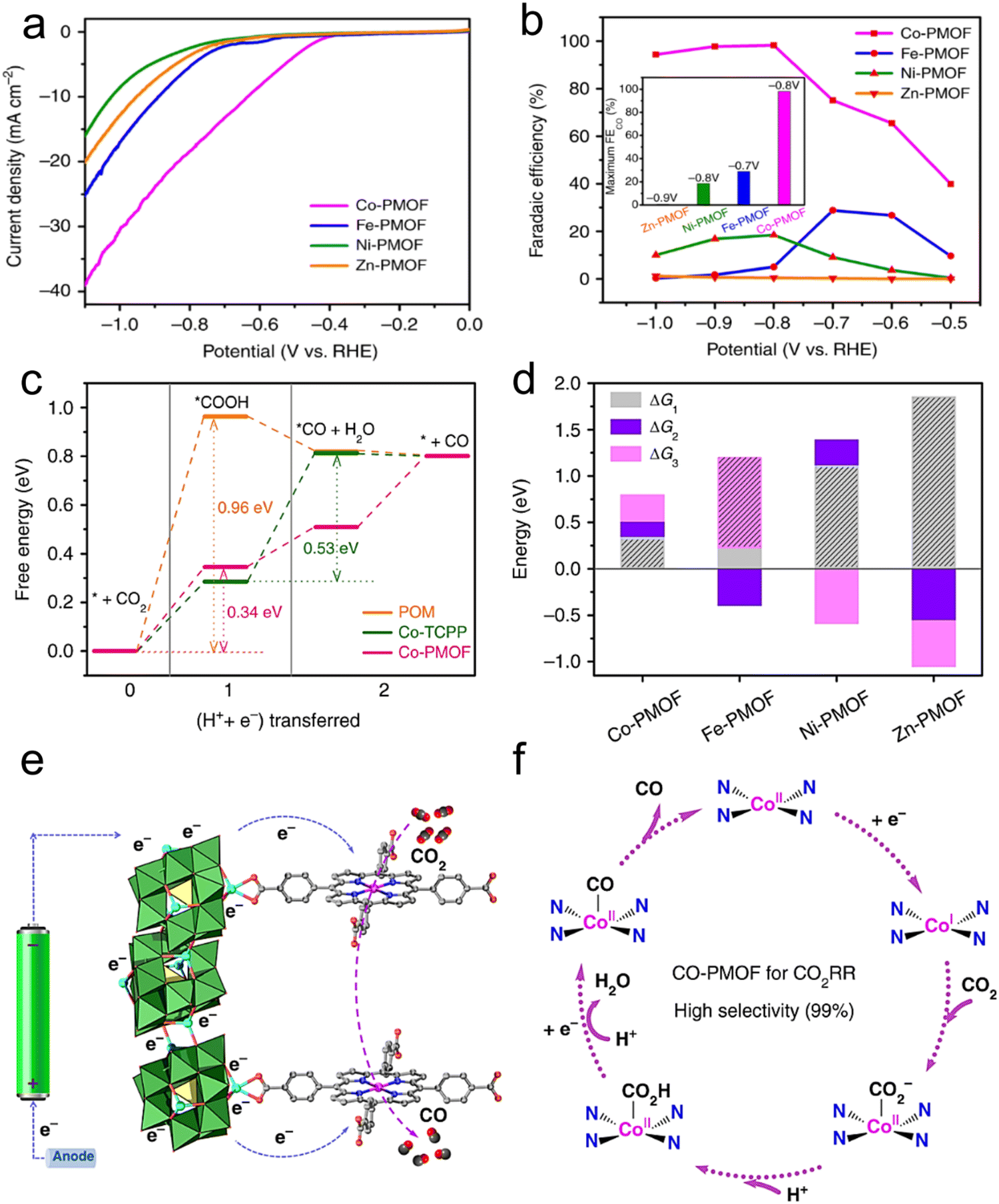 | ||
| Fig. 14 (a) LSV curves of PMOFs. (b) FEs. (c) Free energy diagram. (d) Free energy comparison of elementary reactions. (e and f) Suggested mechanism for the CO2RR on Co-PMOF. Reproduced with permission from ref. 168 Copyright 2018, Springer Nature. | ||
In addition, Wang et al.169 constructed a catalyst (TPY-MOL-CoPP) with pyridine and CoPP center group on an MOL layer, and assumed that TPY and CoPP have a synergistic effect. To verify this view, the authors prepared a catalyst without pyridine (BTB-MOL-CoPP) as a control. The electrochemical performance showed that at −0.86 V, the CO current density (jCO) of BTB-MOL-CoPP was 923 mA mg−1, and its jCO/jH2 ratio was 2.7, while the jCO of TPY-MOL-CoPP was 1314 mA mg−1, the jCO/jH2 ratio was 11.8, and the CO Faraday efficiency of TPY-MOL-CoPP was 92.2%, indicating that TPY-MOL-CoPP containing pyridine groups was more conducive to improving the selectivity and yield in the CO2RR.
By adding Zn2+ or Ca2+ to coordinate TPY sites, it was found that the FECO was significantly reduced from 92.2% to 6% and 8%, respectively. The evaluation of the CO2RR performance of various porphyrin-based catalysts showed that the activity of TPY-MOL-CoPP was higher than that of the other catalysts. Even at low potentials, the FECO of TPY-MOL-CoPP was higher than that of BTB-MOL-CoPP. It was observed that TPY promoted the CO2RR at all the measured potentials. The control experiment of BTB-MOL and TPY-MOL with CoPP-free functionalization showed that the only reaction was the HER, and all the results indicate that CoPP is the catalytic center and TPY ligand plays a role in promoting the CO2RR. In the CV test, the performances of TPY-MOL-CoPP in a saturated CO2- and N2-atmosphere were tested, respectively. It was found that when the scanning rate was 200 mV s−1 in an N2 atmosphere, the CV of TPY-MOL-CoPP with NaH2PO4/Na2HPO4 buffer solution displayed a reduction peak at 0.28 V vs. RHE, suggesting that [CoPP]0 becomes [CoPP]−. This means that the negative starting potential of the CO2RR and HER should be greater than −0.28 V. The above-mentioned tests show that CoPP is the catalytic center of TPY-MOL-CoPP and the TPY ligand plays a role in promoting the CO2RR. Based on the porphyrin MOF-525 (Fe-MOF-525) film as the active catalyst, Hod et al.123 anchored a large number of active molecular catalysts on the conductive electrode for the CO2RR. MOF-525 was chosen to immobilize the catalyst because it has good molecular porosity and chemical stability. In addition, the use of MOFs to fix molecular catalysts can effectively avoid polymerization and deactivation of the catalyst because each catalyst molecule is covalently connected with the frame node. This method generated (1015 active sites cm−2) a high electrochemical surface area to synthesize CO gas (FECO = 50%). Here, although the efficiency of the CO2RR was low, the results showed that molecular catalysts with MOF structures can be used for improved CO2RR. Therefore, it is expected that the accumulation of molecular catalysts in MOFs will be further conducive to the design of advanced CO2RR catalysts.
6.5 Metal nanoparticle@MOF
Noticeably, many metal nanoparticles (NPs) have been explored and used as electrocatalysts for the CO2RR. However, in the catalytic process, the accumulation of these small NPs in macromolecules is always challenging. Thus, to address this problem and improve the practicality of extra catalytic active centers, several methods have been applied, including size reduction and shape control of NPs.170,171 Compared to macro-molecule/large-size catalysts, MOFs having a porous structure with large specific surface area provide an improved platform for efficient CO2 reduction and production functional materials. They can not only be used as a support material for metal molecular coatings, but also as an ideal material to instigate the uniform distribution of nanoparticles to reduce CO2.For example, Hupp et al.172 prepared a catalyst for the CO2RR by decorating Cu on the surface of NU-1000-MOF (Fig. 15a and b). The prepared catalyst exhibited catalytic ability for products of two phases, i.e., liquid-phase formate (HCOO) and gaseous-phase products (H2 and CO). With reference to RHE, formate production with an FE of 31% as the main product was achieved at −0.82 V. Overall, this Cu-NU-1000 MOF displayed a distinct CO2RR performance compared to its individual components (Fig. 15c). Jiang and co-workers designed a composite of Ag2O and ZIF via a refluxing method (Fig. 15d).173 The prepared composite catalyst, Ag2O/ZIF-7, presented improved CO2RR results with an FE of 80% for CO compared to its individual components of ZIF-7 and Ag/C. The improvement in performance was attributed to be the synergistic combination of Ag2O and ZIF, while the mass transfer was promoted due to the large specific surface area of the MOF. The electrocatalysis results showed that the Ag2O nanoparticles were homogeneously dispersed on the ZIF surface. In addition, it was observed that when the Ag2O/ZIF-7 composite reduced CO2, it had a more positive onset potential of −0.6 V than the ZIF-8/Ag2O composite in 0.25 M K2SO4. The Ag2O/ZIF-8 composite displayed a higher FECO of 80.6% at −1.2 V vs. RHE than that of its individual components of Ag/C and ZIF-8. This exceptional catalytic activity can be ascribed to the synergistic effect between the larger surface area of the MOF and significantly small size of the metal NPs (Fig. 15e–g). According to the above discussion, it is clear that MOF composites are also active catalysts.
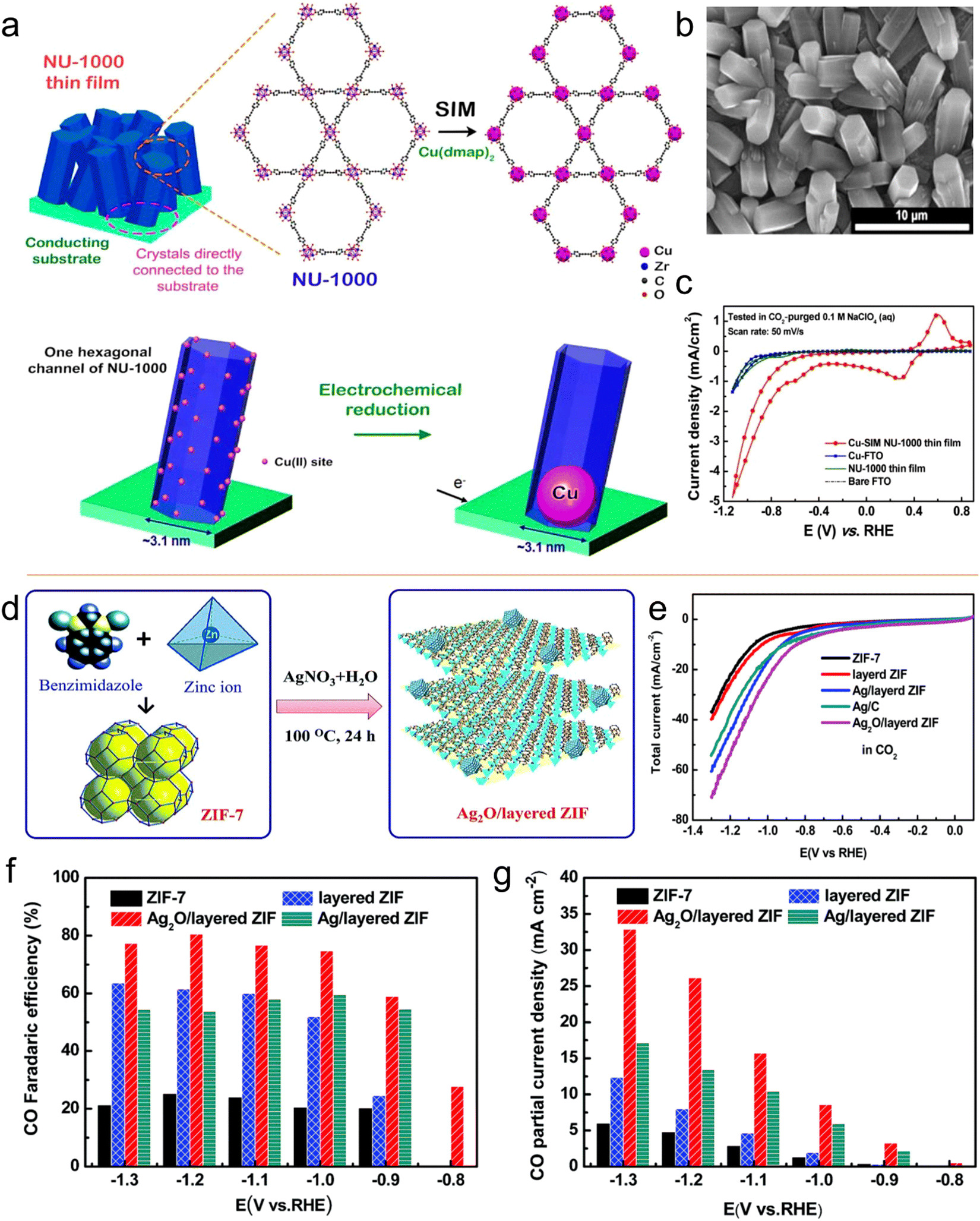 | ||
| Fig. 15 (a) Schematic representation of installation of SIM in Cu(II) single site. (b) SEM image of NU-1000 film. (c) CV curves. Reproduced with permission from ref. 172 Copyright 2017, ACS. (d) Schematic synthesis of Ag2O/layered ZIF. (e) LSV curves and (f) FEs for CO. (g) CO partial current densities vs. V. Reproduced with permission of ref. 173 Copyright 2017, RSC. | ||
7. Confinement effect of MOF-derived carbon materials for CO2RR
7.1 MOF-derived atomic-dispersed carbon materials
Atomic-level catalysts refer to metal/non-metallic active centers that are dispersed at the atomic level on a carrier, avoiding their accumulation. Nanocrystals exhibit excellent activity and selectivity for the CO2RR when their size decreases to the atomic level and they are dispersed atomically because of the significant reduction in their energy with a decrease in their size.174,175 However, during the sintering process, metal atoms easily migrate and accumulate to form clusters, resulting in a decrease in the density of active centers, and thus decreased catalytic activity.176 Therefore, various strategies, such as spatial confinement,177 coordination design178 and defect engineering,179 have been adopted to prevent the agglomeration of metal atoms.An effective way to construct single-atomic catalysts is with the assistance of MOFs to prevent atomic agglomeration. MNC materials are extensively studied catalysts for the CO2RR. Zhao et al.180 reported the decoration of ZIF-8 with Ni atoms, and after high-temperature treatment at 1000 °C, the Zn atom was replaced with Ni, finally forming Ni active centers. This single-atomic catalyst could efficiently reduce CO2 to CO with 70% FE. The FECO of the Co monatomic catalyst prepared using a similar method was up to 94%, and at the overpotential of 520 mV, the current density was 18 mA cm−2 with a TOF of 18200 h−1 for CO. Under the applied voltage of −0.63 V, the current density attenuation of Co–N2 during 60 h operation was negligible, and the FECO remained almost unchanged (Fig. 16a–c).181 The theoretical and experimental results displayed that the decrease in the coordination number of Co–N (4 to 2) supports the initiation of CO2 to the CO2˙− intermediate, thus improving the CO2RR performance. Jiao et al.182 designed several single-atomic electrocatalysts (e.g., M1–N–C, M =,Co Fe, Cu and Ni) using a porphyrin polymetallic framework structure under the confinement effect. These single-atom metal catalysts possessed physical characteristics (specific surface area, particle size, and pore structure). Importantly, when the CO2RR performance of Ni1–N–C was compared with that of the other M1–N–C, the former exhibited a lower COOH production energy and lower desorption energy for CO. Hence, Ni–N–C with single-atom Ni displayed the best performance for the CO2RR as the active center with high selectivity for CO (FE up to 97%, Fig. 16d–g). Furthermore, Ni1–N–C had high selectivity and feasibility in industrial production at the CO2 concentration of 30% and 15% (volume fraction), which further showed its superior and excellent selectivity for the CO2RR. Besides, Hao et al.183 systematically studied the selectivity and activity of transition metal and nitrogen doped-carbon for the CO2RR to elaborate its significance. Using different metal nitrogen-doped carbon materials (e.g., M–N–C, M = Ni, Mn, Cu, Fe, Co) as model catalysts, they systematically studied the selectivity and activity of these catalyst in the CO2RR to CO process through experiments and calculations, and found that the highly dispersed metal–nitrogen coordination center is the active site, which has guiding significance for the development of this type of materials (Fig. 17a). This series of M–N–C materials has a multi-stage pore structure, and the pore size of the micropores is uniformly distributed at 8 Å, showing strong CO2 affinity. Under normal pressure, the CO2 adsorption capacity reached 4–5 mmol g−1 (Fig. 17b). Furthermore, the catalyst precursor is a metal N coordination polymer crystal. After thermal decomposition, the metal–nitrogen coordination center was uniformly dispersed at the molecular level. The surface chemistry of this series of M–N–C materials was analyzed, confirming the existence of the metal–N coordination structure, and the content and distribution of other forms of nitrogen-containing functional groups.
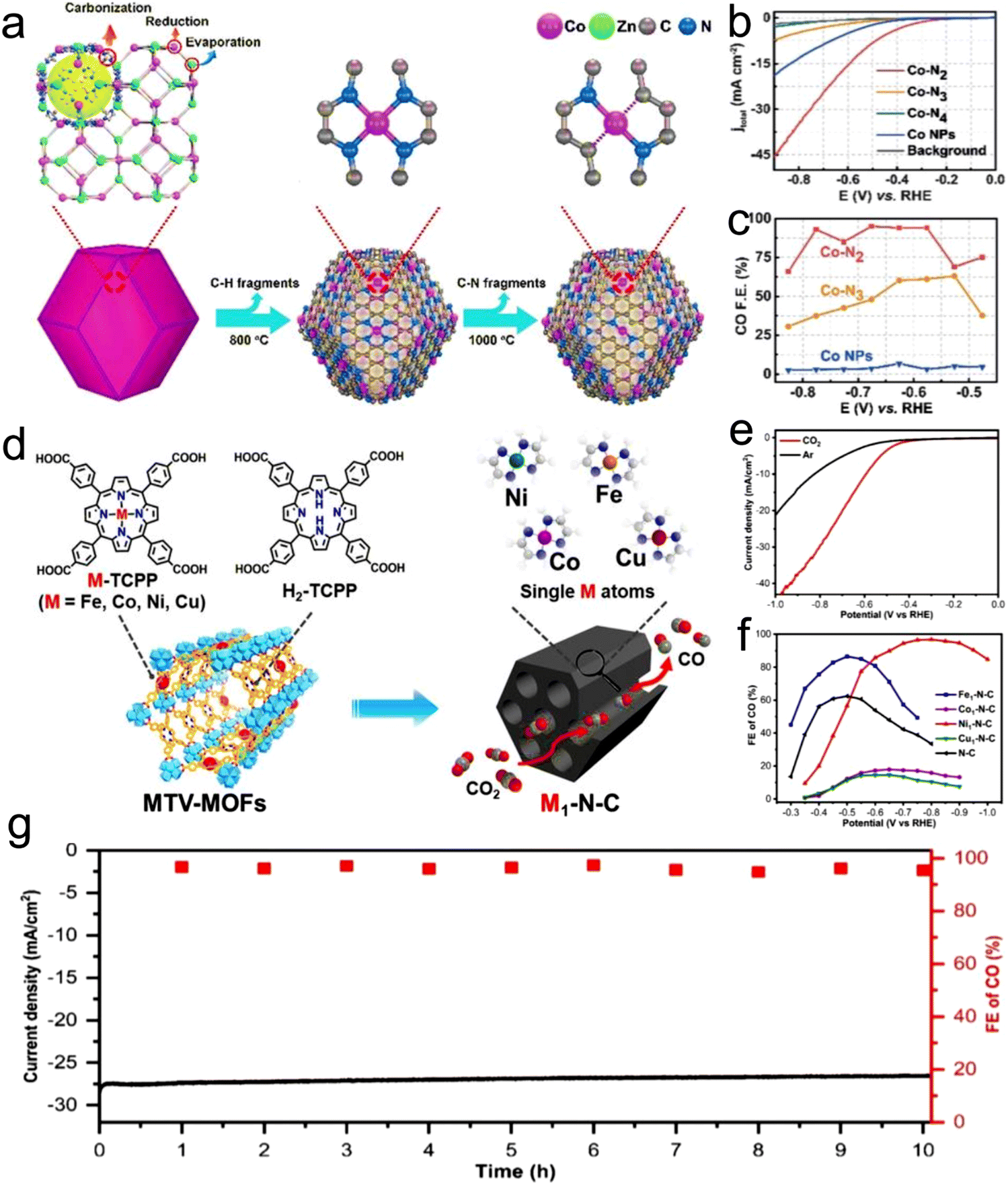 | ||
| Fig. 16 (a) Co–N4 synthesis process. (b) LSV curves of all the materials and (c) FEs. Reproduced with permission from ref. 181 Copyright 2021, Wiley. (d) Synthesis of M1–N–C single-atom catalysts. (e) LSV curves of Ni1–N–C. (f) FEs and (g) durability test of Ni1–N–C. Reproduced with permission from ref. 182 Copyright 2020, Wiley-VCH. | ||
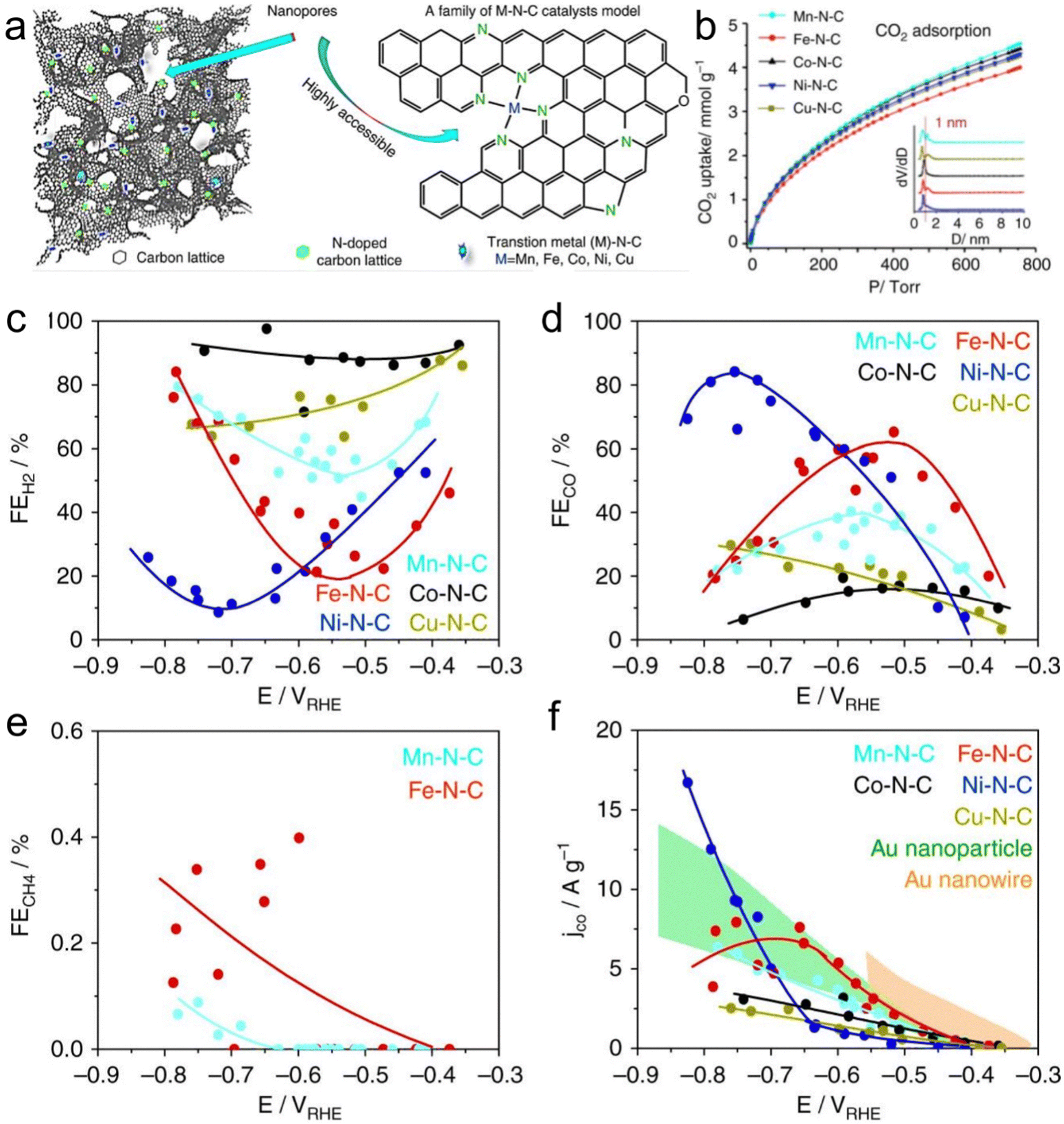 | ||
| Fig. 17 (a) M–N–C model and structure. (b) Adsorption isotherm curves for CO2 and inset is the pore size distribution. (c–e) FEs of H2, CO and CH4, respectively. (f) Mass-normalized partial currents for CO. reproduced with permission from ref. 183 Copyright 2017, Springer Nature. | ||
Here, the different samples did not show significant differences. Firstly, the activity of these catalysts was analyzed by linear sweep voltammetry (LSV). Comparing the LSV curves in the presence and absence of CO2, it was found that Mn–, Fe–, Ni– and Cu–N–C exhibited CO2 reducing activity and showed a lower overpotential compared with that for the HER. However, the HER activity of the Co–N–C sample dominated the Faraday process. The FE of the product was analyzed, and it was found that CO and H2 accounted for the transferred charge of more than 95%. It is worth noting that Mn–N–C and Fe–N–C catalyzed the reaction to produce a detectable amount of methane (CH4). Although the density of metal–nitrogen active centers was low, its mass-normalized CO2 conversion yield is equivalent to the best Au catalyst at present, especially in the high current range with practical application significance (Fig. 17c–f). This shows that metal- and nitrogen-doped carbon catalytic materials have great application significance for the CO2RR.
Recently, Liao et al.184 designed a reasonable active site structure and regulated and integrated its local chemical environment to realize the highly selective C2+ product under neutral conditions. The Cu-HITP MOF was used for the CO2RR, and the FE of the C2+ product was 45.59%, and then the FE of the C2+ products significantly increased to 75.09% when polydopamine (PDA) was coated on the surface of Cu-HITP to provide a chemical microenvironment conducive to the CO2RR to multi-carbon products (Fig. 18a, b and d). In addition, Cu-HITP@PDA has good electrocatalytic stability, where its current density and product selectivity remained basically unchanged in the stability test of 10 h. The CO2RR mechanism was inspected using the in situ FTIR technique.
 | ||
| Fig. 18 (a) Synthesis of Cu-HITP and Cu-HITP@PDA. (b) Comparison of CO2RR performances of Cu-HITP with different PDA coating amounts. Reproduced with permission from ref. 184 Copyright 2022, ACS. (c) Synthesis of In/NC single atom. (d) FEs of different products. Reproduced with permission from ref. 184 Copyright 2022, ACS. (e) Preparation of Fe1N2O2/NC catalyst. Reproduced with permission from ref. 186 Copyright 2022, RSC. (f) TOF and FEs of formate. Reproduced with permission from ref. 185 Copyright 2020, Wiley. | ||
By comparison, it was found that the key intermediates *COH and *OCCOH in the process of CO2RR red-shifted during in situ FTIR testing after the catalyst was coated with PDA due to the presence of hydrogen bond interactions. It was suggested that amino groups can be used as hydrogen bond donors to stabilize some basic intermediates for the formation of multi-carbon products, thus promoting the formation of C–C bonds. Zhang et al.185 also reported a significant development in the design and synthesis of monatomic catalysts for the CO2RR. They designed a nitrogen-doped carbon carrier anchored on an MOF-derived carbon carrier with Inδ+–N4. The monoatomic catalyst possessed an N4 atomic interface structure (Fig. 18c and f). The catalyst exhibited an excellent CO2RR performance. At −0.65 V, In SAS/NC exhibited the maximum FE of 96% and optimal conversion frequency (TOF) of 12500 h−1, which is superior to most similar catalysts. In addition, the XAFS study based on synchrotron radiation showed that the Inδ+–N4 atomic interface structure has high catalytic activity for the CO2RR. The DFT simulation results showed that the Inδ+–N4 atomic interface has low free energy for the formate intermediate (HCOO*), which is conducive to advancing the catalyst activity for the CO2RR. This study provides a novel approach for the rational design of main group catalysts for energy applications.
Recently, Chen et al.186 reported that the Fe–Nx coordination structure is the common structure of Fe–N–C in various M–N–C catalysts, which is expected to replace the traditional noble metal-based CO2RR catalysts in the near future. For Fe–N–C-type electrocatalysts, the energy barrier is higher to produce *COOH. Also, given that the binding of *CO on the active site is relatively strong, CO desorption is difficult.
Therefore, the key to improving the CO2-to-CO conversion efficiency of Fe–N–C electrocatalysts is to support the formation of *COOH (protonation) and optimize the binding strength of *CO (desorption). Zn-MOF-74 was selected as the precursor of oxygen enrichment for synthesis regulation, and then Fe/Zn-MOF-74 was obtained by doping Fe ions. After introducing an N source, it was calcined, finally producing the Fe1N2O2/NC catalyst with a special coordination structure (Fig. 18e). The FECO of the catalyst was maintained above 95% in the large potential window of −0.4 to −0.8 V. It is worth noting that the FECO even reached 99.7% at −0.5 V, which is almost 100%. In addition, the formation mechanism of Fe1N2O2/NC was revealed by analyzing the synchrotron radiation results of the calcined products at different temperatures. The theoretical calculation showed that compared with the traditional Fe–N4 catalyst, the introduction of O and N together to regulate the interface of Fe atoms has a significant advantage in electrocatalytic carbon dioxide reduction to produce CO. The unique coordination structure of Fe1N2O2 showed a small free energy change in the generation of the COOH* intermediate and CO desorption, thus accelerating the reaction rate and improving the catalytic performance. Moreover, recently, carbon-based catalysts having bimetallic sites derived from MOFs have attracted extensive attention due to their bimetal atoms centers and synergistic effect, which enhance the density of active centers and improve the performance. For example, Ren et al.187 designed Fe/Ni bimetal carbon catalysts (Ni/Fe N–C) via the calcination the precursor of ZIF-8. It exhibited a high density of sites and activity and good catalytic performance for the CO2RR. It is worth noting that due to the weak binding energy of *COOH and *CO on the surface of Ni/Fe–N–C, Fe/Ni displayed a decreased energy barrier, while increased CO2RR performance. The reduction selectivity of CO2 to CO was quite good given that the FE reached 98%. In the CO2RR catalytic process, atomically dispersed active centers display excellent activity and selectivity toward CO production. Nevertheless, there are still many limitations in understanding the internal factors and structural influences that affect the performance of monatomic catalysis. Wu et al.188 used the Fe–N–C as a standard catalyst to divide three key parameters, i.e., particle size, Fe percentage and structure of the Fe–N bond, and they completely illustrates the factors affecting the CO2RR performance. Importantly, the particle size and doped iron percentage are crucial to regulate the exterior morphology, porosity, surface area, electrochemical activity and graphitization degree of carbon in FeN4.
Firstly, the Fe cluster catalyzes the formation of the surrounding graphite carbon shell. Secondly, it may promote the charge transfer to the central site of FeN4. It also acts as a catalytic promoter. Reasonable control of the surface coordination number of copper clusters can offer active centers and optimize the activity, selectivity and efficiency for the CO2RR. Sargent et al.190 used a special structure to regulate the surface structure of copper clusters in the deformed HKUST-1 MOF. The results illustrated that the Cu clusters formed at the position of the deformed Cu dimer increased the ethylene FE from 10% to 45%. Additionally, the yield of H2 decreased to less than 7%, which proved that MOFs can be used to alter the surface of copper clusters in the CO2RR catalytic process.
MOF-derived monoatomic-dispersed carbon materials (M–N–C) are designed with the help of the limiting strategy, which can limit the metal atom dispersion, enhance the active center density, reduce the desorption–absorption energy barrier of the intermediates for the CO2RR, speed up the reaction process, and advance the product selectivity.
7.2 MOF-derived carbon-based nanoparticles
Other metal materials derived from MOFs for the CO2RR are metal alloys, metal nanoparticles and metal oxides. Among them, metal oxides and alloys are extensively studied for the CO2RR. Alloys mostly show better or totally diverse catalytic output than monatomic materials, which improve the CO2 conversion effectiveness and purposely reduce CO2 to specific target products. To realize the goal of adjusting the ratio of products, metal oxides usually form alloy oxides with multiple metals to increase the CO2RR performance. For example, Guo et al.191 designed a series of carbon-based catalysts derived from an MOF, which contained different proportions of Cu/In bimetallic oxides. The as-prepared In/Cu bimetallic oxide catalysts exhibited a good CO2RR electrocatalysis performance. It is notable that by regulating the proportion of Cu/In, different CO/H2 compositions could be obtained. When the current density was 11.2 mA cm−2, the maximum FECO reached 92.1%, and the catalyst was stable for more than 24 h. The high activity for the CO2RR is attributed to the fact that the addition of Cu to In2O3 can improve the interaction between electrons, and consequently increase the density of active centers, improve the adsorption capacity of CO2 and accelerate the rate of electron transfer.Zhong and co-workers prepared a layered bimetal conjugated MOF (PcCu–O8–Zn) via a solvothermal method using a phthalocyanine linker and ZnO4 (Fig. 19a and b). The selectivity for the CO2RR to CO was promoted by PcCu–O8–Zn and the FE was high as 88%; furthermore, the TOF was 0.4 s−1. Additionally, PcCu–O8–Zn showed good stability (>10 h) at −0.7 V (Fig. 19c and e). The results showed that the coordination of ZnO4 in PcCu–O8–Zn catalyst has high activity toward CO2 to CO conversion, while the coordination of CuN4 in PcCu–O8–Zn MOF promotes the protonation of *CO2 adsorbed on the ZnO4 complex (Fig. 19d). Thus, the bimetal active centers in the catalytic materials show a worthy synergistic effect on the CO2RR, thus accelerating the whole CO2RR kinetics.189 Reddy et al.192 obtained a Co/Ce0.8Zr0.2O2 catalyst using an MOF as a template, which showed excellent activity and stability for the CO2RR to methane. Alternatively, metal nanoparticles are also widely used as catalysts due to their high conductivity, high stability, and large surface area. Zhang et al.193 used H2O2 as the only reductant and seed-mediated growth method to synthesize sea–urchin–needle-shaped gold nanoparticles as a catalyst for the CO2RR, effectively improving current density and CO selectivity. There is an obvious tip electric field effect at Au needles, which enriches cations and increases the CO2 concentration at the tip during the catalytic process. Kim et al.194 used MOF-74 derivatives to prepare highly dispersed Cu nanoparticles to increase the yield of CO2RR to CH4. The experimental results showed that the Cu NPs derived from the MOF had low aggregation and excellent electrocatalytic properties due to the existence of a stable eutectic structure, high selectivity and activity for mono-carbon products.
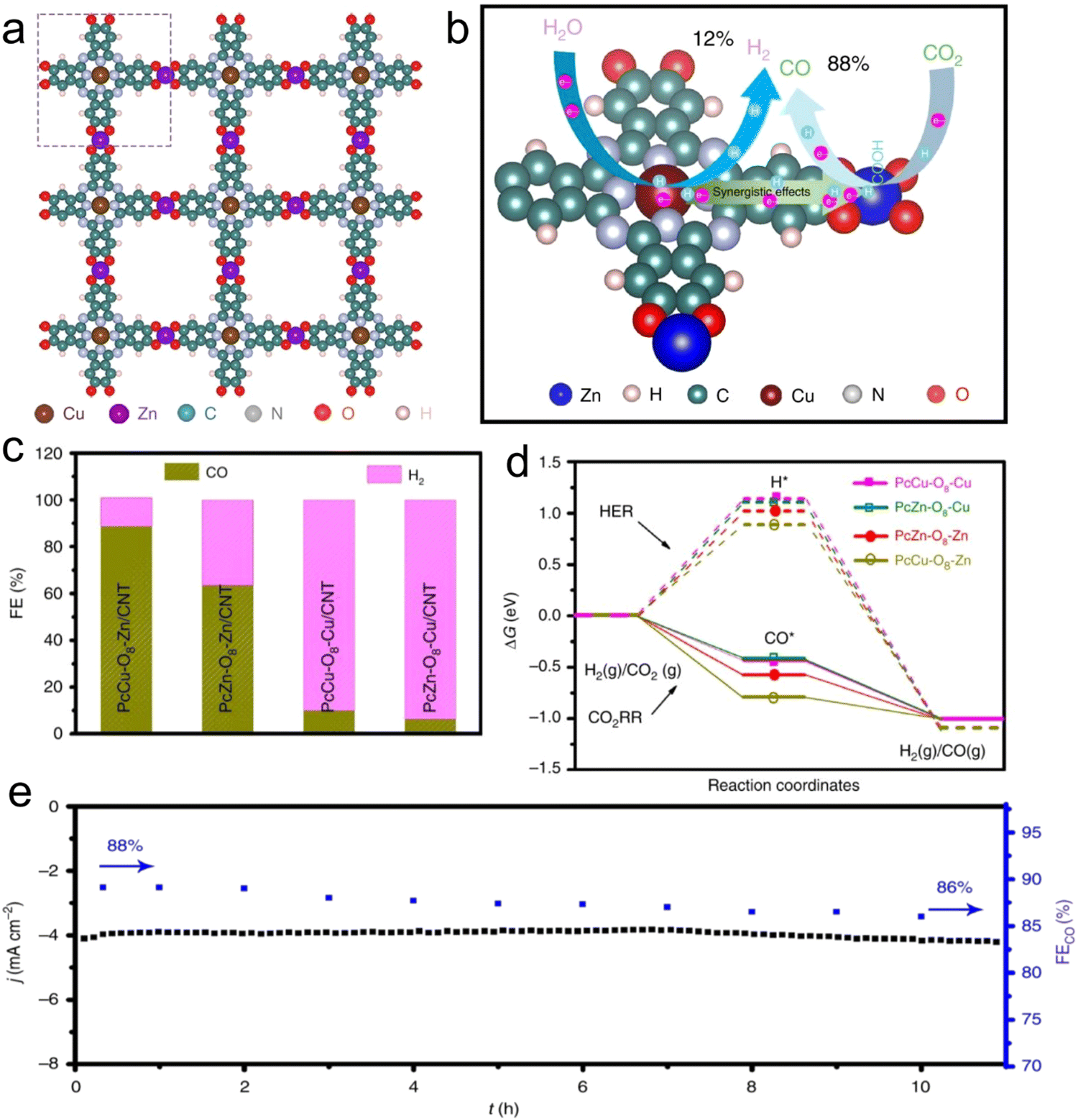 | ||
| Fig. 19 (a) Illustration of the crystal structure of Zn–PcCu–O8. (b) Depiction of CO2RR and HER over Zn–PcCu–O8. (c) FEs of H2 and CO for different catalysts. (d) Possible free energy diagram for different catalysts. (e) Amperometry (i ∼ t) stability test for Zn–PcCu–O8/CNT. Reproduced with permission from ref. 189 Copyright 2020, Springer Nature. | ||
The high selectivity of Cu NP catalysts for the CH4 was attributed to the inhibition of C–C bond coupling by the isolated NPs. Recently, Wang et al.195 reported the design of Cu nano-ribbons via the in situ electrochemical reduction of a Cu-based organometallic skeleton for high selectivity for the production of C2+ compounds (Fig. 20a). The experimental results showed that the FE for C2+ was 82% and the partial current density was 348 mA cm−2 in the flow electrolytic cell when the mesoporous structure was used for CO2 electroreduction (Fig. 20b). Compared with Cu nanoleaves and nanorods without a porous structure, the Cu nanobelts significantly improved the selectivity for C2+ products in CO2 electroreduction. The finite domain (FDTD) outcomes showed that electric field on the surface of catalyst can be increased using a mesoporous structure, thereby increasing the concentration of K ions and OH− and promoting the reduction of CO2 to C2+ products.
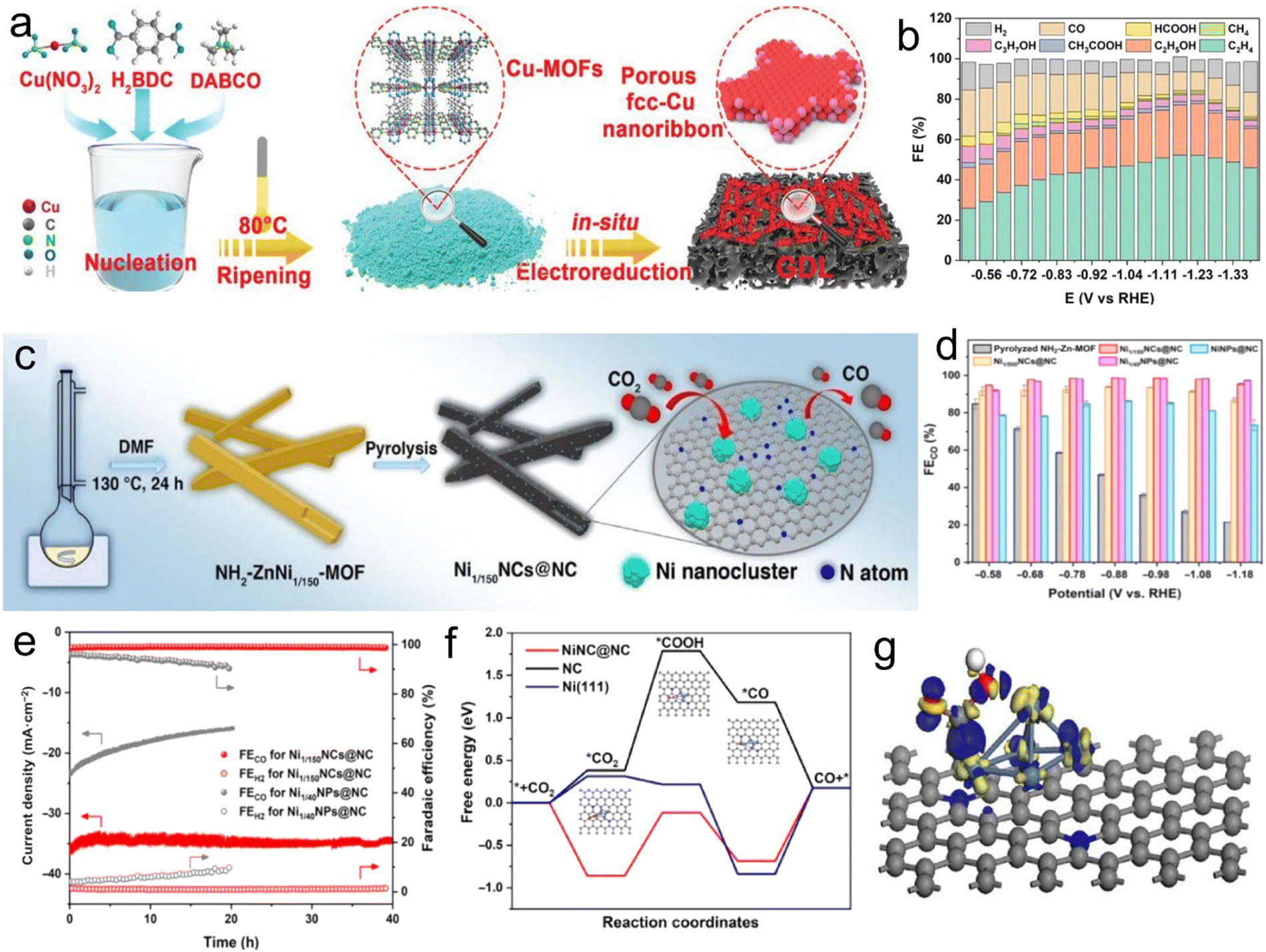 | ||
| Fig. 20 (a) Synthesis scheme of Cu-MOFs and porous Cu nanoribbons. (b) FEs for Cu nanoribbons. Reproduced with permission from ref. 195 Copyright 2021, Wiley. (c) Synthesis of Ni1/150NCs@NC catalyst. (d) FEs. (e) Stability tests. DFT calculation for CO2 electrocatalytic reduction: (f) free-energy diagram for different models. (g) Depiction of electron density difference between Ni cluster and *COOH intermediate. Reproduced with permission from ref. 68 Copyright 2022, Springer. | ||
Recently, Xu et al.68 synthesized highly dispersed Ni nanoclusters with a size of less than 2 nm on NC using Zn/Ni bimetallic MOF precursors (Fig. 20c). The contents and size of the material were successfully controlled by changing the proportion of Zn:Ni in the MOF precursor. The amino functional group on the MOF linker has an important influence on the Ni catalyst size and the performance of the carbon-based catalysts. At the best possible ratio of 150:1, Ni nanoclusters with a particle size of 2 nm were obtained by pyrolysis of the MOF. The synthesized catalyst exhibited a high FECO of 98% and partial current density (JCO, −40 mA cm−2) at −0.88 V (Fig. 20d). The design principle and discovery of the catalyst may have a certain guiding significance for the preparation of highly efficient electrocatalysts for various reactions using other metals. Using bimetallic Ni/Zn MOF precursors with different Ni:Zn ratios, Ni particles with a diameter of 1.8–200 nm were prepared on a nitrogen-doped carbon matrix without any post-processing steps. In the case of an ultra-low Zn:Ni ratio of 150:1 and the existence of amino functional groups in the MOF linker, vastly dispersed 1.9 nm Ni nanoclusters were obtained on NC. The results showed that catalytic performance was significantly enhanced by decreasing the size of Ni on NC. Ni1 was synthesized at the optimum Ni:Zn ratio of 1:150/150NCs@NC. At −0.88 V potential, it exhibited an efficient, selective and stable ECR for CO production with FE of 98.6% and JCO of −40 mA cm−2. In the 40 h stability test (Fig. 20e), the JCO was maintained at 96% and the FECO was stable. Furthermore, the models calculated by DFT showed that compared with the original NC and big-sized Ni particles, the Ni nanoclusters anchored on the NC substrate are more likely to form *COOH intermediates (Fig. 20f and g), and the electron transfer rate on the reaction interface is faster.
7.3 MOF-derived non-metallic carbon materials
Metal-free carbon materials derived from MOFs mainly include intrinsically defective materials and metal-free heteroatom-doped carbon materials. The heteroatom doping can improve the electronic property of materials; accordingly it causes confined charge polarization, spin state redistribution and local charge density, finally enhancing the desorption/adsorption process on the active sites for the reaction intermediates.198–200 In addition, the conductivity of carbon materials is increased by doping heteroatoms, which is also beneficial to activate the carbon atoms, and thus improve the catalytic performance of carbon materials.201 Nevertheless, Jia et al.202 recently found that the inherent defects of a five-membered ring showed greater activity than that of carbon doped with nitrogen atoms. With the deepening of research, it has been found that reasonably designed inherent defects (such as edges, holes, vacancies and topological defects) in the skeleton of carbon can regulate the electronic of materials, offering more active centers for catalysis. Especially, the presence of topological defects can efficiently increase the activity of catalysts for the CO2RR.203 The key technique to create topological defects in the carbon skeleton is denitrification. The original nitrogen atoms are removed from the graphene-like structure (six-membered ring) through the pyrolysis carbonization process to obtain five-membered rings and seven-membered rings. The carbon atoms with dangling bonds connect with each other to form topological defects.204 Kang et al.205 synthesized the Zn NP@ZIF-8 hierarchical porous carbon-rich catalyst with defects (DHPC) through pyrolysis. The mesoporous structure and carbon defects improved the CO2 adsorption and activation efficiency of DHPC. In 0.5 M KHCO3 medium, the FECO reached 99.5% at −0.5 V. The simulation results displayed that the CO2 adsorption energy barrier of the topologically defected five-membered ring was negative, demonstrating that the adsorption of CO2 is a spontaneous process. The N-free DHPC catalyst had a lower energy barrier to reduce CO2 to CO than the N-pyridine-doped carbon catalyst. In addition, the *CO desorption process on the surface of DHPC produced heat. Consequently, the MOF-derived inherent carbon defects had an appropriate adsorption/desorption energy for CO2 and the reaction intermediate. In turn, the CO2 molecules were excited and the electrocatalytic CO2 reduction performance was improved. Chen et al.196 removed pyrrole-N and pyridine-N from porous carbon materials, while they were doped with N heteroatoms through annealing at high temperature under an NH3 atmosphere using an MOF, resulting in extra topological defected carbon (Fig. 21a). The results from NEXAFS characterization of carbon illustrated that with an increase in the annealing temperature, the ratio of pyrrolic and pyridinic-N in the carbon skeleton decreased, with the topological defect density progressively increasing, and the CO2RR performance also improved accordingly. Specifically, DPC-950-NH3 exhibited significant catalytic activity and selectivity for CO2RR to CO. DFT calculations showed that the reaction intermediate has more suitable adsorption and desorption free energy on the carbon material containing defects than that without defects (Fig. 21b). In general, the activity of N-doped carbon materials depends on the content and type of N-doped elements. Zheng et al.197 reported that N-doped mesoporous carbon materials were obtained by metal–organic skeleton transformation and controlled by the secondary treatment of N,N-dimethylformamide (DMF). The carbon materials obtained from metal–organic skeleton conversion had numerous electron-rich defects due to N-doping (Fig. 21c). However, due to the high-temperature treatment, these electron-rich defects have no CO2RR activity. Based on this, when DMF is used for secondary treatment of carbon materials obtained from metal–organic skeleton conversion, the porous and defective characteristics of the carbon materials are conducive to the entry of small molecules of DMF, which can form more pyridine–nitrogen catalytic active sites and increase the CO2RR efficiency of the catalyst. The electrochemical test outcomes displayed that the N-doped mesoporous carbon structure (MNC-D) after DMF secondary treatment had high catalytic activity for the CO2RR in 0.1 M KHCO3 electrolyte. Under the voltage of −0.58 V vs. RHE, the reduction current of carbon dioxide reached −6.8 mA cm−2, and the selectivity for carbon monoxide reached ∼92%. Simultaneously, after 16 h reaction at −0.58 V vs. RHE, its current density and selectivity were 84% and 83% of the initial value, respectively, showing excellent cycle stability (Fig. 21d and e). Thus, this method provides a feasible idea for preparing carbon-based CO2RR materials.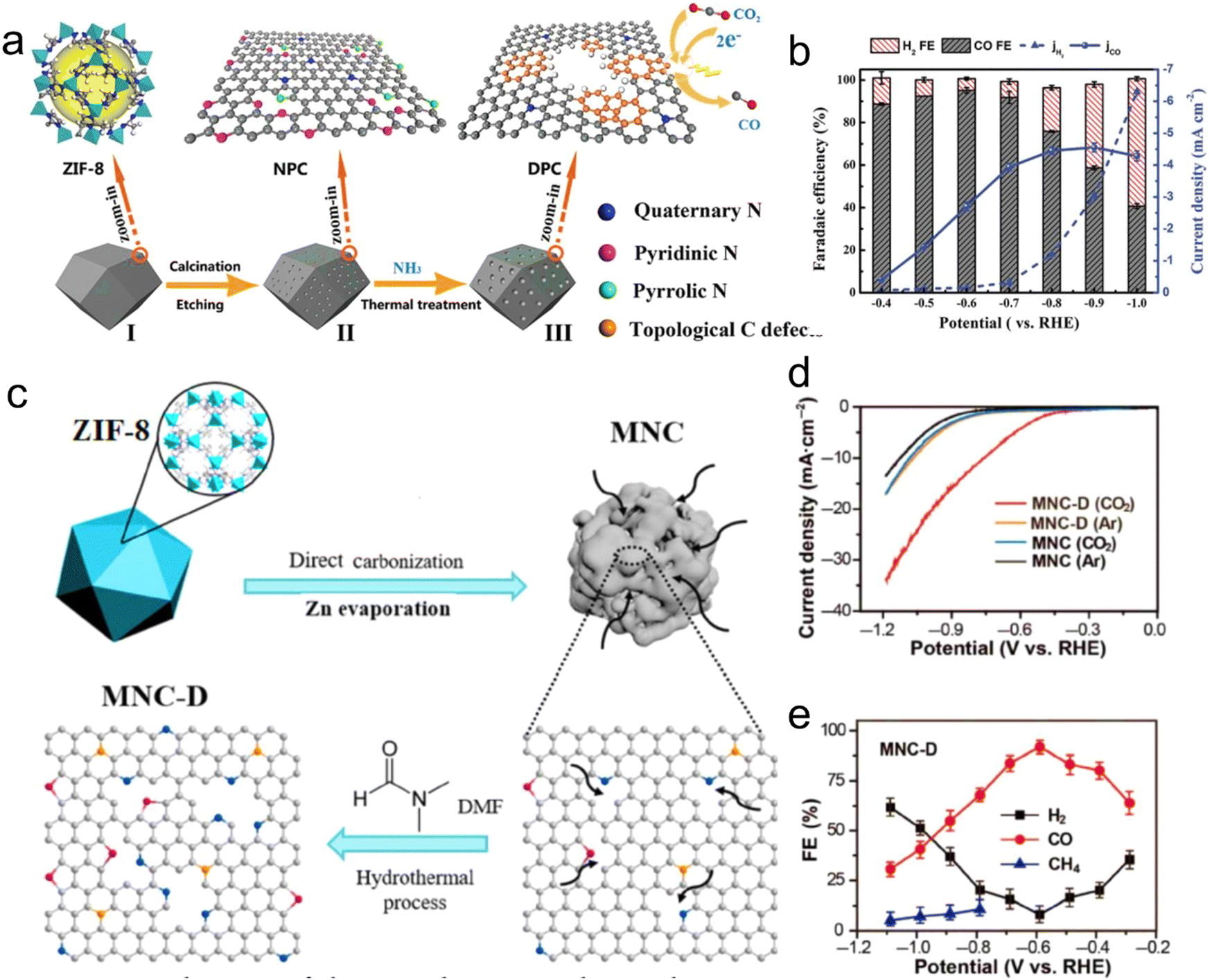 | ||
| Fig. 21 (a) Synthesis route of defected porous carbon. (b) FEs of CO and H2 and the partial current densities over DPC. Reproduced with permission from ref. 196 Copyright 2020, Wiley. (c) Synthetic procedure from MNC-D. (d) LSV curves of MNC-D and MNC. (e) FEs over MNC-D. Reproduced with permission from ref. 197 Copyright 2019, Springer. | ||
In addition, the catalytic performance of carbon materials without doped heteroatoms can be significantly enhanced by suitably regulating the inherent carbon defects. Inherent defects will certainly be introduced in the synthesis of carbon materials. Thus, it will be significant to deeply understand the influence of inherent defects of catalysts for CO2RR outputs using different advanced techniques to interpret their mechanism, particularly in situ techniques and theoretical calculations to obtain further new effective and robust electrocatalysts. In most studies, compared with non-metal carbon materials derived from MOFs, pre-designed reaction centers and monatomic catalysts having porous structures of can enable desorption/adsorption for CO2RR. Nevertheless, because of the high surface structure of single-atom metal catalysts, their catalytic activity significantly decreases in the electrolysis process. Moreover, increasing the interactions between the support and metal is of great importance to improve the durability of monoatomic catalysts in practical applications.
7.4 MOF-derived single-atom catalysts
Catalysts based on transition metals have been studied intensively in the field of CO2RR. The electronic structure of valence electrons of transition metals changes when they are specifically adsorbed with CO2 reduction intermediates due to the existence of d orbitals that are not occupied by electrons. The CO2RR is a multi-step PCET process, and the inherent activity of the catalyst can be evaluated by the electronic behavior of the active centers on the its surface. In this case, the change in the valence electron structure of transition metals is beneficial to improve the reduction efficiency and selectivity in the CO2RR.Given that iron (Fe) is the second most abundant metal on Earth, it is desirable to develop Fe-based SACs to realize the CO2RR.207 Because of their large abundance, it is convenient to study the mechanism of CO2RR on these materials with the most common FeNx structure. However, in the preparation of SACs, there is a common problem that single atoms and nanoparticles coexist, which have a noticeable influence on the catalysis performance.208,209 The proportion of CO/H2 products can be adjusted by altering the ratio of Fe–N4 sites to Fe nanoparticles. For example, an Fe–N–C catalyst (Fe0.5d) with no Fe nanoparticles and 1.5% Fe mass loading showed the best performance, indicating that Fe–N4 is the active site for catalyzing CO2 to CO.210 Some studies also demonstrated that the Fe site bound to pyridine–nitrogen is the real active center for the CO2RR by poisoning the active center through specific binding with Fe–N4 without affecting the N–C site by using a reactant with good affinity to the Fe site (such as SCN−).211 At present, the structure of FeNx is usually based on MOFs as the carbon framework (such as ZIF-8), and then iron ions are further adsorbed using the vacancy generated by the volatilization of Zn at high temperature to capture Fe to obtain the structure of FeNx.137 Under the overpotential of 340 mV, the partial Id reached 94 mA cm−2 with FECO = 90%.
The Fe3+ site is the key to the catalytic activity. It was also be proven that the activity of the catalyst decreases due to the reduction of Fe3+ to Fe2+ at −0.5 V. Moreover, Li et al.213 stated that the Fe(I) site generated in situ is the active site for the CO2RR. The strong interactions between the dz2 orbital with a single electron of Fe(I) and the π* orbital occupied by a single electron in [COOH] is the key to excellent CO2RR activity. For the production of reduction products requiring more than 2e−, the larger desorption free energy of CO at the Fe–N4 site may play a positive role. Simultaneously, the proper adsorption of *H on the surrounding sites can just combine with CO bound on the Fe center, which promotes the production of more than 2e− reduction products.214,215
Recently, He et al.206 designed an efficient method to prepare copper-SA and proposed the concept of effective single atoms. Through the construction of network interpenetrating and self-supporting structures, the probability of single atoms at the interface is greatly improved to avoid the loss of their catalytic activity due to embedding (Fig. 22a). Because a large number of effective single atoms is obtained, a high current density can be achieved at a low metal single-atom content, which has a certain practical application prospect. Carbon nanofiber films (CuSAs/TCNFs) can be prepared with a size of ∼300 cm2 under laboratory conditions, which have good conductivity and mechanical strength. After cutting, they can be directly used as working electrodes for the CO2RR without any adhesive agents. The self-supporting through hole structure of the catalyst can prevent the active site from being embedded inside, forming an enormous copper-single atoms, which can participate in the CO2RR (Fig. 22b). CuSA/TCNF films could be used for the CO2RR in aqueous solution systems to obtain methanol with 44% FE and the partial Id of the C1 product reached −93 mA cm−2 (Fig. 22c). In addition, the CuSA/TCNF film could be used stably for more than 50 h without significant degradation in its performance. DFT calculation showed that Cu–N4–C structure can effectively reduce the Gibbs free energy of the *COOH intermediate and improve the catalytic activity of CuSAs/TCNFs. Simultaneously, Cu–N4–C has good adsorption ability for the *CO intermediate, which can further reduce the *CO intermediate to methanol (Fig. 22d and e). In addition, the hierarchical pore structure of CuSAs/TCNFs can improve the electrochemical active area, enhance the CO2 adsorption capacity, and increase the effective active sites for the CO2RR, leading to a higher current density. Zu et al.216 realized the synthesis of positive-charged single atoms of Sn (Snδ+). The structure and morphology of the catalyst were confirmed by XAFS and HAADF-STEM characterization. In situ FTIR showed that Snδ+ activated CO2 and realized the subsequent protonation process by stabilizing the CO2˙− and HCOO−* intermediates. Simultaneously, nitrogen doping also promotes the desorption process of the decisive HCOO− step. Therefore, the initial potential of HCOO− generation was only −60 mV and the TOF of HCOO− at −1.8 V reached 11930 h−1vs. SCE. Furthermore, its electroreduction activity could be maintained for 200 h. They also synthesized an Sn1/CuO composite single-atomic catalyst with oxygen vacancies through metal support interaction. Zhang et al.217 obtained different morphologies at different pyrolysis temperatures and the structural characterization of the different materials is shown in Fig. 23. Fig. 23f shows that Bi in BiSAs/NC exists in the form of single-atomic dispersion. The catalyst achieved 97% FECO at a low overpotential and the TOF reached 5525 h−1. DFT calculation showed that due to the low reaction energy barrier, the Bi–N4 structure promotes the rapid formation of the CO intermediate *COOH. The nitrogen-doped porous carbon-supported Sb monoatomic catalyst synthesized by the calcination of urea and carbon black achieved efficient CO2RR to CO, and the TOFCO reached 16500 h−1.218 In addition, another single-atom catalyst with Sb–N4 structure exhibited selectivity of 94% for the CO2RR to formate.219 DFT showed that the Sb–N4 site is more conducive to the formation of HCOO*, and thus the product is mainly formate rather than CO. This is different from the product reported in the previous Sb single-atomic catalyst. This may be due to the different preparation methods, resulting in a change in the electronic structure of the single-atomic site, and the intermediate conducive to adsorption changes from *COOH to HCOO*. In terms of catalyst preparation method, Sb–N4 on nitrogen-doped carbon is mainly obtained by co-pyrolysis of SbCl3 with dicyandiamide and pyromellitic acid. In situ XAFS and DFT confirmed the presence of Sbδ+–N4. It is favorable for the CO2RR to HCOO− to occur at the N4 site. For these reported catalysts, their FE reached a relatively high level, but the current density is still low and far away from the requirement for industrial use (1 A cm−2).
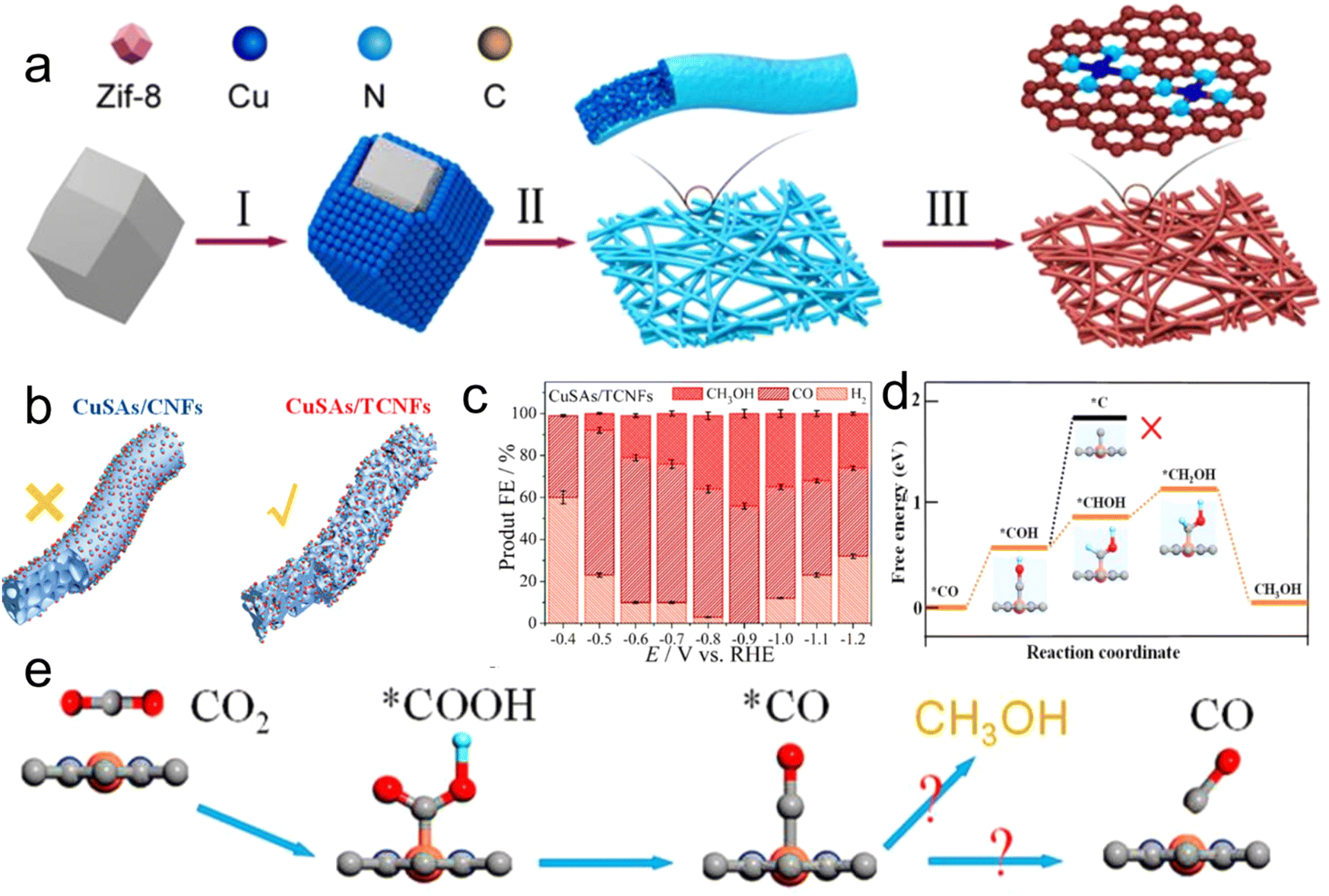 | ||
| Fig. 22 (a) Synthesis of Cu single atom. (b) Illustration of CO2 diffusion on two samples. (c) FEs of products. (d) Free energy model for the conversion of *CO to methanol. (e) Optimized atomic structures of CuSAs/TCNFs and proposed reaction paths for CO2 electroreduction. Reproduced with permission from ref. 206 Copyright 2019, ACS. | ||
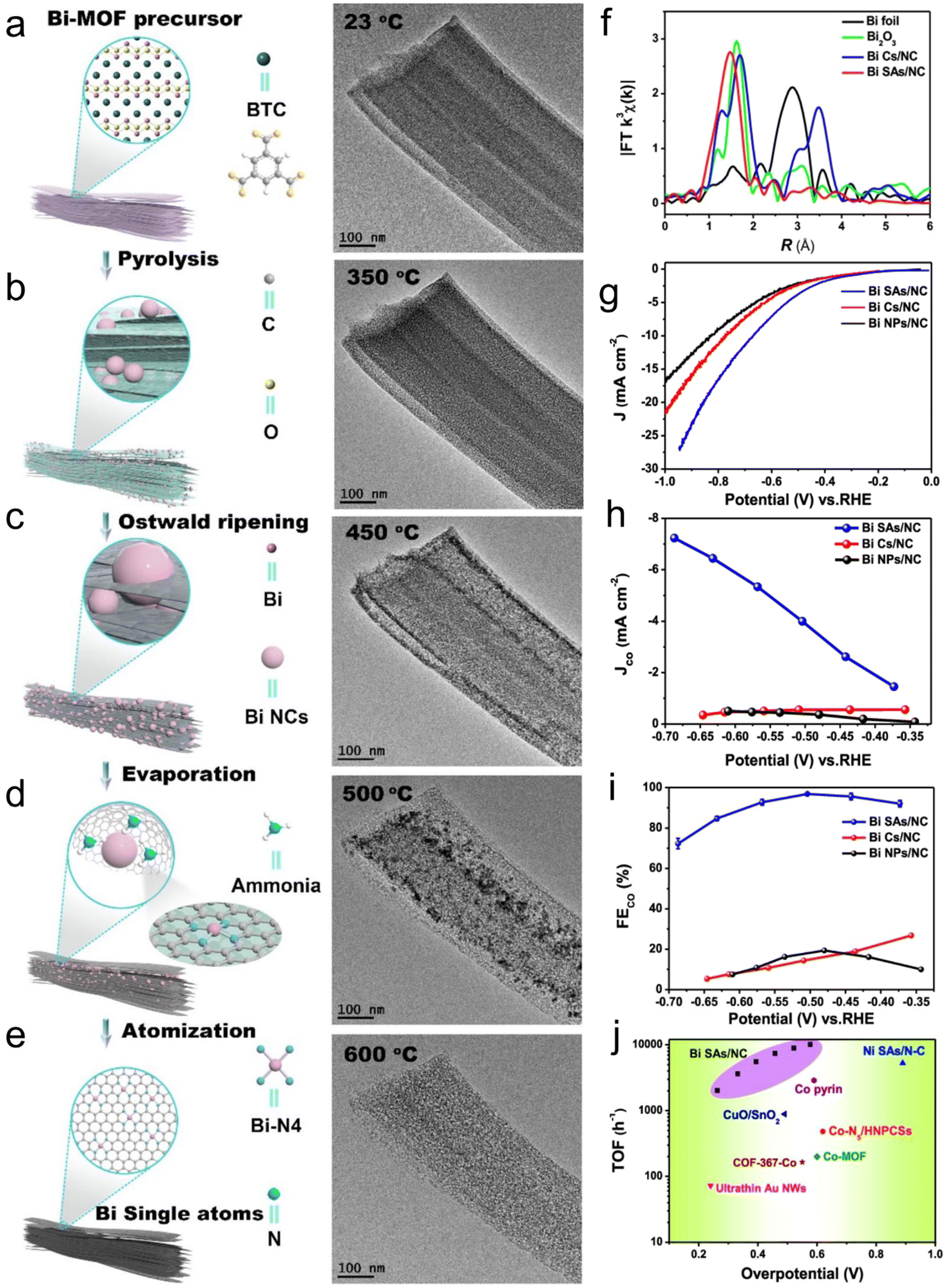 | ||
| Fig. 23 (a–e) Illustration of the transformation of Bi-MOF to Bi-SA together with corresponding TEM images. (f) EXAFS spectra with k3χ(k) function. (d) LSV curves. (h) Partial current densities of CO. (i) FECO and (j) comparison of TOF values. Reproduced with permission from ref. 212 Copyright 2019, ACS. | ||
Furthermore, Wang et al.33 reported the preparation of a type of N-heterocyclic carbene-linked MOF and copper single-atomic center (2Bn–Cu@UiO-67, where Bn is di-benzyl imidazole). Under the condition of Id of 420 mA cm−2, at −1.5 V the FE of CH4 was promoted to as high as 81%. It is worth noting that the FECH4 of the catalyst remained above 70% in a wide potential range and reached an unprecedented 16.3 s−1 TOF, which is almost the best CH4 electrosynthesis catalyst thus far. The researchers chose UiO-67 as the substrate material to load the SAS active center connected by NHC, which can well match the molecular size of NHC and remain stable for several hours in aqueous solution. Simultaneously, the porosity of the catalyst promoted the diffusion of CO2 to 2Bn–Cu, thus significantly improving the utilization of each catalytic site. The design of this NHC-linked Cu-SAS catalyst has great advantages in methane electrosynthesis, providing a new idea for the industrial production of methane. To compare the CO2RR performance of MOF-derived catalysts, a summary of the latest results of the CO2RR is given in Table 3.
| Material | Precursor (MOF) | Main product | Electrolyte | faradaic efficiency [%] | Potential | Current density [mA cm−2] | Ref. |
|---|---|---|---|---|---|---|---|
| NC | ZIF-8 | CO | 0.1 M KHCO3 | 78 | −0.93 | −1.1 | 220 |
| Fe–N–C | ZIF-8 | CO | 0.1 M NaHCO3 | 91 | −0.6 | 4.5 | 221 |
| Fe–N–C | ZIF-8 | CO | 0.5 M KHCO3 | 93.5 | −0.5 | NA | 222 |
| Fe–N–C | Zn–Fe ZIFs | CO | 0.5 M KHCO3 | 86.9 | −0.47 | NA | 223 |
| Ni–N–C | ZIF-8 | CO | 0.5 M KHCO3 | 71.9 | −0.9 | NA | 180 |
| Zn Ni–N–C | Zn Ni–MOF | CO | 1 M KHCO3 | NA | -1.03 | 71.5 ± 2.9 | 224 |
| NiSA–Nx–C | Mg–Ni–MOF-74 | CO | 0.5 M KHCO3 | 98 | −0.8 | NA | 225 |
| Co–N–C | Zn–Co MOF | CO | 0.1 M KHCO3 | 82 | −0.8 | NA | 226 |
| Co–N–C | Zn–Co ZIF | CO | 0.5 M KHCO3 | 94 | −0.63 | 18.1 | 227 |
| Bi–N–C | Bi MOF | CO | 0.1 M NaHCO3 | 97 | −0.5 | 3.9 | 217 |
| Cu–N–C | Cu-doped ZIF-8 | CH3COCH3 | 0.1 M KHCO3 | 36.7 | −0.36 | NA | 228 |
| Ni/Fe–N–C | Zn/Ni/Fe ZIF | CO | 0.5 M KHCO3 | 98 | −0.7 | 7.4 | 187 |
| Fe–N–C/CNTs | ZIF-8 | CO | 0.1 M KHCO3 | 97 | −0.56 | 1.9 | 229 |
| Ni–NPC/CNTs | ZIF-8 | CO | 0.5 M KHCO3 | 94 | −0.77 | 11.2 | 230 |
| Cu/Cu2O/NPC | ZIF-L | C2H5OH | 0.5 M KHCO3 | 70.52 | −0.87 | 10.4 | 231 |
7.5 MOF-derived diatomic catalyst
The development of diatomic catalysts (DACs) for the CO2RR has become a promising frontier research field due to its efficient atom utilization and complex functions. However, the design of DACs at the atomic level and the understanding of the two-site synergistic mechanism are still challenging. Recently, He et al.232 reported that with the help of an MOF, a novel N4Ni/CuN4 bi-site catalyst was developed (Fig. 24a). The crystal structure of the MOF provides ordered and periodic space to classify positions and adjust the relative space distance between the Cu and Ni precursors, which were doped in the form of metal nodes and encapsulated in the cavity of ZIF-8. Therefore, ordered Cu and Ni precursors are conducive to the formation of Ni/Cu adjacent sites after one-step thermal activation. | ||
| Fig. 24 (a) Schematic description of Ni/Cu–N–C fabrication. (b) FECO. Reproduced with permission from ref. 233 Copyright 2022, ACS. (c) Method for the synthesis of different metal-based catalysts. Reproduced with permission from ref. 234 Copyright 2021, RSC. (d) Free energy diagram for CO2 conversion. Reproduced with permission from ref. 233 Copyright 2022, ACS. (e) FE of H2 for various catalysts. Reproduced with permission from ref. 234 Copyright 2021 RSC. | ||
The experimental results showed that the catalyst containing N4Ni/CuN4 has excellent catalytic activity and selectivity for the CO2RR. At −0.79 V (vs. RHE), the FECO reached up to 99.2%, and in the range of −0.39 to −1.09 V (vs. RHE), it reached more than 95% (Fig. 24b). In addition, at −1.09 V, the TOF value was 6895 h−1, while that of Ni–N–C and Cu–N–C was 5119 and 2500 h−1, respectively. Further DFT calculations showed that the synergistic effect between heteronuclear monomers can also be achieved by metal atoms that exhibit similar bonding interactions with the intermediates such as Ni and Cu. Among them, the charge redistribution of the Ni centers caused by adjacent CuN4 sites promotes the adsorption of *COOH, thereby promoting the formation of CO (Fig. 24d). Although the FEs of most Ni-based monoatomic catalysts (Ni–N–C) have achieved satisfactory results in the CO2RR to CO (>90%), their high overpotential (>600 mV vs. RHE) still limits their practical application, which means that the energy consumption required to catalyze the CO2RR is high. Combining the benefits of Fe–N- and Ni–N–C-based catalysts, Jordi et al.234 described the design of a quasi-binary Ni/Fe catalyst to achieve high selectivity and low overpotential in the CO2RR process (Fig. 24c). Here, due to the weak CO bonding, the Ni–N–C catalyst has the ability to quickly desorb *CO (CO* → CO+*), while the Fe–N active center usually shows a low overpotential for the CO2RR due to the fast transfer rate of electrons from the first proton coupling. Simultaneously, due to the synergy between the closely located Fe and Ni active centers, they act as a nanoreactor, considerably affecting the reaction steps of both active centers, thereby improving the activity and selectivity of the CO2RR. In addition, in a specific environment, the presence of Fe near the Ni center will affect the electron density and conformation of the two active centers, consequently promoting the adsorption/desorption of the intermediates in the CO2RR. The researchers prepared ternary MOFs via a one-pot solvothermal method as an alternative to the complex and multi-step process, and by reasonably controlling the amount of Fe and Ni added to IrMOF-3, which is Zn based. Then, the adjacent Fe and Ni double-active centers are prepared by calcination. The experimental results showed that the as-synthesized Ni7/Fe3–N–C material displayed a good FECO of 98% at a low overpotential of −390 mV vs. RHE, which it is greater than that of the single metal catalysts of Fe and Ni. DFT calculations showed that this bimetallic catalyst with adjacent Ni and Fe centers is more favourable for the production of COOH* than the Ni–N–C catalyst. In addition, the Ni/Fe–N–C catalytic material not only promoted the desorption of CO*, but also avoided the undesirable HER side reactions compared with the Fe–N–C catalyst, achieving unexpected catalytic activity for the CO2RR (Fig. 24e). Therefore, the exceptional high catalytic activity can be ascribed to the Fe and Ni centers, which play a synergistic role in regulating the binding energy of the various intermediates throughout the desorption and adsorption process and affecting the different reaction steps of the CO2RR. Recently, Liang et al.235 reported the preparation of novel Bi–In alloy nanoparticles from bimetallic MOFs, considered as an active method to solve the environmental and energy crises. The FEHCOOH of 92.5% at Id of 300 mA cm−2 with 5170 μmol h−1 yield was achieved. In the membrane electrode assembly system, the stability of Bi–In material was also very good. Under the Id of 120 mA cm−2, the nanoparticles with an Fe content of less than 10% showed excellent stability for more than 25 h. In situ spectra and theoretical results showed that the BiIn bimetallic center can provide the best binding energy for *OCHO intermediates, thus accelerating the conversion of CO2 to HCOOH.
The use of organic framework-based catalytic materials in the CO2RR to valuable products has the potential to adjust the catalytic performance by tuning the characteristics of the framework chemically and physically with atomic distribution accuracy. Nevertheless, the adjustable performance of these functional materials is still greatly hindered by their poor conductivity. Mirica et al.236 demonstrated that four structural analogues of 2D cMOF made of metal phthalocyanine (MPc) ligands connected by copper phthalocyanine have a conductivity in the range of 2.73 × 10−3 to 1.04 × 10−1 S cm−1 and can used for CO2RR to CO (Fig. 25a). The catalytic performance (including activity and selectivity) of the MOF was hierarchically controlled via two key factors, i.e., the metal (M = Ni and Co) subunits in MPc and heteroatom cross linkers (X = NH vs. O) between these subunits. The catalytic performance dominantly depended on the selected metal, and additionally regulated by heteroatom bonds. CoPc–Cu–O displayed high selectivity (FECO = 85%) with Id of 17 mA cm−2 under an overpotential of −0.63 V. Without the addition of carbon additive, the current density of −9.5 mA cm−2 was achieved by using CoPc–Cu–O purely as the catalytic electrode, and the FECO was 79% (Fig. 25b–e).
 | ||
| Fig. 25 (a) Synthesis scheme of MPc–Cu–X MOFs. (b–e) FEs of H2 product for different (catalysts). (f) Suggested mechanism for catalytic electrochemical reduction of CO2 to CO on MPc–Cu–XH MOFs. Reproduced with permission from ref. 236 Copyright 2020, ACS. | ||
The reaction mechanism was determined through comparative experiments with phthalocyanine MOF having no metal atom analogues, which supported the main catalytic action of the phthalocyanine central metal on the Cu node (Fig. 25f). DFT calculation further showed that compared to analogues based on NiPc and -NH linkage, the MOFs based on CoPc and O linkage have inferior activation energy in the production of carboxyl intermediates, which is consistent with their higher selectivity and activity. This study demonstrated that 2D-MPc with conductive frame materials has great hope to achieve effective carbon dioxide fixation through tactical linker engineering with multi-level adjustability. These works emphasize the important role of synergetic adjacent bimetallic materials as promising CO2RR electrocatalysts and reveal that these bimetallic centers can well serve as a double-site nanoreactors, affecting different reaction steps of different active centers with electronic and atomic engineering structures, which probably support the application potential of DACs in various fields.
8. Regulation of catalyst microenvironment
It is also one of the key aspects that is widely studied in current research to regulate the microenvironment of single atoms to change the intrinsic catalytic properties of the original single atoms. The regulation of the coordination environment of single atoms of non-noble metals can further improve the catalytic performance, even to a level equivalent to that of noble catalysts. This method avoids the use of expensive noble metal catalysts, and instead selects abundant metal elements, which has potential to realize large-scale industrial applications.8.1 Heteroatom coordination
000 h−1, Fig. 26a–d). This method of adjusting the single-atom coordination environment to change the selectivity of the products lays the foundation for further exploring the single-atom structure-performance relationship. Considering the low current density of the CO2RR at present, Chen et al.239 proposed a novel amination strategy to improve the current density of Ni monatomic catalysts for the CO2RR. Ni–N4/C formed by pyrolytic Ni-doped ZIF-8 was co-heated with urea, and then reacted with ammonia via the hydrothermal method to achieve the amination of the Ni monatomic catalyst. FTIR and XPS both proved the successful modification of -NH2. Tests in a flow-cell showed that the FECO was close to 90% at −0.89 V, and the Id significantly increased to 450 mA cm−2, which can be ascribed to the fact that -NH2 adjusted the electronic structure of the catalyst and enhanced the adsorption and charge transfer efficiency of the *CO2 and *COOH intermediates.
 | ||
| Fig. 26 (a) LSV curves for InA/NC catalyst. (b) FEs and (c) partial current densities over for CO. (d) FEs of formate, H2 and CO. Reproduced with permission from ref. 238 Copyright, 2021 ACS. (e) Schematic of the synthesis of Co–N5/HNPCSs. Reproduced with permission from ref. 241 Copyright 2018, ACS. | ||
The catalyst achieved an FECO of and partial current density of 4.7 mA cm−2 at −0.53 V. The coordination effect of N in FexN on the Fe sites effectively solved the problem of catalyst poisoning caused by the excessive combination of CO and Fe sites.222 The catalyst with a clear Fe–N5 site was obtained via the co-pyrolysis of Fe, melamine and graphene, and subsequently leaching with sulfuric acid. Without the addition of graphene, only an SAC with Fe–N4 site structure could be obtained. During the pyrolysis of melamine, the introduced graphene is mixed with nitrogen heteroatoms and the Fe–N4 sites dispersed on this support will coordinate with the additional N atoms axially, forming Fe–N5 sites. Based on this, the FECO of the catalyst was about 97% at the overpotential of 0.35 V. Theoretical calculation showed that the additional axial coordination further reduces the electrons in the Fe 3d orbital, thus reducing the feedback π bond between Fe and CO, and thus CO is easier to desorb from the Fe site.242 The Co–N site can also enhance the selectivity of the CO2RR by introducing additional N coordination atoms. Chen et al.241 prepared hollow nitrogen-doped carbon spheres via a templating method and obtained electrocatalysts with a Co–N5 coordination configuration by impregnating and adsorbing cobalt phthalocyanine (CoPc). The nitrogen atom in the hollow nitrogen-doped carbon ball was used as an additional nitrogen source to bond with the Co–N4 site in CoPc. A schematic diagram of its synthesis is shown in Fig. 26e. In situ XANES characterization determined that the catalytic active site is Co–N5. The catalyst maintained an FECO of more than 90% in a wide voltage window (−0.57 to −0.88 V), and an FECO of greater than 99% in the window of −0.73 to −0.79 V. Simultaneously, after 10 h testing, the current density and FE exhibited almost no change, indicating that the catalyst has good stability. Theoretical calculations further explained that the formation of *COOH on the Co–N5 site requires a lower free energy, and CO desorption is easier. A similar trend can be found by comparing Co–N4 site with Co–N4−x–Cx. At −0.8 V vs. RHE, the former exhibited an FECO of 82%, while that of the latter was 47%.226 In addition, by comparing the influence of the Ni–C/Ni–N coordination environment on the electrocatalysis performance, the results showed that a change in the coordination microenvironment will lead to a change in performance, which further confirms the above-mentioned conclusion.243 It is notable that in their early work on studying the effect of the coordination number on catalytic performance, they presented different rules. With a reduction in the coordination number of the N-atom, the selectivity of CO increased. By adjusting the annealing temperature, Co–Nx with different coordination numbers was obtained, where upon increasing the temperature, the value of x also decreased gradually. Among them, Co–N2 showed the most outstanding performance. At 520 mV overvoltage, 94% CO FE was achieved. The corresponding partial J was −18.1 mA cm−2. Theoretical research showed that the reduction of the coordination number of the N atom near the Co atom produces more unfilled orbitals of 3d, which is conducive to CO2˙− adsorption. Simultaneously, Co–N2 has the lowest charge transfer impedance, that is, it promotes the activation of CO2, accelerates the charge rate, and leads to an improvement in the CO2RR performance.181 Obviously, the coordination modes of the N atom and central Co atom in these two studies are different. The former increases the axial coordination, while the latter decreases the in-plane coordination, and the reduced N atom is replaced by a C atom. Moreover, these outcomes further show that changing the coordination mode of the coordination atom will significantly affect the catalytic performance. Similarly, by changing the pyrolysis temperature, Jiang et al.225 prepared several NiSA–N4-2–C catalysts. Among them, NiSA–N2–C exhibited better catalytic activity than NiSA–N3–C and NiSA–N3–C. Its highest FECO reached 98% and its TOFCO was 1622 h−1. The catalyst with Ni–N2 site was successfully obtained through plasma-assisted method and nitrogen vacancy-induced coordination reconstruction strategy. Due to its special electronic structure, the free energy barrier for CO2 molecule adsorption and activation was lower.244 Thus, this catalyst could achieve 96% FECO at a low overpotential (590 mV), and J reached 33 mA cm−2 at an overpotential of 890 mV. In addition, considering the charge capacity and hydrogen bond interaction, theoretical calculation further indicated that Ni–N1C3 has higher activity and selectivity than its counterparts in the series (Ni–NxC4−x).245 Recently, Zheng et al.245 reported a plasma activation strategy for the synthesis of Cu-based MOF single-atomic catalyst. The oxygen atoms in the catalyst that were previously coordinated with Cu were partially removed by plasma treatment, thus forming a low-coordination Cu single-atomic site rich in oxygen vacancies. Simultaneously, plasma treatment also led to the porous nature of the material. The low-coordination Cu single-atomic and hierarchical porous structures jointly promoted the CO2RR. The electrochemical performance showed that the maximum FE of CH4 reached 75.3%. The selectivity for the CO2RR products reached 96.5% with a J of 48 mA cm−2. In addition to the vacancies generated by the reduction in the number of N-coordination atoms described above being replaced by C atoms, it has also been reported that Ni–NxV4−x catalysts (where V represents defects, i.e., no coordination atoms) can inhibit the HER.224 This SAC containing coordination unsaturated Ni was obtained via the calcination of ZIF-8 containing Ni, and the mass loading of Ni reached 5.44%, which is higher than that reported in the literature. Another study showed that the Ni–N3 site promoted CO production from the CO2RR with FECO exceeding 90%, and the conversion frequency reached up to 12000 h−1 with the mass activity of 10600 mA mg−1.246 Based on the weak interaction between Ni–O, Lu et al.247 directly pyrolyzed the precursor containing Ni(II)–N/O. With the removal of O, vacancy V was generated, and the structure of Ni–N3–V was obtained. The catalyst showed a much better performance than the Ni–N4 single atom, with a TOF of 1.35 × 105 h−1, which is 4 times larger than that of Ni–N4. In conclusion, these results prove the advantages of the Ni–N(S)xV4−x configuration.
Besides the coordination number, exchange of the coordination atom is another strategy for regulating the electrocatalytic activity of single atoms. The introduction of atoms with lower electronegativity can decrease the energy graph of the intermediates in the CO2RR process, and ultimately accelerate the reaction kinetics. On the carbon-based plane, the asymmetric Cu–S1N3 atomic interface has an optimized *COOH binding energy compared with the Cu–N4 site. In addition, the nearby Cu nanoparticles (Cux) promote the protonation process. Therefore, Cu–S1N3/Cux showed nearly 100% FECO at the voltage of −0.65 V, and maintained more than 90% FECO in the voltage range of −0.55 to −0.75 V, which is superior to Cu–N4 (FECO = 54% at the voltage of −0.7 V) and Cu–S1N3 (FECO = 70% in the potential range of 0.7 to 3 V).251 Zhang et al.252 obtained a Cu–O4 site catalyst (Cu–DBC) by connecting unit point Cu at the MOF node. The methane selectivity of this catalyst was close to 80%, and under the same condition, the partial Id reached −203 mA cm−2 at −0.9 V (vs. RHE), where the Cu–O4 site has a lower CO2RR reaction barrier than the Cu–N4 site Ni–N2–S obtained by S doping. Ni–N2 will generate S-vacancy under the condition of high overpotentials, and both the S-atom and S-vacancy can reduce the reaction energy graph of CO2 to CO. Consequently, the FECO of the catalyst reached 97% under the condition of −0.8 V vs. RHE, with the CO partial Id of 40.3 mA cm−2 under the condition of −0.9 V (vs. RHE).253 Furthermore, changing the type of coordination heteroatom can also affect the reaction path and generate different products. For example, SnN3O1 shows unique properties compared with the main products of Sn–N4, i.e., HCOOH and H2, and the main product CO. Theoretical calculation showed that the SnN3O1 configuration can reduce the energy barrier of *COO activation and *COOH formation.254 In Cd–N4S1, the S atom is axially coordinated with the Cd atom, while the Cd atom is anchored on the graphite carbon plane of Cd–N4.248 The structure diagram is shown in Fig. 27a, where the axial S atom coordination not only reduces the energy graph of CO2RR, but also inhibits the HER. Consequently, Cd–N4S1 showed a superior CO2RR performance, reaching a CO partial Id of 182.2 mA cm−2 with FE of more than 95% in an H-type electrolytic cell. Also, the TOF reached 73000 h−1, which is the best performance reported to date in an H-type electrolytic cell. Theoretical calculation showed that the Cd–N4S1 single atom has an influence on reduction of the energy graph of the CO2RR compared with Cd–N4. Sn–Ox mononuclear species were anchored in carbonized ZIF-8 by chemical vapour deposition. Then, further fluorination was achieved by etching ZIF-8 with C2F4 generated from the pyrolysis of polytetrafluoroethylene, and finally a single-atom electrocatalyst with Sn–C2O2F structure was synthesized. It could maintain an FECO of 90% in a wide potential range (−0.2 to −0.6 V). Under the same conditions, formate (up to 95.2%) was the main product of Sn–N4. The adsorption of the intermediates was optimized through the coordination regulation of the C and O atoms, while the introduction of axial F atoms inhibited the HER side reaction and promoted the conversion of CO2 to CO.249 However, as shown in Fig. 27b–e, compared with Sn–C2O2, the formation energy barrier of *COOH + H+ also increased to a certain extent when the HER is inhibited. Based on this, under the overpotential of 490 mV, the FECO of the catalyst was 90.5% and Id was 10.8 mA cm−2, with the TOF reaching 1566 h−1. Zhang et al.250 clarified through DFT calculations and in situ XAS that the introduction of axial Cl coordination atoms can effectively decrease the energy of formation barrier of key intermediates at the Mn–N4 site. The structural characterization results are shown in Fig. 27f and g. Under the overpotential of 0.49 V, the CO Id was about 10 mA cm−2, and the maximum FE was 97%. The above-mentioned study showed that the introduction of axial coordination heteroatoms shows different properties for different metal single-atomic sites. In addition, Chen et al.255 synthesized an Fe1N4–O1 catalyst through the molten-salt assisted strategy, rapid pyrolysis and controllable activation strategy. Although its oxygen atom was also located in the axial coordination position, this oxygen atom is located in another graphite carbon plane. It is different from the former in that, it is independent of the out-of-plane oxygen atom. This oxygen atom can localize the electrons, promote the desorption of CO, and improve the energy barrier of HER. Therefore, the catalyst could obtain nearly 100% FECO in the voltage range of −0.56 to −0.87 V. Heteroatoms that do not directly coordinate with the metal will also affect the electronic structure of metal single atoms. For example, in the third coordination shell, the P atoms in the form of P–C bonds will enhance the electronic localization of the Fe single atom.256 Thus, the stability of the *COOH intermediate is enhanced, showing 97% CO selectivity at a low overvoltage (320 mV).
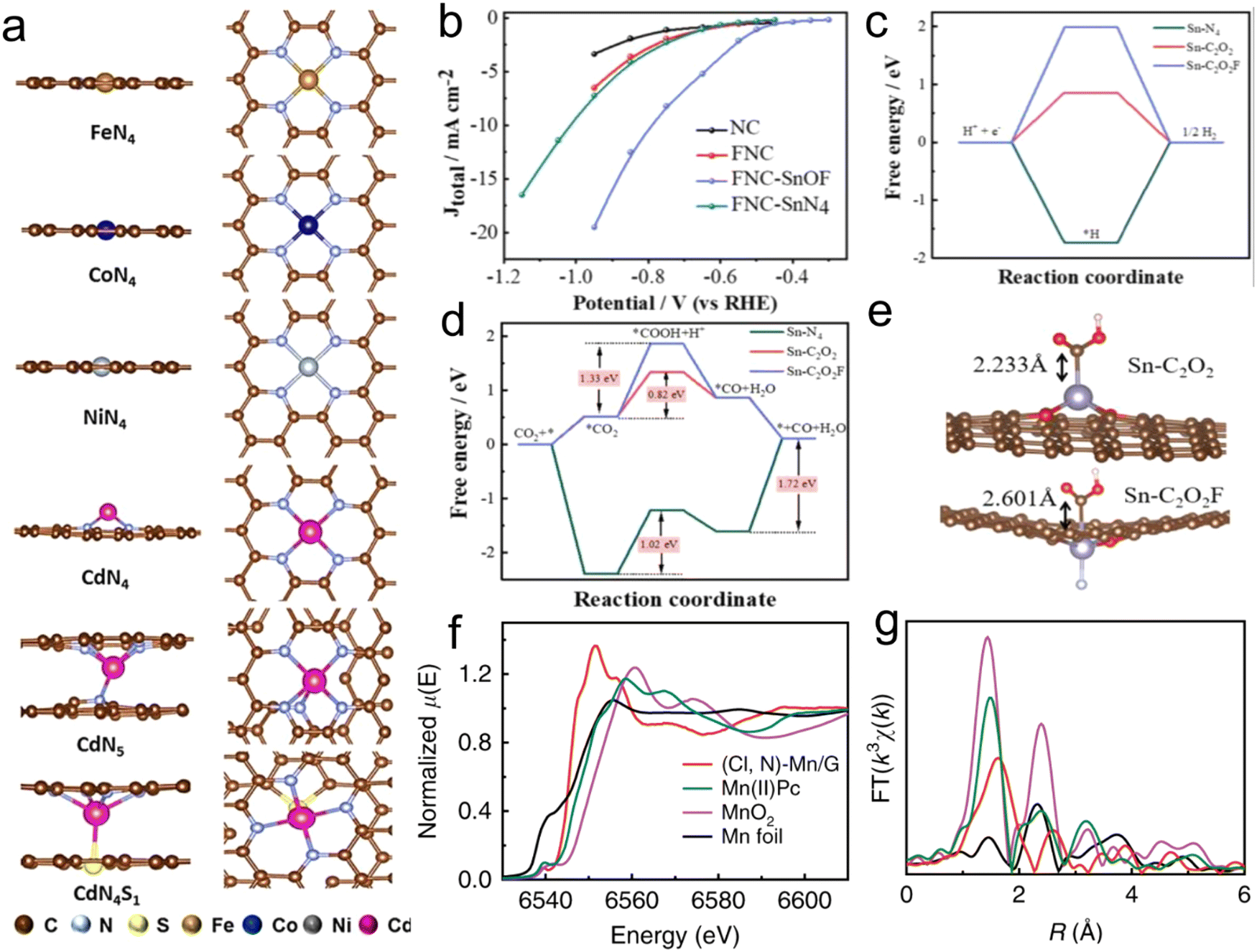 | ||
| Fig. 27 (a) Top and side views of different metal nitride catalysts, Reproduced with permission from ref. 248 Copyright 2021, Wiley. (b) JTotal of NC, FNC, FNC–SnOF, and FNC–SnN4 measured by the controlled potential electrolysis with a carbon paper electrode. (c) Free energy diagram for HER. (d) Free energy diagrams for CO2 conversion to CO. (e) Adsorption configurations of *COOH on Sn–C2O2 (top) and Sn–C2O2F (bottom). Reproduced with permission from ref. 249 Copyright 2021, RSC. (f and g) EXAFS and XANES spectra at Mn K-edge. Reproduced with permission from ref. 250 Copyright 2019, Nature Springer. | ||
In summary, most of the research focused on optimizing the local electronic structure, adjusting the adsorption and binding strength of intermediates, and achieving high selectivity of target products by finely regulating the coordination number of metal single atoms, the types of heteroatoms in coordination, and the coordination structure. Thus, further research is needed on the impact of this strategy on the overall activity of SACs.
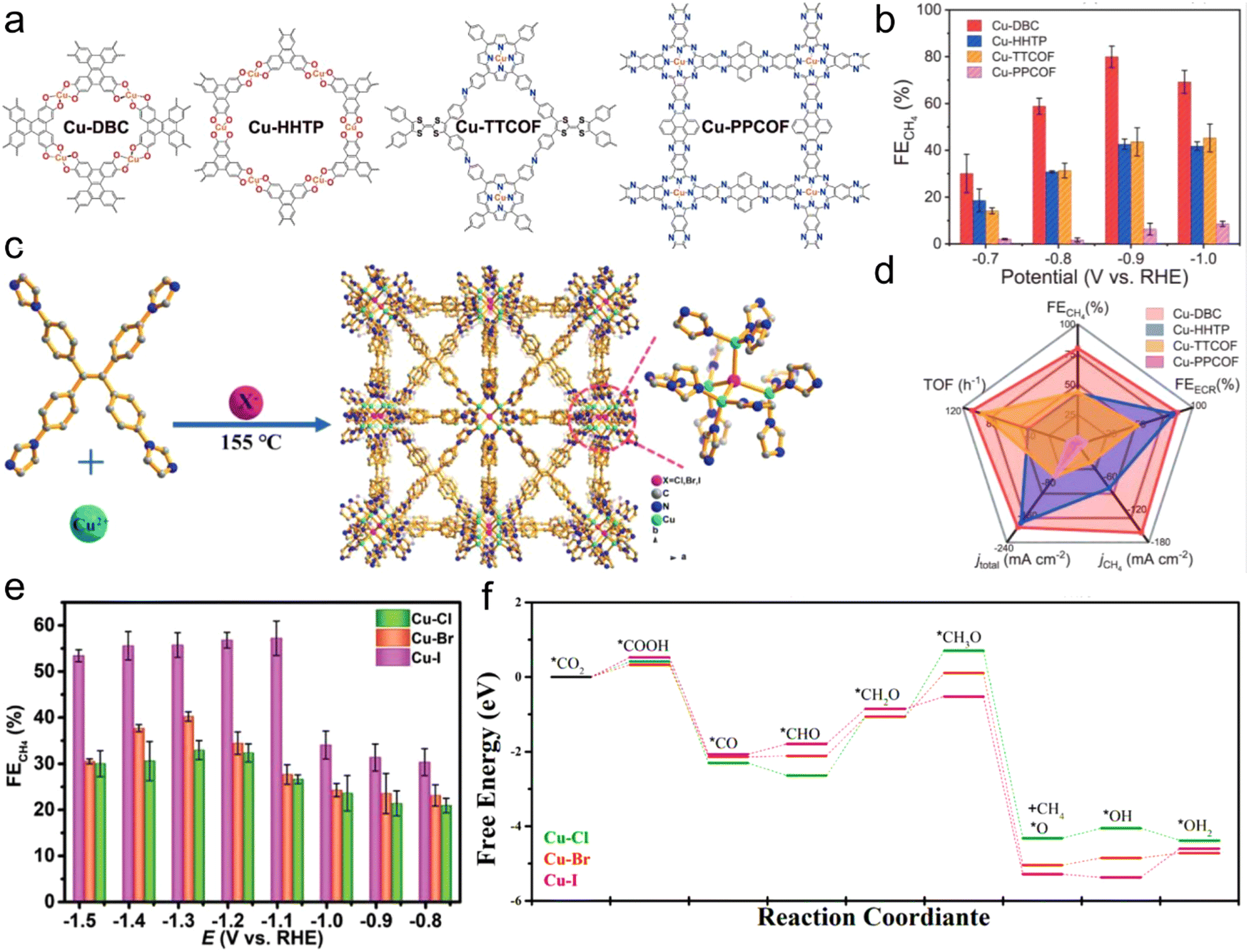 | ||
| Fig. 28 (a) Formula diagrams. (b) FEs of CH4 product. Reproduced with permission from ref. 252 Copyright 2017, Nature Springer. (c) Synthesis and crystal structure of Cu–X. Reproduced with permission from ref. 257 Copyright 2022, Wiley. (d) Inclusive electrochemical reduction performance assessment. Reproduced with permission from ref. 252 Copyright 2017, Nature Springer. (e) Average FEs of CH4 at different potentials over different halogen-based catalysts. (f) Free energy profiles. Reproduced with permission from ref. 257 Copyright 2022, Wiley. | ||
It has been found that the use of single-center catalysts (SSCs) is a favourable approach to attain high selectivity and true catalysis, but most reported SAC materials tend to form more than 2e− product instead of multi-carbons. Simultaneously, except for the single-atom sites of nitrogen coordination, few studies have been conducted on the single metal sites of other heteroatom coordination. Although crystalline SSCs can achieve controllable structure and metal site coordination, low conductivity still a serious issue for crystalline catalytic materials. The cMOF materials are assembled with transition metals with conjugated organic linkers, having unique redox, conductivity and MOF-based characteristics. Although some progress has been made in the application of cMOF in the CO2RR, the research on cMOF electrocatalysts is still rare. Zhang et al.252 reported the preparation of a Cu-based cMOF (Cu-DBC) using graphene-like highly conjugated linkers and Cu sites, and used it as a highly effective electric catalyst for the CO2RR. It was found that highly conjugated organic ligands endow unique redox characteristics and conductivity to MOFs, while the rich and evenly dispersed Cu–O4 centers help to achieve a highly selective and efficient CO2RR to methane (CH4) conversion process. At a low voltage of −0.9 V, Cu-DBC showed about 80% FECH4, and the local Id was −162.5 mA cm−2, which is most efficient catalyst for CO2RR to CH4 (Fig. 28c, e and f). In addition, depending on the crystalline and porous structure of the catalyst, the relation between the coordination environment of the Cu single-atom center and electrochemical reduction catalytic selectivity was also studied. The specificity and detailed catalytic mechanism of the Cu-DBC catalyst for CO2RR to CH4 conversion was further studied through electrolysis measurement and calculation. This study offers a strategy to design CO2RR catalysts with a clear structure and lays a foundation for building accurate structure–reaction correlations of efficient CO2RR catalysts.
The relationship between the CO2RR environment of coordination and selectivity of single-atom Cu site was studied by DFT calculations and electrocatalytic measurements. Compared with the nitrogen-coordination Cu site, the Cu–O4 site in Cu-DBC with lower energy graph has good CO2RR catalysis. Specifically, the design strategy in these studies can lay an experimental and theoretical foundation to understand the relationship among the structure of the catalyst, its microenvironment and CO2RR performance, facilitating further research.
8.2 Bimetal mono-dispersed catalysts
Tandem catalysts are a type of catalyst that exhibit unique catalytic properties by combining two materials with different catalytic functions. Because different active sites have different properties, they can achieve the goal of multi-electron (>2e) reduction products of the CO2RR.263,264 In SACs, the CoPc@Zn–N–C tandem catalyst could achieve much higher performance than its single components CoPc and Zn–N–C,20 that its CH4/CO production ratio is greater than that of the single-component catalysts. Theoretical calculation revealed that CO is first generated on CoPc, while the CoPc site also promotes *H to transfer to Zn–N–C, and CO diffuses on the Zn–N–C center and converted to CH4. The specific DFT calculation results are shown in Fig. 29a, where *COOH is easier to form on CoPc, However, Zn–N4 is more inclined to convert CO to CH4.
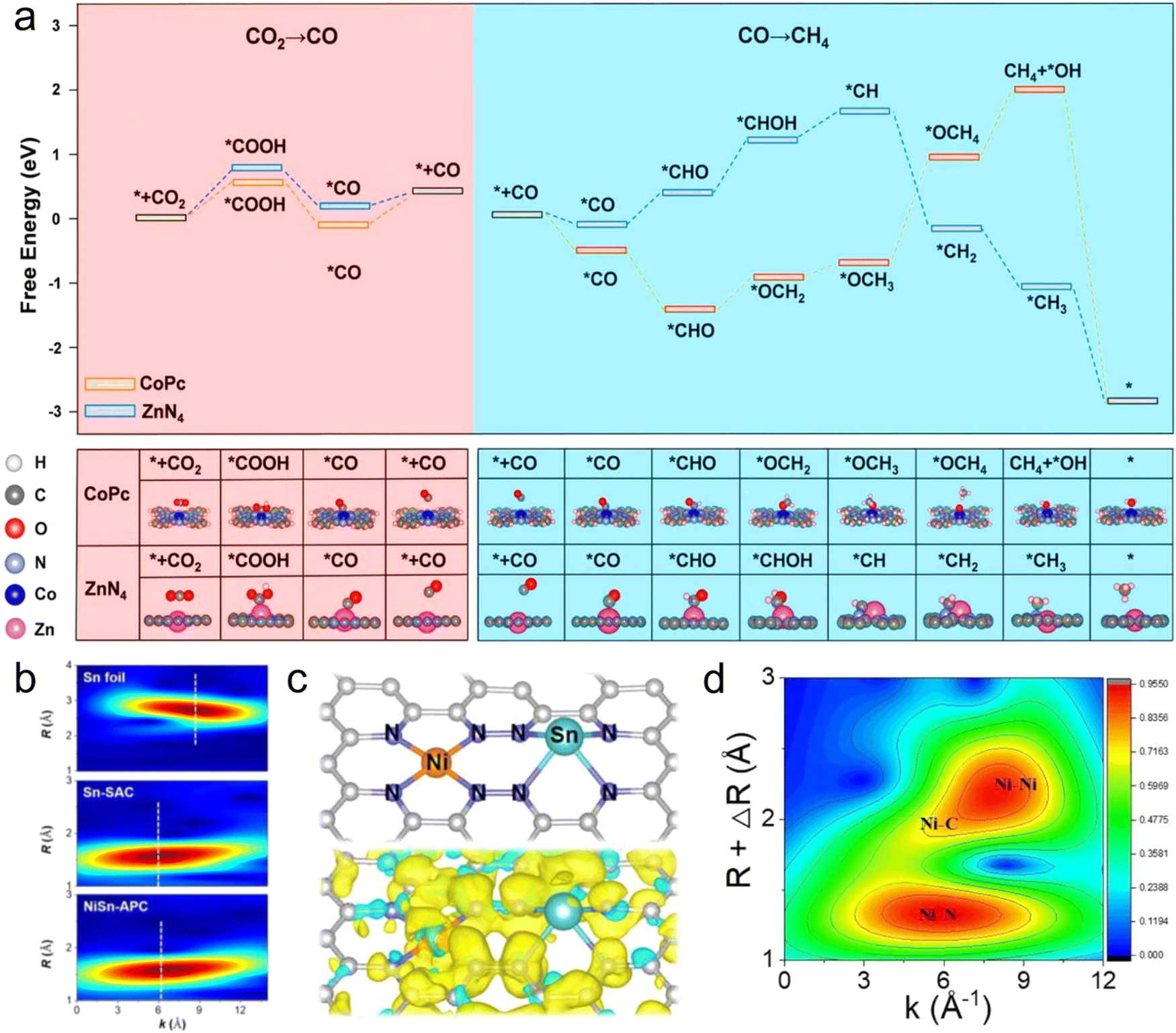 | ||
| Fig. 29 (a) Free energy profiles and intermediate configuration over CoPc and ZnN4. Reproduced with permission from ref. 20 Copyright 2020, Wiley. (b) WT analysis of NiSn-APC, Sn-SAC and Sn foil. (c) Spin electron density model proposed for NiSn-APC (yellow; spin up and green; spin down). Reproduced with permission from ref. 261 Copyright 2021, Wiley. (d) WT-EXAFS plot for Ni-CNC-1000. Reproduced with permission from ref. 262 Copyright 2022, Wiley. | ||
Among the bimetallic catalysts, homonuclear bimetallic catalysts for the CO2RR have also been reported by Li et al.,269 where Cu monoatomic pairs were loaded on Pd10Te3 alloy. In the Cu atom pair, i.e., Cu01–Cux+1, Cu01 tends to adsorb CO2 molecules, while Cu1+x tends to adsorb H2O molecules. Together, they promoted the activation of CO2 molecules. At about −0.78 V, the CO selectivity reached 92%. This reflects the property of “two atoms activate two molecules”, which single atoms do not possess. In addition, Ding et al.270 constructed a double Ni1–N4 site connected by two nitrogen atoms via the pyrolysis of a metal–organic complex occupied by adjacent Ni atoms. Theoretical calculation showed that under the condition of electrocatalysis, the two Ni1–N4 sites bridged by oxygen atoms significantly reduced the CO2 activation barrier, showing high CO selectivity. Cao et al.262 used electrospinning pyrolysis technology to prepare an Ni2–N4–C2 site catalyst. The structural characterization results are shown in Fig. 29d, which confirm the results. The experimental results and DFT calculations displayed that the binuclear Ni biatomic catalyst bonded with N and C atoms adjusts the electronic characteristics of the d-band, optimizes the adsorption of CO2 and its intermediates, enhances the charge and material transport, and promotes the reduction of CO2.
At present, bimetallic catalysts are mainly concentrated on C1 products, and only some tandem catalysts realize the production of multi-carbon products. Considering the advantages of bimetallic mono-dispersed atomic catalysts with different active centers, the generation of multi-carbon products is one of the main reasons for the development of bimetallic mono-dispersed atomic catalysts.
8.3 Metal carrier interactions
The electronic structure of metal atoms is regulated by covalent or ionic bond connection between the linker and metal atoms to change the structure–activity relationship of the metal atoms.237,271 The combination effect between the Cu single atomic site and the C site on C3N4 promoted the formation of C2+ products including C2H4, C2H6 and C2H5OH. The Cu single-atomic site is the active site for adsorbing *COOH, *CO and *CHO. The C site near C3N4 is the active site for *OCH2 and *OCH3.272 The coordination among the Sn single-atomic sites, oxygen vacancies and CuO substrates enables catalyst materials to have extraordinary double layer capacitance, high CO2 adsorption ability and low charge transfer interfacial impedance.273 Systematic experimental findings and theoretical calculation proved that Sn1/V0-CuO-90 promotes the formation of *CO by reducing the energy barrier of *COOH dissociation. Then, it adsorbs on the CuO substrate and further reduces *CO to CH3OH. HAADF-STEM and XAFS further confirmed the structure of Sn1/V0-CuO. Electrochemical experimental results showed that the catalyst methanol FE reached 88.6% at 67 mA cm−2Id. Its FE for ethanol and j at different potentials are shown in Fig. 30a and b, and Sn single atom and CuO rich in oxygen vacancy can synergistically promote the formation of C2H4. Theoretical calculation showed that Sn doping in CuO reduces the free energy of C–C coupling between the *CO intermediates, thereby promoting the formation of C2H4.274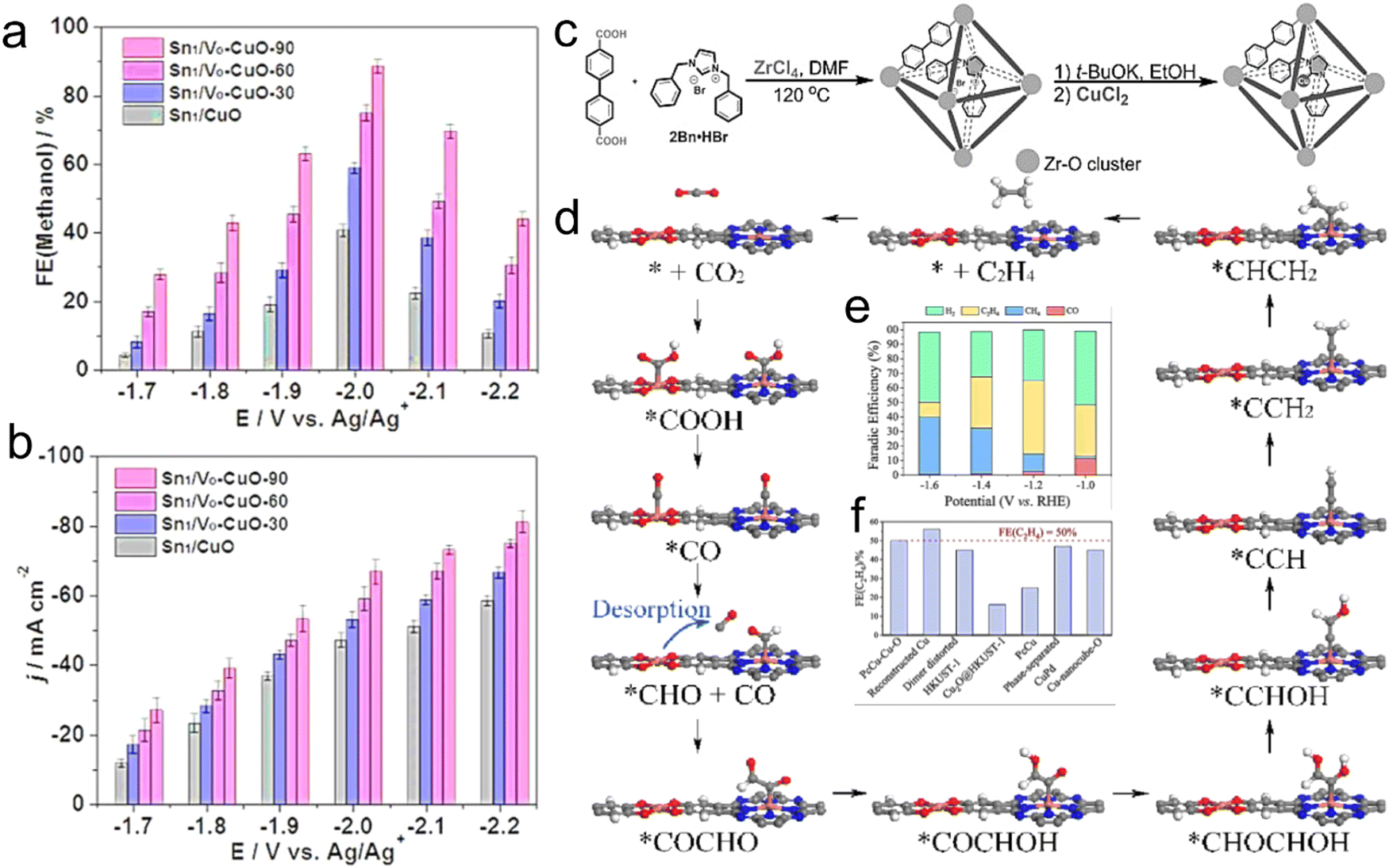 | ||
| Fig. 30 (a) FE(methanol) and (b) partial current densities (j). Reproduced with permission from ref. 273 Copyright 2021, Wiley. (c) Synthesis scheme of 2Bn–Cu@UiO-67. Reproduced with permission from ref. 33 Copyright 2022, Wiley. (d) Proposed CO2RR mechanism of PcCu–Cu–O. (e) FEs of CH4, C2H4, H2 and CO products over PcCu–Cu–O. (f) Comparison of FEs (C2H4) of PcCu–Cu–O and some previously reported catalysts. Reproduced with permission from ref. 278 Copyright 2021, ACS. | ||
The loading of Ir single atoms on an α-Co(OH)2 substrate can effectively stabilize the adsorbed CO2 species and promote rapid electron transport to generate CO.275 The Sn single atom loaded on Cu nanoparticles also showed similar properties, reaching the maximum FECO (95.3%) at −1.0 V.276 The single-atom Pb-doped copper substrate adjusted the electronic structure of copper, and thus adjusted the first protonation process of the CO2RR from the generation of *COOH path to generation of HCOO*. Finally, the FE of formic acid reached 96% under an industrial current density (1 A cm−2). It should be pointed out that the catalytic active site is the Cu substrate rather than the Pb single atom.277 Thus, all the above results indicate that interaction with metal support is a potential strategy to improve the catalyst performance of SACs.
8.4 Space confinement
The space-confinement strategy can promote the multi-electron reduction of CO2 by creating a small reaction space. Recently, a catalyst containing a copper single-atomic site with nitrogen heterocyclic carbene coordination (2Bn–Cu@UiO-67, Bn is a ligand: di-benzyl imidazole) was synthesized. It achieved an unprecedented CH3OH conversion frequency (TOF CH3OH = 58680 h−1) and FECH4 of 81% at −1.5 V vs. RHE. The J was 420 mA cm−2, and it could still maintain an excellent performance in a wide potential range.33 A schematic diagram of its synthesis is shown in Fig. 30c. Due to the electron-donating effect of the nitrogen heterocyclic carbene ligand on the Cu single-atomic site, the metal surface charge density of the Cu site increased. Therefore, the adsorption of the *CHO intermediate was greatly optimized, and *CHO is the main intermediate in the production of CH4. Simultaneously, the pore structure of UiO@67 also has strong CO2 capture capacity. The narrow space between the pores is conducive to the desorption of the intermediates from one 2Bn–Cu site and diffusion to another 2Bn–Cu site in the process of CO2 reduction, realizing further the reduction of the intermediates, which is conducive to the generation of CH4, and the interconnection reaction micro-space provided by UiO-67 in space cooperatively catalyzes the electroreduction of CO2. This work shows that the space-limited strategy has broad prospects.
8.5 Molecular bridging
Covalent organic frameworks (COFs) can separate single metal sites, which is one of the ideal models for building monodispersed metal SACs. This avoids the high-temperature pyrolysis required by traditional methods and can achieve the dispersion of single-atomic sites at a relatively mild temperature. For example, the common way is to directly use four nitrogen atoms on phthalocyanine ligands to coordinate with metal ions. The metal phthalocyanine structural unit is constructed, and then the connection between the structural units is realized through covalent organic chemical reaction to obtain a metal covalent organic network structure with super conjugated system. The COFbpy catalyst (COFbpyMn) anchored at the Mn unit point has a clear and recognizable coordination center, and the reduction of CO2 in water had a low overpotential (190 mV), and the current density reached 12 mA cm−2 at 550 mV overpotential. The TOFCO and TONCO (turnover number) were 1100 and 5800 h−1 (after 16 h), respectively, almost 10 times that of the equivalent Mn-based molecular catalyst.279 Lu et al.280 realized the construction of a series of single-atomic site catalysts by connecting metal phthalocyanines with dioxins. NiPcTFPN and CoPcTFPN achieved (99.8 ± 1.24)% and (96.1 ± 1.25)% FECO, respectively. In addition, the catalyst containing Cu–N4 and Cu–O4 sites (PcCu–Cu–O) constructed by COFs also achieved high selectivity from CO2 to multi-carbon products. In 0.1 mol L−1 KHCO3 solution, the ethylene selectivity reached 50% under the test conditions of −1.2 V (vs. RHE). The in situ infrared spectroscopy experiment also further verified the combination effect between the Cu–N4 site and Cu–O4 site in CuPc,278 as shown in Fig. 30d–f. CO desorbed from the Cu–O4 site can undergo C–C coupling reaction with *CO adsorbed by Cu–N4. MOF materials constructed of metal phthalocyanine also show superior performance. 2D metal phthalocyanine-based MOF nanosheets (NiPc–NiO4) overcome the shortcomings of poor conductivity of traditional MOFs,59 showing 98.4% CO selectivity and JCO as high as 34.5 mA cm−2. The COF composed of metalloporphyrins realized the formation of multi-electron reduction products. In addition, the ultra-thin Cu-Tph COF-Dct (about 3.8 nm) showed an FECH4 of about 80%, and the flow cell exhibited the performance of −220.0 mA cm−2 current density at −0.9 V.281 This is almost twice that of the unexposed COF. DFT calculation revealed that the improved performance is ascribed to the fixation on the COF, and the integration of amino and triazine groups in the functionalized stripping reagent enhances the activation and adsorption of CO2, stabilizes the reaction intermediates and increases the CO concentration near the Cu site. For the metal porphyrin MOF material, the multilayer 2D porphyrin MOF was prepared into a single layer, which is also conducive to the generation of C2 products.282 This is because the monolayer porphyrin-based MOF has a reconstructed Cu–O4 site, while the main products with more than two layers are mainly CO and HCOO−. However, when only a certain potential is applied, only 11.9% FEC2 was achieved. However, upon exposure to light, the FE of C2 increased to 41.1%.9. Summary and perspectives
According to this review, the design of MOF-based catalytic materials is mainly carried out depending on several important physical and chemical properties, including the metal-center characteristics, ligand and coordination environment, and the morphology and particle size of materials. In different types of materials, the research focus on these properties will be slightly different. The metal centers of active MOFs mostly exist in the form of atoms or clusters, with excellent dispersion. Currently, different types of metals and their different coordination environments tend to exhibit completely different bonding capabilities, resulting in different affinities with CO2 or differences in the types and quantities of active-free radicals, which ultimately affect the selectivity and activity of the CO2RR. For the composite catalysts of MOF and metal, the dispersion degree, morphology, particle size, defect state of the supported metal particles and the contact between the two materials usually affect the transfer of electrons and reactant CO2 between the two phases, thus becoming the focus of composite catalyst design. Similarly, the research on MOF derivatives attaches great importance to the characteristics of their metal centers, such as whether they can maintain or introduce metal centers in monatomic state, the types of metal non-metal bonds generated and their catalytic activities.The design of ligand types and coordination environment is mainly to increase the activation and adsorption of CO2 by utilizing the synergistic effect between specific functional groups of ligand molecules and metals. In addition, the design of ligands has different aims in photo- and electro-catalytic reduction. In photocatalysis, ligands can adjust the energy band structure to enhance the utilization of visible light. In electrocatalysis, ligands can communicate charge transport pathways and increase the working efficiency of the active centers by virtue of their electron-donating ability or the intrinsic catalytic activity of molecules. Finally, the control of the morphology and particle size of materials aims to greatly optimize their size in one or several dimensions to obtain structures such as ultra-small nanoparticles, two-dimensional layered materials, and dendritic crystals. These special morphologies can usually reveal more active centers and shorten the transfer path of electrons, thus showing higher catalytic performance. In addition to the common focus presented above, in electrocatalysis systems, the electrode preparation process needs to be optimized to promote the interaction between the catalytic material and electrolyte, improve the density and openness of the active catalytic sites, and achieve more efficient charge transfer. An ideal electrocatalyst needs to produce a high J at a low overpotential, which requires higher conductivity, active site density and CO2 affinity in the material.
Catalytic materials based on MOFs have witnessed significant development in the field of catalytic CO2RR, which will receive even more attention. However, the promotion and application of these materials still face many challenges. Firstly, their catalytic yield is too low, but the energy level required to be input is high, which does not have practical application value. Simultaneously, the cost of the preparation of MOF materials is usually high and their stability is not very prominent. Therefore, it is a future vision to solve the actual carbon emission problem through photo- and electro-catalytic CO2 reduction. It can be seen from the development history of zeolite molecular sieves, which are porous materials, that after the catalytic performance of the material has made a substantial breakthrough, its preparation cost and service life are expected to make considerable progress with the development of large-scale production. Therefore, at present, the urgent problem to be solved for MOF-based catalysts is how to obtain the reaction conversion rate with commercial value through a simple, green and practical process. The pursuit of catalytic performance leads to another big problem, that is, the research on the reaction mechanism of MOF materials for photo- and electro-reduction of CO2 is not deep enough and research in this area generally focuses on the analysis of several common physical and chemical properties of materials by trial and error, instead of forming a system, thus failing to fully utilize the various excellent properties of MOFs and other materials and far from reaching the level of designing high-performance catalysts according to demand. This is mainly because of the complexity of MOF materials, where it is difficult to carry out systematic and comprehensive theoretical calculations and pre-design for multiple influencing factors such as the bonding ability of their active site, the degree of spatial structure openness, charge-carrier transfer efficiency, and the diffusion efficiency of reactants and products.
Also, these complex factors are more difficult to track and grasp after the MOF is compounded or derived. This leads to the fact that most of the current theoretical calculations are supplementary explanations of experimental phenomena, and there are few pre-designed reports based on advanced computing theories. With the rapid development of research and the continuous promotion of the macro policy of carbon neutrality, the research on MOF-based CO2RR catalysts in the future will gradually emphasize the greening and practicality of materials, strengthen the optimization of catalyst preparation technology and the in-depth exploration of the reaction mechanism, and the involvement of advanced computing and the exploration of catalyst design theory will also be more common. With the continuous improvement of catalytic performance, the diversification and selectivity of reduction products will also be gradually emphasized. In improving the catalytic performance, as the key issue, the current hot strategies, such as preparation of single atomic or ultra-small particle size catalysts, design of advantageous coordination environment, development of ultra-thin 2D MOFs, fabrication of efficient heterostructures, application of conductive MOFs, and in situ electrosynthesis, are expected to achieve a significant increase in conversion. At present, the main product of SACs (including bimetallic SCAs) is CO, and the selectivity and current density of CO can maintain a high level in a wide potential window. However, how to improve the performance of the catalyst to next level so that it can operate stably for a long time under industrial conditions is still a problem to be solved at present. In addition, for the generation of multi-electron (>2e) reduction products and C2+ products, SACs with a single active component are difficult to achieve. This is because the metal loading of single-atom catalysts is generally low (<5%), and the distance between active sites is far, CO generated after CO2 reduction is difficult to diffuse from the active site to another single atomic site after desorption, and the possibility of carbon–carbon coupling is low, leading to C1 products dominated by CO, HCOOH and CH4. Accordingly, introducing single metal atoms in tandem catalysts or multi-component multi-site catalysts is one of the strategies that is expected to generate multi-electron products and C2+ products. Additionally there are many challenges faced by SACs to be solved, as follows: (1) exploring ways to replace the traditional competing heterogeneous reactions. (2) Improving the stability of SACs, particularly with a high metal loading or at high temperature or high voltage, to fulfil the industrial requirements. (3) Solving the issues of catalyst carbon deposition and poisoning of the metal active center in heterogeneous catalysis. (4) Screening monatomic catalytic system suitable for industrial application and developing production technology with large-scale application prospect. (5) Developing monatomic catalysis to accelerate its application at the industrial level and promote its wide application in various research fields as soon as possible. Furthermore, controlling the local microenvironment of single metal atoms systematically and accurately through experimental means remains to be further explored. In addition, the in-depth study of the mechanism of the CO2RR on SACs is also extremely challenging. Clarifying the reaction mechanism can design the configuration of SACs in a targeted way to achieve higher catalytic activity and selectivity. In the future, we can expect to deeply explore the mechanism and path of the CO2RR with the help of clearly defined catalytic active sites of SACs, combined with quasi in situ and ex situ characterization methods, and conduct auxiliary guidance and verification through theoretical calculation simulation, thus exploiting the organic unity of material structure, catalytic activity and reaction mechanism. Finally, this should be based on a deep understanding of the reaction system to efficiently use the characteristics of the MOF metal active centers, ligands and crystal morphology and structure and through the rational design of molecular scale to build a more efficient new generation of catalytic materials.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (NSFC 52006105, 51888103 and 92163124), National Key R&D Program of China (2021YFF0500700), Jiangsu Natural Science Foundation (No. BK20190460), Fundamental Research Funds for the Central Universities (30921013103 and 30920041113), and Jiangsu Innovative/Entrepreneurial Talent Program.References
- S. Abernethy and R. B. Jackson, Environ. Res. Lett., 2022, 17, 024019 CrossRef.
- W. Li, D. Wang, Y. Zhang, L. Tao, T. Wang, Y. Zou, Y. Wang, R. Chen and S. Wang, Adv. Mater., 2020, 32, 1907879 CrossRef CAS PubMed.
- T. Zhan, Y. Zou, Y. Yang, X. Ma, Z. Zhang and S. Xiang, ChemCatChem, 2022, 14, e202101453 CAS.
- L. Tang, X. Meng, D. Deng and X. Bao, Adv. Mater., 2019, 31, 1901996 CrossRef CAS PubMed.
- S. Jamil, M. Fasehullah, B. Jabar, P. Liu, M. K. Aslam, Y. Zhang, S. Bao and M. Xu, Nano Energy, 2022, 94, 106961 CrossRef CAS.
- M. K. Aslam, T. Hussain, H. Tabassum, Z. Wei, W. Tang, S. Li, S.-J. Bao, X. S. Zhao and M. Xu, Chem. Eng. J., 2022, 429, 132389 CrossRef CAS.
- N. Farooq, A. u Rehman, A. M. Qureshi, Z. u Rehman, A. Ahmad, M. K. Aslam, H. M. A. Javed, S. Hussain, M. A. Habila, N. AlMasoud and T. S. Alomar, Surf. Interfaces, 2021, 26, 101399 CrossRef CAS.
- Y. Sun, S. Ding, S. Xu, J. Duan and S. Chen, J. Power Sources, 2021, 494, 229733 CrossRef CAS.
- Y. Sun, T. Jiang, J. Duan, L. Jiang, X. Hu, H. Zhao, J. Zhu, S. Chen and X. Wang, ACS Catal., 2020, 10, 11371–11379 CrossRef CAS.
- J. Duan, Y. Sun, S. Chen, X. Chen and C. Zhao, J. Mater. Chem. A, 2020, 8, 18810–18815 RSC.
- D. Banham, T. Kishimoto, Y. Zhou, T. Sato, K. Bai, J.-I. Ozaki, Y. Imashiro and S. Ye, Sci. Adv., 2018, 4, eaar7180 CrossRef PubMed.
- H. Gharibi, N. Dalir, M. Jafari, M. J. Parnian and M. Zhiani, Appl. Surf. Sci., 2022, 572, 151367 CrossRef CAS.
- H. Wang, W. Wang, H. Yu, Q. Mao, Y. Xu, X. Li, Z. Wang and L. Wang, Appl. Catal., B, 2022, 307, 121172 CrossRef CAS.
- S. G. Han, M. Zhang, Z. H. Fu, L. Zheng, D. D. Ma, X. T. Wu and Q. L. Zhu, Adv. Mater., 2022, 34, 2202830 CrossRef CAS PubMed.
- Q. Jiang, Z. Chen, J. Tong, M. Yang, Z. Jiang and C. Li, Chem. Commun., 2017, 53, 1188–1191 RSC.
- Q. Jiang, J. Tong, G. Zhou, Z. Jiang, Z. Li and C. Li, Sol. Energy, 2014, 103, 425–437 CrossRef CAS.
- A. Demont and S. Abanades, RSC Adv., 2014, 4, 54885–54891 RSC.
- S. Dey, B. Naidu and C. Rao, Chem. – Eur. J., 2015, 21, 7077–7081 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed.
- X. Li, S. You, J. Du, Y. Dai, H. Chen, Z. Cai, N. Ren and J. Zou, J. Mater. Chem. A, 2019, 7, 25853–25864 RSC.
- S. S. A. Shah, T. Najam, M. Wen, S.-Q. Zang, A. Waseem and H.-L. Jiang, Small Structures, 2022, 3, 2100090 CrossRef CAS.
- Z. Song, L. Zhang, K. Doyle-Davis, X. Fu, J. L. Luo and X. Sun, Adv. Energy Mater., 2020, 10, 2001561 CrossRef CAS.
- K. Srinivas, Y. Lu, Y. Chen, W. Zhang and D. Yang, ACS Sustainable Chem. Eng., 2020, 8, 3820–3831 CrossRef CAS.
- X. Ma, F. Sun, L. Qin, Y. Liu, X. Kang, L. Wang, D.-E. Jiang, Q. Tang and Z. Tang, Chem. Sci., 2022, 13, 10149–10158 RSC.
- Y. Xu, C. Li, Y. Xiao, C. Wu, Y. Li, Y. Li, J. Han, Q. Liu and J. He, ACS Appl. Mater. Interfaces, 2022, 14, 11567–11574 CrossRef CAS PubMed.
- Y. Zou, T. Zhan, Y. Yang, Z. Fan, Y. Li, Y. Zhang, X. Ma, Q. Chen, S. Xiang and Z. Zhang, J. Mater. Chem. A, 2022, 10, 3216–3225 RSC.
- S. Li, A. V. Nagarajan, X. Du, Y. Li, Z. Liu, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2022, e202211771 CAS.
- X. Zhou, E. Song, Z. Kuang, Z. Gao, H. Zhao, J. Liu, S. Sun, C.-Y. Mou and H. Chen, Chem. Eng. J., 2022, 430, 133035 CrossRef CAS.
- T. Tsujiguchi, Y. Kawabe, S. Jeong, T. Ohto, S. Kukunuri, H. Kuramochi, Y. Takahashi, T. Nishiuchi, H. Masuda, M. Wakisaka, K. Hu, G. Elumalai, J.-I. Fujita and Y. Ito, ACS Catal., 2021, 11, 3310–3318 CrossRef CAS.
- T. Tang, W.-J. Jiang, X.-Z. Liu, J. Deng, S. Niu, B. Wang, S.-F. Jin, Q. Zhang, L. Gu and J.-S. Hu, J. Am. Chem. Soc., 2020, 142, 7116–7127 CrossRef CAS PubMed.
- C. He, Y. Zhang, Y. Zhang, L. Zhao, L. P. Yuan, J. Zhang, J. Ma and J. S. Hu, Angew. Chem., Int. Ed., 2020, 132, 4944–4949 CrossRef.
- S. Chen, W.-H. Li, W. Jiang, J. Yang, J. Zhu, L. Wang, H. Ou, Z. Zhuang, M. Chen, X. Sun, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202114450 CAS.
- J. Zhang, W. J. Jiang, S. Niu, H. Zhang, J. Liu, H. Li, G. F. Huang, L. Jiang, W. Q. Huang and J. S. Hu, Adv. Mater., 2020, 32, 1906015 CrossRef CAS PubMed.
- E. Vijayakumar, S. Ramakrishnan, C. Sathiskumar, D. J. Yoo, J. Balamurugan, H. S. Noh, D. Kwon, Y. H. Kim and H. Lee, Chem. Eng. J., 2022, 428, 131115 CrossRef CAS.
- Y. Zhou, R. Abazari, J. Chen, M. Tahir, A. Kumar, R. R. Ikreedeegh, E. Rani, H. Singh and A. M. Kirillov, Coord. Chem. Rev., 2022, 451, 214264 CrossRef CAS.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabres i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- L. B. Vilhelmsen, K. S. Walton and D. S. Sholl, J. Am. Chem. Soc., 2012, 134, 12807–12816 CrossRef CAS PubMed.
- Y. Ma, H. Peng, J. Liu, Y. Wang, X. Hao, X. Feng, S. U. Khan, H. Tan and Y. Li, Inorg. Chem., 2018, 57, 4109–4116 CrossRef CAS PubMed.
- J. Jiang and O. M. Yaghi, Chem. Rev., 2015, 115, 6966–6997 CrossRef CAS PubMed.
- H. Kobayashi, Y. Mitsuka and H. Kitagawa, Inorg. Chem., 2016, 55, 7301–7310 CrossRef CAS PubMed.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- B. D. McCarthy, A. M. Beiler, B. A. Johnson, T. Liseev, A. T. Castner and S. Ott, Coord. Chem. Rev., 2020, 406, 213137 CrossRef CAS PubMed.
- J. Wen, Y. Li and J. Gao, Chem. Res. Chin. Univ., 2020, 36, 662–679 CrossRef CAS.
- X. Liu, G. Verma, Z. Chen, B. Hu, Q. Huang, H. Yang, S. Ma and X. Wang, The Innovation, 2022, 3, 100281 CrossRef CAS PubMed.
- J. Lei, M. Zeng and L. Fu, Chem. Res. Chin. Univ., 2020, 36, 504–510 CrossRef CAS.
- X. Han, X. Ling, Y. Wang, T. Ma, C. Zhong, W. Hu and Y. Deng, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed.
- Y. He, Q. Shi, W. Shan, X. Li, A. J. Kropf, E. C. Wegener, J. Wright, S. Karakalos, D. Su and D. A. Cullen, Angew. Chem., Int. Ed., 2021, 60, 9516–9526 CrossRef CAS PubMed.
- S. Fang, X. Zhu, X. Liu, J. Gu, W. Liu, D. Wang, W. Zhang, Y. Lin, J. Lu and S. Wei, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- Y. Wang, Y. Pan, L. Zhu, H. Yu, B. Duan, R. Wang, Z. Zhang and S. Qiu, Carbon, 2019, 146, 671–679 CrossRef CAS.
- M. K. Aslam, S. S. A. Shah, S. Li and C. Chen, J. Mater. Chem. A, 2018, 6, 14083–14090 RSC.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao and S. Wei, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang and D. Wang, Angew. Chem., Int. Ed., 2017, 129, 7041–7045 CrossRef.
- J. Li, M. Chen, D. A. Cullen, S. Hwang, M. Wang, B. Li, K. Liu, S. Karakalos, M. Lucero and H. Zhang, Nat. Catal., 2018, 1, 935–945 CrossRef CAS.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS PubMed.
- X.-J. Kong, T. He, J. Zhou, C. Zhao, T.-C. Li, X.-Q. Wu, K. Wang and J.-R. Li, Small, 2021, 17, 2005357 CrossRef CAS PubMed.
- L. Qin, Z. Xu, Y. Zheng, C. Li, J. Mao and G. Zhang, Adv. Funct. Mater., 2020, 30, 1910257 CrossRef CAS.
- J. D. Yi, D. H. Si, R. Xie, Q. Yin, M. D. Zhang, Q. Wu, G. L. Chai, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 133, 17245–17251 CrossRef.
- Q. Dang, H. Huang, L. Li, X. Lyu, S. Zhong, Y. Yu and D. Xu, Chem. Mater., 2021, 33, 5690–5699 CrossRef CAS.
- W. Kai, X. Li, C. Hengze, Q. Huiying, C. Chao and Z. Ning, Chem. J. Chin. Univ., 2016, 37, 723–727 Search PubMed.
- C. Park, W. T. Koo, S. Chong, H. Shin, Y. H. Kim, H. J. Cho, J. S. Jang, D. H. Kim, J. Lee and S. Park, Adv. Mater., 2021, 33, 2101216 CrossRef CAS PubMed.
- M. Usman, M. Ali, B. A. Al-Maythalony, A. S. Ghanem, O. W. Saadi, M. Ali, M. A. Jafar Mazumder, S. Abdel-Azeim, M. A. Habib and Z. H. Yamani, ACS Appl. Mater. Interfaces, 2020, 12, 49992–50001 CrossRef CAS PubMed.
- Y. Liu, D. Deng and X. Bao, Chem, 2020, 6, 2497–2514 CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- Z. Tao and H. Wang, Chem. Res. Chin. Univ., 2020, 36, 1145–1146 CrossRef CAS.
- X. Fu, A. Zhu, X. Chen, S. Zhang, M. Wang and M. Yuan, Chem. Res. Chin. Univ., 2021, 37, 1328–1333 CrossRef CAS.
- H. Wang, X. Wu, G. Liu, S. Wu and R. Xu, Nano Res., 2022 DOI:10.1007/s12274-022-4199-4.
- J. Xiangyuan, Z. Libing, S. Xiaofu, B. Han, L. Zhang, X. Jin and X. Sun, Chem. J. Chin. Univ., 2022, 43, 20220035 Search PubMed.
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS PubMed.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- W. Ni, Y. Xue, X. Zang, C. Li, H. Wang, Z. Yang and Y.-M. Yan, ACS Nano, 2020, 14, 2014–2023 CrossRef CAS PubMed.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, ACS Catal., 2019, 9, 7270–7284 CrossRef CAS.
- J. Li, P. Yan, K. Li, J. You, H. Wang, W. Cui, W. Cen, Y. Chu and F. Dong, J. Mater. Chem. A, 2019, 7, 17014–17021 RSC.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
- H. Zhang, X. Chang, J. G. Chen, W. A. Goddard, B. Xu, M.-J. Cheng and Q. Lu, Nat. Commun., 2019, 10, 3340 CrossRef PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed.
- I. Ledezma-Yanez, E. P. Gallent, M. T. M. Koper and F. Calle-Vallejo, Catal. Today, 2016, 262, 90–94 CrossRef CAS.
- L. Wang, S. Nitopi, A. B. Wong, J. L. Snider, A. C. Nielander, C. G. Morales-Guio, M. Orazov, D. C. Higgins, C. Hahn and T. F. Jaramillo, Nat. Catal., 2019, 2, 702–708 CrossRef CAS.
- Y. Momose, K. Sato and O. Ohno, Surf. Interface Anal., 2002, 34, 615–618 CrossRef CAS.
- X. Zhi, Y. Jiao, Y. Zheng, A. Vasileff and S.-Z. Qiao, Nano Energy, 2020, 71, 104601 CrossRef CAS.
- M. A. Tekalgne, H. H. Do, A. Hasani, Q. Van Le, H. W. Jang, S. H. Ahn and S. Y. Kim, Mater. Today Adv., 2020, 5, 100038 CrossRef.
- X. Su, Y. Sun, L. Jin, L. Zhang, Y. Yang, P. Kerns, B. Liu, S. Li and J. He, Appl. Catal., B, 2020, 269, 118800 CrossRef CAS.
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed.
- Y. Feng, W. An, Z. Wang, Y. Wang, Y. Men and Y. Du, ACS Sustainable Chem. Eng., 2020, 8, 210–222 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- Q. Hu, Z. Han, X. Wang, G. Li, Z. Wang, X. Huang, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, Angew. Chem., Int. Ed., 2020, 59, 19054–19059 CrossRef CAS PubMed.
- D. H. Won, H. Shin, J. Koh, J. Chung, H. S. Lee, H. Kim and S. I. Woo, Angew. Chem., Int. Ed., 2016, 55, 9297–9300 CrossRef CAS PubMed.
- H. S. Jeon, I. Sinev, F. Scholten, N. J. Divins, I. Zegkinoglou, L. Pielsticker and B. R. Cuenya, J. Am. Chem. Soc., 2018, 140, 9383–9386 CrossRef CAS PubMed.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- G. Keerthiga and R. Chetty, J. Electrochem. Soc., 2017, 164, H164–H169 CrossRef CAS.
- J. Albo, A. Sáez, J. Solla-Gullón, V. Montiel and A. Irabien, Appl. Catal., B, 2015, 176–177, 709–717 CrossRef CAS.
- S. Sarfraz, A. T. Garcia-Esparza, A. Jedidi, L. Cavallo and K. Takanabe, ACS Catal., 2016, 6, 2842–2851 CrossRef CAS.
- M. Morimoto, Y. Takatsuji, R. Yamasaki, H. Hashimoto, I. Nakata, T. Sakakura and T. Haruyama, Electrocatalysis, 2018, 9, 323–332 CrossRef CAS.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS PubMed.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- H. Song, M. Im, J. T. Song, J.-A. Lim, B.-S. Kim, Y. Kwon, S. Ryu and J. Oh, Appl. Catal., B, 2018, 232, 391–396 CrossRef CAS.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- Z. Pan, K. Wang, K. Ye, Y. Wang, H.-Y. Su, B. Hu, J. Xiao, T. Yu, Y. Wang and S. Song, ACS Catal., 2020, 10, 3871–3880 CrossRef CAS.
- J.-J. Lv, M. Jouny, W. Luc, W. Zhu, J.-J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS PubMed.
- F. Li, G. H. Gu, C. Choi, P. Kolla, S. Hong, T.-S. Wu, Y.-L. Soo, J. Masa, S. Mukerjee, Y. Jung, J. Qiu and Z. Sun, Appl. Catal., B, 2020, 277, 119241 CrossRef CAS.
- P. Gotico, Z. Halime and A. Aukauloo, Dalton Trans., 2020, 49, 2381–2396 RSC.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- A. Liu, M. Gao, X. Ren, F. Meng, Y. Yang, L. Gao, Q. Yang and T. Ma, J. Mater. Chem. A, 2020, 8, 3541–3562 RSC.
- H. Cui, Y. Guo, L. Guo, L. Wang, Z. Zhou and Z. Peng, J. Mater. Chem. A, 2018, 6, 18782–18793 RSC.
- F. Calle-Vallejo and M. T. Koper, Angew. Chem., Int. Ed., 2013, 125, 7423–7426 CrossRef.
- Q. Zhu, D. Yang, H. Liu, X. Sun, C. Chen, J. Bi, J. Liu, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 132, 8981–8986 CrossRef.
- X. Kang, L. Li, A. Sheveleva, X. Han, J. Li, L. Liu, F. Tuna, E. J. McInnes, B. Han and S. Yang, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- X. Kang, B. Wang, K. Hu, K. Lyu, X. Han, B. F. Spencer, M. D. Frogley, F. Tuna, E. J. McInnes and R. A. Dryfe, J. Am. Chem. Soc., 2020, 142, 17384–17392 CrossRef CAS PubMed.
- S. Dou, J. J. Song, S. B. Xi, Y. H. Du, J. Wang, Z. F. Huang, Z. C. J. Xu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 4041–4045 CrossRef CAS PubMed.
- C.-J. Chang, S.-F. Hung, C.-S. Hsu, H.-C. Chen, S.-C. Lin, Y.-F. Liao and H. M. Chen, ACS Cent. Sci., 2019, 5, 1998–2009 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- L. Majidi, A. Ahmadiparidari, N. Shan, S. N. Misal, K. Kumar, Z. Huang, S. Rastegar, Z. Hemmat, X. Zou and P. Zapol, Adv. Mater., 2021, 33, 2004393 CrossRef CAS PubMed.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- X. Zhang, Y. Zhang, Q. Li, X. Zhou, Q. Li, J. Yi, Y. Liu and J. Zhang, J. Mater. Chem. A, 2020, 8, 9776–9787 RSC.
- Y. Wang, P. Hou, Z. Wang and P. Kang, ChemPhysChem, 2017, 18, 3142–3147 CrossRef CAS PubMed.
- X. Jiang, H. Li, J. Xiao, D. Gao, R. Si, F. Yang, Y. Li, G. Wang and X. Bao, Nano Energy, 2018, 52, 345–350 CrossRef CAS.
- S. Dou, J. Song, S. Xi, Y. Du, J. Wang, Z. F. Huang, Z. J. Xu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 4041–4045 CrossRef CAS PubMed.
- M. Perfecto-Irigaray, J. Albo, G. Beobide, O. Castillo, A. Irabien and S. Pérez-Yáñez, RSC Adv., 2018, 8, 21092–21099 RSC.
- C.-W. Kung, C. O. Audu, A. W. Peters, H. Noh, O. K. Farha and J. T. Hupp, ACS Energy Lett., 2017, 2, 2394–2401 CrossRef CAS.
- X. Tan, C. Yu, C. Zhao, H. Huang, X. Yao, X. Han, W. Guo, S. Cui, H. Huang and J. Qiu, ACS Appl. Mater. Interfaces, 2019, 11, 9904–9910 CrossRef CAS PubMed.
- X. Jiang, H. Wu, S. Chang, R. Si, S. Miao, W. Huang, Y. Li, G. Wang and X. Bao, J. Mater. Chem. A, 2017, 5, 19371–19377 RSC.
- P. De Luna, W. Liang, A. Mallick, O. Shekhah, F. P. García de Arquer, A. H. Proppe, P. Todorović, S. O. Kelley, E. H. Sargent and M. Eddaoudi, ACS Appl. Mater. Interfaces, 2018, 10, 31225–31232 Search PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu and A. Epshteyn, ACS Catal., 2014, 4, 3682–3695 CrossRef CAS.
- L. Ye, J. Liu, Y. Gao, C. Gong, M. Addicoat, T. Heine, C. Wöll and L. Sun, J. Mater. Chem. A, 2016, 4, 15320–15326 RSC.
- R. Senthil Kumar, S. Senthil Kumar and M. Anbu Kulandainathan, Electrochem. Commun., 2012, 25, 70–73 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- I. Choi, Y. E. Jung, S. J. Yoo, J. Y. Kim, H.-J. Kim, C. Y. Lee and J. H. Jang, J. Electrochem. Sci. Technol., 2017, 8, 61–68 CrossRef CAS.
- Y. Wang, P. Hou, Z. Wang and P. Kang, ChemPhysChem, 2017, 18, 3142–3147 CrossRef CAS PubMed.
- X. Jiang, H. Li, J. Xiao, D. Gao, R. Si, F. Yang, Y. Li, G. Wang and X. Bao, Nano Energy, 2018, 52, 345–350 CrossRef CAS.
- Y. Wu, J. Jiang, Z. Weng, M. Wang, D. L. J. Broere, Y. Zhong, G. W. Brudvig, Z. Feng and H. Wang, ACS Cent. Sci., 2017, 3, 847–852 CrossRef CAS PubMed.
- E. S. Donovan, B. M. Barry, C. A. Larsen, M. N. Wirtz, W. E. Geiger and R. A. Kemp, Chem. Commun., 2016, 52, 1685–1688 RSC.
- R. Hinogami, S. Yotsuhashi, M. Deguchi, Y. Zenitani, H. Hashiba and Y. Yamada, ECS Electrochem. Lett., 2012, 1, H17–H19 CrossRef CAS.
- J. Albo, D. Vallejo, G. Beobide, O. Castillo, P. Castaño and A. Irabien, ChemSusChem, 2017, 10, 1100–1109 CrossRef CAS PubMed.
- M. Hammouche, D. Lexa, M. Momenteau and J. M. Saveant, J. Am. Chem. Soc., 1991, 113, 8455–8466 CrossRef CAS.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- G. Zhu, Y. Li, H. Zhu, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2016, 6, 6294–6301 CrossRef CAS.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- S. R. Ahrenholtz, C. C. Epley and A. J. Morris, J. Am. Chem. Soc., 2014, 136, 2464–2472 CrossRef CAS PubMed.
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC.
- J.-D. Yi, R. Xie, Z.-L. Xie, G.-L. Chai, T.-F. Liu, R.-P. Chen, Y.-B. Huang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS PubMed.
- M. Duguet, A. Lemarchand, Y. Benseghir, P. Mialane, M. Gomez-Mingot, C. Roch-Marchal, M. Haouas, M. Fontecave, C. Mellot-Draznieks and C. Sassoye, Chem. Commun., 2020, 56, 10143–10146 RSC.
- P. Mialane, C. Mellot-Draznieks, P. Gairola, M. Duguet, Y. Benseghir, O. Oms and A. Dolbecq, Chem. Soc. Rev., 2021, 50, 6152–6220 RSC.
- J. Gu, W. Chen, G.-G. Shan, G. Li, C. Sun, X.-L. Wang and Z. Su, Mater. Today Energy, 2021, 21, 100760 CrossRef CAS.
- H. Yang, D. Yang, Y. Zhou and X. Wang, J. Am. Chem. Soc., 2021, 143, 13721–13730 CrossRef CAS PubMed.
- D. Yang, B. Ni and X. Wang, Adv. Energy Mater., 2020, 10, 2001142 CrossRef CAS.
- D. Yang, S. Zuo, H. Yang and X. Wang, Adv. Energy Mater., 2021, 11, 2100272 CrossRef CAS.
- S. Zhang, N. Liu, H. Wang, Q. Lu, W. Shi and X. Wang, Adv. Mater., 2021, 33, 2100576 CrossRef CAS PubMed.
- D. Yang, S. Zuo, H. Yang, Y. Zhou and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 18954–18959 CrossRef CAS PubMed.
- H. Yang, D. Yang and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 15527–15531 CrossRef CAS PubMed.
- D. Yang, H. Yu, T. He, S. Zuo, X. Liu, H. Yang, B. Ni, H. Li, L. Gu and D. Wang, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- M.-L. Sun, Y.-R. Wang, W.-W. He, R.-L. Zhong, Q.-Z. Liu, S. Xu, J.-M. Xu, X.-L. Han, X. Ge, S.-L. Li, Y.-Q. Lan, A. M. Al-Enizi, A. Nafady and S. Ma, Small, 2021, 17, 2100762 CrossRef CAS PubMed.
- Y. Benseghir, A. Lemarchand, M. Duguet, P. Mialane, M. Gomez-Mingot, C. Roch-Marchal, T. Pino, M. H. Ha-Thi, M. Haouas, M. Fontecave, A. Dolbecq, C. Sassoye and C. Mellot-Draznieks, J. Am. Chem. Soc., 2020, 142, 9428–9438 CrossRef CAS PubMed.
- J.-X. Wu, S.-Z. Hou, X.-D. Zhang, M. Xu, H.-F. Yang, P.-S. Cao and Z.-Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
- L. Ye, J. Liu, Y. Gao, C. Gong, M. Addicoat, T. Heine, C. Wöll and L. Sun, J. Mater. Chem. A, 2016, 4, 15320–15326 RSC.
- Y. Benseghir, A. Lemarchand, M. Duguet, P. Mialane, M. Gomez-Mingot, C. Roch-Marchal, T. Pino, M.-H. Ha-Thi, M. Haouas, M. Fontecave, A. Dolbecq, C. Sassoye and C. Mellot-Draznieks, J. Am. Chem. Soc., 2020, 142, 9428–9438 CrossRef CAS PubMed.
- Y. T. Guntern, J. R. Pankhurst, J. Vávra, M. Mensi, V. Mantella, P. Schouwink and R. Buonsanti, Angew. Chem., Int. Ed., 2019, 58, 12632–12639 CrossRef CAS PubMed.
- Y.-R. Wang, Q. Huang, C.-T. He, Y. Chen, J. Liu, F.-C. Shen and Y.-Q. Lan, Nat. Commun., 2018, 9, 4466 CrossRef PubMed.
- Y. Guo, W. Shi, H. Yang, Q. He, Z. Zeng, J.-Y. Ye, X. He, R. Huang, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 17875–17883 CrossRef CAS PubMed.
- L. Jiao and H.-L. Jiang, Chem, 2019, 5, 786–804 CAS.
- Y. Wu, S. Cao, J. Hou, Z. Li, B. Zhang, P. Zhai, Y. Zhang and L. Sun, Adv. Energy Mater., 2020, 10, 2000588 CrossRef CAS.
- C.-W. Kung, C. O. Audu, A. W. Peters, H. Noh, O. K. Farha and J. T. Hupp, ACS Energy Lett., 2017, 2, 2394–2401 CrossRef CAS.
- X. Jiang, H. Wu, S. Chang, R. Si, S. Miao, W. Huang, Y. Li, G. Wang and X. Bao, J. Mater. Chem. A, 2017, 5, 19371–19377 RSC.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- R. Qin, P. Liu, G. Fu and N. Zheng, Small Methods, 2018, 2, 1700286 CrossRef.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng and A. I. Rykov, Nat. Commun., 2018, 9, 1–12 CrossRef PubMed.
- S. Wei, Y. Wang, W. Chen, Z. Li, W.-C. Cheong, Q. Zhang, Y. Gong, L. Gu, C. Chen and D. Wang, Chem. Sci., 2020, 11, 786–790 RSC.
- M. Yoo, Y.-S. Yu, H. Ha, S. Lee, J.-S. Choi, S. Oh, E. Kang, H. Choi, H. An and K.-S. Lee, Energy Environ. Sci., 2020, 13, 1231–1239 RSC.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- L. Jiao, W. Yang, G. Wan, R. Zhang, X. Zheng, H. Zhou, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 20589–20595 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- Z.-H. Zhao, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 7986–7993 CrossRef CAS.
- H. Shang, T. Wang, J. Pei, Z. Jiang, D. Zhou, Y. Wang, H. Li, J. Dong, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 22465–22469 CrossRef CAS PubMed.
- D. Zhao, K. Yu, P. Song, W. Feng, B. Hu, W.-C. Cheong, Z. Zhuang, S. Liu, K. Sun, J. Zhang and C. Chen, Energy Environ. Sci., 2022, 15, 3795–3804 RSC.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- N. Mohd Adli, W. Shan, S. Hwang, W. Samarakoon, S. Karakalos, Y. Li, D. A. Cullen, D. Su, Z. Feng and G. Wang, Angew. Chem., Int. Ed., 2021, 133, 1035–1045 CrossRef.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech, E. Brunner, I. M. Weidinger, J. Zhang, A. V. Krasheninnikov, S. Kaskel, R. Dong and X. Feng, Nat. Commun., 2020, 11, 1409 CrossRef CAS PubMed.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- W. Guo, X. Sun, C. Chen, D. Yang, L. Lu, Y. Yang and B. Han, Green Chem., 2019, 21, 503–508 RSC.
- D. Jampaiah, D. Damma, A. Chalkidis, P. Venkataswamy, S. K. Bhargava and B. M. Reddy, Catal. Today, 2020, 356, 519–526 CrossRef CAS.
- L. Xiong, X. Zhang, X. Xie and Y. Peng, Chem. J. Chin. Univ., 2021, 42, 2824–2831 Search PubMed.
- M. K. Kim, H. J. Kim, H. Lim, Y. Kwon and H. M. Jeong, Electrochim. Acta, 2019, 306, 28–34 CrossRef CAS.
- H. Huo, J. Wang, Q. Fan, Y. Hu and J. Yang, Adv. Energy Mater., 2021, 11, 2102447 CrossRef CAS.
- Y. Dong, Q. Zhang, Z. Tian, B. Li, W. Yan, S. Wang, K. Jiang, J. Su, C. W. Oloman, E. L. Gyenge, R. Ge, Z. Lu, X. Ji and L. Chen, Adv. Mater., 2020, 32, 2001300 CrossRef CAS PubMed.
- M. Kuang, A. Guan, Z. Gu, P. Han, L. Qian and G. Zheng, Nano Res., 2019, 12, 2324–2329 CrossRef CAS.
- H. Dong, L. Zhang, L. Li, W. Deng, C. Hu, Z. J. Zhao and J. Gong, Small, 2019, 15, 1900289 CrossRef PubMed.
- Z. Pei, H. Li, Y. Huang, Q. Xue, Y. Huang, M. Zhu, Z. Wang and C. Zhi, Energy Environ. Sci., 2017, 10, 742–749 RSC.
- Z. Pei, Q. Meng, L. Wei, J. Fan, Y. Chen and C. Zhi, Energy Storage Mater., 2020, 28, 55–63 CrossRef.
- J. Zhao, H. Lai, Z. Lyu, Y. Jiang, K. Xie, X. Wang, Q. Wu, L. Yang, Z. Jin and Y. Ma, Adv. Mater., 2015, 27, 3541–3545 CrossRef CAS PubMed.
- Y. Jia, L. Zhang, L. Zhuang, H. Liu, X. Yan, X. Wang, J. Liu, J. Wang, Y. Zheng and Z. Xiao, Nat. Catal., 2019, 2, 688–695 CrossRef CAS.
- Y. Jia, J. Chen and X. Yao, Mater. Chem. Front., 2018, 2, 1250–1268 RSC.
- Q. Wang, Y. Lei, D. Wang and Y. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC.
- Q. Wu, J. Gao, J. Feng, Q. Liu, Y. Zhou, S. Zhang, M. Nie, Y. Liu, J. Zhao and F. Liu, J. Mater. Chem. A, 2020, 8, 1205–1211 RSC.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- C. Cao, S. Zhou, S. Zuo, H. Zhang, B. Chen, J. Huang, X.-T. Wu, Q. Xu and Q.-L. Zhu, Research, 2023 DOI:10.34133/research.0079.
- X. Li, S.-G. Han, W. Wu, K. Zhang, B. Chen, S.-H. Zhou, D.-D. Ma, W. Wei, X.-T. Wu, R. Zou and Q.-L. Zhu, Energy Environ. Sci., 2023, 16, 502–512 RSC.
- M. Zhang, S. Zhou, W. Wei, D.-D. Ma, S.-G. Han, X. Li, X.-T. Wu, Q. Xu and Q.-L. Zhu, Chem. Catal., 2022, 2, 3528–3545 CrossRef CAS.
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, ACS Catal., 2017, 7, 1520–1525 CrossRef CAS.
- F. Pan, W. Deng, C. Justiniano and Y. Li, Appl. Catal., B, 2018, 226, 463–472 CrossRef CAS.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- X. Li, Y. Zeng, C.-W. Tung, Y.-R. Lu, S. Baskaran, S.-F. Hung, S. Wang, C.-Q. Xu, J. Wang and T.-S. Chan, ACS Catal., 2021, 11, 7292–7301 CrossRef CAS.
- W. Ju, A. Bagger, X. Wang, Y. Tsai, F. Luo, T. Möller, H. Wang, J. Rossmeisl, A. S. Varela and P. Strasser, ACS Energy Lett., 2019, 4, 1663–1671 CrossRef CAS.
- A. S. Varela, M. Kroschel, N. D. Leonard, W. Ju, J. Steinberg, A. Bagger, J. Rossmeisl and P. Strasser, ACS Energy Lett., 2018, 3, 812–817 CrossRef CAS.
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, e1808135 CrossRef PubMed.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji and X. Zheng, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- M. Jia, S. Hong, T.-S. Wu, X. Li, Y.-L. Soo and Z. Sun, Chem. Commun., 2019, 55, 12024–12027 RSC.
- Z. Jiang, T. Wang, J. Pei, H. Shang, D. Zhou, H. Li, J. Dong, Y. Wang, R. Cao and Z. Zhuang, Energy Environ. Sci., 2020, 13, 2856–2863 RSC.
- R. Wang, X. Sun, S. Ould-Chikh, D. Osadchii, F. Bai, F. Kapteijn and J. Gascon, ACS Appl. Mater. Interfaces, 2018, 10, 14751–14758 CrossRef CAS PubMed.
- T. Huan and N. Ranjbar, ACS Catal., 2017, 7, 1520–1525 CrossRef CAS.
- X. Qin, S. Zhu, F. Xiao, L. Zhang and M. Shao, ACS Energy Lett., 2019, 4, 1778–1783 CrossRef CAS.
- X. Chen, D.-D. Ma, B. Chen, K. Zhang, R. Zou, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2020, 267, 118720 CrossRef CAS.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie and F. Yang, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- Y. N. Gong, L. Jiao, Y. Qian, C. Y. Pan, L. Zheng, X. Cai, B. Liu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 132, 2727–2731 CrossRef.
- Z. Geng, Y. Cao, W. Chen, X. Kong, Y. Liu, T. Yao and Y. Lin, Appl. Catal., B, 2019, 240, 234–240 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2018, 130, 1962–1966 CrossRef.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- Y. Guo, H. Yang, X. Zhou, K. Liu, C. Zhang, Z. Zhou, C. Wang and W. Lin, J. Mater. Chem. A, 2017, 5, 24867–24873 RSC.
- S. Ye, G. Fan, J. Xu, L. Yang and F. Li, Electrochim. Acta, 2020, 334, 135583 CrossRef CAS.
- Y. Zhang, K. Li, M. Chen, J. Wang, J. Liu and Y. Zhang, ACS Appl. Nano Mater., 2019, 3, 257–263 CrossRef.
- H. Cheng, X. Wu, M. Feng, X. Li, G. Lei, Z. Fan, D. Pan, F. Cui and G. He, ACS Catal., 2021, 11, 12673–12681 CrossRef CAS.
- H. Shi, H. Wang, Y. Zhou, J. Li, P. Zhai, X. Li, G. G. Gargik, J. Hou, H. Yang and X. Guo, Angew. Chem., Int. Ed., 2022, 61, e202208904 CAS.
- T. Zhang, X. Han, H. Liu, M. Biset-Peiró, X. Zhang, P. Tan, P. Tang, B. Yang, L. Zheng, J. R. Morante and J. Arbiol, Energy Environ. Sci., 2021, 14, 4847–4857 RSC.
- K. Yao, H. Wang, X. Yang, Y. Huang, C. Kou, T. Jing, S. Chen, Z. Wang, Y. Liu and H. Liang, Appl. Catal., B, 2022, 311, 121377 CrossRef CAS.
- Z. Meng, J. Luo, W. Li and K. A. Mirica, J. Am. Chem. Soc., 2020, 142, 21656–21669 CrossRef CAS PubMed.
- X. Li, L. Liu, X. Ren, J. Gao, Y. Huang and B. Liu, Sci. Adv., 2020, 6, eabb6833 CrossRef CAS PubMed.
- W. Guo, X. Tan, J. Bi, L. Xu, D. Yang, C. Chen, Q. Zhu, J. Ma, A. Tayal and J. Ma, J. Am. Chem. Soc., 2021, 143, 6877–6885 CrossRef CAS PubMed.
- Z. Chen, X. Zhang, W. Liu, M. Jiao, K. Mou, X. Zhang and L. Liu, Energy Environ. Sci., 2021, 14, 2349–2356 RSC.
- Q. Cheng, K. Mao, L. Ma, L. Yang, L. Zou, Z. Zou, Z. Hu and H. Yang, ACS Energy Lett., 2018, 3, 1205–1211 CrossRef CAS.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong and Y. Wang, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang and J. Lu, Angew. Chem., Int. Ed., 2019, 131, 15013–15018 CrossRef.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty and D. Su, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- C. Jia, S. Li, Y. Zhao, R. K. Hocking, W. Ren, X. Chen, Z. Su, W. Yang, Y. Wang and S. Zheng, Adv. Funct. Mater., 2021, 31, 2107072 CrossRef CAS.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS PubMed.
- Q. Fan, P. Hou, C. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2020, 10, 1903068 CrossRef CAS.
- X. Rong, H. J. Wang, X. L. Lu, R. Si and T. B. Lu, Angew. Chem., Int. Ed., 2020, 132, 1977–1981 CrossRef.
- Y. Wu, C. Chen, X. Yan, X. Sun, Q. Zhu, P. Li, Y. Li, S. Liu, J. Ma and Y. Huang, Angew. Chem., Int. Ed., 2021, 133, 20971–20978 CrossRef.
- W. Ni, Y. Gao, Y. Lin, C. Ma, X. Guo, S. Wang and S. Zhang, ACS Catal., 2021, 11, 5212–5221 CrossRef CAS.
- B. Zhang, J. Zhang, J. Shi, D. Tan, L. Liu, F. Zhang, C. Lu, Z. Su, X. Tan and X. Cheng, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean and R. E. Winans, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- Y. Zhang, L.-Z. Dong, S. Li, X. Huang, J.-N. Chang, J.-H. Wang, J. Zhou, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2021, 12, 6390 CrossRef CAS PubMed.
- C. Jia, X. Tan, Y. Zhao, W. Ren, Y. Li, Z. Su, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2021, 60, 23342–23348 CrossRef CAS PubMed.
- J. Guo, W. Zhang, L. H. Zhang, D. Chen, J. Zhan, X. Wang, N. R. Shiju and F. Yu, Adv. Sci., 2021, 8, 2102884 CrossRef CAS PubMed.
- Z. Chen, A. Huang, K. Yu, T. Cui, Z. Zhuang, S. Liu, J. Li, R. Tu, K. Sun and X. Tan, Energy Environ. Sci., 2021, 14, 3430–3437 RSC.
- X. Sun, Y. Tuo, C. Ye, C. Chen, Q. Lu, G. Li, P. Jiang, S. Chen, P. Zhu and M. Ma, Angew. Chem., Int. Ed., 2021, 60, 23614–23618 CrossRef CAS PubMed.
- Y. Zhang, Q. Zhou, Z. F. Qiu, X. Y. Zhang, J. Q. Chen, Y. Zhao, F. Gong and W. Y. Sun, Adv. Funct. Mater., 2022, 32, 2203677 CrossRef CAS.
- W. R. Leow, Y. Lum, A. Ozden, Y. Wang, D.-H. Nam, B. Chen, J. Wicks, T.-T. Zhuang, F. Li and D. Sinton, Science, 2020, 368, 1228–1233 CrossRef CAS PubMed.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- L. Lin, H. Li, C. Yan, H. Li, R. Si, M. Li, J. Xiao, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1903470 CrossRef CAS PubMed.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 133, 7458–7464 CrossRef.
- X. Cao, L. Zhao, B. Wulan, D. Tan, Q. Chen, J. Ma and J. Zhang, Angew. Chem., Int. Ed., 2022, 134, e202113918 Search PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- D. Chen, L. H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CrossRef CAS PubMed.
- G. Luo, Y. Jing and Y. Li, J. Mater. Chem. A, 2020, 8, 15809–15815 RSC.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou and S.-H. Yu, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS PubMed.
- Z. Zeng, L. Y. Gan, H. Bin Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang and W. Cai, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- Z. Liang, L. Song, M. Sun, B. Huang and Y. Du, Sci. Adv., 2021, 7, eabl4915 CrossRef CAS PubMed.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu and S.-F. Hung, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang and B. Pang, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS PubMed.
- M. Jia, Q. Fan, S. Liu, J. Qiu and Z. Sun, Curr. Opin. Green Sustain. Chem., 2019, 16, 1–6 CrossRef.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- W. Guo, S. Liu, X. Tan, R. Wu, X. Yan, C. Chen, Q. Zhu, L. Zheng, J. Ma and J. Zhang, Angew. Chem., Int. Ed., 2021, 133, 22150–22158 CrossRef.
- Y. Jiang, C. Choi, S. Hong, S. Chu, T.-S. Wu, Y.-L. Soo, L. Hao, Y. Jung and Z. Sun, Cell Rep. Phys. Sci., 2021, 2, 100356 CrossRef CAS.
- X. Sun, C. Chen, S. Liu, S. Hong, Q. Zhu, Q. Qian, B. Han, J. Zhang and L. Zheng, Angew. Chem., Int. Ed., 2019, 131, 4717–4721 CrossRef.
- M. Zhang, Z. Zhang, Z. Zhao, H. Huang, D. H. Anjum, D. Wang, J.-H. He and K.-W. Huang, ACS Catal., 2021, 11, 11103–11108 CrossRef CAS.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li and C.-W. Pao, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed.
- X.-F. Qiu, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed.
- G. C. Dubed Bandomo, S. S. Mondal, F. Franco, A. Bucci, V. Martin-Diaconescu, M. A. Ortuno, P. H. van Langevelde, A. Shafir, N. Lopez and J. Lloret-Fillol, ACS Catal., 2021, 11, 7210–7222 CrossRef CAS.
- M. Lu, M. Zhang, C. G. Liu, J. Liu, L. J. Shang, M. Wang, J. N. Chang, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2021, 133, 4914–4921 CrossRef.
- Y. R. Wang, H. M. Ding, X. Y. Ma, M. Liu, Y. L. Yang, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202114648 CAS.
- J. Han, N. Li, D. Chen, Q. Xu and J. Lu, Appl. Catal., B, 2022, 317, 121724 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |