
Visible light-mediated C (sp3)–H bond functionalization inside an all-organic nanocavity†
Debojyoti
Roy
,
Sunandita
Paul
and
Jyotishman
Dasgupta
 *
*
Department of Chemical Sciences, Tata Institute of Fundamental Research, 1-Homi Bhabha Road, Mumbai-400005, India. E-mail: dasgupta@tifr.res.in
First published on 10th October 2023
Abstract
Ultrafast C–H bond activation and functionalization in confinement using visible light will enable engineering chemical reactions with extraordinary speed and selectivity. To provide a transition metal-free route, here we demonstrate C–H bond activation reactions on poly-aromatic hydrocarbons (PAH) in all-organic cationic nanocage ExBox4+ for the first time. Visible light excitation in the host–guest charge transfer (CT) state allows the formation of oxidized photoproducts with high selectivity. Mechanistic understanding of this CT-mediated photoreaction using femtosecond broadband transient absorption revealed a few ∼100 ps timescale for C–H bond breaking on the attached –CH3 group via sequential electron transfer and proton transfer steps. We envision that our photosensitizer-free method will open up new avenues to pursue organic reactions using cavities that could serve both as photoredox catalysts and hosts for reactive reaction intermediates.
Selective C–H bond activation has remained a grand synthetic challenge due to the low bond polarity and high bond dissociation energy of a typical sp3 C–H bond.1 Transition metal catalysts are often employed industrially to execute C–H activation and functionalization reactions although metal remediation on a large scale is an environmental problem.2 Natural enzymes on the other hand carry out these transformations in their active sites efficiently in water. Cytochrome P450 is a classic example of a class of enzymes that contain the heme co-factor to carry out C–H bond activation and functionalization under physiological conditions.3 Contrary to free solution reactions in such enzymes, the substrates are pre-organized inside hydrophobic cavities to render selectivity and specificity during reaction turnover. Supramolecular cavities that mimic active site confinement of enzymes have been used to carry out a large number of catalytic transformations. Recently Noto et al. used an organic photocatalyst encapsulated inside confinement to carry out pinacol coupling with blue light,4 whereas Mkrtchayn et al. showed that aromatic nitro compounds can be reduced to their corresponding amines in the presence of a UV light source.5 A fundamental challenge in the field is to carry out C–H bond activation without the use of visible light-absorbing photoredox catalysts6 or with UV light excitation.7 To address this problem, Das et al. have used emergent donor–acceptor charge transfer (CT) interactions inside a Pd6L412+ hybrid metal–organic nanocavity to do benzylic sp3 C–H activation.8 However, the presence of six Pd2+-metal centers makes this host cost ineffective for organic synthesis.
In order to completely report a metal-free photoactivation and functionalization protocol via a supramolecular host–guest CT paradigm, here we use an organic cavity ExBox4+ for C–H bond activation using host–guest CT interactions. Here we show for the first time that these all-organic cages can carry out C–H bond activation reactions on benzylic carbon centers of PAH's having different degrees of aromaticity with visible light illumination (Fig. 1). Using broadband transient absorption (TA) spectroscopy, we provide a mechanistic view of the entire photoactivation process which invokes the neutral radical intermediates. We followed the exact literature protocol to synthesize DB·2PF6 (Fig. S1, ESI†) in 65% yield. However, the challenging part was the cyclization step in the presence of a template (pyrene) to give ExBox.4PF6 which took 17 days to complete.9 To do it much more efficiently we tried the synthesis in a microwave chamber,10 and got the desired product within 2 hours at 80 °C (Fig. 2a). After the successful workup steps, we got crude material with 34% yield. The solid material was recrystallized and characterized by 1H NMR and ESI-MS (Fig. S2, ESI†).
 | ||
| Fig. 1 Conceptual representation of visible light-mediated C–H activation and functionalization inside an all organic nanocavity ExBox.4PF6. | ||
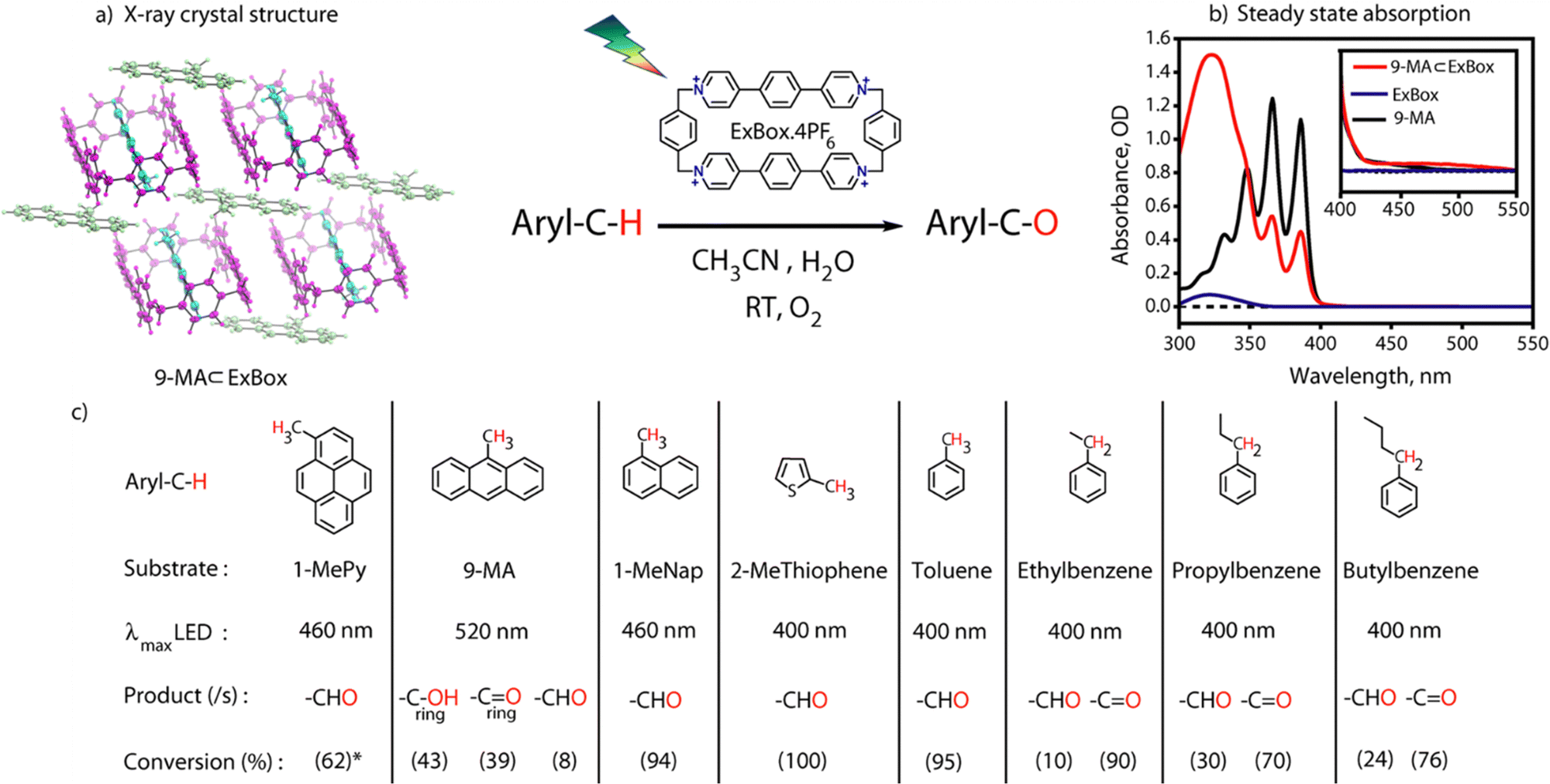 | ||
| Fig. 2 (a) Single crystal structure of 9-MA ⊂ ExBox4+; (b) steady state absorption spectra of 9-MA, ExBox4+and 9-MA ⊂ ExBox4+ showing formation of CT states; (c) visible light mediated photoreaction scheme for sp3 C–H bond functionalization, where 2.5 mM ExBox and 2–3 equiv. of guests are used, photoreaction time = 24 hours (72 hours for Toluene), LED light intensity: 50–175 mW cm−2, and product conversions are obtained from GCMS, * from 1H NMR. | ||
Incarceration of the guest was confirmed from 1H NMR spectra of the host–guest complex where an upfield shift of the guest molecule protons was observed due to hydrophobicity and aromatic ring current of the cage (Fig. S3, ESI†). Integration of proton peak intensities shows a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex for the (1MePy: ExBox4+), (9-MA: ExBox4+) host–guest complex (Fig. S4 and S5, ESI†). The 2:1 complexation is further confirmed by a single crystal x-ray structure (Fig. 2b). Host–guest complex characterizations of other aromatic guests are also given in Fig. S6 and S7, ESI.† The proof of confinement was further established by a displacement assay with Pyrene as a guest (Fig. S8, ESI†). Steady-state absorption spectra (125 μM) of 9-MA ⊂ Exbox4+ in CH3CN (ACN) show an additional broad absorption feature having maxima around 500 nm for the host–guest complex, which has an absorption tail up to 550 nm (Fig. 2c). This feature has been assigned as the CT band originating due to the interaction of the electron donor guest and electron acceptor host molecule.11 From absorption titrations we observed a moderately strong binding of guests (∼103 M−1) (Fig. S9, ESI†). It is noted that there could be an evident confusion regarding the space inside the ExBox cavity for the reported 2:1 complex. We would like to explicitly state that the 2:1 complex here reported is similar to previous work reported by Stoddart and co-workers9 with one inside the cavity while the other in fast exchange residing outside.
:1 complex for the (1MePy: ExBox4+), (9-MA: ExBox4+) host–guest complex (Fig. S4 and S5, ESI†). The 2:1 complexation is further confirmed by a single crystal x-ray structure (Fig. 2b). Host–guest complex characterizations of other aromatic guests are also given in Fig. S6 and S7, ESI.† The proof of confinement was further established by a displacement assay with Pyrene as a guest (Fig. S8, ESI†). Steady-state absorption spectra (125 μM) of 9-MA ⊂ Exbox4+ in CH3CN (ACN) show an additional broad absorption feature having maxima around 500 nm for the host–guest complex, which has an absorption tail up to 550 nm (Fig. 2c). This feature has been assigned as the CT band originating due to the interaction of the electron donor guest and electron acceptor host molecule.11 From absorption titrations we observed a moderately strong binding of guests (∼103 M−1) (Fig. S9, ESI†). It is noted that there could be an evident confusion regarding the space inside the ExBox cavity for the reported 2:1 complex. We would like to explicitly state that the 2:1 complex here reported is similar to previous work reported by Stoddart and co-workers9 with one inside the cavity while the other in fast exchange residing outside.
Visible light excitation (LEDs) in this CT regime allows photochemical reactions inside the cavity. The products of all the CT-mediated photochemical transformations are shown in Fig. 2d. We observe oxidation photoproducts for different guest molecules. 1-MePy and 1-MeNap show selective aldehyde formation (Fig. S10 and S11, ESI†). For 9-MA, a distribution of photoproducts was observed due to the inherent reactivity in the 9 and 10 positions of 9-MA12 (Fig. S12 and S13, ESI†). Under inert conditions with the most reactive guest molecule 9-MA, only the starting material was observed after photoreaction which hints at the involvement of O2 in the process (Fig. S14, ESI†). Removal of the cavity and light (Fig. S15 and S16, ESI†) from the reaction conditions does not show aldehyde product formation. Use of an uncyclized ligand (DB.2PF6) in the photoreaction instead of ExBox4+ shows negligible photoproduct formation, which supports that the photoreaction occurs under a confined region (Fig. S17, ESI†). These control experiments show the role of the cavity as well as the role of CT states in the formation of oxidation photoproducts. We are able to trap cumene hydroperoxide after photoreaction, which suggests in situ generation of benzylic radicals (Fig. S18, ESI†). For toluene, and 2-MeThiophene we do observe the formation of aldehyde functionalized products with ∼ 95–100% conversion from GCMS (Fig. S19 and S20, ESI†). For other alkyl benzene derivatives (ethyl, propyl, butyl) also we do observe oxidation products with high selectivity (Fig. S21, ESI†). To understand the mechanistic pathway of the photoreaction, we carried out broadband femtosecond TA measurements. Fig. 3(a) shows the TA spectra of the 1-MePy ⊂ ExBox4+ host–guest complex at different pump–probe delays after 520 nm pump excitation. An excited state absorption (ESA) feature having maxima around 460 nm and a weak absorption feature up to 780 nm was observed in the ΔA spectra. As we move in time from 100 ps to 500 ps the absorption intensity decreases and a red-shift of the spectra is observed at 2 ns. The 460 nm feature was assigned to the radical cation signature of 1-MePy.13 A double-humped ESA feature was observed in the NIR region (Fig. 3b) having maxima around 990 and 1150 nm, which is a characteristic feature of ExBox4+ anion radical resulting from D1 ← D0 transition from the ExBox3+ unit.14 After varying our guest molecules with more π-electron density to lesser ones i.e., from 1-MePy to 9-MA and 1-MeNap we do see a gradual blue shift of the double hump ESA feature in the NIR region. This spectral shift hints at the interaction of the guest molecules with the ExBox3+ unit (Fig. S22, ESI†). After the singular value decomposition (SVD) analysis (Fig. 3c), we found EAS spectra with 1.9 ps, ∼203 ps, and non-decaying lifetimes, respectively. The short component spectra have been assigned as the spectrum of the CT state, and the spectra with 203 ps lifetime have been assigned as the radical cation state. The assignment of the CT state was based on spectral shift of the CT state with solvent polarity and charge separation after excitation in the CT regime (Fig. S23, ESI†). So, after photoexcitation to the CT state, a single electron transfer (ET) event occurs within ∼1.9 ps, which generates a 1-MePy radical cation and ExBox4+ radical anion. Similar to 1-MePy, we can follow the ET step for 9-MA,16 and 1-MeNap derivatives that occur within ∼1 ps and ∼560 fs (Fig. S24 and S25, ESI†). These radical cation intermediates have acidic protons (pKa < −10),15 which can be pulled off by nearby solvents to generate neutral radicals. So, to check the fate of radical cation species, we carried out secondary kinetic isotope effect (KIE) measurements in which we changed the solvent CH3CN to CD3CN and looked into TA dynamics (Fig. 3d). After monitoring the dynamics at 461 nm, we observed slower decay of the radical cation state of 1-MePy in CD3CN at ∼232 ps, whereas in CH3CN it is around ∼207 ps (KIE = 1.12 for 1-MePy and KIE = 1.38 for 1-MeNap). We have also found some changes in the early timescales of electron transfer (CT event) which could be due to the reorganization of solvent molecules.16 Acetonitrile usually contains trace amounts of water, and we thought to check if these water molecules act as a source of proton acceptor from the radical cations, then additional water content could lead to faster decay of radical cation species.17,18 Indeed, when we added 20% water into the solution mixture and recorded the TA, we observed a faster decay of 1-MePy+ within ∼194 ps shown in Fig. S23 and S26, ESI.† From SVD we assign the 3rd component, which is red-shifted from the radical cation state in Fig. 3c to be the spectral signature of the 1-MePy neutral radical. The assignment of these intermediates has been also verified from TD-DFT calculations (Fig. S27 and S28, ESI†).19 Previous reports on ExBox4+ have shown charge-separated (CS) state formation from the initial Franck–Condon active state via local excitation or CT excitation; however, within a few ps, this CS state recombines.11,20 Here after CS, the presence of these proton transfer coordinates allows the system to bypass the recombination pathway, and generate neutral radicals on benzylic carbon centers with nanosecond lifetimes. These neutral radicals react with O2 in the diffusion-limited timescale and give rise to oxidation photo products.
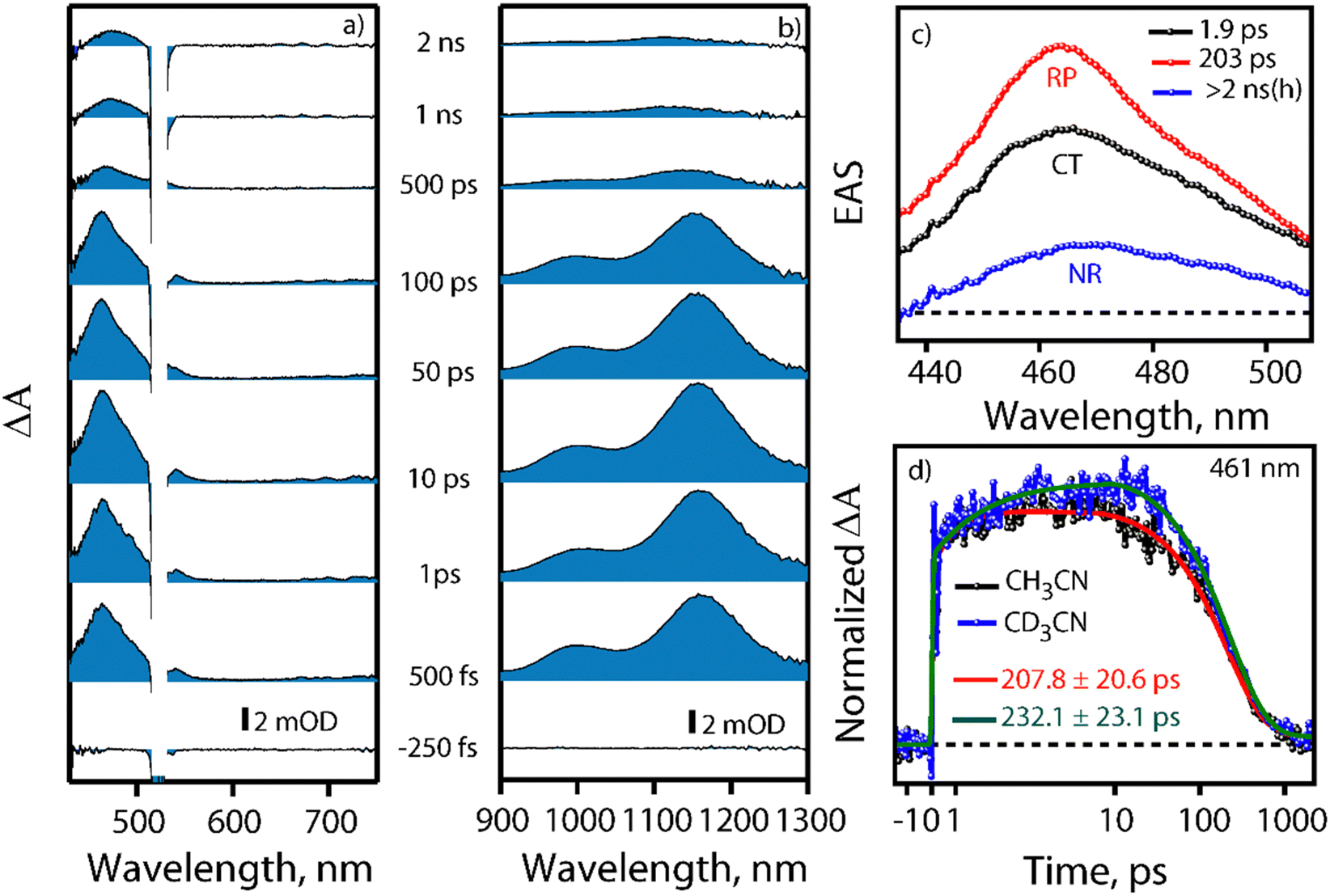 | ||
| Fig. 3 Transient absorption (TA) data of 1-MePy ⊂ ExBox4+ in CH3CN; (a) visible region and (b) NIR region. (c) SVD of TA data with 3-state sequential model: CT-Charge Transfer, RP: Radical Pair, NR: Neutral Radical state. (d) Normalized kinetics of TA data at 461 nm for the 1-MePy ⊂ ExBox4+ complex in CH3CN and CD3CN. | ||
We have done primary KIE measurements with isotopically labelled d12-9-MA where all C–H bonds are replaced by C–D bonds. The 710 nm radical cation feature showed a faster decay for regular 9-MA as compared to isotopically labelled d12-9-MA in ACN (Fig. S29, ESI†). We observed a primary KIE value around 1.16 and hence further prove the existence of the proton transfer step.21 We have done wavelength-dependent TA measurement to check the selectivity of CT excitation with the 9-MA ⊂ Exbox4+ host–guest complex. Along with CS state we can see a shoulder around 590 nm with 400 nm CT or local excitation, which is absent in 520 nm excitation (Fig. S30, ESI†). Hence, we confirm that with 520 nm CT excitation, we are exciting selectively the host–guest complex eliminating any contribution from the free guest molecules in solution. After carrying out photoreaction with 1-MeNap as a guest molecule inside ExBox4+ in a 40% water–ACN ratio we can observe the formation of 1-naphthaldehyde as the major product with selectivity up to 91%; however, when we go to more water content (60%) the product selectivity goes down to 71% due to solubility issues (Fig. S31, ESI†). In summary, we report the first visible light mediated sp3 C–H bond activation and subsequent functionalization inside an all-organic confinement in aqueous and polar solvents, thus generalizing the host–guest CT paradigm. We use well-known cavity ExBox4+ while also providing a facile synthesis route using a microwave chamber. The mechanism provided in Fig. 4 shows that after CT excitation, a single ET event occurs within ∼1 ps which generates a radical cation on the guest and anion radical on the host. Subsequently, the solvent molecule (water) takes the proton from the radical cation within ∼100–200 ps, thereby generating neutral carbon-centred radicals having ns lifetimes. These neutral radicals then react with oxygen to give the corresponding C–O functionalized products. Meanwhile, the host anion radicals can be quenched by O2, thereby regenerating the neutral catalyst. We envision that subsequent steps of making the cavity exclusively water-soluble along with changing of the cavity electronics will reduce the catalytic loading of the ExBox4+ as well as the light-intensity requirement further by engineering the CT molar extinction coefficients.
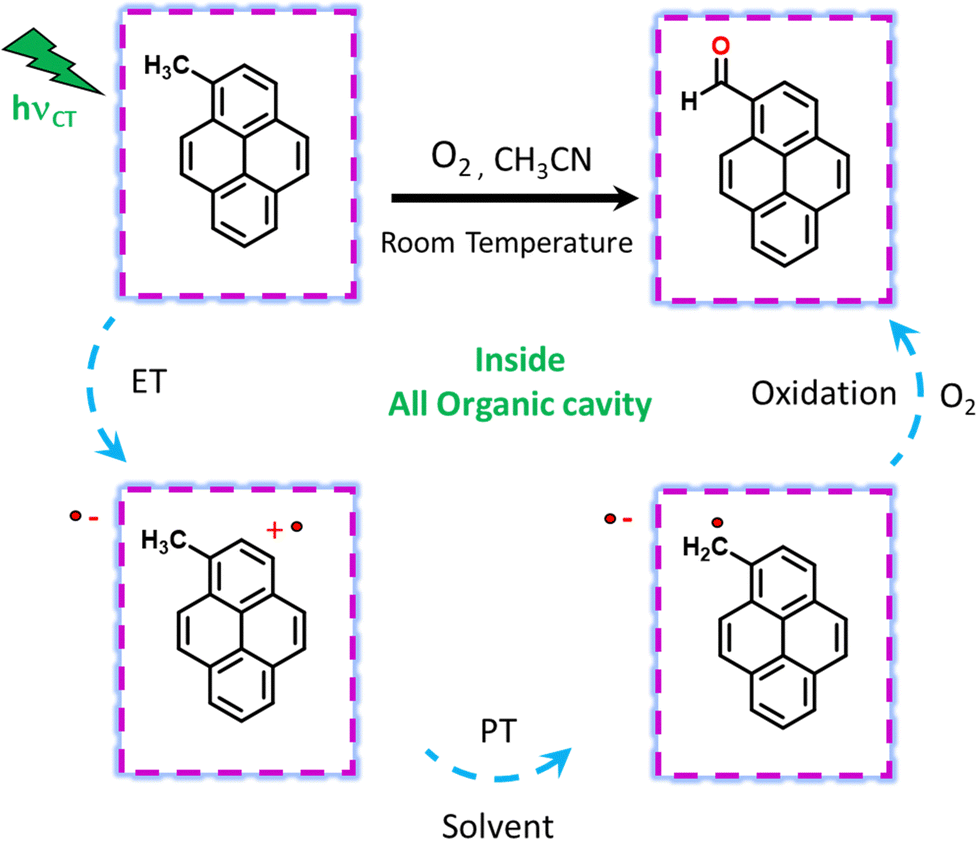 | ||
| Fig. 4 Photoreaction scheme of CT-mediated C–H functionalization inside ExBox4+. CT excitation leads to ET, after which the PT step generates neutral radicals on the benzylic carbons, which undergo oxidation. | ||
D. R., S. P. and J. D. acknowledge support from the Department of Atomic Energy (DAE), Government of India, under Project no. 12-R&D-TFR-5.10-0100.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885–898 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- N. Noto, Y. Hyodo, M. Yoshizawa, T. Koike and M. Akita, ACS Catal., 2020, 10, 14283–14289 CrossRef CAS.
- S. Mkrtchyan, V. B. Purohit, S. Sarfaraz, M. Yar, K. Ayub and V. O. Iaroshenko, ACS Sustainable Chem. Eng., 2023, 11, 8406–8412 CrossRef CAS.
- K. Ohkubo, K. Suga, K. Morikawa and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 12850–12859 CrossRef CAS PubMed.
- H. Wang, C. Cao, D. Li, Y. Ge, R. Chen, R. Song, W. Gao, X. Wang, X. Deng, H. Zhang, B. Ye, Z. Li and C. Li, J. Am. Chem. Soc., 2023, 145, 16852–16861 CrossRef CAS PubMed.
- A. Das, I. Mandal, R. Venkatramani and J. Dasgupta, Sci. Adv., 2019, 5, eaav4806 CrossRef CAS PubMed.
- J. C. Barnes, M. Juricek, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 183–192 CrossRef CAS PubMed.
- D. Dallinger and C. O. Kappe, Chem. Rev., 2007, 107, 2563–2591 CrossRef CAS PubMed.
- R. M. Young, S. M. Dyar, J. C. Barnes, M. Juricek, J. F. Stoddart, D. T. Co and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 12438–12448 CrossRef CAS PubMed.
- P. Anzenbacher, T. Niwa, L. M. Tolbert, S. R. Sirimanne and F. P. Guengerich, Biochemistry, 1996, 35, 2512–2520 CrossRef CAS PubMed.
- N. Getoff, S. Solar, U. B. Richter and M. W. Haenel, Radiat. Phys. Chem., 2003, 66, 207–214 CrossRef CAS.
- S. M. Dyar, J. C. Barnes, M. Juricek, J. F. Stoddart, D. T. Co, R. M. Young and M. R. Wasielewski, Angew. Chem., Int. Ed., 2014, 53, 5371–5375 CrossRef CAS PubMed.
- Y. Qin, L. H. Zhu and S. Z. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS PubMed.
- R. Jimenez, G. R. Fleming, P. V. Kumar and M. Maroncelli, Nature, 1994, 369, 471–473 CrossRef CAS.
- K. Kaupmees, I. Kaljurand and I. Leito, J. Phys. Chem. A, 2010, 114, 11788–11793 CrossRef CAS PubMed.
- M. Oyama and J. Matsui, J. Electr. Chem., 2004, 570, 77–82 CrossRef CAS.
- L. Shen, H. Y. Zhang and H. F. Ji, Org. Lett., 2005, 7, 243–246 CrossRef CAS PubMed.
- X. R. Gong, R. M. Young, K. J. Hartlieb, C. Miller, Y. L. Wu, H. Xiao, P. Li, N. Hafezi, J. W. Zhou, L. Ma, T. Cheng, W. A. Goddard, O. K. Farha, J. T. Hupp, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2017, 139, 4107–4116 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2286415. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03987k |
| This journal is © The Royal Society of Chemistry 2023 |