
Enhancing exciton diffusion by reducing energy disorder in organic solar cells†
Peiyao
Xue
a,
Adiel M.
Calascibetta
c,
Kai
Chen
d,
Karen E.
Thorn
d,
Yiting
Jiang
a,
Jiangjian
Shi
e,
Boyu
Jia
a,
Mengyang
Li
f,
Jingming
Xin
g,
Guilong
Cai
 h,
Rui
Yang
i,
Heng
Lu
a,
Sara
Mattiello
c,
Yao
Liu
i,
Zheng
Tang
f,
Wei
Ma
g,
Xinhui
Lu
h,
Qingbo
Meng
e,
Justin M.
Hodgkiss
d,
Luca
Beverina
c,
Ray P. S.
Han
j and
Xiaowei
Zhan
*ab
h,
Rui
Yang
i,
Heng
Lu
a,
Sara
Mattiello
c,
Yao
Liu
i,
Zheng
Tang
f,
Wei
Ma
g,
Xinhui
Lu
h,
Qingbo
Meng
e,
Justin M.
Hodgkiss
d,
Luca
Beverina
c,
Ray P. S.
Han
j and
Xiaowei
Zhan
*ab
aSchool of Materials Science and Engineering, Peking University, Beijing 100871, China. E-mail: xwzhan@pku.edu.cn
bKey Laboratory of Eco-functional Polymer Materials of Ministry of Education College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou 730070, China
cDepartment of Materials Science, State University of Milano-Bicocca, Via Cozzi 55, Milano, Italy
dMacDiarmid Institute for Advanced Materials and Nanotechnology, School of Chemical and Physical Sciences, Victoria University of Wellington, Wellington 6010, New Zealand
eCAS Key Laboratory for Renewable Energy, Beijing Key Laboratory for New Energy Materials and Devices, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
fCenter for Advanced Low-Dimension Materials, State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China
gState Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an 710049, China
hDepartment of Physics, The Chinese University of Hong Kong, New Territories 999077, Hong Kong, China
iBeijing Advanced Innovation Center for Soft Matter Science and Engineering, State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China
jJiangzhong Cancer Research Center, Jiangxi University of Chinese Medicine, Nanchang 330004, China
First published on 21st October 2022
Abstract
A highly crystalline, highly emissive, and wide-bandgap polymer AC174 with an extremely small Stokes shift is designed and synthesized in water, and is used to reduce system energetic disorder and increase the exciton diffusion length of the classical PM6:Y6 blend system. AC174 is incompatible with PM6 and Y6, improves molecular packing, and reduces system energetic disorder. The long-range Förster resonance energy transfer between the donor and acceptor is enhanced, and the exciton diffusion constant and exciton lifetime are increased, leading to a longer exciton diffusion length and more efficient exciton dissociation and charge generation. The addition of AC174 simultaneously improves the open-circuit voltage, short-circuit current density (JSC) and fill factor of PM6:Y6 devices; especially the highest internal quantum efficiency approaches 100%, and the highest JSC is 28.4 mA cm−2. Ternary devices with 5% AC174 in the donors achieve a power conversion efficiency of 17.2%, higher than those of the parent binary devices based on PM6:Y6 (15.9%) and AC174:Y6 (3.24%).
Introduction
Organic solar cells (OSCs) are regarded as a next-generation photovoltaic technology since they have some merits such as facile fabrication, light weight, flexibility, ecofriendliness and short energy payback time.1,2 Fused-ring electron acceptors (FREAs) pioneered by the Zhan group show strong light absorption in visible and near-infrared (NIR) regions, high electron mobility, low energy loss and stable film morphology,3–9 and have been widely used in high-performance OSCs.10–14 Thanks to the invention of FREAs, the maximum power conversion efficiencies (PCEs) of OSCs have exceeded 20%.15Organic semiconductors usually have low dielectric constants (ca. 2–4) and strong phonon–electron coupling, and therefore, light absorption of the photoactive layers generates strongly bound electron–hole pairs (excitons) with high binding energy (ca. 0.1–1 eV) instead of free charges.16 Then, excitons diffuse to donor/acceptor (D/A) heterojunction interfaces and dissociate into free charges driven by the energy offset between D/A.17,18 Because excitons created in organic semiconductors generally have short lifetimes and limited diffusion lengths (ca. 10–20 nm), intimate intermixing of the donor and acceptor for generating more D/A interfacial contacts is necessary for efficient exciton dissociation,19 but this leads to unfavourable charge transport and recombination. The trade-off between exciton dissociation and charge transport in OSCs requires complex morphology optimization of the photoactive layers.20–23 Thus, increasing the exciton diffusion length is essential for improving device performance.
The exciton diffusion length, LD, is given as  where D is the exciton diffusion constant, and τ is the exciton lifetime.24,25 The lifetime of singlet excitons is determined by radiative and non-radiative decay processes and can be expressed as τ = 1/(kR + kNR), where kR is the rate of radiative decay and kNR is the rate of non-radiative decay. According to the energy gap law, kNR correlates with the energy gap between the singlet ground state (S0) and the lowest-energy singlet excited state (S1); a narrower energy gap leads to a higher kNR and shorter exciton lifetime.26–28 In high-efficiency OSCs, narrow-bandgap (Eg) semiconductor materials (Eg < 1.5 eV)29 are commonly used to improve the light absorption in the NIR region. Accordingly, it is challenging to decrease the non-radiative decay and increase the exciton diffusion length of narrow-bandgap photovoltaic materials.
where D is the exciton diffusion constant, and τ is the exciton lifetime.24,25 The lifetime of singlet excitons is determined by radiative and non-radiative decay processes and can be expressed as τ = 1/(kR + kNR), where kR is the rate of radiative decay and kNR is the rate of non-radiative decay. According to the energy gap law, kNR correlates with the energy gap between the singlet ground state (S0) and the lowest-energy singlet excited state (S1); a narrower energy gap leads to a higher kNR and shorter exciton lifetime.26–28 In high-efficiency OSCs, narrow-bandgap (Eg) semiconductor materials (Eg < 1.5 eV)29 are commonly used to improve the light absorption in the NIR region. Accordingly, it is challenging to decrease the non-radiative decay and increase the exciton diffusion length of narrow-bandgap photovoltaic materials.
To solve this problem, understanding the exciton transport mechanism is critical. The dominating exciton transport mechanism in OSCs is Förster resonance energy transfer (FRET), where the FRET radius correlates with system energetic disorder.30,31 In the FRET model, excitons predominantly hop between aligned chromophores as they diffuse. If the energy disorder increases, the fraction of chromophores that are energetically available for energy transfer will decrease, leading to a smaller FRET radius. The reduced FRET caused by the high systematic energetic disorder limits the exciton diffusion constant. Moreover, higher energy disorder leads to increased non-radiative relaxation,32–35 which reduces the exciton lifetime according to the energy gap law. A decrease in the exciton diffusion constant and exciton lifetime leads to a short exciton diffusion length. Therefore, reducing system energetic disorder is the key to promote the FRET and minimize non-radiative decay, and finally increase the exciton diffusion length in OSC devices.
In this work, we designed and synthesized a highly crystalline wide-bandgap polymer donor, AC174 (Fig. 1a), and used it to reduce energetic disorder and enhance exciton diffusion in the representative PM6:Y6-based OSCs. AC174 is incompatible with PM6 and Y6, and tends to form an individual phase in the PM6:AC174:Y6 ternary blend. Adding a small amount of AC174 can not only maintain good D/A phase separation, which is beneficial for charge transport, but can also improve PM6/Y6 molecular packing and decrease system energetic disorder. Due to the reduced energetic disorder, the exciton diffusion ability and exciton lifetime increase, leading to an enhanced exciton diffusion length and exciton dissociation. As a result, the ternary OSCs based on PM6:AC174:Y6 exhibit a higher PCE value of 17.2% compared with the PM6:Y6 devices (15.9%), and achieve a greatly increased short-circuit current density (JSC) over 28 mA cm−2, which is among the highest JSC values in OSC devices.
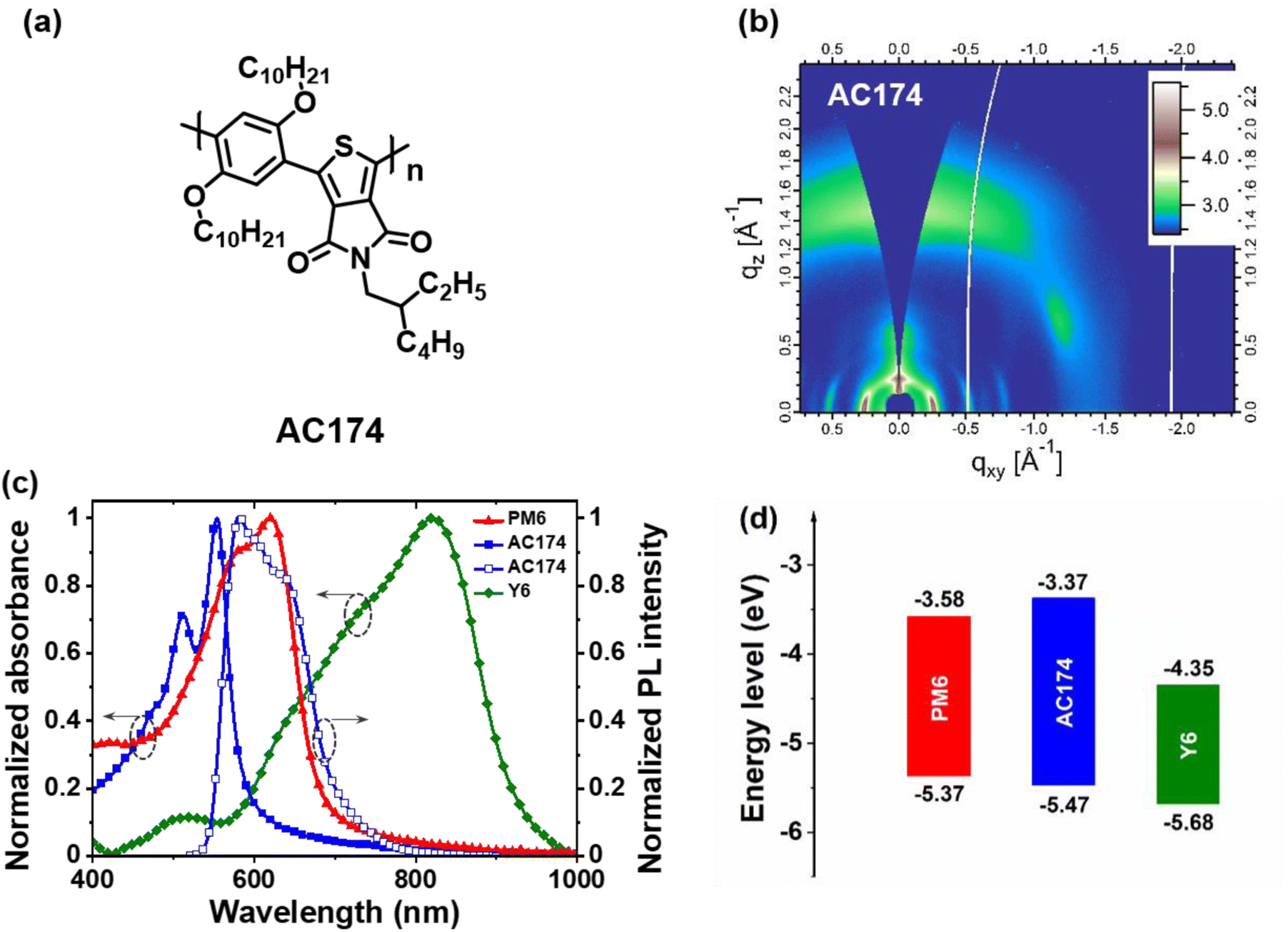 | ||
| Fig. 1 (a) Chemical structure of AC174. (b) GIWAXS pattern of the pure AC174 film. (c) Normalized absorption spectra of PM6, AC174 and Y6 films and the emission spectrum of the AC174 film. (d) Energy level diagram of PM6, AC174 and Y6. | ||
Results and discussion
Materials synthesis and characterization
AC174 was synthesized through Suzuki–Miyaura micellar cross-coupling in water. This particular protocol enables a large reduction in the amount of organic wastes generated by the manufacturing of organic semiconductors.36 The detailed synthesis procedure is described in the ESI† and the nuclear magnetic resonance spectrum of AC174 is shown in Fig. S1 (ESI†). First, we investigated the basic properties of AC174. AC174 exhibits intense π–π stacking in the out-of-plane direction (qz = 1.45 Å−1, d = 4.32 Å) with a face-on molecular orientation, indicating that AC174 is highly crystalline, as shown by the grazing incidence wide-angle X-ray scattering (GIWAXS) method (Fig. 1b). AC174 in diluted dichloromethane solution shows an intense absorption ranging from 300 to 550 nm with an absorption peak at 488 nm; its solid film exhibits an absorption peak at 554 nm, and the optical bandgap (Eg) is calculated to be 2.10 eV (Fig. S2, ESI†). The AC174 solid film shows a strong fluorescence peak at 589 nm. Quite a small Stokes shift of 35 nm is observed, indicating a rigid backbone and a low reorganization energy of AC174, which facilitates reducing energetic disorder and enhancing charge transport according to the Marcus theory.37 The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of AC174 are calculated to be −5.47 eV and −3.37 eV, respectively, according to the ultraviolet photoelectron spectrometer (UPS) spectra and UV-vis absorption spectra (Fig. S3, ESI†).Then, we investigated optoelectronic properties of the PM6:AC174:Y6 blend (Fig. S4, ESI†). AC174, PM6 and Y6 thin films exhibit strong absorption in the regions of 400–600 nm, 500–700 nm and 600–900 nm, respectively (Fig. 1c), yielding panchromatic absorption. The emission range of AC174 (550–700 nm) is heavily overlapped with the absorption range of PM6 (500–700 nm), which is beneficial for the FRET between AC174 and PM6. The energy level diagram of PM6, AC174 and Y6 is shown in Fig. 1d, where the HOMO energy levels of PM6 and AC174 were both measured by the UPS method (Fig. S3, ESI†), and that of Y6 was adopted from the reference, also measured by the same method.38 The HOMO energy level of AC174 is lower than that of PM6, which may increase open-circuit voltage (VOC) in the ternary device.
Device performance and device physics
In order to study the influence of AC174 on the performance of ternary OSCs, we adopted a conventional sandwich device structure of ITO glass/PEDOT:PSS/active layer/PNDIT-F3N/Ag to fabricate devices based on PM6:Y6 and PM6:AC174:Y6, respectively. Fig. 2a shows representative current density–voltage (J–V) curves of optimized PM6:Y6 and PM6:AC174:Y6 based OSCs measured under the illumination of an AM 1.5G solar simulator, 100 mW cm−2. The PM6:Y6 device affords a VOC of 0.812 V, JSC of 26.6 mA cm−2, fill factor (FF) of 73.5%, and PCE of 15.9%. After adding a small amount of AC174, where the content of AC174 in donors (PM6 + AC174) is 5%, the optimized PM6:AC174:Y6 device achieves simultaneous improvement in VOC, JSC and FF, with a VOC of 0.817 V, JSC of 28.4 mA cm−2, and FF of 74.1%, leading to a greatly improved PCE of 17.2% (Table 1). The device performance data with different AC174 contents are listed in Table S1 (ESI†), and the corresponding J–V curves and external quantum efficiency (EQE) spectra are shown in Fig. S5 (ESI†). As the weight ratio of AC174 in donors increases, the VOC shows small changes, while JSC and FF show the tendency of increasing first and then decreasing (Fig. S6, ESI†). Interestingly, when the content of AC174 in the donors reaches 50%, the PCE can still maintain 86% of the PCE of the control device (Table S1†), which is similar to the results in our previous study,39 where PM6:Y6-based devices with relatively low contents of PM6 still exhibited good device performance. However, when the AC174 content exceeds 50%, the PM6 content is too low, restricting the device performance. Because AC174 exhibits poor miscibility with PM6 or Y6, sufficient D:A interpenetrating networks are damaged, which decreases device performance. The statistic PCE distributions of optimized PM6:Y6 or PM6:AC174:Y6 based OSC devices are shown in Fig. S7a (ESI†), exhibiting good device reproducibility in both OSC devices. We have also measured the heat stability and light stability of PM6:Y6 and PM6:AC174:Y6 based OSC devices. The heat stability tests were carried out at 85 °C under a nitrogen atmosphere (Fig. S8a, ESI†), where PM6:Y6 and PM6:AC174:Y6 devices exhibit similar heat stability. The light stability tests were measured under continuous AM 1.5G illumination under a nitrogen atmosphere (Fig. S8b, ESI†). The PM6:Y6 binary device retains 79% of the original PCE, while the PM6:AC174:Y6 device retains 87% of the original PCE after continuous illumination for 300 min. Clearly, the introduction of AC174 improves the device light stability.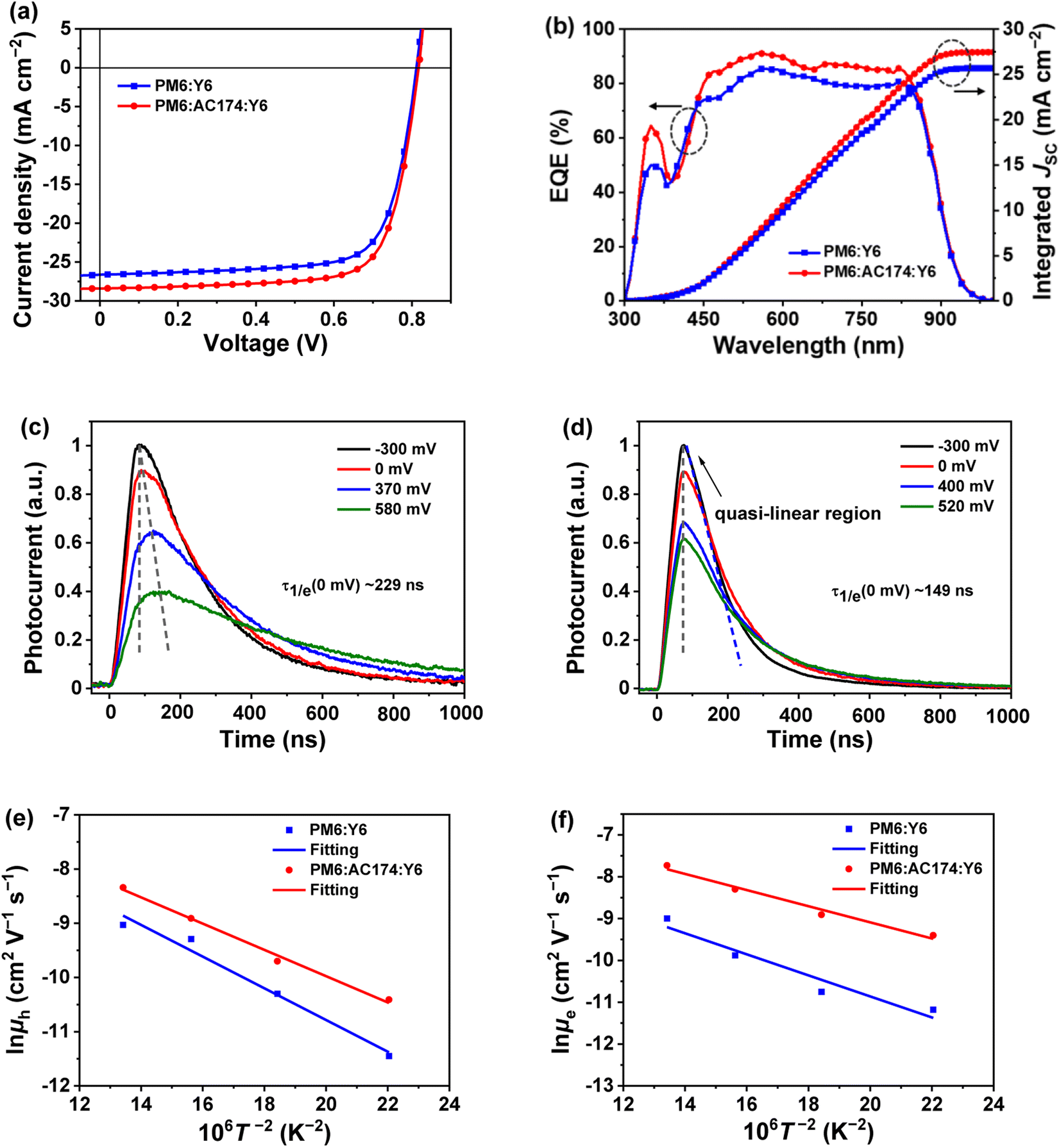 | ||
Fig. 2 (a) J–V curves and (b) EQE and integrated JSC curves of optimized PM6:Y6 and PM6:AC174:Y6 devices. TPC curves of (c) PM6:Y6 and (d) PM6:AC174:Y6 devices at varied external bias voltages. Temperature dependence of hole mobility (e) and electron mobility (f) of PM6:Y6 and PM6:AC174:Y6 films in a ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μ vs. T−2 representation for quantitative analysis of the energy disorder. μ vs. T−2 representation for quantitative analysis of the energy disorder. | ||
| Active layer | V OC (V) | J SC (mA cm−2) | FF (%) | PCE (%) | Calc. JSC (mA cm−2) |
|---|---|---|---|---|---|
| a Average values (in parenthesis) are obtained from 10 devices. | |||||
| PM6:Y6 | 0.812 (0.809 ± 0.004) | 26.6 (26.4 ± 0.5) | 73.5 (72.7 ± 1.3) | 15.9 (15.5 ± 0.2) | 26.0 |
| PM6:AC174:Y6 | 0.817 (0.813 ± 0.007) | 28.4 (28.1 ± 0.6) | 74.1 (73.3 ± 0.8) | 17.2 (16.7 ± 0.3) | 27.9 |
The EQE spectra of optimized PM6:Y6 and PM6:AC174:Y6 devices are shown in Fig. 2b, where in the range of 450–800 nm, the EQE value of the ternary device is apparently higher than that of the device without AC174. The integrated JSC values of PM6:Y6 and PM6:AC174:Y6 devices are 26.0 and 27.9 mA cm−2, respectively, which agrees with the JSC values measured from the J–V curves (less than 3% mismatch). The internal quantum efficiency (IQE) as a function of wavelength was also calculated, using the measured EQE and the active layer absorption determined by optical transfer matrix modelling simulations. As shown in Fig. S7b (ESI†), IQE was found to be higher for the device based on PM6:AC174:Y6, compared to that based on PM6:Y6, in the wavelength range of 580–680 nm. Since the optical properties of the thin film of PM6:Y6 is hardly affected by the addition of AC174 (Fig. S9, ESI†), the higher IQE of the solar cell based on PM6:AC174:Y6 is most likely due to the more efficient dissociation of excitons generated in the polymer donor, with an absorption peak at 620 nm.
To explore the origin of JSC and IQE enhancement in the ternary device, the modulated transient photocurrent (TPC) method was used to characterize the exciton dissociation of the devices.40,41 We adopted a series of external bias voltages (from negative to positive) for the device to regulate the internal electric field. The TPC results of PM6:Y6 and PM6:AC174:Y6 devices are shown in Fig. 2c and d. TPC curves of both devices exhibit a fast rise and a relatively slow decay process. Nonetheless, differences in the dynamic characteristics between these two devices can be clearly seen. Firstly, under the condition of no external bias voltage (0 mV), the photocurrent of the PM6:AC174:Y6 device takes 76 ns to reach the peak, which is 22 ns faster than the PM6:Y6 device. At the same time, the ternary device exhibits a faster decay time of 149 ns, which is 80 ns faster than the device without AC174. The shortened photocurrent peak time and decay time of the ternary device indicate better exciton dissociation. Secondly, when weakening the internal electric field by applying a positive bias voltage, the PM6:Y6 device needs a longer time to reach the photocurrent peak, while the TPC peak time for the PM6:AC174:Y6 device remains unchanged at all voltages. These differences in the TPC results reflect difference in the exciton dissociation. The PM6:Y6 device exhibits relatively slow exciton dissociation; hence, it takes a longer time to reach the photocurrent peak and decay in a low internal electric field because of dynamics equilibrium between excitons and free carriers.42 For the ternary device, the improved exciton dissociation helps to weaken the dependence of the exciton dissociation dynamics on the internal electric field, thus keeping the TPC peak time unchanged at all the bias voltages. Thirdly, a quasi-linear region is observed in the TPC curve of the PM6:AC174:Y6 device at an external bias voltage of −300 mV. This indicates that most of excitons in the ternary device have been dissociated into free carriers to provide a saturated charge concentration.43 In contrast, such a phenomenon is not observed in the PM6:Y6 device, which implies that even under a strong internal electric field, the exciton dissociation in the PM6:Y6 device is still insufficient. The TPC results reveal that the exciton dissociation in the PM6:AC174:Y6 device is more efficient than that in the device without AC174, which is responsible for the higher JSC in the ternary device.
The relationship between JSC and light intensity (Plight) can be adopted to describe charge recombination (Fig. S10, ESI†). The JSC follows a power-law relationship with Plight (JSC ∝ Plightα), where when α reaches 1, it means that all the charges have been collected by electrodes and no bimolecular recombination exists.44 The α values of PM6:Y6 and PM6:AC174:Y6 devices are 0.939 and 0.946, respectively, indicating less bimolecular recombination in the PM6:AC174:Y6 ternary device.
Charge mobility was measured to investigate the charge transport behaviour. We employed the organic field-effect transistor (OFET) method to measure charge mobilities of PM6, AC174 and PM6:AC174 blend films in the horizontal direction (Fig. S11, ESI†). The hole mobilities (μh) of PM6, AC174 and PM6:AC174 blend films are 4.9 × 10−2 cm2 V−1 s−1, 3.5 × 10−3 cm2 V−1 s−1 and 6.3 × 10−2 cm2 V−1 s−1, respectively. Compared with pristine PM6, the PM6:AC174 blend films exhibit a better charge transport in the horizontal direction. We next used the space charge limited current (SCLC) method to measure charge mobilities of PM6:Y6, PM6:AC174:Y6 and AC174:Y6 blend films in the vertical direction (Fig. S12 and Table S2, ESI†).45 The μh of PM6:Y6, PM6:AC174:Y6, and AC174:Y6 blends are 9.1 × 10−4 cm2 V−1 s−1, 1.1 × 10−3 cm2 V−1 s−1, and 6.1 × 10−4 cm2 V−1 s−1, respectively. The electron mobilities (μe) of the above three blends are 8.3 × 10−4 cm2 V−1 s−1, 1.1 × 10−3 cm2 V−1 s−1, and 5.1 × 10−4 cm2 V−1 s−1, respectively. Compared with the PM6:Y6 binary blend, the optimized ternary blend exhibits higher and more balanced charge mobilities, which can reduce charge recombination and improve the FF.
Exciton and charge transport in amorphous or weak crystalline organic semiconductors is regarded as a hopping process between localized sites, following the extended Gaussian disorder model (EGDM). The distribution of electronic density of states (DOS) in LUMO and HOMO energy levels follows a Gaussian curve (denoted as ΔHOMO for the HOMO energy level and ΔLUMO for the LUMO energy level) and the width of the Gaussian curve reflects the energy disorder of the material.46,47 According to the EGDM, charge mobility is a function of temperature and material energy disorder in low electric fields, following the formula:
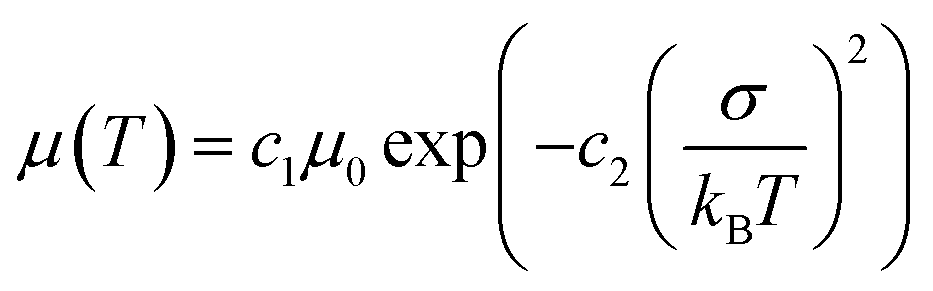 | (1) |
Here, we fabricated space-charge-limited hole-only devices with the structure of ITO/PEDOT:PSS/blend films/Au and electron-only devices with the structure of ITO/ZnO/blend films/BCP/Ag, and investigated the temperature-dependent charge mobility. When the temperature decreases from 273 K to 213 K, the μh of the PM6:Y6 binary device decreases from 1.2 × 10−4 cm2 V−1 s−1 to 1.1 × 10−5 cm2 V−1 s−1, and that of the PM6:AC174:Y6 ternary device decreases from 2.4 × 10−4 cm2 V−1 s−1 to 3.0 × 10−5 cm−2 V−1 s−1 (Fig. S13a and b, ESI†); the μe of the PM6:Y6 binary device decreases from 1.3 × 10−4 cm2 V−1 s−1 to 1.4 × 10−5 cm2 V−1 s−1, while that of the PM6:AC174:Y6 ternary device decreases from 4.4 × 10−4 cm2 V−1 s−1 to 8.3 × 10−5 cm−2 V−1 s−1 (Fig. S13c and d, ESI†). We calculated the σ values of PM6:Y6 and PM6:AC174:Y6 blends alongside the ΔHOMO and ΔLUMO using eqn (1) (Fig. 2e and f). Regarding the energy disorder in the HOMO energy levels (σHOMO), the PM6:AC174:Y6 blend film exhibits a smaller σ value of 107.7 meV compared with the PM6:Y6 blend film (115.1 meV). Furthermore, a lower σ in the LUMO energy levels (σLUMO) was found for the PM6:AC174:Y6 blend film (93.5 meV) compared with the PM6:Y6 blend film (106.8 meV). The reduction in σ values along with increased charge mobility is beneficial for exciton diffusion/dissociation and charge transport.
Device photophysics
Steady-state photoluminescence (PL) and transient gating ultrafast photoluminescence (TG-UFPL) were applied to investigate exciton diffusion and exciton dissociation in different films. As shown in Fig. 1c, the AC174 film exhibits a strong PL emission in the range of 550–700 nm, with a relatively high PL quantum yield (PLQY) of 11.4%. The emission range of AC174 is highly overlapped with the absorption range of PM6, supporting the Förster energy transfer between AC174 and PM6. The PL spectra of pure PM6 and PM6:AC174 (5%) blend films were measured under excitation at 600 nm (Fig. S14, ESI†). Neat AC174 and PM6 films exhibit emission peaks at 580 nm and 673 nm, respectively, while the PM6:AC174 (5%) blend film also exhibits an emission peak at 673 nm, with stronger intensity and higher PLQY (2.85%) compared with PM6 (2.16%), indicating efficient energy transfer from AC174 to PM6. Compared with pure PM6, stronger PL intensity of the PM6:AC174 blend is favourable for enhancing the long-range FRET between the donor and acceptor and therefore, the exciton diffusion length.To investigate the photo-excitation dynamics in more detail, we applied TG-UFPL and transient absorption spectroscopy (TAS). The experimental details of these methods are included in the ESI.† Firstly, we used TG-UFPL to selectively probe the singlet exciton behaviour of the PM6 donor in sub-picoseconds to tens of picoseconds, the critical time scale for the dissociation of PM6 excitons in PM6:Y6 blends.51Fig. 3a shows the PL emission of pure PM6 and PM6:AC174 blend films at a pump fluence of 2.44 μJ cm−2 with an excitation wavelength of 515 nm. We observed that the PM6:AC174 blend has a longer emission decay of ca. 550 fs (amplitude weighted half-intensity) and pure PM6 has a shorter decay of ca. 400 fs, indicating a longer exciton lifetime in the donor blend. The TG-UFPL's ultrashort gate time window (<200 fs) is only sensitive to singlet excitons, which have a high emissive rate, and not to late generated excimers.51 The observed fast PL decays are consistent with the kinetics of Frenkel-type local excitation in PM6 measured by TAS,51 suggesting that we are probing the key excitation species on a critical time scale for photocurrent generation.
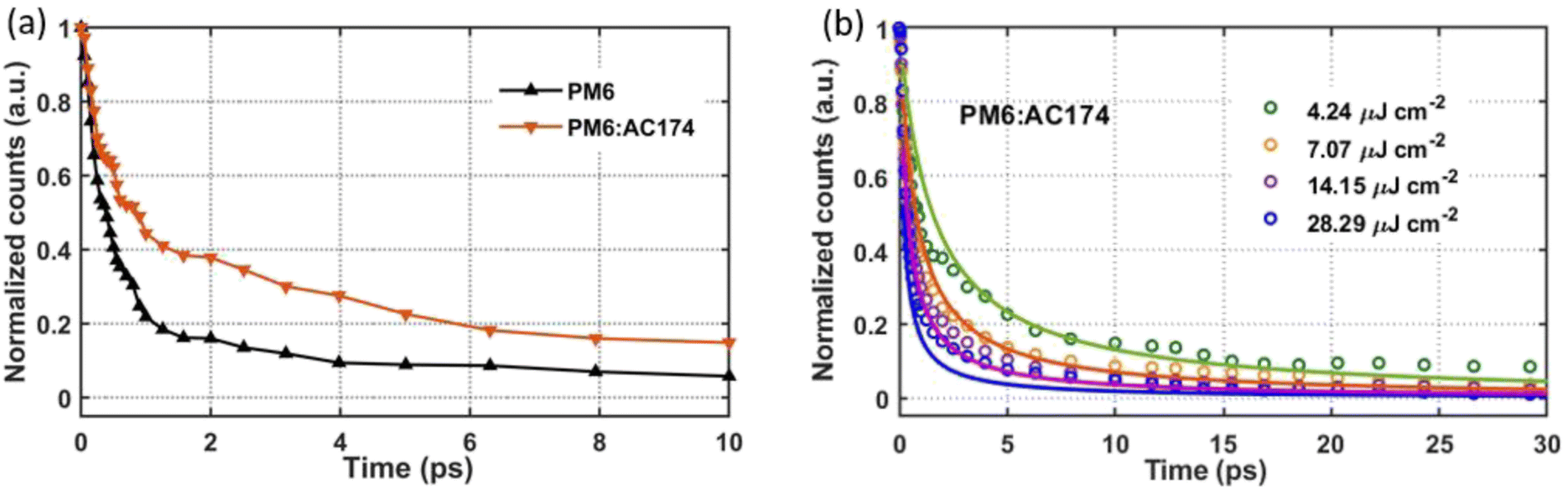 | ||
| Fig. 3 (a) Integrated photoluminescence emission kinetics of PM6 and PM6:AC174 (5%) films. (b) Global fitting of fluence dependent decays for PM6:AC174 (5%) films. | ||
Then, we compared the fluence-dependent PL emission kinetics of the pure PM6 film and PM6:AC174 (5%) blend film with a pump fluence ranging from 4.24 μJ cm−2 to 28.29 μJ cm−2 (Fig. 3b and S15, ESI†). It can be seen that pure PM6 has no clear fluence dependence in the range of our experimental conditions, while the PM6:AC174 blend does. The lack of fluence dependence in PM6, which is consistent with previous transient experimental measurement at similar excitation densities,31 indicates that the excitons are not very mobile on this timescale. However, for the PM6:AC174 blend, exciton annihilation becomes stronger with an increase in the excitation intensity, which indicates that the exciton diffusion ability considerably increases.
Furthermore, the bimolecular exciton recombination (exciton annihilation) decay rate (δ) can be determined according to the following eqn (2),
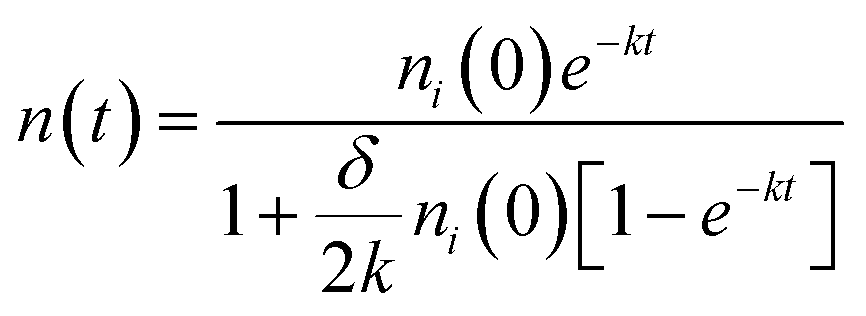 | (2) |
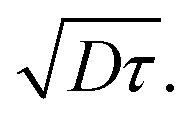 After calculation, the PM6:AC174 blend exhibits a higher D value (0.573 cm2 s−1) and a longer LD value within the relevant charge generation timescale of 10 ps (25 nm) compared with pure PM6 (0.239 cm2 s−1, 15 nm), which indicates a better ability to facilitate rapid exciton diffusion across the PM6 domain in the PM6:AC174 blend.
After calculation, the PM6:AC174 blend exhibits a higher D value (0.573 cm2 s−1) and a longer LD value within the relevant charge generation timescale of 10 ps (25 nm) compared with pure PM6 (0.239 cm2 s−1, 15 nm), which indicates a better ability to facilitate rapid exciton diffusion across the PM6 domain in the PM6:AC174 blend.
The longer exciton diffusion length is beneficial for exciton dissociation in D/A blend systems. We compared the ultrafast PL emission of pure PM6, PM6:Y6 binary blend and PM6:AC174:Y6 ternary blend films at a pump fluence of 7.07 μJ cm−2 (Fig. S16, ESI†). Although 515 nm excitation excites both PM6 and Y6 in the blend thin films, our ultrafast PL, which cannot detect the emission from Y6 due to the sensitivity of the camera, selectively probes PM6 singlet exciton dynamics to avoid the complicated data interpretations involving multiple excitation species. We can estimate the exciton quenching efficiency by comparing the exciton decay time of the donor in the presence and absence of the acceptor. As shown in Fig. S16 (ESI†), the PM6 singlet excitons in PM6:Y6 binary and PM6:AC174:Y6 ternary blends have similar decay times (ca. 300 fs); however, considering that the PM6 exciton in PM6:AC174 has a longer decay time than the pure PM6 (550 fs versus 400 fs), we can conclude that the exciton quenching is more efficient in the ternary blends, indicating faster D/A energy transfer31 and exciton diffusion accompanied with better exciton dissociation and charge generation.
We next employ TAS measurement to observe the charge dynamics when the Y6 molecules are excited. Measurements were conducted with an 800 nm excitation wavelength to selectively excite Y6 with pump fluences ranging from 5 μW to 20 μW. Fig. S17 (ESI†) shows the TAS spectra of the binary and ternary blends with a pump fluence of 20 μW. The photo induced absorption (PIA) band is shown as a negative feature at around 955 nm. Two positive peaks are observed at around 860 nm and 610 nm which can be assigned as an overlapping contribution of the Y6 bleach signal and PM6 bleach signal. The positive ground state bleaching (GSB) signal in the 575–625 nm region of PM6 gauges the charge generation process. Since PM6 is not excited at 800 nm, PM6 bleaching comes about when photoexcitation in Y6 undergo hole transfer to PM6.52 Therefore, PM6 bleaching is proportional to the total charge population. As shown in Fig. 4, the charge formation kinetics shows no distinct difference between the binary and ternary thin films, indicating similar charge formation/exciton dissociation dynamics. However, after the charge formation reaches a peak, when the pump–probe delay time is about 50 ps, it can be seen that the PM6:AC174:Y6 ternary blend has more pronounced non-geminate charge recombination at high excitation density (substantially higher than that encountered under solar excitation conditions). This, in turn, suggests more efficient generation of more mobile charges in the ternary blend, in accordance with the SCLC results. Overall, the PM6:AC174:Y6 ternary OSC exhibits a longer exciton diffusion length, a better exciton dissociation ability and a better charge transport ability. This is likely owing to its lower system energetic disorder, benefitting from its better nanostructure morphology, which is studied in the following section.
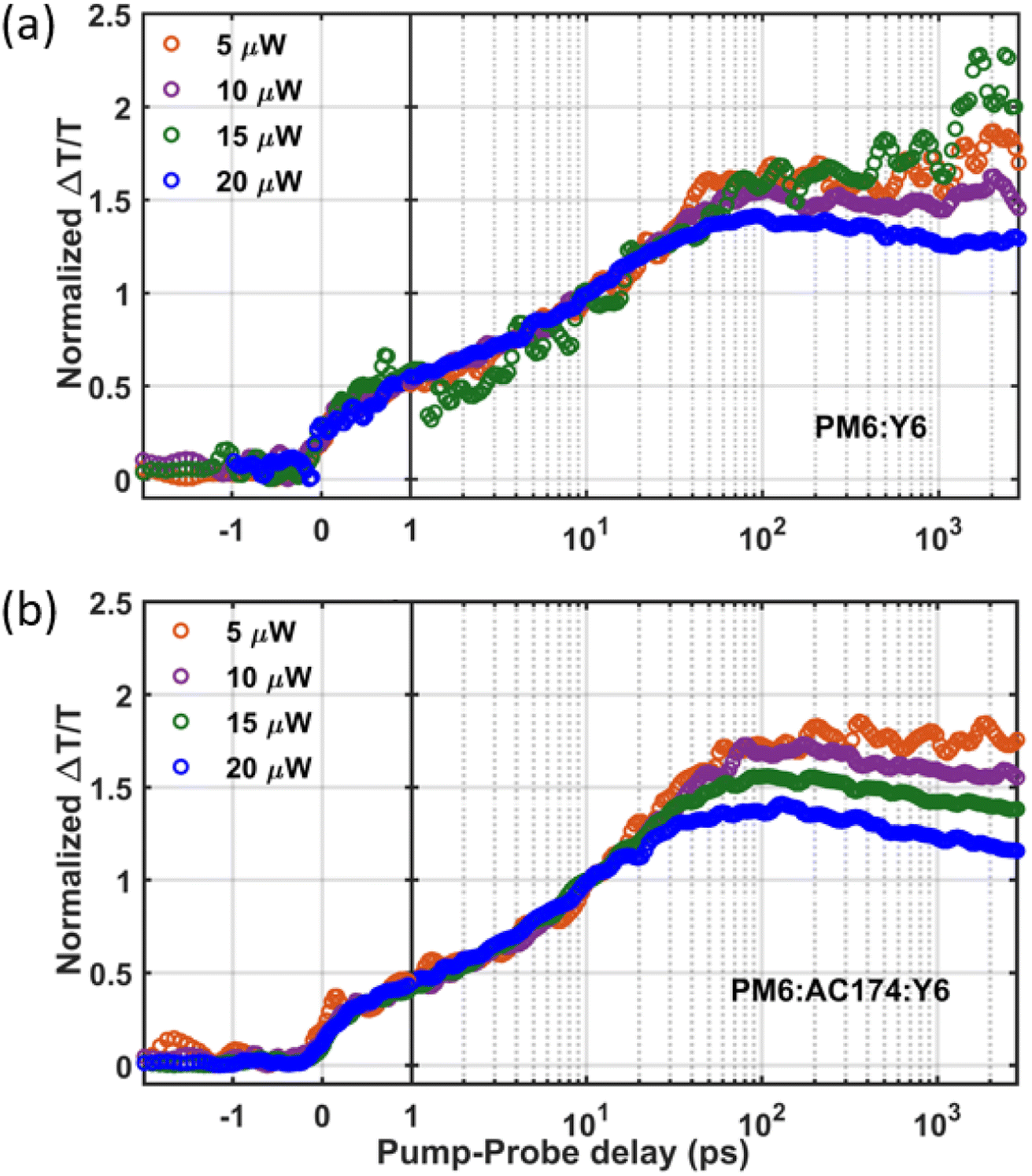 | ||
| Fig. 4 Fluence dependent kinetics of the GSB signal for (a) PM6:Y6 and (b) PM6:AC174:Y6 films under an 800 nm excitation pump, intensity normalized at 10 ps. | ||
Film morphology
The contact angle measurements were used to study the surface tension of materials and evaluate the miscibility between materials. The surface tensions of materials were calculated from the measured contact angles on water and diiodomethane using the Owens–Wendt equation.53 The results are shown in Table S4 and S18 (ESI†). The Flory–Huggins interaction parameters (χ) were calculated applying the equation of 54 According to the Flory–Huggins model, a smaller χ indicates stronger interaction between the two components, namely, better miscibility. The interaction parameter between PM6 and Y6 is relatively low (χPM6:Y6 = 0.045), indicating remarkable miscibility between them. The interaction parameter between PM6:AC174 (5%) blend donors and Y6 is 0.095, which is a little larger than that of the PM6:Y6 system. On one hand, pretty good miscibility still exists between the blend donors and acceptor. On the other hand, slightly decreased D/A miscibility is beneficial for Y6 molecular packing, which is favourable for decreasing energetic disorder and improving exciton diffusion/dissociation and charge transport. In contrast, the χ values between AC174 and PM6 or Y6 are considerably larger (χAC174:PM6 = 0.442; χAC174:Y6 = 0.770), indicating that AC174 has poor miscibility with PM6 or Y6.
54 According to the Flory–Huggins model, a smaller χ indicates stronger interaction between the two components, namely, better miscibility. The interaction parameter between PM6 and Y6 is relatively low (χPM6:Y6 = 0.045), indicating remarkable miscibility between them. The interaction parameter between PM6:AC174 (5%) blend donors and Y6 is 0.095, which is a little larger than that of the PM6:Y6 system. On one hand, pretty good miscibility still exists between the blend donors and acceptor. On the other hand, slightly decreased D/A miscibility is beneficial for Y6 molecular packing, which is favourable for decreasing energetic disorder and improving exciton diffusion/dissociation and charge transport. In contrast, the χ values between AC174 and PM6 or Y6 are considerably larger (χAC174:PM6 = 0.442; χAC174:Y6 = 0.770), indicating that AC174 has poor miscibility with PM6 or Y6.
Transmission electron microscopy (TEM) was employed to study the film morphology of PM6:Y6, PM6:AC174:Y6, AC174:Y6 and PM6:AC174 blend films (Fig. S19, ESI†). PM6:Y6 and PM6:AC174:Y6 films exhibit smooth morphology. The AC174:Y6 blend film exhibits a large phase domain size, which can be attributed to the poor miscibility between AC174 and Y6. As for the PM6:AC174 film, because we added a small amount of AC174 (5%), phase separation cannot be clearly seen. Furthermore, we used atomic force microscopy (AFM) to investigate the surface morphology of PM6:Y6, PM6:AC174:Y6, and AC174:Y6 blend films (Fig. S20, ESI†). The root-mean-square roughness (Rq) of PM6:Y6, PM6:AC174:Y6, and AC174:Y6 blend films are 1.22 nm, 1.23 nm, and 6.61 nm, respectively. The AC174:Y6 blend film exhibits rough surface morphology, owing to the poor miscibility between them.
GIWAXS characterization studies were performed to evaluate molecular packing and the orientation of pure and blend films.55,56 The two-dimensional GIWAXS (2D-GIWAXS) patterns and corresponding 1D line-cuts along the in-plane and out-of-plane directions of pure films and PM6:AC174 blend films are shown in Fig. S21 (ESI†) and 1b. The pure Y6 film exhibits high crystallinity with a preferential face-on orientation indicated by the π–π peak along the qz axis (q = 1.74 Å−1, d = 3.62 Å). The pure PM6 film was reported to be preferentially face-on oriented with the π–π peak at qz = 1.69 Å−1 (d = 3.72 Å), and the coherence length (CL) is 1.86 nm. The PM6:AC174 (5%) blend film exhibits a π–π peak along the qz axis (qz = 1.69 Å−1, d = 3.72 Å), and the CL increases to 1.99 nm. A stronger π–π stacking of PM6 along the qz axis is beneficial for decreasing energetic disorder and improving charge transport in the vertical direction.
The 2D-GIWAXS patterns and corresponding 1D line-cuts along the in-plane and out-of-plane directions of PM6:Y6, PM6:AC174:Y6 and AC174:Y6 blend films are shown in Fig. 5a–c and g. For the AC174:Y6 blend film, π–π peaks located at qz = 1.49 Å−1 and qz = 1.75 Å−1 are attributed to the scattering from face-on oriented AC174 and Y6 crystalline domains, respectively, without forming a co-crystal structure, which indicates poor miscibility between the two materials. For PM6:Y6 and PM6:AC174:Y6 blend films, π–π peaks are located at qz = 1.75 Å−1 (d = 3.60 Å) and qz = 1.74 Å−1 (d = 3.61 Å), respectively, and the corresponding CL values slightly increase from 2.86 nm to 2.87 nm. Both blend films exhibit evident Y6 ordering in the out-of-plane direction, leading to good charge transport.
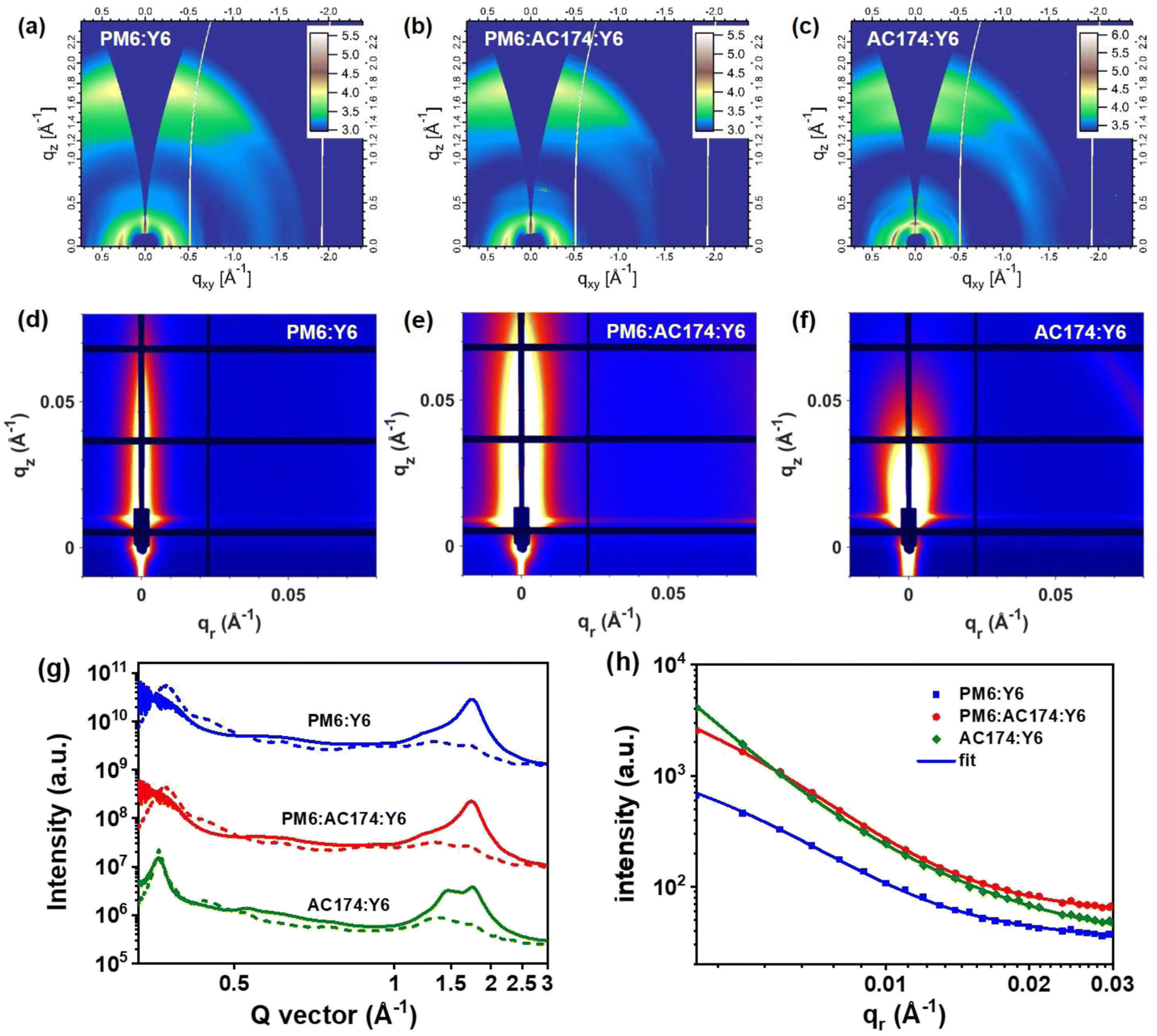 | ||
| Fig. 5 (a–c) 2D GIWAXS patterns and (d–f) 2D GISAXS patterns of PM6:Y6, PM6:AC174:Y6, and AC174:Y6 thin films, respectively. (g) Corresponding GIWAXS profiles in the out-of-plane (solid lines) and in-plane (dashed lines) directions. (h) Corresponding GISAXS fitted scattering intensity along the in-plane direction. | ||
Grazing incidence small-angle X-ray scattering (GISAXS) measurements were conducted to characterize the phase separation of the binary and ternary blends (Fig. 5d–f and h).57 The Debye–Anderson–Brumberger (DAB) model and the fractal-like network model were used to evaluate the intermixing of amorphous phases and pure acceptor domains, respectively, in GISAXS measurements.58 The correlation lengths of the intermixing region (ξ) of PM6:Y6, PM6:AC174:Y6, and AC174:Y6 blend films are 21.2 nm, 25.4 nm, and 50.5 nm, respectively. The corresponding pure acceptor (2Rg) domain sizes are found to be 13.2 nm, 34.2 nm, and 46.8 nm, respectively. The domain size of the pure acceptor region for AC174:Y6 is much larger compared with that of PM6:Y6 and PM6:AC174:Y6, which is consistent with the poor miscibility of AC174:Y6. Compared with the PM6:Y6 blend, the relatively larger intermixing region length of the ternary blend film may contribute to exciton dissociation in this region, consistent with the TPC and TG-UFPL results, leading to higher JSC values in devices. The great increase of pure acceptor domain sizes in the ternary blend film is related to the higher crystallinity of Y6, which facilitates charge transport, confirmed by the SCLC measurements. Moreover, an increased Y6 domain size is beneficial for reducing system energetic disorder, leading to a longer exciton diffusion length, consistent with the TG-UFPL data.
Conclusions
In summary, a highly crystalline, highly emissive, and wide-bandgap polymer AC174 with an extremely small Stokes shift was synthesized in water and was added into the PM6:Y6 blend to fabricate PM6:AC174:Y6 ternary OSC devices. AC174 exhibits poor compatibility with PM6 and Y6, which tends to form an individual phase in ternary films, improves the molecular packing, and reduces system energetic disorder, which is beneficial for FRET and exciton transport. The heavily overlapping of AC174 emission and PM6 absorption spectra as well as a high PLQY of AC174 facilitate FRET between AC174 and PM6, leading to better exciton diffusion ability, a longer exciton lifetime and a longer exciton diffusion length in the PM6:AC174 blend. Relative to the PM6:Y6 binary devices, the PM6:AC174:Y6 ternary OSCs exhibit a longer exciton diffusion length, and more efficient exciton dissociation, charge generation and charge transport. As a result, the ternary OSCs based on PM6:AC174:Y6 yield a PCE of 17.2%, which is higher than that of the PM6:Y6 binary devices (15.9%). It is worth noting that the ternary devices exhibit the highest IQE approaching 100% and an impressive JSC value over 28 mA cm−2. This study provides a facile strategy for morphology optimization, reducing system energetic disorder, and enhancing the exciton diffusion length by introducing a polymer semiconductor with high crystallinity, a high PLQY, and a small Stokes shift, and finally improving the performance of the OSCs.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
X. Z. thanks the NSFC (No. U21A20101 and 21734001). W. M. thanks the NSFC (21704082, 21875182), Key Scientific and Technological Innovation Team Project of Shaanxi Province (2020TD-002), and 111 Project 2.0 (BP2018008). GIWAXS data were acquired at beamlines 7.3.3 at the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors thank Dr Eric Schaible and Dr Chenhui Zhu at beamline 7.3.3 for assistance with data acquisition.Notes and references
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- J. Wang, P. Xue, Y. Jiang, Y. Huo and X. Zhan, Nat. Rev. Chem., 2022, 6, 614–634 CrossRef.
- J. Wang and X. Zhan, Acc. Chem. Res., 2021, 54, 132–143 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS PubMed.
- S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed.
- J. Wang, J. Zhang, Y. Xiao, T. Xiao, R. Zhu, C. Yan, Y. Fu, G. Lu, X. Lu, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2018, 140, 9140–9147 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Y. Ma, M. Zhang, S. Wan, P. Yin, P. Wang, D. Cai, F. Liu and Q. Zheng, Joule, 2021, 5, 197–209 CrossRef CAS.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- K. Chong, X. Xu, H. Meng, J. Xue, L. Yu, W. Ma and Q. Peng, Adv. Mater., 2022, 34, 2109516 CrossRef CAS PubMed.
- Z. Zheng, J. Wang, P. Bi, J. Ren, Y. Wang, Y. Yang, X. Liu, S. Zhang and J. Hou, Joule, 2022, 6, 171–184 CrossRef CAS.
- O. G. Reid, R. D. Pensack, Y. Song, G. D. Scholes and G. Rumbles, Chem. Mater., 2014, 26, 561–575 CrossRef CAS.
- R. A. J. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847–1858 CrossRef CAS PubMed.
- O. V. Mikhnenko, P. W. M. Blom and T.-Q. Nguyen, Energy Environ. Sci., 2015, 8, 1867–1888 RSC.
- Y. Tamai, H. Ohkita, H. Benten and S. Ito, J. Phys. Chem. Lett., 2015, 6, 3417–3428 CrossRef CAS PubMed.
- Z. Zhou, S. Xu, J. Song, Y. Jin, Q. Yue, Y. Qian, F. Liu, F. Zhang and X. Zhu, Nat. Energy, 2018, 3, 952–959 CrossRef CAS.
- L. Ye, H. Hu, M. Ghasemi, T. Wang, B. A. Collins, J.-H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. Li, T. McAfee, J. Zhao, X. Chen, J. L. Y. Lai, T. Ma, J.-L. Bredas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253–260 CrossRef CAS PubMed.
- J. Du, K. Hu, J. Zhang, L. Meng, J. Yue, I. Angunawela, H. Yan, S. Qin, X. Kong, Z. Zhang, B. Guan, H. Ade and Y. Li, Nat. Commun., 2021, 12, 5264 CrossRef CAS PubMed.
- T. Liu, T. Yang, R. Ma, L. Zhan, Z. Luo, G. Zhang, Y. Li, K. Gao, Y. Xiao, J. Yu, X. Zou, H. Sun, M. Zhang, T. A. D. Peña, Z. Xing, H. Liu, X. Li, G. Li, J. Huang, C. Duan, K. S. Wong, X. Lu, X. Guo, F. Gao, H. Chen, F. Huang, Y. Li, Y. Li, Y. Cao, B. Tang and H. Yan, Joule, 2021, 5, 914–930 CrossRef CAS.
- M. Theander, A. Yartsev, D. Zigmantas, V. Sundström, W. Mammo, M. R. Andersson and O. Inganäs, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 12957 CrossRef CAS.
- S. M. Menke and R. J. Holmes, Energy Environ. Sci., 2014, 7, 499–512 RSC.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- S. D. Dimitrov, B. C. Schroeder, C. B. Nielsen, H. Bronstein, Z. Fei, I. McCulloch, M. Heeney and J. R. Durrant, Polymers, 2016, 8, 14 CrossRef PubMed.
- S. Karuthedath, J. Gorenflot, Y. Firdaus, N. Chaturvedi, C. S. P. D. Castro, G. T. Harrison, J. I. Khan, A. Markina, A. H. Balawi, T. A. D. Peña, W. Liu, R.-Z. Liang, A. Sharma, S. H. K. Paleti, W. Zhang, Y. Lin, E. Alarousu, S. Lopatin, D. H. Anjum, P. M. Beaujuge, S. D. Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, D. Andrienko and F. Laquai, Nat. Mater., 2021, 20, 378–384 CrossRef CAS PubMed.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photon., 2018, 12, 131–142 CrossRef CAS.
- S. Chandrabose, K. Chen, A. J. Barker, J. J. Sutton, S. K. K. Prasad, J. Zhu, J. Zhou, K. C. Gordon, Z. Xie, X. Zhan and J. M. Hodgkiss, J. Am. Chem. Soc., 2019, 141, 6922–6929 CrossRef CAS PubMed.
- S. Y. Park, S. Chandrabose, M. B. Price, H. S. Ryu, T. H. Lee, Y. S. Shin, Z. Wu, W. Lee, K. Chen, S. Dai, J. Zhu, P. Xue, X. Zhan, H. Y. Woo, J. Y. Kim and J. M. Hodgkiss, Nano Energy, 2021, 84, 105924 CrossRef CAS.
- S. Xie, Y. Xia, Z. Zheng, X. Zhang, J. Yuan, H. Zhou and Y. Zhang, Adv. Funct. Mater., 2018, 28, 1705659 CrossRef.
- Z. Zhang, Y. Li, G. Cai, Y. Zhang, X. Lu and Y. Lin, J. Am. Chem. Soc., 2020, 142, 18741–18745 CrossRef CAS PubMed.
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photon., 2020, 14, 300–305 CrossRef CAS.
- W. Deng, W. Liu, R. Qian and H. Wu, J. Phys. Chem. Lett., 2022, 13, 544–551 CrossRef CAS PubMed.
- A. Sanzone, A. Calascibetta, M. Monti, S. Mattiello, M. Sassi, F. Corsini, G. Griffini, M. Sommer and L. Beverina, ACS Macro Lett., 2020, 9, 1167–1171 CrossRef CAS PubMed.
- C. Li, X. Zhang, N. Yu, X. Gu, L. Qin, Y. Wei, X. Liu, J. Zhang, Z. Wei, Z. Tang, Q. Shi and H. Huang, Adv. Funct. Mater., 2022, 32, 2108861 CrossRef CAS.
- T. Li, K. Wang, G. Cai, Y. Li, H. Liu, Y. Jia, Z. Zhang, X. Lu, Y. Yang and Y. Lin, JACS Au, 2021, 1, 1733–1742 CrossRef CAS PubMed.
- Y. Wang, M. B. Price, R. S. Bobba, H. Lu, J. Xue, Y. Wang, M. Li, A. Ilina, P. A. Hume, B. Jia, T. Li, Y. Zhang, N. J. L. K. Davis, Z. Tang, W. Ma, Q. Qiao, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2022 DOI:10.1002/adma.202206717.
- J. Shi, D. Li, Y. Luo, H. Wu and Q. Meng, Rev. Sci. Instrum., 2016, 87, 123107 CrossRef PubMed.
- Y. Li, J. Shi, B. Yu, B. Duan, J. Wu, H. Li, D. Li, Y. Luo, H. Wu and Q. Meng, Joule, 2020, 4, 472–489 CrossRef CAS.
- X. Zhao, B. Mi, Z. Gao and W. Huang, Sci. China: Phys., Mech. Astron., 2011, 54, 375–387 CAS.
- J. Seifter, Y. Sun and A. J. Heeger, Adv. Mater., 2014, 26, 2486–2493 CrossRef CAS PubMed.
- P. Schilinsky, C. Waldauf and C. J. Brabec, Appl. Phys. Lett., 2002, 81, 3885–3887 CrossRef CAS.
- G. G. Malliaras, J. R. Salem, P. J. Brock and C. Scott, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13411–13414 CrossRef.
- C. Tanase, E. J. Meijer, P. W. M. Blom and D. M. de Leeuw, Phys. Rev. Lett., 2003, 91, 216601 CrossRef CAS PubMed.
- D. Abbaszadeh, A. Kunz, G. A. H. Wetzelaer, J. J. Michels, N. I. Crăciun, K. Koynov, I. Lieberwirth and P. W. M. Blom, Nat. Mater., 2016, 15, 628–633 CrossRef CAS PubMed.
- H. Bässler, Phys. Status Solidi B, 1993, 175, 15–56 CrossRef.
- W. F. Pasveer, J. Cottaar, C. Tanase, R. Coehoorn, P. A. Bobbert, P. W. M. Blom, D. M. de Leeuw and M. A. J. Michels, Phys. Rev. Lett., 2005, 94, 206601 CrossRef CAS PubMed.
- Y. Jiang, J. Wang, H. Zai, D. Ni, J. Wang, P. Xue, N. Li, B. Jia, H. Lu, Y. Zhang, F. Wang, Z. Guo, Z. Bi, H. Xie, Q. Wang, W. Ma, Y. Tu, H. Zhou and X. Zhan, J. Am. Chem. Soc., 2022, 144, 5400–5410 CrossRef CAS PubMed.
- R. Wang, C. Zhang, Q. Li, Z. Zhang, X. Wang and M. Xiao, J. Am. Chem. Soc., 2020, 142, 12751–12759 CrossRef CAS PubMed.
- M. B. Price, P. A. Hume, A. Ilina, I. Wagner, R. R. Tamming, K. E. Thorn, W. Jiao, A. Goldingay, P. J. Conaghan, G. Lakhwani, N. J. L. K. Davis, Y. Wang, P. Xue, H. Lu, K. Chen, X. Zhan and J. M. Hodgkiss, Nat. Commun., 2022, 13, 2827 CrossRef CAS PubMed.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- S. Nilsson, A. Bernasik, A. Budkowski and E. Moons, Macromolecules, 2007, 40, 8291–8301 CrossRef CAS.
- F. Liu, Y. Gu, X. Shen, S. Ferdous, H.-W. Wang and T. P. Russell, Prog. Polym. Sci., 2013, 38, 1990–2052 CrossRef CAS.
- J. Mai, Y. Xiao, G. Zhou, J. Wang, J. Zhu, N. Zhao, X. Zhan and X. Lu, Adv. Mater., 2018, 30, 1802888 CrossRef PubMed.
- J. Mai, T.-K. Lau, J. Li, S.-H. Peng, C.-S. Hsu, U.-S. Jeng, J. Zeng, N. Zhao, X. Xiao and X. Lu, Chem. Mater., 2016, 28, 6186–6195 CrossRef CAS.
- J. Mai, H. Lu, T.-K. Lau, S.-H. Peng, C.-S. Hsu, W. Hua, N. Zhao, X. Xiao and X. Lu, J. Mater. Chem. A, 2017, 5, 11739–11745 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta07113d |
| This journal is © The Royal Society of Chemistry 2022 |