
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot synthesis of structure-controlled temperature-responsive polymer gels†
Tomoki
Sakai
a,
Nagisa
Ito
a,
Mitsuo
Hara
 a,
Takahiro
Seki
a,
Mineto
Uchiyama
a,
Masami
Kamigaito
a,
Kotaro
Satoh
*b,
Taiki
Hoshino
c and
Yukikazu
Takeoka
*a
a,
Takahiro
Seki
a,
Mineto
Uchiyama
a,
Masami
Kamigaito
a,
Kotaro
Satoh
*b,
Taiki
Hoshino
c and
Yukikazu
Takeoka
*a
aGraduate School of Engineering Nagoya University, Furo-cho, Chikusaku, Nagoya 464-8603, Japan. E-mail: ytakeoka@chembio.nagoya-u.ac.jp; Fax: +81-52-789-4669; Tel: +81-52-789-4670
bDepartment of Chemical Science and Engineering School of Material Chemical Technology Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: satoh@cap.mac.titech.ac.jp; Fax: +81-3-5734-2162; Tel: +81-3-5734-2162
cRIKEN Spring-8 Center, Sayo, Hyogo 679-51982, Japan
First published on 17th June 2022
Abstract
The simultaneous use of metal Lewis acids and photo-radical generators for dithioesters, which are the common dormant species for cationic and radical polymerization, made it possible to convert a cationic species into a radical upon photoirradiation. In this study, we attempted one-pot synthesis of temperature-responsive polymer gels with controlled network structure by using temperature-responsive telechelic polymers prepared by cationic polymerization in the dark followed by the mechanistic transformation into radical cross-linking reaction under photoirradiation. Specifically, the polymerization of 2-(2-ethoxy)ethoxyethyl vinyl ether (EO2) in the presence of a crosslinking agent (divinyl adipate) was performed at 4 °C in toluene using a vinyl ether-type bifunctional RAFT reagent as initiator, a Lewis acid (Et3Al2Cl3) and a photo radical generator (TPO). The conditions for gelation by radical polymerization with photoirradiation were investigated in detail by considering the molecular weight control and overlapping concentration of the telechelic polymers obtained by cationic polymerization. As a result, small-angle X-ray scattering revealed that polymer gels with a relatively uniform network structure can be obtained compared to the cross-linked network of polymer gels obtained by conventional free-radical polymerization.
Introduction
In many previous studies of polymer gels prepared by free radical polymerization, relatively simple low molecular weight monomers and cross-linking agents are dissolved in appropriate solvents, and these are then polymerized by reaction initiators to obtain polymer gels. The resulting network structure of polymer gel can contain dangling chains where one end is not connected to a cross-linking point, loop chains where both ends are connected to one cross-linking point and hang down, and entanglement of polymer chains that is not resolved by the constraint of cross-linking points.1 Furthermore, spatial sparsity also occurs in the network structure, and the actual network structure is extremely heterogeneous compared to the ideal homogeneous network structure used in classical theory.2 Polymer gels composed of cross-linked networks with such heterogeneous structures are used by us in electrophoresis gels for the analysis of biological macromolecules, drug release substrates, and optical materials such as soft contact lenses. If the structure of the cross-linked network can be prepared more well-organized, it may lead to the improvement of various functions of polymer gels.3Recent developments in precision synthesis methods have revealed the possibility of synthesizing polymers with well-defined molecular structures and then linking them together under appropriate conditions to obtain cross-linked networks with well-defined network structures.4–13 Work has been done for quite some time on methods for later cross-linking of end-reactive polymers with well-defined molecular weights.14,15 However, the conditions under which these polymers are connected to each other are important for controlling the network structure. The study by Li et al. points out that one good way to form a uniform network structure is to connect polymers together in the semi-dilute state of the polymer solution.16 In our previous studies, we have also found that the structure of cross-linked networks prepared in a semi-dilute state, where the concentration is sufficiently higher than the overlapping concentration of the polymers and no entanglement occurs between them, is highly uniform.7
In order for research on the functions of polymer gels with such well-ordered network structures to flourish in various research fields, it is important that methods for precise polymer synthesis and cross-link formation between polymers can be easily performed. The synthesis of polymer gels consisting of a network structure with a controlled structure simply by mixing the necessary compounds or by using a simpler process may lead to open new applications of polymer gels. We have reported the mechanistic transformation between cationic and radical polymerization by using RAFT agent as the dual mediator for cationic and radical polymerizations, in which the C–S bond at the chain end was activated either into cationic or radical species during the propagation reactions to form a single polymer chain.17–25 Such a transformation would be applicable for the formation of a gel by dividing roles of the two active species into the chain propagation and cross-linking reaction.
Although the combination of different polymerization mechanism has been applied for well-defined polymer networks, it required purification of the precursor polymers before the linking reaction.26,27 In this study, we conducted the simple mechanistic transformation for the one-pot synthesis of polymer gels with a well-defined network structure through two-step polymerizations from cationic polymerization to radical cross-linking reaction, of which the latter reaction was triggered just by light irradiation at the appropriate timing. Here, we employed a diacrylate- or divinyl ester-type compound as the cross-linker in conjunction with a photo-radical generator, which would not affect the cationic polymerization of vinyl ether from difunctional RAFT agents. The cross-linkers and radical generator worked on the formation of polymer gels upon photoirradiation to induce radical linking reaction between the telechelic poly(vinyl ether) at the RAFT termini (Scheme 1). Especially for creating temperature-responsive polymer hydrogels, 2-(2-ethoxy)ethoxyethyl vinyl ether (EO2) was employed as the monomer for cationic polymerization.
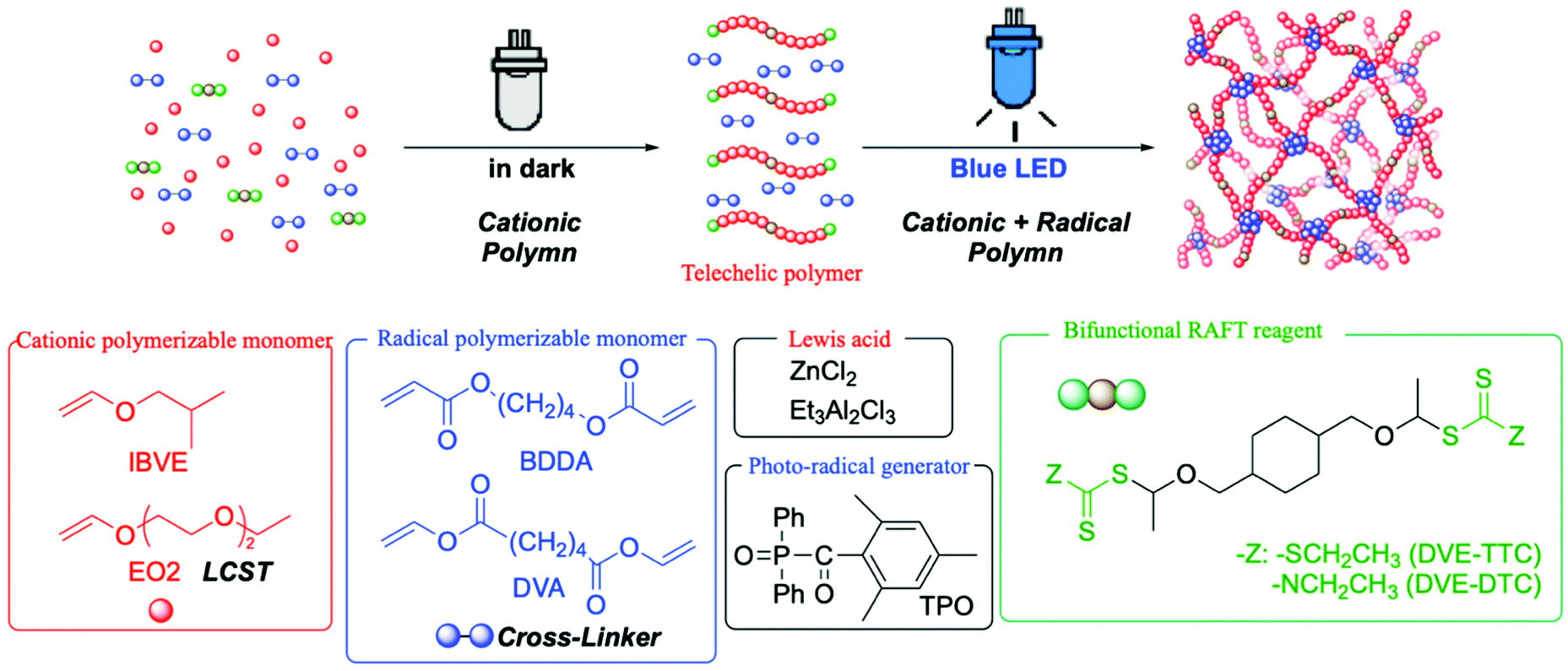 | ||
| Scheme 1 One-pot synthesis of polymer gels by mechanistic transformation from cationic polymerization in the dark into cross-linking radical polymerization under photoirradiation. | ||
The polymer concentration during gelation must be also important for the formation of a highly uniform network structure. In the dilute region, the polymer chains behave as isolated chains, and in this state, either gelation is caused by cluster formation of the resulting polymer, or there is a lower concentration limit at which gelation does not occur. On the other hand, in the concentrated region, entanglement and mutual penetration of polymer chains occur, and there can be a lot of topological heterogeneity in the network structure of the resulting polymer gel. In other words, for the formation of a highly uniform network structure, it is necessary to consider a concentration range based on the overlapping concentration at which polymer chains begin to overlap each other. To this end, the overlapping concentration of the resultant telechelic polymer was first calculated from viscosity measurements of the polymer solution with different concentrations to be used in order to know the appropriate polymer concentration for gelation. And then, the synthesis of polymer gels was examined by changing the monomer concentration and cross-linker concentration in the preparation in consideration of the overlapping concentration. Then, we also observed the swelling behavior of the obtained polymer gels and analyzed the network structure using SAXS.
Experimental section
Materials
Ethyl aluminium sesquichloride (Et3Al2Cl3) (1.5 mol L−1 in toluene), calcium hydride (CaH2), and diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) were purchased from Nippon Aluminium Alkyls, Kanto Chemical, and Sigma-Aldrich, respectively. 2-(2-Ethoxy)ethoxyethyl vinyl ether (EO2) was kindly provided by Maruzen Petrochemical. Divinyl adipate and 1,2-dichlorobenzene were commercially obtained from Tokyo Kasei Kogyo. These reagents were distilled and purified with CaH2 before use. Toluene (Dehydrated) was purified in an organic solvent purifier (Nikko Hansen) before use. Ethyl acetate (EtOAc) was purchased from Kishida Chemical and distilled and purified with CaH2 before use.Analysis equipment
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to an open system. The polymerization solution was diluted with chloroform, and the polymer was purified by washing the organic layer three times each with aqueous HCl solution and distilled water using a separatory funnel. The polymer was then obtained by removing the solvent from the resulting organic layer using an evaporator. After light irradiation, the sample that became gel was also cut out and quenched by immersing it in a methanol solution with triethylamine-methanol mixture for 1 day to quench the catalyst, and then purified by replacing it with a methanol solution. The gel was then soaked in various solvents to achieve equilibrium swelling by the various solvents.
:1) to an open system. The polymerization solution was diluted with chloroform, and the polymer was purified by washing the organic layer three times each with aqueous HCl solution and distilled water using a separatory funnel. The polymer was then obtained by removing the solvent from the resulting organic layer using an evaporator. After light irradiation, the sample that became gel was also cut out and quenched by immersing it in a methanol solution with triethylamine-methanol mixture for 1 day to quench the catalyst, and then purified by replacing it with a methanol solution. The gel was then soaked in various solvents to achieve equilibrium swelling by the various solvents.
Results and discussion
Polymer gel synthesis by mechanistic transformation from cationic polymerization to radical linking reaction
The one-pot gel forming reaction was investigated by using mechanistic transformation from cationic polymerization into radical linking reaction.17–22 For this, the living cationic polymerization of isobutyl vinyl ether (IBVE) was examined using a trithiocarbonate-type difunctional RAFT agent (DVE-TTC) as the initiator in the presence of ZnCl2, 1,4-butanediol diacrylate (BDDA), and a photo-radical generator [diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide: TPO], in which the charge ratio of [IBVE]0/[BDDA]0 was 1/10 with various [DVE-TTC]0 and catalyst concentrations (Fig. 1). First, IBVE was polymerized by ZnCl2 as the Lewis acid catalyst in dark, where the photo-radical generator and diacrylate were almost inert to cationic polymerization, to afford the homopolymer of IBVE with relatively narrow molecular weight distributions (MWDs) (Mw/Mn ∼1.2). In general, the kinetic rate of cationic polymerization depends on the concentration of the Lewis acid catalyst as well as that of the initiator.28 However, the reaction rates of this system were also dependent on the initial feed ratio of the RAFT agent and ZnCl2, probably because of the strong interaction between the basic sulfur atom and Lewis acid. Thus, the higher amount of Lewis acid catalyst was required for preparation of lower-molecular-weight polymers with higher [DVE-TTC]0. The number-average molecular weight (Mn) of the polymers changed in proportion to the initial feed ratio of [IBVE]0/[DVE-TTC]0. When the monomer conversions reached higher than 50%, the reaction mixture was then exposed to blue light LED with the wavelength of 448 nm, under which TPO is known to cleave to generate racial species. The generated radical reacted with the terminal trithiocarbonate, which has been the dormant species for the cationic polymerization of IBVE. As in the case of RAFT radical polymerization, the growing radical species of IBVE could be generated from the trithiocarbonate through addition–fragmentation chain-transfer process to react with diacrylate BDDA. The kinetic rate of the radical reaction depended on the initial concentration of TPO. Especially, the lower concentration caused some induction periods for the mechanistic transformation, as clearly seen in the case for [IBVE]0/[BDDA]0/[DVE-TTC]0 = 200/20/1. After a certain period of time, however, the viscosity of the reaction solution gradually increased and became gel in all cases irrespective of the initial feed ratio.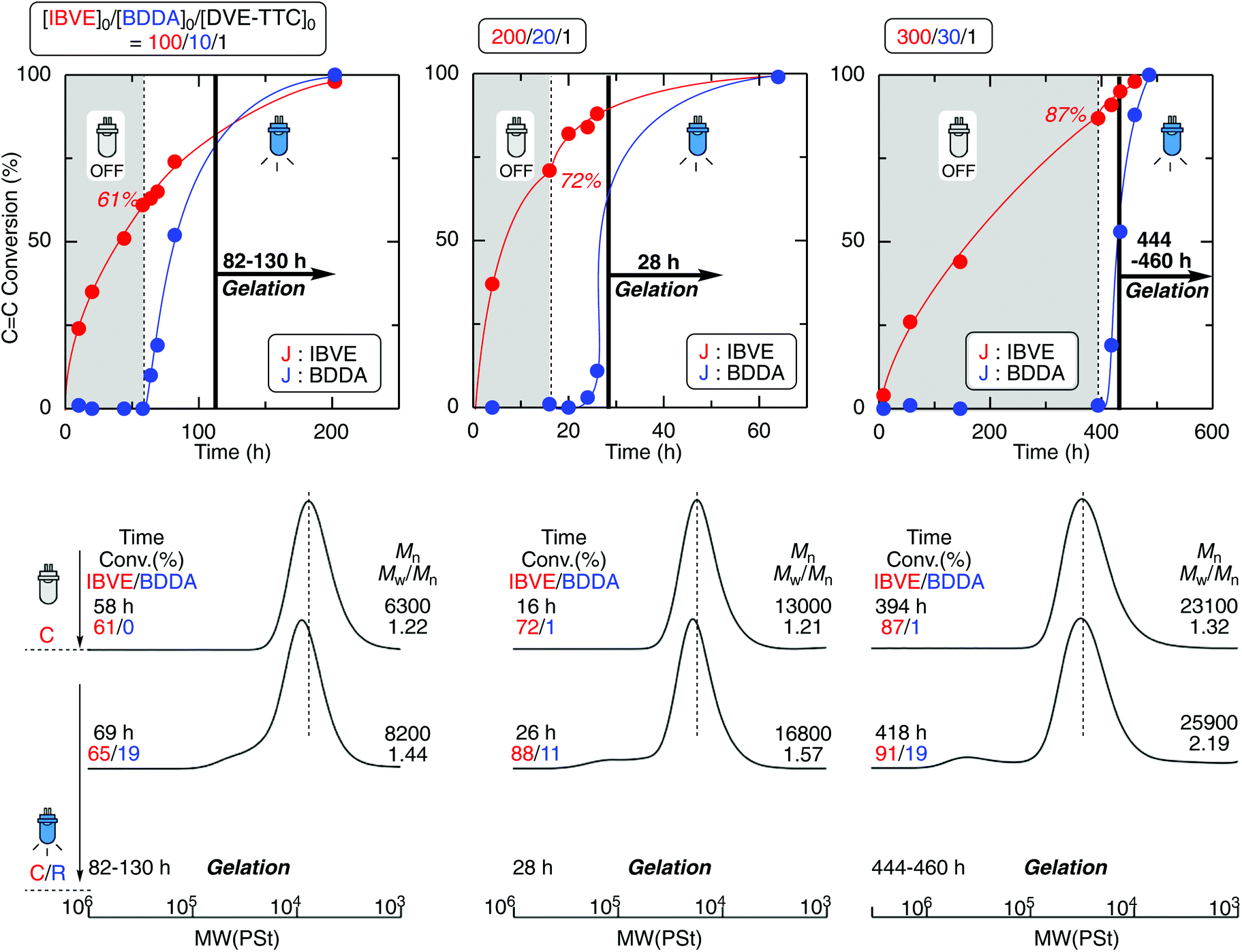 | ||
| Fig. 1 Mechanistic transformation from cationic polymerization in the dark into cross-linking radical polymerization under photoirradiation: [IBVE]0/[BDDA]0/[DVE-TTC]0/[ZnCl2]0/[TPO]0 = 3000/300/30/7.5/15 mM (for 100/110/1), 3000/300/15/5.0/5.0 mM (for 200/20/1), and 2250/225/7.5/2.5/2.5 mM (for 300/30/1) in toluene/Et2O (9/1) at 0 °C. | ||
As a control experiment, the polymerizations were conducted just under the blue light irradiation under the same condition except for the dark periods (Fig. S2†). Under the blue light irradiation in the presence of Lewis acid, both cationic and radical reaction can proceed at the same time.19,20,23 In sharp contrast to the reaction in dark, as the crosslinker of BDDA reacted from the initial stage of polymerization, the molecular weight of the produced polymers became higher than the calculated values and the gelation occurred at the moderate conversions of BDDA in the first tens of hours. Thus, DVE-TTC as the difunctional initiator generated telechelic living IBVE polymer chains by cationic polymerization in dark, and then upon the light irradiation, the growing terminals were transformed into the radical species to react with the cross-linker of BDDA to result in the polymer gel (Scheme 2).
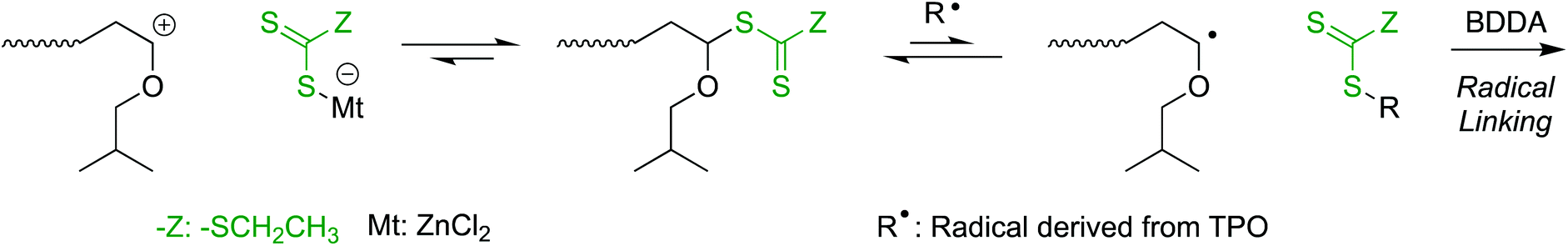 | ||
| Scheme 2 Mechanistic transformation of RAFT terminal from cationic into radical species. | ||
The molecular weight of the cross-linked polymer chain obtained in the off-on reaction was evaluated by cleaving ester bonds at the linking points after transesterification by methanolysis (Fig. 2). The obtained gel was treated with NaOCH3 in the mixture of CH3OH and THF at 60 °C. Since THF is a good solvent for the IBVE homopolymer, the gel swelled first, and then became a homogeneous solution, suggesting that transesterification successfully proceeded and the structure at the linking points changed from BDDA units to methyl acrylate units. The Mns of the product after transesterification were evaluated by SEC. As in the case of the first half of polymerizations in dark, the Mns were close to the calculated values on the assumption that one DVE-TTC RAFT molecule generated one polymer chain. The Mn values increased with the feed ratio of [IBVE]0/[DVE-TTC]0, although the MWDs became broader. These results indicate that the living IBVE polymers had been intermolecularly reacted specifically at around the terminus to form cross-linked gel with relatively well-controlled structures.
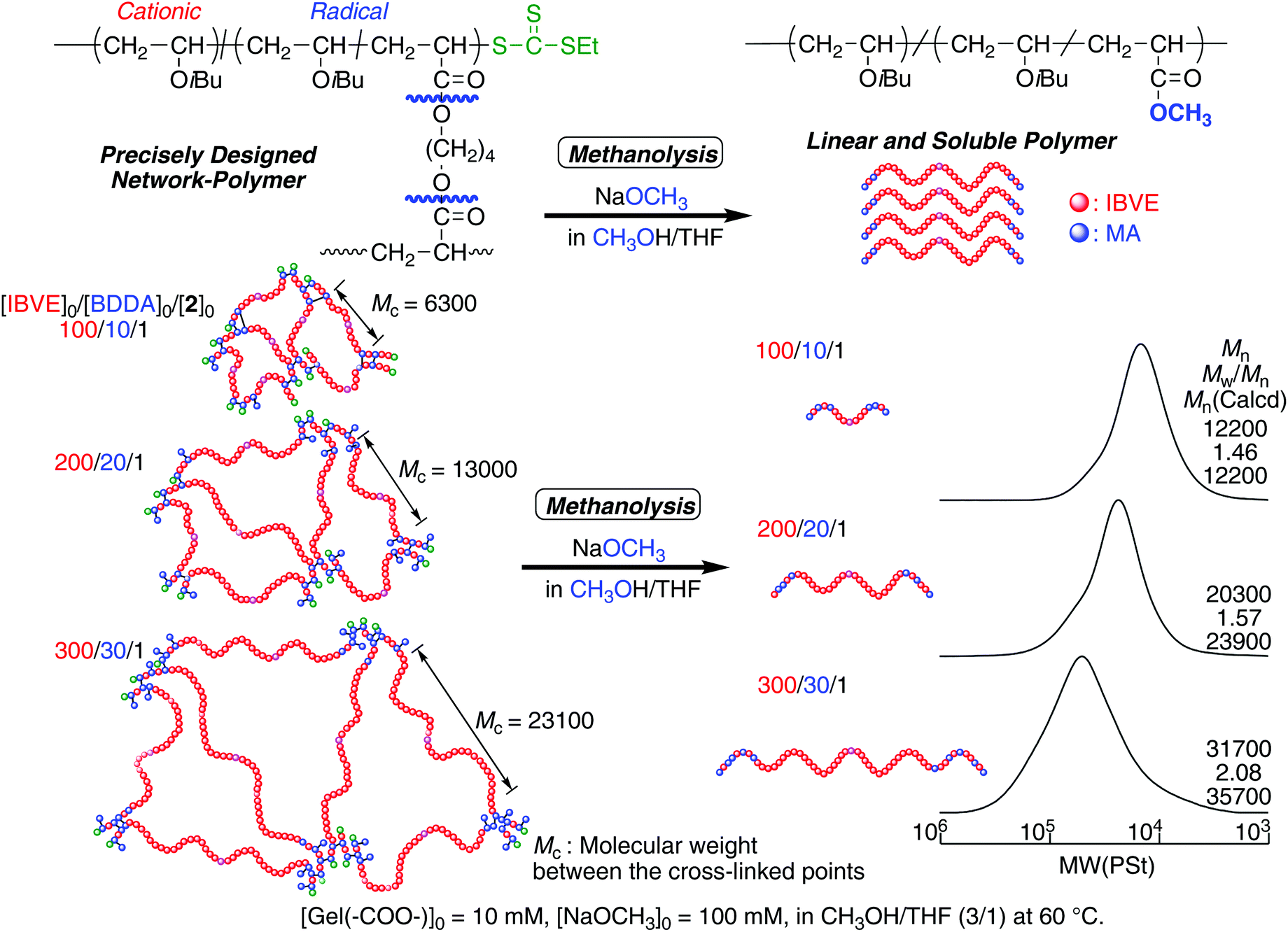 | ||
| Fig. 2 SEC curves of the products after methanolysis of the cross-linked polymer gel prepared in the same experiments as in Fig. 1. | ||
Cationic polymerization of EO2 for temperature-responsive polymer gel by mechanistic transformation
Upon the success on the gel formation by mechanistic transformation, we tried to use 2-(2-ethoxy)ethoxyethyl vinyl ether (EO2) as the monomer for cationic polymerization, of which the homopolymer is known as a temperature-responsive polymer in the aqueous solution with LCST behavior. However, the EO2 polymerization under the same condition as IBVE did not proceed well to generate only low molecular weight oligomer with low monomer conversion (Fig. S3†). This is probably due that the interaction between ZnCl2 and oxyethylene units in EO2 might weaken the Lewis acid's activity. Then, Al-based Lewis acids (EtAlCl2) were employed as the catalyst combined DVE-TTC. Although EO2 was able to be polymerized with EtAlCl2 to reach almost complete conversion, the MWDs of the obtained poly(EO2) were broader than those of poly(IBVE) above-mentioned and the Mns became lower than the calculated values as the polymerization proceeded (Fig. S4†). These should be also attributed to the interaction between Lewis acid and monomer, in which the bidentate- or tridentate-coordination might disturb the activation-deactivation process of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond by the Lewis acid, indicating slow equilibrium between cationic active species and dormant RAFT terminal that also may cause frequent irreversible chain-transfer reaction by β-proton elimination.
S bond by the Lewis acid, indicating slow equilibrium between cationic active species and dormant RAFT terminal that also may cause frequent irreversible chain-transfer reaction by β-proton elimination.
Therefore, we decided to explore another RAFT terminal for the cationic polymerization of EO2 (Fig. S5 and S6†). As the result, the living cationic polymerization of EO2 was achieved using dithiocarbamate-type RAFT agent, which was reported as an effective terminal in cationic polymerization,17 in conjunction with EtAlCl2. Fig. 3 shows the cationic polymerization of EO2 with difunctional dithiocarbamate (DVE-DTC) catalyzed by EtAlCl2 in toluene at 0 °C. The polymerization of EO2 proceeded well to afford telechelic living polymers, of which the Mn increased in direct proportion to the monomer conversion. The deviation of Mn from the calculated value is most probably because of the differences in the hydrodynamic volumes between polystyrene standard and poly(EO2). The chain-end fidelity of dithiocarbamate was also confirmed almost unity by 1H NMR. In addition, as the Lewis acid, Et3Al2Cl3 with a tuned Lewis acidity gave living polymers with slightly narrower MWDs (Mw/Mn ∼1.3) (Fig. S7†).
 | ||
| Fig. 3 Living cationic polymerization of EO2 with bifunctional dithiocarbamate (DVE-DTC): [EO2]0/[DVE-DTC]0/[EtAlCl2]0/[THF]0 = 1000/10/15/400 mM in toluene at 0 °C. (a) time-conversion, (b) Mn and Mw/Mn curves, (c) SEC curves, and (d) 1H NMR spectrum of the obtained polymer. | ||
Dithiocarbamate is known to be ineffective in controlling the polymerization of conjugated acrylate monomers but effective on that of unconjugated monomers such as vinyl acetate (VAc), of which the mechanistic transformation was also achieved in the combination with IBVE.20,21 Therefore, a divinyl ester of divinyl adipate (DVA) was used as the cross-linker for the formation of temperature-responsive poly(EO2) gel. The mechanistic transformation was then examined for the polymerization of EO2 in the presence of DVA and TPO in toluene, in which the living cationic polymerization EO2 was initiated with DVE-DTC by Et3Al2Cl3 in dark. And then, the blue LED was irradiated to the reaction mixture to induce the mechanistic transformation from cationic to radical species. Although the linking reaction did not proceed at low EO2 concentration, the polymer gel successfully formed under appropriate condition: [EO2]0 = 1.5 M or higher (Fig. 4 and S8†). This is because the monomer concentration should be above the overlapping concentration (about 0.64 mol L−1) of poly(EO2) (DPn = 100) (Fig. S9†). At lower concentration of EO2, DVA was consumed slowly upon light irradiation and the cross-linked polymer was observed in the SEC curves at higher molecular weight region, but the intermolecular cross-linking is not sufficient to form the polymer gel (Fig. 4a and d). On the other hand, at the higher EO2 concentrations of 1.5 and 2.0 mol L−1, gelation of the entire solution was observed after 150 and 120 hours of reaction, respectively (Fig. 4b and c). Thus, the poly(EO2) did not significantly gelate at 1.56 times the overlapping concentration, but at the higher concentrations such as 2.34 times the overlapping concentration the polymeric gel was successfully formed.
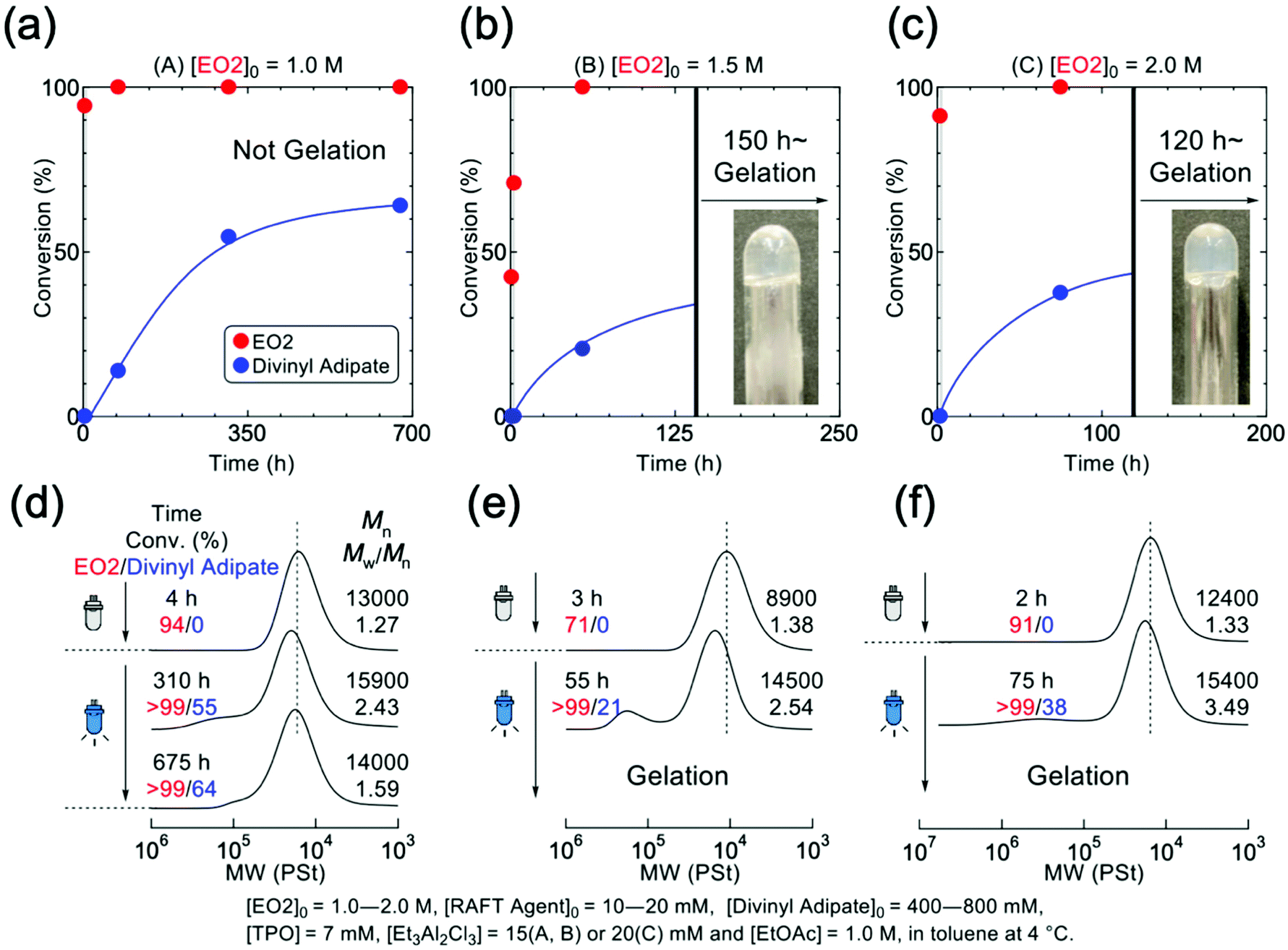 | ||
| Fig. 4 Assuming that the monomer is 100-mer after polymerization, the effect of the concentration of the resulting polymer on gelation is studied based on the concentration of overlapping polymer chains (=0.64 M), (a) time dependence of the reaction in a system with a monomer concentration of 1.0 mol L−1 in the preparation, (b) time dependence of the reaction in a system with a monomer concentration of 1.5 mol L−1 in the preparation, (c) time dependence of the reaction in a system with a monomer concentration of 2.0 mol L−1 in the preparation, (d) time dependence of SEC results for polymers obtained by (a), (e) time dependence of SEC results for polymers obtained by (b), (f) time dependence of SEC results for polymers obtained by (c). (mol L−1 is denoted by “M” in Figure), Blue LED 448 nm (60 mW cm−2). | ||
Polymer gels could also be prepared at fixing the monomer concentration at 1.5 mol L−1 by varying the amount of cross-linker (Fig. S11†). In all cases the reaction resulted in the gels formation, which became faster with higher amounts of cross-linker. After 2 weeks light irradiation, the consumption of vinyl groups inside the polymer gel could be roughly estimated by FT-IR (Fig. S12†). Table 1 summarizes the reaction time and conversion of DVA as the function of the concentration of DVA. Although the gel did not dissolve in any organic solvents, the progress of reaction was estimated from the ratio of the peak intensities of carbonyl and vinyl groups, which roughly concluded that approximately 80% of the vinyl groups derived from the cross-linker reacted under all conditions.
As mentioned above, the monomer concentration was crucial for the gel formation.5,7 If the reactivity of growing terminal is high enough, the telechelic polymer could be gelable as a result of the increase in excluded volume even in dilute solutions below the overlapping concentration. And the intermolecular linking reaction is less likely to occur in a quasi-concentrated solution where the polymers are entangled with each other. However, judging from the slope of the logarithmic plot of the dependence of the reduction concentration on polymer concentration, the system used for gelation in this study is a semi-dilute solution in which no entanglement of polymers occurs (Fig. S9c†). As also seen in the uncomplete conversion of DVA, this might be attributed to that the reactivity of growing radical of EO2 terminal is relatively low toward vinyl ester compared to acrylate. In any case, the polymer gel based on poly(EO2) could be obtained by the mechanistic transformation from cationic polymerization to radical cross-liking reaction by tuning the reaction condition, especially the monomer concentration, although there might be heterogeneity in cross-linking and the presence of uncross-linked chains.
Swelling and shrinkage of polymer gels
The degree of swelling of polymer gels composed of EO2 that had reached equilibrium swelling with various solvents was measured using polymer gels that had been exposed to light for about two more weeks after gelation under conditions in which the amount of cross-linking agent was varied. In most of the solvent systems, the effective cross-link density increased as the degree of swelling decreased with increasing the ratio of the cross-linker amount (Fig. 5).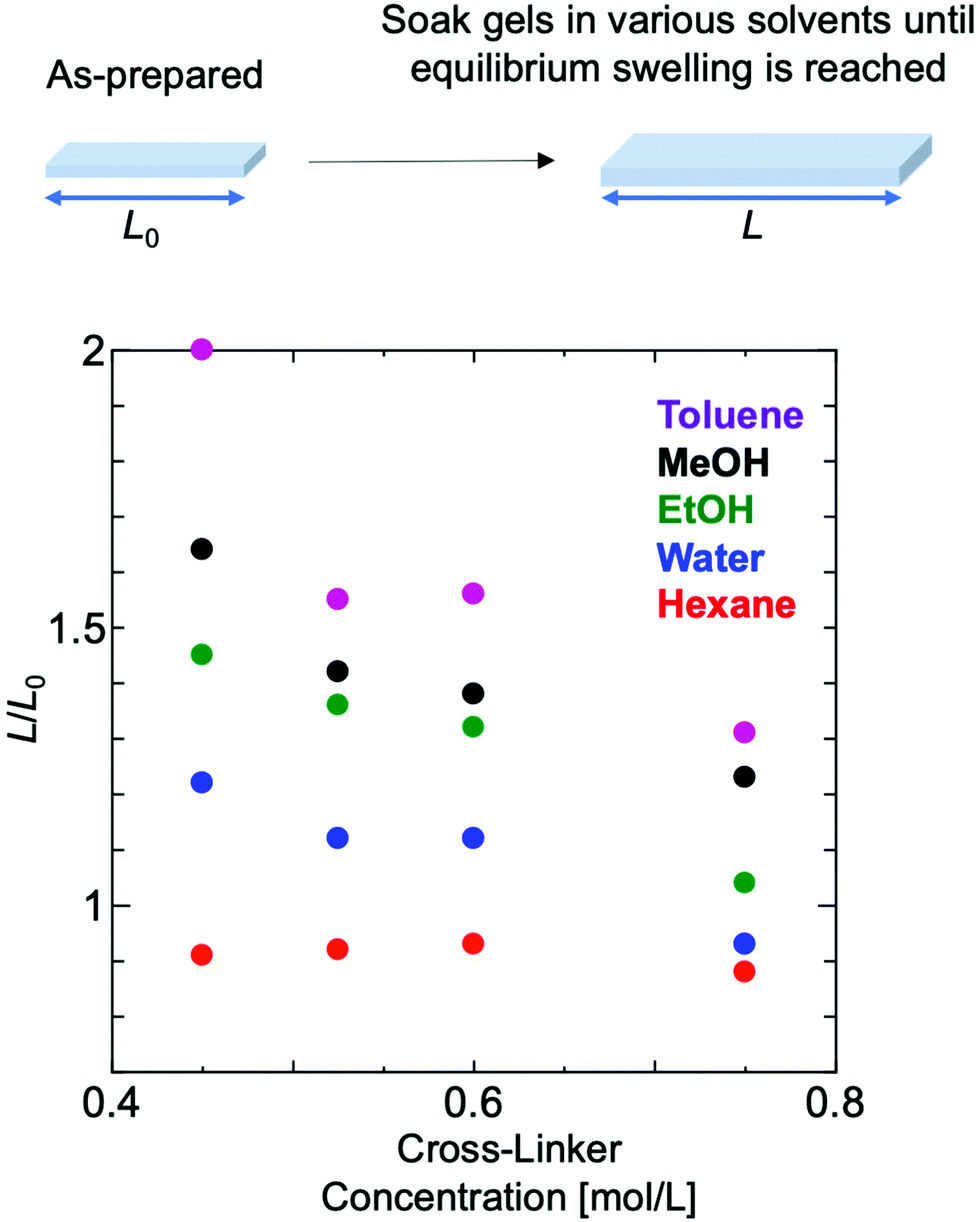 | ||
| Fig. 5 Swelling degree measurement of polymer gels composed of EO2 with different amounts of cross-linker in various solvents. | ||
It was also observed that the polymer swells well in the polymerization solvents toluene and methanol, and shrinks in hexane. When polymer gels with 50 equivalents of cross-linker to initiator were measured in water at varying temperatures, the degree of swelling changed continuously from 30 °C to 45 °C (Fig. 6).
 | ||
| Fig. 6 Temperature dependence of the degree of swelling in water of polymer gels composed of EO2 prepared by adding 50 times the cross-linker to the initiator (750 mM). | ||
Structural analysis of polymer gels
SAXS measurements were performed to evaluate microscopic structural changes in the resulting polymer gels. X-ray scattering is caused by density differences inside the gel.29,30 In order to investigate the detailed structure, we prepared and measured samples in two states in which the density difference was significantly changed: swollen in toluene and shrunken in hexane. Fig. 7a and b show the SAXS profiles obtained from polymer gels synthesized with various amounts of cross-linker (30, 35, 40, 50) in toluene and hexane, respectively. None of the scattering profiles show a dramatic low-q rise in the measured q range (0.069 < q < 3.5 nm−1), suggesting that the formation of significant aggregate structures at this spatial scale is suppressed. The scattering profile of polymer gels is often described by the following equation.31–33| I(q) = IOZ(q) + IDB(q) + IBKG | (1) |
 | (2) |
 | (3) |
 | ||
| Fig. 7 SAXS profiles of gels (a) SAXS scattering profiles of gels prepared at different cross-linking concentrations (the numbers in the figure are the concentration ratios of cross-linker and initiator). These gels were swollen with toluene and was set at 20 °C. (b) SAXS scattering profiles of gels prepared at different cross-linking concentrations (the numbers in the figure are the concentration ratios of cross-linker and initiator). These gels were swollen with hexane and was set at 20 °C. (c) Cross-linker addition amount dependence of IOZ(0)/IDB(0) in toluene, (d) cross-linker addition amount dependence of IOZ(0)/IDB(0) in hexane, (e) cross-linker addition amount dependence of ξ in toluene, (f) cross-linker addition amount dependence of ξ in hexane. | ||
Conclusions
In this study, we addressed the one-pot synthesis of temperature-responsive polymer gels with controlled network structure by switching light irradiation. By mixing all compounds and polymerizing them at more than twice the overlapping concentration, telechelic PEO2 with uniform molecular weight was generated, and subsequent photoirradiation cross-linked the PEO2 to form a gel. The degree of swelling was changed by changing the ratio of cross-linkers, and the polymer gel obtained in this study was confirmed to be temperature-responsive in water. SAXS analysis showed that the excess scattering at low-q was suppressed and the molecular weight between cross-linking points was controlled compared to polymer gels synthesized by free radical polymerization. However, spatial heterogeneity was observed during cross-linking due to the polymerization properties of PEO2, such as the RAFT terminus of PEO2, which is not easily activated radically, and the reactivity of the cross-linking agent.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors thank Maruzen Petrochemical for kindly providing 2-(2-ethoxy)ethoxyethyl vinyl ether (EO2). KS thanks to the financial support by JSPS KAKENHI Grant Number 18KK0158 and 19H00910, and JST-CREST (JPMJCR1992). The SAXS experiments at beam line BL38B1 were performed with the approval of RIKEN (Proposals No. 20210006).References
- Y. W. Gu, J. L. Zhao and J. A. Johnson, Trends Chem., 2019, 1, 318–334 CrossRef CAS.
- M. Shibayama, Polym. J., 2011, 43, 18–34 CrossRef CAS.
- J. Bastide and L. Leibler, Macromolecules, 1988, 21, 2647–2649 CrossRef CAS.
- S. Nakagawa and N. Yoshie, Polym. Chem., 2022, 13, 2074–2107 RSC.
- X. Li, S. Nakagawa, Y. Tsuji, N. Watanabe and M. Shibayama, Sci. Adv., 2019, 5, eaax8647 CrossRef CAS PubMed.
- S. Ida, M. Morimura, H. Kitanaka, Y. Hirokawa and S. Kanaoka, Polym. Chem., 2019, 10, 6122–6130 RSC.
- Y. Okaya, Y. Jochi, T. Seki, K. Satoh, M. Kamigaito, T. Hoshino, T. Nakatani, S. Fujinami, M. Takata and Y. Takeoka, Macromolecules, 2020, 53, 374–386 CrossRef CAS.
- Y. Baba, G. H. Gao, M. Hara, T. Seki, K. Satoh, M. Kamigaito, T. Hoshino, K. Urayama and Y. Takeoka, Macromolecules, 2021, 54, 10468–10476 CrossRef CAS.
- Y. Jochi, T. Seki, T. Soejima, K. Satoh, M. Kamigaito and Y. Takeoka, NPG Asia Mater., 2018, 10, 840–848 CrossRef CAS.
- T. Sakai, M. Kurakazu, Y. Akagi, M. Shibayama and U. Chung, Soft Matter, 2012, 8, 2730–2736 RSC.
- Z. H. Zhang, N. Corrigan, A. Bagheri, J. Y. Jin and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 17954–17963 CrossRef CAS PubMed.
- Y. T. Ma, V. Kottisch, E. A. McLoughlin, Z. W. Rouse, M. J. Supej, S. P. Baker and B. P. Fors, J. Am. Chem. Soc., 2021, 143, 21200–21205 CrossRef CAS PubMed.
- B. W. Zhao, J. J. Li, X. Q. Pan, Z. B. Zhang, G. Q. Jin and J. Zhu, ACS Macro Lett., 2021, 10, 1315–1320 CrossRef CAS PubMed.
- E. J. Kepola, E. Loizou, C. S. Patrickios, E. Leontidis, C. Voutouri, T. Stylianopoulos, R. Schweins, M. Gradzielski, C. Krumm, J. C. Tiller, M. Kushnir and C. Wesdemiotis, ACS Macro Lett., 2015, 4, 1163–1168 CrossRef CAS.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776, 10.1039/b603438a.
- Y. Tsuji, S. Nakagawa, C. I. Gupit, M. Ohira, M. Shibayama and X. Li, Macromolecules, 2020, 53, 7537–7545 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Prog. Polym. Sci., 2022, 124, 101485 CrossRef CAS.
- S. Kumagai, K. Nagai, K. Satoh and M. Kamigaito, Macromolecules, 2010, 43, 7523–7531 CrossRef CAS.
- H. Aoshima, M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2014, 53, 10932–10936 CrossRef CAS PubMed.
- K. Satoh, H. Hashimoto, S. Kumagai, H. Aoshima, M. Uchiyama, R. Ishibashi, Y. Fujiki and M. Kamigaito, Polym. Chem., 2017, 8, 5002–5011 RSC.
- K. Satoh, Y. Fujiki, M. Uchiyama and M. Kamigaito, ACS Symp. Ser., 2018, 1284, 323–334 CrossRef CAS.
- K. Satoh, Z. H. Sun, M. Uchiyama, M. Kamigaito, J. T. Xu and C. Boyer, Polym. J., 2020, 52, 65–73 CrossRef CAS.
- Q. Michaudel, V. Kottisch and B. P. Fors, Angew. Chem., Int. Ed., 2017, 56, 9670–9679 CrossRef CAS PubMed.
- M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer and G. G. Qiao, Adv. Sci., 2020, 7, 2001656 CrossRef CAS PubMed.
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. T. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 3, 1500394 CrossRef.
- T. K. Georgiou, C. S. Patrickios, P. W. Groh and B. Ivan, Macromolecules, 2007, 40, 2335–2343 CrossRef CAS.
- G. Kali, T. K. Georgiou, B. Ivan and C. S. Patrickios, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4289–4301 CrossRef CAS.
- H. Katayama, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 3747–3755 CrossRef CAS.
- D. E. Apostolides, C. S. Patrickios, T. Sakai, M. Guerre, G. Lopez, B. Ameduri, V. Ladmiral, M. Simon, M. Gradzielski, D. Clemens, C. Krumm, J. C. Tiller, B. Ernould and J. F. Gohy, Macromolecules, 2018, 51, 2476–2488 CrossRef CAS.
- X. H. Zhang, K. Kyriakos, M. Rikkou-Kalourkoti, E. N. Kitiri, C. S. Patrickios and C. M. Papadakis, Colloid Polym. Sci., 2016, 294, 1027–1036 CrossRef CAS.
- M. Shibayama, Macromol. Chem. Phys., 1998, 199, 1–30 CrossRef CAS.
- T. Matsunaga, T. Sakai, Y. Akagi, U. Chung and M. Shibayama, Macromolecules, 2009, 42, 1344–1351 CrossRef CAS.
- M. Shibayama, Small Angle Neutron Scattering on Gels, in Soft Matter Characterization, Springer, Dordrecht, 2008 Search PubMed.
- L. S. Z. Ornstein and F. Zernike, Accidental Deviations of Density and Opalescence at the Critical Point of a Single Substance, Amsterdam, 1914 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00554a |
| This journal is © The Royal Society of Chemistry 2022 |