
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal desorption gas chromatography-mass spectrometry for investigating the thermal degradation of polyurethanes
Roland
Becker
*,
Philipp
Scholz
,
Christian
Jung
and
Steffen
Weidner

Federal Institute for Materials Research and Testing, Richard-Willstätter- Strasse 11, D-12489 Berlin, Germany. E-mail: roland.becker@bam.de
First published on 3rd April 2023
Abstract
Thermal Desorption Gas Chromatography-Mass Spectrometry (TD-GC-MS) was used to investigate the thermal degradation of two different polyurethanes (PUs). PU samples were heated at different heating rates and the desorbed products were collected in a cold injection system and thereafter submitted to GC-MS. The prospects and limitations of the detection and quantification of semi-volatile degradation products were investigated. Temperature dependent PU depolymerization was observed at temperatures above 200 °C, proved by an increased release of 1,4-butanediol and methylene diphenyl diisocyanate (MDI) representing the main building blocks of both polymers. Their release was monitored quantitatively based on external calibration with authentic compounds. Size Exclusion Chromatography (SEC) of the residues obtained after thermal desorption confirmed the initial competitive degradation mechanism indicating an equilibrium of crosslinking and depolymerization as previously suggested. Matrix-Assisted Laser Desorption Ionization (MALDI) mass spectrometry of SEC fractions of thermally degraded PUs provided additional hints on the degradation mechanism.
Introduction
Polyurethanes (PUs) represent an important class of synthetic polymers widely used in everyday life.1 Apart from medical,2 automotive3 and various other applications, a main field is its use in thermal insulation.4 This requires materials with high thermal stability, and thus, there is a need for a deeper understanding of thermal degradation processes.The first results regarding the thermal degradation of PUs were published by Dyer et al. in 1958 using so-called model urethanes (carbamates).5 They reported three pathways for the initial low temperature cleavage (200–300 °C). First, depolymerization by dissociation into isocyanate and alcohol was observed, followed by dissociation into a primary amine, olefins and CO2. Finally, CO2 is eliminated, which led to formation of a secondary amine. In 1981 Gaboriaud et al. studied the degradation kinetics of PUs composed of methylene diphenyl diisocyanate (MDI) and propoxylated trimethylol propane.6 Using gas chromatography with flame ionization detection (GC-FID) of directly injected thermally desorbed gas aliquots and thermogravimetric analysis (TGA) and infrared (IR) techniques they were able to determine a random chain scission process at 200 °C leading to isocyanate and polyol. However, they were not able to clearly identify the structure of the released isocyanates. Taking into account some IR band broadening and the absence of one characteristic MDI IR peak, they assumed the release (or formation) of an ‘MDI polymer’. At higher temperatures (600 °C) gaseous products were obtained, which resulted essentially from decomposition of the polyurethane polyol units. Based on these findings, in a second paper a two-step mechanism of PU degradation was formulated for higher temperatures.7 The first step started at 250 °C and consisted of the standard reaction in polyurethane degradation, i.e. scission into a primary amine, carbon dioxide and propenylether, finally releasing propene. The second step (above 400 °C) involved the radical breakdown of the polymer chain.
Since that time, several other, more sophisticated techniques have been applied to gain a deeper insight into the degradation mechanism. Pyrolysis at 500 °C coupled with gas chromatography-mass spectrometry (py-GC/MS) was initially used by Williamson to identify polyurethane formulations where hydrolysis of the isocyanate had occurred immediately prior to curing.8 The same technique was used by Zhang et al. who evaluated the degradation profiles of cast PU films based on isophorone diisocyanate (IPDI).9 They identified more than 15 characteristic volatiles in PU pyrolysates formed between 250 and 700 °C. The kinetics of the thermal degradation of PU elastomers based on poly(ether polyol) soft segments and MDI were investigated by thermogravimetric analysis (TGA) under a nitrogen atmosphere employing four heating rates. The results showed that the first stage of decomposition (depolymerization) correlated with hard-segment (MDI + chain extender) degradation, whereas the second peak (high-temperature degradation) correlated with the degradation of the soft-segments (polyether/polyester). It was also observed that PU elastomers with a higher hard-segment content and a higher soft-segment molecular weight exhibited higher thermal stability.10 Matrix-assisted laser desorption/ionization time of flight (MALDI TOF) mass spectrometry was also utilized to obtain structural information. In a paper by Lattimer et al. segmented PU was degraded at lower temperatures.11 Below 300 °C depolymerization was observed, whereas at higher temperatures cyclic oligomers formed by intramolecular urethane exchange were detected.12 Size Exclusion Chromatography (SEC) was applied by Scholz et al. to monitor the molecular weight loss during degradation of polyether and polyester PUs. They found a different behavior of polyether and polyester based PUs and a temperature dependent competitive mechanism between chain scission (by depolymerization) and crosslinking. Thereby, the presence of oxygen obviously favored crosslinking.13 Py-GC-MS appears to have been used to investigate the potential of waste polymer conversion into volatile fuels at temperatures between 250 and 400 °C.14 While Py-GC-MS, direct pyrolysis MS, MALDI-MS and TG-MS11,15–18 as well as TGA, TGA-MS, and TGA-FTIR19 remain the methods of choice for the characterization of PU, thermal desorption combined with GC-MS (TD-GC-MS) has rarely been employed. Kim et al. used a TD-GC-MS procedure to quantify the content of N-methyl-2-pyrrolidone in polyvinylchloride (PVC), polystyrene (PS) and polymethylmethacrylate (PMMA) matrices.20 Meanwhile, TD-GC-MS has become a standard test method for the determination of low level, regulated phthalates in PVC.21 In a paper by Mitchell et al. volatile organic compounds (VOC) of PU were desorbed at ambient temperature, after thermal (70 °C) and accelerated UV degradation (365 nm), collected on Tenax-TA and submitted to TD-GC-MS analysis.22 Apart from propylene glycol, rather unspecific VOC markers were found. Moreover, differentiation between polyether and polyester PUs failed.
Continuing the work done in previous investigations on the thermal, photo-oxidative, and hydrolytic degradation of various PUs,13 this paper presents a proof-of-concept study. TD-GC-MS was utilized after thermal treatment of two model PUs at 250 °C and collection of the desorbed degradation product in a cold injection system prior to GC-MS analysis. The prospects and limitations regarding PU structure characterization via detection and quantification of low molecular weight degradation products are discussed and correlated with the molecular mass changes monitored by SEC of residues after TD and with results of matrix-assisted laser desorption ionization (MALDI) measurements of SEC fractions.
Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500, 201000, and 1190000 g mol−1) were provided by Polymer Standards GmbH (PSS), Mainz, Germany.
500, 201000, and 1190000 g mol−1) were provided by Polymer Standards GmbH (PSS), Mainz, Germany.
Original polyurethane samples were received in the form of 5 mm plates from BASF company (BASF Polyurethanes, Lemförde, Germany). Their structure (shown in Scheme 1) and composition were confirmed by 1H NMR spectroscopy. For the experiments 3 g of PU was dissolved at ambient temperature under constant stirring in 30 mL DMF. The polymer solution was poured into 100 mL methanol. The precipitate was removed by filtration and dried for 1 h at 50 °C. This solution/precipitation procedure was repeated twice. Finally, the precipitate was dissolved in 20 mL DMF again, poured into a Petri dish and allowed to dry in an oven at 50 °C for 5 d.
 | ||
| Scheme 1 General structure of the investigated polyurethanes. | ||
Thermal desorption gas chromatography mass spectrometry (TD-GC-MS)
For the detection of semi-volatile compounds released from the PU materials by thermal desorption gas chromatography-mass spectrometry (TD-GC-MS) an Agilent 7890A gas chromatograph (GC) equipped with a thermal desorption unit (TDU-2), a CIS 4 cold injection system, an MPS 2 XL autosampler and an Agilent 5975c Inert XL quadrupole mass selective detector (all from Gerstel, Mülheim a. d. Ruhr, Germany) was employed. PU samples (4 mg) were inserted into the TDU by means of m-vials (Gerstel) specifically designed for this purpose. The TDU temperature always started at 35 °C (delay time: 0.5 min) and was raised at a rate of 120 K min−1 to the final temperature (50–250 °C, see individual experiments) and held at this temperature between 1 and 30 min as specified for the respective experiments below. A total helium flow (5.0, Linde, Berlin, Germany) of 104 mL min−1 was maintained (100 mL TD flow, 3 mL septum purge and 1 mL GC column carrier gas flow). The cold injection system started at −50 °C and was raised to 275 °C at a rate of 6 K s−1 or 12 K s−1 respectively, and held at this temperature for 5 min. The GC was equipped with either a DB-1 capillary column (Agilent, 30 m × 0.25 mm × 0.25 mm film thickness) or DB-17ms capillary column (Agilent, 60 m × 0.25 mm × 0.25 mm film thickness). The oven program using the DB-1 column started at 40 °C (held for 2 min) and was raised to 320 °C with a rate of 20 K min−1 (held for 12 min). For the DB-17ms column the same program was used with a final temperature of 310 °C. The mass selective detector (MS) was operated in electron impact mode (70 eV). Transfer line, ion source, and quadrupole temperatures were 320 °C, 230 °C and 150 °C, respectively. Mass spectrometric data were recorded in scan mode with the m/z range between 35 and 1050. The obtained total ion chromatograms were recorded with an Agilent ChemStation and compared with the NIST mass spectral data library (version 14).External calibration for the quantification of 1,4-butanediol was performed by adding between 0.1 and 7 μL of a solution of butanediol in ethanol (24.92 μg μL−1) into the micro vials of the TD unit to give eight equidistantly concentrated calibration points.
For MDI calibration a stock solution in dry acetone (2.03 μg μL−1) was prepared under argon sheet gas. Six calibration solutions between 0.203 and 2.03 μg μL−1 were prepared manually in 2 mL GC vials. 1 μL of each calibration solution was transferred to the micro vials for the TD unit.
Gel permeation chromatography (GPC)
A modular system consisting of an isocratic pump (Bischoff Chromatography, Leonberg, Germany), a column oven (HV-125, Schreiber, Rheinbach, Germany), a UV detector (SPD-M20A, 254 nm, Shimadzu GmbH, Germany) and a refractive index detector (Optilab reX, Wyatt Technology Europe GmbH, Dernbach, Germany) was used. The system was kept at 50 °C. DMF with 1 wt% LiBr was used as eluent. Samples were dissolved in DMF and manually injected (Rheodyne valve) at a flow rate of 1.0 mL min−1. For separation the system was equipped with 2 GRAM columns (10 μm, 8 × 300 mm) and a GRAM pre-column (10 μm, 8 × 50 mm, GRAM, Polymer Standard Service – PSS, Mainz, Germany). Calibration was performed using a set of PMMA standards (2200, 23500, 201000, and 1190000 g mol−1). Typically, the injected sample volume was 100 μL with a concentration of 2–3 mg mL−1. All samples were filtered before injection using a syringe filter with an average pore size of 0.25 μm. Elugrams and molecular mass distribution curves were evaluated by means of WinGPC software (PSS, Mainz, Germany).
Matrix-assisted laser desorption ionization-time of flight mass spectrometry
The MALDI-TOF mass spectra were measured on a Bruker Autoflex maX mass spectrometer equipped with a Smartbeam laser (355 nm). The spots were prepared by dropping 1 μL of a 5:2 (v/v) solution of matrix (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile, 20 mg mL−1 in tetrahydrofuran (THF)) and analyte onto the stainless-steel target. The analyte solutions were prepared by dissolving the polymer residues in dimethylformamide (DMF, Roth, Germany). Potassium trifluoroacetate (3 mg mL−1 in THF) was added to the premixed solution to achieve potassium adduct ion formation.
Results and discussion
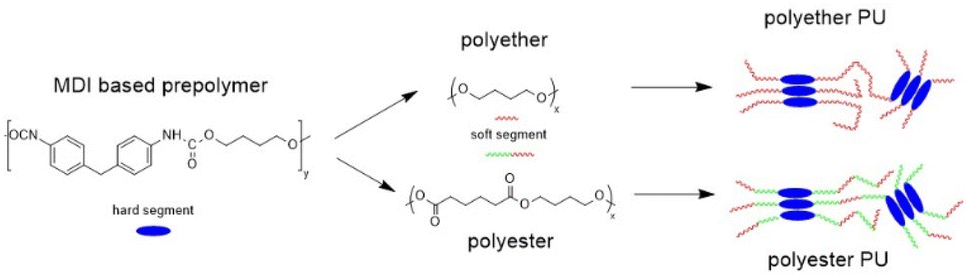
Fig. 1 shows TD-GC-MS chromatograms of the original polyether and polyester PUs (Fig. 1a and b) after artificial weathering (40 °C, 90% humidity, 340 nm, 36 W m−2). For more details see elsewhere.13 Using the nonpolar DB-1 column sharp peaks were detected in a retention time range between 8 and 18 min, which could be attributed to butylhydroxytoluene (BHT) (2) and Metilox (3), isocyanate monomers (7–9) and bio-based plasticizers (4–6) along with several peaks to which no structure could be assigned. This observation, that not only semi-volatile additives but also isocyanate monomers were formed, was the reason for employing TD-GC-MS in addition to size exclusion chromatography to investigate the behavior of PU esters and ethers under thermal stress. The TD-GC-MS chromatograms (a and b) additionally showed a very broad peak at a retention time of 5.5 min, which could be attributed to 1,4-butanediol (indicated by an asterisk) by mass spectra and retention time comparison using the reference compound. Based on these results it became obvious that for a reliable investigation of PU degradation products by TD-GC-MS neat polymer material without any stabilizers and other additives has to be used. The TD-GC-MS chromatograms of purified (by precipitation, see the Experimental) polyether and polyester PUs are presented in Fig. 1c and d. Again, a very broad, but now more intense, peak at a retention time of 5.5 min (corresponding to 1,4 butanediol) along with a few more intense peaks at higher retention times were observed. The latter could be identified as nonspecific degradation products for PU using the spectra library. However, other than in the technical PUs (Fig. 1a and b), isocyanates could not be detected by thermal desorption of the purified PUs under the applied conditions. A closer look at the isocyanate structures shown in Fig. 1b reveals that the NCO-moieties in the technical PUs are attached to isopropyl side chains rather than directly to the benzene ring as is the case in MDI.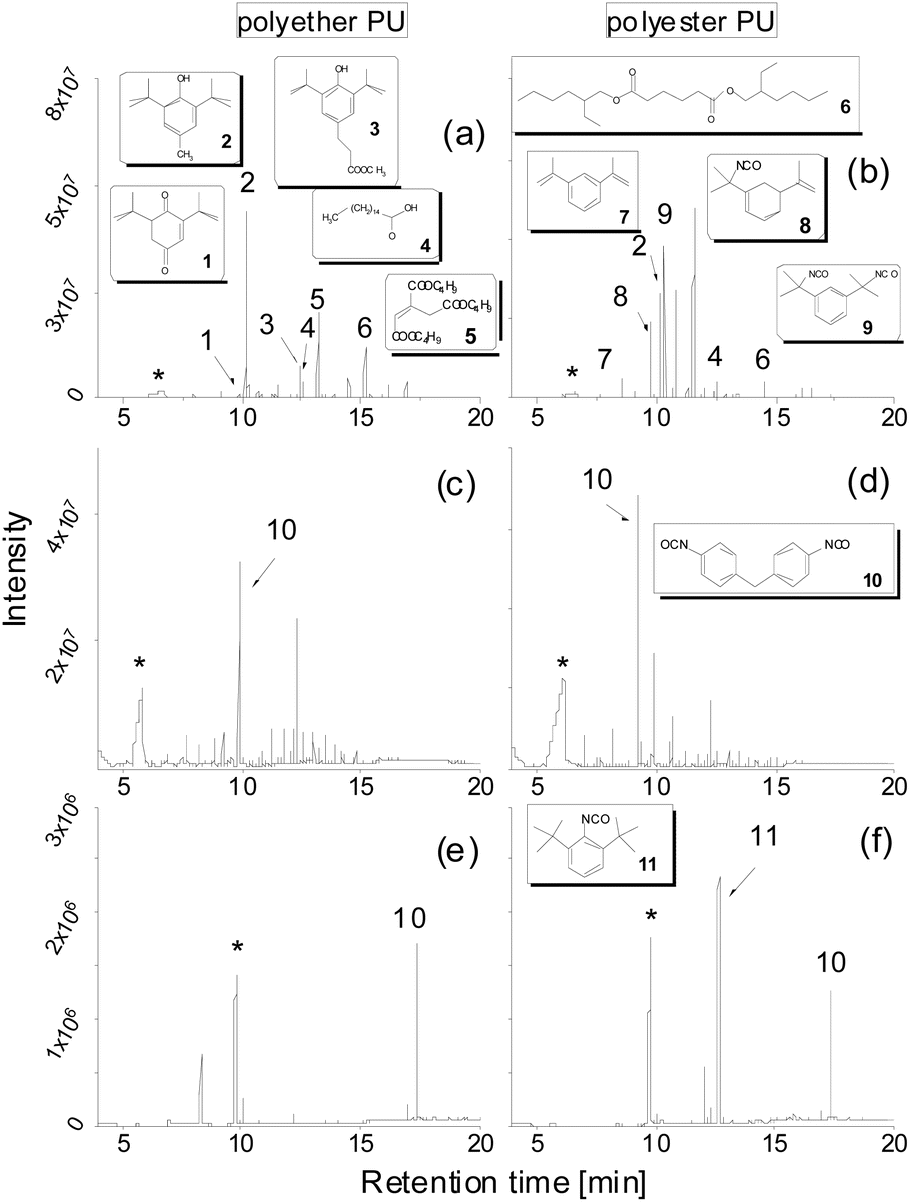 | ||
| Fig. 1 TD-GC-MS chromatograms of the original polyether and polyester PUs with determined structures (a and b); after purification by precipitation in MeOH using a DB-1 column and thermal desorption for 1 min at 250 °C and 12 K min−1 (c and d), and using a polar DB-17ms column and thermal desorption for 10 min at 250 °C and 12 K min−1 (e and f) (the peak of 1,4-butanediol is indicated by an asterisk). | ||
A change to the more polar DB-17ms column enabled the detection of MDI as a distinct chromatographic peak (Fig. 1e and f).
Moreover, 1,4-butanediol also eluted in a sharp peak at higher retention time (9.8 min). In addition, an isocyanate moiety directly bound to the aromatic ring was released from PU, confirmed by the excellent match of its fragmentation pattern with the spectral library. Along with findings referred to in Fig. 1a this underpins the capability of this modified TD approach to characterize PUs regarding their relevant components.
These improved chromatographic conditions were the basis for quantification of the two degradation products of the PU backbone. For this, an external calibration was performed, where defined amounts of both compounds were separately placed into the TDU, submitted to desorption, collected in the CIS and subsequently transferred to a gas chromatograph (see the Experimental). In case of 1,4-butanediol the linearity of the detector response enabled quantification on the basis of a linear regression with a coefficient of determination (R2) of 0.9990. MDI displays a slightly non-linear dependency of its detector response. A quadratic fit was used for quantification (R2 = 0.9968). The respective calibration curves are shown in Fig. 2a and b. The respective limits of quantification were determined by the regression method to be 3.43 μg (1,4-butanediol) and as 9-fold the background noise, i.e. 0.178 μg (MDI).
 | ||
| Fig. 2 Calibration curves of (a) MDI and (b) 1,4-butanediol (see the Experimental). | ||
As previously discussed by Gaboriaut et al. the typical degradation of PU proceeds in 3 steps: (i) dissociation to the original polyol and isocyanate, (ii) formation of a primary amine, alkene, and carbon dioxide, and (iii) formation of a secondary amine and carbon dioxide.6,7 However, in our experiments formation of amine structures could not be observed by mass spectrometry. Moreover, according to Gaboriaut and other authors the formation of carbon dioxide from isocyanate and/or urethane degradation was found to start at temperatures > 350 °C, which is far higher than the investigated temperature range here.5–8 Thus, we have to assume the dissociation into polyol and MDI to be the predominant degradation process at 250 °C. This was also proved by our TD-GC/MS investigations, which show the predominant formation of butanediol and MDI. It should be noted that the final amount of substance quantified by the TD-GC-MS procedure is that portion of 1,4-butanediol or MDI that is released into the MS after the following consecutive steps: formation by thermal degradation of the PU, transport with the helium gas flow, collection in the CIS and release from the CIS into the MS. The formation as well as the desorption kinetics in the TDU may be influenced by the phase composition of the respective polymer and, in parallel, side reactions especially of the isocyanate may occur.
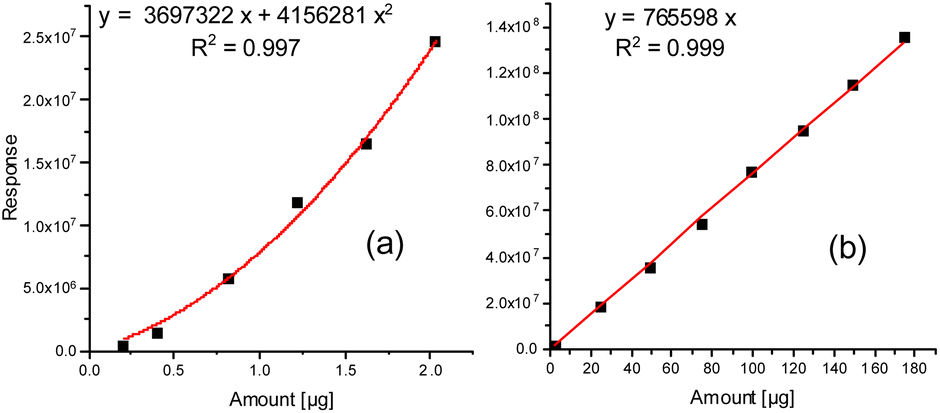
Possibly, partial back-reaction of diol and isocyanate may also take place in the CIS. Therefore, from Fig. 3 it is obvious that the initial desorption of both 1,4-butanediol and MDI proceeds in a largely linear manner with time and has a tendency to be faster at a higher heating rate. For the 1,4 butanediol desorption (especially from polyether PU, Fig. 3a) this linear behavior reveals a slight tendency towards flattening at longer desorption times. It remains unclear whether this was a result of a reduced desorption due to partial 1,4-butanediol loss caused by side reactions becoming more relevant during prolonged heating or due to diffusion kinetics within the molten PU samples.
 | ||
| Fig. 3 Time-dependent thermal desorption at 250 °C of 1,4 butanediol and MDI from polyether PU (a and c) and polyester PU (b and d) at heating rates of 6 (red) and 12 K s−1 (black). | ||
Likewise, the desorption of MDI starts linearly, but with increasing exposure time the amount collected in the CIS and released to the GC tends to reach a plateau in case of the polyether PU (Fig. 3c) and even decreases in case of the polyester PU (Fig. 3d). It remains open whether this was a result of a higher instability due to the reactive isocyanate moieties or due to a possible back reaction of MDI with the excess 1,4-butanediol present in the cold injection system prior to gas chromatography.
Preliminary experiments using matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) of fractions taken from size exclusion chromatography (SEC) of purified (by precipitation in MeOH) PUs revealed that a significant amount of neat oligo-butanediol was generated during thermal treatment of a polyether PU even at 150 °C, but at a significantly longer incubation time of 7 d (Fig. 4a). 1,4-Butanediol structures (D = 72 Da) can also be found in the low mass region of a MALDI spectrum recorded from higher mass SEC fractions (e.g. SEC fraction 3, Fig. 4b). However, there these units are still connected to smaller parts of the polymer backbone that also contain MDI units (284 Da).
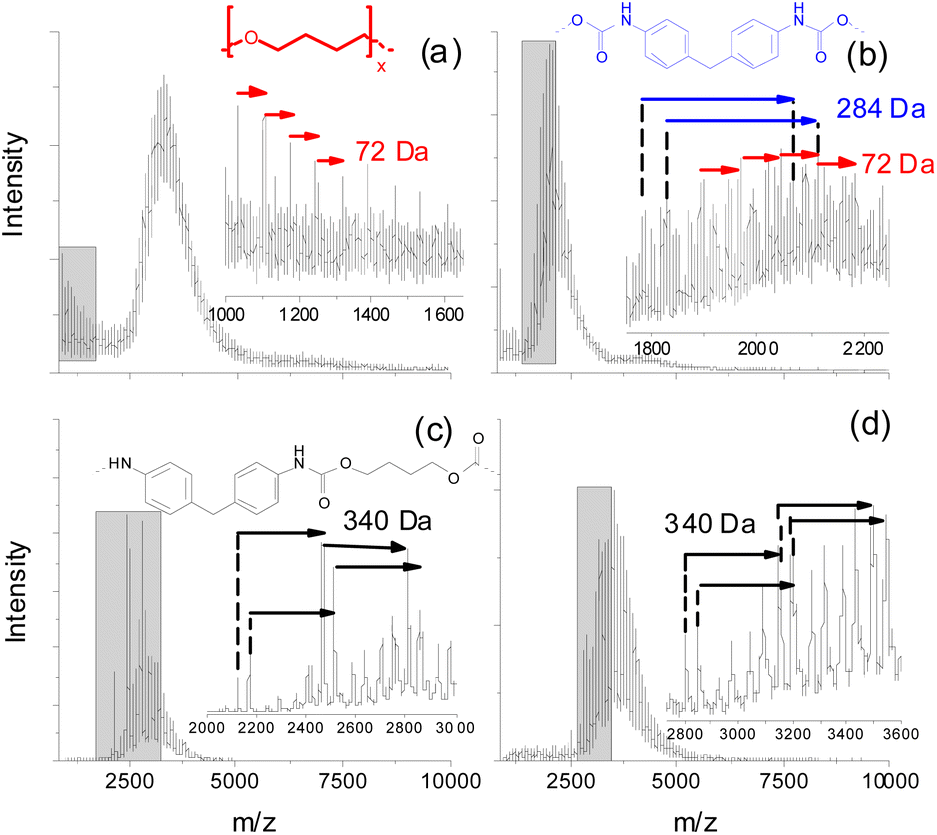 | ||
| Fig. 4 MALDI TOF mass spectra of lower mass SEC fractions of purified (by precipitation in MeOH) polyether and polyester PUs after thermal treatment at 150 °C (7 d): (a and b) polyether PU, (c and d) polyester PU. | ||
Polyester urethanes thermally treated under identical conditions showed a slightly different result. Oligo-butanediol structures could not be found. Instead of that, in two exemplarily shown spectra of lower mass SEC fractions (Fig. 4c and d) various series of peaks were found, which can be attributed to MDI units still bound to aliphatic residues having possible masses of 340 Da. Unfortunately, these MALDI experiments could not be done at higher degradation temperatures since the process of crosslinking (see the next clause) led to insoluble residues with only minor soluble parts, which could not be fractionated into fractions having reasonable concentrations for MALDI. Thus, the presented MALDI mass spectrometric data may not be directly compared to TD-MS data. Moreover, both methods cover completely different mass ranges. But this might help to explain the differences in the MDI desorption of polyether and polyester PUs. Polyether PUs seem to degrade to oligo-butanediol and lower mass polymer fragments showing repeat units of MDI, whereas the degradation of polyester PUs at the same temperature results in a significantly higher amount of such MDI containing polymer fragments. This indicates a lower tendency of polyester PUs to completely release all bonded MDI. Neat MDI cannot be detected in MALDI due to its low molecular mass.
Size exclusion chromatography
Recently, it was shown that PU degradation under thermo-oxidative conditions is characterized by a competitive mechanism of depolymerization accompanied by crosslinking.13 However, these investigations were performed after treatment at elevated temperatures (100–200 °C) under oxidative (air) as well as non-oxidative (nitrogen) conditions for several days (up to 7 days). In contrast to that, exposure times in TD-GC-MS were much shorter (in a time frame of minutes), whereas temperatures were slightly higher (250 °C). For this purpose, polymer residues were collected after the thermal desorption process and dissolved in DMF. The results are shown as molar mass distribution (MD) curves in Fig. 5a and b. | ||
| Fig. 5 Molar mass distribution curves of a polyester PU (a) and a polyether PU (b) and the course of the molar mass changes monitored by SEC over time (c) with y0 = 18950 (24220), xc = 10 (8), w = 19 (12) and A = 9.43 × 106 (7.86 × 106) for the polyether (polyester) PU. | ||
Surprisingly, the shape of these two curves was quite similar to our previous results,13 but on a different time scale. The degradation process started after a few minutes, clearly visible by an increase of the maximum of the Mp values (Mp = mass at peak maximum) after 5 min exposure. Simultaneously, the distribution curves began to broaden indicating an increase of the polymer dispersity Ð (Ð = Mw/Mn, where Mw represents the weight average molar mass and Mn represents the number average molar mass). At 10 min a maximum of the molar mass increase was reached. At even longer exposure times a significant decrease of mass values was found. The data compiled in Fig. 5c were fitted using a Gauss function (for fitting parameters see the figure caption). Since in SEC only soluble parts of the polymer could be investigated, the intensity of the curves shown in Fig. 5a and b can serve as a rough estimate of its amount. A quantitative explanation cannot be given, because this would presume a similar refractive index of all degradation products, which must be excluded. After 5 min the intensity of the MD curves slightly decreases for polyether PU (Fig. 5b) and remains almost constant for polyester PU (Fig. 5a). There, the peak area at 1 and 5 min is almost identical. This indicates the beginning of branching of polymer chains, which leads to larger macromolecules that are still soluble. After 10 minutes both polymers show a significant loss of the intensity of the mass distribution curves. This indicates a dramatic loss of soluble PU molecules within the TD residues and might serve as evidence for more intense crosslinking. This assumption is also supported by the shape of the MD curves which are characterized by a long tailing towards extremely high molar masses up to 2000000 g mol−1. Simultaneously, the masses of soluble polymer residues start to decrease, revealing decomposition. Since the lower exclusion limit of this type of column was around 1000 g mol−1, lower mass depolymerization products were not detectable by SEC.
Conclusions
Based on the presented results one could conclude that the presented TD-GC-MS method shows prospects as an additional tool for rapid characterization of polyurethanes complementary to SEC and MALDI, which cover completely different molar mass regions. Its unique capability to detect semi-volatiles such as additives and isocyanates closes a gap for the comprehensive monitoring of thermal degradation processes. This allows for differentiating different types of PU and may help during material optimization or in the simulative investigation of thermal recycling procedures.Author contributions
R. Becker – Conceptualization, Data curation, Supervision, Formal analysis, Writing – original draft; P. Scholz – Investigation, Methodology, Writing – review & editing; C. Jung – Investigation, Methodology, Data curation, Formal analysis; S. Weidner – Conceptualization, Data curation, Supervision, Formal analysis, Writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Federal Institute for Materials Research and Testing (BAM) for technical support and BASF AG, Lemförde for supplying the PU material.Notes and references
- J. O. Akindoyo, M. Beg, S. Ghazali, M. Islam, N. Jeyaratnam and A. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- S. Wendels and L. Avérous, Bioact. Mater., 2021, 6, 1083–1106 CrossRef CAS PubMed.
- M. Groenewolt, Polym. Int., 2019, 68, 843–847 CrossRef CAS.
- D. Naldzhiev, D. Mumovic and M. Strlic, Build. Environ., 2020, 169, 106559 CrossRef.
- E. Dyer and G. E. Newborn Jr, J. Am. Chem. Soc., 1958, 80, 5495–5498 CrossRef CAS.
- F. Gaboriaud and J. Vantelon, J. Polym. Sci. Polym. Chem. Ed., 1981, 19, 139–150 CrossRef CAS.
- F. Gaboriaud and J. Vantelon, J. Polym. Sci. Polym. Chem. Ed., 1982, 20, 2063–2071 CrossRef CAS.
- J. Williamson, M. Cocksedge and N. Evans, J. Anal. Appl. Pyrolysis, 1980, 2, 195–205 CrossRef CAS.
- Y. Zhang, Z. Xia, H. Huang and H. Chen, J. Anal. Appl. Pyrolysis, 2009, 84, 89–94 CrossRef CAS.
- A. Agić and E. G. Bajsić, J. Appl. Polym. Sci., 2007, 103, 764–772 CrossRef.
- R. Lattimer, M. Polce and C. Wesdemiotis, J. Anal. Appl. Pyrolysis, 1998, 48, 1–15 CrossRef CAS.
- R. P. Lattimer and R. C. Williams, J. Anal. Appl. Pyrol., 2002, 63, 85–104 CrossRef CAS.
- P. Scholz, V. Wachtendorf, U. Panne and S. M. Weidner, Polym. Test., 2019, 77, 105881 CrossRef.
- L. Jiang, D. Zhang, M. Li, J.-J. He, Z.-H. Gao, Y. Zhou and J.-H. Sun, Fuel, 2018, 222, 11–20 CrossRef CAS.
- P. Rizzarelli and S. Carroccio, in Reactions and Mechanisms in Thermal Analysis of Advanced Materials, Scrivener Publishing WILEY, 2015, vol. 10, pp. 221–251 Search PubMed.
- T. M. Crescentini, J. C. May, J. A. McLean and D. M. Hercules, Polymer, 2019, 181, 121624 CrossRef CAS PubMed.
- K. Aou, A. K. Schrock, V. V. Ginzburg and P. C. Price, Polymer, 2013, 54, 5005–5015 CrossRef CAS.
- J. T. Mehl, R. Murgasova, X. Dong, D. M. Hercules and H. Nefzger, Anal. Chem., 2000, 72, 2490–2498 CrossRef CAS PubMed.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34, 1068–1133 CrossRef CAS.
- Y.-M. Kim, J. W. Kim, H. M. Moon, M.-J. Lee, A. Hosaka, A. Watanabe, N. Teramae, Y.-K. Park and S.-W. Myung, Anal. Sci., 2017, 33, 821–824 CrossRef CAS.
- ASTM D7823-20: Standard Test Method for Determination of Low Level Phthalates in Poly (Vinyl Chloride) Plastics by Thermal Desorption—Gas Chromatography/Mass Spectrometry, American Society for Testing and Materials, 2020 Search PubMed.
- G. Mitchell, C. Higgitt and L. T. Gibson, Polym. Degrad. Stabil., 2014, 107, 328–340 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |