
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating the coordination environment of a metal–organic framework for an efficient electrocatalytic oxygen evolution reaction†
Enjun
Lv
a,
Jiayi
Yong
a,
Jinguli
Wen
a,
Zhirong
Song
a,
Yi
Liu
 b,
Usman
Khan
*a and
Junkuo
Gao
*a
b,
Usman
Khan
*a and
Junkuo
Gao
*a
aInstitute of Functional Porous Materials, School of Materials Science and Engineering, Zhejiang Sci-Tech University, Hangzhou 310018, China. E-mail: usman.cssp@hotmail.com; jkgao@zstu.edu.cn
bInstitute for Composites Science Innovation, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, P. R. China
First published on 12th August 2022
Abstract
The oxygen evolution reaction (OER) plays a vital role in fuel cells, water splitting and metal–air batteries. Efficient electrocatalysts could overcome the higher overpotential of the OER, improve electron transfer efficiency, and promote water decomposition. In this work, a novel metal–organic framework (MOF) with efficient OER electrocatalytic performance (defined as FeCo-L1L2) was successfully prepared by a free assembly of metal ions (Fe and Co), 2,5-dihydroxyterephthalic acid (defined as L1) and 4,6-dihydroxyisophthalic acid (defined as L2). The overpotential was only 283 mV at the current density conditions of 10 mA cm−2 with a Tafel slope of 31.6 mV dec−1. Its excellent OER performance is attributed to the synergistic effect of the bimetals of FeCo-L1L2 and the coordination environment optimization created by the dual ligands. This work not only improved the catalytic performance of MOFs in the OER but also proposed a new strategy for the structural design of MOFs.
Introduction
With the rapid increase in environmental pollution and energy crisis, it is essential to advance eco-friendly and efficient energy storage and conversion systems. Various electrochemical devices, including fuel cells, water electrolyzers and metal–air batteries, have achieved considerable attention because of their environmental friendliness and high energy conversion efficiency. Hydrogen energy is considered to be the most efficient green energy with development potential in the future. However, large-scale production and storage of hydrogen energy through low-cost and high-yield industrial production is still a considerable challenge.1 Although hydrogen energy can be obtained from non-renewable fossil fuels through traditional petrochemical methods, at the same time, inefficient conversion methods and resource crisis of non-renewable fossil fuels are inevitable.2 In contrast, electrochemical water splitting to produce H2 has been considered to be the most promising approach.3–5 Electrochemical water splitting mainly includes the anodic oxygen evolution reaction (OER) and the cathodic hydrogen evolution reaction (HER).6 These are the two most crucial core half reaction steps in electrochemical water splitting.7 To overcome the overpotential (the theoretical overpotential ∼1.23 V) caused by the activation energy barrier, a higher potential is required to trigger the initial reaction.8 At present, electrocatalysts based on precious metals, such as IrO2/RuO2 (OER) and Pt (HER), have higher catalytic performance. However, precious metals have poor durability and high cost as catalysts,9 which significantly limits the development and application of precious metal-based electrocatalysts.10 Therefore, to improve the efficiency of electrocatalysis and reduce costs, researchers are actively seeking efficient non-noble metal electrocatalysts.11–13The reports in recent years have proved conclusively that earth-abundant 3d transition metals (3d TMs) (e.g., Ni, Co, and Fe) have enormous catalytic potential.14–16 For example, the catalytic performance of transition metal-based oxides is comparable to that of IrO2, and transition metal-based carbides have extremely excellent catalytic stability.17,18 In addition, there are transition metal oxyhydroxides,19 sulfides,20,21 phosphides,22–24etc., and their good catalytic performance shows the possibility of replacing precious metal catalysts.25 Metal–organic frameworks (MOFs) have emerged as unique porous materials, and have been applied to numerous fields.26–31 The coordination of pore structure and functional sites32 can accelerate the reaction kinetics,33 and makes them an ideal platform for excellent electrocatalysts.34–39 However, in the structure of MOFs, the metal nodes are surrounded by organic linking groups,40 which limits their catalytic activity and conductivity;41 thereby, it is vital to construct these structures with a rational design.42–45 In terms of macrostructure control,46 Lou et al. typically construct a unique core–shell structure through chemical etching methods and other methods,47 which could provide a larger catalytic interface area,48 thereby significantly improving the electrocatalytic performance.49 Li et al. used carbonization to precisely regulate Hofmann-type MOFs into different forms (including nanosheets, nanoflowers,50 nanotubes and aggregates), which OER catalytic performance far exceeds that of commercial RuO2 catalysts.51 In terms of microstructure design, doping heterogeneous metal atoms and different ligands into the crystal lattice can improve the electronic structure.52–54 The latter is more challenging,55 and the electrocatalytic performance of MOFs needs to be enhanced to the next level.56
To deal with the above challenges, we have synthesized a bimetal–organic framework (FeCo-L1L2) using a simple and cost-effective solvothermal method using the free assembly of Fe and Co ions with 2,5-dihydroxyterephthalic acid (defined as L1) and 4,6-dihydroxyisophthalic acid (defined as L2). The synthetic strategy is shown in Scheme 1. The synthesis process is simple and does not require additional modification and processing. In the structure of FeCo-L1L2, the isomeric ligand L2 replaces the partial ligand L1 to coordinate with the metal ion, causing the coordination environment of the metal center to change. Moreover, the synergistic effect of the bimetal also provides abundant metal active sites for the FeCo-L1L2 structure. Therefore, FeCo-L1L2 shows excellent OER activity in 1.0 M KOH. Under the ultra-low current density conditions of 10 mA cm−2, the overpotential is only 283 mV, the Tafel slope is 31.6 mV dec−1, and the long-lasting stability was excellent. This work not only improved the catalytic performance of MOFs in OER, but also proposed a new strategy for the structural design of MOFs.
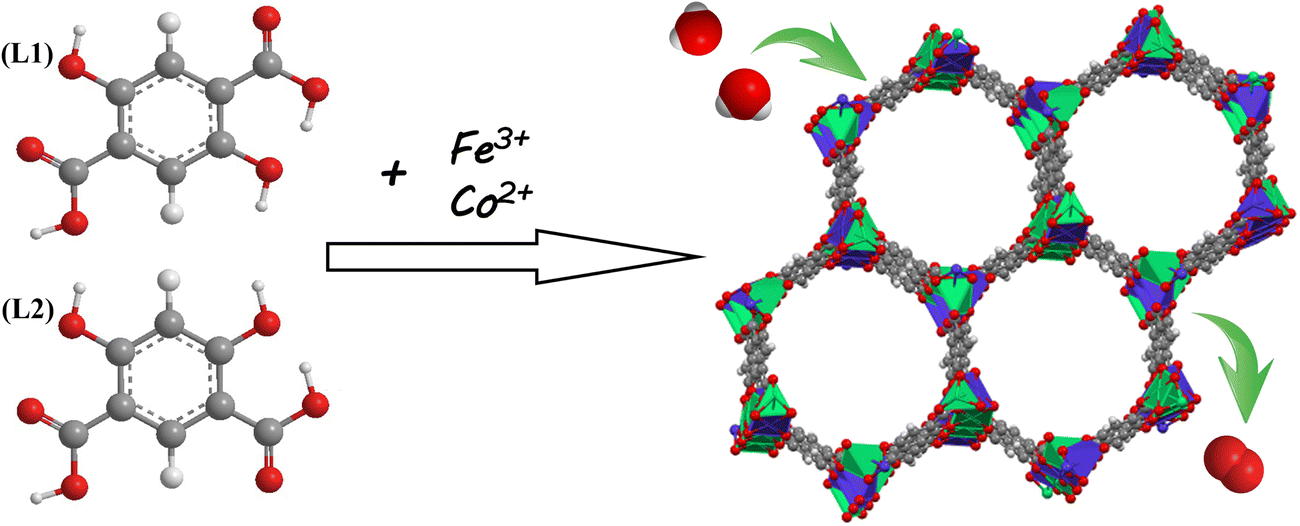 | ||
| Scheme 1 Schematic illustration of the synthesis of FeCo-L1L2 electrocatalysis. | ||
Results and discussion
The scanning electron microscopy (SEM) images in Fig. 1 show that FeCo-L1, FeCo-L2, and FeCo-L1L2 have different surface morphologies. As shown in Fig. 1, the morphology of FeCo-L1 comprises about 1 μm sized uniform spindle-shaped particles. FeCo-L2 comprises nano-particles with no specific morphology, and it is easy to aggregate into clusters. In particular, FeCo-L1L2 is a nano-acicular cluster. To further explore the microstructure of FeCo-L1L2, we conducted a detailed study using transmission electron microscopy (TEM). Fig. 1d shows a low-magnification image of the nano-acicular structure from FeCo-L1L2, which demonstrated that the nano-acicular whisker constructed together. The longer the nano-acicular structure, the sharper the top. It is judged by Fig. 1e that the top size of the nano-acicular structure is less than 20 nm. Furthermore, from the corresponding energy dispersive spectrometer (EDS) elemental mapping images (Fig. S1, ESI†), it can be observed that Fe and Co elements are uniformly distributed. It indicates that the metal ions Fe and Co are uniformly incorporated into the structure of FeCo-L1L2. In addition, the atomic ratio of Fe and Co in the FeCo-L1L2 sample was analyzed using the EDS spectrum (Fig. S2, ESI†), and the result showed that the exact atomic ratio of Fe/Co was 4.79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.93, which was close to 1:1. Since the morphology and microstructure of materials have a significant influence on their properties, we designed different atomic ratios of Fe, Co or ligands L1 and L2, and synthesized different ratios of FeCo-L1L2 by similar solvothermal synthesis. According to SEM images of FeCo-L1L2 with different metal atom ratios or different ligand ratios (Fig. S3, S4, ESI†), it could be found that the morphology of the crystallite changed with the ratio.57 While maintaining a specific ratio of dual ligands, the more Co it contains, the product is more to be like a globular sea urchin.58,59 In contrast, the more Fe it contains, the more the product resembles a nano-particle. When maintaining a specific ratio of bimetallic atoms, the more ligand L1 it contains, the more likely the product is to resemble a spindle-shaped particle. However, the more isomeric ligand L2 it contains, the more amorphous the product is. Therefore, when the atomic ratio of the metal ions Fe and Co and the ratio of ligands L1 and L2 both are close to 1:1, the reaction product is a uniformly nano-acicular cluster.60 Compared with the morphology of other ratios, the nano-acicular structure of FeCo-L1L2 possesses the tip-enhanced effect, which enhances the local electric field and is conducive to promoting mass transfer.61 The nano-acicular structure of FeCo-L1L2 possesses a greater tip-enhanced effect than the morphology of other ratios, which enhances the local electric field and is conducive to promoting mass transfer. The tip-enhanced effect is contributes to improving the OER performance. Thus, the nano-acicular structure does have advantages for enhancing the OER performance of FeCo-L1L2.
:4.93, which was close to 1:1. Since the morphology and microstructure of materials have a significant influence on their properties, we designed different atomic ratios of Fe, Co or ligands L1 and L2, and synthesized different ratios of FeCo-L1L2 by similar solvothermal synthesis. According to SEM images of FeCo-L1L2 with different metal atom ratios or different ligand ratios (Fig. S3, S4, ESI†), it could be found that the morphology of the crystallite changed with the ratio.57 While maintaining a specific ratio of dual ligands, the more Co it contains, the product is more to be like a globular sea urchin.58,59 In contrast, the more Fe it contains, the more the product resembles a nano-particle. When maintaining a specific ratio of bimetallic atoms, the more ligand L1 it contains, the more likely the product is to resemble a spindle-shaped particle. However, the more isomeric ligand L2 it contains, the more amorphous the product is. Therefore, when the atomic ratio of the metal ions Fe and Co and the ratio of ligands L1 and L2 both are close to 1:1, the reaction product is a uniformly nano-acicular cluster.60 Compared with the morphology of other ratios, the nano-acicular structure of FeCo-L1L2 possesses the tip-enhanced effect, which enhances the local electric field and is conducive to promoting mass transfer.61 The nano-acicular structure of FeCo-L1L2 possesses a greater tip-enhanced effect than the morphology of other ratios, which enhances the local electric field and is conducive to promoting mass transfer. The tip-enhanced effect is contributes to improving the OER performance. Thus, the nano-acicular structure does have advantages for enhancing the OER performance of FeCo-L1L2.
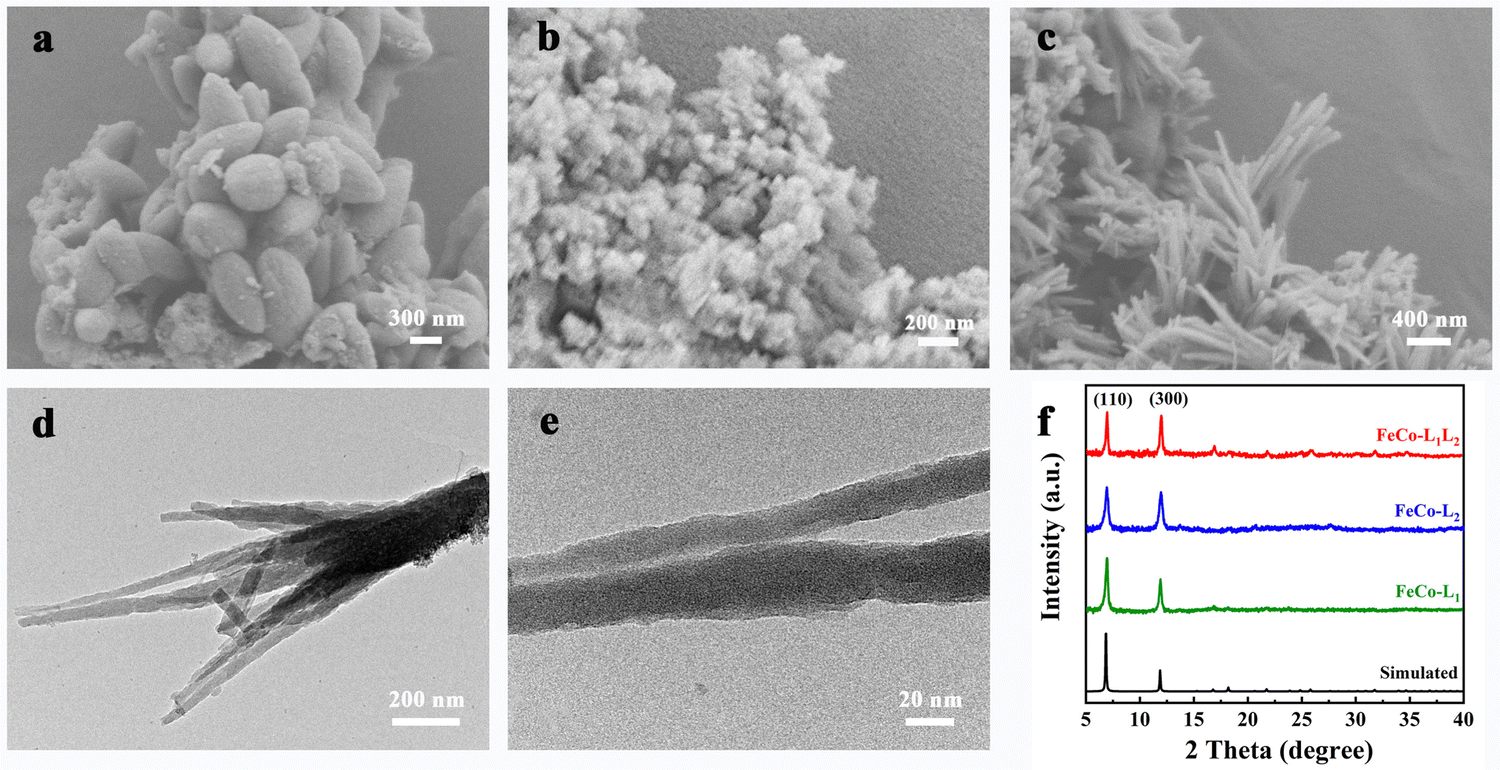 | ||
| Fig. 1 (a) SEM image of FeCo-L1, (b) SEM image of FeCo-L2, (c) SEM images and (d) (e) TEM images of FeCo-L1L2, (f) XRD patterns of the as-synthesized FeCo-L1, FeCo-L2, and FeCo-L1L2 with the corresponding simulated patterns. | ||
The crystal structures of the prepared FeCo-L1, FeCo-L2 and FeCo-L1L2 were analyzed by powder X-ray diffraction (XRD). As shown in Fig. 1f, the diffraction peaks of FeCo-L1, FeCo-L2 and FeCo-L1L2 at 7.0° and 12.0° correspond to the main peak positions of the corresponding simulated MOF-74. The PXRD results confirmed that FeCo-L1, FeCo-L2 and FeCo-L1L2 all were the isomorphs of MOF-74. Comparing the XRD patterns of FeCo-L1L2 with FeCo-L1 and FeCo-L2, it is found that adding isomeric ligand L2 did not change the original crystal structure. However, the diffraction peak intensity of FeCo-L1L2 at 7.0° and 12.0° decreased slightly. It might because that ligand L2 replaced the position of partial body L1, which caused some structural defects in the original crystalline form, resulting in a decrease in crystal crystallinity. Comparing Fe-L1L2 and Co-L1L2 with the corresponding simulated MOF-74 (Fig. S5a, ESI†), shows that the structure composed of single metal and double ligand coordination is the same as the structure consisting of double metals and double ligands coordination. It proves that no matter the incorporation of a metal or a ligand, a simple coordination substitution has occurred, and the original crystal structure of MOF-74 has not been changed. The presence of surface functional groups in FeCo-L1, FeCo-L2 and FeCo-L1L2 was demonstrated via Fourier transform infrared (FT-IR) spectroscopy (Fig. S5b, ESI†). The peak observed at 3700–3000 cm−1 can be ascribed to the O–H vibration. Due to the hydrogen bond formed between the carbonyl group and the hydroxyl group, the O–H stretching band range is enlarged. The νC–H peak appears at the tail of the νO–H broad peak, and the O–H and C–H stretching bands are superimposed. When the carbonyl group is conjugated with the benzene ring, the π electron cloud density in the system tends to be more uniform, making the single bond shorter and the double bond elongated, and the characteristic frequency appears to shift to a lower band. Therefore, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration showed a strong absorption band at 1750–1480 cm−1. FeCo-L1 has a strong absorption band at 1580 cm−1. FeCo-L2 showed two similar symmetrical rabbit ear peaks at 1625 cm−1 and 1560 cm−1. It may be due to the difference in the carboxyl substitution position on the benzene ring, which makes the peak shape different. The shape of the corresponding peak in the FeCo-L1L2 band combined the characteristics of FeCo-L1 and FeCo-L2. The symmetrical rabbit ear peak is weakened, but it still maintains a good peak intensity. It indicates that both ligand L1 and ligand L2 exist in the structure of FeCo-L1L2, further proving that FeCo-L1L2 was successfully synthesized.
O vibration showed a strong absorption band at 1750–1480 cm−1. FeCo-L1 has a strong absorption band at 1580 cm−1. FeCo-L2 showed two similar symmetrical rabbit ear peaks at 1625 cm−1 and 1560 cm−1. It may be due to the difference in the carboxyl substitution position on the benzene ring, which makes the peak shape different. The shape of the corresponding peak in the FeCo-L1L2 band combined the characteristics of FeCo-L1 and FeCo-L2. The symmetrical rabbit ear peak is weakened, but it still maintains a good peak intensity. It indicates that both ligand L1 and ligand L2 exist in the structure of FeCo-L1L2, further proving that FeCo-L1L2 was successfully synthesized.
To gain insight into the valence engineering of FeCo-L1, FeCo-L2 and FeCo-L1L2, the structural and electronic properties of FeCo-L1, FeCo-L2 and FeCo-L1L2 were compared by X-ray photoelectron spectroscopy (XPS) (Fig. S6, ESI†). The full range XPS spectra proved the presence of Fe, Co, C, O and N elements in FeCo-L1, FeCo-L2 and FeCo-L1L2. Moreover, the chemical states of Fe and Co in FeCo-L1, FeCo-L2 and FeCo-L1L2 are very similar in Fig. 2. The Fe 2p spectrum of FeCo-L1L2 shows two main peaks at 710.87 and 724.37 eV.62,63 Compared with FeCo-L1 and FeCo-L2, the binding energy of Fe 2p3/2 in FeCo-L1L2 is reduced by about 0.3 eV (Fig. S7a, ESI†). In particular, the Co 2p spectrum of FeCo-L1L2 shows two main peaks at 781.08 and 796.80 eV.64 Compared with FeCo-L1 and FeCo-L2, the binding energy of Co 2p3/2 in FeCo-L1L2 is also reduced by 0.16 eV (Fig. S8b, ESI†). The isomeric ligand L2 replaces the partial ligand L1 to coordinate with the metal ion, which causes the coordination environment of the metal center to change.65 For further confirmation, the Fe 2p spectrum and the Co 2p spectrum of Fe-L1L2, Co-L1L2 and FeCo-L1L2 were compared. It was found that the binding energy of Fe 2p3/2 and Co 2p3/2 did not decrease. Therefore, incorporating isomeric ligands can change the coordination environment of metal centers and enhance the coupling of Fe and Co in the bimetal FeCo-L1L2. It is worth noting that the synergistic effect of bimetals has been recognized by the researchers as having a vital role in improving the catalytic performance of the OER,66–68 and the substitution coordination of isomeric ligands still needs theoretical analysis.69 On the other hand, quantitative analyses of FeCo-L1, FeCo-L2 and FeCo-L1L2 from XPS are consistent with the results of EDS spectrum atomic analysis, which all prove that the atomic ratio of Fe and Co is approximately 1:1 (Table S1, ESI†).
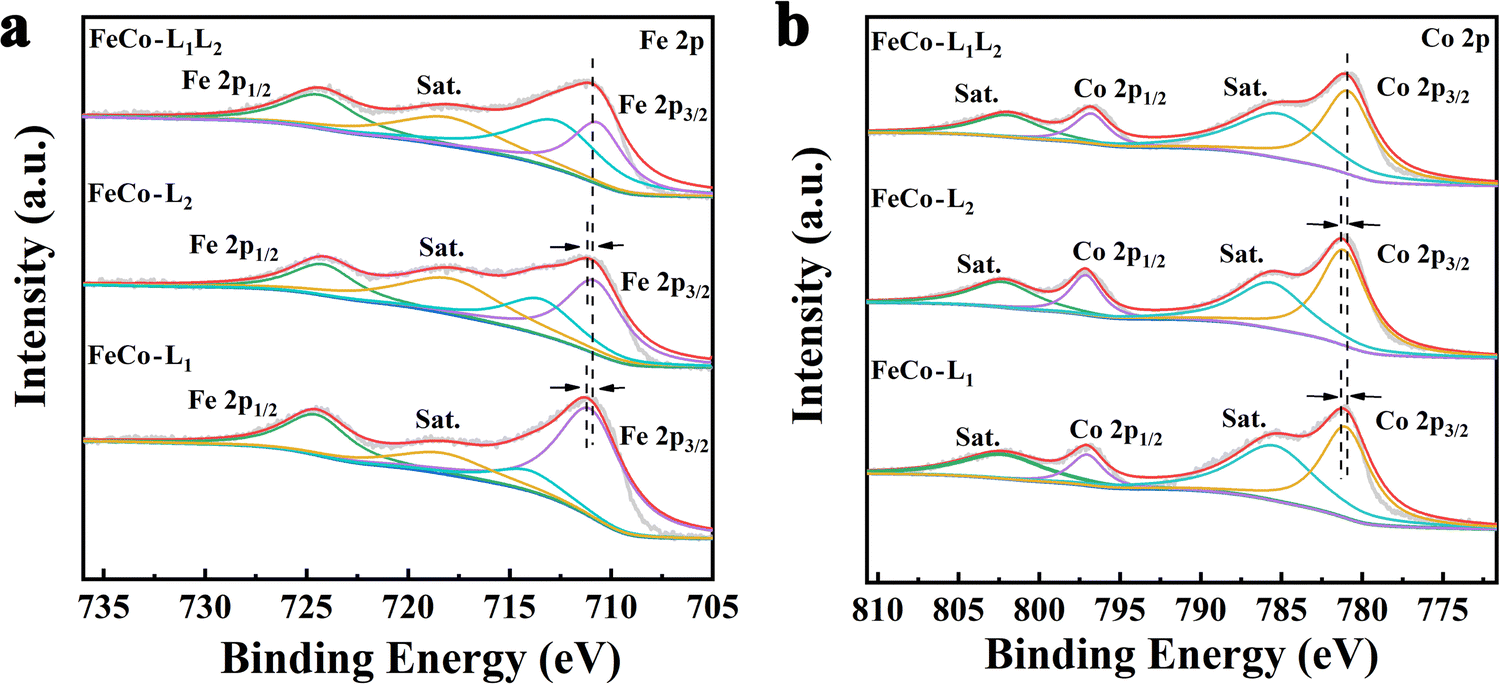 | ||
| Fig. 2 X-Ray photoelectron spectroscopy (XPS) spectra of (a) Fe 2p and (b) Co 2p for FeCo-L1, FeCo-L2 and FeCo-L1L2. | ||
Electrochemical measurements were carried out in a standard three-electrode system in an alkaline medium (1.0 M KOH solution), and the samples were utilized as catalysts directly by depositing onto a glassy-carbon electrode (GCE, geometric area: 0.07 cm−2). Linear sweep voltammetry (LSV) tests were performed on the prepared samples and commercial precious metal catalysts (IrO2) at a scanning speed of 10 mV s−1 to characterize the OER activity. Fig. 3a and b show that the overpotentials for FeCo-L1, FeCo-L2 and FeCo-L1L2, Co-L1L2, and commercial IrO2 at 10 mA cm−2 are 366 mV, 353 mV, 283 mV, 321 mV and 309 mV, respectively. In particular, the FeCo-L1L2 displayed the best OER performance, which is 26 mV better than commercial IrO2. Moreover, the overpotential at 50 mV cm−2 for FeCo-L1L2 is only 314 mV, which is far below those overpotentials of FeCo-L1, FeCo-L2, and Co-L1L2 at 50 mV cm−2. Fig. S9 (ESI†) further supplements the LSV curves of a single metal and a single ligand. The comparison found that after the introduction of Co ions in Fe-L1 and Fe-L2, a heterogeneous bimetal FeCo-L1 and FeCo-L2. With the synergistic effect of mixed metals, its OER catalytic activity is optimized. Interestingly, after adding an isomeric ligand L2 to Co-L1, the substitution of ligand L2 connects some metal ions to enhance the catalytic effect of Co-L1L2. Therefore, we found that whether it is the introduction of Co ions into Fe-L1L2 or the addition of ligand L2 to FeCo-L1, FeCo-L1L2 can enhance the OER catalytic performance.
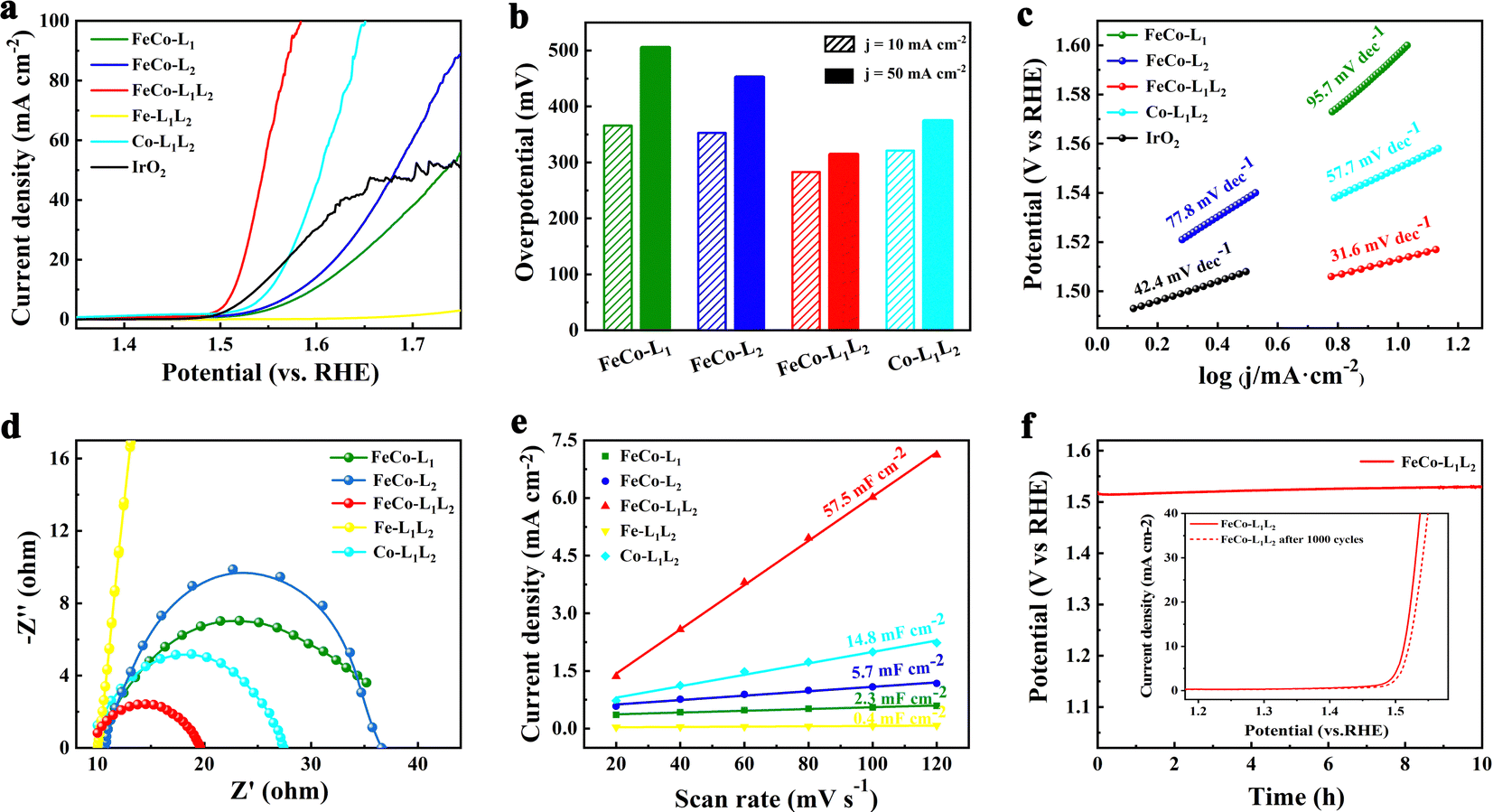 | ||
| Fig. 3 OER performance of various samples. (a) Polarization curves (5 mV s−1) in 1.0 M KOH solution and (b) overpotentials required for 10 mA cm−2 and 50 mA cm−2. (c) Tafel plots. (d) Electrochemical impedance plots. (e) Capacitive currents as a function of the scan rate to give the double-layer capacitance (Cdl). (f) Chronopotentiometry curves of FeCo-L1L2 for 10 h at 10 mA cm−2 in 1.0 M KOH solution. Inset: LSV curves before and after 1000 cycles for FeCo-L1L2. | ||
The Tafel slope further evaluated the OER kinetics of electrocatalysts, as shown in Fig. 3c. The results of the Tafel slope are consistent with LSV, and FeCo-L1L2 has a superior Tafel slope (31.6 mV dec−1), which is lower than that of most of the reported OER catalysts. Electrochemical impedance spectroscopy (EIS) tests were performed to explore the charge transport dynamics further. Fig. 3d shows that the Rct is about 10 Ω for FeCo-L1L2, which the Nyquist semicircle is much smaller than other catalyst samples. It indicates that FeCo-L1L2 has a lower mass transfer resistance and a faster reaction rate. The double-layer capacitance (Cdl) was calculated through the measured cyclic voltammogram (CV) to explore the electrochemical active surface area (ECSA) of the samples. Each sample was scanned at 20, 40, 60, 80, 100 and 120 mV s−1 to get different CV plots (Fig. S10, ESI†). As shown in Fig. 3e, it is clear that FeCo-L1L2 has the largest Cdl (57.5 mF cm−2). The addition of another ligand could significantly improve the Cdl of FeCo-L1L2. It shows that under a specific catalytic area, the number of active sites in FeCo-L1L2 increases, which means the density of active sites increases. Fig. 3f shows the chronopotential curve tested at a constant current density (10 mA cm−2) to evaluate the durability and stability of FeCo-L1L2. After FeCo-L1L2 worked stably for 10 hours, the crystal structure and morphology showed very limited changes (Fig. S11, ESI†) and retained 98.9% of the initial potential. In addition, the LSV curve of FeCo-L1L2 only slightly changed after 1000 CVs, which depicts the superior and long-term stability of the obtained catalyst.
To optimize the ratio of bimetals to the dual ligands and design FeCo-L1L2 with the best catalytic performance, we performed linear sweep voltammetry (LSV) tests on FeCo-L1L2 samples with different metal ratios and ligand ratios (Fig. S12, ESI†). To more intuitively demonstrate the relationship in metal ratio, ligand ratio and catalytic performance, volcano-type plots of overpotential with different metal atom ratios and different ligand ratios at 10 mA cm−2 were obtained, as shown in Fig. 4. When a single metal or ligand FeCo-L1L2 is incorporated with heterogeneous metal atoms or isomeric ligands, the OER catalytic performance of FeCo-L1L2 changes significantly. The incorporation of heterogeneous metal atoms improves the electronic structure, so the catalytic activity of FeCo-L1L2 OER, which has the synergistic effect of bimetal, is significantly enhanced. Unexpectedly, adding isomeric ligands replaced part of the in situ ligands, optimized the coordination environment, and further improved the OER catalytic performance of FeCo-L1L2. According to Fig. 4, it can be found that the OER performance of FeCo-L1L2 is the best when the ratio of bimetal and the ratio of double ligand are close to 1:1. At the ratio of 1:1, the morphology and electronic structures of the electrocatalysts were optimized, resulting to the best OER performance.
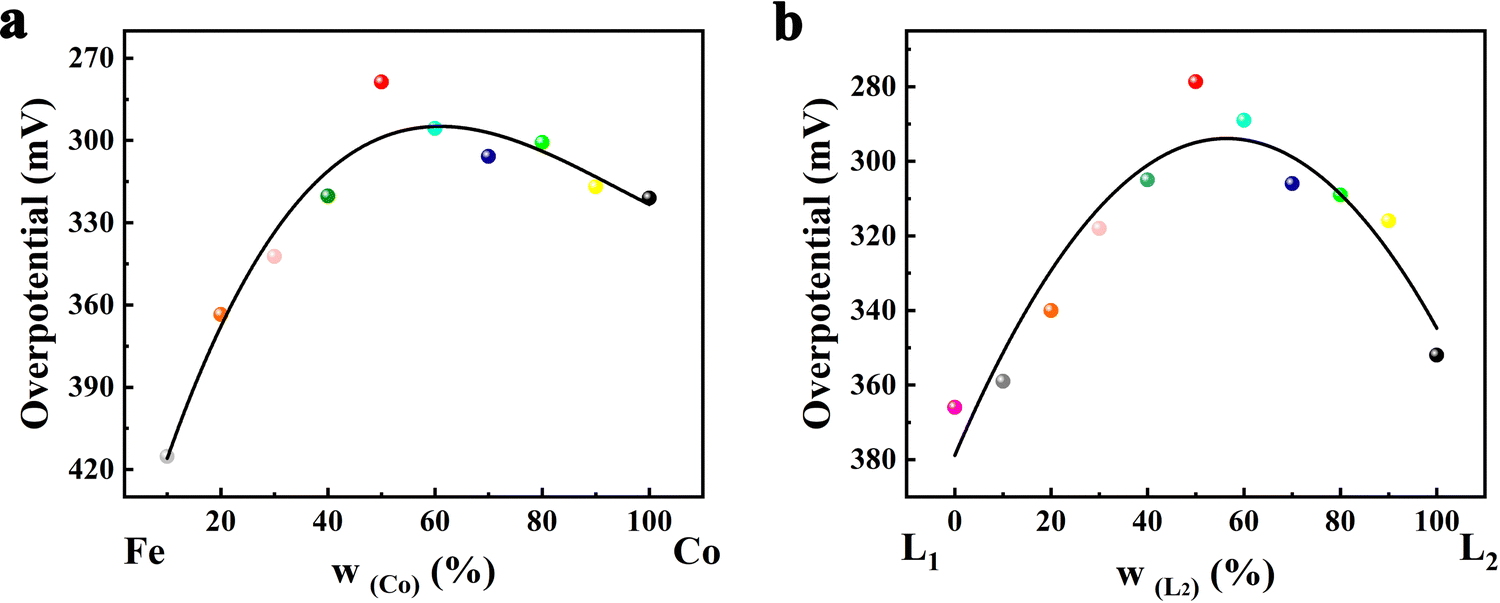 | ||
| Fig. 4 Volcano-type plots of overpotential with (a) different metal atom ratios and (b) different ligand ratios at 10 mA cm−2. | ||
Conclusions
We successfully prepare a novel bimetal MOF with high-efficiency OER catalytic performance by the free assembly of metal ions Fe, Co, 2,5-dihydroxyterephthalic acid (L1), and 4,6-dihydroxyisophthalic acid (L2). In the structure of FeCo-L1L2, the isomeric ligand L2 replaces the partial ligand L1 to coordinate with the metal ion, causing the coordination environment of the metal center to change. Moreover, the synergistic effect of the bimetals also provides abundant metal active sites for the FeCo-L1L2 structure. Therefore, FeCo-L1L2 shows excellent OER activity in 1.0 M KOH. Under the ultra-low current density conditions of 10 mA cm−2, the overpotential is only 283 mV, the Tafel slope is 31.6 mV dec−1, and the long-lasting stability is excellent. This work not only improved the catalytic performance of MOFs in OER, but also proposed a new strategy for the structural design of MOFs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (LY20E020001) and the National Natural Science Foundation of China (51602301 and 51672251).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- N. Hanikel, M. S. Prevot and O. M. Yaghi, Nat. Nanotechnol., 2020, 15, 348–355 CrossRef CAS PubMed.
- X. Fang, L. Jiao, R. Zhang and H.-L. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 23852–23858 CrossRef CAS PubMed.
- G. Cai, P. Yan, L. Zhang, H.-C. Zhou and H.-L. Jiang, Chem. Rev., 2021, 121, 12278–12326 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- C. Chen, Y. Tuo, Q. Lu, H. Lu, S. Zhang, Y. Zhou, J. Zhang, Z. Liu, Z. Kang, X. Feng and D. Chen, Appl. Catal., B, 2021, 287, 119953 CrossRef CAS.
- S. Sultan, J. N. Tiwari, A. N. Singh, S. Zhumagali, M. Ha, C. W. Myung, P. Thangavel and K. S. Kim, Adv. Energy Mater., 2019, 9, 1900624 CrossRef.
- H. Liu, X. Lu, Y. Hu, R. Chen, P. Zhao, L. Wang, G. Zhu, L. Ma and Z. Jin, J. Mater. Chem. A, 2019, 7, 12489–12497 RSC.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, M. Parlinska-Wojtan, X. Pei, O. M. Yaghi and R. J. Behm, J. Am. Chem. Soc., 2019, 141, 5201–5210 CrossRef CAS PubMed.
- K. Chakarawet, M. Atanasov, J. Marbey, P. C. Bunting, F. Neese, S. Hill and J. R. Long, J. Am. Chem. Soc., 2020, 142, 19161–19169 CrossRef CAS PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2019, 32, 1806326 CrossRef PubMed.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef PubMed.
- G. Chen, H. Wan, W. Ma, N. Zhang, Y. Cao, X. Liu, J. Wang and R. Ma, Adv. Energy Mater., 2019, 10, 1902535 CrossRef.
- Y. Liu, Z. Sun, X. Sun, Y. Lin, K. Tan, J. Sun, L. Liang, L. Hou and C. Yuan, Angew. Chem., Int. Ed., 2020, 59, 2473–2482 CrossRef CAS.
- T. Liu, P. Li, N. Yao, T. Kong, G. Cheng, S. Chen and W. Luo, Adv. Mater., 2019, 31, 1806672 CrossRef.
- S. Gao, M. Yang, S. Li, J. Xia, J. Mei, S. Xie and H. Liu, Sustainable Energy Fuels, 2019, 3, 823–830 RSC.
- T. Liu, P. Li, N. Yao, G. Cheng, S. Chen, W. Luo and Y. Yin, Angew. Chem., Int. Ed., 2019, 58, 4679–4684 CrossRef CAS PubMed.
- L. Guo, X. Bai, H. Xue, J. Sun, T. Song, S. Zhang, L. Qin, K. Huang, F. He and Q. Wang, Chem. Commun., 2020, 56, 7702–7705 RSC.
- J. Gao, Q. Huang, Y. Wu, Y.-Q. Lan and B. Chen, Adv. Energy Sustainability Res., 2021, 2, 2100033 CrossRef.
- W. Xu and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1348–1354 CrossRef CAS PubMed.
- Z. Ji, H. Wang, S. Canossa, S. Wuttke and O. M. Yaghi, Adv. Funct. Mater., 2020, 30, 2000238 CrossRef CAS.
- N. Hanikel, M. S. Prevot, F. Fathieh, E. A. Kapustin, H. Lyu, H. Wang, N. J. Diercks, T. G. Glover and O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1699–1706 CrossRef CAS PubMed.
- Z. L. Ji, T. Li and O. M. Yaghi, Science, 2020, 369, 674–680 CrossRef CAS PubMed.
- M. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- C. A. Trickett, T. M. Osborn Popp, J. Su, C. Yan, J. Weisberg, A. Huq, P. Urban, J. Jiang, M. J. Kalmutzki, Q. Liu, J. Baek, M. P. Head-Gordon, G. A. Somorjai, J. A. Reimer and O. M. Yaghi, Nat. Chem., 2019, 11, 170–176 CrossRef CAS PubMed.
- K. Ge, S. Sun, Y. Zhao, K. Yang, S. Wang, Z. Zhang, J. Cao, Y. Yang, Y. Zhang, M. Pan and L. Zhu, Angew. Chem., Int. Ed., 2021, 60, 12097–12102 CrossRef CAS PubMed.
- X. Ling, F. Du, Y. Zhang, Y. Shen, W. Gao, B. Zhou, Z. Wang, G. Li, T. Li, Q. Shen, Y. Xiong, X. Wang, Y. Zhou and Z. Zou, J. Mater. Chem. A, 2021, 9, 13271–13278 RSC.
- X. F. Lu, B. Y. Xia, S. Q. Zang and X. W. D. Lou, Angew. Chem., Int. Ed., 2020, 59, 4634–4650 CrossRef CAS PubMed.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 RSC.
- W. Li, S. Watzele, H. A. El-Sayed, Y. Liang, G. Kieslich, A. S. Bandarenka, K. Rodewald, B. Rieger and R. A. Fischer, J. Am. Chem. Soc., 2019, 141, 5926–5933 CrossRef CAS PubMed.
- L. Jiao, Y. Wang, H. L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- V. Y. Mao, P. J. Milner, J. H. Lee, A. C. Forse, E. J. Kim, R. L. Siegelman, C. M. McGuirk, L. B. Porter-Zasada, J. B. Neaton, J. A. Reimer and J. R. Long, Angew. Chem., Int. Ed., 2020, 59, 19468–19477 CrossRef CAS PubMed.
- J. S. Derrick, M. Loipersberger, R. Chatterjee, D. A. Iovan, P. T. Smith, K. Chakarawet, J. Yano, J. R. Long, M. Head-Gordon and C. J. Chang, J. Am. Chem. Soc., 2020, 142, 20489–20501 CrossRef CAS PubMed.
- M. Zhao, T. Guo, W. Qian, Z. Wang, X. Zhao, L. Wen and D. He, Chem. Eng. J., 2021, 422, 130055 CrossRef CAS.
- M. G. C. Lei Sun and M. Dinca, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef PubMed.
- U. Khan, A. Nairan, J. Gao and Q. Zhang, Small Struct., 2022 DOI:10.1002/sstr.202200109.
- X. Xiao, C.-T. He, S. Zhao, J. Li, W. Lin, Z. Yuan, Q. Zhang, S. Wang, L. Dai and D. Yu, Energy Environ. Sci., 2017, 10, 893–899 RSC.
- H. L. Nguyen, C. Gropp, Y. Ma, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 20335–20339 CrossRef CAS PubMed.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y. S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- W. Huang, J. Li, X. Liao, R. Lu, C. Ling, X. Liu, J. Meng, L. Qu, M. Lin, X. Hong, X. Zhou, S. Liu, Y. Zhao, L. Zhou and L. Mai, Adv. Mater., 2022, 34, 2200270 CrossRef CAS PubMed.
- L. Vallez, S. Jimenez-Villegas, A. T. Garcia-Esparza, Y. Jiang, S. Park, Q. Wu, T. M. Gill, D. Sokaras, S. Siahrostami and X. Zheng, Energy Adv., 2022, 1, 357–366 RSC.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CAS.
- J. Nai and X. W. Lou, Adv. Mater., 2018, 31, 1706825 CrossRef PubMed.
- Z. Li, S. Deng, H. Yu, Z. Yin, S. Qi, L. Yang, J. Lv, Z. Sun and M. Zhang, J. Mater. Chem. A, 2022, 10, 4230–4241 RSC.
- Y. Li, M. Lu, Y. Wu, Q. Ji, H. Xu, J. Gao, G. Qian and Q. Zhang, J. Mater. Chem. A, 2020, 8, 18215–18219 RSC.
- Z. Ji, J. Liu, Y. Deng, S. Zhang, Z. Zhang, P. Du, Y. Zhao and X. Lu, J. Mater. Chem. A, 2020, 8, 14680–14689 RSC.
- M. J. Wang, X. Zheng, L. Song, X. Feng, Q. Liao, J. Li, L. Li and Z. Wei, J. Mater. Chem. A, 2020, 8, 14145–14151 RSC.
- A. L. Jaffe and J. R. Long., Nature, 2020, 578, 222–223 CrossRef CAS PubMed.
- J. Zhou, Y. Hu, Y.-C. Chang, Z. Hu, Y.-C. Huang, Y. Fan, H.-J. Lin, C.-W. Pao, C.-L. Dong, J.-F. Lee, C.-T. Chen, J.-Q. Wang and L. Zhang, ACS Catal., 2022, 12, 3138–3148 CrossRef CAS.
- X. Zhou, J. Gao, Y. Hu, Z. Jin, K. Hu, K. M. Reddy, Q. Yuan, X. Lin and H.-J. Qiu, Nano Lett., 2022, 22, 3392–3399 CrossRef CAS PubMed.
- W. Zhou, D. D. Huang, Y. P. Wu, J. Zhao, T. Wu, J. Zhang, D. S. Li, C. Sun, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2019, 58, 4227–4231 CrossRef CAS PubMed.
- Y. Pan, R. Abazari, Y. Wu, J. Gao and Q. Zhang, Electrochem. Commun., 2021, 126, 107024 CrossRef CAS.
- Y. Wu, Y. Li, J. Gao and Q. Zhang, SusMat, 2021, 1, 66–87 CrossRef.
- H. Xu, F. Zhong, F. Chen, T.-X. Luan, P. Li, S. Xu and J. Gao, J. Mater. Chem. C, 2022, 10, 7469–7475 RSC.
- P. Liu, B. Chen, C. Liang, W. Yao, Y. Cui, S. Hu, P. Zou, H. Zhang, H. J. Fan and C. Yang, Adv. Mater., 2021, 33, 2007377 CrossRef CAS PubMed.
- S. Zhao, M. Li, M. Han, D. Xu, J. Yang, Y. Lin, N.-E. Shi, Y. Lu, R. Yang, B. Liu, Z. Dai and J. Bao, Adv. Funct. Mater., 2018, 28, 1706018 CrossRef.
- Q. Wang, F. Wei, D. Manoj, Z. Zhang, J. Xiao, X. Zhao, F. Xiao, H. Wang and S. Wang, Chem. Commun., 2019, 55, 11307–11310 RSC.
- T. Tang, W.-J. Jiang, S. Niu, N. Liu, H. Luo, Y.-Y. Chen, S.-F. Jin, F. Gao, L.-J. Wan and J.-S. Hu, J. Am. Chem. Soc., 2017, 139, 8320–8328 CrossRef CAS PubMed.
- Z. Xue, Y. Li, Y. Zhang, W. Geng, B. Jia, J. Tang, S. Bao, H.-P. Wang, Y. Fan, Z.-W. Wei, Z. Zhang, Z. Ke, G. Li and C.-Y. Su, Adv. Energy Mater., 2018, 8, 1801564 CrossRef.
- Y. Duan, S. Sun, S. Xi, X. Ren, Y. Zhou, G. Zhang, H. Yang, Y. Du and Z. J. Xu, Chem. Mater., 2017, 29, 10534–10541 CrossRef CAS.
- X. Zhao, B. Pattengale, D. Fan, Z. Zou, Y. Zhao, J. Du, J. Huang and C. Xu, ACS Energy Lett., 2018, 3, 2520–2526 CrossRef CAS.
- W. Li, W. Fang, C. Wu, K. N. Dinh, H. Ren, L. Zhao, C. Liu and Q. Yan, J. Mater. Chem. A, 2020, 8, 3658–3666 RSC.
- J. Gao, Y. Cai, X. Qian, P. Liu, H. Wu, W. Zhou, D.-X. Liu, L. Li, R.-B. Lin and B. Chen, Angew. Chem., Int. Ed., 2021, 60, 20400–20406 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00140c |
| This journal is © The Royal Society of Chemistry 2022 |