
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControllable electrostatic manipulation of structure building blocks in noble metal aerogels†
Wei
Wei
 ab,
René
Hübner
c,
Maximilian
Georgi
a,
Cui
Wang
a,
Xiaodong
Wu
d and
Alexander
Eychmüller
*a
ab,
René
Hübner
c,
Maximilian
Georgi
a,
Cui
Wang
a,
Xiaodong
Wu
d and
Alexander
Eychmüller
*a
aPhysical Chemistry, Technische Universität Dresden, Andreas-Schubert-Bau, Zellescher Weg 19, 01069 Dresden, Germany. E-mail: alexander.eychmueller@tu-dresden.de
bCenter of Analysis and Test, School of Chemistry and Chemical Engineering, Jiangsu University, Xuefu Road 301, 212013 Zhenjiang, China
cHelmholtz-Zentrum Dresden-Rossendorf, Institute of Ion Beam Physics and Materials Research, Bautzner Landstrasse 400, 01328 Dresden, Germany
dCollege of Materials Science and Engineering, Nanjing Tech University, Puzhu South Road 30, 210009 Nanjing, China
First published on 6th May 2022
Abstract
The important role of structure homogeneity in three-dimensional network nanostructures serving as noble metal aerogels (NMAs) has attracted extensive attention in the field of electrochemistry in the last two decades, whereas a comprehensive study of tailoring skeleton units and element distributions in NMAs is still lacking. Herein, a new modulation strategy to easily prepare multiscale NMAs with tunable composition is developed by utilizing the electrostatic interaction between oppositely charged colloidal metal nanoparticles. The modulation rule of the chemical distribution in bimetallic aerogels leads to the construction of the as-tailored double skeleton aerogels for the first time. Considering their specific structures, the intrinsic and exceptional catalytic and electrocatalytic performances of NMAs were investigated. This study optimizes the structure homogeneity of noble metal aerogels by investigating nanoparticle–ligand interactions and provides further proof of their exceptional electrocatalytic capabilities.
Introduction
Aerogels are self-supporting lightweight materials that contain highly porous multidimensional interconnected networks.1–3 Due to the aerogel structure, these materials retain nanoscale properties at a macroscale and have extremely low density. In the past decades, tremendous advances have been made in synthesizing and developing novel aerogel materials from nanoparticles of oxides, semiconductors, carbon, polymers, and metals acting as building blocks for assembled networks.4–8 In particular, noble metal aerogels (NMAs) have emerged as unique and attractive material platforms for multiple applications (e.g., catalysis, sensing, and surface-enhanced Raman scattering). The advantages of noble metals include a high electrical conductivity with a filled electron d-band. Additionally, due to the aerogel structure, they have a self-supported architecture with abundant active sites and electron/mass transfer pathways.9–11 However, NMAs, as one of the newest members of the aerogel family, were only discovered in 2009;12 they are still in their infancy and will be extensively studied in the twenty-first century.3,11The development of synthesis methodologies and the disclosure of the formation mechanisms of NMAs are a top priority. The sol–gel process is the typical method for NMA production. Specifically, this process refers to the fabrication procedure where wet hydrogels are formed by destabilization of building blocks to allow the gradual formation of continuous networks using steady nanoparticle solutions or molecular precursors. The gels are then dried by non-destructive drying methods (e.g., supercritical CO2 drying or freeze drying) to become homogeneous aerogels. The triggering destabilization process is an important and difficult step in the fabrication of a broad range of NMA systems.11 In previous studies, additional additives (such as oxidants, salts, and dopamine) were used to obtain gels.13–15 Additionally, heating or freezing procedures as gelation-initiating operations have also been introduced in the NMA preparation.16–19 However, many fundamental and operating difficulties remain, such as long gelation times in aqueous systems, time-consuming washing steps, and costly drying procedures in the NMA preparation. Recently, our group studied the NMA formation and developed the freeze–thaw strategy, the disturbance-promoted gelation method, and the excessive-reductant-directed gelation strategy.20–23 Inspired by these significant advancements in the NMA preparation, it is necessary to regulate the structure formation of the building blocks or the skeleton units in the gel to further enhance the catalytic properties of NMAs.
Using the self-assembly of oppositely charged particles via electrostatic interactions, different nanoparticles (NPs) were arranged to form macroscopic structures of well-arrayed superparticles.24–27 For example, a simple one-step approach was developed to obtain Au/CdS crystalline superlattices by mixing negatively charged gold nanocrystals and positively charged cadmium sulfide particles.25 Lin's group synthesized novel Pt–shell–Au–core nanocomposites by the electrostatic self-assembly of positively charged Pt NPs and negatively charged Au NPs.26 Recently, this method has been extended to NMA synthesis, where Pd gels were fabricated by a calcium–ion-induced electrostatic crosslinking process. However, this method still required a time-consuming ultracentrifugation procedure for the Pd NP precursor.28 Hence, the electrostatic manipulation or the homo-/heteroaggregation of oppositely charged colloids was determined to be important in the preparation of advanced materials and has received increasing attention.24–28 According to the DLVO theory, named after Derjaguin, Landau, Vervey, and Overbeek, an electrostatic aggregation occurs by the combined action of attractive electrostatic forces (repulsive interactions are weaker than attractive interactions) and van der Waals forces. Due to the attractive electrostatics, the rate of electrostatic homo-/heteroaggregation can be higher than that expected based on the monotonous colloidal particle arrangements alone. Because of this higher rate, a simple, rapid, and general synthesis method for NMAs by electrostatic manipulation with structure controllability and corresponding application studies is urgently needed.
In developing a feasible synthesis based on electrostatic interactions, the first step was to investigate oppositely charged noble metal particles as precursors. Due to the ligand chemistry in colloid solutions, an extensive range of ligand-stabilized gold NPs with different surface charges were obtained in prior studies.22 The next objective was to develop a better understanding of electrostatic manipulation to prepare and modulate NMAs in a simple and novel process. Inspired by these empirical considerations, a simple and rapid method for the preparation of Au gels with tunable inner structures (ligament size = 10–30 nm) was developed by mixing oppositely charged gold NP solutions (CM = 0.2–0.5 mM) without further addition of the reagent, thus verifying the electrostatic homoaggregation approach. Next, an array of Au–Pd bimetallic aerogels with controlled ligament sizes were prepared by electrostatic heteroaggregation by mixing separately synthesized, oppositely charged gold and palladium NPs. Taking advantage of the non-reagent-triggered destabilization of this method than previous reports (Table S6, ESI†), we obtained bimetallic aerogels with double-skeleton structures and high specific surface areas (22.5–97.3 m2 g−1) by adjusting the Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd ratio. In addition, we found that, by replacing dopamine with polydopamine as a ligand for noble metal nanoparticles, the gold nanoparticle precursor mixed with other charged noble metal particles results in NMAs with smaller ligament sizes. Compared with the above-mentioned electrostatic manipulation, the average ligament sizes of Au in the NMAs decrease from 15.8 nm to 6.3 nm, and those of Pd in the NMAs decrease from 5.6 nm to 4.0 nm in the case of the dopamine assisted method. Notably, the catalytic reduction of p-nitrophenol and the ethanol oxidation reaction (EOR) of NMAs were exploited as model reactions for the optimization of composition-tuned aerogels in a three-dimensional (3D) nanowire-based network. Hence, this study not only provides a fast and effective method to obtain double-skeleton-structured NMAs directly from noble metal particle solutions without subsequent addition of the reagent but also presents a strategy to adjust the element distribution and hence the chemical or electrocatalytic performance of NMAs. These findings also illustrate a new synthesis mechanism of bimetallic aerogels with double-skeleton structures for particular applications.
:Pd ratio. In addition, we found that, by replacing dopamine with polydopamine as a ligand for noble metal nanoparticles, the gold nanoparticle precursor mixed with other charged noble metal particles results in NMAs with smaller ligament sizes. Compared with the above-mentioned electrostatic manipulation, the average ligament sizes of Au in the NMAs decrease from 15.8 nm to 6.3 nm, and those of Pd in the NMAs decrease from 5.6 nm to 4.0 nm in the case of the dopamine assisted method. Notably, the catalytic reduction of p-nitrophenol and the ethanol oxidation reaction (EOR) of NMAs were exploited as model reactions for the optimization of composition-tuned aerogels in a three-dimensional (3D) nanowire-based network. Hence, this study not only provides a fast and effective method to obtain double-skeleton-structured NMAs directly from noble metal particle solutions without subsequent addition of the reagent but also presents a strategy to adjust the element distribution and hence the chemical or electrocatalytic performance of NMAs. These findings also illustrate a new synthesis mechanism of bimetallic aerogels with double-skeleton structures for particular applications.
Results and discussion
Fabrication of gold aerogels by electrostatic co-aggregation
A variety of gold NP solutions with different zeta potentials were obtained by the application of different ligands. Gold NPs were synthesized by reducing tetrachloroaurate with a specific amount of NaBH4 in the presence of a defined amount of the respective ligand. Based on previous research on ligand-assisted gold NPs, several ligands were found (six cationic ligands and eight anionic ligands) to stabilize gold NPs after repeated screening experiments (Table S1, ESI†). The UV-vis absorption spectra of these gold NP solutions are shown in Fig. S1 (ESI†). Subsequently, we matched oppositely charged colloidal gold NP solutions (equivalent volume and gold concentration) to increase the attractive electrostatic forces between the colloidal gold NPs. A series of trial-and-error experiments showed the possibility of obtaining a metallic gel by mixing oppositely charged gold NP solutions. These results, summarized in Fig. 1a and Fig. S2 (ESI†), show different variations in the final product (solutions, powders, or gels), thus implicating different microstructures (stable colloids without precipitation, metastable colloids with precipitation, or typical 3D gel networks) in the final products. | ||
| Fig. 1 Fabrication process of gold gels by electrostatic interactions. (a) Summary of the status of gold gels induced by mixing of different ligand-stabilized gold NP solutions. The inverted triangle, diffused black circle, and pink circle represent the formation of a gel, powder, and stable colloidal solution, respectively. (b) Photographs of the synthesis of a gold aerogel using oppositely charged Au NP solutions (CM = 0.2 mM) by applying two different ligands (red: NaCA and purple: CTAB). (c) UV-vis absorption spectra of the Au NP solutions before and after mixing (inset: the proposed schematic gelation process). (d) TEM images and ligament size distributions (insets) of Au NPs fabricated in the presence of different ligands and the obtained gold aerogel. The scale bar in (d) is 50 nm. | ||
In the next step, we focused on one pair of oppositely charged colloidal gold NP solutions (NaCA and CTAB) that successfully formed a gel (Fig. 1b). The details of the synthesis procedure are provided in the supplementary information. Before mixing, the trisodium citrate (NaCA) ligand-protected (negatively charged n-Au NPs) and cetyltrimethylammonium bromide (CTAB) ligand-protected (positively charged p-Au NPs) Au NP solutions were brownish reddish and purple, respectively, in color. These solutions turned black immediately after mixing. Accordingly, the characteristic absorption peak (∼502 nm) of the gold NPs in the ultraviolet-visible (UV-vis) spectrum (Fig. 1c) disappeared, and the broad-band absorption curve that appeared after mixing suggested the formation of large nanostructured gold aggregates.22 The representative time-sequential in situ optical images of the Au NP mixtures show the aggregation of the Au NPs into oligomers and confirm the formation of large branched-nanowire (NW)-like gold gel networks (Fig. S3 and Movie S1, ESI†). This formation process is similar to the salt-induced gelation reported in the literature.22
The morphologies of the ligand-stabilized Au NPs and the final aerogels (obtained after freeze-drying) were examined using a transmission electron microscopy (TEM) (Fig. 1d). It was confirmed that there is no significant difference in the morphology and size distribution of the separately synthesized gold NPs. The negatively charged NaCA-stabilized (4.3 ± 1.2 nm) and positively charged CTAB-stabilized (6.7 ± 2.7 nm) gold NPs (with a quasi-spherical shape) form a porous gel structure consisting of interconnected Au NWs with an average ligament size of 17.2 ± 4.9 nm. The larger diameter of the Au gel compared to that of the initial Au NPs can result from “less controlled” reaction conditions. Several Au NPs get in contact with each other more easily and they directly melt together to form bigger NPs due to the fusion of gold. Combining previous experience (freeze–thaw strategy, disturbance-promoted gelation method, etc.) with the present results, we conclude that the final porous gold gel assembles by gradual Au NP aggregation caused by attractive electrostatic interactions. The fusion of the soft Au oligomers is due to a competition between the growth and sedimentation of the Au aggregates.20–22
To understand the self-assembly behavior of the oppositely charged gold NPs by electrostatic co-aggregation, mixtures of negatively charged NaCA-stabilized gold NPs and positively charged CTAB-stabilized gold NPs at different volume ratios (P/N) were characterized by zeta potential measurements. Fig. 2a shows the numerical results of the zeta potential in this mixed system. As the P/N ratio increases from 0.09 to 1.0, the zeta potential decreases and approaches almost zero, suggesting that the mixture is converted to a gold gel or aggregate. After the addition of abundant p-Au NPs into n-Au NPs (the P/N value increases from 2.0 to 11.0), the zeta potential becomes more positive again and the NP mixture remains stable as a colloidal solution. In particular, at P/N = 1, the zeta potential (ζ) of the CTAB-stabilized gold NPs (+27.7 mV) decreases to approximately 0 mV (measured value +1.5 mV) after adding the NaCA-stabilized gold NPs (−37.5 mV), indicating the neutralization of the surface charges. For P/N < 1, the zeta potential of the final solutions varies from +3 to +6 mV. For P/N > 1, the zeta potential increases again. Based on these observations, we investigated in more detail how different volume ratios affect the stability of the solution and the formation of the gel. To evaluate this mechanism, we performed density functional theory (DFT) calculations to determine the binding energy (Ebinding) of the protonated forms of the ligands on the Au(111) surface (Fig. S4 and S5, ESI†), i.e. for cetyltrimethylammonium ions (CTA+) and citrate ions (CA−). As shown in Table S2 (ESI†), the binding energy of Au(111)–CTA+ (−0.67 eV) is smaller than the binding energy of Au(111)–CA− (−0.36 eV), indicating a stronger interaction of the gold particle with CTA+ compared to that with CA−. This suggests that the CTAB-stabilized gold NPs (p-Au NPs) are more stable than the NaCA-stabilized NPs (n-Au NPs). Therefore, if the amount of CTAB-stabilized gold NPs is high, the ligand can prevent the aggregation of the gold NPs due to the gold particles interacting with the ligand molecules. In contrast to the usual preparation of gold aerogels, the gelation time and BET surface values of the gold aerogels can be easily adjusted by this simple and convenient strategy of applying oppositely charged Au NP solutions of various volume ratios (Fig. 2b). By increasing the amount of the slightly smaller NaCA-stabilized Au NPs, the ligament size increases and consequently, the specific surface area of the gels decreases. One explanation for the increase in the ligament size is the higher mobility of smaller Au NPs, which causes them to aggregate in a less-ordered manner. Further studies are necessary to gain a deeper understanding of this aggregation behavior.
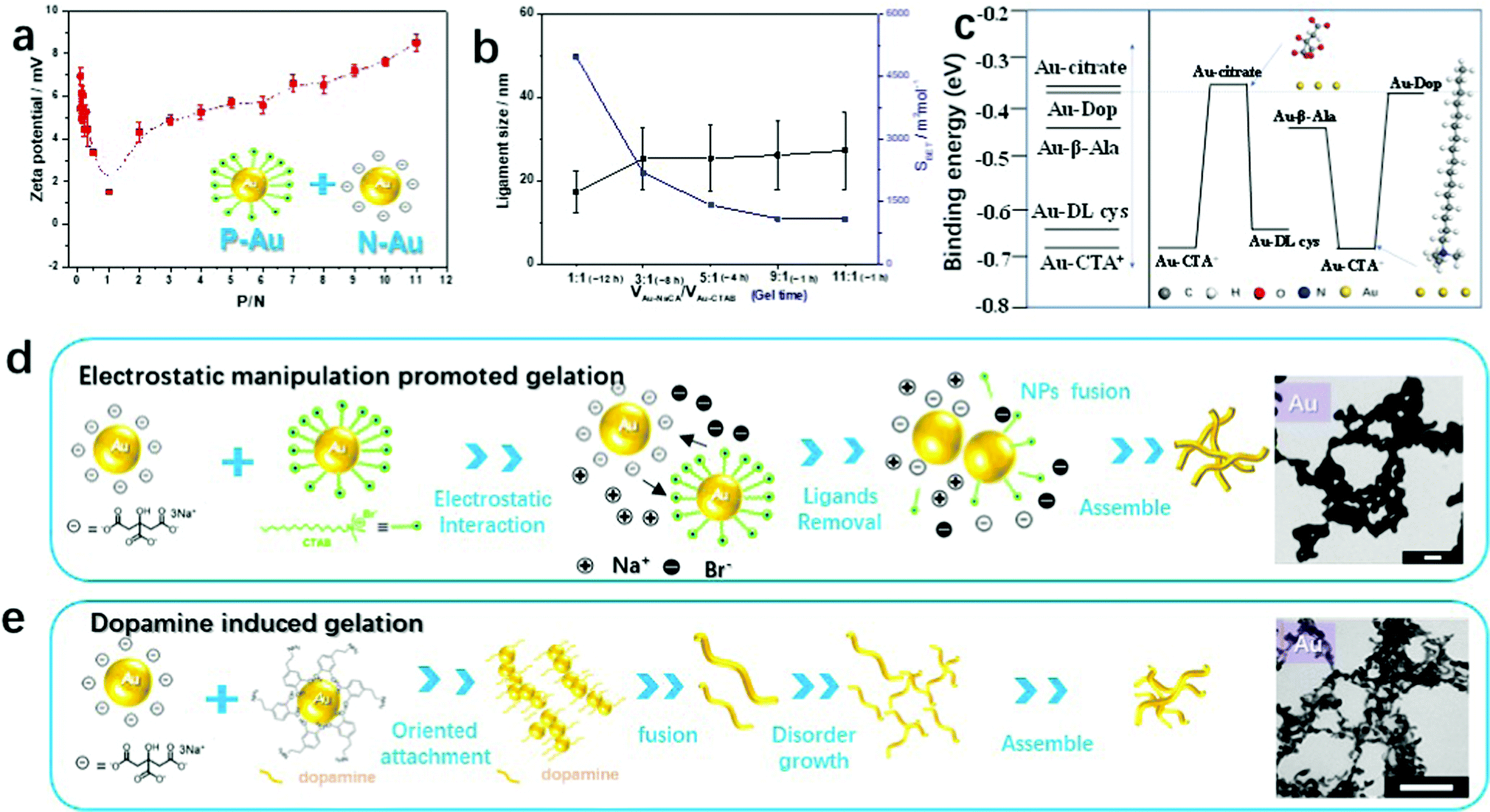 | ||
| Fig. 2 Electrostatic manipulation and analysis of the Au gelation process. (a) Zeta potential of p-Au/n-Au mixtures in aqueous solution at different volume mixing ratios (P/N). (b) Ligament size and BET surface area values of Au aerogels prepared from different volume ratios of negatively and positively charged Au NPs. (c) DFT calculations. Left: Calculated binding energies (Ebinding) of different capping ligands adsorbed on the Au(111) surface. Right: A typical mixing process based on different capping ligands. Inset: Optimized structures of citrate and CTA+ on the Au(111) surface. (d) Proposed schematic gelation mechanism induced by the electrostatic interaction of oppositely charged Au NPs. (e) Proposed schematic gelation mechanism induced by dopamine. The scale bars in (d) and (e) are 100 nm. | ||
Deeper insights into the electrostatic-manipulation-promoted gelation
To obtain a better understanding of the interaction of the charged nanoparticles and their ligands, we investigated the mixed nanoparticle solutions and their products as shown in Fig. 1a and Fig. S2 (ESI†). From the experimental observations mentioned above, the mixing results of most ligands contain polymer functional groups, such as poly(diallyldimethylammonium chloride) solution (PDDA), polyvinylpyrrolidone (PVP), and poly(sodium-4-styrenesulfonate) (PSS), leading to stable colloidal gold NP solutions without precipitation. Due to their large size, the polymer ligands stabilize the gold nanoparticle solutions due to steric effects.22 Most gold NP solutions containing thiolate-based ligands mixed with negatively charged gold NPs have undesirable effects on gelation. A few precipitates were found at the bottom of the vessel. The final products of these mixtures are shown in Fig. S6b, d, and e (ESI†). The high binding energy of a single thiol group on the Au NP surface (Au–S) prevents destabilization by oppositely charged particles. Due to the strong bonding, thiol based functionalized Au NPs remain in the colloidal state even when the oppositely charged Au NPs are added. Consequently, the color of the particle solution does not change and remains reddish.29,30 Meanwhile, dopamine, as an initiator, triggers the formation of gold gels efficiently via electrostatic interactions. More than half of the different gold NP solutions are transformed into gels after mixing with dopamine-functionalized gold NP solutions (Fig. S6f, ESI†). This is similar to previous reports of a dopamine-based method that can construct NMAs with a special oriented attachment mechanism.15,31 Due to the complex and diverse roles of dopamine during the gelation, we will discuss this special case in the next chapter.Similar to the behavior of thiol-based ligands, quaternary ammonium surfactants, such as CTAB and cetyltrimethylammonium chloride (CTAC), protect most gold NPs from aggregation and precipitation. Only a few specific n-Au NPs stabilized with NaCA force the particles to aggregate. Changing the concentration of the CTAC ligand before mixing with different n-Au NPs leads to the same results, and aggregation is rarely observed (Fig. S7, ESI†). The corresponding zeta potential of each mixture does not show any significant changes (Fig. S8, ESI†). Except for the NaCA-stabilized Au NPs (∼1.2 mV), all mixtures display positive (>10 mV) or very negative (PSS −40 mV) zeta potentials. A high zeta potential value implies highly charged particles, which prevents the aggregation of the particles due to electrostatic repulsion. We can infer that, when the zeta potential value is high, the stability of the colloid is high, and the probability of generating aggregates is low. In contrast to thiolates, CTAB/CTAC does not form covalent bonds with the Au NPs. The long and branched chain and the steric functional groups of these ligands may be effective tools to prevent the aggregation of gold NPs. Most of the mixtures were still present as a colored solution without aggregation. Furthermore, the role of halides, which will be discussed later, may be important. Overall, changing the concentration of the CTAB/CTAC ligands leads to the same results as changing the concentration of the initial ligands. Similar results are found for the adjustment of the thiolate bond ligand concentrations in the mixture system (Fig. S9, ESI†). The aforementioned results suggest that the mixture of mutually attractive colloidal species (i.e. negatively and positively charged colloids) is not the only factor in obtaining metal hydrogels. Metal bonds are formed by the electrostatic attraction between the metal nuclei and free electrons, and surface-capping ligands complicate the gelation process.3 In particular, the charge density or the interaction between the ligands plays an important role and needs further investigation to understand their influence.
To further investigate the mechanism of the electrostatic-manipulation-promoted gelation and support our assumptions, we carried out DFT calculations to evaluate the binding energy (Ebinding) of the protonated form of CTAB on the Au surface. We compared these results with other capping ligands such as NaCA, β-Ala, and L-cysteine (Fig. S10–S12, ESI†). As shown in Fig. 2c and Table S1 (ESI1), the Ebinding increases in the order of Au(111)–CTA+ (−0.67 eV) < Au(111)–L-Cys (−0.65 eV) < Au(111)–β-Ala (−0.43 eV) < Au(111)–CA− (−0.36 eV). Based on this comparison, the long-branched chain of CTA+ and the Au–S bonds from the thiolate-bond ligands such as cysteamine (Cys), cysteamine hydrochloride (Cys·HCl), and L-cysteine hydrochloride (L-Cys) prevent the formation of aggregated structures and thus maintain the gold NP stability. As described above, the CTAB-stabilized gold NPs behaved differently in the actual experiment. They can be easily destabilized by mixing with NaCA-stabilized gold NPs. We speculate that another component of the surfactant CTAB, in particular the halide ion (Br−), could play a significant role. Our recent work disclosed detailed mechanisms regarding the specific ion effects to explain the gelation process and showed a cation-directed manipulation mechanism for controlling the ligament size of the resulting noble metal gels.29 Zhang et al. demonstrated a facile approach for the template-free and environmentally friendly synthesis of sponge-like gold structures by the halide-induced aggregation of Au NPs.32 The Br− ion replaces some citrate ions from the NaCA-stabilized gold NPs and finally produces nanowires in aggregated network structures that undergo fusion by salting-out effects.
Combining the above results and previous studies, we suggest the gelation mechanism of oppositely charged colloidal gold NP solutions as follows (Fig. 2d): (i) the negative gold NPs instantly approach the positive gold NPs because of the electrostatic interaction. (ii) The Br− ions from the CTAB-stabilized gold NPs replace a few citrate ions from the NaCA-stabilized gold NPs. The ligand-replaced gold particles, especially with halide ions Br−, are unstable compared with the original gold NPs that are characterized by oppositely charged ions. (iii) Metal aggregation continuously occurs by fusing gold NPs with free surface gold NPs to achieve a lower surface energy. (iv) The aggregation process is repeated and more Au NWs are successively formed to assemble randomly in a three-dimensional network. (v) Hydrogel precipitation at the bottom of the reaction flask is caused by the gravitational settlement of the particles and network multi-aggregates. This gelation process is similar to previously suggested gelation mechanisms, such as the salting-out-assisted mechanism for NMAs.29 The significant difference lies in the addition of a salt to obtain oppositely charged particles in the latter case. In the gelation process reported here, the addition of a salt to a colloidal solution is no longer necessary, since the mixing of oppositely charged solutions directs and initiates gelation. In addition to fewer reaction steps and a short gelation time leading to the cost reduction of the gel preparation, the follow-up washing time for hydrogel cleaning is also reduced compared to the excessive-reductant-directed gelation strategy.
Dopamine-induced gelation in electrostatic manipulation
In the following, we focus on the strong and effective gelation support by the dopamine molecule, which was observed in the above-mentioned gelation overview of oppositely charged Au NPs (Fig. 1a and Fig. S3, ESI†). In previous studies, we demonstrated the synthesis of Au aerogels via a dopamine-induced route, where the Au aerogels are composed of 3D nanowire networks with diameters of 5–6 nm.15 Zhu et al. introduced a facile synthetic strategy to form polydopamine (PDA)-capped AuPt hydrogels using a one-step method.31 Meanwhile, PDA-coated gold nanoparticles could be synthesized with diverse morphologies depending on the dopamine concentration including aggregated NPs, almost monodisperse NPs, and NP chains.38 Dopamine acts not only as a ligand stabilizer but also as a gelation trigger depending on the reaction environment. Interestingly, we found an analogous gelation phenomenon for ligand-stabilized gold NPs mixed with a gold NP solution in the presence of dopamine (e.g. Au-CA + Au-Dop), as revealed by real-time observations. The gelation process was observed by in situ optical microscopy (Movie S2 and Fig. S13c, ESI†) and was similar to the above-mentioned gelation process. According to UV-vis absorption spectroscopy (Fig. S13a, ESI†), dopamine shows an absorption maximum at 280 nm and the synthesized ligand-protected Au NPs show an absorption maximum at 519 nm. After mixing, the absorption peak at 519 nm turned to a broad-band absorption curve, similar to the previous descriptions. The morphologies and ligament sizes of the resulting gold gels (NaCA-stabilized Au NPs and dopamine-stabilized Au NPs) were investigated using a TEM (Fig. S13b (ESI†) and the inset photograph of Fig. 2e). The gel structures are composed of interconnected networks of ultrathin wire-like strands and exhibit a small ligament size (5.7 ± 1.1 nm) with open pores. These results agree with similar experiments where dopamine acts as a gelation trigger in the dopamine-induced route, resulting in similar gold-branched-nanowire networks with an average diameter of 5 nm.15,31In the theoretical calculation analysis, there is a small difference in the binding energies of Au(111)–β-Ala, Au(111)-Dop, and Au(111)-CA−. The Ebinding lies in the order of Au(111)–β-Ala (−0.43 eV) < Au(111)–Dop (−0.39 eV) < Au(111)–CA− (−0.36 eV). We found that dopamine shows intermediate binding ability to the gold surface with different ligands. Additionally, this reagent shows improved gelation performance. We believe that the strong gelation phenomena are due to the chemical and adhesive properties of dopamine. The chemical properties of dopamine mainly focus on self-polymerization and vinyl polymerization in aerated alkaline solutions.38 Adhesion is the most important property of dopamine as a biological material.15 Given these two properties of dopamine, we conclude that the presence of dopamine in a mixture of noble metal NPs can form interlaced branched nanowires to obtain aerogels.35,36 Based on previous and current research studies, a mechanism is shown in Fig. 2e, where small noble metal nanoparticles are first formed by reducing the corresponding metal ions with sodium borohydride and stabilizing them with different ligands (one of them is dopamine). After mixing, the noble metal NPs fuse together to form branched-nanowire chain structures assisted by dopamine, which serves as a structure director due to the oriented-attachment growth mechanism.31,36 Additionally, the dopamine in the basic solution oxidizes and polymerizes into polydopamine to form a coating layer on the surface of the hydrogel.35,36 The aggregation process is a multistep reaction as nanochains grow disorderly to assemble into three-dimensional networks. The final hydrogel forms at the bottom of the reaction flask by the gravitational settlement of the multi-aggregates.
Further characterization of the gold aerogels
The crystalline structure of the gold aerogels obtained by the electrostatic interaction and dopamine-induced gelation was further characterized by X-ray diffraction (XRD). As shown in Fig. S14 (ESI†), the dominant peaks at 38.2°, 44.3°, 64.6°, 77.5°, and 81.7° demonstrate the crystalline nature of the gold aerogels and correspond to the diffraction from the (111), (200), (220), (311), and (222) planes of face-centered-cubic metallic gold (PDF#04-0784). The difference in the final ligament size of the gels was further confirmed by XRD for the homologous samples, where the diffraction peaks broaden with the decreasing ligament size following the Scherrer equation and the nanoparticular structure of the aerogels.37,39Both the morphology and ligament size of the gold gels are strongly influenced by the P/N ratio in the CTAB/NaCA hybrid system via electrostatic manipulation, as previously demonstrated (Fig. S15, ESI†). When the concentration of CTAB in the final mixture decreases (Fig. S15a and b, ESI†), the ligament size increases by ca. 10 nm. Because the Br− ions from CTAB partially coat the Au NPs, forming close-packed aggregates with little fusion, high Br− ion concentrations can prevent the fusion of the aggregates.32 If dopamine is involved in the synthesis of the gold aerogel (Fig. S15c and d, ESI†), it plays a critical role in controlling the metal structure. The results of the comparative study show that the dopamine-assisted method yields a smaller ligament size (5.7 ± 1.1 nm) of gold aerogels compared to the CTAB/NaCA hybrid system method (Table S3, ESI†). This causes the gold aerogel to have a high surface area of 57.4 m2 g−1. Because the ligament size also affects other porosity-related properties such as the specific surface area, the pore volume, and the pore size distribution, N2-physisorption experiments were performed for the samples (Fig. S16 and Table S3, ESI†). The measured isotherms are similar to each other and exhibit a typical NMA-like behavior,20–22i.e. a mixture of type II and IV isotherms, which is caused by the broad pore size distributions. The aerogels are composed of micro-, meso-, and macropores. As expected, both the specific surface area and total pore volume decrease with the increasing ligament size. In summary, the simple mixing of oppositely charged NP solutions is a promising approach for achieving controlled Au aerogel architectures (ligament sizes ranging from ∼6 nm to 30 nm). It is possible to convert nanoparticles to gels by simply mixing different ligand-stabilized gold NPs.
Fabrication of Au–Pd aerogels by two gelation processes and their characterization
Bimetallic Au–Pd aerogels were extensively investigated in our previous publications because of the combination of the high electrical conductivity of gold and the high catalytic activity of palladium.20–23 Optimizing the synthesis of NMAs is a key issue for large-scale applications. On this account, bimetallic AuPd aerogels were also prepared using the new synthetic routes presented here: (a) electrostatic-manipulation-promoted gelation and (b) dopamine-induced gelation. The Au–Pd aerogels were synthesized from NaCA-stabilized gold NPs and CTAB-stabilized palladium NPs or dopamine-stabilized palladium NPs. The gel compositions and properties are summarized in Table S4 (ESI†). Compared to the Au NPs, the Pd NPs formed stable, dark-grey colloidal solutions (Fig. S17a and S18a, ESI†). After mixing both types of NPs, the color immediately turned black and a gel precipitated. From in situ optical microscopy observations (Fig. S17c and S18c, ESI†), a similar aggregation and gelation process was observed for the monometallic gels: Au and Pd NPs start to agglomerate into oligomers and form a gel network. This was also observed in the UV-vis spectra of the solutions after mixing and the TEM images of the final products (Fig. S17b and S18b, ESI† and Movies S3 and S4, ESI†) recorded in situ during gelation.The negatively charged gold NPs and positively charged palladium NPs were mixed to obtain unordered, gel-like three-dimensional networks. To modulate the ligament size of the Au–Pd aerogels, mixtures of NaCA-stabilized gold NPs and CTAB-stabilized palladium NPs at different volume mixing ratios were investigated. Since the TEM images of the Au–Pd gels (Fig. S17b, ESI†) show two different ligament sizes in the gel structure, a strong indication for the separation of both metals and the element distribution of gold and palladium were analyzed. From the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and the corresponding energy-dispersive X-ray spectroscopy (EDX)-based element distributions in Fig. S19 (ESI†), the formation of a double-skeleton structure in the Au–Pd aerogel is deduced. The two metals do not form an alloy, which was typically observed in previous studies.23 More details about the double-skeleton structure are provided by the EDX data in Fig. 3a and b. The separation of the Au and Pd NPs is observed for both synthesis routes: electrostatic manipulation-promoted gelation and dopamine-induced gelation. Both types of metal nanoparticles form separated gel networks, which are intertwined. Nevertheless, the separated gels share crosslinks at which they are interconnected. If one of the metals is added to a small percentage of the second metal, these metal NPs tend to become incorporated into the 3D network metal gel as a respective metal part. As in the case of the monometallic gold gels, the mixing ratio of the n-Au NPs and p-Pd NPs plays a crucial role in the bimetallic gel morphology and influences the ligament size of the gels. If the concentration of the gold solution is high (e.g. for Au1Pd1), the Au part forms large ligaments with a size of ca. 15 nm. This is in contrast with the small ligament sizes of ca. 8 nm for high concentrations of palladium NPs (e.g. for Pd9Au1). Additionally, the morphological features of the Au–Pd aerogel with a molar ratio of 1:1 in NaCA/CTAB (Fig. 3a) and NaCA/Dop (Fig. 3b) can be compared in detail. Under the influence of dopamine, smaller ligaments (Au ligament size: 6.3 ± 1.8 nm and Pd ligament size: 4.0 ± 1.2 nm) are observed in bimetallic aerogels compared to the electrostatic-manipulation-promoted synthesis route (Au ligament size: 15.7 ± 4.6 nm and Pd ligament size: 5.5 ± 1.4 nm). Similar to the synthesized Au gels, different mixing ratios of the noble metal NPs correlate with the specific surface area of the bimetallic aerogels. Again, the measured N2 isotherms show the typical behavior for metal gels (Fig. S20, ESI†).20–23 The isotherm types agree with those for the Au aerogels. The BET surface areas of the Au–Pd aerogels increase with the increasing palladium content, which leads to smaller ligament sizes in the respective gel samples (Fig. 3c). This demonstrates an element-composition-dependent morphology evolution of the resulting bimetallic Au–Pd gels, which was not only indicated by EDX-based element distribution maps, but also revealed by conventional bright-field TEM imaging (Fig. S21, ESI†). It is worth noting that both metals form branched structures of their individual elements so that a double-skeleton structure evolves rather than an alloyed structure. Regarding the Au–Pd aerogels, at high concentrations of the gold solution, the gold content yielded large ligament sizes and the palladium resulted in small-sized gels attached to the large-sized gold gels. Instead, at high concentrations of the palladium solution, the palladium content yielded small ligament sizes and palladium wires may have adhered to the large-sized gold gels. The hierarchically structured pores, which are mainly composed of mesopores and macropores, are found to afford large BET surface areas (41.6 and 97.3 m2 g−1) and pore volumes (0.13 and 0.14 cm3 g−1) for the Au1Pd1 and Pd9Au1 aerogels, respectively (Fig. S20, ESI†). The full data set for all the Au:Pd ratios is summarized in Table S4 (ESI†). We also used ICP-AES and SEM-EDX measurements to verify the molar ratio of the two elements in the bimetallic aerogels (Table S5, ESI†). The measured compositions of the gels fit the initial precursor ratios. Slight differences, especially for the EDX measurements, are present because this method is based only on local determinations of the elemental ratios.
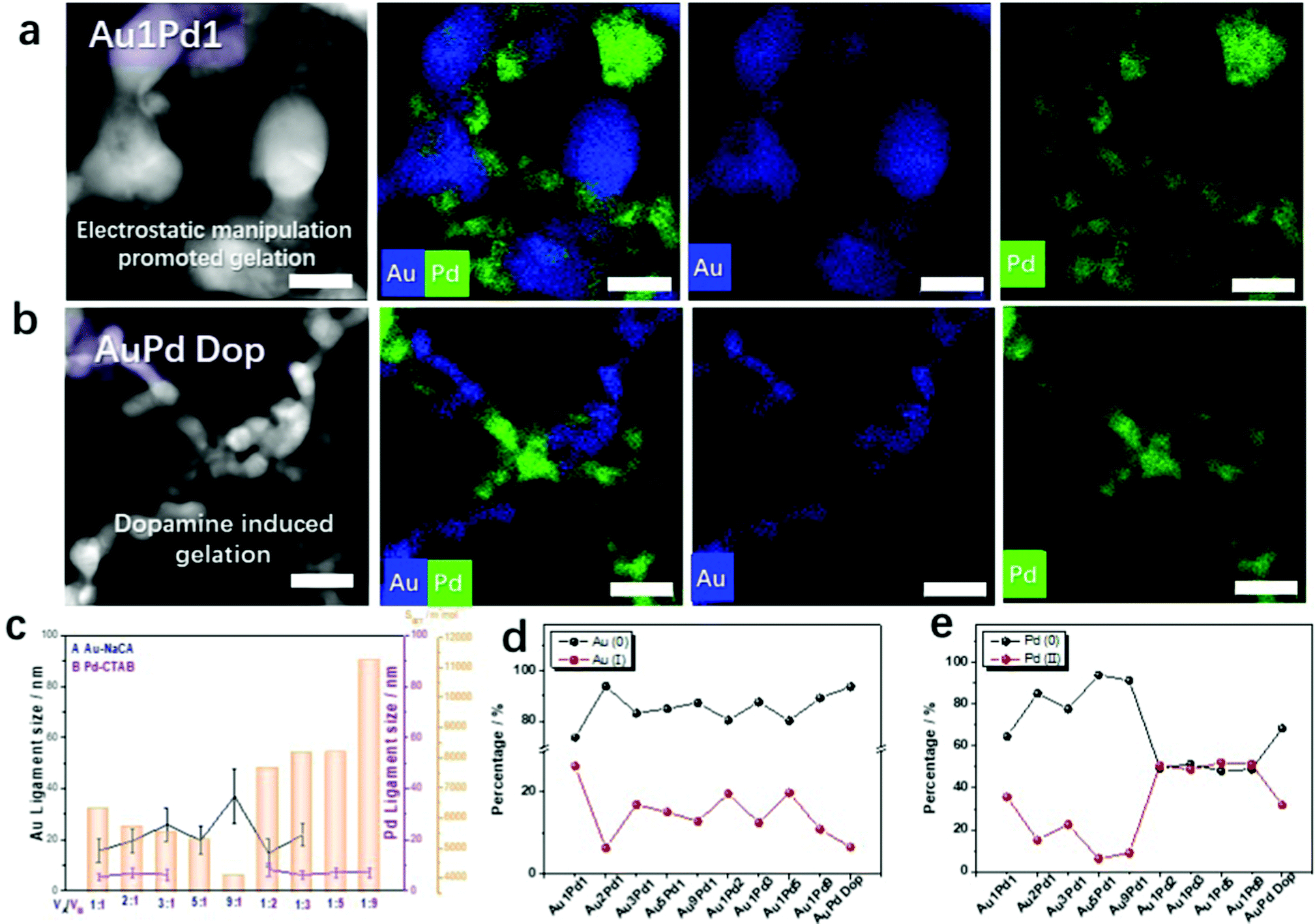 | ||
| Fig. 3 Electrostatic manipulation and analysis of Au–Pd gels. (a and b) HAADF-STEM images and EDX-based element distributions of Au–Pd aerogels from two gelation processes: electrostatic-manipulation-promoted gelation (a) and dopamine-induced gelation (b). The scale bars in the HAADF-STEM images and element distributions are both 20 nm. (c) Ligament sizes and BET surface area values of the Au–Pd aerogels prepared from different volume ratios of negatively charged Au NPs and positively charged Pd NPs. (d and e) Valence-state fractions of gold and palladium for Au–Pd aerogels synthesized by the two gelation processes as derived by deconvolution of the corresponding XPS spectra. | ||
The separation of gold and palladium in the gel structure was also observed by XRD measurements. The XRD patterns indicate the crystallographic phases of the corresponding noble metals rather than those of mixed or alloyed crystals (Fig. S22, ESI†). In particular, for high palladium contents in the bimetallic Au–Pd aerogel, separate gold and palladium diffraction peaks are observed in the XRD patterns, proving that the two metals are phase-separated. These findings are also different from those of previously reported Au–Pd aerogels prepared in one-step or two-step processes, where our group used a co-reduction approach.29,33,34 Furthermore, the dependence of the ligament size on the aerogel composition was confirmed by XRD, where the diffraction peaks get broadened with the decreasing ligament size of both Au and Pd network structures.39
X-Ray photoelectron spectroscopy (XPS) was used to determine the valence states of the synthesized Au–Pd aerogels. As shown in Fig. S23–S25 (ESI†), both Au and Pd feature various valence states, which can be proven by the presence of oxygen in the spectra and the deconvolution of the high-resolution XPS spectra. By integrating the corresponding peak areas of the high-resolution XPS spectra,40 the relative contents of the Au and Pd species in the Au–Pd aerogels were calculated and are summarized in Fig. S3d and e (ESI†). With the increasing Au amount in the bimetallic system, the fractions of Au(I) complexes and Pd(II) complexes decrease from 26.3% Au(I) and 35.6% Pd(II) for Au1Pd1 to 6.3% Au(I) and 15.1% Pd(II) for Au2Pd1; 16.9% Au(I) and 22.5% Pd(II) for Au3Pd1; 15.1% Au(I) and 6.3% Pd(II) for Au5Pd1; and 12.8% Au(I) and 9.0% Pd(II) for Au9Pd1, which are higher than those in Au–Pd Dop (Au1:Pd1 ratio 6.5% Au(I) and 31.8% Pd(II)). Meanwhile, in terms of increasing the Pd content in the bimetallic system, the fraction of the Au(I)-complex decreases from 26.3% for Au1Pd2 to 19.6% for Au1Pd3, 12.5% for Au1Pd3, 19.8% for Au1Pd5, and 10.9% for Au1Pd9. Correspondingly, the fraction of the Pd(II)-complex increases from 35.6% for Au1Pd1 to 50.7% for Au2Pd1, 48.7% for Au3Pd1, 52% for Au5Pd1, and 51.2% for Au9Pd1. XPS analysis shows that the high Pd content in the bimetallic system displays a larger fraction of the high-valence-state Pd(II) because Pd is more susceptible to oxidation compared to Au. Previous research has indicated that a higher oxidation state of the Au–Pd aerogel catalysts promotes the electrocatalytic properties.23 Hence, these aerogels were used as a platform for further chemical and electrocatalytic tests.
Applications of Au and Au–Pd aerogels for chemical and electrocatalytic tests
Due to their intriguing physicochemical properties, NMAs have been extensively employed in diverse applications. However, the effects of the ligament size and the element distribution on catalytic performance have been scarcely studied. To evaluate the catalytic activity of the Au and Au–Pd aerogels with different ligament sizes and element compositions, the chemical reduction of p-nitrophenol (p-NP) to p-aminophenol (p-AP) and the electrocatalytic alcohol electro-oxidation reaction (EOR) were selected as model reactions. For the reduction of p-NP, typical reaction-time-dependent UV-vis absorption spectra are shown in Fig. 4a, b, and Fig. S26 (ESI†). For this catalytic reaction, we used Au aerogels prepared by dopamine-induced gelation and Au1PdX (X = 1, 3, 5, 9) aerogels obtained by electrostatic-manipulation-promoted gelation. These aerogels were chosen due to their good dispersion in solution, smallest ligament sizes and highest specific surface areas, which are important factors for the catalytic activity. A small amount of Au or Au1PdX (20 μL, 0.5 mg mL−1) converts p-NP to p-AP within 200 s. This is shown by the decreasing intensity of the peak maximum of p-NP and the increasing intensity of the peak maximum of p-AP (Fig. 4a and b). Over 95% of the p-NP is reduced by Au and Au–Pd aerogels with ligament sizes below 8 nm, while the Au aerogel using β-alanine-stabilized NPs has low activity because of the excessive polymer ligands on the metal surface, inhibiting the interaction between the substrates and reactive sites (Fig. S15d, ESI†). The catalytic performance of the Au–Pd aerogels with different compositions was also investigated. The catalytic conversion efficiency of the Au–Pd aerogels progressively increases with the increasing Pd content, revealing the high catalytic activity of palladium. In particular, the efficiency after 60 s was relatively low for Au1Pd1 (72.6%) and the highest for Au1Pd9 (98.2%). Apart from the influence of the chemical composition on the catalytic activity, the ligament size is another important factor in the reduction reaction. For bimetallic aerogels, larger apparent rate constants (kapp) were obtained for gels with smaller ligament sizes (Fig. 4c). The kapp values were calculated to be 5.57 s−1 and 17.57 s−1 in the case of Au1PdX (X = 1 and 9, respectively). Intriguingly, increasing the Pd content in bimetallic aerogels indicated a larger ligament size of palladium, but an obvious decrease in the gold ligament size was still observed, reflecting that these smaller particles still dominated the catalysis process. Similarly, the catalytic performance of the corresponding plots of −ln(At/A0) versus time is shown in Fig. S26d–h (ESI†). The −ln(At/A0) values show a linear correlation, revealing that the chemical reduction follows a first-order reaction law. More importantly, the AuPd dopamine-promoted (Dop) aerogel shows the highest catalytic activity (efficiency 98.4%), which is three times higher than that of the Au1Pd1 aerogel using oppositely charged NP solutions of the same ratio. This observation further confirms the high catalytic activity of aerogels having a small ligament size.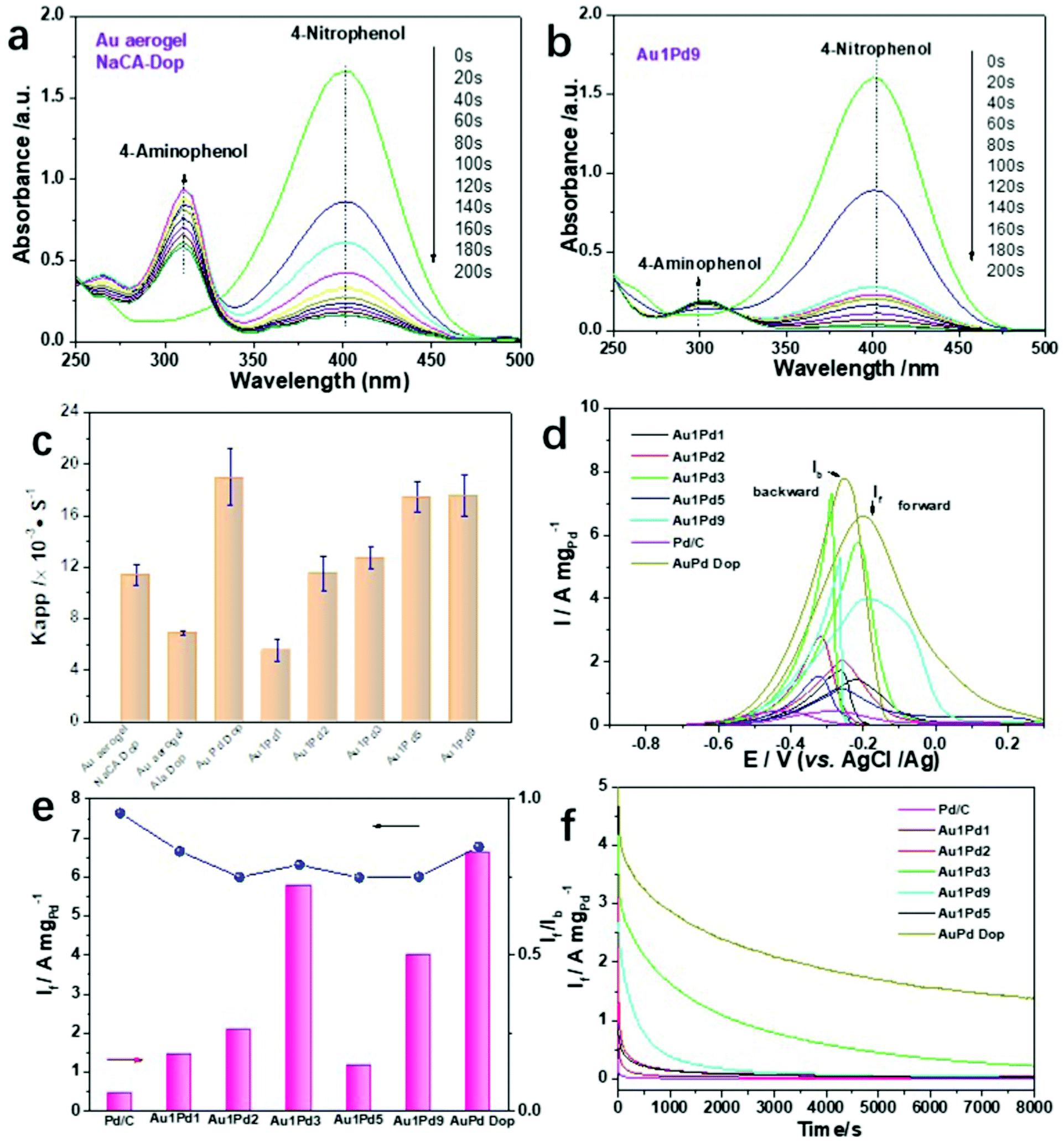 | ||
| Fig. 4 Various applications of Au–Pd aerogels in chemical catalysis and electrocatalysis. (a and b) Time-dependent UV-vis spectra of the reduction of p-nitrophenol for the Au and Au1Pd9 aerogels. (c) Comparison of the apparent rate constant of Au and Au–Pd aerogels with different Au/Pd molar ratios. (d) Cyclic voltammetry curves of the ethanol electro-oxidation with various Au–Pd aerogel catalysts and commercial Pd/C. (e) Summarized If and If/Ib of various Au–Pd aerogels and Pd/C. (f) Chronoamperometry curves of different Au–Pd aerogel catalysts and commercial Pd/C. | ||
To explore the performance benefits of the catalysts, we additionally investigated the EOR reaction. As illustrated in Fig. 4d, cyclic voltammetry (CV) measurements of commercial Pd/C and various Au–Pd aerogels were performed in the aqueous solution containing 1.0 M KOH and 1.0 M ethanol probing the voltage range from −0.9 to 0.3 V at a scan rate of 50 mV s−1. The corresponding forward current density (If) and the ratio of the forward/backward current density (If/Ib) are summarized in Fig. 4e. The highest mass activity of 6.60 A mgPd−1 was attained for the Au1Pd1 Dop aerogel, which is 4.6 and 15.3 times higher than that of Au1Pd1 prepared by electrostatic manipulation and commercial Pd/C, respectively.
In general, all bimetallic aerogels demonstrate a higher If compared to commercial Pd/C, indicating their high EOR activity; lower If/Ib ratios were obtained for the aerogels in comparison to Pd/C. Moreover, we make a summary of aerogels toward ethanol oxidation in the alkaline environment in Table S7 (ESI†), further exhibiting the excellent performance of AuPd gels. This also indicates that a smaller ligament size of the aerogels plays a major role in electrocatalysis and a higher oxidation state of the catalysts promotes the catalytic activity toward the EOR. Moreover, the electrocatalytic oxidation performance of Pd is much higher than that of Au. However, further efforts need to focus on the variation trend of the catalytic properties of Au–Pd in different ratios. They are not consistent with the increasing trend of the composition. Finally, the durability of various Au–Pd aerogels for the EOR was evaluated by the current response with time chronoamperometry at −0.23 V for 8000 s in an aqueous solution containing 1.0 M KOH and 1.0 M ethanol (Fig. 4f). After 8000 s, the residual current response of the Au–Pd Dop aerogel was higher than those of all other gel compositions and commercial Pd/C, suggesting its considerable tolerance and excellent durability toward the EOR. Moreover, the presented electrostatic interaction method is theoretically quite general and easy to implement, which may open a potential way to improve the electrocatalytic performance of various double-skeleton aerogels.
Summary
Diverse challenges (such as long gelation times, time-consuming washing operations, and costly procedures) in the synthesis of NMAs hinder their dissemination and the optimization of their potential applications. Utilizing the surface charge neutralization, the self-healing behavior of noble metal aggregates and the effect of destabilization in the mixture, an electrostatic manipulation method has been presented by directly mixing oppositely charged noble metal nanoparticle solutions to prepare the corresponding noble metal hydrogels (Au and Au–Pd, with metal salt concentrations CM = 0.2 to 0.5 mM) without introducing additional reagents. Additionally, mixing ligand-stabilized gold NPs with dopamine-coated gold NPs is a convenient way to obtain hydrogels. The differently charged metal NPs were obtained by simple functionalization with different ligands during the reduction of the metal salts. The positive and negative charges of the metal NPs (Au and Pd) were characterized by measuring their zeta potentials. Notably, these methods produce monometallic gold aerogels with widespread ligament sizes (5.7 to 27.2 nm) and corresponding specific surface areas (5.5 to 57.4 m2 g−1). Bimetallic aerogels with extensively tunable compositions (e.g., Au–Pd), variable ligament sizes (5.6 to 37.0 nm), and specific surface areas (22.5 to 97.3 m2 g−1) were also obtained. Furthermore, these new synthetic approaches (electrostatic-manipulation-promoted gelation or dopamine-induced gelation) led to the formation of double-skeleton-like twined gel structures, where both elements form separated gel networks. Finally, utilizing the unique chemical properties and element adjustability of nanostructured noble metals, the chemical reduction of p-nitrophenol to p-aminophenol and the electrocatalytic ethanol oxidation were studied. The small ligament sizes and large specific surface areas increased the catalytic performance of the gels due to the presence of more active sites in the nanostructured network. Therefore, the present work not only establishes a novel and simple method to synthesize various single-/bimetallic noble metal aerogels with artificially controlled element distribution (e.g., double-skeleton structure) but also offers a new level for developing high-performance (electro)-catalysts by making the most favorable and size distribution.Experimental procedures
Synthesis procedures
NMAs were synthesized at ambient temperature (∼293 K). A specific amount of ligand, noble metal salts, and NaBH4 were sequentially added to deionized water under stirring. The molar ratios of the metal salt (M), ligand (L), and reductant (R) were fixed at 1/10/4.21,22,29 The as-prepared noble metal nanoparticle solution was mixed for 10 min. Colloidal hydrogels were formed by mixing positively charged and negatively charged gold NP solutions in a volume ratio of 1:1. After grinding for ∼12 h, a monolithic gel was obtained. The fabrication process can be readily scaled up to 800 mL using the same procedure. The as-prepared hydrogel was washed with water five times before solvent exchange with tert-butanol. After being flash-frozen and freeze-dried for 24 h, the corresponding aerogel was obtained. For AuPd gels, the procedure was analogous to the synthesis described above; only a Pd NP solution was used instead of a second Au NP solution. Further experimental details can be found in the ESI.†
Catalytic reduction
The catalytic performance of the NMAs was evaluated by the reduction of p-nitrophenol at room temperature. All aqueous solutions were freshly prepared. A standard catalytic test reaction was carried out in a quartz glass cuvette. For the catalytic reduction of p-NP, 30 μL of p-NP (10 mM) and 150 μL of NaBH4 (2 M) were added to a quartz glass cell containing 2.97 mL of deionized water. Thereafter, 20 μL of an aqueous solution containing the aerogel catalyst (0.5 mg mL−1) were injected into the cell to start the reaction. The additional details are provided in the ESI.†Electrocatalysis
All electrochemical tests were performed using an electrochemical workstation in a three-electrode system. A platinum foil and an Ag/AgCl (saturated KCl aqueous solution) electrode were used as the counter and reference electrodes, respectively. A glassy carbon electrode (GCE, diameter = 3 mm) was chosen as the substrate for holding the catalysts. For the electro-oxidation of ethanol, the performance was measured in an N2-saturated 1.0 M KOH aqueous solution containing 1.0 M ethanol. Further details regarding the procedure can be found in the ESI.†Author contributions
Wei: writing – original draft, methodology, data curation, and conceptualization. René Hübner: data curation, investigation, and writing – review and editing. Maximilian Georgi: data curation, conceptualization, and writing – review and editing. Cui Wang: validation, investigation, and writing – review and editing. Xiaodong Wu: software, investigation, and writing – review and editing. Alexander Eychmüller: supervision, funding acquisition, and writing – review and editing.Data and software availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.† Additional data related to this paper can be requested from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Nelli Weiß, Susanne Goldberg, and Ankita Bora for TEM measurements; Linlin Wang, Dr Juliane Simmchen, and Sharan Priyanka for optical imaging and zeta potential measurements; Daniel Spittel for electrochemical measurements and Dr Ran Du and Dr Xuelin Fan for helpful discussions. The use of the HZDR Ion Beam Center TEM facilities and the funding of TEM Talos by the German Federal Ministry of Education and Research (BMBF; grant No. 03SF0451) in the framework of HEMCP are acknowledged. The authors would like to thank Weiwei Wang from Shiyanjia Lab (https://www.shiyanjia.com) for the XPS analysis. This work was supported by the Swiss National Science Foundation (SNF), the German Research Foundation (DFG EY 16/18-2), the National Natural Science Foundation of China (21607063), the Jiangsu Overseas Visiting Scholar Program for University Prominent Young & Middle-aged Teachers and Presidents, the Outstanding Chinese and Foreign Youth Exchange Program of the China Association for Science and Technology (CAST), the Youth Talent Training Program of Jiangsu University, and the High-Performance Computing Center of Nanjing Tech University.References
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 37, 22–45 CrossRef.
- C. Ziegler, A. Wolf, W. Liu, A.-K. Herrmann, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2017, 56, 13200–13221 CrossRef CAS PubMed.
- X. Jiang, R. Du, R. Hübner, Y. Hu and A. Eychmüller, Matter, 2021, 4, 54–94 CrossRef CAS.
- S. Kistler, Nature, 1931, 127, 741 CrossRef CAS.
- J. Mohanan, I. Arachchige and S. Brock, Science, 2005, 307, 397–400 CrossRef CAS PubMed.
- H. Sun, X. Zhen and C. Gao, Adv. Mater., 2013, 25, 2554–2560 CrossRef CAS PubMed.
- B. Cai, V. Sayevich, N. Gaponik and A. Eychmüller, Adv. Mater., 2018, 30, 1707518 CrossRef PubMed.
- Q. Zheng, L. Fang, H. Guo, K. Yang, Z. Cai, M. Meador and S. Gong, Adv. Funct. Mater., 2018, 28, 1706365 CrossRef.
- R. Du, X. Fan, X. Jin, R. Hübner, Y. Hu and A. Eychmüller, Matter, 2019, 1, 39–56 CrossRef.
- B. Cai and A. Eychmüller, Adv. Mater., 2019, 31, 1804881 CrossRef PubMed.
- R. Du, X. Jin, R. Hübner, X. Fan, Y. Hu and A. Eychmüller, Adv. Energy Mater., 2020, 10, 1901945 CrossRef CAS.
- N. C. Bigall, A. K. Herrmann, M. Vogel, M. Rose, P. Simon, W. Carrillo-Cabrera, D. Dorfs, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2009, 48, 9731–9734 CrossRef CAS PubMed.
- W. Liu, A. K. Herrmann, D. Geiger, L. Borchardt, F. Simon, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2012, 51, 5743–5747 CrossRef CAS PubMed.
- K. G. Ranmohotti, X. Gao and I. U. Arachchige, Chem. Mater., 2013, 25, 3528–3534 CrossRef CAS.
- D. Wen, W. Liu, D. Haubold, C. Zhu, M. Oschatz, M. Holzschuh, A. Wolf, F. Simon, S. Kaskel and A. Eychmüller, ACS Nano, 2016, 10, 2559–2567 CrossRef CAS PubMed.
- B. Cai, D. Wen, W. Liu, A. K. Herrmann, A. Benad and A. Eychmüller, Angew. Chem., Int. Ed., 2015, 54, 13101–13105 CrossRef CAS PubMed.
- C. Zhu, Q. Shi, S. Fu, J. Song, H. Xia, D. Du and Y. Lin, Adv. Mater., 2016, 28, 8779–8783 CrossRef CAS PubMed.
- H. L. Gao, L. Xu, F. Long, Z. Pan, Y. X. Du, Y. Lu, J. Ge and S. H. Yu, Angew. Chem., Int. Ed., 2014, 53, 4561–4566 CrossRef CAS PubMed.
- A. Freytag, S. Sánchez-Paradinas, S. Naskar, N. Wendt, M. Colombo, G. Pugliese, J. Poppe, C. Demirci, I. Kretschmer, D. W. Bahnemann, P. Behrens and N. C. Bigall, Angew. Chem., Int. Ed., 2016, 55, 1200–1203 CrossRef CAS PubMed.
- R. Du, J.-O. Joswig, R. Hübner, L. Zhou, W. Wei, Y. Hu and A. Eychmüller, Angew. Chem., Int. Ed., 2020, 59, 8293–8300 CrossRef CAS PubMed.
- R. Du, J.-O. Joswig, X. Fan, R. Hübner, D. Spittel, Y. Hu and A. Eychmüller, Matter, 2020, 2, 908–920 CrossRef PubMed.
- R. Du, J. Wang, Y. Wang, R. Hübner, X. Fan, I. Senkovska, Y. Hu, S. Kaskel and A. Eychmüller, Nat. Commun., 2020, 11, 1590 CrossRef CAS PubMed.
- X. Fan, S. Zerebecki, R. Du, R. Hübner, G. Marzum, G. Jiang, Y. Hu, S. Barcikowki, S. Reichenberger and A. Eychmüller, Angew. Chem., Int. Ed., 2020, 59, 5706–5711 CrossRef CAS PubMed.
- M. E. Leunissen, C. G. Christova, A. P. Hynninen, C. P. Royall, A. I. Campbell, A. Imhof, M. Dijkstra, R. Van Roij and A. Van Blaaderen, Nature, 2005, 437, 235–240 CrossRef CAS PubMed.
- J. Kolny, A. Kornowski and H. Weller, Nano Lett., 2002, 2, 361–364 CrossRef CAS.
- S. Zhang, Y. Shao, G. Yin and Y. Lin, Angew. Chem., Int. Ed., 2010, 49, 2211–2214 CrossRef CAS PubMed.
- S. Wintzheimer, T. Granath, M. Oppmann, T. Kister, T. Thai, T. Kraus, N. Vogel and K. Mandel, ACS Nano, 2018, 12, 5093–5120 CrossRef CAS PubMed.
- D. Wen, A.-K. Herrmann, L. Borchardt, F. Simon, W. Liu, S. Kaskel and A. Eychmüller, J. Am. Chem. Soc., 2014, 136, 2727–2730 CrossRef CAS PubMed.
- R. Du, Y. Hu, R. Hübner, J.-O. Joswig, X. Fan and A. Eychmüller, Sci. Adv., 2019, 5, eaaw4590 CrossRef PubMed.
- J. Beurton, P. Lavalle, A. Pallotta, T. Chaigneau, I. Clarot and A. Boudier, Int. J. Pharm., 2020, 580, 119244 CrossRef CAS PubMed.
- Y. Wu, L. Jiao, W. Xu, W. Gu, C. Zhu, D. Du and Y. Lin, Small, 2019, 15, 1900632 CrossRef PubMed.
- Z. Zhang, H. Li, F. Zhang, Y. Wu, Z. Guo, L. Zhou and J. Li, Langmuir, 2014, 30, 2648–2659 CrossRef CAS PubMed.
- L. Kühn, A. K. Herrmann, B. Rutkowski, M. Oezaslan, M. Nachtegaal, M. Klose, L. Giebeler, N. Gaponik, J. Eckert and T. J. Schmidt, Chem. – Eur. J., 2016, 22, 13446–13450 CrossRef PubMed.
- M. Oezaslan, A. K. Herrmann, M. Werheid, A. I. Frenkel, M. Nachtegaal, C. Dosche, C. L. Bonnaud, H. C. Yilmaz, L. Kühn, E. Rhiel, N. Gaponik, A. Eychmüller and T. J. Schmidt, ChemCatChem, 2017, 9, 798–808 CrossRef CAS.
- L. Jiao, W. Xu, H. Yan, Y. Wu, W. Gu, H. Li, D. Du, Y. Lin and C. Zhu, Chem. Commun., 2019, 55, 9865–9868 RSC.
- Z. Liao, W. Zhang, Z. Qiao, J. Luo, A. Erpuding, A. Niwaer, X. Meng, H. Wang, X. Li, F. Zuo and Z. Zhao, J. Colloid Interface Sci., 2020, 562, 81–90 CrossRef CAS PubMed.
- A. Freytag, M. Colombo and N. C. Bigall, Z. Phys. Chem., 2017, 231, 63–75 CrossRef CAS.
- K. H. Samuel, Sy, L. W. Cola, Ho, W. C. Yu Lau, H. Ko and C. H. Jonathan Choi, Langmuir, 2018, 34(46), 14033–14045 CrossRef PubMed.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- R. Du, W. Jin, R. Hübner, L. Zhou, Y. Hu and A. Eychmüller, Adv. Energy Mater., 2020, 10, 1903857 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00213b |
| This journal is © The Royal Society of Chemistry 2022 |