
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, theoretical study, antioxidant, and anticholinesterase activities of new pyrazolo-fused phenanthrolines†
Efraín Polo-Cuadrado *a,
Cristian Rojas-Peñaab,
Karen Acosta-Quirogaab,
Lorena Camargo-Ayalac,
Iván Britod,
Jonathan Cisternad,
Félix Moncadae,
Jorge Trillerasf,
Yeray A. Rodríguez-Núñezg and
Margarita Gutierrez*a
*a,
Cristian Rojas-Peñaab,
Karen Acosta-Quirogaab,
Lorena Camargo-Ayalac,
Iván Britod,
Jonathan Cisternad,
Félix Moncadae,
Jorge Trillerasf,
Yeray A. Rodríguez-Núñezg and
Margarita Gutierrez*a
aLaboratorio Síntesis Orgánica y Actividad Biológica (LSO-Act-Bio), Instituto de Química de Recursos Naturales, Universidad de Talca, Casilla 747, Talca 3460000, Chile. E-mail: epolo@utalca.cl; mgutierrez@utalca.cl
bDoctorado en Química, Departamento de Química Orgánica y Fisicoquímica, Universidad de Chile, Santiago, Chile
cDoctorado en Ciencias Mención I + D de Productos Bioactivos, Instituto de Química de Recursos Naturales, Laboratorio de Síntesis Orgánica (LSO-Act-Bio), Universidad de Talca, Casilla 747, Talca 3460000, Chile
dDepartamento de Química, Facultad de Ciencias Básicas, Universidad de Antofagasta, Avda., Campus Coloso, Antofagasta 02800, Chile
eDepartamento de Química, Universidad Nacional de Colombia, Av. Cra 30 # 45-03, Bogotá, Colombia
fGrupo de Investigación en Compuestos Heterocíclicos, Universidad del Atlántico, Puerto Colombia 081007, Colombia
gDepartamento de Química, Facultad de Ciencias Exactas, Universidad Andrés Bello, Republica 275, Santiago, 8370146, Chile
First published on 17th November 2022
Abstract
Pyrazole-fused phenanthroline compounds were obtained through several synthetic routes. NMR, HRMS, and IR techniques were used to characterize and confirm the chemical structures. Crystal structures were obtained from compounds 3a, 5b, 5j, 5k, and 5n and analyzed using X-ray diffraction. Compounds were evaluated as acetyl (AChE) and butyrylcholinesterase (BChE) inhibitors, and the results showed a moderate activity. Compound 5c presented the best activity against AChE (IC50 = 53.29 μM) and compound 5l against BChE enzyme (IC50 = 119.3 μM). Furthermore, the ability of the synthetic compounds to scavenge cationic radicals DPPH and ABTS was evaluated. Compound 5e (EC50 = 26.71 μg mL−1) presented the best results in the DPPH assay, and compounds 5e, 5f and 5g (EC50 = 11.51, 3.10 and <3 μg mL−1, respectively) showed better ABTS cationic radical scavenging results. Finally, in silico analyses indicated that 71% of the compounds show good oral availability and are within the ranges established by the Lipinski criteria.
1. Introduction
Many compounds of interest in the pharmacological field usually contain 5 and 6-membered heteroatom rings with nitrogen, sulfur, or oxygen atoms. The synthesis of fused heterocycles offers new opportunities to create compounds with improved biological activity and has significant relevance in medicinal chemistry.1,2 Within this context, fused heterocycles such as pyrazolo[3,4-b]pyridine have been reported as antimicrobial,3 anti-inflammatory,4 cytotoxic,5 and antitumoral6 compounds. Likewise, phenanthroline derivatives have chemotherapeutic, antimicrobial, and anticancer potential, highly valued biological activities in pharmacology.4,5,7–9There are various synthetic strategies for obtaining a phenanthroline scaffold. One strategy is the formation of the pyridine ring from an existing aminopyrazole in reaction with a dicarbonyl species or equivalents and aldehydes through a Hantzsch-type reaction.10–12
Similarly, the reaction can be initiated from a pyridine precursor that has a leaving group in the C-2 position, and an electrophilic group (carbonyl or cyano group) in the C-3 position that a hydrazine hydrate can attack to form the fused five-membered pyrazole ring (see Fig. 1).13
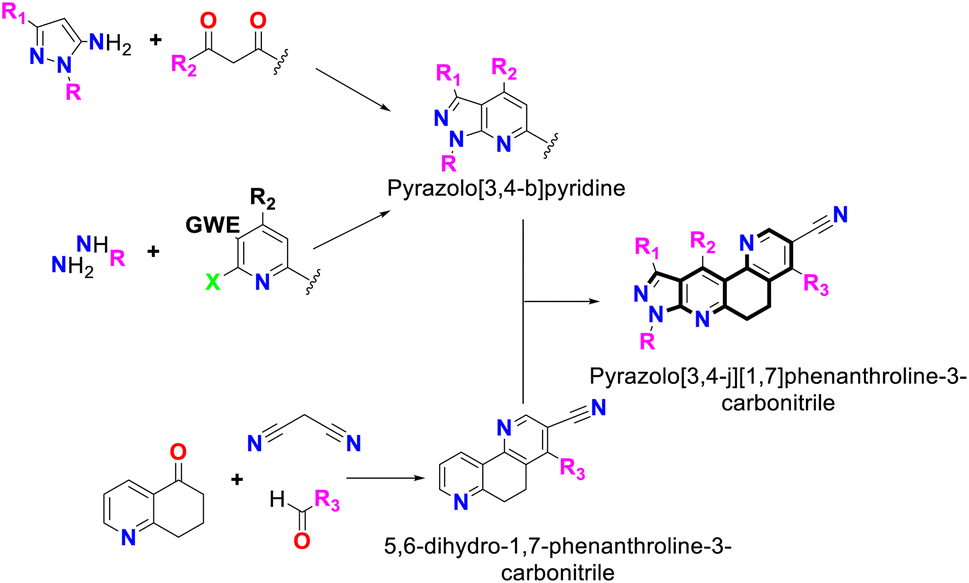 | ||
| Fig. 1 Synthesis of pyrazolo[3,4-b]pyridine-6,8-dihydro-1,7-phenanthroline and its combined-fused derivatives. | ||
On the other hand, phenanthrolines can be synthesized using the classic Skraup reaction from nitroanilines and glycerol in a strongly acid medium.14 The phenanthroline derivatives1–4,6,7,15 have been reported as scaffolds for the preparation of biologically active metal complexes.16–18 Likewise, its 5,6-dihydro phenanthrolinic analogs are of chemical interest and can be obtained through a Michael-type addition reaction among a dihydro-5-quinolinone system, malononitrile and diverse aldehydes.19
Compounds derived from pyrazolo[3,4-b]pyridine and phenanthroline have demonstrated promising inhibitory capacity against cholinesterase enzymes (ChEs) involved in Alzheimer disease (AD) development and antioxidant capacities (AO).20–24 In recent years, replacement therapies with AO drugs have been evaluated and investigated and have shown significant potential to reduce the symptoms and incidence of AD, characterized by a progressive deterioration of cognitive functions.25
One of the characteristics described in patients with AD is the accumulation of extracellular plaques of the β-amyloid peptide (Aβ), and the neurofibrillary tangles of the hyperphosphorylated tau protein.26,27 This reduces the number of functional monomers and contributes to the loss of homeostasis of metal ions.28,29 Furthermore, it has been found that Aβ can generate free radicals that have been strongly correlated with episodes of oxidative stress and neurotoxicity,28,30,31 which has generated a growing interest in the relationship between oxidative stress and neurodegenerative diseases.
Likewise, it is known that with physiological aging the production of radicals or highly oxidizing species increases, and the protection capacity against these species decreases, which generates an increase in an oxidative cellular environment.32 Several studies have been able to link increased brain oxidative stress with cognitive decline caused by normal aging and neurodegenerative diseases.19,33 Oxidative species contribute to neuronal damage in various dementia diseases, including AD, in which oxidative damage is one of the first markers of cognitive dysfunction.34–36
Thus, the present work aimed to synthesize and evaluate the inhibition capacity toward the AChE and BChE enzymes and the AO capacity of a new group of compounds derived from the pyrazolo-fused phenanthroline system, in addition to computationally analyzing their pharmacokinetic properties.
2. Results and discussion
2.1 Chemistry
Initially, compound 5a was synthesized by method A, which consisted of two continuous steps with a possible mechanism displayed in Scheme 2. Here, reagents 1a and 2 combine to generate the (E)-6-benzylidene-3-methyl-1-phenyl-1,6,7,8-tetrahydro-5H-pyrazole[3,4-b]quinolin-5-one chalcone (3a),37 which reacts with ammonium acetate to produce the respective imine, and subsequently undergoes a Michael-type reaction with malononitrile to produce compound 8. This was followed by cyclization, isomerization, and aromatization to generate the final product (5a) in low yield (7%). Therefore, the mechanism suggests that compound 5a was obtained by domino reactions.38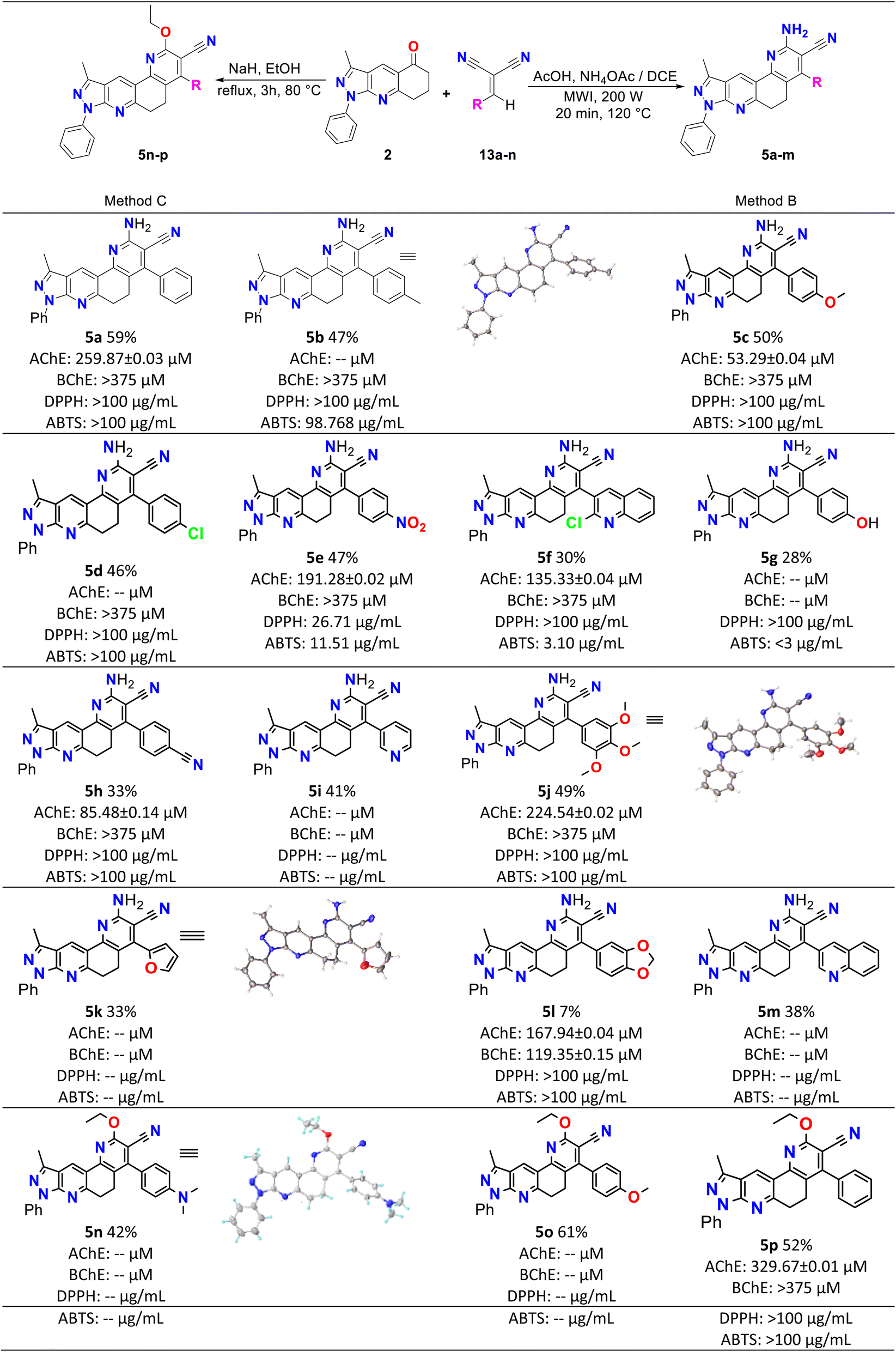 | ||
| Scheme 1 Synthesis, yields, biological and antioxidant activities of pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile derivatives (5a–p). | ||
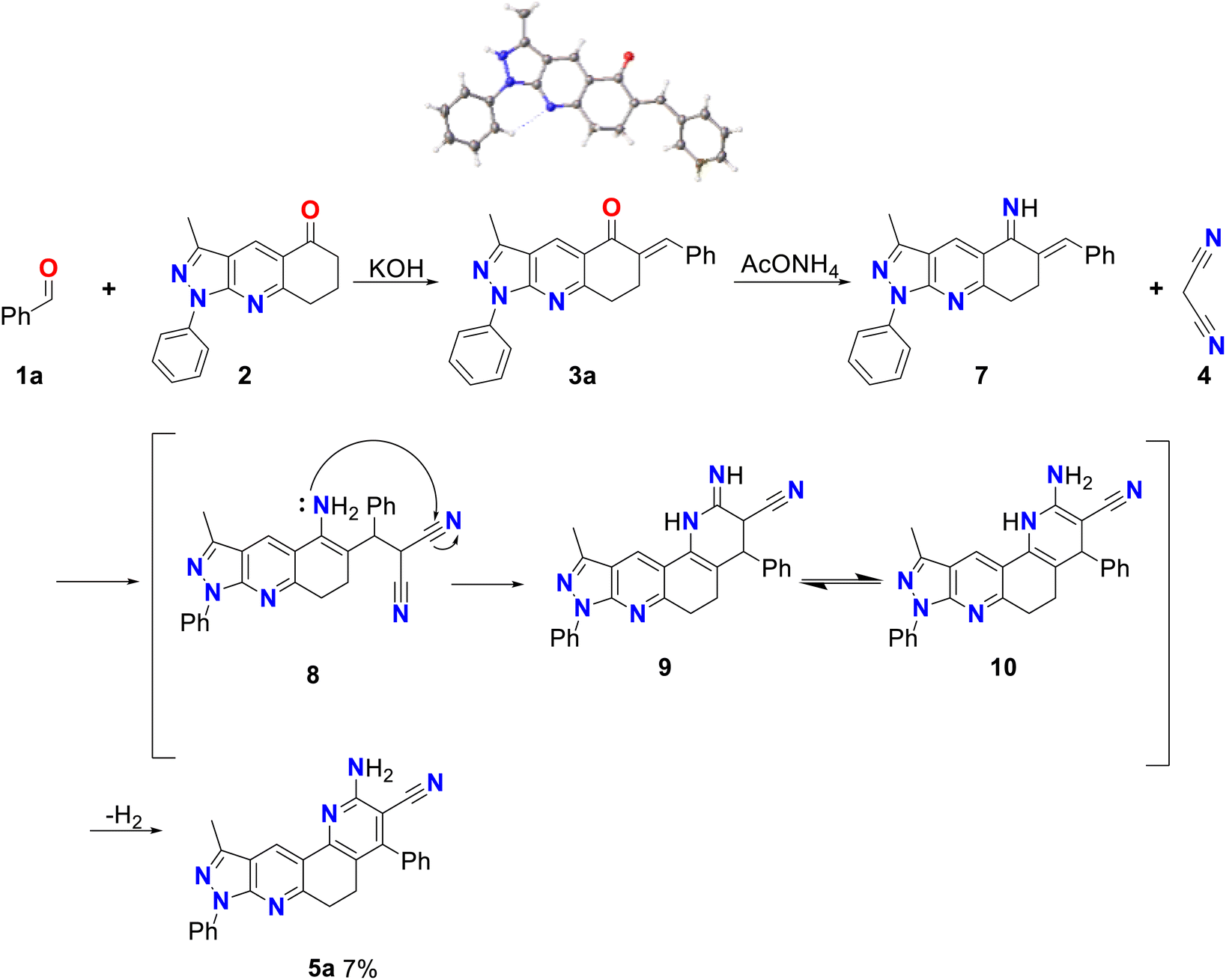 | ||
| Scheme 2 Mechanism by method A of 2-amino-10-methyl-4,8-diphenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5a). | ||
To obtain better yields, method B was proposed (see Scheme 3A), which, like method A, consisted of two successive stages. A possible mechanism for this method begins with a Knoevenagel reaction between an aromatic aldehyde (1a–m) and malononitrile (4), using L-proline as the catalyst, which in its zwitterion form extracts the proton of 4, forming the respective carbanion (4′), which then attacks the protonated aldehyde (1′), forming the corresponding intermediate (1′′) that loses water to produce molecule 13 (see Scheme 3B).39 Simultaneously, compound 2 reacts with ammonium acetate to produce imine 11, which by isomerization is transformed into enamine 12, which subsequently undergoes a Michael reaction with compound 13 to produce molecule 14, which by isomerization transforms into compound 8, which after cyclization, isomerization and aromatization generates the final product 5a–m with yields of up to 59%.38 Structurally, these final compounds have a 2-amino-3-cyanoquinoline system that is of great interest for both reactivity and biological applications. Analogues to this system have been obtained through ionic liquid assisted synthesis,40 which may be used as an alternative synthetic strategy in future works.
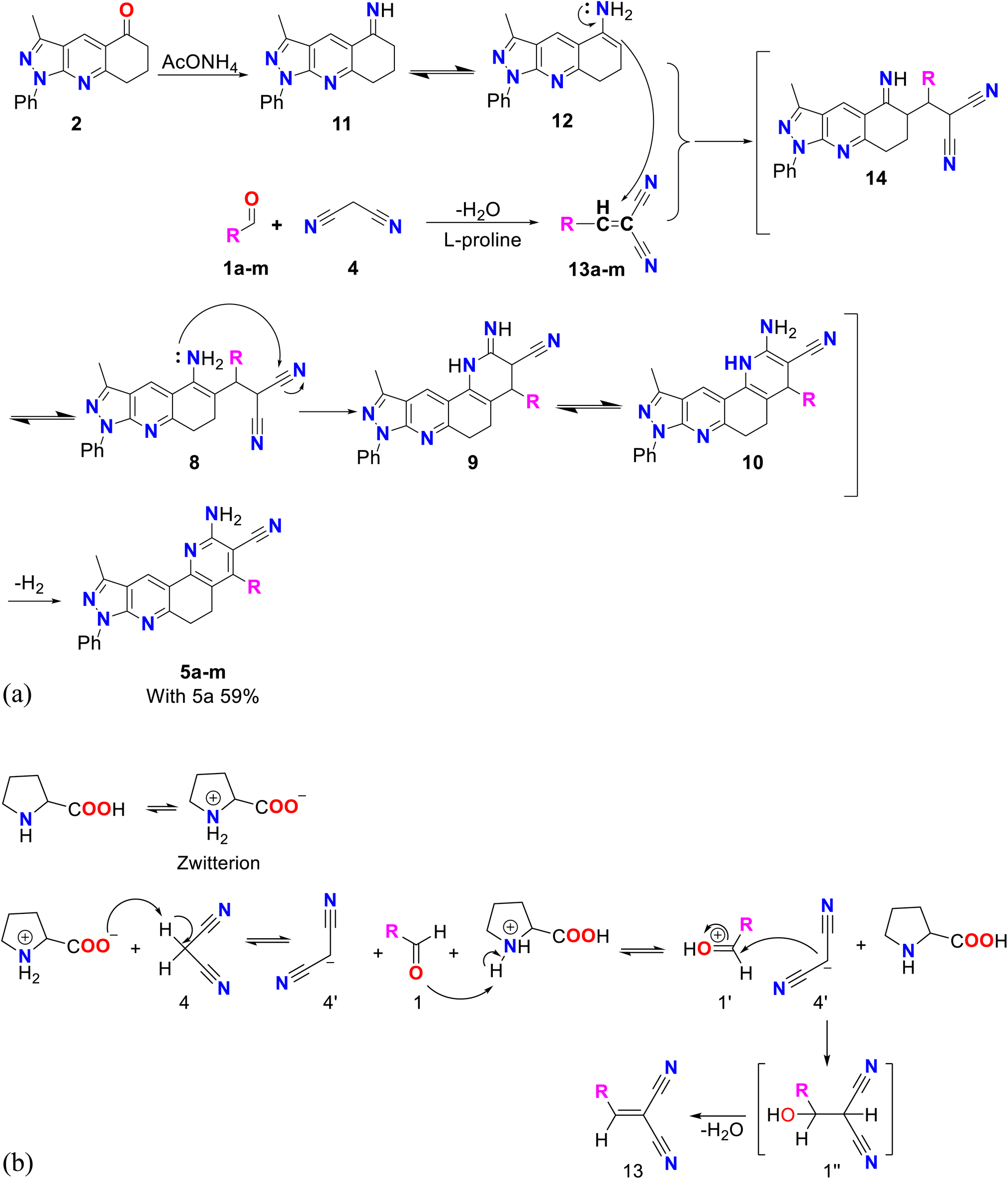 | ||
| Scheme 3 (A) Mechanism by method B of pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5a–m). (B) Mechanism of Knoevenagel reaction catalyzed by L-proline. | ||
Finally, method C was used to obtain compounds 5n–p. In Scheme 4, a possible reaction mechanism is proposed for this method, which, like the previous two, consisted of two successive stages. Initially, a Knoevenagel reaction occurs between the aromatic aldehyde (1n–p) and malononitrile 4, using L-proline as a catalyst to produce molecule 13, in the same way as in methodology B (see Scheme 3B). Simultaneously, compound 2 reacts with sodium hydride to produce the enolate 15, which undergoes a Michael reaction with compound 13 to produce molecule 16. This molecule is cyclized, and by the action of ethanol in the medium compound 17 is formed, which undergoes dehydration and finally aromatization to generate compound 5n–p.41
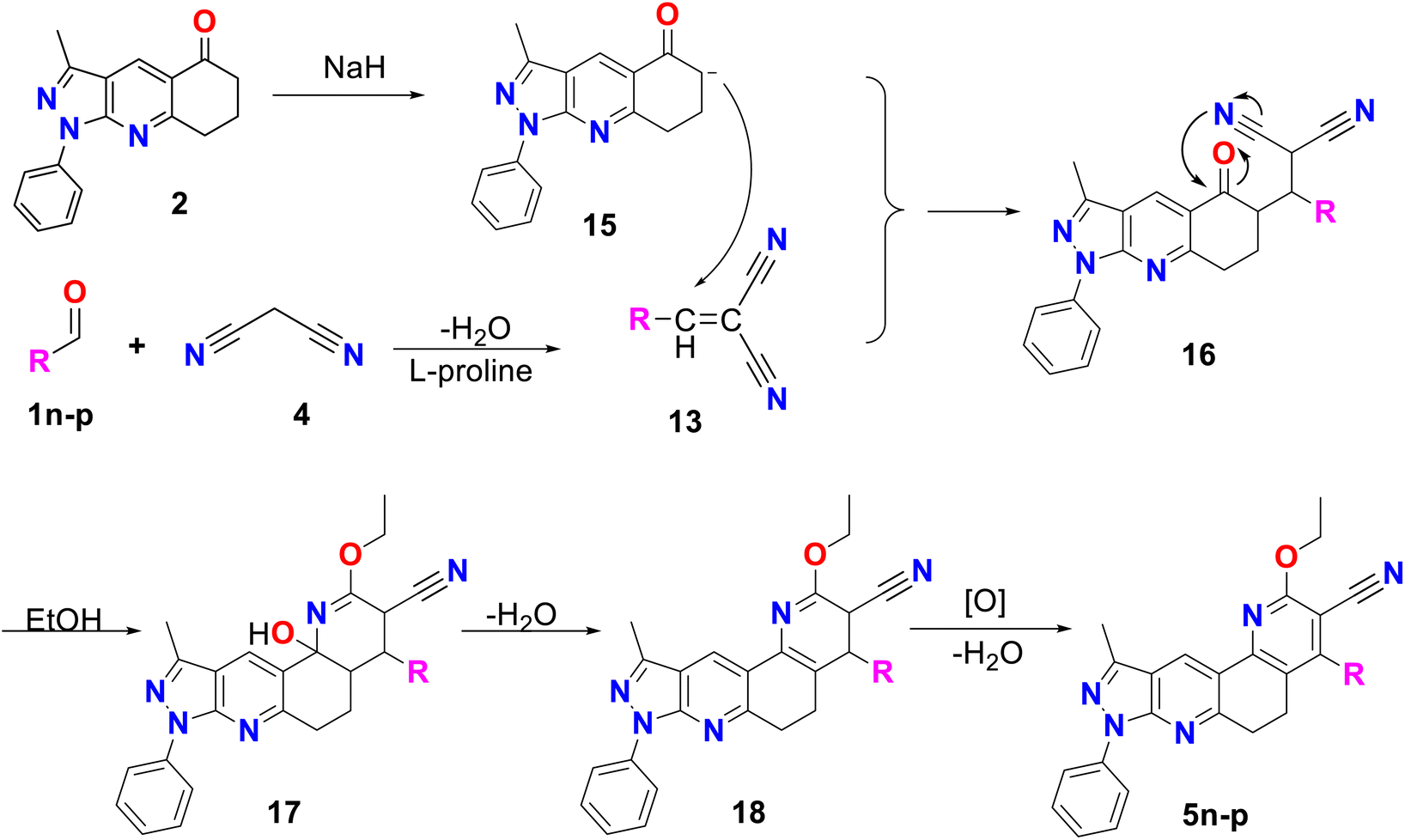 | ||
| Scheme 4 Mechanism by method C of pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile derivatives (5n–p). | ||
On the other hand, to provide relevant information when characterizing the type of phenanthrolines obtained in this work, the vibrational frequencies of molecules 5a and 5p were calculated computationally and compared with those obtained experimentally by infrared spectroscopy. Molecules 5a and 5p are selected because molecule 5a is the base structure of phenanthrolines 5a–m, while 5p is the base structure of compounds 5n–p.
2.2 FT-IR spectra
Experimental and simulated (B3LYP/6-31G*) infrared (IR) spectra of molecules 5a and 5p are shown in Fig. 2. The resulting vibrational frequencies for the optimized geometries, the proposed vibrational assignments, and the IR intensities are given in Table 1. The modes are numbered from the lowest to highest frequency. Comparisons of theoretical and experimental IR spectra indicate that strong vibrations in the experimental spectrum are also strong in the theoretical spectrum.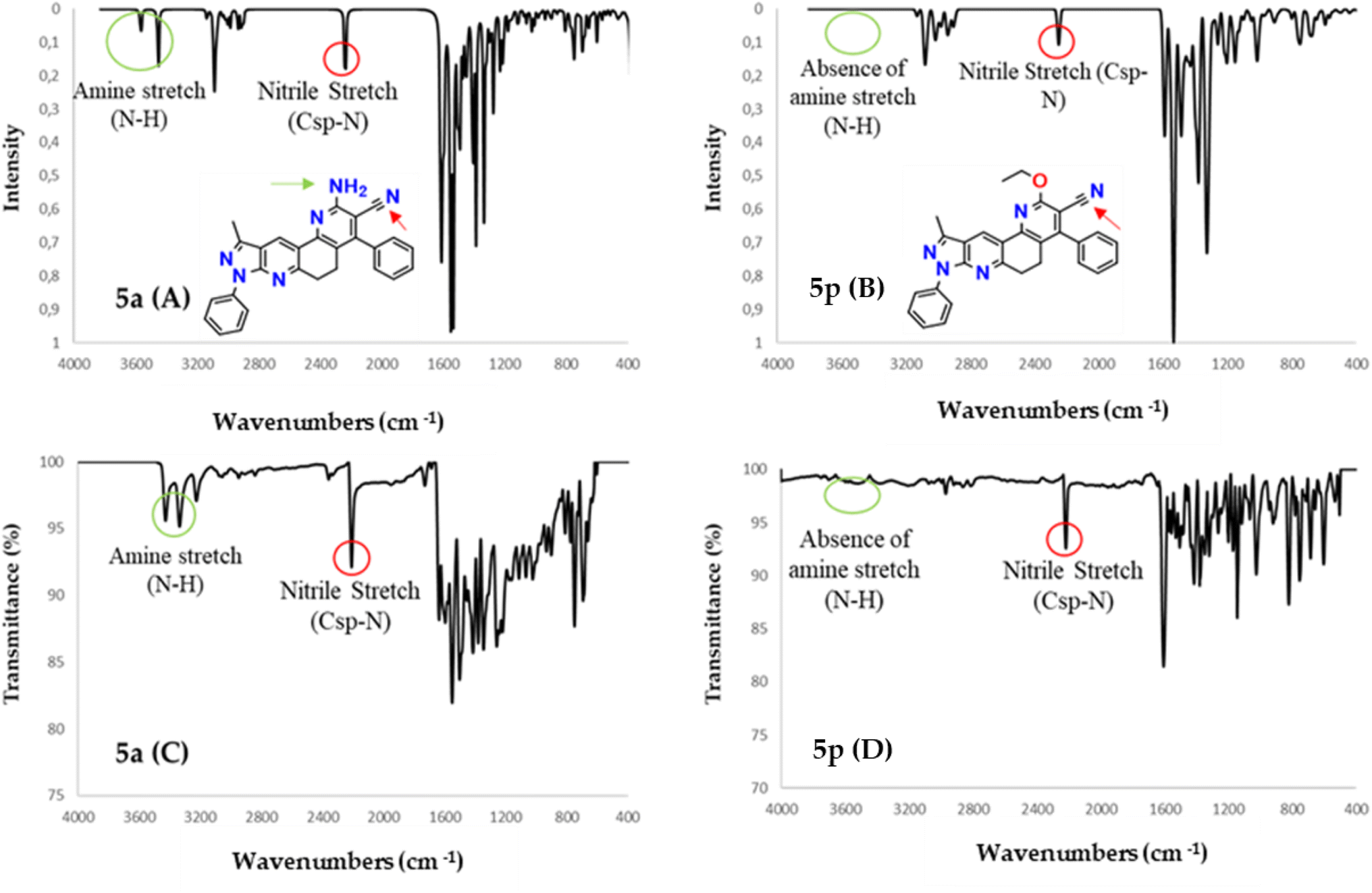 | ||
| Fig. 2 IR spectra calculated with DFT (B3LYP/6-31G*) (A and B) along with experimental (C and D) IR spectra for molecules 5a and 5p. | ||
| Normal mode | B3LYP/6-31G(d) | Experimental in this study | Approximate assignments | |
|---|---|---|---|---|
| Freqa (cm−1) | Intensity (km mol−1) | Freq (cm−1) | ||
| a Scaling factor: 0.963. | ||||
| 5a | ||||
| 1 | 1033 | 0.93 | 1034 | Balancing methyl (C–H) |
| 2 | 1450 | 7.73 | 1452 | Methyl twist (C–H) |
| 3 | 1386 | 143.00 | 1346 | Methyl symmetric bending (C–H) |
| 4 | 1457 | 0.01 | 1466 | Scissoring methylene (C–H) |
| 5 | 1533 | 201.84 | 1593 | Stretch C–C |
| 6 | 1606 | 354.99 | 1610 | Scissor amine (N–H) |
| 7 | 2229 | 114.71 | 2210 | Nitrile stretch (Csp–N) |
| 8 | 2890 | 9.78 | 2898 | Asymmetric stretching methylene (C–H) |
| 9 | 2903 | 29.20 | 2908 | Symmetric stretching methylene (C–H) |
| 10 | 2927 | 24.77 | 2941 | Symmetrical stretching methyl (C–H) |
| 11 | 2975 | 15.05 | 2979 | Asymmetric methyl stretching (C–H) |
| 12 | 3081 | 42.14 | 3078 | Alkene stretch (Csp2–H) |
| 13 | 3445 | 74.54 | 3334 | Symmetric stretching amine (N–H) |
| 14 | 3559 | 35.48 | 3427 | Asymmetric stretching amine (N–H) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| 5p | ||||
| 1 | 1019 | 33.43 | 1026 | Balancing methyl (C–H) |
| 2 | 1155 | 63.7 | 1146 | Tension alkyl ether C–O |
| 3 | 1202 | 79.2 | 1174 | Tension alkyl vinyl ether C–O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) |
| 4 | 1327 | 170.4 | 1321 | Methyl symmetric bending (C–H) |
| 5 | 1533 | 455.77 | 1606 | Stretch CC |
| 6 | 2251 | 62.2 | 2217 | Nitrile stretch (Csp–N) |
| 7 | 2906 | 27.99 | 2908 | Symmetric stretching methylene (C–H) |
| 8 | 2944 | 19.51 | 2966 | Symmetrical stretching methyl (C–H) |
| 9 | 3081 | 41.96 | 3074 | Alkene stretch (Csp2–H) |
As seen in Table 1, there are some useful strong frequencies in the IR to characterize the 5a and 5p molecules, along with their respective derivatives, such as the CC stretching band, around 1600 cm−1. The nitrile stretching (Csp–N) was observed around 2210 cm−1, while the symmetric stretching of methylene (Csp3–H) was observed at about 2905 cm−1. On the other hand, the symmetric stretching of methyl (Csp3–H) and the Csp2–H stretching were observed at about 3080 cm−1.
In addition, bands are observed for fundamental frequencies in identifying the 5a molecule and its derivatives, such as the amine (N–H) snipping signal at 1610 cm−1. The N–H symmetrical stretching signal was seen at approximately 3334 cm−1, and the asymmetric stretching signal of N–H at about 3427 cm−1. Likewise, there are important frequencies for identifying the 5p molecule and its derivatives, such as the alkyl ether (O–C) tension signal at 1146 cm−1 and the alkyl vinyl ether (C–O–C) tension signal at 1174 cm−1.
As can be seen in Table 1, the frequencies calculated with the B3LYP/6-31G*method presented a good agreement with the experimental data. Therefore this computational method is a helpful tool for the structural characterization of the compounds studied.42
2.3 Biological activity
Table 2 shows the results of the biological activity tests. The values obtained in the anticholinesterase tests were expressed as half of the maximum inhibitory concentration (IC50). The results reveal that most of the analyzed compounds presented inhibitory activity of ChEs enzymes. IC50 values were between 53.29 and 329.67 μM for AChE and between 119.3 and >875.17 μM for BChE. Among the compounds synthesized, 5c (IC50 = 53.29 μM) presented the best inhibitory activity against AChE; however, its potency was lower than that of the positive control galantamine (IC50 = 0.57 μM). Furthermore, 5l presented the best inhibition result against the BChE enzyme, with an IC50 = 119.3 μM that was 15 times less potent than galantamine (IC50 = 8.08 μM).| Compound | Anticholinesterase assay | Antioxidant assay | Folde | ||
|---|---|---|---|---|---|
| AChEa (μM) | BChEa (μM) | DPPHb (μg mL−1) | ABTSb (μg mL−1) | ||
| a IC50 ± SEM.b EC50.c Reference compound of the anticholinesterase assay.d Reference compound of the antioxidant assay.e Fold = IC50 of ascorbic acid/IC50 of compounds 5a–p. | |||||
| 5a | 259.87 ± 0.03 | >875.17 | >100 | >100 | — |
| 5b | nd | >847.41 | >100 | 98.77 | 0.28 |
| 5c | 53.29 ± 0.04 | >817.84 | >100 | >100 | — |
| 5d | nd | >810.04 | >100 | >100 | — |
| 5e | 191.28 ± 0.015 | ≥375 | 26.71 | 11.51 | 2.40 |
| 5f | 135.33 ± 0.04 | ≥375 | >100 | 3.10 | 8.91 |
| 5g | nd | nd | >100 | <3 | |
| 5h | 85.48 ± 0.14 | ≥375 | >100 | >100 | — |
| 5i | nd | nd | nd | nd | — |
| 5j | 224.54 ± 0.018 | ≥375 | >100 | >100 | — |
| 5k | nd | nd | nd | nd | — |
| 5l | 167.94 ± 0.04 | 119.35 ± 0.15 | >100 | >100 | — |
| 5m | nd | nd | nd | nd | — |
| 5n | nd | nd | nd | nd | — |
| 5o | nd | nd | nd | nd | — |
| 5p | 329.67 ± 0.01 | ≥375 | >100 | >100 | — |
| Galantaminec | 0.57 ± 0.05 | 8.08 ± 0.02 | — | — | — |
| Ascorbic acidd | — | — | 1.5 ± 0.2 | 27.62 ± 3.5 | 1 |
Molecular docking was used to understand the binding mode of compound 5c, which presented the best inhibitory activity against AChE. As observed in Fig. 3, the ligand did not present any interaction with constituent residues of the active site and, due to its rigidity, it does not fully enter the catalytic cavity, being located at approximately 8 Å from the catalytic triad (residues in yellow). It is found by obstructing access to the stabilized pocket through an aromatic hydrogen bond type interaction between residue D276 and a hydrogen from the phenol ring linked to the pyrazole scaffold. This interaction is observed at the periphery of the active site cavity and is energetically weaker than a classical hydrogen bond.
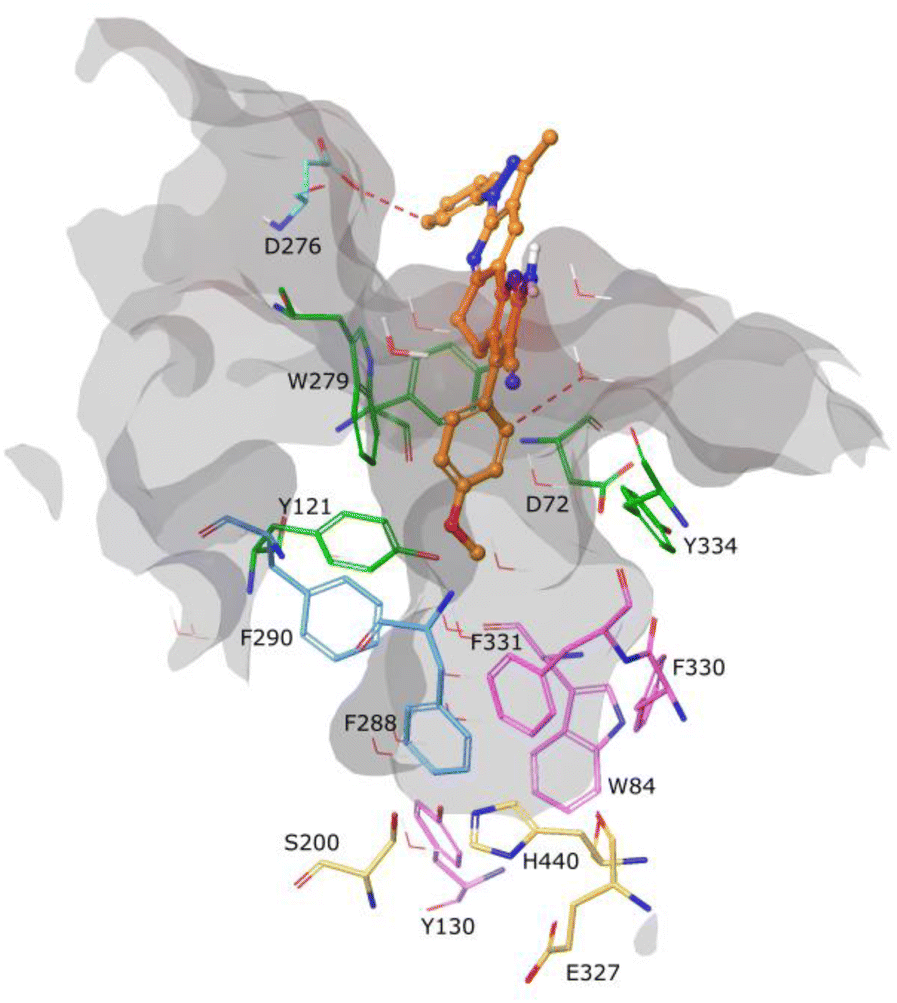 | ||
| Fig. 3 Binding molecular interactions for compound 5c (faded orange) within AChE active sites. Residues of AChE are shown in stick representation. Catalytic triad (yellow), peripheral anionic (orange), anionic site (magenta), and oxyanion hole (blue). Red dotted lines represent aromatic H-bonds. Relevant amino acids are shown in a thin tube. Compound 5c is shown in ball-and-stick representation. The active site cavity is represented in gray. | ||
Similarly, the results of the antioxidant tests were reported as the mean maximum effective concentration (EC50). The results show that all the analyzed compounds presented antioxidant activity in the tests carried out (DPPH and ABTS). The EC50 ranges obtained were 26.71 and >100 μg mL−1 for the DPPH assay and <3 and >100 μg mL−1 for the ABTS assay. Among the compounds analyzed, 5e (EC50 = 26.71 μg mL−1) presented better results in the DPPH test. However, its antioxidant potency did not exceed that of ascorbic acid (EC50 = 1.5 μg mL−1), used as a positive control. Compounds 5e, 5f, and 5g (see Scheme 1) presented EC50 values of 11.51, 3.10, and <3 μg mL−1, respectively, in the ABTS test, being more effective in stabilizing the ABTS radical 2, 9 and more than nine times compared to ascorbic acid (EC50 = 27.62 μg mL−1).
Both ABTS and DPPH radicals can be stabilized by hydrogen atom transfer (HAT) or electron transfer (ET) mechanisms, but the reactivity patterns and mechanism are difficult to interpret.43 With these considerations, it is possible to infer that the antioxidant activity presented by the compounds is due to the presence of nitrogen atoms, which can act on radicals through ET. The “NH2” groups present in compounds 5a–k can contribute to stabilizing both radicals through both mechanisms.44
On the other hand, the results for molecules 5e, and 5f in the ABTS test could be mainly attributed to the presence of the substituents “4-NO2 and 2-chloroquinoline”, respectively. The presence of electron withdrawing groups such as “Cl” and “NO2” has been shown to improve the antioxidant activity of the molecules that contain them.45 In contrast, the molecules that contain electron donor groups such as “Me” or “OMe” tend to impair the antioxidant capacity, which could justify the results for molecules 5e, 5c, 5j, and 5k. Compounds containing quinoline rings have been reported to have significant antioxidant properties, especially if these quinolines contain halogen atoms.46–48
Furthermore, some secondary metabolites are phenolic compounds, whose natural function is to act as antioxidants for plants.49 Numerous studies show that compounds containing phenolic rings tend to show results comparable to or better than positive controls such as ascorbic acid.50–53 It has even been reported that there is a direct relationship between the number of free hydroxyl groups of phenolic compounds and their antioxidant activity, and it has been reported that the position of said hydroxyl groups in aromatic rings also plays an important role in antioxidant capacity.50,53 The good results presented by compound 5g suggest considering further studies to analyze the influence of the number and position of the free hydroxyl group in the phenolic ring on the antioxidant capacity of pyrazolo-phenanthroline derivatives.
Physicochemical properties and drug-likeness (Lipinski's rules54), along with gastrointestinal absorption and brain permeability (Table S1 and Fig. S48†), of the synthesized compounds were determined using the open access software SwissADME (https://www.swissadme.ch).55 The ranges exhibited by the synthesized compounds for each of the parameters that make up Lipinski's criteria are molecular weight MW values (<500) from 428.49 to 518.57, hydrogen bond donor HBD (<5) from 0 to 2, hydrogen bond acceptor HBA (<10) from 4 to 7 and logP (<5) from 3.10 to 4.79. Fig. 4 shows the drug-likeness distribution graphs of synthesized compounds. Thus, 82% of the compounds did not violate Lipinski's rules, and the rest of them only showed one violation corresponding to MW above the established range. Likewise, 71% of the compounds presented high gastrointestinal absorption, while none of them presented permeability of the blood–brain barrier. It is important to highlight that compound 5f is not a substrate for P-glycoprotein (P-gp), which could indicate that its bioavailability would not be reduced (Fig. S1†). According to the biological results presented, the compounds obtained are biologically active and promising to be used as oral drugs.
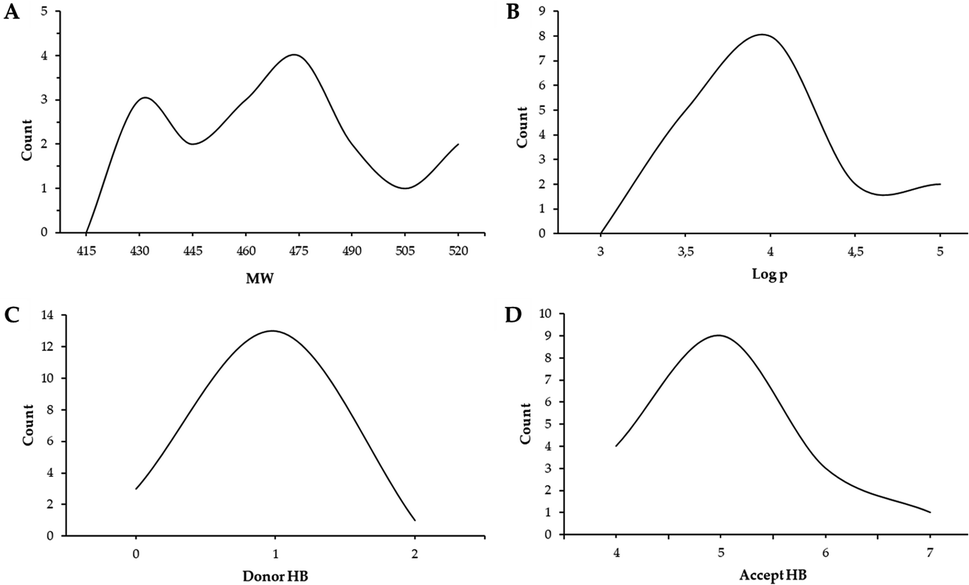 | ||
| Fig. 4 Distribution graphs of synthesized compounds' molecular properties. | ||
DFT calculations were performed for compounds 5a–p to better understand the relationship between their structure and their antioxidant activity. Table S2† displays the HOMO and LUMO energies obtained from those calculations along with the computed electronegativity, chemical hardness, smoothness, and electronic affinity. The results obtained from the computational analysis show that molecule 5e presents the highest LUMO energy, electronic affinity, softness and electronegativity. The first three properties provide information on the ease to accept electrons of a compound along with its susceptibility to nucleophilic attacks. In addition, it has been shown that molecules with high electronegativity display high antioxidant capacity as they tend to form stable radical species. These results are consistent with the biological assays since the 5e molecule presented the best values in the DPPH test and was one of the three molecules that presented antioxidant capacity exceeding the ABTS test positive control. The significant correlation between the electronic properties evaluated and the antioxidant capacity for the 5e molecule suggests that the interaction with the studied radicals is through electronic transfer. No other correlation between the electronic properties and the antioxidant activities was observed.
2.4 X-Ray diffraction measurements
Suitable diffraction-quality single crystals for X-ray structural studies were obtained for compounds 3a, 5b, 5j, 5k and 5n. All compounds crystallize in centrosymmetric space groups (see Table 3 for more details). Molecular and crystal structures of each compound are displayed in Fig. 5. All compounds show normal bond angles and distances.56| Compound | 3a | 5b | 5j | 5k | 5n |
|---|---|---|---|---|---|
| Empirical formula | C24H19N3O | C28H22N6 | C30H26N6O3 | C25H18N6O | C31H28N6O |
| Formula mass, g mol−1 | 365.42 | 442.51 | 518.57 | 418.45 | 500.59 |
| CCDC | 2200163 | 2200166 | 2200164 | 2200165 | 2203148 |
| Collection T, K | 296.49 | 296.86 | 295.94 | 296.33 | 294.94 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/c | P21/c | P21/n | P21/c | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 14.714(2) | 10.4257(14) | 10.0883(8) | 11.971(14) | 8.156(2) |
| b (Å) | 16.279(3) | 13.8104(17) | 12.4670(11) | 14.236(19) | 9.356(2) |
| c (Å) | 7.7182(12) | 16.570(2) | 20.6411(18) | 12.823(15) | 19.728(5) |
| α (°) | 90 | 90 | 90 | 90 | 101.682(5) |
| β (°) | 94.980(2) | 101.663(3) | 100.785(2) | 101.08(4) | 92.413(5) |
| γ (°) | 90 | 90 | 90 | 90 | 114.101(5) |
| V (Å3) | 1841.8(5) | 2336.6(5) | 2550.2(4) | 2144(5) | 1332.6(6) |
| Z | 4 | 4 | 4 | 4 | 2 |
| ρcalcd (g cm−3) | 1.318 | 1.258 | 1.351 | 1.296 | 1.248 |
| Crystal size (mm) | 0.19 × 0.15 × 0.12 | 0.21 × 0.18 × 0.15 | 0.18 × 0.16 × 0.11 | 0.17 × 0.15 × 0.10 | 0.17 × 0.15 × 0.09 |
| F(000) | 768.0 | 928.0 | 1088.0 | 872.0 | 528.0 |
| Abs coeff (mm−1) | 0.082 | 0.078 | 0.090 | 0.084 | 0.079 |
| 2θ range (°) | 17.8 to 59.458 | 5.746 to 52.09 | 5.892 to 54.016 | 5.724 to 56.056 | 5.616 to 53.064 |
| Range h,k,l | −20/20, −22/22, −10/10 | −12/12, −16/17, −20/20 | −12/11, −15/15, −26/26 | −15/15, −18/18, −16/16 | −10/10, −11/10, −24/24 |
| No. total refl. | 29908 |
18109 |
25717 |
20098 |
25207 |
| No. unique refl. | 5043 | 4602 | 5498 | 5099 | 5509 |
| Comp. θ max (%) | 94.5 | 99.9 | 99.8 | 99.5 | 99.7 |
| Max/min transmission | 0.746/0.676 | 0.7453/0.6121 | 0.7454/0.6773 | 0.7457/0.6316 | 0.7454/0.6337 |
| Data/restraints/parameters | 5043/0/254 | 4602/0/336 | 5498/0/323 | 5099/0/295 | 5509/3/376 |
| Final R [I > 2σ (I)] | R1 = 0.0665 wR2 = 0.1614 | R1 = 0.0604 wR2 = 0.1327 | R1 = 0.0574 wR2 = 0.1366 | R1 = 0.0810, wR2 = 0.2074 | R1 = 0.0797 wR2 = 0.2035 |
| R Indices (all data) | R1 = 0.1016 wR2 = 0.1941 | R1 = 0.0963 wR2 = 0.1641 | R1 = 0.0913 wR2 = 0.1633 | R1 = 0.1335 wR2 = 0.2649 | R1 = 0.1508 wR2 = 0.2631 |
| Goodness of fit/F2 | 1.073 | 1.053 | 1.035 | 1.053 | 1.014 |
| Largest diff. Peak/hole (eÅ−3) | 0.36/−0.46 | 0.40/−0.26 | 0.45/−0.30 | 1.15/−0.63 | 0.26/−0.27 |
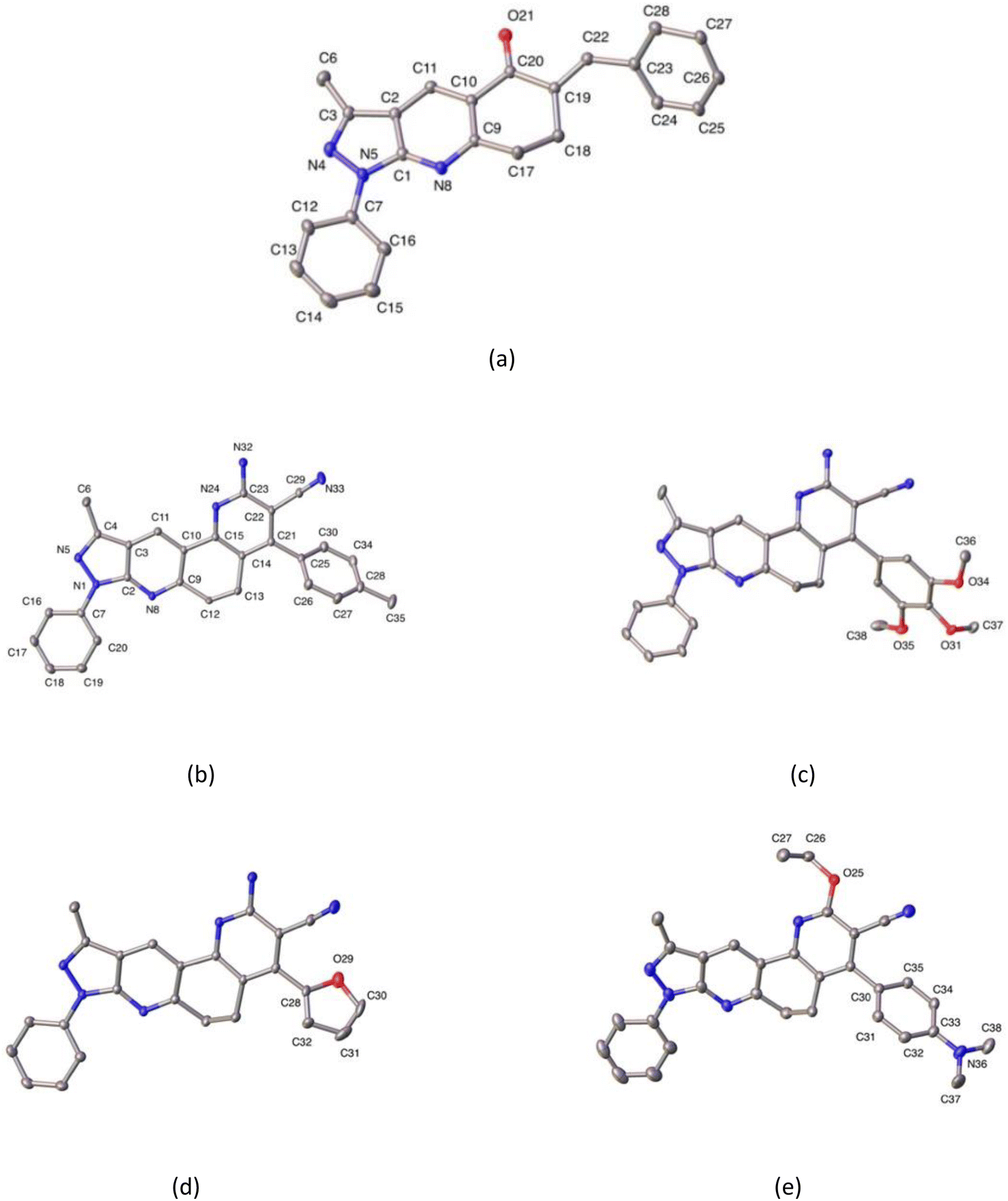 | ||
| Fig. 5 ORTEP plots of compounds 3a (a), 5b (b) and, partially labeled 5j (c), 5k (d), and 5n (e). Hydrogen atoms are omitted for the sake of clarity. Thermal ellipsoids were drawn with 30% of probability. | ||
Additionally, crystal data are summarized in Table 3, while selected bond distances and angles, as well as intra- and intermolecular hydrogen bonds, are provided in Tables 4 and 5. The molecular structure of compound 3a corresponds to three-fused rings of 3-methyl-1H-pyrazole (3MP), and 4,5,6,7,8-tetrahydro-5-quinolinone (THQ) moieties with benzyl and phenyl groups attached to the THQ and 3MP rings, respectively. Meanwhile, the compounds 5b, 5j, 5k, and 5n are four-fused rings composed of 3MP and functionalized 5,6-dihydro-1,7-phenanthroline (DHPhen), with tolyl (5b), 1-(2,3,4-trimethoxy)phenyl (5j), furyl (5k), and 1-(4-N,N′-aminomethyl)phenyl (5n) groups (see Fig. 5 for more details).
| Compound | QT (Å) | θ (°) | φ (°) | Conformation |
|---|---|---|---|---|
| 3a | 0.380(3) | 121.5(5) | 85.4(4) | Screw-boat |
| 5b | 0.287(7) | 90.0(2) | 4.00(2) | Twist-boat |
| 5j | 0.418(3) | 116.1(4) | 93.4(4) | Screw-boat |
| 5k | 0.380(3) | 61.7(5) | 276.9(5) | Screw-boat |
| 5n | 0.467(4) | 67.6(5) | 274.1(6) | Screw-boat |
| Atoms | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H···A (°) |
|---|---|---|---|---|
| a x, 3/2 − y, −1/2 + z.b x, 3/2 − y,1/2 + z.c 1 − x, 1 − y,−z.d 2 − x, 1/2 + y,1/2 − z.e −x, 1 − y, 1 − z.f 1 − x, 2 − y, 1 − z.g −x, 1/2 − y, 1/2 − z. | ||||
| 3a | ||||
| C(17)–H(17B)⋯O(21)a | 0.97 | 2.52 | 3.389 | 150.0 |
| C(18)–H(18B)⋯O(21)b | 0.97 | 2.58 | 3.407 | 143.0 |
|
||||
| 5b | ||||
| N(32)–H(32A)⋯N(33A)c | 0.93 | 2.28 | 3.100 | 158.0 |
| N(32)–H(32A)⋯N(33B)c | 0.88 | 2.28 | 3.10 | 159.0 |
| N(32)–H(32B)⋯N(5)d | 0.93 | 2.29 | 3.201 | 170.0 |
|
||||
| 5j | ||||
| N(32)–H (32B)⋯N(33)e | 0.87 | 2.49 | 3.167 | 136.0 |
|
||||
| 5k | ||||
| N(32)–H (32A)⋯N(33)f | 0.96 | 2.27 | 3.164 | 155.0 |
| N(32)–H(32B)⋯N(5)g | 0.94 | 2.40 | 3.310 | 165.0 |
| C(32)–H(32)⋯N(32)b | 0.93 | 2.55 | 3.410 | 154.0 |
All compounds show dihedral angles between phenyl and fused 3MP rings. The values of these dihedral angles are 20.22(9), 11.45(9), 12.19(11), and 1.84(14)°, respectively. In compound 5n, this small value is caused by an intramolecular hydrogen bond between the phenyl and the fused pyridyl ring, the latter being an almost coplanar fragment.
In the case of compound 3a, this shows a dihedral angle of 44.85(12)° between the benzyl group and the THQ ring. Additionally, all compounds show dihedral angles between their respective substituent ring, and fused DPH in ring of 57.25(9), 73.70(8), 133.25(12)/68.0(9), and 121.18(13)° for 5b, 5j, 5k, and 5n, respectively. Additionally the saturated six-membered ring has two different conformations, according to Cremer & Pople parameters.57 In the cases of compounds 3a, 5j, 5k, and 5n this conformation is screw-boat, and in the case of compound 5b is twist-boat (see Table 4 for more details).
In the crystal structure, the molecules in each compound are stabilized by intra- and intermolecular hydrogen bond interactions C–H⋯N, C–H⋯O, and N–H⋯N. The details of these interactions are depicted in Table 5. In compounds 5b, and 5k N–H⋯N interactions generate a supramolecular 2D aggregate with base vectors [0 1 0], and [2 0 1], and plane (1 0–2); meanwhile, in compounds 5j and 5n, C–H⋯N, and N–H⋯N generate centrosymmetric rings with a graph-set notation R22(12), and R22(16), respectively.58 In compound 3a, the crystal packing is further stabilized by π–π stacking interaction between the pyridyl ring with a distance of 3.890 Å, angle of 6.309°, and slippage of 1.436 Å.
2.5 Conformational analyses
Finally, all compounds show positional disorder over different fragments. In the case of compound 3a, the phenyl ring was modeled over two positions with 0.5 occupancy ratio. In compound 5b, the non-planar ring and nitrile fragment are disordered over three and two positions, respectively. These were modeled using a rigid model with 0.33 and 0.5 occupancy ratios on each part, respectively. In compound 5j the phenyl ring is disordered over two positions. Modeling was done using FVAR in a 66:34 occupancy ratio, and in compound 5n, the ethoxy fragment is disordered over two positions. Modeling was carried out using FVAR in a 70:30 occupancy ratio.
To determine which configuration of compounds 5b, 5j, 5k, and 5n is most stable, DFT energies were computed at different dihedral angle values between atoms 14, 21, 25, and 30 (compounds 5b, 5j, and 5k) or 14, 21, 25, and 35 (compound 5n) (see Fig. 6). Table 6 and Fig. 7 show the conformation energies obtained for the analyzed compounds. These results reveal that for all compounds 5b, 5j, and 5n in addition to the experimental angles determined by the crystallographic study other energy minima with nearly the same energy exist at angles around −90° and 120°. In addition, high rotational barriers of approximately 2.4 eV at angles of −180, 0, and 180° prevent the conversion between rotamers. On the other hand, for compound 5k the experimental conformation is 0.07 eV more stable than the other possible rotamers, and it has a much lower rotational barrier of 0.14 eV at 0°. These observations are in qualitative agreement with the positional disorder observed in the X-ray analysis.
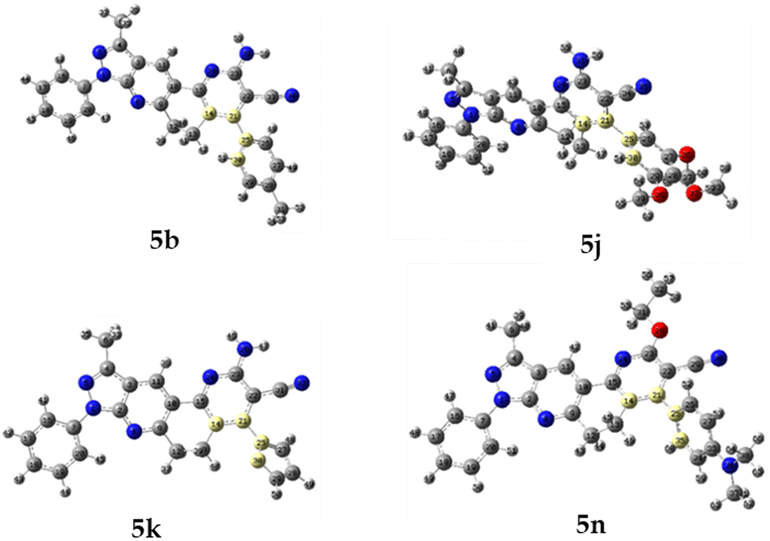 | ||
| Fig. 6 Modified dihedral angle for molecules 5b, 5j, 5k, and 5n. | ||
| Dihedral angle | E relative (eV) | |||
|---|---|---|---|---|
| 5b | 5j | 5k | 5n | |
| a Dihedral angle obtained in crystallographic analysis. | ||||
| −180 | 2.48 | 2.37 | 0.32 | 2.39 |
| −120 | 0.01 | 0.20 | 0.07 | 0.01 |
| −90 | 0.01 | 0.00 | 0.13 | 0.03 |
| 0 | 2.45 | 2.11 | 0.14 | 2.43 |
| 90 | 0.01 | 0.01 | 0.13 | 0.03 |
| 120 | 0.00 | 0.00 | 0.07 | 0.00 |
| 180 | 2.48 | 2.37 | 0.32 | 2.39 |
| −59.1a | 0.00 | — | — | — |
| 67.8 | 0.06 | — | — | — |
| −57.6 | — | 0.12 | — | — |
| 71.3a | — | 0.00 | — | — |
| −47.5a | — | — | 0.00 | — |
| 41 | — | — | 0.08 | — |
| −59.6a | — | — | — | 0.01 |
| 62.8 | — | — | — | 0.09 |
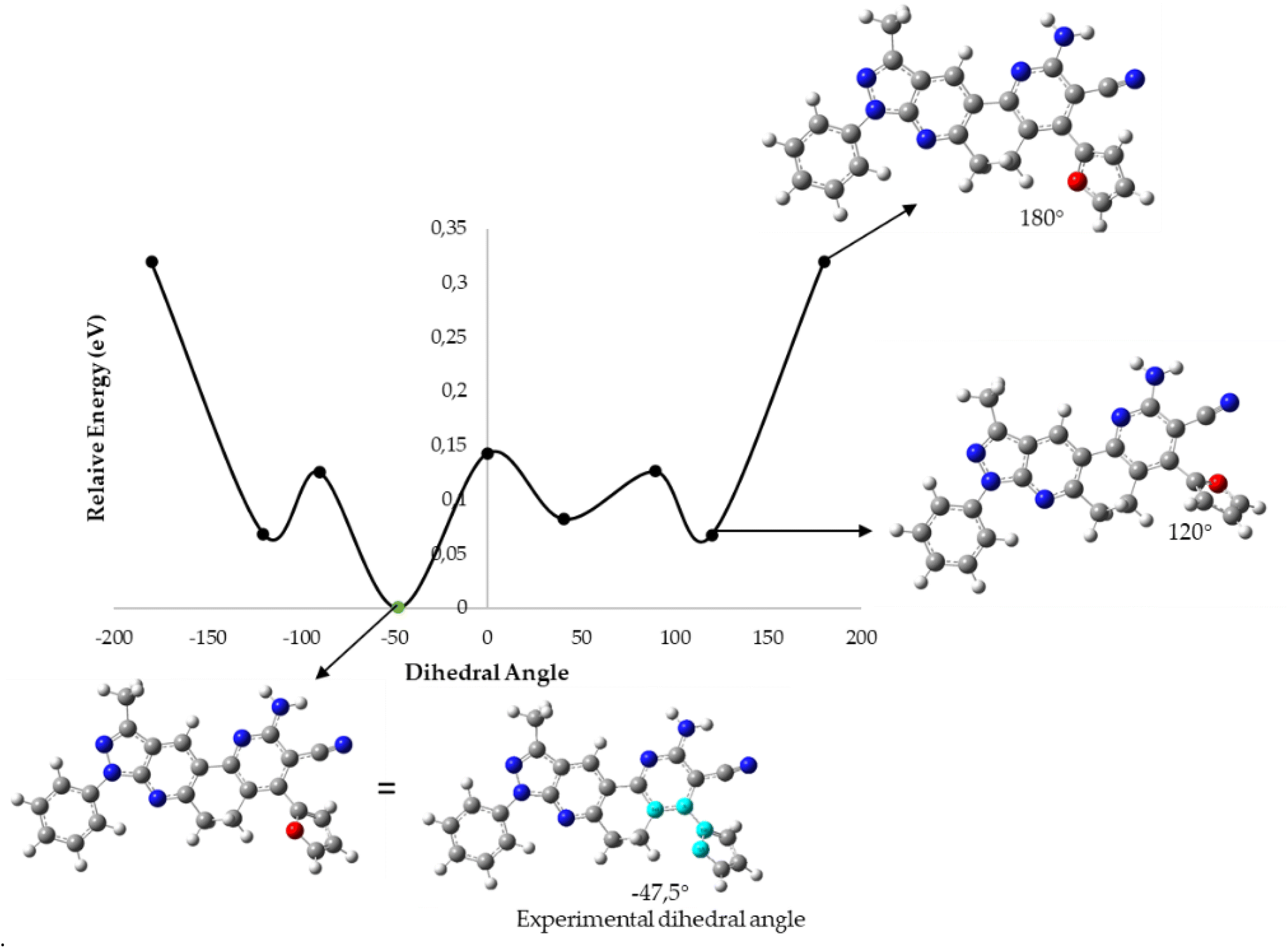 | ||
| Fig. 7 B3LYP/6-31G* potential energy curve along the dihedral angle between the substituent ring and the fused DPH rings for the 5k molecule. | ||
3. Experimental section
3.1 General information
The experiments were performed in Discover microwave apparatus (CEM Corporation, Matthews, NC, USA), while the ultrasonic irradiation was performed using a Branson Model 1510, 115v, 1.9 L Ultrasonic Bath with a mechanical timer (60 min with continuous hold), and a heater switch, 47 kHz. The 1H and 13C NMR spectra (400 MHz for proton and 100 MHz for carbon) were recorded on an AM-400 spectrometer (Bruker, Rheinstetten, Germany) using CDCl3 and DMSO-d6 as solvents. Tetramethylsilane (TMS) was used as an internal standard. IR spectra (KBr granules, 500–4000 cm−1) were recorded on a NEXUS 670 FT-IR spectrophotometer (Thermo Nicolet, Madison, WI, USA). ESI-MS and ESI-MS/MS high-resolution mass spectrometry was carried out on a high resolution (Q) hybrid quadrupole time of flight (TOF) mass spectrometer (Waters/Micromass Q-TOF micro, Manchester, UK) with a constant nebulizer temperature of 100 °C. Melting points (uncorrected) were measured on an IA9100 electrothermal melting point device (Stone, Staffs, UK). The progress of the reaction was monitored by thin-layer chromatography (TLC) using silica gel 60 (Merck, Darmstadt, Germany). All reagents were purchased from Merck or Sigma Aldrich (St. Louis, MO, USA) and used without further purification. The final purification of all the products for the analysis was carried out by column chromatography, with the use of silica gel 60 (Merck 7734) with a particle size that fluctuates between 0.063 and 0.200 mm, with the eluent phase of ether mixtures of petroleum:ethyl acetate in increasing polarity.
3.2 Chemistry
3.2.2.1 2-Amino-10-methyl-4,8-diphenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5a). Yield 59%; orange solid; mp 258–260 °C; IR (KBr, cm−1): 3421, 3328, 3224, 2949, 2839, 2363, 2203, 1633, 1555, 1499, 1340, 1218, 1022, 747; 1H NMR (400 MHz, DMSO-d6); 2.60–2.64 (m, 5H), 3.03–3.07 (m, 2H), 6.86 (m, 2H), 7.28 (t, J = 8.0 Hz, 1H), 7.39 (dd, J = 8.0, 1.6 Hz, 2H), 7.50–7.56 (m, 6H), 8.26 (d, J = 8.0 Hz, 2H), 8.88 (s, 1H); 13C NMR (400 MHz, DMSO-d6); 12.2 (CH3), 23.3 (CH2), 31.5 (CH2), 89.0 (C), 116.1 (C), 116.6 (C), 117.3(C), 119.9 (2 × CH), 123.6 (CH), 125.4 (CH), 127.3 (CH), 128.3 (2 × CH), 128.6 (C), 128.8 (CH), 129.0 (2 × CH), 129.1 (CH), 135.8 (C), 139.0 (C), 143.9 (C), 149.9 (C), 153.1 (C), 153.4 (C), 158.9 (C), 160.0 (C); HRMS (ESI, m/z): calcd for C27H21N6 [M + H]+ 429.1828 found 429.1830.
3.2.2.2 2-Amino-10-methyl-8-phenyl-4-(p-tolyl)-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5b). Yield 47%; green solid; mp 218–220 °C; IR (KBr, cm−1): 3494, 3473, 3354, 2952, 2214, 1612, 1601, 1546, 1502, 1375, 1245, 1111, 822; 1H NMR (400 MHz, CDCl3); 2.45 (s, 3H), 2.71 (s, 3H), 2.78–2.82 (m, 2H), 3.12–3.15 (m, 2H), 3.88 (s, 3H), 5.21 (s, 2H), 7.25–7.30 (m, 3H), 7.33–7.35 (m, 2H), 7.51 (t, J = 8.0 Hz, 2H), 8.30 (d, J = 8.0 Hz, 2H), 8.94 (s, 1H); HRMS (ESI, m/z): calcd for C28H23N6 [M + H]+ 443.1984 found 443.1989.
3.2.2.3 2-Amino-4-(4-methoxyphenyl)-10-methyl-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthr-oline-3-carbonitrile (5c). Yield 50%; yellow solid; mp 253–255 °C; IR (KBr, cm−1): 3485, 3355, 2949, 2212, 1598, 1508, 1245, 1178, 1028; 1H NMR (400 MHz, CDCl3); 2.70 (s, 3H), 2.79–2.83 (m, 2H), 3.11–3.15 (m, 2H), 3.88 (s, 3H), 5.19 (s, 2H), 7.03–7.06 (m, 3H), 7.27–732 (m, 2H), 7.50 (t, J = 8.0 Hz, 2H), 8.30 (d, J = 8.0 Hz, 2H), 8.91 (s, 1H); HRMS (ESI, m/z): calcd for C28H23N6O [M + H]+ 459.1933 found 459.1930.
3.2.2.4 2-Amino-4-(4-chlorophenyl)-10-methyl-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5d). Yield 46%; orange solid; mp 282–284 °C; IR (KBr, cm−1): 3467, 3349, 3236, 2961, 2214, 1630, 1598, 1497, 1433, 1245, 1328, 1082, 753; 1HNMR (400 MHz, DMSO-d6); 2.61–2.65 (m, 5H), 3.06–3.09 (m, 2H), 6.91 (m, 2H), 7.30 (t, J = 7.5 Hz, 1H), 7.46 (d, J = 8.6 Hz, 2H), 7.52–7.56 (m, 2H), 7.61–763 (m, 2H), 8.27 (d, J = 8.8 Hz, 2H), 8.91 (s, 1H); HRMS (ESI, m/z): calcd for C27H20ClN6 [M + H]+ 463.1438 found 463.1441.
3.2.2.5 2-Amino-10-methyl-4-(4-nitrophenyl)-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5e). Yield 46%; orange solid; mp 282–284 °C; IR (KBr, cm−1): 3467, 3349, 3236, 2961, 2214, 1630, 1598, 1497, 1433, 1245, 1328, 1082, 753; 1HNMR (400 MHz, DMSO-d6); 2.45–2.46 (m, 2H), 2.65 (m, 3H), 2.97–2.98 (m, 2H), 6.70 (m, 2H), 7.31 (t, J = 7.0 Hz, 1H), 7.44–7.46 (m, 2H), 7.53–7.57 (m, 2H), 7.63 (d, J = 8.3 Hz, 2H), 8.26 (d, J = 8.6 Hz, 2H), 9.01 (s, 1H); HRMS (ESI, m/z): calcd for C27H20N7O2 [M + H]+ 474.1678 found 474.1683.
3.2.2.6 2-Amino-4-(2-chloroquinolin-3-yl)-10-methyl-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5f). Yield 30%; yellow solid; mp 295–297 °C; IR (KBr, cm−1): 3410, 3320, 3050,2960, 2900, 2200, 1560, 1500, 1410, 1330, 1140, 1030, 756; 1HNMR (400 MHz, DMSO-d6); 2.60 (t, J = 7.3 Hz, 2H), 2.64 (s, 3H), 3.09–3.12 (m, 2H), 7.11 (s, 2H), 7.03–7.06 (m, 3H), 7.29 (t, J = 7.3 Hz, 1H), 7.51–7.56 (m, 2H), 7.77 (t, J = 7.5 Hz, 1H), 7.93–7.97 (m, 1H), 8.08–8.15 (m, 2H), 8.26 (t, J = 9.3 Hz, 2H), 8.67 (s, 1H), 8.95 (s, 1H); HRMS (ESI, m/z): calcd for C30H21ClN7 [M + H]+ 514.1547 found 514.1548.
3.2.2.7 2-Amino-4-(4-hydroxyphenyl)-10-methyl-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5g). Yield 28%; red solid; mp > 300 °C; IR (KBr, cm−1): 3390, 3336, 3217, 3064,2937, 2870, 2204, 1583, 1510, 1441, 1176, 758; NMR (400 MHz, DMSO-d6); 2.15–2.17 (m, 2H), 2.60 (s, 3H), 2.89–3.00 (m, 2H), 6.78 (d, J = 8.8 Hz, 2H), 7.23–7.29 (m, 4H), 7.53 (t, J = 7.1 Hz, 2H), 7.66 (bs, 2H), 8.26 (d, J = 7.6 Hz, 2H), 8.31 (s, 1H), 9.72 (s, 1H); HRMS (ESI, m/z): calcd for C27H23N6O [M + 3H]+ 447.1933 found 447.2064.
3.2.2.8 2-Amino-4-(4-cyanophenyl)-10-methyl-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5h). Yield 33%; yellow solid; mp > 300 °C; IR (KBr, cm−1): 3426, 3355, 3056, 2927, 2208, 1590, 1546, 1411, 1334, 1251, 904, 746; 1H NMR (400 MHz, DMSO-d6); 2.60–2.64 (m, 5H), 3.06–3.10 (m, 2H), 6.98 (m, 2H), 7.30 (t, J = 6.7 Hz, 1H), 7.53 (t, J = 7.9 Hz, 2H), 7.65 (t, J = 7.6 Hz,H), 8.04 (d, J = 8.3 Hz, 2H), 8.26 (t, J = 8.8 Hz, 2H), 8.92 (s, 1H); HRMS (ESI, m/z): calcd for C28H20N7 [M + H]+ 454.1780 found 454.1808.
3.2.2.9 2-Amino-10-methyl-8-phenyl-4-(pyridin-3-yl)-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5i). Yield 41%; yellow solid; mp 245–247 °C; 1H NMR (400 MHz, CDCl3); 2.70 (s, 3H), 2.99–3.04 (m, 2H), 3.11–3.15 (m, 2H), 5.26 (s, 2H), 7.27–7.32 (m, 1H), 7.48–7.52 (m, 3H), 7.72–7.76 (m, 1H), 8.28 (t, J = 8.9 Hz, 2H), 8.62 (dd, J = 1.5, 12.5 Hz, 1H), 8.76 (d, J = 4.9 Hz, 1H), 8.93 (s, 1H); HRMS (ESI, m/z): calcd for C26H20N7 [M + H]+ 430.1780 found 430.1781.
3.2.2.10 2-Amino-10-methyl-8-phenyl-4-(3,4,5-trimethoxyphenyl)-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5j). Yield 49%; yellow solid; mp 273–275 °C; IR (KBr, cm−1): 3460, 3348, 3055, 2936,2833, 2212, 1589, 1417, 1240, 1125, 1001, 758; 1H NMR (CDCl3, 400 MHz): 2.60 (s, 3H), 3.20 (s, 4H), 3.83 (s, 9H), 6.64 (s, 2H), 7.20–7.25 (m, 2H), 7.44 (t, J = 7.5 Hz, 2H), 7.81 (m, 1H), 8.23 (d, J = 7.8 Hz, 2H), 8.74 (s, 1H); HRMS (ESI, m/z): calcd for C30H26N6O3 [M + H]+ 519.2145 found 519.2164.
3.2.2.11 2-Amino-4-(furan-2-yl)-10-methyl-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5k). Yield 33%; red crystal; mp 219–221 °C; IR (KBr, cm−1): 1H NMR (400 MHz, CDCl3); 2.67 (s, 3H), 3.31 (t, J = 6.4 Hz, 2H) 3.40 (t, J = 6.2 Hz, 2H), MHz, CDCl3); 2.67 (s, 3H), 3.31 (t, J = 6.4 Hz, 2H) 3.40 (t, J = 6.2 Hz, 2H), 6.54 (s, 1H), 6.75–6.76 (m, 1H), 7.26–7.31 (m, 2H), 7.51 (t, J = 7.5 Hz, 2H), 7.60 (s, 1H), 7.65 (s, 1H), 8.29 (d, J = 8.3 Hz, 2H), 8.79 (s, 1H); HRMS (ESI, m/z): calcd for C25H18N6NaO [M + Na]+ 441.1440 found 441.1260.
3.2.2.12 2-Amino-4-(benzo[d][1,3]dioxol-5-yl)-10-methyl-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5l). Yield 7%; yellow solid; mp > 300 °C; IR (KBr, cm−1): 3431, 3336, 3221,3053, 2937, 2854, 2210, 1720, 1593, 1493, 1355, 1248, 1093, 1038, 926, 818; 1H NMR (400 MHz, CDCl3); 2.71 (s, 3H), 3.29 (bs, 2H), 6.06 (s, 2H), 6.92 (d, J = 7.8 Hz, 1H), 7.02–7.06 (m, 2H), 7.31–7.35 (m, 1H), 7.54 (t, J = 7.6 Hz, 2H), 7.90 (s, 1H), 8.32 (t, J = 8.1 Hz, 2H), 8.85 (s, 1H); HRMS (ESI, m/z): calcd for C28H20N6NaO2 [M + Na]+ 495.1545 found 495.1547.
3.2.2.13 2-Amino-10-methyl-8-phenyl-4-(quinolin-3-yl)-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5m). Yield 38%; orange solid; mp 258–260 °C; IR (KBr, cm−1): 3419, 3062, 2958 2927, 2202, 1599, 1462, 1277, 1122, 1072, 744; 1H NMR (400 MHz, DMSO-d6); 2.65 (s, 3H), 2.69–2.75 (m, 2H), 3.08–3.13 (m, 2H), 7.01 (s, 2H), 7.29 (t, J = 7.5 Hz, 1H), 7.52–7.55 (m, 2H), 7.73 (t, J = 7.6 Hz, 1H), 7.87–7.91 (m, 1H), 8.12 (dd, J = 8.1, 12.9 Hz, 2H), 8.28 (d, J = 7.6 Hz, 2H), 8.54 (s, 1H), 8.9–8.96 (m, 2H); HRMS (ESI, m/z): calcd for C30H21N7 [M]+ 479.1858 found 479.2041; calcd for C30H20N7 [M − H]+ 478.1780 found 478.2022.
:7) as the eluent, resulting in compounds 5n–p.
3.2.3.1 4-(4-(Dimethylamino)phenyl)-2-ethoxy-10-methyl-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5n). Yield 42%; orange crystal; mp > 300 °C IR (KBr, cm−1): 3039, 2970, 2870, 2212, 1649, 1556, 1254, 1349, 1107, 835; 1H NMR (400 MHz, CDCl3); 1.56 (t, J = 7.1 Hz, 3H), 2.72 (s, 3H), 3.04 (s, 3H), 3.12–3.14 (m, 2H), 3.36–3.38 (m, 2H), 4.69 (q, J = 7.1 Hz, 2H), 6.81 (d, J = 8.8 Hz, 2H), 7.24–7.26 (t, J = 8.0 Hz, 3H), 7.43 (m, 2H), 8.26 (d, J = 8.0 Hz, 2H), 8.8.84 (s, 1H).
3.2.3.2 2-Ethoxy-4-(4-methoxyphenyl)-10-methyl-8-phenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5o). Yield 61%; yellow solid; mp > 300 °C; 1H NMR (400 MHz, CDCl3); 1.52 (t, J = 7.0 Hz, 3H), 2.65 (s, 3H), 2.79 (t, J = 7.2 Hz, 2H), 3.08 (t, J = 7.2 Hz, 2H), 3.83 (s, 3H), 4.63 (q, J = 7.0 Hz, 2H), 7.00 (d, J = 8.0 Hz, 2H), 7.23 (t, J = 8.0 Hz, 3H), 7.43 (t, J = 8.0 Hz, 2H), 8.26 (d, J = 8.0 Hz, 2H), 8.72 (s, 1H); 13C NMR (100 MHz, CDCl3); 12.5 (CH3), 14.4 (CH3), 24.1 (CH2), 31.8 (CH2), 55.2 (CH3), 63.0 (CH2), 95.1 (C), 114.1 (2 × CH), 115.5 (C), 116.6 (C), 120.4 (2 × CH), 121.9 (C), 123.6 (C), 125.4 (CH), 127.0 (C), 127.1 (CH), 128.8 (2 × CH), 129.9 (2 × CH), 139.3 (C), 143.8 (C), 150.4 (C), 152.5 (C), 155.2 (C), 159.4 (C), 160.2 (C), 162.9 (C); HRMS (ESI, m/z): calcd for C30H25N5O2 [M]+ 487.2008 found 487.2138.
3.2.3.3 2-Ethoxy-10-methyl-4,8-diphenyl-6,8-dihydro-5H-pyrazolo[3,4-j][1,7]phenanthroline-3-carbonitrile (5p). Yield 52%; yellow solid; mp > 300 °C; IR (KBr, cm−1): 3049, 2968, 2864, 2218, 1606, 1506, 1381, 1254, 1146, 1024, 822, 754; 1H NMR (400 MHz, CDCl3); 1.44 (t, J = 7.0 Hz, 3H), 2.59 (s, 3H), 2.69 (t, J = 7.2 Hz, 2H), 3.01 (t, J = 7.2 Hz, 2H), 4.57 (q, J = 7.0 Hz, 2H), 7.14 (t, J = 8.0 Hz, 1H), 7.22 (d, J = 8.0 Hz, 2H), 7.35–7.41 (m, 5H), 8.17 (d, J = 8.0 Hz, 2H), 8.70 (s, 1H); 13C NMR (100 MHz, CDCl3); 12.5 (CH3), 14.5 (CH3), 24.1 (CH2), 31.8(CH2), 63.1 (CH2), 95.1 (C), 115.3 (C), 116.7 (C), 120.6 (2 × CH), 121.8 (C), 123.6 (C), 125.6 (CH), 127.3 (CH), 128.3 (2 × CH), 128.8 (2 × CH), 128.9 (2 × CH), 129.2 (CH), 135.1 (C), 139.3 (C), 143.9 (C), 150.5 (C), 152.8 (C), 155.4 (C), 159.6 (C), 162.9 (C); HRMS (ESI, m/z): calcd for C29H24N5O [M + H]+ 458.1981 found 458.2120.
3.3 Biological activity
| Percentage of free radical removal DPPH˙+ = 100 × (1 − AE/AD) | (1) |
| Percentage of free radical removal ABTS˙+ = [(A0 − As)/A0] × 100 2 |
3.4 X-Ray diffraction measurements
Crystals of compounds 3a, 5b, 5j, 5k and 5n were measured and their diffraction data were collected at ca. 296 K with a D8 Venture diffractometer equipped with a bidimensional CMOS Photon 100 detector, using graphite monochromated Mo-Kα (λ = 0.71073 Å). The diffraction frames were integrated using the APEX3 package62 and were corrected for absorptions with SADABS. The structure of the title compound was solved by intrinsic phasing63 using the OLEX 2 program.64 All the structures were then refined with the full-matrix least-squares method based on F2 (SHELXL-2014).63 X-ray crystal structure determinations, CIF files containing tables of crystallographic parameters, bond lengths, bond angles, and a list of structure factors have been deposited in the Cambridge Crystallographic Data Centre (CCDC no. 2200163–2200166 and 2203148).3.5 Computational methods
Density functional theory (DFT) was employed to optimize the ground state geometries and to compute the vibrational frequencies of the 5a–5p molecules. These calculations were performed with the Gaussian 16 computational package using the B3LYP functional and the 6-31G* basis set.65–70 The calculated vibrational frequencies were scaled by 0.9627.The ionization potential (IP) and the electronic affinity (EA) were estimated from the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies as IP = −εHOMO and as EA = −εLUMO. These estimates were employed to compute the electronegativity (χ), chemical hardness (η) and softness (S) parameters as χ = (IP + EA)/2, η = (IP − EA)/2 and S = 1/η.53
Molecular docking was performed using Glide71 (Schrödinger Release 2021-2: 2021d. Glide. Schrödinger, LLC New York, NY.) software within the Schrödinger Small-Molecule Drug Discovery Suite. The atomic coordinates for human AChE protein were extracted from the Protein Data Base (PDB ID: 4EY7). LigPrep (Schrödinger Release 2021-2: 2021c. LigPrep. Schrödinger, LLC New York, NY.) and Preparation Wizard (Schrödinger Release 2021-2:, 2021b. Protein Preparation Wizard. Epik, Schrödinger, LLC, New York, NY.) module were used to obtain the minimized energy structure of the ligand and optimize the AChE enzyme, respectively, under the OPLS-AA force field.72 The absorption, distribution, metabolism, and excretion properties of the compounds were predicted using SwissADME (https://www.swissadme.ch) open access software.
4. Conclusions
In conclusion, a total of 16 new pyrazol[3,4-j][1,7]phenanthroline-3-carbonitrile derivatives were synthesized, with amino (5a–m) and ethoxy (5n–p) groups in position 2. Two synthetic methodologies were used to obtain derivatives 5a–m, where method B presented better results with respect to reaction times, yields and easy workability. Likewise, a third methodology was used to efficiently obtain the 5n–p compounds.The synthesized compounds were evaluated for their antioxidant activity with the aid of DPPH and ABTS assays. Compound 5e stood out for having the best results of DPPH assay, presenting an EC50 value of 25 μg mL−1. Additionally, in the ABTS test, compounds 5e, 5f, and 5g presented the best results with EC50 values of 11.51, 3.1 and <3 μg mL−1, evidencing even better results than the ascorbic acid that was used as a positive control. Structure-antioxidant activity relationship studies indicate that compound 5e has a strong ability to accept electrons, supporting a radical state, and it can also react with soft species such as DPPH and ABTS radicals. Additionally, the compounds showed moderate activity as cholinergic inhibitors, with compounds 5c and 5l being the most active on AChE and BChE, respectively. In addition, the pharmacokinetic and pharmacodynamic properties analyzed in silico demonstrate that the new molecular entities are as oral drugs promising to be used. Finally, DFT studies reasonably predicted the conformations with lower relative energy of the crystallized compounds 5b, 5j, 5k, and 5n.
Author contributions
Efraín Polo-Cuadrado: investigation, methodology, and writing – original draft; Cristian Rojas-Peña: investigation, methodology, and writing – original draft; Karen Acosta-Quiroga: investigation, methodology, and writing – original draft; Lorena Camargo-Ayala: investigation, methodology, and writing – original draft; Iván Brito: writing – original draft, visualization, and software; Jonathan Cisterna: writing – original draft, visualization, and software; Félix Moncada: data curation, software, visualization, conceptualization, and writing – original draft; Jorge Trilleras: writing – review & editing and supervision; Yeray A. Rodríguez-Núñez: conceptualization, methodology, and writing – original draft; Margarita Gutiérrez: project administration, resources, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Research Group of the Laboratory of Organic Synthesis and Biological Activity of the University of Talca. E. P-C. thanks FONDECYT Post-Doctoral Fellowship No. 3220681. Fondecyt Project 1200531 and the ANID National Doctorate Scholarship 2019 Folio No. 21190020. The authors also acknowledge FONDEQUIP program (EQM 130021, 160063 and 180024).Notes and references
- T. Shiro, T. Fukaya and M. Tobe, Eur. J. Med. Chem., 2015, 97, 397–408 CrossRef CAS PubMed.
- F. Salehian, H. Nadri, L. Jalili-Baleh, L. Youseftabar-Miri, S. N. Abbas Bukhari, A. Foroumadi, T. Tüylü Küçükkilinç, M. Sharifzadeh and M. Khoobi, Eur. J. Med. Chem., 2021, 212, 113034 CrossRef CAS PubMed.
- M. S. Salem and M. A. M. Ali, Biol. Pharm. Bull., 2016, 39, 473–483 CrossRef CAS PubMed.
- L. W. Mohamed, M. A. Shaaban, A. F. Zaher, S. M. Alhamaky and A. M. Elsahar, Bioorg. Chem., 2019, 83, 47–54 CrossRef CAS PubMed.
- R. Lin, P. J. Connolly, Y. Lu, G. Chiu, S. Li, Y. Yu, S. Huang, X. Li, S. L. Emanuel, S. A. Middleton, R. H. Gruninger, M. Adams, A. R. Fuentes-Pesquera and L. M. Greenberger, Bioorg. Med. Chem. Lett., 2007, 17, 4297–4302 CrossRef CAS PubMed.
- M. A. El-borai, H. F. Rizk, M. F. Abd-Aal and I. Y. El-Deeb, Eur. J. Med. Chem., 2012, 48, 92–96 CrossRef CAS PubMed.
- T. El-Sayed Ali, Eur. J. Med. Chem., 2009, 44, 4385–4392 CrossRef CAS PubMed.
- M. S. Nafie, A. M. Amer, A. K. Mohamed and E. S. Tantawy, Bioorg. Med. Chem., 2020, 28, 115828 CrossRef CAS PubMed.
- R. Aggarwal, S. Kumar, R. Sadana, A. Guzman and V. Kumar, Synth. Commun., 2021, 51, 3308–3324 CrossRef CAS.
- D. M. Volochnyuk, S. V. Ryabukhin, A. S. Plaskon, Y. V. Dmytriv, O. O. Grygorenko, P. K. Mykhailiuk, D. G. Krotko, A. Pushechnikov and A. A. Tolmachev, J. Comb. Chem., 2010, 12, 510–517 CrossRef CAS PubMed.
- S. A. Ibrahim, H. F. Rizk, M. A. El-Borai and M. E. Sadek, J. Iran. Chem. Soc., 2021, 18, 1391–1404 CrossRef CAS.
- G. Pandey, S. Bhowmik and S. Batra, Org. Lett., 2013, 15, 5044–5047 CrossRef CAS PubMed.
- B. Orlikova, W. Chaouni, M. Schumacher, M. Aadil, M. Diederich and G. Kirsch, Eur. J. Med. Chem., 2014, 85, 450–457 CrossRef CAS PubMed.
- H. Saggadi, D. Luart, N. Thiebault, I. Polaert, L. Estel and C. Len, RSC Adv., 2014, 4, 21456–21464 RSC.
- S. M. H. Sanad and A. E. M. Mekky, Chem. Biodivers., 2021, 19, e202100500 Search PubMed.
- M. McCann, A. Kellett, K. Kavanagh, M. Devereux and A. L. S. Santos, Curr. Med. Chem., 2012, 19, 2703–2714 CrossRef CAS PubMed.
- N. L. Stevanović, T. P. Andrejević, A. Crochet, T. Ilic-Tomic, N. S. Drašković, J. Nikodinovic-Runic, K. M. Fromm, M. I. Djuran and B. Đ. Glišić, Polyhedron, 2019, 173, 114112 CrossRef.
- N. D. Savić, S. Vojnovic, B. Glišić, A. Crochet, A. Pavic, G. V. Janjić, M. Pekmezović, I. M. Opsenica, K. M. Fromm, J. Nikodinovic-Runic and M. I. Djuran, Eur. J. Med. Chem., 2018, 156, 760–773 CrossRef PubMed.
- M. A. Smith, K. Hirai, K. Hsiao, M. A. Pappolla, P. L. R. Harris, S. L. Siedlak, M. Tabaton and G. Perry, J. Neurochem., 1998, 70, 2212–2215 CrossRef CAS PubMed.
- E. H. Edinsha Gladis, K. Nagashri and J. Joseph, Inorg. Chem. Commun., 2020, 122, 108232 CrossRef CAS.
- L. Tabrizi, D. Q. Dao and T. A. Vu, RSC Adv., 2019, 9, 3320–3335 RSC.
- H. Mutlu Gençkal, J. Mol. Struct., 2020, 1209, 127917 CrossRef.
- T. Pan, S. Xie, Y. Zhou, J. Hu, H. Luo, X. Li and L. Huang, Bioorg. Med. Chem. Lett., 2019, 29, 2150–2152 CrossRef CAS PubMed.
- T. Umar, S. Shalini, M. K. Raza, S. Gusain, J. Kumar, P. Seth, M. Tiwari and N. Hoda, Eur. J. Med. Chem., 2019, 175, 2–19 CrossRef CAS PubMed.
- C. Bancher, C. Brunner, H. Lassmann, H. Budka, K. Jellinger, G. Wiche, F. Seitelberger, I. Grundke-Iqbal, K. Iqbal and H. M. Wisniewski, Brain Res., 1989, 477, 90–99 CrossRef CAS PubMed.
- M. P. Murphy and H. Levine, J. Alzheimer's Dis., 2010, 19, 311–323 Search PubMed.
- D. S. Eisenberg and M. R. Sawaya, Nature, 2017, 547, 170–171 CrossRef CAS PubMed.
- M. G. Savelieff, S. Lee, Y. Liu and M. H. Lim, ACS Chem. Biol., 2013, 8, 856–865 CrossRef CAS PubMed.
- K. P. Kepp, Prog. Neurobiol., 2016, 143, 36–60 CrossRef CAS PubMed.
- A. C. Andorn and R. N. Kalaria, J. Alzheimer's Dis., 2000, 2, 69–78 CAS.
- M. Rosini, E. Simoni, A. Milelli, A. Minarini and C. Melchiorre, J. Med. Chem., 2014, 57, 2821–2831 CrossRef CAS PubMed.
- J. Apelt, M. Bigl, P. Wunderlich and R. Schliebs, Int. J. Dev. Neurosci., 2004, 22, 475–484 CrossRef CAS PubMed.
- F. Collin, C. Cheignon and C. Hureau, Biomarkers Med., 2018, 12, 201–203 CrossRef CAS PubMed.
- J. M. Carney, P. E. Starke-Reed, C. N. Oliver, R. W. Landum, M. S. Cheng, J. F. Wu and K. A. Floyd, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3633–3636 CrossRef CAS PubMed.
- A. Kontush, Free Radical Biol. Med., 2001, 31, 1120–1131 CrossRef CAS PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed.
- E. Polo, K. Ferrer-Pertuz, J. Trilleras, J. Quiroga and M. Gutiérrez, RSC Adv., 2017, 7, 50044–50055 RSC.
- J. Wang, Z. Li, X. Wang, Y. Zhou and C. Guo, Heterocycles, 2015, 91, 49–63 CrossRef CAS.
- M. Venkatanarayana and P. K. Dubey, Synth. Commun., 2012, 42, 1746–1759 CrossRef CAS.
- R. Chatterjee, S. Bhukta and R. Dandela, J. Heterocycl. Chem., 2022, 59, 633–654 CrossRef CAS.
- S. M. Roopan, A. Bharathi, J. Palaniraja, K. Anand and R. M. Gengan, RSC Adv., 2015, 5, 38640–38645 RSC.
- M. Karabacak, M. Çinar and M. Kurt, J. Mol. Struct., 2008, 1–3, 28–35 CrossRef.
- D. C. Christodouleas, C. Fotakis, A. Nikokavoura, K. Papadopoulos and A. C. Calokerinos, Food Anal. Methods, 2015, 8, 1294–1302 CrossRef.
- M. P. Rigobello, R. Stevanato, F. Momo, S. Fabris, G. Scutari, R. Boscolo, A. Folda and A. Bindoli, Free Radical Res., 2004, 38, 315–321 CrossRef CAS PubMed.
- W. B. Li, X. P. Qiao, Z. X. Wang, S. Wang and S. W. Chen, Bioorg. Chem., 2020, 105, 104427 CrossRef CAS PubMed.
- M. Orhan Puskullu, B. Tekiner and S. Suzen, Mini-Rev. Med. Chem., 2013, 13, 365–372 CAS.
- E. A. Wilhelm, A. T. Ferreira, M. P. Pinz, A. S. Dos Reis, A. G. Vogt, A. L. Stein, G. Zeni and C. Luchese, An. Acad. Bras. Cienc., 2017, 89, 457–467 CrossRef CAS PubMed.
- M. Sankaran, C. Kumarasamy, U. Chokkalingam and P. S. Mohan, Bioorg. Med. Chem. Lett., 2010, 20, 7147–7151 CrossRef CAS PubMed.
- S. Dall'Acqua, R. Cervellati, M. C. Loi and G. Innocenti, Food Chem., 2008, 106, 745–749 CrossRef.
- D.-O. Kim, Y. L. Chang and C. Y. Lee, Crit. Rev. Food Sci. Nutr., 2004, 44, 253–273 CrossRef CAS PubMed.
- K. Acosta-Quiroga, C. Rojas-Peña, L. S. Nerio, M. Gutiérrez and E. Polo-Cuadrado, RSC Adv., 2021, 11, 21926–21954 RSC.
- I. Gülçin and A. Dastan, J. Enzyme Inhib. Med. Chem., 2007, 22, 685–695 CrossRef PubMed.
- F. Sonmez, Z. Gunesli, B. Z. Kurt, I. Gazioglu, D. Avci and M. Kucukislamoglu, Mol. Diversity, 2019, 23(4), 829–844 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- D. Cremer, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 498–500 CrossRef.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- G. L. Ellman, K. D. Courtney, V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, LWT-Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS.
- S. Khwaja, K. Fatima, M. Hasanain, C. Behera, A. Kour, A. Singh, S. Luqman, J. Sarkar, D. Chanda, K. Shanker, A. K. Gupta, D. M. Mondhe and A. S. Negi, Eur. J. Med. Chem., 2018, 151, 51–61 CrossRef CAS PubMed.
- Bruker AXS INC, APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 2002, 98, 11623–11627 CrossRef.
- W. J. Hehre, K. Ditchfield and J. A. Pople, J. Chem. Phys., 2003, 56, 2257 CrossRef.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.08, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2200163–2200166 and 2203148. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra05532e |
| This journal is © The Royal Society of Chemistry 2022 |