
Bifunctional atomically dispersed ruthenium electrocatalysts for efficient bipolar membrane water electrolysis†
Zhipeng
Yu‡
abc,
Chaowei
Si‡
d,
Francisco Javier
Escobar-Bedia
 e,
Alec P.
LaGrow
a,
Junyuan
Xu
f,
Maria J.
Sabater
e,
Isilda
Amorim
a,
Ana
Araujo
abc,
Juliana P. S.
Sousa
a,
Lijian
Meng
g,
Joaquim Luis
Faria
bc,
Patricia
Concepcion
e,
Bo
Li
*d and
Lifeng
Liu
*a
e,
Alec P.
LaGrow
a,
Junyuan
Xu
f,
Maria J.
Sabater
e,
Isilda
Amorim
a,
Ana
Araujo
abc,
Juliana P. S.
Sousa
a,
Lijian
Meng
g,
Joaquim Luis
Faria
bc,
Patricia
Concepcion
e,
Bo
Li
*d and
Lifeng
Liu
*a
aClean Energy Cluster, International Iberian Nanotechnology Laboratory (INL), Avenida Mestre Jose Veiga, 4715-330 Braga, Portugal. E-mail: lifeng.liu@inl.int
bLSRE-LCM – Laboratory of Separation and Reaction Engineering – Laboratory of Catalysis and Materials, Faculdade de Engenharia, Universidade do Porto, Rua Dr Roberto Frias, 4200-465 Porto, Portugal
cALiCE – Associate Laboratory in Chemical Engineering, Faculdade de Engenharia, Universidade do Porto, Rua Dr Roberto Frias, 4200-465 Porto, Portugal
dShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang 110016, China. E-mail: boli@imr.ac.cn
eInstituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas (UPV-CSIC), Avenida de los Naranjos s/n, 46022 Valencia, Spain
fLaboratory of Advanced Spectro-electrochemistry and Li-on Batteries, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, China
gCentre of Innovation in Engineering and Industrial Technology, Instituto Superior de Engenharia do Porto, Instituto Politécnico do Porto, 4249-015 Porto, Portugal
First published on 24th June 2022
Abstract
Atomically dispersed catalysts (ADCs) have recently drawn considerable interest for use in water electrolysis to produce hydrogen, because they allow for maximal utilization of metal species, particularly the expensive and scarce platinum group metals. Herein, we report the electrocatalytic performance of atomically dispersed ruthenium catalysts (Ru ADCs) with ultralow Ru loading (0.2 wt%). The as-obtained Ru ADCs (Ru (0.2)-NC) are active for both hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), which only require a low overpotential (η) of 47.1 and 72.8 mV to deliver 10 mA cm−2 for HER in 0.5 M H2SO4 and 1.0 M KOH, respectively, and of 300 mV for OER in 1.0 M KOH, showing favorable bifunctionality. Density functional theory (DFT) calculations reveal that the Ru–N bonding plays an important role in lowering the energy barrier of the reactions, boosting the HER and OER activities. Furthermore, the bipolar membrane (BPM) water electrolysis using the bifunctional Ru (0.2)-NC as both HER and OER catalysts can afford 10 mA cm−2 under a low cell voltage of only 0.89 V, and does not show any performance decay upon 100 h continuous operation, showing great potential for energy-saving hydrogen production.
Introduction
Hydrogen (H2) has now been broadly accepted to be an important alternative to the conventional fossil fuels, and its widespread usage is crucial for achieving global carbon neutrality.1 Water electrolysis, powered by renewable energy like solar and wind, is proposed to be the most promising approach to “green” hydrogen production,2 and many countries have recently launched initiatives aiming to accelerate the deployment of electrolyzers to produce green hydrogen.3 However, for large-scale deployment of electrolyzers on the gigawatt (GW) scale or beyond, it is essential to address some major challenges in terms of, for example, the availability of key materials and components, costs and energy demand, such that massive production of green hydrogen will become technically viable and economically competitive.Electrocatalysts are key components of water electrolyzers and critically determine their electrochemical performance toward the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER). In the last two decades, considerable efforts have been made to developing efficient and stable electrocatalysts with reduced utilization of metal species taken into account.4–11 In particular, atomically dispersed catalysts (ADCs) have drawn significant attention in recent years and were demonstrated to be promising electrocatalysts for water electrolysis.12–14 ADCs allow for maximal utilization of metal species and can help markedly reduce the metal loading in the catalyst layers without compromising the electrocatalytic performance. To some extent, this will enable high-performance platinum group metal (PGM) catalysts to be used in electrolyzers, but meanwhile not notably increase the materials costs, especially for the relatively less expensive PGM such as ruthenium (Ru). Indeed, Ru-based materials have recently been extensively studied for use to catalyze the HER and OER.4,15–18 They were long known to be active for the OER in both acidic and alkaline solutions, and were lately also demonstrated to be good electrocatalysts for the HER.19–21 Particularly, Ru ADCs with a Ru loading of 0.2–1.0 wt% have shown electrocatalytic performance comparable to that of the commercial benchmark catalysts (e.g., Pt/C for HER and RuO2 for OER).19–25 It has been generally accepted that the RuNx species in Ru ADCs are electrocatalytically active sites and play an important role in improving the catalytic performance.21,23,26 The nitrogen groups on the catalyst support can not only impart uniform dispersion of Ru species,7 but also alter the electronic structure of Ru, prompting the electrocatalytic reaction.21,23 Notwithstanding some progress, the intrinsic catalytic performance and stability of Ru ADCs still need to be further improved.
While ADCs with a reduced amount of metal species in electrocatalysts may contribute to cost reduction, for large-capacity electrolyzers lowering the energy demand is also an effective approach to saving system costs. To this end, the recently developed bipolar membrane water electrolysis (BPMWE) seems promising.27 A bipolar membrane (BPM) consists of a cation exchange membrane (CEM) permselective to cations adjoining with an anion exchange membrane (AEM) permselective to anions. It enables the HER to occur in kinetically favorable acidic solution in the cathodic compartment and the OER to simultaneously take place in kinetically favorable alkaline solution in the anodic compartment. Moreover, when used in the “forward-bias” configuration (e.g., CEM side facing the cathode and AEM side facing the anode),27,28 the external electrical energy needed to drive water electrolysis can be significantly diminished due to the assistance of electrochemical neutralization of acid and alkaline. BPMWE has been recently reported with a number of earth-abundant electrocatalysts,27–29 and has showed preferable energy-saving feature in comparison to the conventional proton exchange membrane water electrolysis (PEMWE) or anion exchange membrane water electrolysis (AEMWE).
Herein, we report the electrocatalytic performance of Ru ADCs with an ultralow Ru loading of only 0.2 wt% (Ru (0.2)-NC) synthesized through a two-step wet chemical impregnation – pyrolysis method. We show that the ammonium salt treated activated carbon may offer abundant pyrrolic and pyridinic nitrogen sites that can bind Ru species during thermal pyrolysis. The as-prepared Ru (0.2)-NC exhibits outstanding HER and OER performance in 1.0 M KOH, with a high turnover frequency (TOF) of 11.5 s−1 at η = 100 mV for HER and 4.89 s−1 at η = 300 mV for OER as well as good catalytic stability of at least 100 h at a current density of 10 mA cm−2. Density functional theory (DFT) calculations confirm that the Ru atoms bonding with either pyrrolic or pyridinic nitrogen indeed markedly decrease the energy barrier to the catalytic reactions, boosting the activity. Furthermore, we demonstrate that the Ru (0.2)-NC can be used as bifunctional electrocatalysts in BPMWE in the forward-bias configuration, where the cell only demands a low voltage of 0.89 V to deliver 10 mA cm−2 and can stably operate for 100 h without any performance decay, showing great promise for low-cost, energy-saving production of green hydrogen.
Results and discussion
Fig. 1a shows the XRD patterns of as-prepared Ru (0.2)-NC and the pristine N-doped carbon (NC) control sample. There are two broad bumps appearing at ca. 23.6° and 44.4°, which are ascribed to the diffraction of graphitic carbon. No diffraction peaks from any metallic ruthenium or ruthenium compounds can be resolved for the Ru (0.2)-NC, suggesting that ruthenium may either be atomically dispersed on NC or consist of tiny crystallites that significantly widen the diffraction peaks. The surface chemical states of Ru (0.2)-NC and pristine NC were investigated by X-ray photoelectron spectroscopy (XPS). As displayed in Fig. S1 and Table S1 (ESI†), the XPS survey spectra clearly show the presence of C (93.7 at%), O (5.2 at%), and N (1.1 at%) in pristine NC, and C (93.0 at%), O (5.5 at%), N (1.3 at%), and Ru (0.2 at%) in Ru (0.2)-NC. The high-resolution N1s spectra of both samples can be deconvoluted into several peaks (Fig. 1b), corresponding to pyridinic-N (398.0 eV), pyrrolic-N (399.8 eV), graphitic-N (400.9 eV) and oxidized-N (402.9 eV), respectively.30,31 Notably, different from the pristine NC, the Ru (0.2)-NC shows a component arising from metal−nitrogen (M−N) bonding at 399.0 eV, indicating that Ru atoms are anchored by uncoordinated nitrogen species. Further quantitative XPS analysis manifests that the content of the M−N bond is 10.7% for Ru (0.2)-NC (Table S2, ESI†). Furthermore, the high-resolution Ru3p spectrum of Ru (0.2)-NC shows two peaks at 462.3 and 484.9 eV (Fig. S2a, ESI†), indicating that Ru in Ru (0.2)-NC carries positive charges with an oxidation state of Ruδ+ (0 < δ < 2). This was also confirmed by X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy measurements of the same sample in a recent work,32 where the average formal oxidation state of Ru in Ru (0.2)-NC was found to be +1.2 and the mean coordination number of Ru with N is about 4. In addition, the high-resolution O1s spectra of pristine NC and Ru (0.2)-NC were also compared. The O1s XPS spectrum of pristine NC can be deconvoluted into three peaks located at 530.5, 532.5 and 536.4 eV (Fig. S2b, ESI†), corresponding to C–OH, C–O–C, and H2O, respectively. After loading Ru onto NC, a new peak located at 529.5 eV appears, which can be attributed to Ru−O bonding (Fig. S2c, ESI†).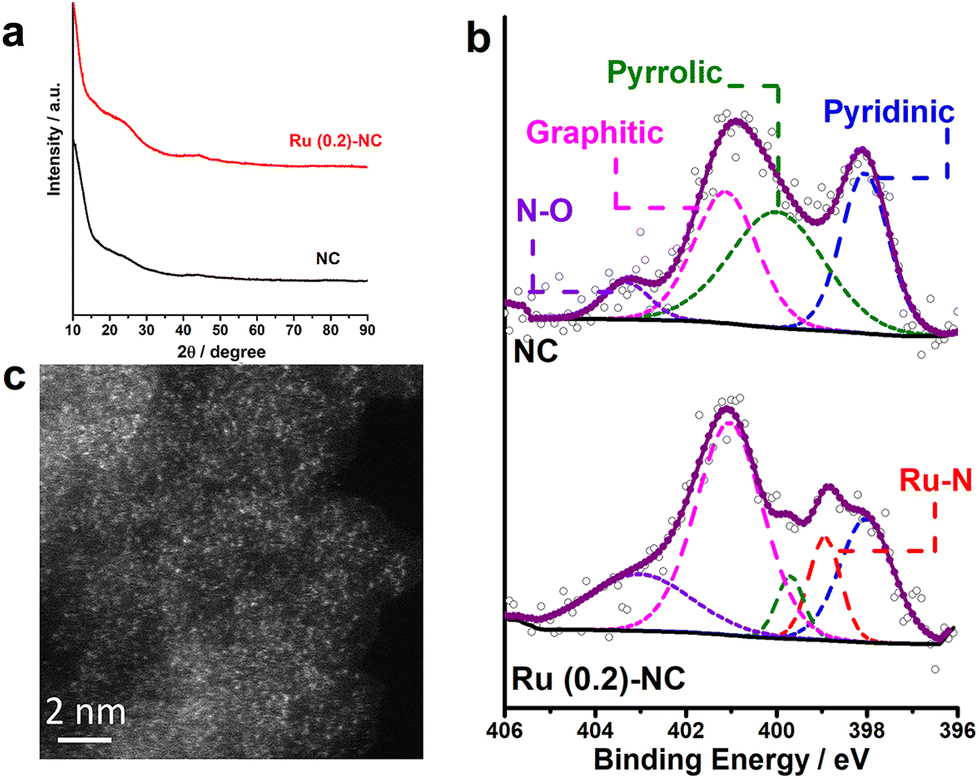 | ||
| Fig. 1 (a) XRD patterns and (b) high-resolution N1s spectra of Ru (0.2)-NC and pristine NC. (c) HAADF-STEM image of Ru (0.2)-NC. | ||
The morphology and microstructure of Ru (0.2)-NC catalysts were further examined by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Loading Ru on the NC support did not markedly alter its morphology, as revealed by SEM examination (Fig. S3, ESI†). Fig. S4a and b (ESI†) show high-resolution TEM (HRTEM) images of Ru (0.2)-NC, where only amorphous microstructure is observed and no large Ru nanoparticles are visible. Further high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging revealed a high degree of atomic dispersion of Ru on the NC support (Fig. 1c). However, some ultrafine clusters with sub-nanometric sizes were also occasionally found along with the ADCs (Fig. S4c and S4d, ESI†). The nitrogen adsorption/desorption isotherm of Ru (0.2)-NC was further measured and compared to that of pristine NC. Both samples show a type IV isotherm, indicating a mesoporous nature of materials (Fig. S5a, ESI†). The Brunauer–Emmett–Teller (BET) specific surface area of the Ru (0.2)-NC catalysts is 782 m2 g−1, substantially higher than that of the pristine NC (546 m2 g−1). However, the pore size distribution of these two samples is similar, both showing a maxima at 3.7 nm (Fig. S5b, ESI†), which suggests that loading Ru atoms onto NC does not alter the mesoporous structure of the NC support. The enlarged specific surface area of Ru (0.2)-NC likely results from the introduced Ru species and associated local atomic structure changes, which is beneficial to expose more active sites and promote mass transport.
The electrocatalytic HER performance of the Ru (0.2)-NC and other control catalysts including pristine NC and commercial Pt/C was investigated in both 0.5 M H2SO4 and 1.0 M KOH in a three-electrode configuration at room temperature. The pristine NC support exhibits inferior HER performance in both acidic and alkaline solutions and only delivers a current density of −5.56 and −6.76 mA cm−2 in 0.5 M H2SO4 and 1.0 M KOH (Fig. 2a and b), respectively, when an overpotential (η) of 250 mV is applied. Upon loading Ru, the HER catalytic current density is significantly enhanced and becomes favorably comparable to that of the commercial Pt/C benchmark, merely requiring a low overpotential of 47.1 and 72.8 mV to deliver a cathodic current density of 10 mA cm−2 in 0.5 M H2SO4 and 1.0 M KOH, respectively. This indicates that the atomically dispersed Ru will be a good low-cost alternative to Pt/C for HER, particularly for PEM water electrolysis in acidic environment where non-PGM catalysts, though existing, do not show sufficiently good catalytic performance.33–35 The reaction kinetics of all catalysts was studied by the Tafel analysis (Fig. S6, ESI†). The Ru (0.2)-NC exhibits a Tafel slope of 72 mV dec−1 in 0.5 M H2SO4, higher than that of Pt/C (34 mV dec−1), indicating that the HER proceeds on Ru (0.2)-NC through the Volmer–Heyrovsky mechanism. In 1.0 M KOH, the Ru (0.2)-NC shows a Tafel slope of 74 mV dec−1, close to that of Pt/C (77 mV dec−1). Similar trend for HER kinetics was also confirmed by the electrochemical impedance spectroscopy (EIS) studies, where Ru (0.2)-NC shows the same and even smaller charge transfer resistance (Rct) than that of Pt/C in 0.5 M H2SO4 and 1.0 M KOH, respectively (Fig. S7, ESI†). For PGM electrocatalysts, mass activity is a critical performance indicator toward practical applications reflecting the effectiveness of PGM utilization.7,13 Ru (0.2)-NC can deliver an exceptionally large mass activity of 18.0 and 13.8 A mg−1 at η = 50 mV in 0.5 M H2SO4 and 1.0 M KOH, respectively (Fig. S8, ESI†), which is 92 and 153 times higher than that of commercial Pt/C, suggesting that dispersing metal catalysts on atomic scale is indeed an effective strategy of maximizing metal utilization without compromising the catalytic activity.
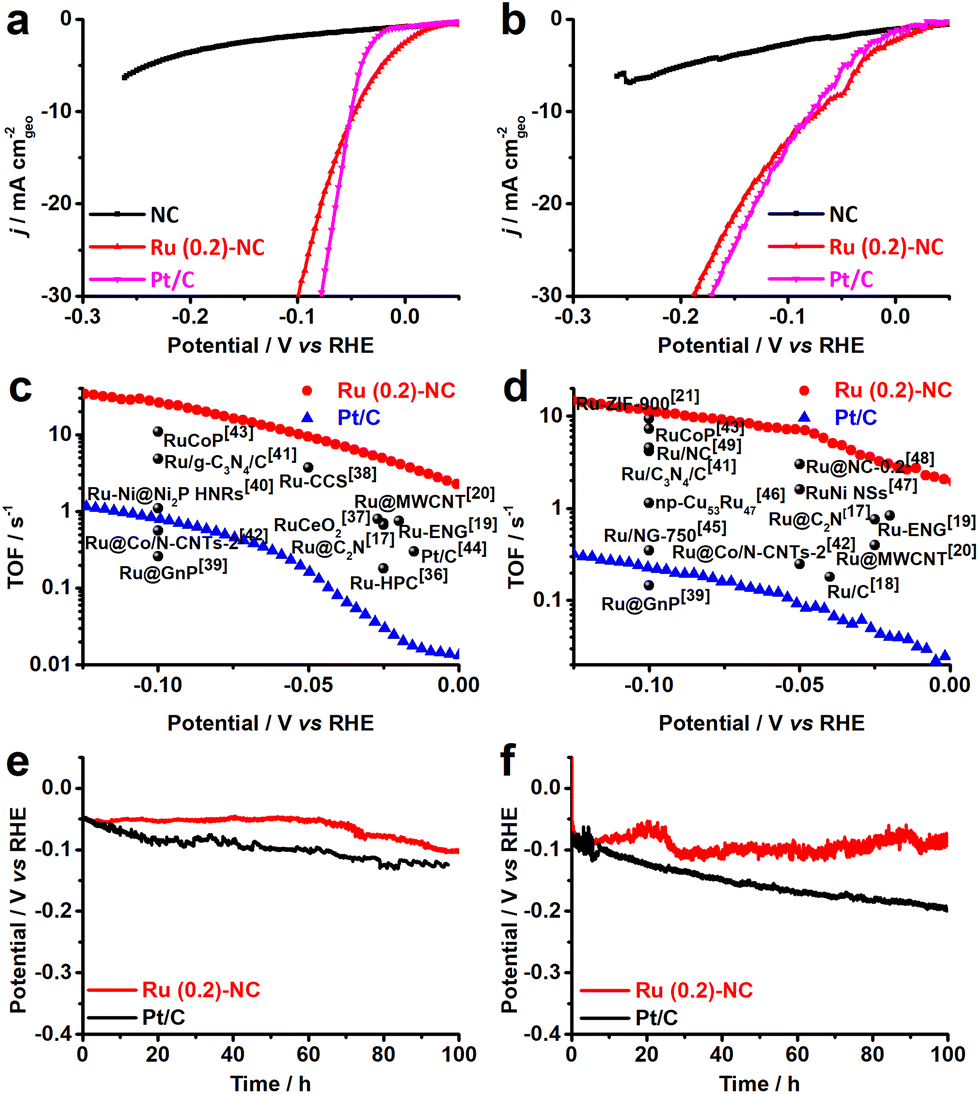 | ||
| Fig. 2 Electrocatalytic HER performance of Ru (0.2)-NC and other control catalysts. (a and b) LSV curves. Scan rate: 5 mV s−1. (c and d) Comparison of the TOF values with other start-of-the-art Ru-based HER catalysts. (e and f) Long-term catalytic stability test at −10 mA cm−2. Data were acquired in (a, c and e) 0.5 M H2SO4 and (b, d and f) 1.0 M KOH, respectively. | ||
To gain more insight into the good activity, the electrochemically active surface area (ECSA) of all catalysts was measured. As revealed in Fig. S9 (ESI†), Ru (0.2)-NC shows an ECSA value of 334.6 cm2, higher than that of the commercial Pt/C (258.9 cm2), which indicates that the highly dispersed Ru provides more active sites for the HER. The intrinsic HER catalytic activity of Ru (0.2)-NC was assessed by turnover frequency (TOF) and compared to that of some other Ru-based electrocatalysts reported recently (Fig. 2c and d). The Ru (0.2)-NC shows a TOF value of 26.2 and 11.5 s−1 at η = 100 mV in 0.5 M H2SO4 and 1.0 M KOH, respectively, substantially outperforming commercial Pt/C and many other Ru-based electrocatalysts17–21,36–49 (Tables S3 and S4, ESI†), which corroborates that Ru (0.2)-NC is intrinsically more active for the HER. Moreover, the Ru (0.2)-NC catalysts reveal outstanding catalytic stability for the HER activity in both acidic and alkaline electrolytes (Fig. 2e and f), able to sustain at −10 mA cm−2 for 100 h continuous electrolysis without significant performance degradation. The slight potential increase might stem from the active site loss.50,51 In contrast, Pt/C exhibits a notable performance decay, consistent with previous reports in the literature, which may result from weak interactions between the Pt NPs and the carbon support.43,52,53
The electrocatalytic activity of Ru (0.2)-NC and other control catalysts toward the OER was further studied in 1.0 M KOH. Ru (0.2)-NC shows a comparatively low η10 value of 300 mV (Fig. 3a), outperforming the commercial RuO2 nanoparticle benchmark catalysts (η10 = 310 mV). Moreover, Ru (0.2)-NC exhibits a Tafel slope of 62 mV dec−1 (Fig. 3b), much smaller than that of commercial RuO2 nanoparticles (86 mV dec−1), indicating more favorable OER kinetics. The EIS measurements also confirmed the faster reaction kinetics of Ru (0.2)-NC compared to the control sample, as evidenced by its smaller Rct (Fig. S10, ESI†). The Ru (0.2)-NC can deliver a superior mass activity of 16.9 A mg−1 at η = 300 mV, which is 563 times higher than that of commercial RuO2 in 1.0 M KOH (Fig. S11, ESI†). Meanwhile, the TOF values of Ru (0.2)-NC and other start-of-the-art Ru-based OER catalysts are compared in Fig. 3c. The Ru (0.2)-NC exhibits an impressive TOF value of 4.89 s−1 at η = 300 mV, remarkably higher than that of commercial RuO2 and many other Ru-based OER catalysts reported recently54–59 (see details in Table S5, ESI†). The long-term stability of Ru (0.2)-NC and the RuO2 reference catalyst were evaluated by chronopotentiometry (CP) at 10 mA cm−2 (Fig. 3d). The potential required to maintain 10 mA cm−2 for Ru (0.2)-NC does not show an obvious increase after the 100 h continuous test, revealing very good durability. In comparison, RuO2 needs a higher potential to deliver 10 mA cm−2 and the potential needed continues to increase over time, due probably to the gradual dissolution of high-valence Ru species.60,61
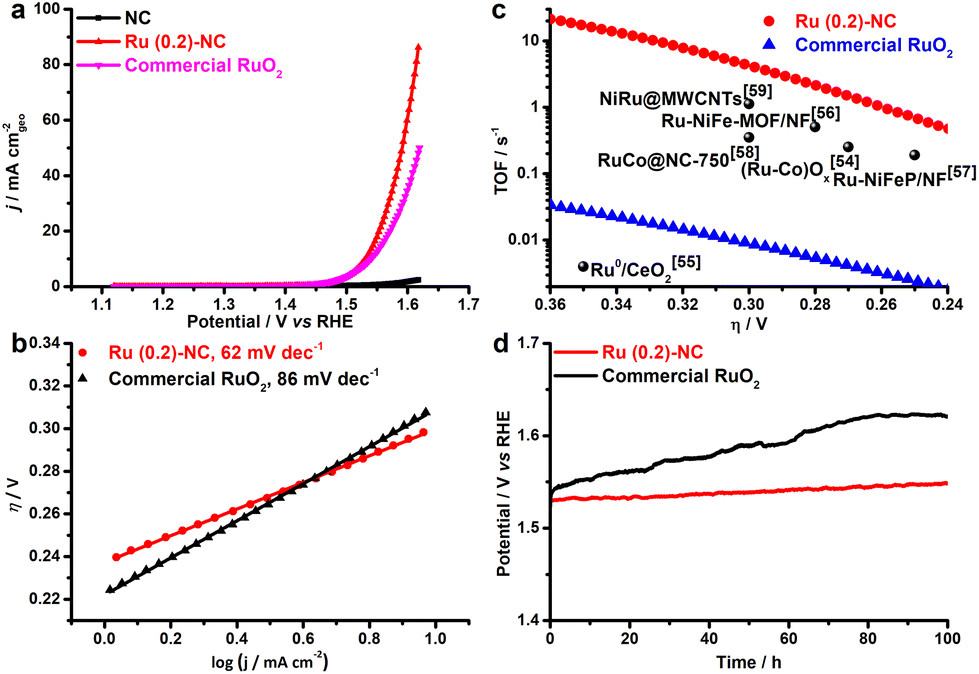 | ||
| Fig. 3 Electrocatalytic OER performance of Ru (0.2)-NC and other control catalysts. (a) LSV curves. Scan rate: 5 mV s−1. (b) Tafel slopes. (c) Comparison of the TOF values with other start-of-the-art OER catalysts. (d) Long-term catalytic stability test at 10 mA cm−2 in 1.0 M KOH. | ||
In order to gain insight into the catalytically active sites and reaction mechanisms of the Ru (0.2)-NC catalyst, we performed DFT calculations and obtained the Gibbs free energy diagrams for the OER and the HER. A RuN4 model was used for the computation, considering the atomic structure of Ru (0.2)-NC confirmed by EXAFS.32Fig. 4a shows the adsorption configurations of the reaction intermediates on the pyrrole-type Ru (0.2)-NC model catalysts during the OER, which involves successive electron transfer steps with the related oxygenated intermediates of OH*, O* and OOH*. The OER Gibbs free energy diagrams of Ru (0.2)-NC and RuO2 were calculated at the potential of U = 1.23 V (Fig. 4b). For RuO2, the calculations reveal that the elementary reaction step *O + OH− → *OOH + e− is the rate determining step (RDS) with a high energy barrier of 2.10 eV. In comparison, for Ru (0.2)-NC, the RDS has been altered to the last elementary step OOH* + OH− → * + O2 (g) + H2O (l) + e− with a much lower energy barrier of 0.86 eV, revealing a more favorable OER kinetics. The Gibbs free energy diagrams of Ru (0.2)-NC and Pt/C for the HER were also derived from the DFT calculations (Fig. 4c). The Ru (0.2)-NC exhibits a ΔGH value (−0.38 eV) comparable to Pt (−0.19 eV), indicating appropriate H adsorption on the Ru sites of Ru (0.2)-NC. The band-order and Bader charge analyses confirm that there is 0.89 e charge transferred from the Ru site to the N atoms when Ru forms chemical bonds with the N atoms (Fig. 4d and e). This indicates that the Ru sites lose their electrons and thus are positively charged, which are expected to be able to regulate their interaction with the reaction intermediates, promoting the activity. Furthermore, the crystal orbital Hamilton populations (COHP) analysis reveals that the peaks of the antibonding orbital are close to the Fermi level, suggesting that the Ru atoms in Ru (0.2)-NC catalysts are easy to form bonds with the adsorbed species, reducing the energy barrier of the reaction (Fig. 4f).
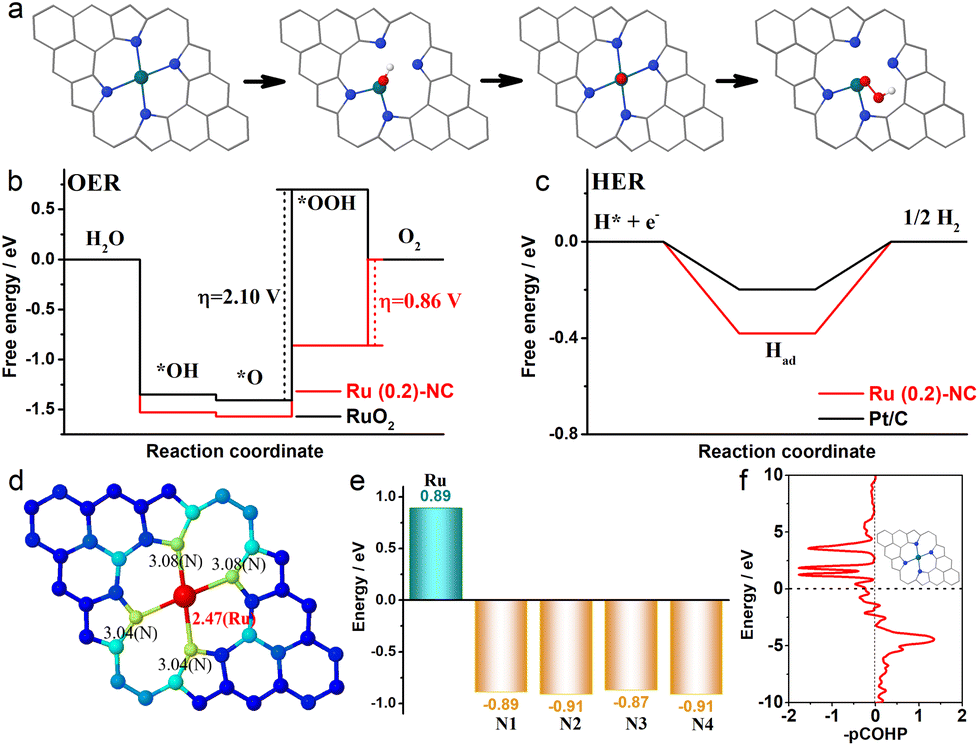 | ||
| Fig. 4 (a) Adsorption configurations of the intermediates during the OER process on pyrrole-type Ru (0.2)-NC (the balls in cyan, blue, red and white represent Ru, N, O and H atoms, respectively). Gibbs free-energy diagrams for (b) the four steps of OER and (c) the two steps of HER on pyrrole-type Ru (0.2)-NC. The dotted lines in panel (b) denote the RDS. (d) The band-order, (e) Bader charge and (f) COHP analyses of pyrrole-type Ru (0.2)-NC. | ||
While the above calculations were made based on the pyrrole-type Ru (0.2)-NC catalyst model, it was also reported that the pyridinic-type metal moieties are active sites toward the electrocatalysis.62 Therefore, we further calculated the Gibbs free energy of the reaction intermediates on pyridinic-type Ru (0.2)-NC at U = 1.23 V. As shown from the free energy diagrams for the OER and HER (Fig. S12, ESI†), our Ru (0.2)-NC catalyst exhibits the lower energy barrier for both model reactions relative to RuO2 and comparable with Pt/C. Overall, the Gibbs energy calculations agree well with our experimental observation, demonstrating that the atomically dispersed Ru sites on Ru (0.2)-NC indeed help decrease the energy barrier in the RDS step and enhance the electrocatalytic activity for both OER and HER.
On the basis of the excellent electrocatalytic performance of Ru (0.2)-NC illustrated in the HER and OER, we further used Ru (0.2)-NC as the bifunctional electrocatalysts to perform overall water electrolysis in a two-electrode configuration in 1.0 M KOH in the presence of an anion exchange membrane (AEM). The result shows that the Ru (0.2)-NC electrode pair needs a cell voltage of 1.67 V to achieve 10 mA cm−2, outperforming the electrode pair comprising commercial Pt/C and RuO2 catalysts (Fig. 5a). Given that the electrical energy demand is high in this case, we further employed a bipolar membrane (BPM) to improve the overall water electrolysis performance. Working in the “forward-bias” configuration, a BPM allows the HER and OER to be accomplished in kinetically favorable acidic and alkaline environments, respectively, at a markedly lowered cell voltage thanks to the assistance of electrochemical neutralization energy.27–29 Such asymmetric acid-alkaline BPMWE using Ru (0.2)-NC as both cathode and anode catalysts can operate under a low cell voltage (V10) of only 0.89 V to deliver 10 mA cm−2 (Fig. 5b). Moreover, a high current density of 100 mA cm−2 can be achieved at a cell voltage of merely 1.12 V. The BPMWE based on the commercial Pt/C || RuO2 also exhibits a dramatic negative shift in the cell voltage to reach a given current density (V10 = 0.91 V), but it is not as good as the BPMWE based on the Ru (0.2)-NC || Ru (0.2)-NC electrode pair. The faradaic efficiency of the HER and OER in the BPMWE was measured (Fig. 5c), and the volumes of the H2 and O2 gases collected matched well with those calculated, showing an efficiency of close to 100%. This indicates that there was no side reaction occurring during the BPMWE. Stability is a critically important indicator of electrocatalysts for practical applications in water electrolyzers. We examined the catalytic stability of the Ru (0.2)-NC || Ru (0.2)-NC electrode pair and commercial Pt/C || RuO2 electrode pair in the AEM water electrolysis (AEMWE) and BPMWE at a constant current density of 10 mA cm−2 (Fig. 5d). In both cases, the Pt/C || RuO2 electrode pair suffered a notable performance decay in the course of 100 h water splitting. In contrast, the Ru (0.2)-NC || Ru (0.2)-NC pair exhibited outstanding stability and was able to sustain continuous AEMWE and BPMWE at 10 mA cm−2 for 100 h without degradation. In particular, the BPMWE was accomplished at a low, stable voltage of 0.89 V, which shows great potential for energy-saving hydrogen production. Furthermore, the operational stability of the Ru (0.2)-NC electrode pair was tested at a higher current density of 50 mA cm−2. The electrolysis can be accomplished stably under a low voltage of 1.08 V for at least 50 h in the forward-bias BPM configuration (Fig. S13, ESI†), which shows remarkably better performance than in AEMWE.
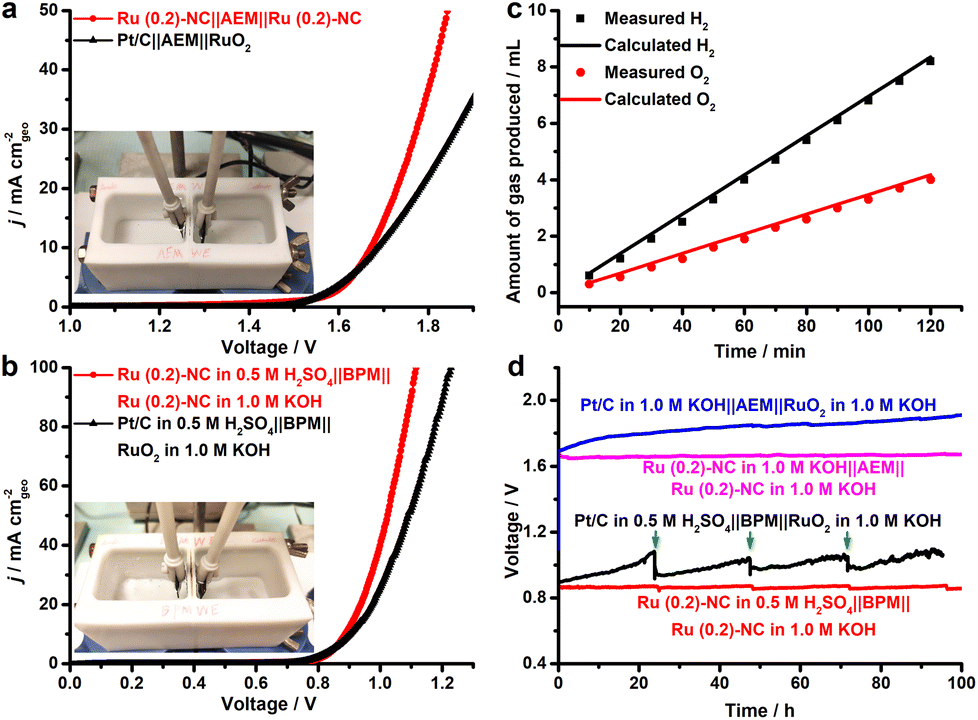 | ||
| Fig. 5 Overall water electrolysis tests of Ru (0.2)-NC and other control catalysts, performed in a two-compartment Teflon cell separated by (a) the anion exchange membrane (AEM) and (b) the bipolar membrane (BPM). Inset of (a) and (b): a digital photograph showing the testing cell used. (c) Faradaic efficiency of Ru (0.2)-NC in the BPM-based water electrolysis measured a fixed current density of 10 mA cm−2. (d) Operational stability of AEMWE and BPMWE for Ru (0.2)-NC and other control catalysts at 10 mA cm−2. The arrows in (d) indicate the fluctuation arising from the replenishment of electrolyte. | ||
We further examined the morphology, atomic structure and chemical state changes of Ru (0.2)-NC after the stability test. HAADF-STEM investigation revealed that most atomically dispersed Ru still retained upon the HER at the cathode and the OER at the anode (Fig. S14a and S14b, ESI†), though clustering happened in some places. Similar to the as-prepared Ru (0.2)-NC, there were no XRD diffraction peaks observed from both cathode and anode after the extended BPMWE (Fig. S14c, ESI†), indicating that the amount of Ru clusters formed is so little and/or the size of formed nanoclusters is so small that the presence of such clusters, if any, cannot be detected by XRD. Furthermore, XPS analyses of the post-stability test samples also demonstrated that the chemical state of Ru in Ru (0.2)-NC was barely changed (Fig. S14d, ESI†). All these post-mortem examination results illustrate that Ru (0.2)-NC is both microstructurally and chemically stable at the BPMWE conditions under investigation.
Conclusions
In summary, we have successfully proved the high electrocatalytic performance of atomically dispersed Ru supported on nitrogen-doped carbon with an ultralow Ru loading (0.2 wt%) obtained through a two-step deposition-pyrolysis method. The as-prepared Ru (0.2)-NC catalysts exhibit superior electrocatalytic activity and better stability for both HER and OER in comparison to the state-of-the-art commercial Pt/C and RuO2 catalysts as well as many other Ru-based HER and OER catalysts reported in the literature. Comprehensive DFT calculations confirm that the chemical bonding formed between Ru and N atoms can effectively lower the energy barrier of the rate determining step of the OER and that the Ru–N4 moieties can more easily absorb the reaction intermediates, both contributing to the enhanced OER performance. Furthermore, we accessed the suitability of using Ru (0.2)-NC as the bifunctional catalysts in AEM and BPM water electrolysis. A significant reduction in the applied external cell voltage can be achieved in the “forward-bias” BPM configuration, owing to the assistance of electrochemical neutralization energy. In particular, the BPM electrolyzer using Ru (0.2)-NC as the bifunctional catalysts is able to afford a current density of 10 mA cm−2 at a low cell voltage of merely 0.89 V and can continuously and stably produce hydrogen at this voltage up to 100 hours without performance decay, holding substantial potential for use in low-cost hydrogen production.Conflicts of interest
There are no conflicts to declare.Author contributions
Z. P. Y. and L. L conceived the experiments and wrote the manuscript. F. J. E., M. J. S. and P. C. synthesized the catalysts. Z. P. Y. performed XRD, SEM, XPS and electrocatalytic tests. C. W. S. and B. L. performed DFT calculations. A. P. L carried out the TEM characterization. J. P. S. S. conducted N2 adsorption/desorption isometry measurements. J. Y. X., I. A., A. A., L. J. Meng and J. L. F. contributed to the discussion. All co-authors read and agreed the manuscript. L. L. coordinated the project.Acknowledgements
L. L. acknowledges the financial support from the National Innovation Agency of Portugal through the Mobilizador Programme (Baterias 2030, Grant No. POCI-01-0247-FEDER-046109). B. L. acknowledges the Natural Science Foundation of LiaoNing Province, China (Grant No. 20180510014) for funding. Z. P. Y. is grateful for the scholarship offered by the China Scholarship Council (Grant No. 201806150015). This work was also in part financially supported by: LA/P/0045/2020 (ALiCE), UIDB/50020/2020 and UIDP/50020/2020 (LSRE-LCM) funded by national funds through FCT/MCTES (PIDDAC); project 2DMAT4FUEL (POCI-01-0145-FEDER-029600 - COMPETE2020 – FCT/MCTES - PIDDAC, Portugal). In addition, this work was carried out in part through the use of the INL Advanced Electron Microscopy, Imaging and Spectroscopy (AEMIS) Facility.Notes and references
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, The role of hydrogen and fuel cells in the global energy system, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- S. E. Hosseini and M. A. Wahid, Hydrogen production from renewable and sustainable energy resources: Promising green energy carrier for clean development, Renewable Sustainable Energy Rev., 2016, 57, 850–866 CrossRef CAS.
- S. Furfari and A. Clerici, Green hydrogen: the crucial performance of electrolysers fed by variable and intermittent renewable electricity, Eur. Phys. J. Plus, 2021, 136, 509 CrossRef CAS.
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, Recent Advances and Prospective in Ruthenium-Based Materials for Electrochemical Water Splitting, ACS Catal., 2019, 9, 9973–10011 CrossRef CAS.
- S. Sultan, J. N. Tiwari, A. N. Singh, S. Zhumagali, M. Ha, C. W. Myung, P. Thangavel and K. S. Kim, Single Atoms and Clusters Based Nanomaterials for Hydrogen Evolution, Oxygen Evolution Reactions, and Full Water Splitting, Adv. Energy Mater., 2019, 9, 1900624 CrossRef.
- Z. Yu, J. Xu, Y. Li, B. Wei, N. Zhang, Y. Li, O. Bondarchuk, H. Miao, A. Araujo, Z. Wang, J. L. Faria, Y. Liu and L. Liu, Ultrafine oxygen-defective iridium oxide nanoclusters for efficient and durable water oxidation at high current densities in acidic media, J. Mater. Chem. A, 2020, 8, 24743–24751 RSC.
- J. Xu, J. Li, Z. Lian, A. Araujo, Y. Li, B. Wei, Z. Yu, O. Bondarchuk, I. Amorim, V. Tileli, B. Li and L. Liu, Atomic-Step Enriched Ruthenium–Iridium Nanocrystals Anchored Homogeneously on MOF-Derived Support for Efficient and Stable Oxygen Evolution in Acidic and Neutral Media, ACS Catal., 2021, 11, 3402–3413 CrossRef CAS.
- J. Xu, Z. Lian, B. Wei, Y. Li, O. Bondarchuk, N. Zhang, Z. Yu, A. Araujo, I. Amorim, Z. Wang, B. Li and L. Liu, Strong Electronic Coupling between Ultrafine Iridium–Ruthenium Nanoclusters and Conductive, Acid-Stable Tellurium Nanoparticle Support for Efficient and Durable Oxygen Evolution in Acidic and Neutral Media, ACS Catal., 2020, 10, 3571–3579 CrossRef CAS.
- Y. Dong, Z. Fang, W. Yang, B. Tang and Q. Liu, Integrated Bifunctional Electrodes Based on Amorphous Co–Ni–S Nanoflake Arrays with Atomic Dispersity of Active Sites for Overall Water Splitting, ACS Appl. Mater. Interfaces, 2022, 14, 10277–10287 CrossRef CAS PubMed.
- Q. Shi, H. Liu, J. Liang, Y. Zhang, Y. Dong, W. Yang and Q. Liu, Spatially confined growth of ultrathin NiFe layered double hydroxide nanosheets within carbon nanofibers network for highly efficient water oxidation, Int. J. Hydrogen Energy, 2022, 47, 16047–16055 CrossRef CAS.
- Q. Liu, Q. Shi, Y. Ma, Z. Fang, Z. Zhou, G. Shao, H. Liu and W. Yang, ZIF-derived two-dimensional Co@Carbon hybrid: Toward highly efficient trifunctional electrocatalysts, Chem. Eng. J., 2021, 423, 130313 CrossRef CAS.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Single-Atom Electrocatalysts, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- Z. Yu, J. Xu, S. Feng, X. Song, O. Bondarchuk, J. L. Faria, Y. Ding and L. Liu, Rhodium single-atom catalysts with enhanced electrocatalytic hydrogen evolution performance, New J. Chem., 2021, 45, 5770–5774 RSC.
- T. Sun, L. Xu, D. Wang and Y. Li, Metal organic frameworks derived single atom catalysts for electrocatalytic energy conversion, Nano Res., 2019, 12, 2067–2080 CrossRef CAS.
- S. B. Scott, R. R. Rao, C. Moon, J. E. Sørensen, J. Kibsgaard, Y. Shao-Horn and I. Chorkendorff, The low overpotential regime of acidic water oxidation part I: the importance of O2 detection, Energy Environ. Sci., 2022, 15, 1977–1987 RSC.
- Q. Shi and H. Liu, Ruthenium quantum dots supported on carbon nanofibers as an efficient electrocatalyst for hydrogen evolution reaction, Int. J. Hydrogen Energy, 2021, 46, 36763–36770 CrossRef CAS.
- J. Mahmood, F. Li, S.-M. Jung, M. S. Okyay, I. Ahmad, S.-J. Kim, N. Park, H. Y. Jeong and J.-B. Baek, An efficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
- Q. Wang, M. Ming, S. Niu, Y. Zhang, G. Fan and J.-S. Hu, Scalable Solid-State Synthesis of Highly Dispersed Uncapped Metal (Rh, Ru, Ir) Nanoparticles for Efficient Hydrogen Evolution, Adv. Energy Mater., 2018, 8, 1801698 CrossRef.
- Y. Yang, J. Kim, C. Kim, A. Seong, O. Kwon, J. H. Lee, I. Kristanto, L. Zhang, J. Zhou, J.-Q. Wang, J.-B. Baek, S. K. Kwak and G. Kim, Edge-selective decoration with ruthenium at graphitic nanoplatelets for efficient hydrogen production at universal pH, Nano Energy, 2020, 76, 105114 CrossRef CAS.
- D. H. Kweon, M. S. Okyay, S.-J. Kim, J.-P. Jeon, H.-J. Noh, N. Park, J. Mahmood and J.-B. Baek, Ruthenium anchored on carbon nanotube electrocatalyst for hydrogen production with enhanced Faradaic efficiency, Nat. Commun., 2020, 11, 1278 CrossRef CAS PubMed.
- L. Xing, H. Gao, G. Hai, Z. Tao, J. Zhao, D. Jia, X. Chen, M. Han, S. Hong, L. Zheng, X. Huang, W. Dong, G. Wang and X. Shu, Atomically dispersed ruthenium sites on whisker-like secondary microstructure of porous carbon host toward highly efficient hydrogen evolution, J. Mater. Chem. A, 2020, 8, 3203–3210 RSC.
- X. Peng, S. Zhao, Y. Mi, L. Han, X. Liu, D. Qi, J. Sun, Y. Liu, H. Bao, L. Zhuo, H. L. Xin, J. Luo and X. Sun, Trifunctional Single-Atomic Ru Sites Enable Efficient Overall Water Splitting and Oxygen Reduction in Acidic Media, Small, 2020, 16, 2002888 CrossRef CAS PubMed.
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Dynamic oxygen adsorption on single-atomic Ruthenium catalyst with high performance for acidic oxygen evolution reaction, Nat. Commun., 2019, 10, 4849 CrossRef PubMed.
- D. Wang, Q. Li, C. Han, Z. Xing and X. Yang, Single-atom ruthenium based catalyst for enhanced hydrogen evolution, Appl. Catal., B, 2019, 249, 91–97 CrossRef CAS.
- Z. Yu, Y. Li, A. Torres-Pinto, A. P. LaGrow, V. M. Diaconescu, L. Simonelli, M. J. Sampaio, O. Bondarchuk, I. Amorim, A. Araujo, A. M. T. Silva, C. G. Silva, J. L. Faria and L. Liu, Single-atom Ir and Ru anchored on graphitic carbon nitride for efficient and stable electrocatalytic/photocatalytic hydrogen evolution, Appl. Catal., B, 2022, 310, 121318 CrossRef CAS.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- Y. Ding, P. Cai and Z. Wen, Electrochemical neutralization energy: from concept to devices, Chem. Soc. Rev., 2021, 50, 1495–1511 RSC.
- I. Amorim, J. Xu, N. Zhang, Z. Yu, A. Araújo, F. Bento and L. Liu, Dual-phase CoP−CoTe2 nanowires as an efficient bifunctional electrocatalyst for bipolar membrane-assisted acid-alkaline water splitting, Chem. Eng. J., 2021, 420, 130454 CrossRef CAS.
- J. Xu, I. Amorim, Y. Li, J. Li, Z. Yu, B. Zhang, A. Araujo, N. Zhang and L. Liu, Stable overall water splitting in an asymmetric acid/alkaline electrolyzer comprising a bipolar membrane sandwiched by bifunctional cobalt-nickel phosphide nanowire electrodes, Carbon Energy, 2020, 2, 646–655 CrossRef CAS.
- K. Artyushkova, B. Kiefer, B. Halevi, A. Knop-Gericke, R. Schlogl and P. Atanassov, Density functional theory calculations of XPS binding energy shift for nitrogen-containing graphene-like structures, Chem. Commun., 2013, 49, 2539–2541 RSC.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Isolated Diatomic Ni-Fe Metal–Nitrogen Sites for Synergistic Electroreduction of CO2, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- F. J. Escobar-Bedia, M. Lopez-Haro, J. J. Calvino, V. Martin-Diaconescu, L. Simonelli, V. Perez-Dieste, M. J. Sabater, P. Concepción and A. Corma, Active and Regioselective Ru Single-Site Heterogeneous Catalysts for Alpha-Olefin Hydroformylation, ACS Catal., 2022, 12, 4182–4193 CrossRef CAS.
- X. Wang, Y. V. Kolen'ko, X.-Q. Bao, K. Kovnir and L. Liu, One-Step Synthesis of Self-Supported Nickel Phosphide Nanosheet Array Cathodes for Efficient Electrocatalytic Hydrogen Generation, Angew. Chem., Int. Ed., 2015, 54, 8188–8192 CrossRef CAS PubMed.
- W. Li, X. Wang, D. Xiong and L. Liu, Efficient and durable electrochemical hydrogen evolution using cocoon-like MoS2 with preferentially exposed edges, Int. J. Hydrogen Energy, 2016, 41, 9344–9354 CrossRef CAS.
- W. Li, D. Xiong, X. Gao, W.-G. Song, F. Xia and L. Liu, Self-supported Co-Ni-P ternary nanowire electrodes for highly efficient and stable electrocatalytic hydrogen evolution in acidic solution, Catal. Today, 2017, 287, 122–129 CrossRef CAS.
- T. Qiu, Z. Liang, W. Guo, S. Gao, C. Qu, H. Tabassum, H. Zhang, B. Zhu, R. Zou and Y. Shao-Horn, Highly exposed ruthenium-based electrocatalysts from bimetallic metal-organic frameworks for overall water splitting, Nano Energy, 2019, 58, 1–10 CrossRef CAS.
- E. Demir, S. Akbayrak, A. M. Önal and S. Özkar, Nanoceria-Supported Ruthenium(0) Nanoparticles: Highly Active and Stable Catalysts for Hydrogen Evolution from Water, ACS Appl. Mater. Interfaces, 2018, 10, 6299–6308 CrossRef CAS PubMed.
- D. Luo, B. Zhou, Z. Li, X. Qin, Y. Wen, D. Shi, Q. Lu, M. Yang, H. Zhou and Y. Liu, Biomimetic organization of a ruthenium-doped collagen-based carbon scaffold for hydrogen evolution, J. Mater. Chem. A, 2018, 6, 2311–2317 RSC.
- F. Li, G.-F. Han, H.-J. Noh, I. Ahmad, I.-Y. Jeon and J.-B. Baek, Mechanochemically Assisted Synthesis of a Ru Catalyst for Hydrogen Evolution with Performance Superior to Pt in Both Acidic and Alkaline Media, Adv. Mater., 2018, 30, 1803676 CrossRef PubMed.
- Y. Liu, S. Liu, Y. Wang, Q. Zhang, L. Gu, S. Zhao, D. Xu, Y. Li, J. Bao and Z. Dai, Ru Modulation Effects in the Synthesis of Unique Rod-like Ni@Ni2P–Ru Heterostructures and Their Remarkable Electrocatalytic Hydrogen Evolution Performance, J. Am. Chem. Soc., 2018, 140, 2731–2734 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S.-Z. Qiao, High Electrocatalytic Hydrogen Evolution Activity of an Anomalous Ruthenium Catalyst, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- Z. Liu, X. Yang, G. Hu and L. Feng, Ru Nanoclusters Coupled on Co/N-Doped Carbon Nanotubes Efficiently Catalyzed the Hydrogen Evolution Reaction, ACS Sustainable Chem. Eng., 2020, 8, 9136–9144 CrossRef CAS.
- J. Xu, T. Liu, J. Li, B. Li, Y. Liu, B. Zhang, D. Xiong, I. Amorim, W. Li and L. Liu, Boosting the hydrogen evolution performance of ruthenium clusters through synergistic coupling with cobalt phosphide, Energy Environ. Sci., 2018, 11, 1819–1827 RSC.
- J. N. Hansen, H. Prats, K. K. Toudahl, N. Mørch Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, Is There Anything Better than Pt for HER?, ACS Energy Lett., 2021, 6, 1175–1180 CrossRef CAS PubMed.
- R. Ye, Y. Liu, Z. Peng, T. Wang, A. S. Jalilov, B. I. Yakobson, S.-H. Wei and J. M. Tour, High Performance Electrocatalytic Reaction of Hydrogen and Oxygen on Ruthenium Nanoclusters, ACS Appl. Mater. Interfaces, 2017, 9, 3785–3791 CrossRef CAS PubMed.
- Q. Wu, M. Luo, J. Han, W. Peng, Y. Zhao, D. Chen, M. Peng, J. Liu, F. M. F. de Groot and Y. Tan, Identifying Electrocatalytic Sites of the Nanoporous Copper–Ruthenium Alloy for Hydrogen Evolution Reaction in Alkaline Electrolyte, ACS Energy Lett., 2020, 5, 192–199 CrossRef CAS.
- G. Liu, W. Zhou, B. Chen, Q. Zhang, X. Cui, B. Li, Z. Lai, Y. Chen, Z. Zhang, L. Gu and H. Zhang, Synthesis of RuNi alloy nanostructures composed of multilayered nanosheets for highly efficient electrocatalytic hydrogen evolution, Nano Energy, 2019, 66, 104173 CrossRef CAS.
- Z.-L. Wang, K. Sun, J. Henzie, X. Hao, C. Li, T. Takei, Y.-M. Kang and Y. Yamauchi, Spatially Confined Assembly of Monodisperse Ruthenium Nanoclusters in a Hierarchically Ordered Carbon Electrode for Efficient Hydrogen Evolution, Angew. Chem., Int. Ed., 2018, 57, 5848–5852 CrossRef CAS PubMed.
- J. Zhang, P. Liu, G. Wang, P. P. Zhang, X. D. Zhuang, M. W. Chen, I. M. Weidinger and X. L. Feng, Ruthenium/nitrogen-doped carbon as an electrocatalyst for efficient hydrogen evolution in alkaline solution, J. Mater. Chem. A, 2017, 5, 25314–25318 RSC.
- S. M. Alia, S. Stariha and R. L. Borup, Electrolyzer Durability at Low Catalyst Loading and with Dynamic Operation, J. Electrochem. Soc., 2019, 166, F1164–F1172 CrossRef CAS.
- S. Siracusano, V. Baglio, N. Van Dijk, L. Merlo and A. S. Aricò, Enhanced performance and durability of low catalyst loading PEM water electrolyser based on a short-side chain perfluorosulfonic ionomer, Appl. Energy, 2017, 192, 477–489 CrossRef CAS.
- P. Paciok, M. Schalenbach, M. Carmo and D. Stolten, On the mobility of carbon-supported platinum nanoparticles towards unveiling cathode degradation in water electrolysis, J. Power Sources, 2017, 365, 53–60 CrossRef CAS.
- Z. Pu, I. S. Amiinu, Z. Kou, W. Li and S. Mu, RuP2-Based Catalysts with Platinum-like Activity and Higher Durability for the Hydrogen Evolution Reaction at All pH
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Values, Angew. Chem., Int. Ed., 2017, 56, 11559–11564 CrossRef CAS PubMed.
Values, Angew. Chem., Int. Ed., 2017, 56, 11559–11564 CrossRef CAS PubMed. - C. Wang and L. Qi, Heterostructured Inter-Doped Ruthenium–Cobalt Oxide Hollow Nanosheet Arrays for Highly Efficient Overall Water Splitting, Angew. Chem., Int. Ed., 2020, 59, 17219–17224 CrossRef CAS PubMed.
- E. Demir, S. Akbayrak, A. M. Önal and S. Özkar, Ceria supported ruthenium(0) nanoparticles: Highly efficient catalysts in oxygen evolution reaction, J. Colloid Interface Sci., 2019, 534, 704–710 CrossRef CAS PubMed.
- Y. Lin, L. Zhao, L. Wang and Y. Gong, Ruthenium-doped NiFe-based metal–organic framework nanoparticles as highly efficient catalysts for the oxygen evolution reaction, Dalton Trans., 2021, 50, 4280–4287 RSC.
- Y. Lin, M. Zhang, L. Zhao, L. Wang, D. Cao and Y. Gong, Ru doped bimetallic phosphide derived from 2D metal organic framework as active and robust electrocatalyst for water splitting, Appl. Surf. Sci., 2021, 536, 147952 CrossRef CAS.
- C. Gao, H. Wang, S. Li, B. Liu, J. Yang, J. Gao, Z. Peng, Z. Zhang and Z. Liu, Enhanced cobalt-based catalysts through alloying ruthenium to cobalt lattice matrix as an efficient catalyst for overall water splitting, Electrochim. Acta, 2019, 327, 134958 CrossRef CAS.
- Z. Peng, J. Liu, B. Hu, Y. Yang, Y. Guo, B. Li, L. Li, Z. Zhang, B. Cui, L. He and M. Du, Surface Engineering on Nickel–Ruthenium Nanoalloys Attached Defective Carbon Sites as Superior Bifunctional Electrocatalysts for Overall Water Splitting, ACS Appl. Mater. Interfaces, 2020, 12, 13842–13851 CrossRef CAS PubMed.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- N. Hodnik, P. Jovanovič, A. Pavlišič, B. Jozinović, M. Zorko, M. Bele, V. S. Šelih, M. Šala, S. Hočevar and M. Gaberšček, New Insights into Corrosion of Ruthenium and Ruthenium Oxide Nanoparticles in Acidic Media, J. Phys. Chem. C, 2015, 119, 10140–10147 CrossRef CAS.
- L. Bai, Z. Duan, X. Wen, R. Si, Q. Zhang and J. Guan, Highly Dispersed Ruthenium-Based Multifunctional Electrocatalyst, ACS Catal., 2019, 9, 9897–9904 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi00892k |
| ‡ These authors contribute equally to this work. |
| This journal is © the Partner Organisations 2022 |