
Dehydrogenative coupling of methane over Pt/Al2O3 catalysts: effect of hydrogen co-feeding†
Tatsuki
Tomono
a,
Riku
Takamura
a,
Miru
Yoshida-Hirahara
a,
Tomokazu
Yamamoto
b,
Syo
Matsumura
bc,
Hideki
Kurokawa
 a and
Hitoshi
Ogihara
*a
a and
Hitoshi
Ogihara
*a
aGraduate School of Science and Engineering, Saitama University, 255 Shimo-Okubo, Sakura-ku, Saitama 338-8570, Japan. E-mail: ogihara@mail.saitama-u.ac.jp
bThe Ultramicroscopy Research Center, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
cNational Institute of Technology, Kurume College, 1-1-1 Komorino, Kurume 830-8555, Japan
First published on 12th June 2023
Abstract
The dehydrogenative conversion of methane (DCM) is a promising technology for using natural gas as a chemical resource. However, direct methane conversion is challenging owing to the high stability of methane molecules. In this study, we developed a novel DCM system in which a typical dehydrogenation catalyst, Pt/Al2O3, steadily converted methane into C2 hydrocarbons with the aid of H2 co-feeding. The catalytic performance of Pt/Al2O3 in the non-oxidative coupling of methane (NOCM) was significantly affected by the presence of hydrogen. When pure methane was fed over the Pt/Al2O3 catalyst, the catalyst was quickly deactivated via coke deposition. In contrast, when H2 was co-fed with methane, the deactivation of the catalysts was suppressed, and C2 hydrocarbons were stably formed. X-ray photoelectron spectroscopy and thermogravimetric analysis showed that H2 co-feeding suppressed coke deposition on the Pt surface. At a reaction temperature of 600 °C, the Pt/Al2O3 catalyst showed a C2 hydrocarbon formation rate of >8 μmol min−1 gcat−1 over 24 h in the presence of H2. Furthermore, Pt loading significantly affected the DCM reaction. A low Pt loading was effective for producing hydrocarbons. Electron microscopy analysis showed that with increasing Pt loading, the proportion of coarse nanoparticles increased. Fourier transform infrared spectroscopy suggested that the well-coordinated Pt sites were likely to form coke and deactivate, whereas the highly under-coordinated Pt sites were less likely to form coke. Because Pt/Al2O3 with a low Pt loading contains under-coordination sites, the catalyst was stable for the NOCM.
Introduction
Natural gas production has increased owing to improvements in shale gas extraction technology. The role of natural gas in the chemical industry is expected to expand because oil, which is the main raw material in the chemical industry, is on the verge of gradual depletion. The main component of natural gas is methane (CH4). Thus, if CH4 is converted into basic chemicals, such as lower olefins and aromatics, natural gas can be used as an alternative to oil resources. However, the direct conversion of CH4 into chemicals is challenging because CH4 is a highly stable molecule.1–3 CH4 molecules have strong C–C bonds and highly symmetric structures, which hinder its activation.For the conversion of CH4, several strategies have been developed. Dehydrogenative conversion of CH4 (DCM) is a promising approach for the coupling and aromatization of CH4. Mo/HZSM-5 is a well-known DCM catalyst that enables the aromatization of CH4, and related Mo-based catalysts have been vigorously investigated.4–10 Except for Mo/HZSM5, various catalysts have also been developed for DCM (e.g., single iron sites embedded in a silica matrix,11 Fe/HZSM-5,12 liquid indium metal13 and Ni–P alloys14).
Recently, several Pt-based catalysts have been reported to be effective for DCM. For example, PtSn/HZSM-5 is effective for the coupling and aromatization of CH4, where ethylene is formed on highly dispersed PtSn nanoparticles and then converted to aromatics on Brønsted acid sites of HZSM-5.15 The Pt/CeO2 catalyst was effective for CH4 coupling to form C2 hydrocarbons, where the active site is assumed as a single-atom Pt16 and Pt–Ce interface.17,18 Furthermore, Pt–Bi alloy catalysts enhanced CH4 coupling to C2 hydrocarbons,19,20 and recently, atomically thin Pt nanolayer on two-dimensional metal carbide was reported as an effective CH4 coupling catalyst.21
DCM requires cleavage of the strong C–H bonds in the CH4 molecule. However, continuous cleavage of the C–H bond of the CH4 molecule frequently incurs coke formation, which covers the catalyst surface and leads to deactivation. As mentioned above, Pt alloys and single-atom Pt are effective Pt-based catalysts for DCM. In these reaction systems, the deactivation of Pt catalysts is suppressed by precisely controlling the structure of the Pt catalysts; for example, the formation of alloy structure and single-atom sites. In other words, a simple supported Pt catalyst would excessively cleave the C–H bonds of the CH4 molecule and be deactivated by coke formation. In this study, we demonstrated that a simple Pt catalyst (Pt/Al2O3) promotes DCM by co-feeding of hydrogen. In the dehydrogenation of lower alkanes, such as ethane and propane, it is known that hydrogen co-feeding inhibits catalyst deactivation due to coke deposition.22–25 For CH4 conversion, the effect of hydrogen co-feeding on aromatization using Mo/HZSM-5 has been investigated.26–29 Furthermore, Kim et al. reported that hydrogen co-feeding improves C2 selectivity for the radical-based conversion of CH4 at high temperatures (1080 °C) using SiO2-based Fe catalysts.30 However, for dehydrogenative CH4 coupling at low temperatures, the positive effect of hydrogen co-feeding has not been reported. In this study, we revealed that hydrogen co-feeding is effective for CH4 coupling and inhibits the deactivation of Pt/Al2O3 catalysts, thereby facilitating the stable formation of C2 hydrocarbons. Furthermore, the catalytic activity of Pt/Al2O3 is influenced by the Pt loading. We discuss the catalytically active Pt sites for DCM based on various characterisations.
Experimental section
Preparation of the Pt/Al2O3 catalysts
Pt/Al2O3 catalysts were prepared using an impregnation method. H2PtCl6·6H2O (Kanto Chemical Co., Inc.) and Al2O3 (AKP-G07; Sumitomo Chemical Co., Ltd.) were added to deionised water (approximately 40 mL). The water was evaporated on a hot plate under stirring. The sample was dried at 100 °C overnight and calcined in air at 500 °C for 2 h. The Pt loadings were 1, 3, 5, and 10 wt%, which were denoted as Pt(1)/Al2O3, Pt(3)/Al2O3, Pt(5)/Al2O3, and Pt(10)/Al2O3, respectively.Catalytic performance
DCM reactions were performed in a fixed-bed reactor. Pt/Al2O3 (0.10 g) and quartz wool (50 mg) were placed in a quartz reactor. Before the DCM reaction, Ar (40 mL min−1) flowed into the reactor, and then the catalyst bed was heated to reaction temperatures (600, 700, or 800 °C). The catalyst was reduced with H2 (10 mL min−1) at 600 °C for 1 h. After purging the reactor with Ar, CH4 or a CH4/H2 mixture was introduced. The flow rate of CH4 and H2 was 20 and 1–10 mL min−1, respectively. The outlet gas was analysed using gas chromatography. For H2 and CH4, a gas chromatograph (Shimadzu GC-8A, TCD) equipped with an active carbon column (Ar carrier gas) was used. For the hydrocarbons, a gas chromatograph (Shimadzu GC-2014, FID) equipped with an SH-Rt-Q-BOND column was used (N2 carrier gas). The calculations for CH4 conversion, product formation rate, and product selectivity can be found in the ESI.†Characterization
Powder X-ray diffraction (XRD) patterns of the catalysts were recorded with a D2 phaser (Bruker) using Cu Kα radiation. Thermogravimetric (TG) analysis was performed using a DTG-60 instrument (Shimadzu). The spent catalyst was placed in a Pt cell and heated to 800 °C at 10 °C min−1 under flowing air (100 mL min−1). The amount of coke was calculated by the weight loss between 300 °C and 600 °C, which was attributed to coke combustion (C + O2 → CO2). X-ray photoelectron spectroscopy (XPS) was performed using an AXIS-NOVA (Shimadzu) with a monochromatic Al Kα source at 15 kV and 20 mA. The C 1s binding energy (285.0 eV) was used as a reference for charge correction. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and energy-dispersive X-ray (EDX) mappings were obtained using an aberration-corrected transmission electron microscope (JEM-ARM 200CF, JEOL) operated at 200 kV. X-ray absorption fine structure spectroscopy (XAFS) measurements of the Pt L3-edge were performed on the BL5S1 beamline at the Aichi Synchrotron Radiation Center (Aichi, Japan) using a Si(111) monochromator in the transmission mode. CO chemisorption analysis was performed using a BP-1 (Hemmi Slide Rule Co., Ltd). The Pt/Al2O3 was pre-treated with H2 at 600 °C for 1 h. The catalyst was placed in a glass cell, and H2 flowed at 400 °C for 1 h. After cooling to room temperature under flowing He, several CO pulses were introduced until the CO adsorption was saturated. The amount of CO chemisorbed on Pt, and the dispersion of Pt were calculated by assuming that a CO molecule was adsorbed on a Pt site. Using the number of Pt sites, the turnover number (TON) and turnover frequency (TOF) were calculated based on the yield of the hydrocarbon products. Fourier transform infrared (FT-IR) spectra were recorded with an FT/IR 4100 (JASCO) with a resolution of 2 cm−1. Pt/Al2O3 catalyst powder (10 mg) was pressed into a disk (diameter: 10 mm). The disk was placed in an IR cell and reduced under H2 flow (50 mL min−1) at 300 °C for 1 h. After evacuating the cell at 300 °C, the background spectrum was measured at room temperature. CO (25 mm Hg) was introduced into the cells and maintained for 20 min. After evacuating the cell to room temperature, the spectrum of the chemisorbed CO was measured at room temperature. Using the background spectrum, FT-IR difference spectra of adsorbed CO species on Pt/Al2O3 were obtained.Results and discussion
Effect of H2 co-feeding on DCM reaction
In this study, the main products of the DCM reactions were C2 hydrocarbons (eqn (1) and (2)), benzene (eqn (3)), and coke (eqn (4)).| 2CH4 → C2H6 + H2 | (1) |
| 2CH4 → C2H4 + 2H2 | (2) |
| 6CH4 → C6H6 + 9H2 | (3) |
| CH4 → C + 2H2 | (4) |
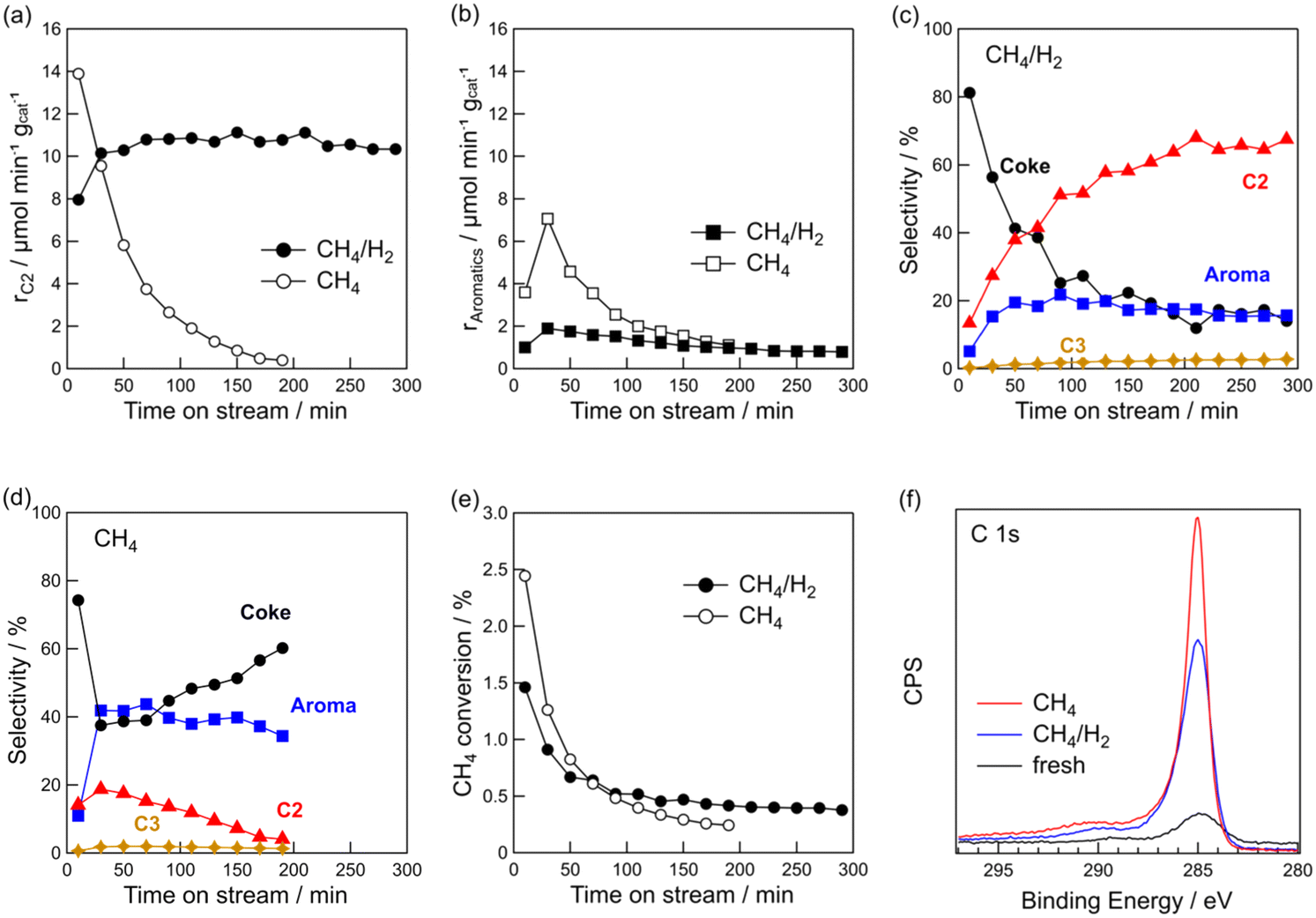 | ||
| Fig. 1 Time course of formation rate of (a) C2 hydrocarbons and (b) aromatics, (c and d) product selectivity, and (e) CH4 conversion for DCM reaction. (f) C 1s XPS of Pt(1)/Al2O3 before and after DCM. Catalyst: Pt(1)/Al2O3, T: 600 °C, flow rate: 20 (CH4) and 20 + 1 (CH4 + H2) mL min−1, and catalyst mass: 0.10 g. | ||
Fig. 1e shows the time course of CH4 conversion. Regardless of the presence of hydrogen, the conversion decreased during the early stages of the reaction. For CH4/H2, the CH4 conversion stabilised at approximately 0.4% after 2 h. It should be noted that CH4 conversion decreased for the DCM of CH4/H2, but C2 hydrocarbons were stably formed during the reaction, thereby indicating that the Pt sites effective for CH4 coupling were not deactivated. This point will be discussed later.
The rapid decrease in the CH4 conversion during the initial stage of the reaction was due to coke deposition on the catalyst. Coke deposition on the catalyst was confirmed by TG and XPS analyses. The TG profiles of the spent catalysts are shown in Fig. S1 in the ESI.† The amounts of coke were 8.2 wt% for pure CH4 and 6.0 wt% for CH4/H2, indicating that coke formation was suppressed by hydrogen co-feeding. C 1s XPS showed that the intensity of the C 1s peak of the spent catalysts was higher than that of the fresh catalyst (Fig. 1f). The peak of C 1s with CH4/H2 was lower than with CH4, indicating that the addition of hydrogen suppressed coke deposition. From the results of the TG and XPS analyses, it was found that the coexistence of hydrogen with CH4 suppressed coke deposition while facilitating the stable formation of C2 hydrocarbons.
CH4 (20 mL min−1)/H2 (1 mL min−1) mixture was used. Next, we studied the effect of the volume of co-fed hydrogen and found that increasing the volume of hydrogen to 3 mL min−1 led to a decreased CH4 conversion (Fig. S2†). Additionally, rC2 decreased by approximately 1/3 when the volume of co-fed hydrogen was increased from 1 to 3 mL min−1. Because CH4 coupling is a dehydrogenation reaction, it is likely that excess hydrogen suppresses the dehydrogenation reaction. We also tested the effect of the reaction temperature (Fig. S3†). When the reaction temperature increased from 600 to 700 °C, more co-fed hydrogen was required to obtain the same rC2 at 600 °C. At 800 °C, the catalyst was deactivated quickly even when hydrogen co-existed. At higher temperatures, more hydrogen co-feeding was required because coke deposition was more likely to occur thermodynamically. Based on the results, we concluded that a volume fraction of CH4/H2 = 20/1 and reaction temperature = 600 °C are suitable for the stable formation of C2 hydrocarbons via DCM on Pt(1)/Al2O3.
Effect of Pt particle size
To investigate the effect of Pt particle size, Pt(1, 3, 5, and 10)/Al2O3 were prepared. Table 1 shows the results of CO chemisorption analyses of the catalysts. The dispersion of Pt decreased, and the average particle size became larger with increasing Pt loading. The particle sizes of Pt(1, 3, 5, and 10)/Al2O3 were 3.4, 5.3, 11, and 14 nm, respectively. In the XRD patterns of Pt/Al2O3 (Fig. S4†), diffraction peaks of the θ-Al2O3 support (PDF 00-035-0121) are observed for all catalysts. Peaks due to Pt metal (PDF 00-004-0802) increased in intensity with increasing Pt loading. In addition, peaks corresponding to Pt(111) and Pt(200) were observed for Pt(5 and 10)/Al2O3. The absence of peaks due to Pt in the Pt(1)/Al2O3 catalyst suggests that Pt was highly dispersed on the catalyst.| Pt loading/wt% | Amount of chemisorbed CO/μmol gcat−1 | Dispersiona/% | Average particle size (CO)b/nm | Average particle size (STEM)c/nm |
|---|---|---|---|---|
| a Dispersion was estimated by CO chemisorption analysis. b Average particle size was calculated by CO chemisorption analysis. c Average particle size was calculated by STEM analysis. | ||||
| 1 | 23 | 44 | 3.4 | 2.6 ± 0.8 |
| 3 | 43 | 28 | 5.3 | 2.5 ± 1.7 |
| 5 | 33 | 13 | 11 | 3.1 ± 4.3 |
| 10 | 53 | 10 | 14 | 6.6 ± 1.4 |
Fig. 2a shows the HAADF-STEM image and particle size distribution of the fresh Pt(1)/Al2O3 catalysts. Pt(1)/Al2O3 exhibited dispersed Pt nanoparticles of approximately 2 nm in size. Based on the size distribution, the mean particle size was 2.6 nm. With increasing Pt loading, larger particles ranging from 10 to 20 nm were observed, in addition to small 2 nm nanoparticles (Fig. S5†). Therefore, as the Pt loading increased, the proportion of coarse nanoparticles increased as well. The particle sizes evaluated from STEM images were 2.6 ± 0.8, 2.5 ± 1.7, 3.1 ± 4.3, and 6.6 ± 1.4 nm, respectively (Table 1). The HAADF-STEM images were consistent with the XRD patterns and CO chemisorption measurements. Fig. 2b shows a HAADF-STEM image of the spent catalyst. There, the Pt particles appeared to be finer than those in the fresh catalyst. It is not entirely clear why the particle size changed after contact with CH4; however, it is possible that structural changes, such as the formation of quasi-stable carbides, caused a change in the Pt particle size. Fig. 2c–h show the EDX mapping and the corresponding HAADF-STEM image of the spent Pt(1)/Al2O3. The signal of Pt overlapped with that of the Al2O3 support (i.e., the signals of Al and O), indicating that Pt was supported on Al2O3. In contrast, the signal of C did not completely overlap in the Al region. The presence of carbon was confirmed even in areas where Al was not present. EDX mapping analysis suggested that the coke formed on the Pt particles moved toward the Al2O3 support and grew away from the Al2O3 support.
 | ||
| Fig. 2 HAADF-STEM images and particle size distribution of (a) fresh and (b) spent Pt(1)/Al2O3. EDX mapping images for (c) Al, (d) O, (e) C, (f) Pt, (g) overlay of (c–f), and (h) HAADF-STEM image of the spent catalyst. Spent catalyst: DCM was performed on Pt(1)/Al2O3 for 5 h under CH4/H2 (20/1). | ||
The Fourier transforms of the k3-weighted extended X-ray absorption fine structure (EXAFS) oscillation at the Pt L3-edge for the fresh Pt/Al2O3 catalysts, and the curve-fitting results are shown in Fig. S6 and Table S1.† As the Pt loading increased, the peak intensity of the Pt–Pt bond increased. The presence of Pt–O bonds is considered to be due to the oxidation of the Pt surface by air exposure based on ex situ XAFS measurements.31,32 For Pt(1)/Al2O3, the Pt–Pt coordination number was 4.2, while it was 10.2 for Pt(10)/Al2O3. The increase in the Pt–Pt coordination number reflects an increase in the Pt particle size with increasing Pt loading. This result is consistent with the HAADF-STEM and CO chemisorption measurements.
The above characterizations show that Pt particle size can be controlled by Pt loading. Thus, the effect of Pt particle size on the DCM reaction was investigated. Fig. 3a–d show the time course of rC2, raromatics, formation rate of coke (rcoke), and CH4 conversion for DCM on the Pt(1, 3, 5, and 10)/Al2O3 catalysts. Fig. 3a shows that C2 hydrocarbons were formed on all the catalysts, and rC2 increased with decreasing Pt loading; i.e., decreasing Pt particle size. At 290 min, rC2 for Pt(1)/Al2O3 and Pt(10)/Al2O3 were 10 and 8 μmol min−1 gcat−1, respectively. In contrast, rcoke increased with increasing Pt loading (Fig. 3c). At 290 min, rcoke for Pt(1)/Al2O3 and Pt(10)/Al2O3 were 4 and 100 μmol min−1 gcat−1, respectively. In other words, rcoke of Pt(10)/Al2O3 was 25 times higher than that of Pt(1)/Al2O3, thereby indicating that Pt(10)/Al2O3 produced almost exclusively coke during DCM. The result suggested that large Pt particles tend to form coke dominantly. Also, CH4 conversion increased with increasing Pt loading (Fig. 3d); however, the increase in CH4 conversion was thought to be due to the enhancement of coke formation. The raromatics were lower than rC2 and rcoke, and were hardly affected by the Pt loading (Fig. 3b).
Fig. 3e shows the product selectivity. The product distribution was influenced by the Pt loading. Obviously, the selectivity for hydrocarbons (C2, C3, and aromatics) was higher for Pt/Al2O3 with lower Pt loading. It must be emphasised that hydrocarbons are more likely to be formed on low Pt loading catalysts, whereas coke is more likely to be formed on catalysts with high Pt loadings. The time course of product selectivity is shown in Fig. S7† and supports the above findings.
The increase in coke formation with increasing Pt loading was confirmed using TG analysis. The TG profiles of the spent catalysts (Fig. 3f) showed that the amount of coke deposited on Pt(1)/Al2O3 was 6.0 wt%, whereas that on Pt(10)/Al2O3 was 43 wt%.
As shown in Table 1, the amount of adsorbed CO on Pt(10)/Al2O3 (53 μmol gcat−1) was higher than that on Pt(1)/Al2O3 (23 μmol gcat−1). Hence, it is assumed that the number of Pt atoms affects the product distribution, that is, excess dehydrogenation of CH4 (i.e. coke formation) might occur when a larger amount of exposed Pt is present. Therefore, the weight of Pt(10)/Al2O3 was reduced to the same value as the number of Pt sites exposed on Pt(1)/Al2O3, and provided for the DCM reaction (Fig. S8†). However, Pt(10)/Al2O3 predominantly formed coke (the selectivity for coke was 77%), indicating that coke formation was not attributed to the number of exposed Pt sites.
XPS analysis
Fig. 4 shows the Pt 4f and Al 2p XPS profiles of the fresh and spent Pt(1, 3, 5, and 10)/Al2O3 catalysts. For the fresh catalysts (Fig. 4a), Pt0 (71.1 and 74.5 eV) and Al3+ peaks (74.7 eV) were observed.33 The intensity of the Pt0 peak increased with increasing Pt loading, indicating that the number of exposed Pt sites increased at large Pt particles. Fig. 4b shows XPS profiles of the spent catalysts. The contact of CH4/H2 with the catalysts changed their XPS profiles, and a significant reduction in the intensity of the Pt0 and Al3+ peaks for the spent catalysts was observed. This suggests that the surface of Pt/Al2O3 was covered with coke. Indeed, the results from C 1s XPS show that the carbon peak increased for the spent catalysts compared to the fresh catalysts (Fig. S9†). Also, the carbon peak intensified upon increasing Pt loading. This result is consistent with the DCM reaction results shown in Fig. 3, which indicates that an increase in Pt loading leads to an increased coke formation.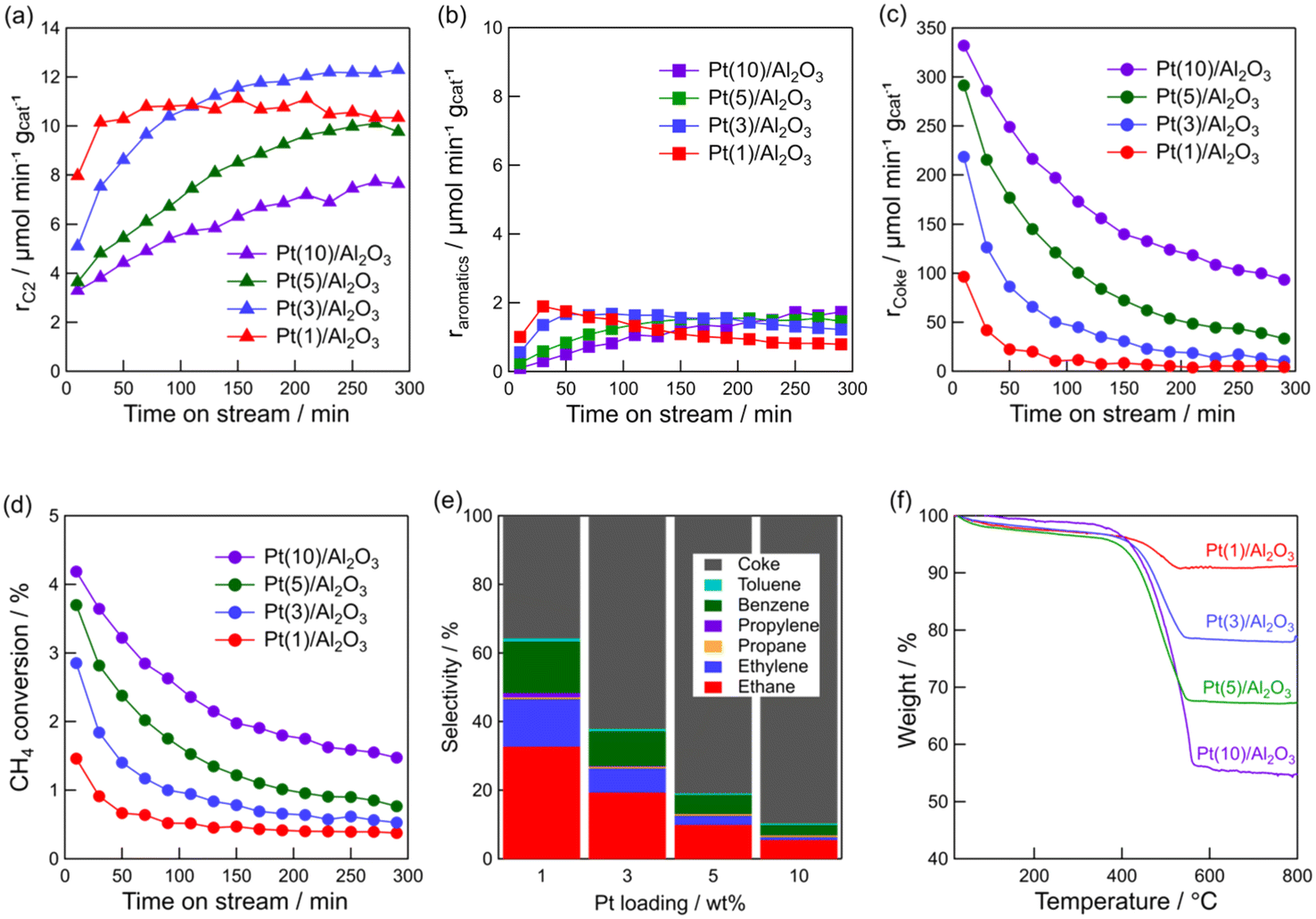 | ||
| Fig. 3 Time course of formation rate of (a) C2 hydrocarbons, (b) aromatics, (c) coke, and (d) CH4 conversion for DCM reaction. (e) Product selectivity for DCM reaction. (f) TG profiles of the spent catalysts. Catalyst: Pt(1, 3, 5, and 10)/Al2O3, T: 600 °C, flow rate: 20 + 1 (CH4 + H2) mL min−1, and catalyst mass: 0.10 g. | ||
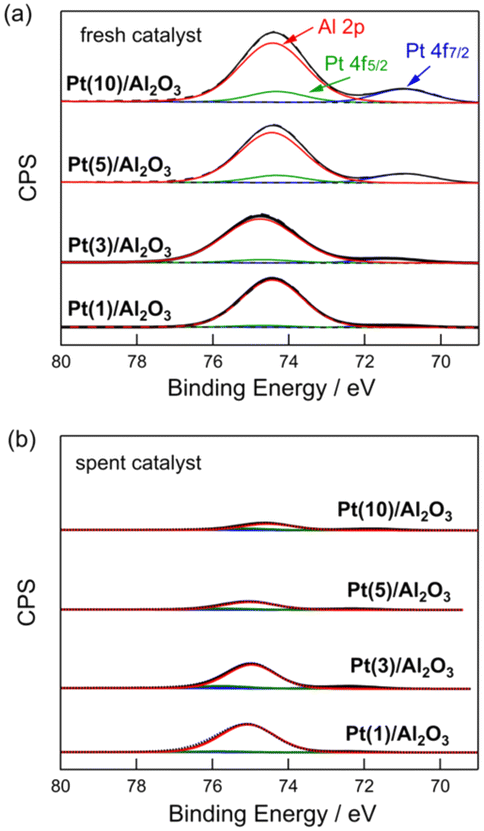 | ||
| Fig. 4 Pt 4f and Al 2p XPS of (a) fresh and (b) spent Pt(1, 3, 5, and 10)/Al2O3 catalysts. DCM reaction conditions: T = 600 °C, flow rate = 20 + 1 (CH4 + H2) mL min−1, and catalyst mass = 0.10 g. | ||
The coverage of Pt/Al2O3 with coke was evaluated by the peak reduction ratios of Pt 4f7/2 and Al 2p between the fresh and spent catalysts. When the surfaces of Pt/Al2O3 are covered by coke formed from CH4, the peaks of Pt 4f7/2 and Al 2p XPS must be reduced. Thus, the peak reduction ratios between the fresh and spent catalysts represent the degree of coke coverage on Pt/Al2O3. Table 2 lists the results of the study. For the Pt 4f7/2 XPS, the reduction in peak intensity was 54–55% for Pt(1 and 3)/Al2O3, and for Pt(5 and 10)/Al2O3, it was 85–87%. This suggests that the coverage of the catalyst surface with coke increased with increasing Pt size. A similar trend was observed for Al 2p XPS, where the reduction in peak intensity was greater for the catalysts with higher Pt loadings. The change in the XPS peak after DCM indicated that coke formation was more favourable at higher Pt loadings (i.e., larger Pt particles), and the coke covered the surface of the catalysts.
| Pt loading/wt% | Peak reduction ratio/% | |
|---|---|---|
| Pt 4f7/2 | Al 2p | |
| 1 | 54 | 50 |
| 3 | 55 | 63 |
| 5 | 85 | 87 |
| 10 | 87 | 92 |
It should be noted that the decrease in peak intensity of Al 2p suggests that coke was deposited on the Al2O3 support, as well as on the Pt surface. Since the Al2O3 support has no catalytic activity for dissociating the C–H bond within the CH4 molecule, the coke deposition on Al2O3 could potentially be due to the migration (spillover) of coke precursors generated on the Pt site towards the Al2O3 support. As shown in Fig. 2, the EDX mapping analysis of the spent Pt(1)/Al2O3 catalyst also supported the spillover of coke.
FT-IR spectra
CO is a typical probe-molecule to characterize the surface structure of Pt metal.34Fig. 5a shows the FT-IR spectra of CO adsorbed on fresh Pt(1, 3, 5, and 10)/Al2O3. The peaks of the CO band for Pt(1, 3, 5, and 10)/Al2O3 were 2065, 2065, 2071, and 2089 cm−1, respectively. With increasing Pt loading, the CO band shifted from 2065 to 2089 cm−1. A similar trend was previously reported for the Pt catalysts with different particle sizes (19, 10, 3.0, and 1.4 nm).34 Generally, it is known that CO adsorbates show different vibrational frequency on Pt metal: 2098–2080 (well-coordinated Pt0 site like terraces), 2075–2060 (under-coordinated Pt0 sites like steps and edges), and 2055–2000 cm−1 (highly under-coordinated Pt0 sites like corners).34–36 Thus, in Fig. 5a, a sharp peak for Pt(10)/Al2O3 at 2089 cm−1 can be assigned to the well-coordinated Pt0 site. The shift of CO band to lower wavenumbers for Pt(1, 3, 5, and 10)/Al2O3 suggested that the fraction of steps and edges varied with the Pt loading (i.e., Pt particle sizes). A similar relationship between surface structure and particle size is previously reported; the increase in nanoparticle size results in an increase in the proportion of well-coordinated sites, and the decrease in the proportion of under-coordinated sites decreases.37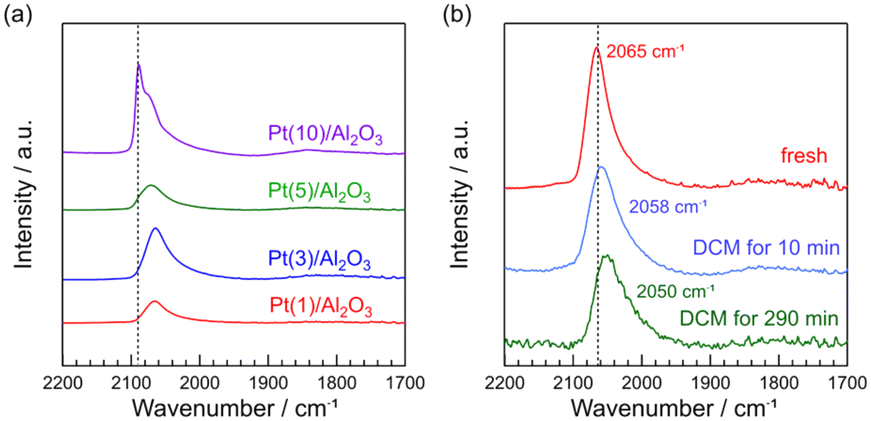 | ||
| Fig. 5 FT-IR spectra of chemisorbed CO on Pt/Al2O3 catalysts. (a) fresh Pt(1, 3, 5, and 10)/Al2O3 and (b) fresh and spent Pt(1)/Al2O3. DCM reaction conditions: T = 600 °C, flow rate = 20 + 1 (CH4 + H2) mL min−1, catalyst mass = 0.10 g. | ||
The FT-IR spectra of CO adsorbed on Pt(1)/Al2O3 before and after DCM are shown in Fig. 5b. We can see that the CO band was shifted from 2065 to 2050 cm−1 during the DCM reaction. Previously, the CO adsorption on under-coordinated Pt0 sites was precisely reported; CO adsorption on 6- and 7-fold Pt0 sites and <6-fold Pt0 sites are attributed to 2075–2060 and 2055–2000 cm−1, respectively.34 Thus, the shift of CO band from 2065 to 2050 cm−1 with DCM reaction indicated that the portion of <6-fold Pt0 sites increased by the contact with CH4. Probably, 6- and 7-fold Pt0 sites are likely to be covered with coke. Consequently, CO was not adsorbed on the 6- and 7-fold Pt0 sites, resulting in the shift of CO band upon contact with CH4.
FT-IR spectra show that Pt(10)/Al2O3 has a large amount of well-coordinated Pt sites. As shown in the reaction results and STEM images, larger Pt particles are more likely to form coke and readily deactivated by coke coverage. Thus, it can be concluded that well-coordinated Pt sites are ineffective for the coupling of CH4, whereas coke is dominantly formed. FT-IR spectra and the DCM reaction results suggested that among the under-coordinated Pt0 sites, <6-fold Pt0 sites like corners are more suitable for CH4 coupling, where the deactivation by the coke formation is unlikely to occur, facilitating CH4 coupling.
Stability test
A stability test was performed using the Pt(1)/Al2O3 catalyst at 600 °C in CH4/H2. The results are shown in Fig. 6. For 24 h, the CH4 conversion and rC2 were stable at about 0.3% and >8 μmol min−1 gcat−1, respectively. The hydrocarbon selectivity remained at approximately 80% over 24 h, whereas the coke selectivity was stable at approximately 20%. These results support the long-term stability of the DCM catalytic system. The TON and TOF of C2 hydrocarbons were 862 and 0.01 s−1, respectively. Compared to previously reported Pt-based DCM catalysts, such as Pt/CeO2 (C2 TOF = 0.006 s−1), and Pt–Bi/ZSM-5 (C2 TOF = 0.05 s−1),16,20 we conclude that the catalytic performance of our system is comparable to previous works. Furthermore, the CH4 conversion to products of this work was comparable to previously reported Pt-based catalysts (PtSn/HZSM-5).15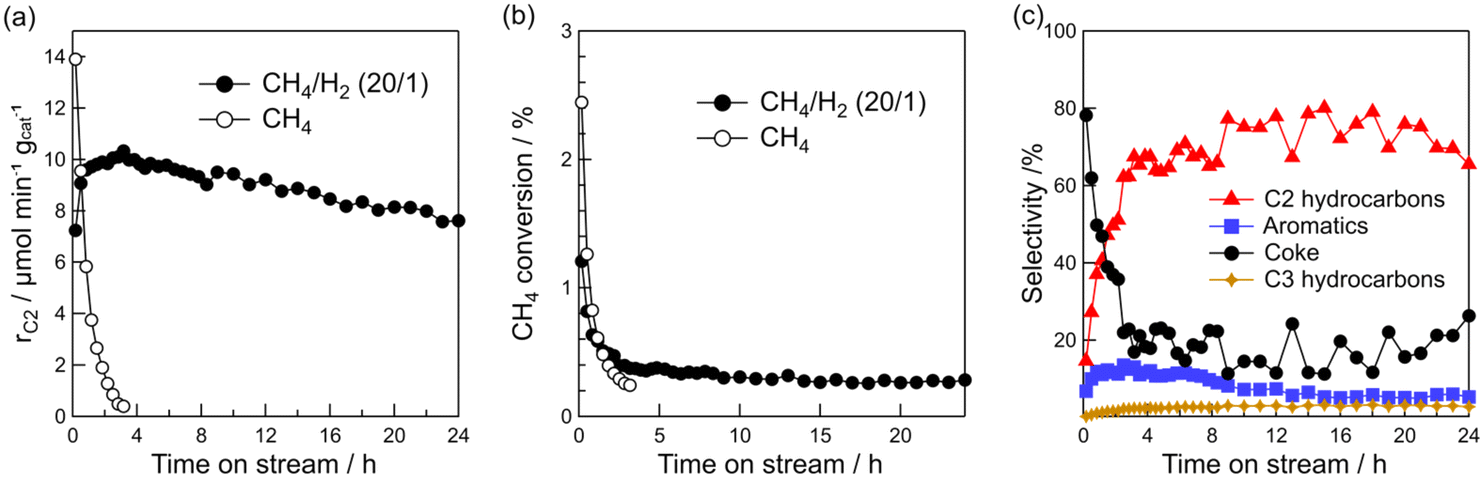 | ||
| Fig. 6 Time course of (a) formation rate of C2 hydrocarbons, (b) methane conversion, and (c) product selectivity for DCM reaction. Catalyst: Pt(1)/Al2O3, T: 600 °C, flow rate: 20 (CH4) and 20 + 1 (CH4 + H2) mL min−1, and catalyst mass: 0.10 g. | ||
Discussion
The findings revealed in this study are as follows:• A simple Pt/Al2O3 catalyst was active for CH4 coupling in the presence of hydrogen.
• Hydrogen co-feeding suppresses catalyst deactivation, facilitating the stable formation of C2 hydrocarbons.
• Pt loading had a significant effect on CH4 coupling in the presence of hydrogen.
• The coordination mode of Pt sites determines CH4 coupling or coke formation.
Previous studies of DCM reactions using Pt-based catalysts have provided insights into the surface structure of Pt. Xie et al. reported that Pt single atoms in Pt/CeO2 catalysts are highly active in the DCM reaction, and Pt nanoparticles with sizes of 3 nm, can convert CH4 to coke instead of C2.16 The result is consistence with the present study; that is, Pt(1)/Al2O3 involving approximately 3 nm Pt particles deactivated rapidly (Fig. 1a). The new insight of this work is that co-feeding H2 suppressed the deactivation of Pt(1)/Al2O3 due to coke deposition.
The formation of coke from CH4 on the Pt surface was previously reported at low temperatures (approximately 300 °C),38 indicating that the Pt surface has the ability to decompose CH4 molecules. In order to produce C2 hydrocarbons, CH4 molecules should not be completely decomposed to coke, whereas adsorbed CH3 intermediates must exist stably and couple to C2 hydrocarbons. Therefore, the stability of intermediates on the Pt surface is crucial for the DCM reaction. Density functional theory calculations performed by Viñes et al. showed that CH3 and CH2 intermediates formed from CH4 were stabilised at sites located at the edges and corners.39 A similar finding was reported by Gerceker et al. Ethylene TOF values were predicted by the microkinetic model, while the step site showed a higher TOF than the terrace site.15 The previous works support our findings. From the reaction results, particle size analysis, XPS, and FT-IR, we proposed that small Pt particles with highly under-coordinated Pt0 sites like corners can produce C2 hydrocarbons stably without deactivation due to coke coverage. As the previous works indicated, the highly under-coordinated Pt0 sites would stably form CH3 and CH2 intermediates, facilitating the production of C2 hydrocarbons. We believe that the theoretically-predicted mechanism can be demonstrated experimentally in the present study. Our study suggests that the stability of intermediates is enhanced under H2 co-feeding. Thus, it can be concluded that both the presence of highly under-coordinated Pt0 sites and H2 co-feeding facilitated the stable formation of CH3 and CH2 intermediates to produce C2 hydrocarbons.
Conclusions
The effect of hydrogen co-feeding on DCM using Pt/Al2O3 was investigated. The addition of hydrogen significantly changed the DCM behaviour. When only CH4 was used, Pt/Al2O3 was deactivated rapidly. In contrast, in the DCM, using a CH4/H2 mixture provided stable C2 hydrocarbons. For 24 h, a rC2 > 8 μmol min−1 gcat−1 and hydrocarbon selectivity ≈ 80% was achieved. TG and XPS analyses indicated that hydrogen co-feeding suppressed the coke formation on Pt/Al2O3. The Pt loading of Pt/Al2O3 affected the DCM behaviour. Pt/Al2O3 with low Pt loading tended to form hydrocarbons predominantly, whereas catalysts with high Pt loading mainly produced coke. HAADF-STEM, CO chemisorption, XAFS, and XRD measurements suggested that the low-Pt-loading catalysts contain Pt nanoparticles of approximately 2 nm in size. As the Pt loading increased, coarse Pt particles of several tens of nanometres in size became conspicuous. The FT-IR spectra suggested that the well-coordinated Pt sites were covered with coke during the DCM and deactivated. In contrast, highly under-coordinated Pt sites, like corners, are less likely to form coke and are more effective for CH4 coupling. Thus, low-Pt-loading catalysts produce C2 hydrocarbons in a stable manner.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JST CREST (grant number: JPMJCR15P4 and JPMJCR17P3). We appreciate the technical support provided by the Comprehensive Analysis Center for Science at Saitama University for XRD and XPS analyses. HAADF-STEM and EDX analyses were performed at the Ultramicroscopy Research Center of Kyushu University, supported by the Advanced Research Infrastructure for Materials and Nanotechnology (ARIM, grant number: JPMXP1222KU0043) of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan. A part of this work was supported by the NIMS microstructural characterization platform as a program of “Nanotechnology Platform” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.Notes and references
- Y. F. Gao, L. Neal, D. Ding, W. Wu, C. Baroi, A. M. Gaffney and F. X. Li, ACS Catal., 2019, 9, 8592–8621 CrossRef CAS.
- A. I. Olivos-Suarez, A. Szecsenyi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- E. E. Wolf, J. Phys. Chem. Lett., 2014, 5, 986–988 CrossRef CAS PubMed.
- R. W. Borry, Y. H. Kim, A. Huffsmith, J. A. Reimer and E. Iglesia, J. Phys. Chem. B, 1999, 103, 5787–5796 CrossRef CAS.
- W. P. Ding, S. Z. Li, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2001, 105, 506–513 CrossRef CAS.
- Z. R. Ismagilov, E. V. Matus and L. T. Tsikoza, Energy Environ. Sci., 2008, 1, 526–541 RSC.
- I. Lezcano-Gonzalez, R. Oord, M. Rovezzi, P. Glatzel, S. W. Botchway, B. M. Weckhuysen and A. M. Beale, Angew. Chem., Int. Ed., 2016, 55, 5215–5219 CrossRef CAS PubMed.
- F. Solymosi, J. Cserenyi, A. Szoke, T. Bansagi and A. Oszko, J. Catal., 1997, 165, 150–161 CrossRef CAS.
- D. J. Wang, J. H. Lunsford and M. P. Rosynek, J. Catal., 1997, 169, 347–358 CrossRef CAS.
- L. S. Wang, L. X. Tao, M. S. Xie, G. F. Xu, J. S. Huang and Y. D. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- X. G. Guo, G. Z. Fang, G. Li, H. Ma, H. J. Fan, L. Yu, C. Ma, X. Wu, D. H. Deng, M. M. Wei, D. L. Tan, R. Si, S. Zhang, J. Q. Li, L. T. Sun, Z. C. Tang, X. L. Pan and X. H. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- B. M. Weckhuysen, D. J. Wang, M. P. Rosynek and J. H. Lunsford, Angew. Chem., Int. Ed. Engl., 1997, 36, 2374–2376 CrossRef CAS.
- Y. Nishikawa, H. Ogihara and I. Yamanaka, ChemistrySelect, 2017, 2, 4572–4576 CrossRef CAS.
- A. L. Dipu, S. Ohbuchi, Y. Nishikawa, S. Iguchi, H. Ogihara and I. Yamanaka, ACS Catal., 2020, 10, 375–379 CrossRef CAS.
- D. Gerceker, A. H. Motagamwala, K. R. Rivera-Dones, J. B. Miller, G. W. Huber, M. Mavrikakis and J. A. Dumesic, ACS Catal., 2017, 7, 2088–2100 CrossRef CAS.
- P. F. Xie, T. C. Pu, A. M. Nie, S. Hwang, S. C. Purdy, W. J. Yu, D. Su, J. T. Miller and C. Wang, ACS Catal., 2018, 8, 4044–4048 CrossRef CAS.
- D. Bajec, A. Kostyniuk, A. Pohar and B. Likozar, Chem. Eng. J., 2020, 396, 125182 CrossRef CAS.
- D. Eggart, X. Huang, A. Zimina, J. Z. Yang, Y. Pan, X. L. Pan and J. D. Grunwaldt, ACS Catal., 2022, 12, 3897–3908 CrossRef CAS.
- J. Z. Chen, Z. W. Wu, X. B. Zhang, S. Choi, Y. Xiao, A. Varma, W. Liu, G. H. Zhang and J. T. Miller, Catal. Sci. Technol., 2019, 9, 1349–1356 RSC.
- Y. Xiao and A. Varma, ACS Catal., 2018, 8, 2735–2740 CrossRef CAS.
- Z. Li, Y. Xiao, P. R. Chowdhury, Z. W. Wu, T. Ma, J. Z. Chen, G. Wan, T. H. Kim, D. P. Jing, P. L. He, P. J. Potdar, L. Zhou, Z. H. Zeng, X. L. Ruan, J. T. Miller, J. P. Greeley, Y. Wu and A. Varma, Nat. Catal., 2021, 4, 882–891 CrossRef CAS.
- V. Galvita, G. Siddiqi, P. P. Sun and A. T. Bell, J. Catal., 2010, 271, 209–219 CrossRef CAS.
- S. Saerens, M. K. Sabbe, V. V. Galvita, E. A. Redekop, M. F. Reyniers and G. B. Marin, ACS Catal., 2017, 7, 7495–7508 CrossRef CAS.
- D. Sanfilippo and I. Miracca, Catal. Today, 2006, 111, 133–139 CrossRef CAS.
- J. J. H. B. Sattler, A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2013, 15, 12095–12103 RSC.
- Z. Liu, M. A. Nutt and E. Iglesia, Catal. Lett., 2002, 81, 271–279 CrossRef CAS.
- H. T. Ma, R. Ohnishi and M. Ichikawa, Catal. Lett., 2003, 89, 143–146 CrossRef CAS.
- T. Osawa, I. Nakano and O. Takayasu, Catal. Lett., 2003, 86, 57–62 CrossRef CAS.
- Y. Song, Y. B. Xu, Y. Suzuki, H. Nakagome, X. X. Ma and Z. G. Zhang, J. Catal., 2015, 330, 261–272 CrossRef CAS.
- S. J. Han, S. W. Lee, H. W. Kim, S. K. Kim and Y. T. Kim, ACS Catal., 2019, 9, 7984–7997 CrossRef CAS.
- Y. Lei, H. Y. Zhao, R. D. Rivas, S. Lee, B. Liu, J. L. Lu, E. Stach, R. E. Winans, K. W. Chapman, J. P. Greeley, J. T. Miller, P. J. Chupas and J. W. Elam, J. Am. Chem. Soc., 2014, 136, 9320–9326 CrossRef CAS PubMed.
- M. A. Ramallo-Lopez, F. G. Requejo, A. F. Craievich, J. Wei, M. Avalos-Borja and E. Iglesia, J. Mol. Catal. A: Chem., 2005, 228, 299–307 CrossRef.
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer Physical Electronic Division, Eden Prairie, MN, 1979 Search PubMed.
- M. J. Kale and P. Christopher, ACS Catal., 2016, 6, 5599–5609 CrossRef CAS.
- M. Siemer, G. Tomaschun, T. Klüner, P. Christopher and K. Al-Shamery, ACS Appl. Mater. Interfaces, 2020, 12, 27765–27776 CrossRef CAS PubMed.
- M. J. Lundwall, S. M. McClure and D. W. Goodman, J. Phys. Chem. C, 2010, 114, 7904–7912 CrossRef CAS.
- D. N. McCarthy, C. E. Strebel, T. P. Johansson, A. den Dunnen, A. Nierhoff, J. H. Nielsen and Ib Chorkendorff, J. Phys. Chem. C, 2012, 116, 15353–15360 CrossRef CAS.
- P. Ferreira-Aparicio, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Appl. Catal., A, 1997, 148, 343–356 CrossRef CAS.
- F. Viñes, Y. Lykhach, T. Staudt, M. P. A. Lorenz, C. Papp, H. P. Steinruck, J. Libuda, K. M. Neyman and A. Görling, Chem. – Eur. J., 2010, 16, 6530–6539 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00612c |
| This journal is © The Royal Society of Chemistry 2023 |