
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining the targeted and untargeted screening of environmental contaminants reveals associations between PFAS exposure and vitamin D metabolism in human plasma†
Henrik
Carlsson
 ,
Akshai Parakkal
Sreenivasan
,
Ida
Erngren
,
Anders
Larsson
and
Kim
Kultima
*
,
Akshai Parakkal
Sreenivasan
,
Ida
Erngren
,
Anders
Larsson
and
Kim
Kultima
*
Department of Medical Sciences, Uppsala University, Akademiska sjukhuset, Entrance 61, floor 3, 751 85 Uppsala, Sweden. E-mail: kim.kultima@medsci.uu.se
First published on 11th May 2023
Abstract
We have developed, validated, and applied a method for the targeted and untargeted screening of environmental contaminants in human plasma using liquid chromatography high-resolution mass spectrometry (LC-HRMS). The method was optimized for several classes of environmental contaminants, including PFASs, OH-PCBs, HBCDs, and bisphenols. One-hundred plasma samples from blood donors (19–75 years, men n = 50, women n = 50, from Uppsala, Sweden) were analyzed. Nineteen targeted compounds were detected across the samples, with 18 being PFASs and the 19th being OH-PCB (4-OH-PCB-187). Ten compounds were positively associated with age (in order of increasing p-values: PFNA, PFOS, PFDA, 4-OH-PCB-187, FOSA, PFUdA, L-PFHpS, PFTrDA, PFDoA, and PFHpA; p-values ranging from 2.5 × 10−5 to 4.67 × 10−2). Three compounds were associated with sex (in order of increasing p-values: L-PFHpS, PFOS, and PFNA; p-values ranging from 1.71 × 10−2 to 3.88 × 10−2), all with higher concentrations in male subjects compared with female subjects. Strong correlations (0.56–0.93) were observed between long-chain PFAS compounds (PFNA, PFOS, PFDA, PFUdA, PFDoA, and PFTrDA). In the non-targeted data analysis, fourteen unknown features correlating with known PFASs were found (correlation coefficients 0.48–0.99). Five endogenous compounds were identified from these features, all correlating strongly with PFHxS (correlation coefficients 0.59–0.71). Three of the identified compounds were vitamin D3 metabolites, and two were diglyceride lipids (DG 24:6;O). The results demonstrate the potential of combining targeted and untargeted approaches to increase the coverage of compounds detected with a single method. This methodology is well suited for exposomics to detect previously unknown associations between environmental contaminants and endogenous compounds that may be important for human health.
Environmental significanceCombining the targeted and untargeted screening of environmental contaminants in human plasma may detect previously unknown associations between environmental contaminants and endogenous compounds with important implications for human health. Here we demonstrate how the methodology may be used to detect previously unknown associations between known environmental contaminants and endogenous metabolites and lipids. Specifically, we found strong associations between the perfluorohexanesulfonic acid and three metabolites of vitamin D3 and two diglyceride lipids. The present method is fast and straightforward and applicable for high-throughput exposome profiling of large cohorts. |
1 Introduction
It has been estimated that environmental exposures likely have a much more significant impact on chronic diseases than genetic factors.1–3 To acknowledge this the exposome concept was introduced to describe the totality of exposures of an individual throughout life, considering both exogenous and endogenous exposures.4–6 To measure the exposome, different techniques are needed,7 and an important group of compounds is environmental contaminants. A subset of environmental contaminants has been described as environmental organic acids, defined as environmental organic compounds utilized in commerce with at least one ionizable proton.8 This group of compounds includes per- and polyfluoroalkyl substances (PFASs),9,10 the phenolic metabolites of polybrominated diphenylethers (OH-BDEs)11 and polychlorinated biphenyls (OH-PCBs),12 and bisphenols.13 There are several exposure sources of these substances ranging from food, drinking water, and consumer products to both indoor and outdoor environments.9,12–15 These compounds are of serious concern because many of them have, or could potentially have, endocrine disrupting effects and other health effects, such as association with an increased risk of cancer and immune, reproductive, and developmental effects, as well as the potential to bioaccumulate.12,16–20 Because of the typically low abundances of environmental contaminants in blood, about 1000 times lower compared to endogenous compounds and drugs,6 highly sensitive methods are required. This may be achieved by using targeted measurements with triple quadrupole mass spectrometry21 or untargeted discovery with high-resolution mass spectrometry instruments coupled to liquid chromatography (LC-HRMS). The benefit of using LC-HRMS in the untargeted screening mode is that the data may be used to discover previously unknown environmental contaminants and to find previously unknown associations with both endogenous and exogenous compounds.21,22We developed a method for high-throughput combined targeted and untargeted screening of environmental contaminants in human plasma using LC-HRMS. The methodology was inspired by our previous work in untargeted metabolomics and lipidomics,23–25 applying fast and straightforward workflows for data analysis not typically used for environmental contaminant screening. The result is a method applicable for high-throughput exposome profiling of large cohorts, giving both quantitative measures of environmental contaminants and the possibility to reveal previously unknown associations between environmental exposures and endogenous compounds in the same experiments.
2 Materials and methods
2.1. Chemicals
Methanol (MeOH, ≥99.9%, MS grade) was purchased from Honeywell (Seelze, Germany), acetic acid (100%, anhydrous for analysis) and ammonium acetate (NH4Ac) from Merck (Darmstadt, Germany), and water was purified using a Milli-Q IQ 7000 system (Merck Millipore, Burlington, MA, USA).All standards and references were purchased from Wellington Laboratories (Guelph, Ontario, Canada). A detailed summary of all references and standards used with molecular formulae, monitored m/z-values, and retention times is given in the ESI (Table S1†). In summary, the following were used, including Wellington product names within parentheses, representing a total of 37 different compounds; native standards: a mixture of 24 common PFASs (PFAC-24PAR), bisphenol A (BPA), and a mixture containing α-, β-, and γ-HBCD (HBCD-MXA); heavy isotope labeled standards: a mixture of 19 common labeled PFASs (MPFAC-24ES), labeled bisphenol A (MBPA), a mixture containing labeled α-, β-, and γ-HBCD (MHBCD-MXA), labeled 6-OH-BDE47 (M6HBDE47), labeled 6-OH-BDE100 (M6PHBDE100), and a mixture containing seven labeled OH-PCBs (MHPCB-MXA).
2.2. Study samples
The use of surplus sample volumes from routine samples was approved by the Uppsala Regional Ethics Committee, Uppsala, Sweden (01/367). The blood donor plasma samples (n = 100) were collected in 2021 and in agreement with the ethical permit, only age and sex were gathered for the subjects. The samples were stored at −80 °C until sample preparation. Prepared samples were stored at −80 °C until analysis. The Declaration of Helsinki and its subsequent revisions were followed. The demographic data of the subjects are summarized in Table 1. Newborn bovine serum (USA) was purchased from Sigma-Aldrich and stored at −20 °C until analysis.| Sex | n | Age in years, mean (±SD) | Median age in years | Age range in years |
|---|---|---|---|---|
| Female | 50 | 44.9 (±14.0) | 47 | 19–71 |
| Male | 50 | 45.6 (±14.0) | 48 | 21–75 |
2.3. Sample preparation
The samples were prepared in randomized order. Fifty μL plasma was added to 140 μL MeOH in 0.6 mL Eppendorf tubes, followed by adding 10 μL internal standards (IS) in MeOH (final concentration 2.5 ng mL−1 of each IS). The samples were then vortexed for 15 s and centrifuged at 4 °C with 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 rpm. One hundred μL of the supernatants were transferred to plastic HPLC vials (Thermo Scientific) with polyimide-lined caps (Macherey-Nagel, Dueren, Germany) and stored at −80 °C until analysis. Samples were prepared in six batches with each batch containing up to 28 samples. Ten μL of each prepared sample were pooled and used for quality control throughout the experiments. For MS/MS (MS2) data collection experiments, a pooled prepared sample was concentrated about four times. The sample was evaporated under a gentle stream of nitrogen and reconstituted in 3:1 MeOH:H2O.
800 rpm. One hundred μL of the supernatants were transferred to plastic HPLC vials (Thermo Scientific) with polyimide-lined caps (Macherey-Nagel, Dueren, Germany) and stored at −80 °C until analysis. Samples were prepared in six batches with each batch containing up to 28 samples. Ten μL of each prepared sample were pooled and used for quality control throughout the experiments. For MS/MS (MS2) data collection experiments, a pooled prepared sample was concentrated about four times. The sample was evaporated under a gentle stream of nitrogen and reconstituted in 3:1 MeOH:H2O.
2.4. Calibration curves and quality controls
Internal standard calibration was performed for all compounds where both native and heavy isotope labeled standards were available. For compounds for which only heavy isotope labeled standards were available, one-point calibration compared with the corresponding internal standard was performed. Calibration curves, quality controls, and blanks were prepared using newborn bovine serum filtered through ENVI-Carb SPE tubes (250 mg, Merck). The used filters contain graphitized non-porous carbon and were used for the purpose of obtaining a calibration matrix with decreased background levels of, e.g., PFASs.26 Calibrators and QCs were prepared the same way as the authentic samples, but the total 150 μL MeOH used for precipitation was made up of 130 μL MeOH, 10 μL IS MeOH solution (the same as for samples described above), and 10 μL native reference MeOH solution (of varying concentrations).The calibration curves were prepared at seven calibrator levels, spanning the plasma concentration range of 0.02–80 ng mL−1. As most of the reference standards used were provided as mixtures, the same concentrations were used for all targeted compounds, i.e., not considering individual reference intervals for each compound. The approximate limit of detection (LOD) and lowest limit of quantification (LLOQ) for each specific compound were estimated by visual evaluation of the corresponding peaks in the calibrator samples, considering a peak with height >2 × 103 that matches the corresponding internal standard as a clearly detected peak. For estimations of LODs and matrix effects, background (blank) subtraction was performed for the compounds for which background levels were observed. The QCs were prepared at two concentration levels: 2 ng mL−1 (QCL) and 20 ng mL−1 (QCH). The calibration curve was prepared once with the first batch of samples. Three of each QC (QCL and QCH) were prepared with all batches. Also prepared with each batch of samples was one blank sample without IS and one blank sample with IS using the same newborn bovine serum used for calibration curves and quality controls.
The calibration curve was also prepared in 3:1 MeOH:H2O (without matrix) to estimate matrix effects. Two corresponding calibration curves were also prepared for the heavy isotope-labeled standards to control their linearity. Solvent blanks (3:1 MeOH:H2O) with and without IS were prepared to control possible carry-over. The bovine serum calibration curves were used for estimating sample concentrations and 1/x2 weighting was used for each curve to acknowledge the large range of concentrations covered.
2.5. Liquid chromatography-mass spectrometry analysis
Samples were injected in a randomized order. After every tenth sample, an injection of the pooled sample followed by a newborn bovine serum blank injection was done. Prior to injecting the study samples, all calibrator samples and QC samples were injected. Six repeated newborn bovine serum blank injections conditioned the column before any runs, and before the injection of samples, six repeated injections of the pooled sample were done.Twenty μL of each sample was injected on a reversed-phase HPLC column (Accucore C18, 100 × 2.1 mm, 2.6 μm, Thermo Scientific) using an Ultimate 3000 HPLC system (Thermo Scientific) interfaced to a high-resolution hybrid quadrupole Q Exactive Orbitrap MS (Thermo Scientific). A 17.5 min long chromatographic program including a gradient was applied using the mobile phases H2O with 0.1% acetic acid and 10 mM NH4Ac (mobile phase A) and MeOH with 0.1% acetic acid and 10 mM NH4Ac (mobile phase B); 5% B for 1 min, 5–100% B for 9.5 min, hold at 100% B for 3 min, return to 5% B over 0.1 min followed by re-equilibration at 5% B for 3.9 min. The flow rate was 0.6 mL min−1, and the column temperature was 55 °C.
HRMS was operated in negative ionization mode with the following source parameters: spray voltage: 2.5 kV, capillary temperature: 275 °C, sheath gas flow rate: 55, auxiliary gas flow rate: 15, sweep gas flow rate: 3, S-lens RF level: 50, auxiliary gas heater: 450 °C. The HRMS analysis was performed in the full scan mode, collecting data in the profile mode with a resolution of 70000 in the range of m/z 212.5–750.
Following the full-scan experiments of all samples, MS2 experiments were performed using a solution of all standards and references (both native and heavy isotope-labeled) at a concentration of 50 ng mL−1 as well as a concentrated pooled sample (four-fold concentration). The MS2 experiments were performed in six m/z windows: (1) 212.5–300 m/z, (2) 290–390, (3) 380–480, (4) 470–570 m/z, (5) 560–660 m/z, (6) 650–750 m/z. For each separate m/z window, ten experiments (with separate injections) were done, both for the reference solution and the pooled sample: one full scan (MS1) followed by nine data-dependent MS2 scans at nine different collision energies (normalized collision energy, NCE): 10, 15, 20, 22.5, 25, 27.5, 30, 35, and 40 (in total 120 experiments). Apart from varying NCEs the settings were the same for all data-dependent MS2 experiments: resolution: 35000, loop count: 10 (TopN), maximum injection time: 200 ms, and isolation window: 1.9 m/z, with data collected in the profile mode. Chromatography was performed in the same way as the main MS1 experiments. Twenty μL was injected for each experiment.
During method development, several parameters were evaluated and optimized. For mobile phase additives, three options were evaluated: (1) 0.1% acetic acid, (2) 2 mM ammonium acetate (NH4Ac), and (3) 0.1% acetic acid with 10 mM NH4Ac, with the third option resulting in optimal responses and peak shapes. The following ion source parameters were evaluated for the targeted compounds, with the tested and optimal values within parentheses: capillary temperature (250 °C, 275 °C, 300 °C, 325 °C, and 350 °C; optimal: 275 °C), S-lens RF level (0, 25, 50, 75, and 100; optimal: 50), and spray voltage (2.00 kV, 2.25 kV, 2.50 kV, 2.75 kV, and 3.00 kV; optimal: 2.50 kV).
2.6. Statistical analysis
The LC-HRMS data for targeted compounds were processed using TraceFinder 4.1 software (Thermo Scientific) using ten ppm mass tolerance to extract ion chromatograms. The concentration of targeted compounds was estimated using 1/x2 weighted calibration curves with internal standard calibration. For some of the compounds, a background was detected in the newborn calf serum used as the matrix for calibration curves and QCs (in order of increasing background level: PFDA, 6:2FTS, PFOS, PFTrDA, PFNA, and PFUdA). For these compounds, the concentration in the calf serum was calculated using the standard addition method, and the estimated concentrations were adjusted using this information. Injections of the precipitation solvent showed negligible background of these compounds, thus identifying the calf serum as the background source.To investigate the associations of the targeted compounds with age and sex, linear models were fitted on log-transformed concentration data from TraceFinder, including compounds present in ≥15% of the samples. The linear models used age, sex and the interaction age: sex as independent variables and compound concentrations as dependent variables, according to the following equation: y = b0 + b1 × age + b2 × sex + b3 × age × sex. The variables were evaluated individually with p < 0.05 as the limit for significance. The correlations of the concentrations between compounds were estimated using Pearson correlation. The resulting correlation coefficients were used for distance calculation between the compounds and the obtained distance matrix was subjected to hierarchical clustering. This is plotted as heatmaps, where the compounds are clustered based on the correlation between them.
For the untargeted data the raw data was converted to mzML using MSConvert, peak picked using FeatureFinder, and linked using FeatureLinker,27 as previously described by us.24 FeatureFinder and FeatureLinker were run using in-house nf-core-metabolinden workflow28 to quantify and link features in this data, using non-default parameters (available at https://github.com/caramba-uu/env-con).24 All statistical analyses and plots were done using python-3.10.4,29 scipy-1.7.3,30 sklearn-1.0.2,31 seaborn-0.11.2,32 numpy-1.23.1 (ref. 33) and pandas-1.4.3.34
2.7. Identification of unknown compounds
In the untargeted evaluation, features present in >90% of the samples and a correlation coefficient of absolutely >0.6 were used to obtain unknown features having at least a moderate correlation with targeted compounds. The m/z of the resulting features was compared against the NORMAN database.35 To improve our identification of unknown compounds, we made a linear model to predict logKow values for the unknown compounds using the retention times and listed logKow values in the NORMAN database for the targeted compounds (estimated by EPISuite36). When multiple matches were found, the candidates were evaluated based on the listed logKow values compared to our predicted logKow values. A similar logKow was deemed to increase the likelihood of the matching compound. The NORMAN database focuses on exogenous environmental compounds. For unknowns deemed to correspond to endogenous substances not found in the NORMAN database, we used HMDB,37 Metlin,38 and Lipid Maps39 to find likely candidates within 5 ppm of the observed m/z. Furthermore, the available MS2 spectra were compared with molecular structure databases using CSI:FingerID.40 Finally, the matching MS2 spectra were manually evaluated to strengthen the identification.
3 Results
We developed a combined targeted and untargeted method for screening environmental contaminants using LC-HRMS. The method was optimized for a total of 37 environmental contaminants across different chemical classes: PFASs (24 compounds), OH-PBDEs (two compounds), OH-PCBs (seven compounds), HBCDs (three isomers), and bisphenol A. One hundred plasma samples from blood donors ranging in age from 19 to 75, with 50 females and 50 males, were screened with the method. Nineteen of the monitored compounds were detected across the samples. Their concentrations were estimated based on calibration curves. Full names and abbreviations for the 19 compounds are given in Table 2. The method was validated by evaluating repeated injections of pooled and spiked QC samples. The MS1 and MS2 data were further investigated for untargeted compounds associated with the detected targeted compounds.| Compound | Abbreviation | Median conc. (ng mL−1) | Min conc. (ng mL−1) | Max conc. (ng mL−1) | CV (%) | Coverage (%) | p age | p sex |
|---|---|---|---|---|---|---|---|---|
| a Bold: p < 0.05. | ||||||||
| 1H,1H,2H,2H-perfluoro-1-decanesulfonic acid (8:2) |
8:2FTS | <LLOQ | <LLOQ | 0.02 | 13 | 53 | 2.64 × 10−1 | 7.42 × 10−1 |
| 1H,1H,2H,2H-perfluoro-1-octanesulfonic acid (6:2) |
6:2FTS | 0.27 | 0.27 | 0.33 | 3 | 100 | 3.95 × 10−1 | 1.09 × 10−1 |
| 2,2′,3,4′,5,5′,6-Heptachloro-4-biphenylol | 4-OH-PCB-187 | <LLOQ | <LLOQ | <LLOQ | 132 | 15 | 7.12 × 10− 4 | 6.90 × 10−1 |
| N-ethylperfluoro-1-octanesulfonamidoacetic acid | N-EtFOSAA | 0.06 | 0.06 | 0.14 | 34 | 10 | 4.08 × 10−1 | 2.83 × 10−1 |
| N-methylperfluoro-1-octanesulfonamidoacetic acid | N-MeFOSAA | 0.08 | 0.08 | 2.15 | 201 | 49 | 2.83 × 10−1 | 5.05 × 10−1 |
| Perfluoro-1-octanesulfonamide | FOSA | <LLOQ | <LLOQ | 0.06 | 46 | 93 | 1.58 × 10− 3 | 2.46 × 10−1 |
| Perfluorobutanesulfonic acid | L-PFBS | <LLOQ | <LLOQ | 0.07 | 72 | 79 | 7.24 × 10−1 | 3.20 × 10−1 |
| Perfluoroheptanesulfonic acid | L-PFHpS | 0.10 | 0.02 | 0.42 | 53 | 100 | 4.13 × 10− 3 | 1.71 × 10− 2 |
| Perfluorohexanesulfonic acid | PFHxS | 1.50 | 0.23 | 31.82 | 140 | 100 | 9.63 × 10−1 | 5.06 × 10−1 |
| Perfluoro-n-decanoic acid | PFDA | 0.19 | 0.05 | 0.83 | 59 | 100 | 2.30 × 10− 4 | 1.11 × 10−1 |
| Perfluoro-n-dodecanoic acid | PFDoA | <LLOQ | <LLOQ | 0.07 | 87 | 61 | 3.30 × 10− 2 | 5.44 × 10−1 |
| Perfluoro-n-heptanoic acid | PFHpA | 0.03 | <LLOQ | 0.13 | 66 | 94 | 4.67 × 10− 2 | 8.61 × 10−1 |
| Perfluoro-n-hexanoic acid | PFHxA | <LLOQ | <LLOQ | 0.02 | 39 | 55 | 5.13 × 10−2 | 6.27 × 10−1 |
| Perfluoro-n-nonanoic acid | PFNA | 0.37 | 0.10 | 2.15 | 70 | 100 | 2.50 × 10− 5 | 3.88 × 10−2 |
| Perfluoro-n-octanoic acid | PFOA | 0.85 | 0.29 | 2.96 | 54 | 100 | 8.83 × 10−2 | 2.56 × 10−1 |
| Perfluoro-n-tridecanoic acid | PFTrDA | <LLOQ | <LLOQ | 0.11 | 121 | 49 | 6.78 × 10− 3 | 8.03 × 10−1 |
| Perfluoro-n-undecanoic acid | PFUdA | 0.15 | <LLOQ | 0.76 | 74 | 98 | 3.96 × 10− 3 | 2.41 × 10−1 |
| Perfluorooctanesulfonic acid | PFOS | 3.39 | 0.82 | 15.16 | 58 | 100 | 2.89 × 10− 5 | 2.36 × 10−2 |
| Perfluoropentanesulfonic acid | L-PFPeS | 0.04 | <LLOQ | 0.45 | 104 | 99 | 1.87 × 10−1 | 5.92 × 10−1 |
3.1. Method validation
The developed method displayed desirable robustness and selectivity for the targeted analytes. The CV in concentration and retention time for the detected targeted analytes was calculated based on the repeated injections of the pooled sample (containing equal volumes of all 100 samples, total number of injections, n = 22). The CV in concentration ranged from 0.9% to 12.3%, with a mean CV of 5.9%. The CV in retention time ranged from 0.1% to 0.4%, with a mean CV of 0.2%. This demonstrates the high instrumental reproducibility of the method. A representative chromatogram from an injection of the pooled sample is presented in the ESI (Fig. S1†). Validation results for spiked QC samples and calibrators are summarized in the ESI (Table S2†).The calibration curves for the targeted analytes, using 1/x2 weighting, generally exhibited good linearity with a mean coefficient of determination (R2) of 0.959 (range 0.594–0.995). Except for 6:2FTS (R2 = 0.594), BPA (R2 = 0.612), and PFUdA (R2 = 0.881), the remaining compounds exhibited R2-values in the range 0.974–0.995. Carry-over was evaluated by investigating the presence of the targeted compounds in the solvent blank injected directly after the highest calibrator (80 ng mL−1). For the majority of compounds, no carry-over was observed, and for the two compounds with observed carry-over (FOSA and PFHpA) the carry-over was very low considering the concentrations observed in actual samples (corresponding to <40% of the lowest calibrator, i.e., <0.01 ng mL−1 after injection of 80 ng mL−1).
For 21 of the 37 targeted compounds, including 16 of the 19 compounds detected in the samples under study (see Section 3.2. below), the response was good with clearly detected peaks in the lowest calibrator sample (0.02 ng mL−1) with average peak heights 7 × 103 (ranging 2 × 103–2 × 104; adjusted for background concentrations when relevant) and matching the corresponding IS (where IS was available). By using 10 ppm filtering all noise was virtually removed from the extracted ion chromatograms, resulting in easily interpretable data. For the 21 compounds with good response at the lowest calibrator, LODs were estimated to be approximately 0.01 ng mL−1, i.e., half of the lowest calibrator with the corresponding LLOQ (lowest limit of quantification) at 0.02 ng mL−1. For these compounds the calibration curves spanned the plasma concentration range of 0.02–80 ng mL−1. For the remaining 16 compounds, the response was not as good with large variations in response between compounds. The majority of these compounds, with the exception of N-MeFOSAA, N-EtFOSAA, and 4-OH-PCB-187 (that had acceptably low LODs), were not detected in the samples under study. By visually evaluating the peak height at the different calibration levels we estimated LODs and LLOQs as follows: 4:2FTS, N-MeFOSAA, N-EtFOSAA, 6-OH-BDE47, 4-OH-PCB-187, and 4-OH-PCB-61, LOD at 0.04 ng mL−1 and LLOQ at 0.08 ng mL−1 (calibration curves spanning the plasma concentration range 0.08–80 ng mL−1); 4-OH-PCB-172, 4-OH-PCB-159, 4-OH-PCB-29, and 4-OH-PCB-12, LOD at 0.16 ng mL−1 and LLOQ at 0.32 ng mL−1 (calibration curves spanning the plasma concentration range 0.3–80 ng mL−1); 6-OH-BDE100, PFBA, and HBCD (including all three isomers), LOD at 0.63 ng mL−1 and LLOQ at 1.25 ng mL−1 (calibration curves spanning the plasma concentration range 1.25–80 ng mL−1); BPA, LOD at 2.5 ng mL−1 and LLOQ at 5 ng mL−1 (calibration curve spanning the plasma concentration range 5–80 ng mL−1). The matrix effect was on average 82% (range 43–168%; adjusted for background concentrations when relevant), indicating low matrix effects with slightly more suppression (<100%) than enhancement (>100%) on average.
Spiked QC samples at two concentration levels (2 ng mL−1 and 20 ng mL−1) were used to evaluate within- and between-batch accuracy and precision (detailed results in ESI†). The precision measured as CVs of the targeted analytes was low, ranging from 1.4% to 9.0% within-batch (mean 3.2%) and 4.8–7.9% between-batch (mean 6.0%). The bias was acceptable, ranging from −8–33.6% within-batch (mean 1.3%) to −4.9–53.7% between-batch (mean 6.0%), compared to the nominal values. Only four compounds had bias values over >10% (6:2FTS, PFNA, PFTrDA, and PFUdA). The high bias values for PFNA, PFTrDA, and PFUdA were caused by the high background concentrations in the calf serum used for calibration curves. Overall, these results demonstrate a generally accurate and precise method.
3.2. Results of targeted analysis
The developed method was applied to analyze 100 plasma samples from blood donors. Out of the 37 targeted compounds, 19 were detected across the samples including 18 PFAS compounds and one hydroxylated PCB metabolite (4-OH-PCB-187) (Table 2).Eleven of the 19 compounds were detected in ≥93% of the studied samples, whereas some were only detected in a subset. The least detected compounds were N-EtFOSAA and 4-OH-PCB-187, which were detected in 10 and 15 samples, respectively. The estimated concentrations for all compounds were within the low end of the calibration curve, with the highest single concentrations observed across the samples for PFHxS at 31.8 ng mL−1 and PFOSs at 15.2 ng mL−1 (Table 2).
Ten compounds (in order of increasing p-values: PFNA, PFOS, PFDA, 4-OH-PCB-187, FOSA, PFUdA, L-PFHpS, PFTrDA, PFDoA, and PFHpA) were positively associated with age (p-values ranging from 2.5 × 10−5 to 4.67 × 10−2) (Table 2). Three compounds (in order of increasing p-values: L-PFHpS, PFOS, and PFNA) were associated with sex (p-values ranging from 1.71 × 10−2 to 3.88 × 10−2), all with higher concentrations in male subjects compared with female subjects (Table 2). No significant age × sex interactions were found. Four illustrative examples (PFNA, PFOS, PFDoA, and 4-OH-PCB-187) of the observed differences regarding age and sex are presented in Fig. 1.
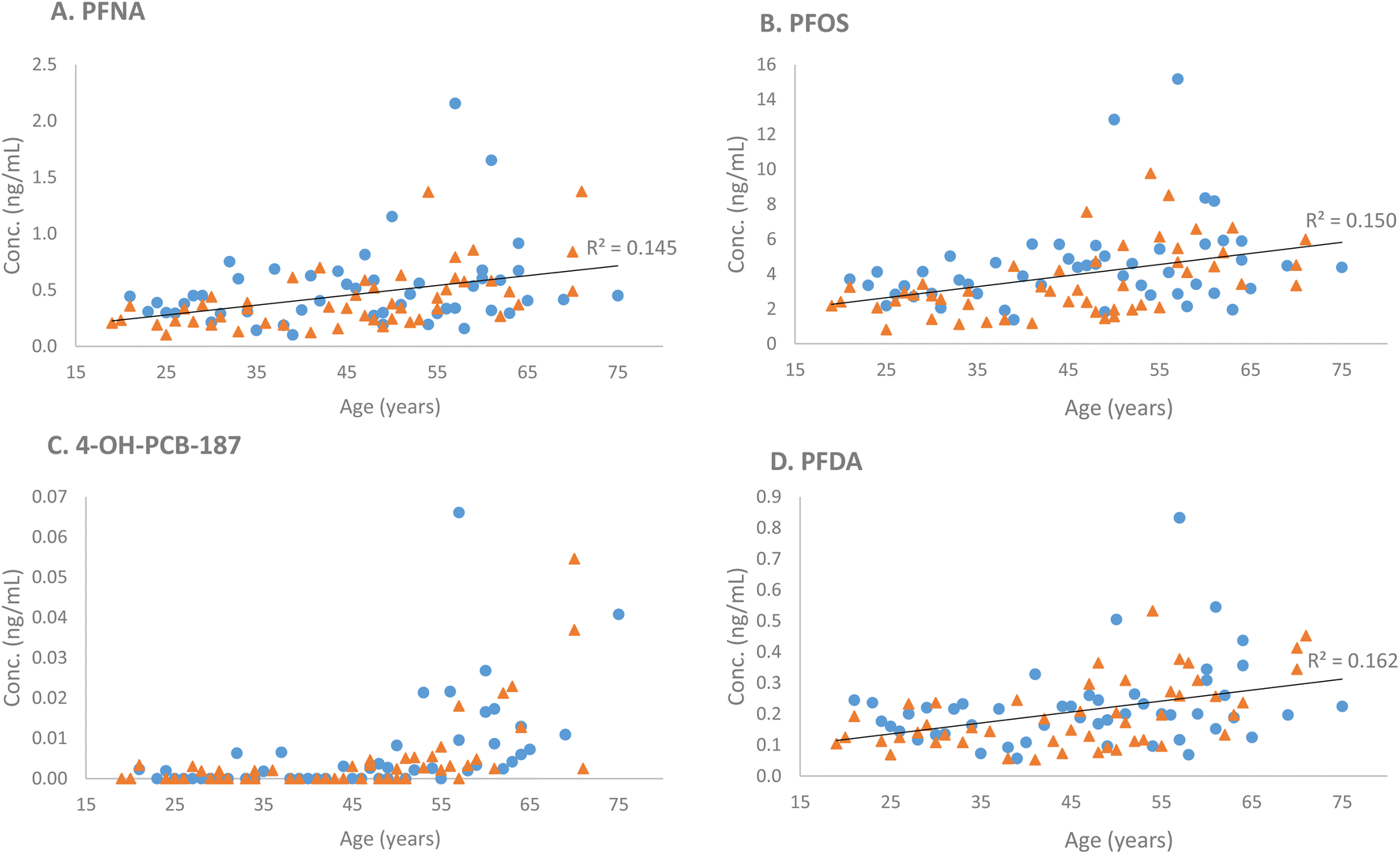 | ||
| Fig. 1 Examples of compounds that exhibited significant differences associated with age: (A) PFNA (p = 2.5 × 10−5); (B) PFOS (p = 2.89 × 10−5); (C) 4-OH-PCB-187 (p = 7.12 × 10−4); (D) PFDA (p = 2.3 × 10−4); and sex: (A) PFNA (p = 3.88 × 10−2) and (B) PFOS (p = 2.36 × 10−2). The regression line is not shown for 4-OH-PCB-187 because of low coverage and concentrations <LLOQ for this compound (estimated LLOQ = 0.08 ng mL−1); the concentrations in the figure were estimated using one-point calibration compared with the corresponding internal standard only considering samples with clearly detected peaks. Male subjects are labeled with blue circles and female subjects with orange triangles. | ||
Moderate to strong correlations (0.56–0.93) were observed between the concentrations of long-chain PFAS compounds with high coverage: PFNA, PFOS, PFDA, PFUdA, PFDoA, and PFTrDA. L-PFHpS correlated with PFOS, PFOA, and PFHxS (0.48–0.69). The structurally similar FOSA and N-MeFOSAA were strongly correlated (0.93). The short-chain L-PFBS correlated with the other short-chain L-PFPeS (0.78) and PFHpA (0.68). PFHxS correlated with L-PFPeS (0.70) and L-PFHpS (0.68). The non-PFAS compound, 4-OH-PCB-187, demonstrated weak to moderate correlations to PFAS compounds (≤0.53). A single compound, the short-chain 6:2FTS, exhibited virtually no correlation with any of the other investigated compounds (≤0.10) (Fig. 2).
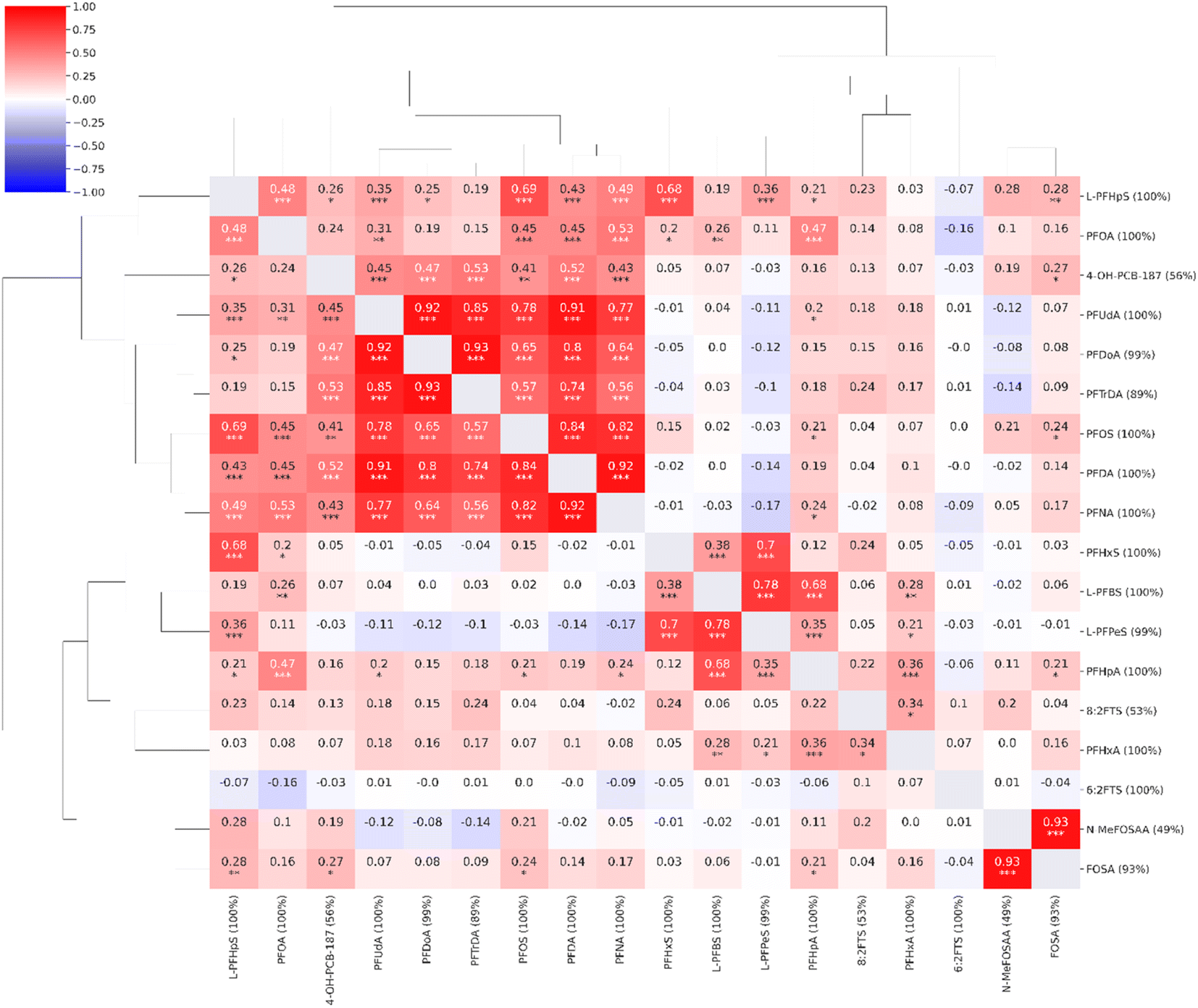 | ||
| Fig. 2 Correlation heatmap for the compounds observed in the targeted analysis (n = 18, coverage cutoff ≥ 15%). The percentage values within parentheses next to compound names indicate compound coverage. The stars (*) below correlation coefficients indicate significance: ***: p ≤ 0.001; **: p ≤ 0.01; *: p ≤ 0.05. A detailed heatmap with the specific p-values is found in the ESI (Fig. S2†). | ||
3.3. Results of untargeted screening
In the untargeted data analysis, 39538 features were detected and quantified. Eleven out of the 18 compounds found in the targeted approach were also quantified in the untargeted data. The compounds not detected in the untargeted data analysis were compounds with low concentrations or low responses, e.g., 4-OH-PCB-187 and 6:2FTS. For the eleven compounds detected using both methods, there was a good correlation between the concentrations estimated using the targeted approach and the same data analyzed using the untargeted approach (ESI, Fig. S5†). The quantitative results of the untargeted approach were similar to the targeted approach with strong correlations between long-chain PFAS compounds (Fig. 3).
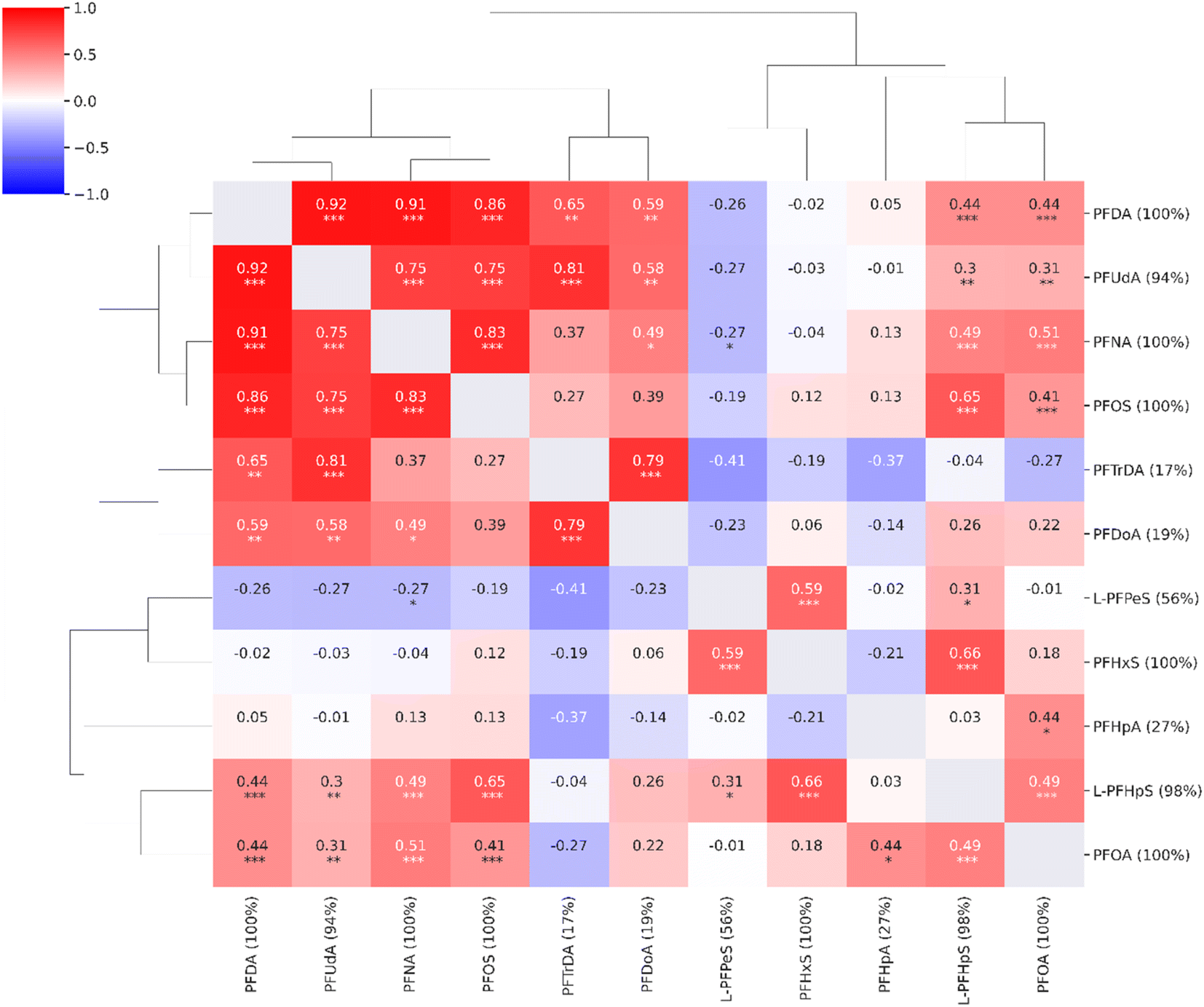 | ||
| Fig. 3 Correlation heatmap for the compounds from the target list observed in the untargeted analysis (n = 11, coverage cutoff ≥ 15%). The percentage values within parentheses next to compound names indicate compound coverage. The stars (*) below correlation coefficients indicate significance: ***: p ≤ 0.001; **: p ≤ 0.01; *: p ≤ 0.05. A detailed heatmap with the specific p-values is found in the ESI (Fig. S3†). | ||
Including all features detected in ≥90% of the samples (4939 features), we found fourteen features with statistically significant associations (correlation coefficients 0.48–0.99) to seven of the confirmed environmental contaminants (Fig. 4). Based on the available MS1 and MS2 data, we identified four compounds at confidence level 2 (putative identifications, supported by MS2 spectra) and three at confidence level 3 (tentative identifications). The confidence levels for identification used here were introduced and described by Schymanski et al. to communicate the confidence of HRMS identifications.41
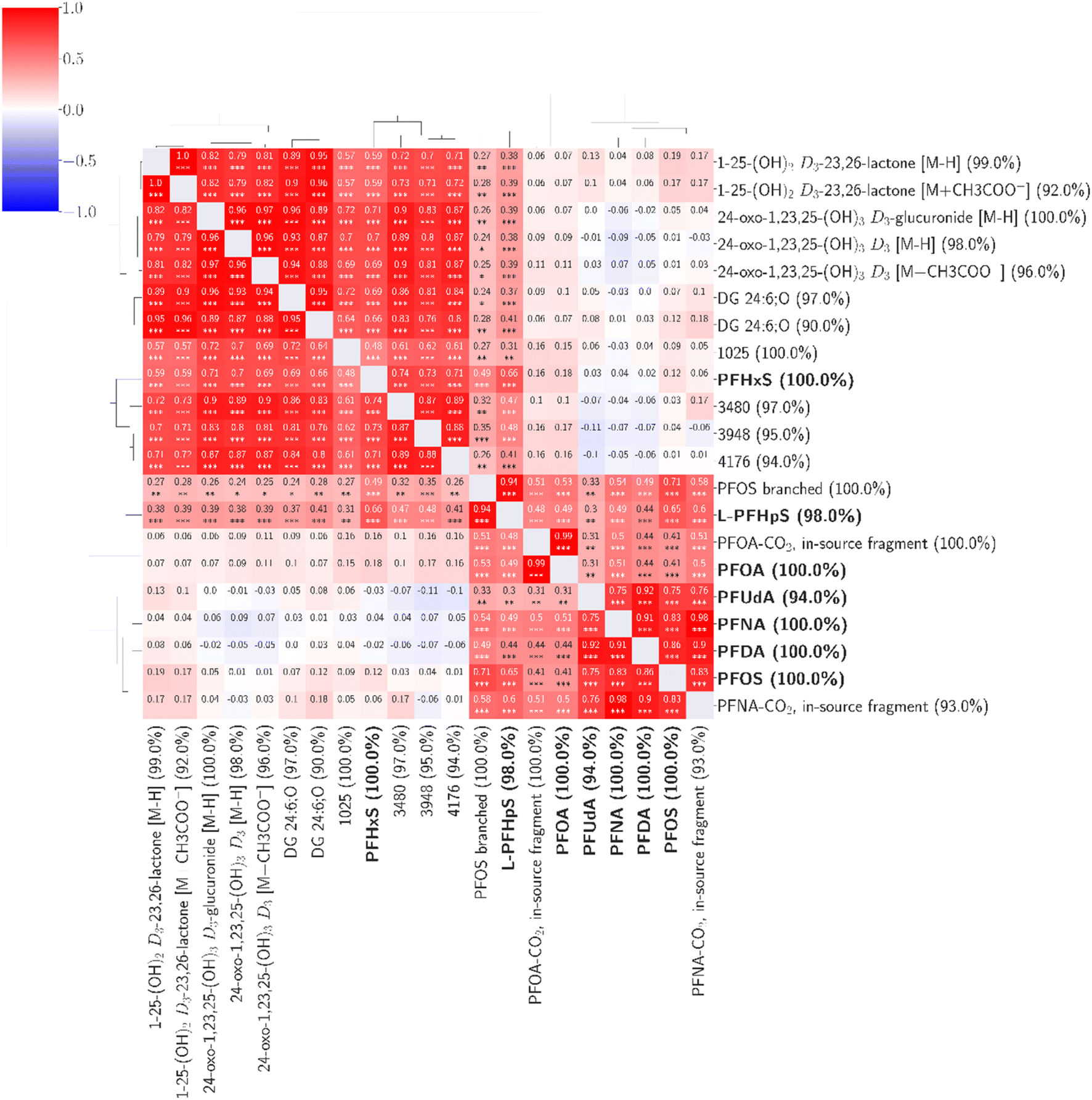 | ||
| Fig. 4 Fourteen unknown features were found to be associated with seven confirmed environmental contaminants. The percentage values within parentheses next to compound names indicate compound coverage. The stars (*) below correlation coefficients indicate significance: ***: p ≤ 0.001; **: p ≤ 0.01; *: p ≤ 0.05. A detailed heatmap with the specific p-values is found in the ESI (Fig. S4†). | ||
Three compounds correlated strongly (correlation coefficients 0.76–0.99) with six targeted PFASs (PFOA, PFNA, PFOS, PFDA, PFUdA, and L-PFHpS). By evaluating the mass spectra and retention times compared to the known targeted compounds, these three were identified at confidence level 2 (Table S3†) as branched PFOS and two in-source fragments of PFOA and PFNA. The branched PFOS was identified based on exhibiting an m/z within 2 ppm of the theoretical m/z and a retention time 0.1 minutes before linear PFOS. Various branched PFOS variants were included in the used reference mix, matching the untargeted compound(s) observed here, although it is not possible to determine which specific branched forms were observed here based on the data. The two in-source fragments can be formed from loss of carboxylic groups from the parent compounds (loss of CO2). They also share the exact retention times and exhibited high correlations (0.98–0.99) with the parent compounds, making in-source fragmentation their most probable source.
Five endogenous compounds, all strongly correlating with PFHxS, were identified (Table 3). The identification process for these endogenous compounds started from the observed m/z of the respective molecular features. Using the databases HMDB,37 Metlin,38 and Lipid Maps39 we searched for compounds within 5 ppm from the observed m/z to find likely candidates with matching molecular formulae. When several possible candidates were present we evaluated and compared the different candidates to select the most likely compound to be observed in human plasma (e.g., excluding plant metabolites). When available we compared the logP values presented in the databases between the different candidates for matching with the observed retention times. Finally, the MS2 spectra (available for four of the five compounds, identified at confidence level 2) were carefully evaluated to identify characteristic fragments to further strengthen the proposed identities. Annotated MS2 spectra for these compounds are presented in the ESI.†
| Unknown ID | Observed m/z | rt (min) | Correlation coefficient towards PFHxS (p-value)a | Identity (confidence level)b | Molecular formula | Mass error (ppm) |
|---|---|---|---|---|---|---|
| a p-values for correlation coefficients. All p-values are presented in the ESI. b Identification confidence levels according to Schymanski et al. 2014.41 | ||||||
| 314 | 621.3294 | 8.07 | 0.71 (p = 2.4 × 10−16) | Vitamin D3 metabolite: 24-oxo-1α,23,25-trihydroxyvitamin D3-(glucuronide) (level 2) | C33H50O11 | 2.25 |
| 3143 & 3842 | 445.2965/505.3175 | 9.07 | 0.7 (p = 7.6 × 10−16)/0.69 (p = 5.4 × 10−15) | Vitamin D3 metabolite: 24-oxo-1α,23,25-trihydroxyvitamin D3 (level 3) | C27H42O5 (loss of hydrogen and acetate adduct respectively) | 1.38 |
| 4534 & 2991 | 443.2803/503.3019 | 8.87 | 0.59 (p = 1.2 × 10−10)/0.59 (p = 4.8 × 10−10) | Vitamin D3 metabolite: 1,25-dihydroxyvitamin D3-23,26-lactone (level 2) | C27H40O5 (loss of hydrogen and acetate adduct respectively) | 0.17 |
| 3613 & 4862 | 459.2753/459.2764 | 8.08/8.24 | 0.69 (p = 9.3 × 10−15)/0.66 (p = 1.7 × 10−12) | Diglyceride lipid: DG 24:6;O (level 3) | C27H40O6 | 0.11 |
Three metabolites of vitamin D3 were strongly positively correlated with PFHxS (correlation coefficients 0.59–0.71) and with moderate correlation towards branched PFOS and L-PFHpS (correlation coefficients 0.24–0.38). The vitamin D3 metabolites are as follows: 1α,25-dihydroxyvitamin D3-26,23-lactone (1,25(OH)2D3-26,23-lactone; level 2), 24-oxo-1α,23,25-trihydroxyvitamin D3 (24(O)-1,23,25(OH)3D3; level 3) and, 24-oxo-1α,23,25-trihydroxyvitamin D3-glucuronide (24(O)-1,23,25(OH)3D3-glucuronide; level 2). The observed m/z for the compounds were all within 2.25 ppm from the theoretical m/z values, giving high confidence to the proposed molecular formulae. For 1,25(OH)2D3-26,23-lactone and 24(O)-1,23,25(OH)3D3 two adducts were observed (with two corresponding features): loss of hydrogen ([M − H]−; the most abundant adduct for both) and acetate adduct ([M + CH3COO−]−). For both compounds, both adducts correlated with PFHxS with highly similar correlation coefficients (Table 3). The observation of two adducts exhibiting the same correlation for these compounds gives further confidence to the proposed identities. For 1,25(OH)2D3-26,23-lactone and 24(O)-1,23,25(OH)3D3-glucuronide, the concentration in the pooled sample used for MS2 data collection was sufficiently high to collect qualitative MS2 spectra supporting the proposed structures (ESI†). For 24(O)-1,23,25(OH)3D3 no useful MS2 spectrum was collected; therefore, this compound is labeled as tentatively identified (confidence level 3). However, the observation of the glucuronide conjugate (24(O)-1,23,25(OH)3D3-glucuronide) of the same compound, also correlated with PFHxS, supports the assigned identity of the non-conjugated form.42 Following the observations of vitamin D3 metabolites correlating with PFHxS, we extracted the mass traces for the central vitamin D metabolites 25-hydroxyvitamin D3 and 1,25-hydroxyvitamin D3 from the data and investigated possible associations with PFHxS and the other targeted PFASs. No statistically significant associations were found (ESI, Fig. S6†).
Finally, two forms of the diglyceride lipid DG 24:6;O (identified at confidence level 3) also strongly correlated with PFHxS (correlation coefficients 0.66 and 0.69) and with moderate correlation towards branched PFOS and L-PFHpS (correlation coefficients 0.24–0.41) were identified. The observed m/z for these lipids deviated only 0.11 ppm from the theoretical m/z, making the assigned molecular formula, typical of diglyceride lipids, highly probable. Annotated MS2 spectra supporting the proposed identities are found in the ESI. Several structural isomers of these lipids are present in plasma, and here two separate peaks (corresponding to two features) were observed and identified. The annotated identity DG 24:6;O refers to the total number of carbons, double bonds and modifications (six double bonds and an OH-group) of the two fatty acids. In the HMBD, 20 different structural isomers for these DG lipids are reported, and authentic references would be required to specifically identify which isomers are found to correlate with PFHxS in the present study.
4 Discussion
As outlined by Vermeulen et al., several challenges lie ahead for exploiting the full potential of exposome research.7 One major point is to improve the available technologies to screen for exogenous chemicals at higher-throughput rates and lower costs and to further develop the chemical and spectral data resources to identify these chemicals in samples. With that in mind, we developed a combined targeted and untargeted LC-HRMS method that is fast, simple, and highly applicable for high-throughput analysis of large cohorts. To our knowledge, the metabolomics-inspired workflows used here have not been previously used for the screening of environmental contaminants.High-resolution mass spectrometry has been described as a prime example of a technique suited for assessing the exposome.7 Using HRMS, thousands to tens of thousands of chemical features may be measured in a single analytical run. Data analysis tools developed for, e.g., metabolome profiling may be used to map the exposome from untargeted HRMS data. A wide range of analytical methodologies are available for different compound classes. For example, several classes of common environmental contaminants, e.g., PCBs and BDEs, typically require gas chromatography-based methods, whereas their hydroxylated metabolites (OH-PCBs and OH-BDEs, respectively) can be measured by LC-based methods with electrospray ionization.43,44 Here we focused our methodology on compounds that form ions during negative electrospray ionization, a group of compounds that have been labeled environmental organic acids and include, e.g., PFASs, OH-PCBs, OH-BDEs, bisphenols, and HBCDs.8 The developed method was optimized with regard to the response and chromatographic performance of all targets from the different compound classes. The targeted compounds were chosen based on environmental relevance and commercial availability. We decided to limit the required sample volume to 50 μL, which is relatively low in environmental analysis26 so the method should apply to the analysis of cohorts where sample volume is limited.
Across the 100 studied samples from blood donors, we detected 19 of the 37 targeted compounds, with 18 of those being PFASs. PFASs are a large group of synthetic compounds with more than 10000 different PFAS chemicals listed by the US Environmental Protection Agency.45 PFASs were detected in all 100 studied samples, with similar concentrations to those reported recently in similar study populations.46,47 The unusually high concentration of PFHxS in a single sample may be related to PFAS contamination of the drinking water in certain areas of Uppsala, which was discovered and mitigated in 2012. The contamination has been sourced to PFHxS-based aqueous film-forming foam used for firefighting during military training at a nearby airport until at least 2003, which keeps leaking to the environment today.48,49
During the last 20 years, the industrial production of PFASs has changed significantly internationally due to increased awareness of the associated risks and regulatory pressures.50 Some specific PFASs have been phased out, most notably PFOS and PFOA, while short-chain alternatives have seen increased use as replacements. Short-chain PFASs are known to be eliminated faster in animals and humans than the long-chain legacy PFASs that bioaccumulate and are therefore expected to be less toxic. However, there is growing evidence that the toxicity of some short-chain PFASs has been severely underestimated, which raises new concerns.51,52 In the present study, nine PFASs showed a positive association with increasing age. Most of these were long-chained,9 except for short-chain PFHpA. The higher concentrations in older subjects could be related to the bioaccumulating properties of these PFAS, and similar associations have been reported previously.53,54 We also found higher concentrations of L-PFHpS, PFOS, and PFNA in male compared to female subjects, which agrees with previous observations for several PFASs.53,55 It has been proposed that this sex difference could be related to occupational exposure to PFASs in male-dominated occupations.53,56
When investigating the correlation between targeted analytes, we found strong correlations of the concentrations between long-chain PFASs across the samples, potentially related to the bioaccumulating properties of these compounds and possibly indicating common sources of exposures.57 Structurally related PFASs such as FOSA and N-MeFOSAA, and short-chained L-PFBS and L-PFPeS were also found to correlate. Among the targeted compounds, short-chained 6:2FTS exhibited virtually no correlation among the targeted compounds. This compound has been increasingly used as a substitute for various industrial applications that formerly required the use of PFOS or PFOA, and is less toxic and bioaccumulative compared to those more fluorinated compounds.9,58,59 Decreased accumulation together with differing manufacturing trends could explain why no correlations were observed for this compound. As could be expected, structurally related compounds with similar chemical properties were found to correlate in concentrations.
The final targeted compound detected in the samples was 4-OH-PCB-187. This OH-PCB had low coverage at 15% and was mainly detected in subjects >50 years old, showing a positive association with increasing age. Hydroxylated PCBs are strongly retained in human blood and may cause endocrine-related toxicity.60,61 The 4-OH-PCB-187 is one of the predominant OH-PCB congeners in human plasma, and in several studies, the most abundant OH-PCB.43,60 That this specific OH-PCB is detected in the studied samples is therefore not surprising, and the association with increasing age is in line with its accumulating properties. PCBs were phased out in the 1970s but are still present in our environment, with the main exposure source being the diet, in Sweden, particularly from the consumption of fatty fish.60,62,63
Having evaluated the method's performance on targeted analytes, we then explored the untargeted data. An initial challenge was optimizing our peak-picking methods to detect the typically low-level environmental contaminants amongst the much more abundant endogenous compounds also detected with the untargeted method. With too liberal criteria for peak-picking, noise will be included as features, and with too strict criteria, low-level compounds will not be detected. Considering this, we found a balanced set of parameters that could extract and integrate most of the targeted compounds from the untargeted data. We compared the output from the untargeted data with that from the targeted approach and found high agreement and the same general trends with strong correlations between several long-chain PFASs. The targeted approach was, however, better for detecting compounds of very low concentrations or with low responses. The best choice may be to use a combined approach, collecting untargeted data that is evaluated using a list of targets or suspects and with a separate evaluation of the untargeted data.
It is challenging to identify features when data is collected in an untargeted mode. To facilitate feature identification, we therefore used a detailed strategy to collect high-quality MS2 spectra. The complete m/z range of the primary full scan method was divided into six overlapping m/z windows, and nine data-dependent MS2 scans, with varying collision energies, were collected using a pooled sample of all individuals included in the study. This improved our success rate for identification. As an example of this, linear perfluorocarboxylic acids are almost fully fragmented even at low collision energies, while perfluoroalkylsulfonates and the vitamin D3 metabolites require high collision energies for sufficient fragmentation (example MS2 spectra in ESI†).
We identified three metabolites of vitamin D3 with strong positive association with PFHxS: 1α,25-dihydroxyvitamin D3-26,23-lactone (1,25(OH)2D3-26,23-lactone), 24-oxo-1α,23,25-trihydroxyvitamin D3 (24(O)-1,23,25(OH)3D3) and 24-oxo-1α,23,25-trihydroxyvitamin D3-glucuronide (24(O)-1,23,25(OH)3D3-glucuronide). Vitamin D3 is a type of vitamin D made in the human skin under UV light, as opposed to vitamin D2, which humans get from the diet.64 Associations between vitamin D3 biomarkers and PFAS have previously been reported,65 and recently, the vitamin D receptor has been identified as a potential target for the toxic effects of PFASs.66 Previously Etzel et al. observed PFHxS to be associated with higher vitamin D concentrations and lower odds of being vitamin D deficient.65 This observed association was not modified by age and sex. In that study, PFAS concentrations were compared with concentrations of the main vitamin D metabolite 25-hydroxyvitamin D3.65 We also found 25-hydroxyvitamin D3 and the additional active vitamin D3 metabolite 1,25-hydroxyvitamin D3 in our untargeted data but found no statistically significant associations with PFHxS or other measured PFAS compounds. It has been reported that other endocrine-disrupting chemicals may affect circulating concentrations of vitamin D,67 and vitamin D status has been associated with several diseases, including cancer, cardiovascular disease, multiple sclerosis,68 and other autoimmune diseases.64,65,69 Two forms of the diglyceride lipid DG 24:6;O were also strongly associated with PFHxS. To our knowledge, no reports on associations between PFASs and diglycerides have been made; however, there are several examples of how PFAS exposures alter the concentrations of other blood lipids.70–73 Based on this, the interaction between the lipidome and PFASs is important to investigate in human health.
5 Conclusions
We here present a combined targeted and untargeted method suited for high-throughput analysis of environmental contaminants in plasma. This application of metabolomics-inspired workflows for the screening of environmental contaminants has a high potential for exposome profiling. Nineteen targeted environmental contaminants were detected in the study population, with ten associated with age and three with sex. In the evaluation of the untargeted data, three vitamin D3 metabolites and two diglyceride lipids were shown to associate with PFHxS strongly. The results demonstrate the potential of combining targeted and untargeted approaches that may be important in understanding the effects of environmental contaminants on human health.Data availability
The parameters and code used are available at https://github.com/caramba-uu/env-con. The raw and processed data is available upon reasonable request to the corresponding author.Author contributions
Henrik Carlsson: conceptualization, methodology, investigation, writing – original draft, funding acquisition. A. Parakkal Sreenivasan: methodology, investigation, formal analysis, visualization, writing – original draft. I. Erngren: methodology, investigation, writing – original draft. A. Larsson: resources, supervision, writing – review & editing. K. Kultima: conceptualization, resources, supervision, writing – original draft, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Abbreviations
| 4-OH-PCB-187 | 2,2′,3,4′,5,5′,6-Heptachloro-4-biphenylol |
| 6:2FTS | 1H,1H,2H,2H-perfluoro-1-octanesulfonic acid (6:2) |
| 8:2FTS | 1H,1H,2H,2H-perfluoro-1-decanesulfonic acid (8:2) |
| FOSA | Perfluoro-1-octanesulfonamide |
| HBCD | Hexabromocyclododecane |
| IS | Internal standard |
| LC-HRMS | High-resolution mass spectrometry |
| L-PFBS | Perfluorobutanesulfonic acid |
| L-PFHpS | Perfluoroheptanesulfonic acid |
| L-PFPeS | Perfluoropentanesulfonic acid |
| MeOH | Methanol |
| MS | Mass spectrometry |
| N-EtFOSAA | N-ethylperfluoro-1-octanesulfonamidoacetic acid |
| NH4Ac | Ammonium acetate |
| N-MeFOSAA | N-methylperfluoro-1-octanesulfonamidoacetic acid |
| OH-BDE | Phenolic metabolites of polybrominated diphenylethers |
| OH-PCB | Phenolic metabolites of polychlorinated biphenyls |
| PFAS | Per- and polyfluoroalkyl substances |
| PFDA | Perfluoro-n-decanoic acid |
| PFDoA | Perfluoro-n-dodecanoic acid |
| PFHpA | Perfluoro-n-heptanoic acid |
| PFHxA | Perfluoro-n-hexanoic acid |
| PFHxS | Perfluorohexanesulfonic acid |
| PFNA | Perfluoro-n-nonanoic acid |
| PFOA | Perfluoro-n-octanoic acid |
| PFOS | Perfluorooctanesulfonic acid |
| PFTrDA | Perfluoro-n-tridecanoic acid |
| PFUdA | Perfluoro-n-undecanoic acid |
| RT | Retention time |
Acknowledgements
This work was supported by FORMAS (2020–01267 and 2022-00488), the Swedish Research Council (2021-02189), Region Uppsala (ALF-grant and R&D funds), Neuro Sweden, Åke Wiberg foundation and Lars Hierta Memorial Foundation. We also acknowledge the anonymous reviewers for their careful reading and constructive criticism, which improved the manuscript.References
- P. Lichtenstein, N. V. Holm, P. K. Verkasalo, A. Iliadou, J. Kaprio, M. Koskenvuo, E. Pukkala, A. Skytthe and K. Hemminki, Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland, N. Engl. J. Med., 2000, 343, 78–85 CrossRef CAS PubMed.
- T. A. Manolio, F. S. Collins, N. J. Cox, D. B. Goldstein, L. A. Hindorff, D. J. Hunter, M. I. McCarthy, E. M. Ramos, L. R. Cardon, A. Chakravarti, J. H. Cho, A. E. Guttmacher, A. Kong, L. Kruglyak, E. Mardis, C. N. Rotimi, M. Slatkin, D. Valle, A. S. Whittemore, M. Boehnke, A. G. Clark, E. E. Eichler, G. Gibson, J. L. Haines, T. F. C. Mackay, S. A. McCarroll and P. M. Visscher, Finding the missing heritability of complex diseases, Nature, 2009, 461, 747–753 CrossRef CAS PubMed.
- S. M. Rappaport, Genetic Factors Are Not the Major Causes of Chronic Diseases, PLoS One, 2016, 11, e0154387 CrossRef PubMed.
- C. P. Wild, Complementing the genome with an ‘exposome’: the outstanding challenge of environmental exposure measurement in molecular epidemiology, Cancer Epidemiol., Biomarkers Prev., 2005, 14, 1847–1850 CrossRef CAS PubMed.
- S. M. Rappaport, Implications of the exposome for exposure science, J. Exposure Sci. Environ. Epidemiol., 2011, 21, 5–9 CrossRef CAS PubMed.
- S. M. Rappaport, D. K. Barupal, D. Wishart, P. Vineis and A. Scalbert, The blood exposome and its role in discovering causes of disease, Environ. Health Perspect., 2014, 122, 769–774 CrossRef PubMed.
- R. Vermeulen, E. L. Schymanski, A.-L. Barabási and G. W. Miller, The exposome and health: Where chemistry meets biology, Science, 2020, 367, 392–396 CrossRef CAS PubMed.
- R. R. Gerona, J. M. Schwartz, J. Pan, M. M. Friesen, T. Lin and T. J. Woodruff, Suspect screening of maternal serum to identify new environmental chemical biomonitoring targets using liquid chromatography-quadrupole time-of-flight mass spectrometry, J. Exposure Sci. Environ. Epidemiol., 2018, 28, 101–108 CrossRef CAS PubMed.
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. J. van Leeuwen, Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins, Integr, Environ. Assess. Manage., 2011, 7, 513–541 CAS.
- Y. Fujii, K. H. Harada and A. Koizumi, Occurrence of perfluorinated carboxylic acids (PFCAs) in personal care products and compounding agents, Chemosphere, 2013, 93, 538–544 CrossRef CAS PubMed.
- E. Parry, A. R. Zota, J.-S. Park and T. J. Woodruff, Polybrominated diphenyl ethers (PBDEs) and hydroxylated PBDE metabolites (OH-PBDEs): A six-year temporal trend in Northern California pregnant women, Chemosphere, 2018, 195, 777–783 CrossRef CAS PubMed.
- R. Tehrani and B. Van Aken, Hydroxylated polychlorinated biphenyls in the environment: sources, fate, and toxicities, Environ. Sci. Pollut. Res. Int., 2014, 21, 6334–6345 CrossRef CAS PubMed.
- J. Corrales, L. A. Kristofco, W. B. Steele, B. S. Yates, C. S. Breed, E. S. Williams and B. W. Brooks, Global Assessment of Bisphenol A in the Environment: Review and Analysis of Its Occurrence and Bioaccumulation, Dose-Response, 2015, 13, 1559325815598308 CrossRef PubMed.
- W. A. Gebbink, A. Glynn and U. Berger, Temporal changes (1997–2012) of perfluoroalkyl acids and selected precursors (including isomers) in Swedish human serum, Environ. Pollut., 2015, 199, 166–173 CrossRef CAS PubMed.
- L. S. Haug, S. Huber, G. Becher and C. Thomsen, Characterisation of human exposure pathways to perfluorinated compounds-comparing exposure estimates with biomarkers of exposure, Environ. Int., 2011, 37, 687–693 CrossRef CAS PubMed.
- M. Kumar, D. K. Sarma, S. Shubham, M. Kumawat, V. Verma, A. Prakash and R. Tiwari, Environmental Endocrine-Disrupting Chemical Exposure: Role in Non-Communicable Diseases, Front. Public Health, 2020, 8, 553850 CrossRef PubMed.
- K. Steenland and A. Winquist, PFAS and cancer, a scoping review of the epidemiologic evidence, Environ. Res., 2021, 194, 110690 CrossRef CAS PubMed.
- J. C. DeWitt, Toxicological Effects of Perfluoroalkyl and Polyfluoroalkyl Substances, Springer International Publishing, 2015 Search PubMed.
- A. A. Jensen and H. Leffers, Emerging endocrine disrupters: perfluoroalkylated substances, Int. J. Androl., 2008, 31, 161–169 CrossRef CAS PubMed.
- E. Brosset and G. Ngueta, Exposure to per- and polyfluoroalkyl substances and glycemic control in older US adults with type 2 diabetes mellitus, Environ. Res., 2022, 216, 114697 CrossRef PubMed.
- Y. Liu, L. A. D'Agostino, G. Qu, G. Jiang and J. W. Martin, High-resolution mass spectrometry (HRMS) methods for nontarget discovery and characterization of poly- and per-fluoroalkyl substances (PFASs) in environmental and human samples, Trends Anal. Chem., 2019, 121, 115420 CrossRef CAS.
- Y. Liu, E. S. Richardson, A. E. Derocher, N. J. Lunn, H.-J. Lehmler, X. Li, Y. Zhang, J. Y. Cui, L. Cheng and J. W. Martin, Hundreds of Unrecognized Halogenated Contaminants Discovered in Polar Bear Serum, Angew. Chem., Int. Ed. Engl., 2018, 57, 16401–16406 CrossRef CAS PubMed.
- H. Carlsson, N. Rollborn, S. Herman, E. Freyhult, A. Svenningsson, J. Burman and K. Kultima, Metabolomics of Cerebrospinal Fluid from Healthy Subjects Reveal Metabolites Associated with Ageing, Metabolites, 2021, 11(2), 126 CrossRef CAS PubMed.
- H. Carlsson, A. Vaivade, P. Emami Khoonsari, J. Burman and K. Kultima, Evaluation of polarity switching for untargeted lipidomics using liquid chromatography coupled to high resolution mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2022, 1195, 123200 CrossRef CAS PubMed.
- S. Herman, T. Åkerfeldt, O. Spjuth, J. Burman and K. Kultima, Biochemical Differences in Cerebrospinal Fluid between Secondary Progressive and Relapsing-Remitting Multiple Sclerosis, Cells, 2019, 8(2), 84 CrossRef CAS PubMed.
- S. Salihović, A. M. Dickens, I. Schoultz, F. Fart, L. Sinisalu, T. Lindeman, J. Halfvarson, M. Orešič and T. Hyötyläinen, Simultaneous determination of perfluoroalkyl substances and bile acids in human serum using ultra-high-performance liquid chromatography-tandem mass spectrometry, Anal. Bioanal. Chem., 2020, 412, 2251–2259 CrossRef PubMed.
- H. L. Röst, T. Sachsenberg, S. Aiche, C. Bielow, H. Weisser, F. Aicheler, S. Andreotti, H.-C. Ehrlich, P. Gutenbrunner, E. Kenar, X. Liang, S. Nahnsen, L. Nilse, J. Pfeuffer, G. Rosenberger, M. Rurik, U. Schmitt, J. Veit, M. Walzer, D. Wojnar, W. E. Wolski, O. Schilling, J. S. Choudhary, L. Malmström, R. Aebersold, K. Reinert and O. Kohlbacher, OpenMS: a flexible open-source software platform for mass spectrometry data analysis, Nat. Methods, 2016, 13, 741–748 CrossRef PubMed.
- nf-core-metabolinden: Metabolomics quaLIty coNtrol anD paramEter optimizatioN, Github Search PubMed.
- G. Van Rossum and F. L. Drake, Python 3 Reference Manual:(Python Documentation Manual Part 2), CreateSpace, Scotts Valley, CA Search PubMed.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. van Mulbregt and SciPy 1.0 Contributors, SciPy 1.0: fundamental algorithms for scientific computing in Python, Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, A. Müller, J. Nothman, G. Louppe, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, arXiv [cs.LG], 2012, preprint, pp. 2825–2830, DOI:10.48550/arXiv.1201.0490.
- M. Waskom, seaborn: statistical data visualization, J. Open Source Softw., 2021, 6, 3021 CrossRef.
- C. R. Harris, K. J. Millman, S. J. van der Walt, R. Gommers, P. Virtanen, D. Cournapeau, E. Wieser, J. Taylor, S. Berg, N. J. Smith, R. Kern, M. Picus, S. Hoyer, M. H. van Kerkwijk, M. Brett, A. Haldane, J. F. Del Río, M. Wiebe, P. Peterson, P. Gérard-Marchant, K. Sheppard, T. Reddy, W. Weckesser, H. Abbasi, C. Gohlke and T. E. Oliphant, Array programming with NumPy, Nature, 2020, 585, 357–362 CrossRef CAS PubMed.
- W. McKinney, in Proceedings of the 9th Python in Science Conference, SciPy, 2010 Search PubMed.
- NORMAN suspect list exchange, https://www.norman-network.com/nds/SLE/, accessed 13 April 2023 Search PubMed.
- EPA United States Environmental Protection Agency, EPI Suite™-Estimation Program Interface, https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface, accessed 13 April 2023 Search PubMed.
- D. S. Wishart, A. Guo, E. Oler, F. Wang, A. Anjum, H. Peters, R. Dizon, Z. Sayeeda, S. Tian, B. L. Lee, M. Berjanskii, R. Mah, M. Yamamoto, J. Jovel, C. Torres-Calzada, M. Hiebert-Giesbrecht, V. W. Lui, D. Varshavi, D. Varshavi, D. Allen, D. Arndt, N. Khetarpal, A. Sivakumaran, K. Harford, S. Sanford, K. Yee, X. Cao, Z. Budinski, J. Liigand, L. Zhang, J. Zheng, R. Mandal, N. Karu, M. Dambrova, H. B. Schiöth, R. Greiner and V. Gautam, HMDB 5.0: the Human Metabolome Database for 2022, Nucleic Acids Res., 2022, 50, D622–D631 CrossRef CAS PubMed.
- C. A. Smith, G. O'Maille, E. J. Want, C. Qin, S. A. Trauger, T. R. Brandon, D. E. Custodio, R. Abagyan and G. Siuzdak, METLIN: a metabolite mass spectral database, Ther. Drug Monit., 2005, 27, 747–751 CrossRef CAS PubMed.
- E. Fahy, M. Sud, D. Cotter and S. Subramaniam, LIPID MAPS online tools for lipid research, Nucleic Acids Res., 2007, 35, W606–W612 CrossRef PubMed.
- K. Dührkop, H. Shen, M. Meusel, J. Rousu and S. Böcker, Searching molecular structure databases with tandem mass spectra using CSI:FingerID, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12580–12585 CrossRef PubMed.
- E. L. Schymanski, J. Jeon, R. Gulde, K. Fenner, M. Ruff, H. P. Singer and J. Hollender, Identifying small molecules via high resolution mass spectrometry: communicating confidence, Environ. Sci. Technol., 2014, 48, 2097–2098 CrossRef CAS PubMed.
- C. Jenkinson, R. Desai, M. D. McLeod, J. Wolf Mueller, M. Hewison and D. J. Handelsman, Circulating Conjugated and Unconjugated Vitamin D Metabolite Measurements by Liquid Chromatography Mass Spectrometry, J. Clin. Endocrinol. Metab., 2022, 107, 435–449 CrossRef PubMed.
- N. Quinete, T. Kraus, V. N. Belov, C. Aretz, A. Esser and T. Schettgen, Fast determination of hydroxylated polychlorinated biphenyls in human plasma by online solid phase extraction coupled to liquid chromatography-tandem mass spectrometry, Anal. Chim. Acta, 2015, 888, 94–102 CrossRef CAS PubMed.
- C. M. Butt, M. L. Miranda and H. M. Stapleton, Development of an analytical method to quantify PBDEs, OH-BDEs, HBCDs, 2,4,6-TBP, EH-TBB, and BEH-TEBP in human serum, Anal. Bioanal. Chem., 2016, 408, 2449–2459 CrossRef CAS PubMed.
- CompTox Chemicals Dashboard, https://comptox.epa.gov/dashboard/chemical-lists/pfasmaster, accessed 13 April 2023 Search PubMed.
- B. Göckener, T. Weber, H. Rüdel, M. Bücking and M. Kolossa-Gehring, Human biomonitoring of per- and polyfluoroalkyl substances in German blood plasma samples from 1982 to 2019, Environ. Int., 2020, 145, 106123 CrossRef PubMed.
- L. T. Miaz, M. M. Plassmann, I. Gyllenhammar, A. Bignert, O. Sandblom, S. Lignell, A. Glynn and J. P. Benskin, Temporal trends of suspect- and target-per/polyfluoroalkyl substances (PFAS), extractable organic fluorine (EOF) and total fluorine (TF) in pooled serum from first-time mothers in Uppsala, Sweden, 1996–2017, Environ. Sci.: Processes Impacts, 2020, 22, 1071–1083 RSC.
- I. Gyllenhammar, A. Glynn, J. Benskin, O. Sandblom, A. Bignert and S. Lignell, Temporal trends of poly- and perfluoroalkyl substances (PFASs) in pooled serum samples from first-time mothers in Uppsala 1997-2016, 2017 Search PubMed.
- Domstolsförhandling PFAS, https://www.uppsalavatten.se/hushall/vatten-och-avlopp/dricksvatten/domstolsforhandling-pfas/, accessed 13 April 2023.
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, An overview of the uses of per- and polyfluoroalkyl substances (PFAS), Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373 RSC.
- P. A. Rice, J. Aungst, J. Cooper, O. Bandele and S. V. Kabadi, Comparative analysis of the toxicological databases for 6:2 fluorotelomer alcohol (6:2 FTOH) and perfluorohexanoic acid (PFHxA), Food Chem. Toxicol., 2020, 138, 111210 CrossRef CAS PubMed.
- S. V. Kabadi, J. W. Fisher, D. R. Doerge, D. Mehta, J. Aungst and P. Rice, Characterizing biopersistence potential of the metabolite 5:3 fluorotelomer carboxylic acid after repeated oral exposure to the 6:2 fluorotelomer alcohol, Toxicol. Appl. Pharmacol., 2020, 388, 114878 CrossRef CAS PubMed.
- E. Obeng-Gyasi, Factors associated with elevated Per- and Polyfluoroalkyl substances serum levels in older adults, Natl. Bur. Econ. Res. Bull. Aging Health, 2022, 2, 100086 CrossRef PubMed.
- Y. Tian, Y. Zhou, M. Miao, Z. Wang, W. Yuan, X. Liu, X. Wang, Z. Wang, S. Wen and H. Liang, Determinants of plasma concentrations of perfluoroalkyl and polyfluoroalkyl substances in pregnant women from a birth cohort in Shanghai, China, Environ. Int., 2018, 119, 165–173 CrossRef CAS PubMed.
- R. B. Jain and A. Ducatman, Serum concentrations of selected perfluoroalkyl substances for US females compared to males as they age, Sci. Total Environ., 2022, 842, 156891 CrossRef CAS PubMed.
- E. M. Tanner, M. S. Bloom, Q. Wu, K. Kannan, R. M. Yucel, S. Shrestha and E. F. Fitzgerald, Occupational exposure to perfluoroalkyl substances and serum levels of perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) in an aging population from upstate New York: a retrospective cohort study, Int. Arch. Occup. Environ. Health, 2018, 91, 145–154 CrossRef CAS PubMed.
- L. P. Burkhard, Literature Review of PFAS Bioaccumulation Data, 2020, https://epa.figshare.com/articles/presentation/Literature_review_of_PFAS_bioaccumulation_data/13120190/1, accessed 13 April 2023 Search PubMed.
- Z. Wang, I. T. Cousins, M. Scheringer and K. Hungerbühler, Fluorinated alternatives to long-chain perfluoroalkyl carboxylic acids (PFCAs), perfluoroalkane sulfonic acids (PFSAs) and their potential precursors, Environ. Int., 2013, 60, 242–248 CrossRef CAS PubMed.
- R. C. Buck, in Toxicological Effects of Perfluoroalkyl and Polyfluoroalkyl Substances, ed. J. C. DeWitt, Springer International Publishing, Cham, 2015, pp. 451–477 Search PubMed.
- F. A. Grimm, D. Hu, I. Kania-Korwel, H.-J. Lehmler, G. Ludewig, K. C. Hornbuckle, M. W. Duffel, Å. Bergman and L. W. Robertson, Metabolism and metabolites of polychlorinated biphenyls, Crit. Rev. Toxicol., 2015, 45, 245–272 CrossRef CAS PubMed.
- Å. Bergman, A. Brouwer, L. Hagmar, I. Meerts and A. Sjödin, Hydroxylated PCBs (OH-PCBs) as potential endocrine disrupters – exposures and toxic effects, APMIS, 2001, 109, S505 CrossRef.
- S. Mikolajczyk, M. Warenik-Bany and M. Pajurek, PCDD/Fs and PCBs in Baltic fish – Recent data, risk for consumers, Mar. Pollut. Bull., 2021, 171, 112763 CrossRef CAS PubMed.
- J. Szlinder-Richert, I. Barska, J. Mazerski and Z. Usydus, PCBs in fish from the southern Baltic Sea: levels, bioaccumulation features, and temporal trends during the period from 1997 to 2006, Mar, Pollut. Bull., 2009, 58, 85–92 CrossRef CAS PubMed.
- D. D. Bikle, Vitamin D metabolism, mechanism of action, and clinical applications, Chem. Biol., 2014, 21, 319–329 CrossRef CAS PubMed.
- T. M. Etzel, J. M. Braun and J. P. Buckley, Associations of serum perfluoroalkyl substance and vitamin D biomarker concentrations in NHANES, 2003-2010, Int. J. Hyg. Environ. Health, 2019, 222, 262–269 CrossRef CAS PubMed.
- E. R. Azhagiya Singam, K. A. Durkin, M. A. La Merrill, J. D. Furlow, J.-C. Wang and M. T. Smith, The vitamin D receptor as a potential target for the toxic effects of per- and polyfluoroalkyl substances (PFASs): An in-silico study, Environ. Res., 2022, 114832 Search PubMed.
- L. E. Johns, K. K. Ferguson and J. D. Meeker, Relationships Between Urinary Phthalate Metabolite and Bisphenol A Concentrations and Vitamin D Levels in U.S. Adults: National Health and Nutrition Examination Survey (NHANES), 2005-2010, J. Clin. Endocrinol. Metab., 2016, 101, 4062–4069 CrossRef CAS PubMed.
- A. Miclea, M. Bagnoud, A. Chan and R. Hoepner, A Brief Review of the Effects of Vitamin D on Multiple Sclerosis, Front. Immunol., 2020, 11, 781 CrossRef CAS PubMed.
- J. Hahn, N. R. Cook, E. K. Alexander, S. Friedman, J. Walter, V. Bubes, G. Kotler, I.-M. Lee, J. E. Manson and K. H. Costenbader, Vitamin D and marine omega 3 fatty acid supplementation and incident autoimmune disease: VITAL randomized controlled trial, BMJ [Br. Med. J.], 2022, 376, e066452 Search PubMed.
- Y. Li, L. Barregard, Y. Xu, K. Scott, D. Pineda, C. H. Lindh, K. Jakobsson and T. Fletcher, Associations between perfluoroalkyl substances and serum lipids in a Swedish adult population with contaminated drinking water, Environ. Health, 2020, 19, 33 CrossRef CAS PubMed.
- C. Canova, G. Barbieri, M. Zare Jeddi, M. Gion, A. Fabricio, F. Daprà, F. Russo, T. Fletcher and G. Pitter, Associations between perfluoroalkyl substances and lipid profile in a highly exposed young adult population in the Veneto Region, Environ. Int., 2020, 145, 106117 CrossRef CAS PubMed.
- G. Liu, B. Zhang, Y. Hu, J. Rood, L. Liang, L. Qi, G. A. Bray, L. DeJonge, B. Coull, P. Grandjean, J. D. Furtado and Q. Sun, Associations of Perfluoroalkyl substances with blood lipids and Apolipoproteins in lipoprotein subspecies: the POUNDS-lost study, Environ. Health, 2020, 19, 5 CrossRef CAS PubMed.
- T. Schillemans, I. A. Bergdahl, K. Hanhineva, L. Shi, C. Donat-Vargas, J. Koponen, H. Kiviranta, R. Landberg, A. Åkesson and C. Brunius, Associations of PFAS-related plasma metabolites with cholesterol and triglyceride concentrations, Environ. Res., 2022, 216, 114570 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00060e |
| This journal is © The Royal Society of Chemistry 2023 |