
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycal mediated synthesis of piperidine alkaloids: fagomine, 4-epi-fagomine, 2-deoxynojirimycin, and an advanced intermediate, iminoglycal†
Hemender R. Chanda,
Mritunjay K. Tiwaric and
Asish K. Bhattacharya *ab
*ab
aDivision of Organic Chemistry, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pashan, Pune, 411008, Maharashtra, India. E-mail: ak.bhattacharya@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, India
cPhysical and Material Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pashan, Pune, 411008, Maharashtra, India
First published on 17th November 2022
Abstract
Glucal and galactal are transformed into 2-deoxyglycolactams, which are important building blocks in the synthesis of biologically active piperidine alkaloids, fagomine and 4-epi-fagomine. In one of the strategies, reduction of 2-deoxyglycolactam-N-Boc carbonyl by lithium triethylborohydride (Super-Hydride®) has been exploited to generate lactamol whereas reduction followed by dehydration was utilized as the other strategy to functionalize the C1–C2 bond in the iminosugar substrate. The strategies provide the formal synthesis of 2-deoxynojirimycin, nojirimycin and nojirimycin B. DFT studies were carried out to determine the reason for the failure of the formation of the 2-deoxygalactonojirimycin derivative. Further, DFT studies suggest that phenyl moieties of protecting groups and lone pairs of oxygen in carbamate group plays a vital role in deciphering the conformational space of the reaction intermediates and transition-state structures through cation–π or cation–lone pair interactions. The influence of these interactions is more pronounced at low temperature when the entropy factor is small.
Introduction
Polyhydroxylated alkaloids are of considerable interest as potential therapeutic agents, they are also used as an important tool to understand biological recognition processes.1 Hence, their synthesis and biological activity studies have assumed significance. These occur in the species of Streptomyces, family Leguminosae, Solanaceae, and Convolvulaceae and possess therapeutic potential.1 Nojirimycin 1 (Fig. 1) was the first natural polyhydroxylated piperidine alkaloid isolated from a Streptomyces filtrate in 1966 by Inouye et al.2 1,2-Dideoxy-iminosugars exemplify a small but essential class of glycosidase inhibitors.3 One of the members of this family, fagomine 5a, was isolated from the seeds of Japanese buckwheat Fagopyrum esculentum australe Moench4 and also from the seeds of Castanospermum australe5 (Leguminosae). It has also been reported that 4-epi-fagomine 5b (Fig. 1) acts as a potent glycosidase inhibitor.6 The batzellasides 6 are a novel class of C-alkylated iminosugars originally isolated from Batzella sp., a marine sponge from Madagascar7a and are active against Staphylococcus epidermidis. Their unique structural features resemble an intriguing extension of the iminosugar frameworks, hence their synthesis7b,c has attracted the attention of researchers who practice contemporary drug discovery.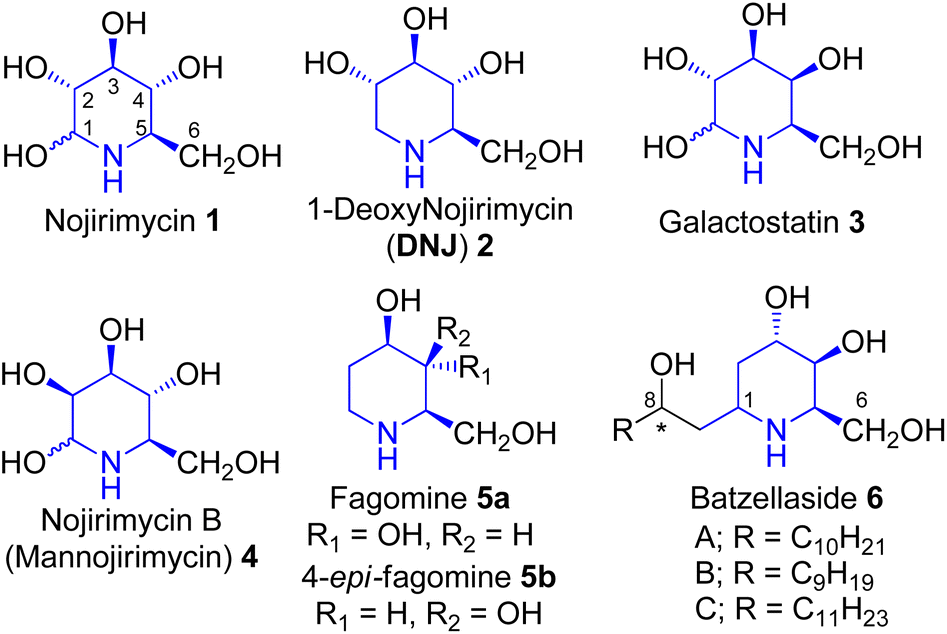 | ||
| Fig. 1 Some common piperidine alkaloids of therapeutic value. | ||
Results and discussion
Reported synthesis of fagomine and 4-epi-fagomine involves either use of carbohydrate building blocks8,9 or non-carbohydrate precursors.10 Although nojirimycin 1 is very active glycosidase inhibitor,2b it has been observed that the deoxy derivatives displays a broad range of activities and are much more stable than nojirimycin 1 itself thereby attracting interest of synthetic chemists on the synthesis of deoxynojirimycin.11We have designed the synthesis of fagomine 5a and 4-epi-fagomine 5b using carbohydrate building blocks as shown in Scheme 1. Fagomine 5a and 4-epi-fagomine 5b could be synthesized by deprotection and reduction of the corresponding 2-deoxyglycolactams 7a/b. By lactamization of δ-hydroxy amides 8a/b, 2-deoxyglycolactams 7a/b could be obtained. δ-Hydroxy amides 8a/b could be accessed by aminolysis of 2-deoxyglycolactones 9a/b which in turn could be obtained from readily available tri-O-benzyl-D-glucal 10a or tri-O-benzyl-D-galactal 10b respectively (Scheme 1). Nitrogen from ammonia is the source of the heteroatom in our planned synthesis of piperidine alkaloids.
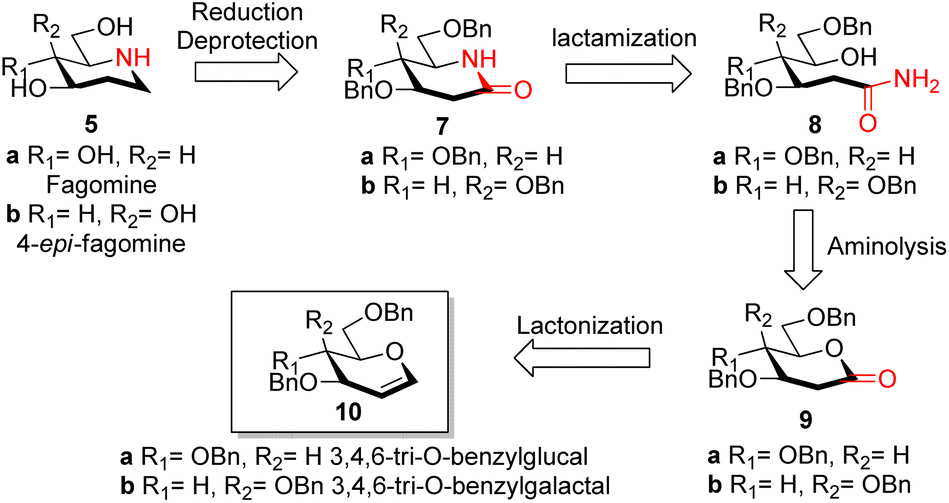 | ||
| Scheme 1 Retrosynthetic plan for the synthesis of fagomine 5a and 4-epi-fagomine 5b. | ||
With our research interests in sesquiterpene lactones12a and sugar-derived 2-deoxy-δ-lactone transformations,12b–d 2-deoxyglycolactones 9a/b were readily synthesized from glycals 10a/b by known literature protocol.13 Further manipulating 2-deoxyglycolactone 9a/b we thought that by using Pandit's method14a we could open the 2-deoxyglycolactone 9a/b with 7 N methanolic ammonia. Hence, the treatment of lactones 9a/b with methanolic ammonia (aminolysis) furnished the ring-opened compound δ-hydroxy amides 8a (82%) and 8b (96%) from 2-deoxyglycolactones 9a and 9b respectively (Scheme 2). With this step nitrogen atom is incorporated in the molecule by cleavage of C–O bond in 2-deoxyglycolactone 9.
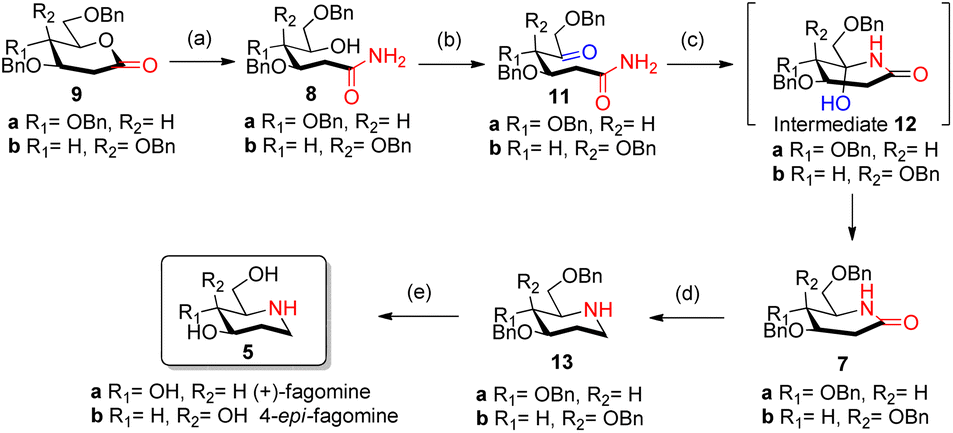 | ||
| Scheme 2 Synthesis of fagomine 5a and 4-epi-fagomine 5b; reagents and conditions: (a) NH3 in CH3OH 7 N solution, (6 h, 82% 8a), (11 h, 96% 8b); (b) AC2O/DMSO, (23 h, 59% 11a), (26 h, 64% 11b); (c) HCOOH/NaBH3CN, CH3CN reflux, (4.5 h, 59% 7a), (4.5 h, 59% 7b). (d) LiAlH4/THF reflux, (4 h, 49% 13a), (2 h, 41% 13b); (e) ref. 8 (H2, Pd/C, EtOH, HCl, 85%). | ||
δ-Hydroxy amides 8a/b under Albright Goldmann oxidation condition i.e. Ac2O and DMSO at rt provided the desired δ-keto amides 11a/b which without purification was carried on for the next step. The crude of the above reaction was then treated with formic acid and NaBH3CN, to furnish the desired 2-deoxyglycolactams 7a/b. This crucial step comprises an intramolecular reductive amination, which involved condensation of the amine with the ketone to furnish the cyclized product. Formic acid complexes with the ketone carbonyl and increases its electrophilicity to facilitate the attack of amine, leading to the formation of a new C–N bond. The iminium ion 14 formed in situ after dehydration from the intermediate 12a/b undergoes NaBH3CN reduction to form the desired 2-deoxyglycolactams 7a/b (Fig. 2).
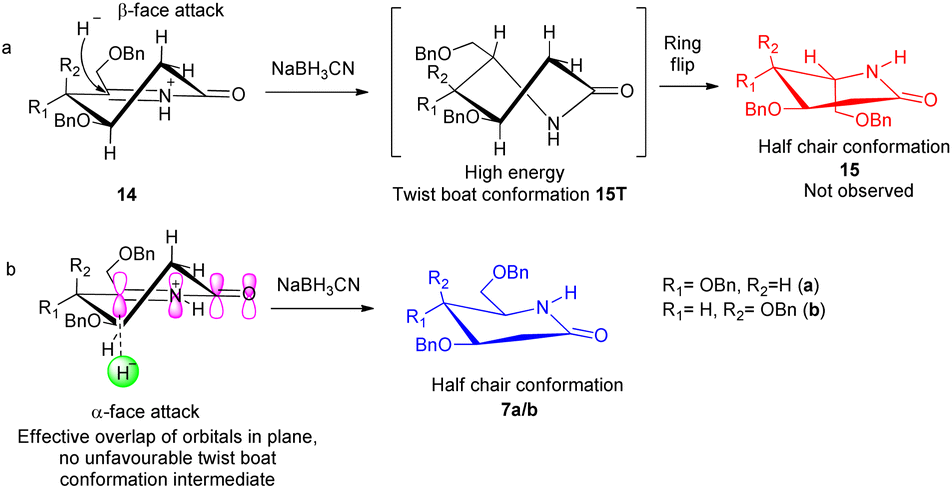 | ||
| Fig. 2 Stereochemical course of the reduction of Schiff base 14; (a) β-face attack of the hydride, (b) α-face attack of the hydride. | ||
It is noteworthy here to describe the stereochemical course of the reduction of Schiff base 14. The mechanism of this step presumably involves a hydride donation by the NaBH3CN reagent to the acyliminium ion 14a/b, initially formed by an acid-catalyzed dehydration of the hydroxy lactam substrates 12a/b. Attack of hydride can take place from β-face as well as from the α-face leading to the formation of 2-deoxyglycolactam.14a The 2-deoxyglycolactam adopts a stable half chair conformation.14b Here the reduction is governed by the stereoelectronically controlled transition states. Attack of hydride from the β-face results in an unfavorable twist boat conformation 15T which then flips to a favorable half chair conformation 15 (Fig. 2(a)). However, as 15a/b was not formed it indicates that hydride approaches from the α-face of the piperidine ring, in which hydride orbital overlaps effectively with the orbitals of the double bond which in turn are in conjugation with the lactam carbonyl. Moreover, during α-face hydride attack there is no formation of twist boat conformation transition state, instead this directly leads to the formation of 2-deoxyglycolactam product in stable half chair conformation 7a/b (Fig. 2(b)). None of the other products being formed and the NMR of the products 7a/b supports the stereoselective reduction step.
Reduction of the carbonyl group of 2-deoxyglycolactam 7a/b shall furnish the desired benzyl protected fagomine 13a and 4-epi-fagomine 13b which was successfully achieved by carrying out the reaction in THF with slow addition of LiAlH4 at 0 °C and then to rt and finally reflux for around 4 h to yield benzyl protected fagomine 13a in 49% and benzyl protected 4-epi-fagomine 13b in 41% respectively from the corresponding 2-deoxyglycolactams 7a/b (Scheme 2). Finally, following the reported procedure by Shipman et al.8a or Vankar et al.8b benzyl deprotection can be carried out to furnish fagomine 5a and 4-epi-fagomine 5b respectively in 12% and 13% overall yields from the corresponding 2-deoxyglycolactones 9a/b.
Intrigued by the amazing chemistry of 2-deoxyglycolactams 7a/b, we envisioned that 2-deoxyglycolactams 7a/b can be utilized to synthesize an advanced intermediate viz. iminoglycal 17 from which the biologically important piperidine alkaloids such as nojirimycin and its analog 2-deoxynojirimycin derivative can be readily synthesized. Iminoglycals were obtained as important reaction products15a,b or were intermediates15c synthesized which are utilized further for C–O15d or C–C bond forming reactions at C-1 of the piperidine nucleus, for the synthesis of natural products like (+)-deoxoprosophylline,15e,f (+)-fagomine,8a (−)-1-epi-adenophorine15g and potential immunosuppressant compounds.15h Although many syntheses of nojirimycin are reported,11a it is noteworthy to mention that synthesis of 2-deoxynojirimycin derivative is less known, so far we came across only two references.7b,c Hence, we formulated a retrosynthetic plan for its synthesis from a common intermediate as shown in Scheme 3.
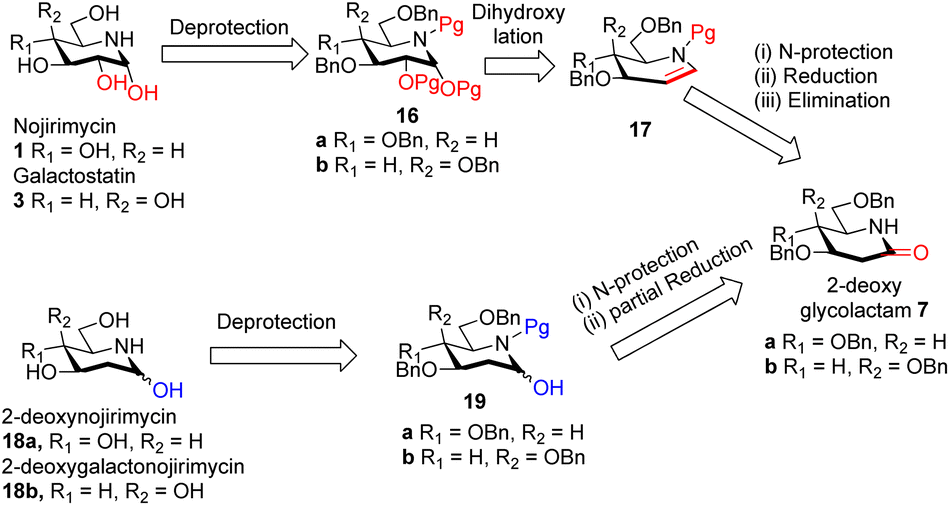 | ||
| Scheme 3 Retrosynthetic plan for the synthesis of nojirimycin 1 galactostatin 3, 2-deoxynojirimycin 18a and 2-deoxygalactonojirimycin 18b. | ||
We visualized that N-protected iminoglycal 17 can be readily obtained in 3 steps from 2-deoxyglycolactam 7 by (i) N-protection, (ii) reduction of the carbonyl (iii) elimination. Appropriate stereoselective dihydroxylation of 17 will furnish protected nojirimycin or galactostatin derivative which on deprotection will give the desired nojirimycin 1 and galactostatin 3. On the other hand, 2-deoxyglycolactamol 19 can be obtained by (i) N-protection and (ii) partial reduction of the carbonyl group of 2-deoxyglycolactam 7. 2-Deoxynojirimycin 18a or 2-deoxygalactonojirimycin 18b can be synthesized from 19a/b by deprotection reaction condition.
To achieve the N-protection of 2-deoxyglycolactam 7a/b various conditions were tried such as Boc2O, Py, DMAP, rt16a (Table 1, ESI†). Finally, by following the reaction condition16b Boc2O, DCM, NEt3, DMAP (cat), 0 °C then rt (Table 1, ESI†) 2-deoxyglycolactam-N-Boc derivative 20a/b formation in 95% and 79% respectively from 7a and 7b was achieved.
Our idea was then to partially reduce the carbonyl group of 2-deoxyglycolactams 7a/b and also to subsequently bring the dehydration of the lactamol hydroxy group. To proceed for that we started with 2-deoxyglycolactams 7a/b and treated with NaBH4 in MeOH following the reported procedure16c carried out on the similar type of substrates (Table 2, ESI†). However, in all the cases even with 2-deoxyglycolactam-N-Boc 20a/b and also under varying conditions and solvents desired product was not formed (Table 2, ESI†).
During our literature survey in the synthesis of fagomine, 4-epi-fagomine, and nojirimycin, we visualized that so far Super-Hydride or LiBHEt3 are used mainly for three purposes (i) regioselective epoxide ring opening10c,17 (ii) reduction of the ester to alcohol18 and (iii) displacement of triflate –OTf by hydride.9a There are hardly any references for the use of Super-Hydride in the reduction of amide carbonyl and which is utilized in the synthesis of piperidine alkaloids. On extensive search we came across only three references using Super-Hydride in the reduction of carbonyl but it is not used on iminosugar substrate.19 This inspired us to use Super-Hydride in our synthetic strategy of iminosugar. As we proposed in our retrosynthetic plan in partial reduction of amide carbonyl and also partial reduction followed by elimination of OH group to introduce a double bond i.e. to generate an iminoglycal 18a/b, we took advantage of Super-Hydride here to play the double role. With fine-tuning of the reaction conditions using Super-Hydride the reaction shall proceed to furnish the desired products 21a and 17a/b (Scheme 4).
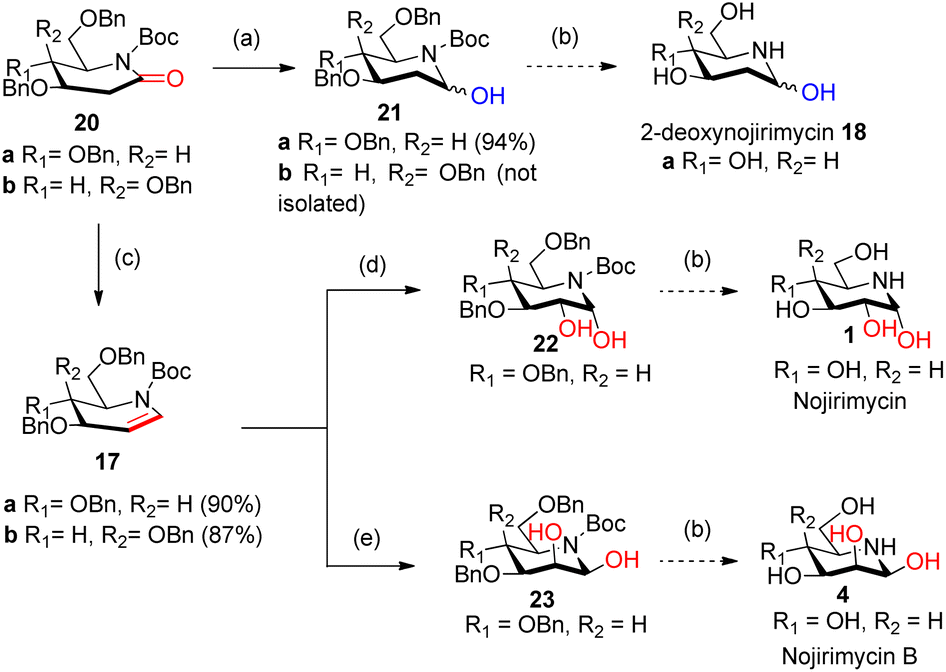 | ||
Scheme 4 Synthesis of 2-deoxynojirimycin 18a, nojirimycin 1 and nojirimycin B 4; reagents and conditions: (a) (i) Super-Hydride, toluene, −76 °C 1 h; (ii) NH4Cl, −76 °C to rt; (b) (i) aq. HCl, MeOH, heat; (ii) H2, Pd–C (10%), AcOH; (c) (i) Super-Hydride, toluene, −76 °C, 30 min; (ii) TFAA, DIPEA, DMAP (cat), −76 °C to rt; (d) (DHQ)2AQN (5 mol%), K3Fe(CN)6, K2CO3, K2OsO2(OH)4 (5.59 mol%) CH3SO2NH2, t-butyl alcohol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1) 0 °C for 66 h; (e) (DHQD)2AQN (5 mol%), K3Fe(CN)6, K2CO3, K2OsO2(OH)4 (5.59 mol%) CH3SO2NH2, t-butyl alcohol:H2O (1:1) 0 °C for 60 h. :H2O (1:1) 0 °C for 66 h; (e) (DHQD)2AQN (5 mol%), K3Fe(CN)6, K2CO3, K2OsO2(OH)4 (5.59 mol%) CH3SO2NH2, t-butyl alcohol:H2O (1:1) 0 °C for 60 h. | ||
2-Deoxygluconolactam-N-Boc 20a was treated with Super-Hydride in toluene19a,b at −76 °C for 1 h, and then with NH4Cl (sat) solution at −76 °C to rt furnished the desired 2-deoxygluconolactamol 21a in 94% yield. The 1H NMR spectrum of 2-deoxygluconolactamol 21a δH 5.64 brs represents H1 protons deshielded due to –OH and also by N atom. Reduction happened chemoselectively at δ-lactam carbonyl only, as the CO group of Boc was observed intact at 156.7 ppm in 13C NMR spectrum. However, when 2-deoxygalacatonolactam-N-Boc 20b was treated with Super-Hydride under similar conditions, desired product 2-deoxygalactonolactamol 21b could not be formed but starting material was recovered (Scheme 4). Interestingly, during our literature survey we observed that 3,4-di-epi-isomer of 21a was utilized by Yoda et al.7b,c for the synthesis of (+)-batzellaside B 6 and its C8-epimer. By known methods viz. aq. HCl, MeOH, 76 °C, and then hydrogenolysis with H2, Pd–C (10%), AcOH can easily furnish the desired product 2-deoxynojirimycin 18a from 21a.
It was then necessary to reduce and dehydrate both in situ the 2-deoxyglycolactam-N-Boc 20 to generate iminoglycal 17a/b. This was achieved when 2-deoxyglycolactam-N-Boc 20a/b was subjected to reaction conditions19a,c by treating with Super-Hydride, in toluene at −76 °C for 30 min, and then with TFAA and base DIPEA in presence of catalytic DMAP, then raising the temperature from −76 °C to rt, starting material was consumed in 12 h, with 90% 17a and 87% 17b respectively from 20a and 20b (Scheme 4). It was noteworthy that carrying out the reaction at −76 °C or −70 °C had no profound effect on the yields of the products, we preferred carrying the reaction at lower temperature (−76 °C) to the reported reaction condition19c at −70 °C. In 1H NMR spectrum of iminoglucal 17a, δH 7.11–6.93 (m, 1H), 5.10–4.90 (m, 1H), corresponds to vinylic protons, 13C NMR δC 101.5 indicates the chemical shift of β-carbon of enamine which is shielded by N atom. The other enamine α-carbon signal is merged with the aromatic carbons which appear in the range 128.6–126.6 ppm. However, in this case by 13C NMR isomeric mixture of products could be predicted possibly due to the presence of a Boc group which attains different conformation, in the plane of piperidine ring and out of the plane of the piperidine ring.
To further extensively understand the reaction step for the selective conversion of 20a to 21a (Scheme 4), we performed the Density Functional Theory (DFT) calculations for this step of reaction involving both the substrates 20a and 20b. We have come up with the following explanation for the above selective conversions. The formation of 2-deoxygalactonolactamol 21b that could not be isolated under the conditions (a) given in Scheme 4 is endergonic. However, the formation of 17b under condition (c) (ii) from 20b further suggests that 21b is in dynamic equilibrium with 20a under condition (a), as the subsequent treatment of 21b formed in situ with TFAA and base DIPEA in the presence of catalytic DMAP condition (c) (ii) gives the desired dehydrated product 17b. To further elaborate the above point, a relative DFT studies involving the substrates 20a and 20b for this particular step of the reaction was performed. The computational details are provided in the ESI.†
Two different pathways corresponding to the approach of the Super-Hydride to the re- and si-faces of the prochiral substrates (20a/b) have been investigated. Both modes of approach are associated with two different conformations (C1 and C2) of reactant and product complexes and the transition state geometry (see, computational details section (ESI†) for more details). Results of the gas-phase calculations are provided in Tables 3 and 4 (see ESI†) for 20a and 20b respectively. Please refer to Fig. S1 and S2 in the ESI† for the optimized gas-phase transition state geometries involving 20a and 20b. The results of gas-phase calculations reveal that the conformation C2 is significantly more stable than the conformation C1. Therefore, the free energy profile diagrams and all the further analyses are exclusively based on conformation C2. Furthermore, the solvent-phase calculations were performed only on conformation C2. The results of the solvent phase calculations are provided in Table 5 (see ESI†) and the optimized solvent-phase transition state geometries are represented in Fig. S3 (see ESI†).
Therefore, two different pathways (each with two different conformations of reactant and product complexes and transition-state geometries) have been investigated for both the substrates, 20a and 20b (Fig. S1 and S2 in ESI†). Results of the gas-phase calculations are provided in Tables 3 and 4 in the ESI† for 20a and 20b, respectively. Tables 3 and 4† reveals that conformation 2 for each mode of approach for both the substrates are energetically favorable. Therefore, the free energy profile diagrams and all the further analyses for both the substrates exclusively involve conformation 2. It is to be noted that the solvent-phase calculations were performed only on conformation 2 for both the pathways for the two substrates. The results of the solvent phase calculations are provided in Table 5, ESI.†
Fig. 3 illustrates the gas-phase free energy profile for the conversion of 2-deoxygluconolactam-N-Boc 20a to 2-deoxygluconolactamol-N-Boc 21a involving both the re- and si-face pathways. A very strong interaction between the Super-Hydride and 20a has been observed, as the reactant complex in the pathway shown in Fig. 3(a) (for the re-face approach) is 25.5 kcal mol−1 lower in energy than the infinitely separated reactants. Besides, the reactant complex for the si-face approach is 2.5 kcal mol−1 less stable as compared to the re-face reactant complex. The subsequent barriers for the conversion to 2-deoxygluconolactamol-N-Boc 21a for the re- and si-face approaches are 15.1 and 12.5 kcal mol−1, respectively. However, the relative energies of two transition state geometries with respect to the infinitely separated reactants are almost the same. Further, the product complex for the si-face pathway was found to be 3.0 kcal mol−1 more stable than the re-face approach (Fig. 3). Notably, both the pathways lead to stable product complexes (the intermediates for the next step of the reaction) with the reaction step being exergonic. Thus, one would expect that both the pathways are feasible for 20a in the given condition, which leads to product 21a that we were able to isolate.
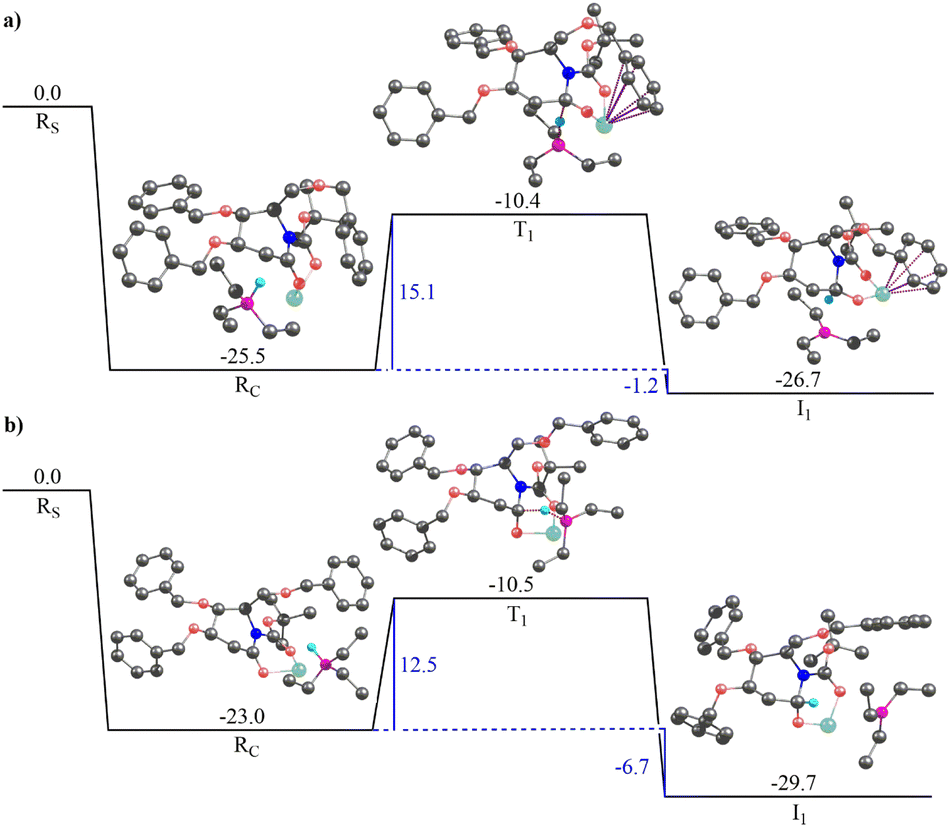 | ||
| Fig. 3 The gas-phase free energy profile for reduction of 2-deoxygluconolactam-N-Boc 20a to 2-deoxygluconolactamol-N-Boc 21a representing approach of the Super-Hydride to: (a) re-face of 20a and (b) si-face of 20a. Rs, Rc, T1, I1 represent infinitely separated reactants, the reactant complex, the transition state geometry, and the product complex, respectively. Hydrogen atoms that are not part of the reaction coordinate have been removed for clarity. All the values are in kcal mol−1. Hydrogen atoms that are not part of the reaction coordinate have been removed for clarity. Color codes: black – carbon, coral – oxygen, cyan – hydrogen, light green – lithium, fuchsia – boron, blue – nitrogen. The dotted pink lines represent cation–π interaction and dotted maroon lines represent bond breaking/bond forming. | ||
The solvent phase calculations (employing toluene as solvent) for 20a reveal a more distinct trend. The re face pathway is kinetically favorable with a lower (by 2.5 kcal mol−1) barrier as compared to the si-face pathway (Fig. S4a and b in ESI†). The reactant complexes for both the pathways are almost of the same energy. However, the product complex obtained for the si-face approach is 3.7 kcal mol−1 lower in energy than the re-face product, depicting the si-face pathway is thermodynamically favorable. Moreover, both the pathways reveal the process being exergonic with thermally stable product complexes, which could be isolated after subsequent hydrolysis.
Fig. 4 illustrates the gas-phase free energy profile for the reduction of 2-deoxygalactonolactam-N-Boc 20b by Super-Hydride. The reactant complex for the re-face pathway, in this case, is 7.8 kcal mol−1 lower in energy than the si-face reactant complex, which according to the Boltzmann distribution law substantiates for more than 99.99% of the population in this microstate. Thus, any further chemical conversion is possible only through the re-face reactant complex, and hence it rules out the si-face pathway for the reduction of the 20b using Super-Hydride. The subsequent barrier for the reduction of the 2-deoxygalactolactam-N-Boc 20b (through re-face approach) is 19.6 kcal mol−1, with the overall reaction step being endergonic by 7.8 kcal mol−1. The solvent-phase calculations also reproduce the gas-phase findings with slightly altered energy values (Fig. S4c and d, ESI†). The reactant complex for the re face approach in the solvent-phase was obtained to be 4.8 kcal mol−1 lower in energy than the si face reactant complex, which accounts for this microstate being occupied by about 99.99% of the total population (as per the Boltzmann distribution law). Thus, the solvent-phase results also indicate the selective reduction through the re face of the substrate 20b. The obtained barrier for the re face approach in the solvent-phase was obtained to be 17.6 kcal mol−1. Importantly, this barrier with respect to the infinitely separated reactants is merely 0.1 kcal mol−1. Thus, it could be deduced that the barrier is easily surmountable at the reaction temperature (−76 °C). However, the product complex obtained for this pathway in the solvent-phase is 3.8 kcal mol−1 higher in energy than the reactant complex, implying that the reaction of 20b with Super-Hydride is endergonic by 3.8 kcal mol−1. This suggests that the product is significantly unstable in the case of 2-deoxygalactolactam-N-Boc, and that is why it could not be isolated experimentally.
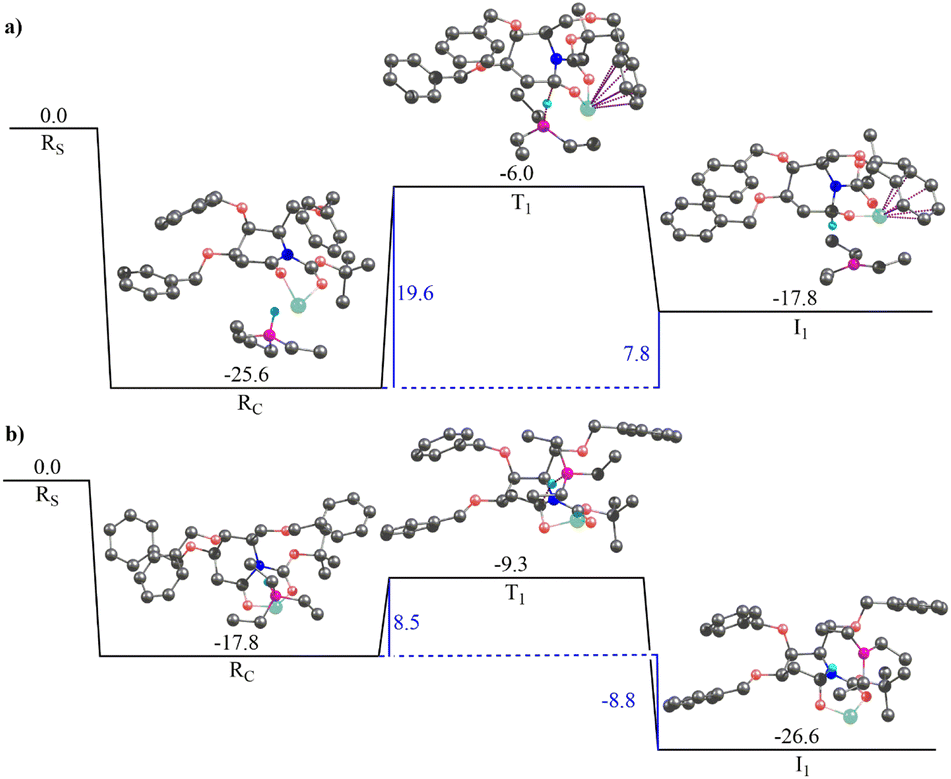 | ||
| Fig. 4 The gas-phase free energy profile for reduction of 2-deoxygalactonolactam-N-Boc 20b to 2-deoxygalactonolactamol-N-Boc 21b representing approach of the Super-Hydride to: (a) re-face of 20b and (b) si-face of 20b. Rs, Rc, T1, I1 represent infinitely separated reactants, the reactant complex, the transition state geometry, and the product complex, respectively. Hydrogen atoms that are not part of the reaction coordinate have been removed for clarity. All the values are in kcal mol−1. Hydrogen atoms that are not part of the reaction coordinate have been removed for clarity. Color codes: black – carbon, coral – oxygen, cyan – hydrogen, light green – lithium, fuchsia – boron, blue – nitrogen. The dotted pink lines represent cation–π interaction and dotted maroon lines represent bond breaking/bond forming. | ||
These results corroborate our original hypothesis that, 20a a relatively stable product under the condition (a) given in Scheme 4, and hence the corresponding 2-deoxyglucolactamol-N-Boc 21a could be isolated experimentally. However, 20b forms an unstable product complex, which converts back into the reactant under condition (a) Scheme 4, and thus could not be separated under the applied experimental conditions. However, the product was not extensively unstable and the barrier height was also surmountable at the given temperature. Therefore, when the conditions for the forthcoming step (TFAA and base DIPEA, catalytic DMAP then to rt) were enforced immediately, a successful conversion of 21b to 17b was achieved. Thereby provides the reason why 2-deoxyglucolactamol-N-Boc 21a could be isolated, whereas 2-deoxygalactolactamol-N-Boc 21b could not be.
By synthesizing the iminoglycal 17a/b we have functionalized the C-1 and C-2 position of iminosugar, which can grant access to the synthesis of various other biologically active piperidine alkaloids. As per our retrosynthetic plan dihydroxylation of iminoglycal can procure nojirimycin 1 and nojirimycin B 4 (mannojirimycin) (Scheme 4). Following the very well-established condition for dihydroxylation20a with AD-mix α and AD-mix β (commercial reagent) in t-butyl alcohol–H2O (1:1) 0 °C for 4 days, resulted in complete recovery of starting material. Strong chelating ligands and using methane sulfonamide are known to accelerate the dihydroxylation reaction which prompted us to try this reaction condition by using strong chelating ligands (DHQ)2AQN and (DHQD)2AQN.20b,21 To our surmise, on treating iminoglucal 17a with (DHQ)2AQN (5 mol%), with K3Fe(CN)6 as oxidant and K2CO3 as a base, K2OsO2(OH)4 (5.59 mol%) CH3SO2NH2 as an additive in t-butyl alcohol:H2O (1:1) 0 °C, was completed in 66 h, furnished the protected derivative of nojirimycin 22. However, even after purification by preparative thin layer chromatography (PTLC) it was difficult to obtain the pure product nojirimycin 22 and it was contaminated with some uncharacterized impurities due to which the peaks in NMR were not very distinct for analysis. But the LC-MS and HRMS were in good agreement with the desired product nojirimycin derivative 22. Similarly, by treating iminoglucal 17a with (DHQD)2AQN (5 mol%), with K3Fe(CN)6, K2CO3, K2OsO2(OH)4 (5.59 mol%) CH3SO2NH2 in t-butyl alcohol:H2O (1:1) 0 °C for 60 h, furnished the protected derivative of nojirimycin 23. Similar to nojirimycin derivative 22 in this case also it was difficult to purify the nojirimycin derivative 23 by PTLC and it was contaminated with some uncharacterized impurities. Nojirimycin derivative 22 and 23 can be readily converted to nojirimycin 1 and nojirimycin B 4 by known methods of Boc deprotection and dehydrogenation as reported in the synthesis of nojirimycin and deoxynojirimycin.
All the attempts for the dihydroxylation of the iminogalactal 17b by following the same conditions as that for iminoglucal 17a with (DHQD)2AQN as well as (DHQ)2AQN could not furnish the desired product. Probably we could reason that, the axial OBn group at C-4 in the case of iminogalactal 17b blocks the approach of Osmium from β-face and α-face is blocked by Boc group, which is already occupying the α-face (Boc group placed trans to C-4 OBn group to minimize steric interaction). The situation is different in iminoglucal 17a where the OBn group at C-4 is in equatorial position doesn't hinder the entry of osmium atom from either of the facial attack, also the Boc group maintains the more stable equatorial position in the plane of the piperidine ring without hampering the dihydroxylation process.
By following the known reaction conditions, generally, Boc deprotection is obtained in quantitative yields10c and debenzylation are obtained in yields of 85%8a utilizing assumption for final deprotection (Boc deprotection and debenzylation) starting from glucolactam 7a 2-deoxynojirimycin 21a can be readily obtained in 76%, and in 22% starting from 2-deoxygluconolactone 9a. Similarly, nojirimycin 1 can be synthesized in 22% from 2-deoxygluconolactam 7a and in 6% starting from 2-deoxygluconolactone 9a. Likewise nojirimycin B or mannojirimycin 4 can be synthesized in 52% from 2-deoxygluconolactam 7a and in 15% starting from 2-deoxygluconolactone 9a. Initially, once again we tried to ascertain the failure of the dihydroxylation reaction with iminogalactal 17b using DFT studies but we observed that incorporating the heavier Os atom and bulky ligands made the calculation process very slow and time-consuming. DFT calculations for this problem still deserves attention for some more interesting results which will be undertaken in the future.
Conclusions
We have successfully synthesized fagomine and 4-epi-fagomine from 2-deoxygluconolactone and 2-deoxygalactonolactone, respectively from chiral pool approach employing less expensive reagents and easy to handle reaction conditions. By slight variation in reaction condition employing Super-Hydride, we have synthesized iminoglycal and functionalized the C-1 and C-2 position of iminosugar, which can serve as a handle for the synthesis of various other biologically active molecules. The partially reduced product by Super-Hydride i.e. 2-deoxynojirimycin derivative can be utilized for the synthesis of epimers of natural products such as batzellasides. Thus, the use of Super-Hydride opens a new route for the synthesis of different iminosugars. Also, formal synthesis of nojirimycin, nojirimycin B, and 2-deoxynojirimycin has been achieved. From the DFT studies, we could reason the failure of the formation of 2-deoxygalactonojirimycin via Super-Hydride reduction of 2-deoxygalactonolactam to 2-deoxygalactonolactamol.Experimental
General methods
The FT-IR spectra were recorded on a FT-IR-8300 Shimadzu spectrometer and are reported in cm−1. NMR spectra were recorded on Bruker ACF 200 or AV200 (200 MHz for 1H NMR and 50 MHz for 13C NMR) or AV400 (400 MHz for 1H NMR and 100 MHz for 13C NMR) or JEOL ECX 400 (400 MHz for 1H NMR and 100 MHz for 13C NMR) or Bruker DRX-500 (500 MHz for 1H NMR and 125 MHz for 13C NMR) spectrometers using CDCl3 as solvent. Tetramethylsilane (0.00 ppm) served as an internal standard in 1H NMR and CDCl3 (77.0 ppm) in 13C NMR, respectively. Chemical shifts are expressed in parts per million (ppm) and coupling constants (J) in hertz (Hz). Mass spectra were recorded on LC-MS/MSTOF API QSTAR PULSAR spectrometer, samples introduced by infusion method using the Electrospray Ionization Technique (ESI). HRMS (ESI) of samples was taken on an Orbitrap (quadrupole plus ion trap) and TOF mass analyzer. Analytical thin-layer chromatography (TLC) were performed on 0.2 mm coated Merck pre-coated silica gel (EM 60-F254) plates. Visualization was accomplished with UV light (254 nm) and exposure to either ethanolic phosphomolybdic acid (PMA) or p-anisaldehyde–acetic acid–sulfuric acid charring reagent followed by heating. PTLC separations were carried out on 0.25 mm E. Merck silica gel plates (60F254). All the melting points reported are recorded in an open capillary using Büchi melting point apparatus B-540 and are uncorrected. Optical rotations were recorded on Jasco P-2000 polarimeter Na-lamp, λ = 589 nm. Flash chromatography was performed with CombiFlash Rf 200i equipped with UV/VIS and ELSD, Isco Teledyne Inc., USA using RediSep® column (SiO2). All other chemicals were of analytical grade. Chemical nomenclature was generated using ChemDraw.(3R,4R)-3,4,6-Tris(benzyloxy)-5-hydroxyhexanamide (8a) and (3R,4S)-3,4,6-tris(benzyloxy)-5-hydroxyhexanamide (8b). 2-Deoxygluconolactone 9a/b (1.0 g, 2.32 mmol) was dissolved in methanolic ammonia solution (7 N, 22 mL) and was stirred at room temperature for 1.5 h. After completion of the reaction (TLC), the reaction mixture was concentrated in vacuo (inside a fume hood, taking necessary precautions for ammonia gas) followed by purification by SiO2 column chromatography (EtOAc–petroleum ether, 6
:4) to afford 8a/b.
(3R,4R)-3,4,6-Tris(benzyloxy)-5-hydroxyhexanamide (8a). Colorless solid; 859 mg, 82%; mp 74–76 °C; Rf 0.26 (EtOAc–petroleum ether, 1
:1); [α]20D +14.27 (c 1.43, CHCl3); IR (CHCl3): νmax 3473, 3374, 3201, 3012, 2869, 1673, 1615, 1404, 1216, 1072, 1028, 908, 747, 698, 667 cm−1; 1H NMR (400 MHz, CDCl3) δ = 7.37–7.23 (m, 15H, 3 Ph-H), 5.65 (brs, 1H, NH), 5.22 (brs, 1H, NH), 4.66–4.60 (m, 2H, CH2Ph), 4.60–4.47 (m, 4H, CH2Ph), 4.31–4.23 (m, 1H, H3), 3.95 (brs, 1H, H5), 3.69–3.65 (m, 1H, H4), 3.65–3.60 (m, 2H, H6), 3.06 (brs, 1H, OH), 2.64–2.56 (m, 1H, H2a), 2.56–2.47 (m, 1H, H2b); 13C NMR (100 MHz, CDCl3) δ = 173.2 (C1), 138.0, 137.8, 137.6 (ArC), 128.5, 128.5, 128.3, 128.3, 128.0, 127.9, 127.8 (ArCH), 78.1 (C4), 76.7 (C3), 73.5 (CH2Ph), 73.3 (CH2Ph), 71.1 (C6), 70.8 (C5), 37.2 (C2); ESI-MS m/z 450.2240 [M + H]+; HRMS (ESI) m/z calcd for C27H31NO5Na+ [M + Na]+ 472.2094, found 472.2087.
(3R,4S)-3,4,6-Tris(benzyloxy)-5-hydroxyhexanamide (8b). Yellowish gum; 1.08 g, 96%; Rf 0.19 (EtOAc–petroleum ether, 1
:1); [α]20D +2.92 (c 1.2, CHCl3); IR (CHCl3): νmax 3660, 3372, 3019, 2872, 1736, 1454, 1216, 1101, 1064, 908, 755, 698, 668 cm−1; 1H NMR (200 MHz, CDCl3): δ = 7.33–7.25 (m, 15H), 6.00 (brs, 1H), 5.51 (brs, 1H), 4.79–4.48 (m, 6H), 4.18–4.10 (m, 1H), 3.95 (brs, 1H), 3.76–3.72 (m, 1H), 3.60–3.44 (m, 2H), 2.83 (brs, 1H), 2.68–2.47 (m, 2H), 1.80 (brs, 1H); 13C NMR (50 MHz, CDCl3): δ = 173.5, 137.7, 137.6, 128.5, 128.4, 128.2, 128.0, 127.9, 127.8, 78.7, 77.2, 74.0, 73.4, 72.9, 71.0, 69.8, 37.6; ESI-MS m/z 450.4348 [M + H]+, 472.4115 [M + Na]+; HRMS (ESI) m/z calcd for C27H31NO5Na+ [M + Na]+ 472.2094, found 472.2088. Data was in good agreement with the previous report.22
(3R,4R)-3,4,6-Tris(benzyloxy)-5-oxohexanamide (11a) and (3R,4S)-3,4,6-tris(benzyloxy)-5-oxohexanamide (11b). To a solution of 8a/b (1 g, 2.2 mmol) in dry dimethyl sulfoxide (8 mL, 0.11 M) and acetic anhydride (5 mL, 0.05 M) was stirred under an inert atmosphere for 23–26 h in a well-ventilated hood. Water (50 mL) was added and the mixture was stirred for another 30 min during which a yellow oil precipitated. The water layer was then removed and the residue was dissolved in dichloromethane and extracted with water (4 × 10 mL). The organic layer was then washed with brine (2 × 10 mL). The organic layer was dried with anhydrous Na2SO4 and concentrated in vacuo. The product [11a (587 mg, 59%) 11b (637 mg, 64%)] was used without further purification for the subsequent reactions.
(5R,6R)-4,5-Bis(benzyloxy)-6-(benzyloxymethyl)piperidin-2-one (7a) and (5S,6R)-4,5-bis(benzyloxy)-6-(benzyloxymethyl)piperidin-2-one (7b). Crude compound 11a/b (582 mg, 1.302 mmol) was dissolved in CH3CN (20 mL) and HCOOH (3.8 mL) was added to the reaction mixture followed by NaBH3CN (177 mg, 2 eq.) and the reaction mixture was refluxed at 85 °C for 4.5 h. The reaction mixture was then cooled in an ice-bath and was quenched by slowly adding aq. HCl solution (0.1 N, 30 mL). After stirring for another 15 minutes, EtOAc (50 mL) and then saturated aq. NaHCO3 solution (50 mL) was added to it slowly taking necessary precautions from the brisk efference. The addition of NaHCO3 solution was continued under cold conditions if the effervescence doesn't cease. The water layer was separated and extracted with EtOAc (2 × 25 mL), the combined organic fractions were pooled and then washed with brine (1 × 30 mL) and dried (anhydrous Na2SO4). After concentration in vacuo, the resulting crude was purified by SiO2 column chromatography (EtOAc–petroleum ether, 4
:6) to afford 7a as a colourless solid and 7b as semi-solid.
(5R,6R)-4,5-Bis(benzyloxy)-6-(benzyloxymethyl)piperidin-2-one (7a). Colorless needles (crystallization by EtOAc–petroleum ether) 329 mg, 59%; m.p. 73–75 °C [Lit23 m.p. 73–75 °C]; Rf 0.32 (EtOAc–petroleum ether, 1
:1); [α]20D +16.78 (c 1.02, CHCl3); [Lit23 [α]20D +17 (c 1, CHCl3)]; IR (CHCl3): νmax 3396, 3019, 2868, 1666, 1455, 1215, 1100, 755, 699, 669 cm−1; 1H NMR (500 MHz, CDCl3): δ = 7.34–7.23 (m, 15H, 3 Ph-H), 6.33 (brs, 1H, NH), 4.79 (d, J = 11.5 Hz, 1H, CH2Ph), 4.65–4.60 (m, 1H, CH2Ph), 4.58–4.51 (m, 2H, CH2Ph), 4.47–4.43 (m, 2H, CH2Ph), 3.88 (dt, J = 5.3, 7.2 Hz, 1H, H3), 3.63–3.51 (m, 3H, H6a, H5, H4), 3.42–3.35 (m, 1H, H6b), 2.79 (dd, J = 5.3, 17.2 Hz, 1H, H2e), 2.48 (dd, J = 7.6, 17.4 Hz, 1H, H2a); 13C NMR (125 MHz, CDCl3): δ = 169.8 (C1), 137.8, 137.7, 137.5 (ArC), 128.5, 128.0, 128.0, 127.9, 127.9, 127.8, 127.8, 127.7, 127.6 (ArCH), 75.7 (C4), 75.5 (C3), 73.6 (CH2Ph), 73.3 (CH2Ph), 71.7 (CH2Ph), 71.0 (C6), 54.9 (C5), 35.2 (C2); ESI-MS m/z 432.78 [M + H]+, 454.57 [M + Na]+, 470.74 [M + K]+; HRMS (ESI) m/z calcd for C27H29NO4+ [M + Na]+ 454.1989, found 454.1993. Data was in good agreement with the previous report.23
(5S,6R)-4,5-Bis(benzyloxy)-6-(benzyloxymethyl)piperidin-2-one (7b). Semi-solid; 332 mg, 59%; Rf 0.21 (EtOAc–petroleum ether, 4
:6); [α]20D +29.43 (c 1.1, CHCl3); IR (CHCl3): νmax 3395, 3017, 2926, 1663, 1454, 1216, 1114, 756, 698, 668 cm−1; 1H NMR (200 MHz, CDCl3): δ = 7.40–7.23 (m, 15H), 6.06 (s, 1H), 4.94 (d, J = 11.6 Hz, 1H), 4.68–4.53 (m, 3H), 4.50–4.38 (m, 2H), 4.00 (brs., 1H), 3.84 (ddd, J = 1.6, 6.3, 10.7 Hz, 1H), 3.62–3.46 (m, 3H), 2.93–2.61 (m, 2H); 13C NMR (50 MHz CDCl3): δ = 170.2, 138.1, 137.7, 137.4, 128.6, 128.5, 128.4, 128.0, 128.0, 127.9, 127.8, 127.5, 75.6, 73.8, 73.6, 71.7, 70.9, 70.6, 54.2, 33.7; ESI-MS m/z 432.39 [M + H]+, 454.39 [M + Na]+; HRMS (ESI) m/z calcd for C27H30NO4+ [M + H]+ 432.2169, found 432.2170. Data was in good agreement with the previous report.22
:1, 100 mL). After stirring for 15 min, 0.5 M aq. NaOH (75 mL) was added and the mixture was stirred for another 10 minutes. The water layer was then separated and extracted with diethyl ether (3 × 50 mL), the organic fractions were pooled, washed with brine, and finally dried (anhydrous Na2SO4) and concentrated in vacuo. The crude product was purified by SiO2 column chromatography (EtOAc–petroleum ether, 1:1) to afford 13a/b.
Tri-O-benzyl fagomine/(2R,3R,4R)-3,4-bis(benzyloxy)-2-(benzyloxy-methyl)piperidine (13a). Yellow syrup; 120 mg, 49%; Rf 0.12 (EtOAc–petroleum ether, 1
:1); [α]20D +21.76 (c 1.1, CHCl3); [Lit8a [α]18D +30.5 (c 1.90, CHCl3)]; IR (CHCl3): νmax 3151, 3017, 2922, 1398, 1220, 1099, 772, 669, 615 cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.40–7.20 (m, 15H, 3 Ph-H), 4.93 (d, J = 10.8 Hz, 1H, CH2Ph), 4.70 (d, J = 11.8 Hz, 1H, CH2Ph), 4.63 (d, J = 11.5 Hz, 1H, CH2Ph), 4.57–4.49 (m, 1H, CH2Ph), 4.49–4.40 (m, 2H, CH2Ph), 3.71 (dd, J = 2.5, 9.0 Hz, 1H, H6a), 3.60–3.46 (m, 2H, H6b, H3), 3.32 (t, J = 9.2 Hz, 1H, H4), 3.09–2.99 (m, 1H, H1e), 2.70 (ddd, J = 2.5, 6.3, 9.3 Hz, 1H, H5), 2.55 (dt, J = 2.3, 12.7 Hz, 1H, H1a), 2.30 (brs., 1H, NH), 2.18–2.06 (m, 1H, H2e), 1.56–1.41 (m, 1H, H2a); 13C NMR (100 MHz, CDCl3): δ = 138.7, 138.6, 138.1 (ArC), 128.4, 128.3, 128.3, 128.0, 127.8, 127.7, 127.6, 127.6, 127.5 (ArCH), 82.4 (C3), 80.7 (C4), 75.1 (CH2Ph), 73.3 (CH2Ph), 71.5 (CH2Ph), 70.6 (C6), 60.0 (C5), 43.5 (C1), 32.0 (C2); ESI-MS m/z 418.42 [M + H]+; HRMS (ESI) m/z calcd for C27H32NO3+ [M + H]+ 418.2377, found 418.2378. Data was in good agreement with the previous report.8a
Tri-O-benzyl-4-epi-fagomine/(2R,3S,4R)-3,4-bis(benzyloxy)-2-(benzyloxymethyl)piperidine (13b). Yellow syrup; 102 mg, 41%; Rf 0.12 (EtOAc–petroleum ether); [α]20D −4.07 (c 1.0, CHCl3); IR (CHCl3): νmax 3302, 3089, 3066, 3019, 2929, 1455, 1365, 1216, 1088, 751, 699, 669 cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.39–7.20 (m, 15H, 3 Ph-H), 4.94 (d, J = 11.5 Hz, 1H, CH2Ph), 4.68–4.55 (m, 3H, CH2Ph), 4.52-4.37 (m, 2H, CH2Ph), 3.93 (brs, 1H, H4), 3.57–3.49 (m, 1H, H6a), 3.49–3.43 (m, 1H, H3), 3.43–3.36 (m, 1H, H6b), 3.27 (brs, 1H, NH), 3.16–3.04 (dd, J = 2.0, 13.3 Hz, 1H, H1e), 2.78 (t, J = 6.8 Hz, 1H, H5), 2.57 (dt, J = 2.9, 12.9 Hz, 1H, H1a), 2.05–1.87 (m, 1H, H2a), 1.79 (d, J = 10.0 Hz, 1H, H2e); 13C NMR (100 MHz, CDCl3): δ = 139.0, 138.6, 138.0 (ArC), 128.3, 128.2, 128.1, 128.1, 127.9, 127.7, 127.4, 127.2 (ArCH), 79.6 (C3), 74.0, 73.3, 73.2 (C4), 70.3 (C6), 70.0, 58.7 (C5), 44.1 (C1), 27.6 (C2); ESI-MS m/z 418.30 [M + H]+; HRMS (ESI) m/z calcd for C27H32NO3+ [M + H]+ 418.2377, found 418.2377.
:2:93) to afford 20a/b.
tert-Butyl(2R,3R,4S)-3,4-bis(benzyloxy)-2-((benzyloxy)methyl)-6-oxopiperidine-1-carboxylate (20a). Pale yellow oily syrup; 175 mg, 95%; Rf 0.76 (EtOAc–petroleum ether, 1
:1); [α]25D −49.53 (c 1.12, CHCl3); IR (CHCl3): νmax 3021, 2978, 2402, 2360, 1767, 1718, 1511, 1220, 1034, 789, 734, 670 cm−1; 1H NMR (500 MHz, CDCl3): δ = 7.37–7.17 (m, 15H), 4.70–4.59 (m, 2H), 4.59–4.47 (m, 3H), 4.45 (s, 2H), 4.07–3.99 (m, 1H), 3.86 (td, J = 5.5, 8.5 Hz, 1H), 3.67 (dd, J = 6.9, 9.3 Hz, 1H), 3.53 (dd, J = 4.1, 9.3 Hz, 1H), 2.86 (dd, J = 4.9, 16.8 Hz, 1H), 2.64 (dd, J = 8.9, 16.5 Hz, 1H), 1.48 (s, 9H); 13C NMR (125 MHz, CDCl3): δ = 169.5, 152.1, 137.7, 137.7, 128.4, 128.4, 127.9, 127.9, 127.8, 127.7, 127.6, 127.4, 83.3, 76.3, 75.4, 73.2, 72.2, 71.6, 70.3, 58.9, 37.5, 27.9; ESI-MS m/z 554.23 [M + Na]+; HRMS (ESI) m/z calcd for C32H37NO6Na [M + Na]+ 554.2513, found 554.2513.
tert-Butyl(2R,3S,4S)-3,4-bis(benzyloxy)-2-((benzyloxy)methyl)-6-oxopiperidine-1-carboxylate (20b). Pale yellow oily syrup; 146 mg, 79%; Rf 0.57 (EtOAc–petroleum ether, 1
:1); flash chromatography (EtOAC–Et3N–petroleum ether, 5:2:93); [α]25D +1.16 (c 1.14, CHCl3); IR (CHCl3): νmax 3014, 2362, 1741, 1707, 1657, 1516, 1265, 1033, 812, 759, 674 cm−1; 1H NMR (200 MHz, CDCl3): δ = 7.44–7.17 (m, 15H), 4.93–4.75 (m, 1H), 4.72–4.56 (m, 3H), 4.47 (d, J = 1.8 Hz, 2H), 4.42–4.27 (m, 1H), 4.14 (dd, J = 1.7, 4.1 Hz, 1H), 3.95–3.82 (m, 2H), 3.81–3.66 (m, 1H), 3.06–2.87 (m, 1H), 2.81–2.64 (m, 1H), 1.45 (s, 9H); 13C NMR (50 MHz, CDCl3): δ = 168.5, 152.4, 138.0, 137.9, 137.8, 128.4, 128.3, 127.8, 127.6, 127.4, 83.7, 73.6, 73.4, 73.2, 73.0, 71.3, 68.9, 57.0, 36.9, 27.7; ESI-MS m/z 554.27 [M + Na]+; HRMS (ESI) m/z calcd for C32H37NO6Na+ [M + Na]+ 554.2513, found 554.2521.
tert-Butyl(2R,3R)-3,4-bis(benzyloxy)-2-((benzyloxy)methyl)-6-hydroxypiperidine-1-carboxylate (21a). N-Boc protected lactam 20a (100 mg, 0.188 mmol) was dissolved in dry toluene (5.0 mL) and cooled to −76 °C under inert atmosphere, and Super-Hydride (1.0 M in THF, 0.21 mL, 1.12 eq.) was added slowly dropwise over a period of 10 min, and stirred at −76 °C for 1 h. Saturated NH4Cl solution (4.0 mL) was added and stirred further for 1.5 h at −76 °C, and then temp was raised to room temperature and stirred at room temperature for 10 h. Reaction mixture was then treated with 10% Na2CO3 solution (4 mL) and DCM (10 mL) was added to the reaction mixture. The organic layer was separated, and the aq. layer was extracted with DCM (3 × 5 mL). All the organic layers were pooled together, dried (anhydrous Na2SO4), concentrated in vacuo and finally purified by SiO2 column chromatography (EtOAC–Et3N–petroleum ether, 5
:1:44) to afford 21a as a viscous oil (94 mg, 94%); Rf 0.38 (EtOAc–petroleum ether, 3:7); [α]25D −47.44 (c 1.21, CHCl3); IR (CHCl3): νmax 3741, 3019, 2362, 2334, 1692, 1531, 1216, 757, 695, 672 cm−1; 1H NMR (200 MHz, CDCl3): δ 7.30–7.25 (m, 15H), 5.64 (brs, 1H), 4.71–4.48 (m, 6H), 4.05–4.01 (m, 2H), 3.85–3.68 (m, 2H), 3.62–3.50 (m, 1H), 2.26–2.15 (m, 1H), 2.04–1.90 (m, 1H), 1.69 (brs, 1H), 1.46 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 156.6, 138.1, 138.0, 137.4, 128.5, 128.3, 127.7, 127.6, 80.7, 77.2, 74.7, 73.2, 72.9, 71.7, 71.4, 30.9, 28.3; ESI-MS m/z 556.27 [M + Na]+; HRMS (ESI) m/z calcd for C32H39NO6Na+ [M + Na]+ 556.2670, found 556.2670.
:2:95) to afford 17a/b.
tert-Butyl(2R,3R)-3,4-bis(benzyloxy)-2-((benzyloxy)methyl)-3,4-dihydropyridine-1(2H)-carboxylate (17a). Viscous oil (119 mg, 90%); Rf 0.57 (EtOAc–petroleum ether, 1
:1); [α]25D −97.97 (c 1.10, CHCl3); IR (CHCl3): νmax 3739, 3426, 2362, 2334, 1645, 1547, 1365, 924, 800, 699 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.35–7.24 (m, 15H), 7.11–6.93 (m, 1H), 5.10–4.90 (m, 1H), 4.74–4.56 (m, 3H), 4.52–4.39 (m, 4H), 4.19–4.13 (m, 1H), 3.86–3.57 (m, 3H), 1.54–1.49 (m, 9H); 13C NMR (125 MHz, CDCl3, mixture of isomers): δ 152.7, 152.2, 138.8, 138.6, 138.3, 138.0, 128.6, 128.5, 128.4, 128.2, 127.7, 127.4, 127.3, 126.9, 126.6, 101.5, 81.4, 81.3, 77.9, 77.8, 75.6, 75.1, 73.1, 72.9, 72.9, 72.8, 72.7, 71.9, 71.5, 71.2, 71.1, 70.9, 70.7, 70.4, 70.2, 68.4, 66.9, 66.8, 66.5, 66.0, 28.2, 28.0, 27.8; ESI-MS m/z 538.27 [M + Na]+; HRMS (ESI) m/z calcd for C32H37NO5Na [M + Na]+ 538.2564, found 538.2564.
tert-Butyl(2R,3S)-3,4-bis(benzyloxy)-2-((benzyloxy)methyl)-3,4-dihydropyridine-1(2H)-carboxylate (17b). Pale yellow viscous oil; 115 mg, 87%; purification by SiO2 column chromatography (EtOAc–Et3N–petroleum ether, 3
:2:95); Rf 0.57 (EtOAc–petroleum ether, 1:1); [α]25D −56.21 (c 1.13, CHCl3); IR (CHCl3): νmax 3740, 3620, 2362, 2334, 1647, 1547, 1367, 921, 821, 678 cm−1; 1H NMR (200 MHz, CDCl3): δ 8.29 (s, 1H), 7.36–7.22 (m, 15H), 4.93–4.73 (m, 3H), 4.73–4.61 (m, 3H), 4.49–4.32 (m, 2H), 4.05–3.92 (m, 2H), 3.75 (dd, J = 3.9, 5.1 Hz, 1H), 3.50 (q, J = 7.2 Hz, 1H), 1.50–1.44 (m, 9H); 13C NMR (50 MHz, CDCl3): δ 150.9, 138.8, 138.3, 137.5, 128.6, 128.5, 128.2, 128.1, 128.1, 127.7, 127.7, 127.6, 127.5, 127.4, 110.5, 84.6, 75.5, 75.1, 72.9, 71.5, 68.4, 67.7, 27.7; ESI-MS m/z 538.08 [M + Na]+; HRMS (ESI) m/z calcd for C32H37NO5Na+ [M + Na]+ 538.2564, found 538.2565.
tert-Butyl(2R,3R,5R,6R)-3,4-bis(benzyloxy)-2-((benzyloxy)methyl)-5,6-dihydroxypiperidine-1-carboxylate (22). (DHQ)2AQN (5.0 mg, 0.0058 mmol, 5 mol%), K3Fe(CN)6 (118 mg, 0.358 mmol, 3 eq.), K2CO3 (114 mg, 0.826 mmol, 70 eq.), and K2OsO2(OH)4 (2.5 mg, 0.0068 mmol, 5.59 mol%) were dissolved in tert-butyl alcohol and water (6 mL each) at room temperature. CH3SO2NH2 (23 mg, 0.242 mmol, 2.0 eq.) was added. The solution was cooled to 0 °C and Boc-iminoglycal 17a was added (61 mg, 0.118 mmol). The mixture was stirred at 0 °C for 66 h. In the work up, Na2SO3 (200 mg) was slowly added and the suspension was warmed to room temperature with vigorous stirring. EtOAc was added and the aq. layer was further extracted with ethyl acetate (2 × 5 mL), the combined organic layers were washed with 2 M NaOH (20 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated in vacuo, which on preparative TLC separation (EtOAc–petroleum ether, 7
:3) furnished 22 (20 mg, 30%), Rf 0.21 (EtOAc–petroleum ether, 7:3); [α]25D −13.33 (c 1.1%, CHCl3); νmax (CHCl3)/cm−1 3443, 3064, 2927, 2859, 2362, 2334, 1690, 1499, 1368, 1086, 757, 699, 669; 1H NMR (200 MHz, CDCl3) δ 7.35–7.26 (m, 15H), 5.65–5.52 (m, 1H), 4.70–4.41 (m, 6H), 4.24–4.04 (m, 1H), 3.95–3.89 (m, 1H), 3.85–3.80 (m, 1H), 3.75–3.63 (m, 2H), 3.58–3.45 (m, 1H), 2.68 (brs, 1H), 1.68 (brs, 1H), 1.48–1.40 (m, 9H); 13C NMR (100 MHz, CDCl3) δ 153.9, 138.0, 137.3, 137.3, 128.6, 128.5, 128.4, 128.4, 128.2, 128.2, 128.1, 128.0, 127.9, 127.8, 127.6, 127.5, 87.9, 81.6, 81.3, 78.5, 77.2, 73.3, 73.0, 72.4, 64.1, 61.3, 28.3; ESI-MS m/z 572.26 [M + Na]+; HRMS (ESI) m/z calcd for C32H39NO7Na+ [M + Na]+ 572.2619, found 572.2619.
tert-Butyl(2R,3R,5S,6S)-3,4-bis(benzyloxy)-2-((benzyloxy)methyl)-5,6-dihydroxypiperidine-1-carboxylate (23). (DHQD)2AQN (4.16 mg, 0.00485 mmol, 5 mol%), K3Fe(CN)6 (96 mg, 0.291 mmol, 3 eq.), K2CO3 (93.7 mg, 0.679 mmol, 70 eq.), and K2OsO2(OH)4 (2 mg, 0.00543 mmol, 5.59 mol%) were dissolved in tert-butyl alcohol and water (5 mL each) at room temperature, CH3SO2NH2 (18.43 mg, 0.194 mmol, 2.0 eq.) was added. The solution was cooled to 0 °C and Boc-iminoglycal 17a was added (50 mg, 0.097 mmol). The mixture was stirred at 0 °C for 60 h. In the work up Na2SO3 (200 mg) was slowly added and the suspension was warmed to room temperature with vigorous stirring. The aq. layer was further extracted with EtOAc (2 × 10 mL), the combined organic layers were washed with 2 M NaOH (20 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated in vacuo, which on preparative TLC separation (EtOAc–petroleum ether, 2
:8) furnished 23 (38 mg, 71%), Rf 0.23 (EtOAc–petroleum ether, 7:3); [α]25D −18.79 (c 1.15%, CHCl3); νmax (CHCl3)/cm−1, 3445, 3060, 2930, 2860, 2365, 2340, 1692, 1490, 1364, 1080, 750, 690, 667; 1H NMR (400 MHz, CDCl3) δ 7.39–7.24 (m, 15H), 5.69–5.56 (m, 1H), 4.69–4.48 (m, 6H), 4.19–4.08 (m, 1H), 3.99–3.93 (m, 1H), 3.87–3.85 (m, 2H), 3.78–3.69 (m, 1H), 3.63–3.55 (m, 1H), 2.68 (brs, 1H), 1.75 (brs, 1H), 1.53–1.47 (m, 9H); 13C NMR (100 MHz, CDCl3): δ 155.4, 138.2, 137.9, 137.7, 128.6, 128.5, 128.4, 128.4, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.9, 127.7, 127.5, 81.2, 81.1, 78.4, 77.2, 77.0, 73.1, 73.0, 72.8, 71.6, 65.7, 28.3; ESI-MS m/z 572.27 [M + Na]+; HRMS (ESI) m/z calcd for C32H39NO7Na+ [M + Na]+ 572.2619, found 572.2621.
Author contributions
H. R. C. performed synthesis of all the compounds; M. K. T. performed DFT analysis of selected compounds; A. K. B. conceptualised the idea as well as critically analysed all experiments as well as corrected the manuscript. All authors searched literature and drafted the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. R. C. and M. K. T. are grateful to the Council of Scientific and Industrial Research (CSIR), New Delhi, India for the award of Senior Research Fellowships (SRF).Notes and references
- A. A. Watson, G. W. J. Fleet, N. Asano, R. J. Molyneux and R. J. Nash, Phytochemistry, 2001, 56, 265–295 CrossRef CAS PubMed.
- (a) S. Inouye, T. Tsuruoka and T. Niida, J. Antibiot., 1966, 19, 288–292 CAS; (b) T. Niwa, S. Inouye, T. Tsuruoka, Y. Koaze and T. Niida, Agric. Biol. Chem., 1970, 34, 966–968 CrossRef CAS.
- (a) J.-Y. Goujon, D. Gueyrard, P. Compain, O. R. Martin, K. Ikeda, A. Kato and N. Asano, Bioorg. Med. Chem., 2005, 13, 2313–2324 CrossRef CAS PubMed , and references cited therein;; (b) M. S. Pearson, M. Mathé-Allainmat, V. Fargeas and J. Lebreton, Eur. J. Org. Chem., 2005, 2159–2191 CrossRef CAS , and references therein..
- M. Koyama and S. Sakamura, Agric. Biol. Chem., 1974, 38, 1111–1112 CrossRef CAS.
- R. J. Molyneux, M. Benson, R. Y. Wong, J. E. Tropea and A. D. Elbein, J. Nat. Prod., 1988, 51, 1198–1206 CrossRef CAS.
- J.-Q. Fan, S. Ishii, N. Asano and Y. Suzuki, Nat. Med., 1999, 5, 112–115 CrossRef CAS PubMed.
- (a) N. L. Segraves and P. Crews, J. Nat. Prod., 2005, 68, 118–121 CrossRef CAS PubMed; (b) J. Wierzejska, M. Ohshima, T. Inuzuka, T. Sengoku, M. Takahashi and H. Yoda, Tetrahedron Lett., 2011, 52, 1173–1175 CrossRef CAS; (c) J. Wierzejska, S. I. Motogoe, Y. Makino, T. Sengoku, M. Takahashi and H. Yoda, Beilstein J. Org. Chem., 2012, 8, 1831–1838 CrossRef CAS PubMed.
- (a) J. Désiré, P. J. Dransfield, P. M. Gore and M. Shipman, Synlett, 2001, 1329–1331 CrossRef; (b) N. Kumari, B. G. Reddy and Y. D. Vankar, Eur. J. Org. Chem., 2009, 160–169 CAS.
- (a) G. W. J. Fleet and P. W. Smith, Tetrahedron Lett., 1985, 26, 1469–1472 CrossRef CAS; (b) H. M. Corkran, S. Munneke, E. M. Dangerfield, B. L. Stocker and M. S. M. Timmer, J. Org. Chem., 2013, 78, 9791–9802 CrossRef CAS PubMed.
- (a) K. A. Kumar, J. S. Rathee, M. Subramanian and S. Chattopadhyay, J. Org. Chem., 2013, 78, 7406–7413 CrossRef CAS PubMed; (b) S. R. Kallam, R. Datrika, S. R. Khobare, V. S. Gajare, N. Rajana, H. R. Mohan, J. M. Babu, V. Siddaiah and T. V. Pratap, Tetrahedron Lett., 2016, 57, 1351–1353 CrossRef CAS; (c) H. Takahata, Y. Banba, H. Ouchi, H. Nemoto, A. Kato and I. Adachi, J. Org. Chem., 2003, 68, 3603–3607 CrossRef CAS PubMed; (d) J. A. Castillo, J. Calveras, J. Casas, M. Mitjans, M. P. Vinardell, T. Parella, T. Inoue, G. A. Sprenger, J. Joglar and P. Clapés, Org. Lett., 2006, 8, 6067–6070 CrossRef CAS PubMed; (e) L. Bartali, D. Scarpi, A. Guarna, C. Prandi and E. G. Occhiato, Synlett, 2009, 913–916 CAS; (f) R. W. Bates and P. S. Ng, Tetrahedron Lett., 2011, 52, 2969–2971 CrossRef CAS; (g) K. Csatayová, S. G. Davies, A. M. Fletcher, J. G. Ford, D. J. Klauber, P. M. Roberts and J. E. Thomson, Tetrahedron, 2015, 71, 7170–7180 CrossRef; (h) K. Csatayová, S. G. Davies, A. M. Fletcher, J. G. Ford, D. J. Klauber, P. M. Roberts and J. E. Thomson, J. Org. Chem., 2014, 79, 10932–10944 CrossRef PubMed.
- (a) P. Compain and O. R. Martin, in Iminosugars: From Synthesis to Therapeutic Applications, Wiley VCH, New York, 2007 CrossRef; (b) H. Iida, N. Yamazaki and C. Kibayashi, J. Org. Chem., 1987, 52, 3337–3342 CrossRef CAS; (c) T. Wennekes, A. J. Meijer, A. K. Groen, R. G. Boot, J. E. Groener, M. van Eijk, R. Ottenhoff, N. Bijl, K. Ghauharali, H. Song, T. J. O'Shea, H. Liu, N. Yew, D. Copeland, R. J. van den Berg, G. A. van der Marel, H. S. Overkleeft and J. M. Aerts, J. Med. Chem., 2010, 53, 689–698 CrossRef CAS PubMed; (d) T.-H. Chan, Y.-F. Chang, J.-J. Hsu and W.-C. Cheng, Eur. J. Org. Chem., 2010, 5555–5559 CrossRef CAS; (e) S. F. Jenkinson, G. W. J. Fleet, R. J. Nash, Y. Koike, I. Adachi, A. Yoshihara, K. Morimoto, K. Izumori and A. Kato, Org. Lett., 2011, 13, 4064–4067 CrossRef CAS PubMed; (f) O. K. Karjalainen and A. M. P. Koskinen, Org. Biomol. Chem., 2011, 9, 1231–1236 RSC.
- (a) H. R. Chand and A. K. Bhattacharya, Asian J. Org. Chem., 2016, 5, 201–206 CrossRef CAS; (b) H. R. Chand and A. K. Bhattacharya, Asian J. Org. Chem., 2019, 8, 2113–2120 CrossRef CAS; (c) A. K. Bhattacharya and H. R. Chand, Process for synthesis of piperidine alkaloids, US Pat. 9834515, 2017; (d) H. R. Chand, Approaches Towards the Synthesis of Polyhydroxylated Alkaloids and Tetrahydropyrans Using Carbohydrate Scaffolds; Chemical Transformations of Abundant Natural Products and Chemical Examination of Polyalthia longifolia var. pendula for Bioactive Molecules, PhD thesis, SP Pune Univ., India, 2016.
- (a) F.-Y. Dupradeau, S.-I. Hakomori and T. Toyokuni, J. Chem. Soc., Chem. Commun., 1995, 221–222 RSC; (b) A. Squarcia, F. Vivolo, H. G. Weinig, P. Passacantilli and G. Piancatelli, Tetrahedron Lett., 2002, 43, 4653–4655 CrossRef CAS; (c) F. X. Jiang, Q. Z. Liu, D. Zhao, C. T. Luo, C. P. Guo, W. C. Ye, C. Luo and H. Chen, Eur. J. Med. Chem., 2014, 77, 211–222 CrossRef CAS PubMed.
- (a) H. S. Overkleeft, J. van Wittenburg and U. K. Pandit, Tetrahedron, 1994, 50, 4215–4224 CrossRef CAS; (b) Y. Nishimura, H. Adachi, T. Satoh, E. Shitara, H. Nakamura, F. Kojima and T. Takeuchi, J. Org. Chem., 2000, 65, 4871–4882 CrossRef CAS PubMed.
- (a) M. Natsume, M. Wada and M. Ogawa, Chem. Pharm. Bull., 1978, 26, 3364–3372 CrossRef CAS; (b) M. Natsume and M. Wada, Chem. Pharm. Bull., 1975, 23, 2567–2572 CrossRef CAS; (c) T. Fuchss, H. Streicher and R. R. Schmidt, Liebigs Ann./Recl., 1997, 1315–1321 CrossRef CAS; (d) V. Di Bussolo, A. Fiasella, M. R. Romano, L. Favero, M. Pineschi and P. Crotti, Org. Lett., 2007, 9, 4479–4482 CrossRef CAS PubMed; (e) P. J. Dransfield, P. M. Gore, M. Shipman and A. M. Z. Slawin, Chem. Commun., 2002, 150–151 RSC; (f) P. J. Dransfield, P. M. Gore, I. Prokeš, M. Shipman and A. M. Z. Slawin, Org. Biomol. Chem., 2003, 1, 2723–2733 RSC; (g) A. Chennaiah, A. Dahiya, S. Dubbu and Y. D. Vankar, Eur. J. Org. Chem., 2018, 6574–6581 CrossRef CAS; (h) G.-L. Zhang, X.-J. Zheng, L.-H. Zhang and X.-S. Ye, Med. Chem. Commun., 2011, 2, 909–917 RSC.
- (a) D. Wang, Y.-H. Li, Y.-P. Wang, R.-M. Gao, L.-H. Zhang and X.-S. Ye, Bioorg. Med. Chem., 2011, 19, 41–51 CrossRef PubMed; (b) D. L. Flynn, R. E. Zelle and P. A. Grieco, J. Org. Chem., 1983, 48, 2424–2426 CrossRef CAS; (c) T. Bach, H. Bergmann, B. Grosch, K. Harms and E. Herdtweck, Synthesis, 2001, 1395–1405 CrossRef CAS.
- (a) H. Takahata, Y. Banba, M. Sasatani, H. Nemoto, A. Kato and I. Adachi, Tetrahedron, 2004, 60, 8199–8205 CrossRef CAS; (b) Y. Banba, C. Abe, H. Nemoto, A. Kato, I. Adachi and H. Takahata, Tetrahedron: Asymmetry, 2001, 12, 817–819 CrossRef CAS.
- (a) J. P. Shilvock, R. J. Nash, J. D. Lloyd, A. L. Winters, N. Asano and G. W. J. Fleet, Tetrahedron: Asymmetry, 1998, 9, 3505–3516 CrossRef CAS; (b) M. Ruiz, T. M. Ruanova, V. Ojea and J. M. Quintela, Tetrahedron Lett., 1999, 40, 2021–2024 CrossRef CAS; (c) M. Ruiz, V. Ojea, T. M. Ruanova and J. M. Quintela, Tetrahedron: Asymmetry, 2002, 13, 795–799 CrossRef CAS; (d) M. Ruiz, V. Ojea and J. M. Quintela, Synlett, 1999, 204–206 CrossRef CAS; (e) J. P. Shilvock, J. R. Wheatley, R. J. Nash, A. A. Watson, R. C. Griffiths, T. D. Butters, M. Müller, D. J. Watkin, D. A. Winkler and G. W. J. Fleet, J. Chem. Soc., Perkin Trans. 1, 1999, 2735–2745 RSC; (f) J. P. Shilvock, K. Y. Hsia, R. J. Nash, J. D. Lloyd, A. L. Winters, N. Asano and G. W. J. Fleet, Tetrahedron: Asymmetry, 1998, 9, 4157–4164 CrossRef CAS.
- (a) C. Pedregal, J. Ezquerra, A. Escribano, M. C. Carreño and J. L. G. Ruano, Tetrahedron Lett., 1994, 35, 2053–2056 CrossRef CAS; (b) D. F. Oliveira, P. C. Miranda and C. R. Correia, J. Org. Chem., 1999, 64, 6646–6652 CrossRef CAS PubMed; (c) J. Yu, V. Truc, P. Riebel, E. Hierl and B. Mudryk, Tetrahedron Lett., 2005, 46, 4011–4013 CrossRef CAS.
- (a) K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K. S. Jeong, H. L. Kwong, K. Morikawa, Z. M. Wang, D. Xu and X.-L. Zhang, J. Org. Chem., 1992, 57, 2768–2771 CrossRef CAS; (b) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS; (c) S. Takano, T. Yoshimitsu and K. Ogasawara, J. Org. Chem., 1994, 59, 54–57 CrossRef CAS.
- H. Becker and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 1996, 35, 448–451 CrossRef CAS.
- Q. H. Fan, J. B. Pickens, S. Striegler and C. D. Gervaise, Bioorg. Med. Chem., 2016, 24, 661–671 CrossRef CAS PubMed.
- M. M. Pichon, F. Stauffert, L. G. Addante-Moya, A. Bodlenner and P. Compain, Eur. J. Org. Chem., 2018, 1538–1545 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05224e |
| This journal is © The Royal Society of Chemistry 2022 |