
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-ion polymer/LLZO hybrid electrolytes with high lithium conductivity†
Marine
Lechartier
ab,
Luca
Porcarelli
 ac,
Haijin
Zhu
c,
Maria
Forsyth
acd,
Aurélie
Guéguen
b,
Laurent
Castro
*b and
David
Mecerreyes
*cd
ac,
Haijin
Zhu
c,
Maria
Forsyth
acd,
Aurélie
Guéguen
b,
Laurent
Castro
*b and
David
Mecerreyes
*cd
aPOLYMAT University of the Basque Country UPV/EHU, Avenida Tolosa 72, Donostia-San Sebastian 2008, Spain
bToyota Motor Europe Research & Development 2, Material Engineering, Battery & Fuel Cell, Hoge Wei 33 B, B-1930 Zaventem, Belgium
cARC Centre of Excellence for Electromaterials Science and Institute for Frontier Materials, Deakin University, Melbourne, Australia
dIkerbasque, Basque Foundation for Science, E-48011, Bilbao, Spain. E-mail: david.mecerreyes@ehu.es
First published on 25th November 2021
Abstract
Hybrid solid electrolytes which combine the properties of inorganic and polymeric ion conductors are being investigated for lithium batteries which use lithium metal anodes. The number of inorganic/polymer compositions and their synergy in ion-conducting properties are limited by the hybrid fabrication method and the limited compatibility between both types of materials. Here we report a hybrid solid electrolyte formed by a poly(ethylene glycol) type single-ion polymer network and ceramic garnet-type nanoparticles of Li7−3XAlXLa3Zr2O12 (LLZO) with very high lithium conductivity. The combination of a lithium-single ion polymer matrix with LLZO inorganic particles results in flexible free-standing films by using a fast UV-photopolymerization process with facile control of its composition. This methodology showed excellent dispersion of the LLZO nanoparticles within the gel polymer network with up to 50 wt% ceramic content, as shown in the enviromental ESEM images. These hybrid electrolytes have high ionic conductivity values (1.4 × 10−4 S cm−1 at 25 °C) and high lithium transference number as compared to previous hybrid electrolytes. The effect of LLZO nanoparticle content on the lithium transport was investigated in detail using solid-state nuclear magnetic resonance spectroscopy (NMR). Finally, determination of the critical current density (CCD) before lithium dendrites are initiated has been carried out on both pristine and hybrid electrolytes, so as to assess their potential as solid electrolytes for lithium metal batteries.
Introduction
The application of lithium ion batteries, while very popular for small electronics, may be limited in devices such as electric vehicles (EV), in which a high energy density is desired. Replacing the graphite anode with a lithium metal anode is among the most promising strategies for obtaining high energy density batteries because of its high theoretical capacity and low reductive potential.1 However, lithium metal batteries present some technical and safety problems in particular those arising from the non-uniform electrochemical deposition of lithium and also due to the flammability of current liquid electrolytes.2 In this sense, solid-state batteries are promising options for achieving high energy and power densities thereby limiting the risk of liquid electrolyte leakage and flammability while controlling the growth of dendrites through mechanical means.Solid electrolytes have gained considerable attention due to their various advantages over liquid electrolytes such as potential dendrite prevention, better safety and battery downsizing.3,4 Currently, research on new solid electrolytes is divided between new inorganic super ionic conducting materials, such as perovskites,5 NASICON6 and garnets,7 and organic polymer electrolytes.8 Within this battle between inorganic and organic materials, inorganic materials present high ionic conductivity values at room temperature, from 10−4 to 10−2 S cm−1.9 Nevertheless, two main issues concerning their use as potential solid electrolytes are the high grain boundary resistance towards electrodes10 and their brittleness.11,12 On the other hand, polymer solid electrolytes, which are composed of a polymer host and a dissolved salt, can be easily processed and display better interfaces thanks to their flexibility. Nonetheless, their poor ionic conductivity (below 10−4 S cm−1) restricts their use at room temperature.13 One approach to improve this is by the addition of additives such as ionic liquids or new lithium salts and through the development of ionogel polymer electrolytes.14,15 It is worth remarking that, while inorganic materials mostly show lithium-ion mobility, in typical polymer electrolytes both the anion and lithium cation participate in the ionic conductivity process. In the last few years, the so called lithium single-ion conducting polymer electrolytes have become popular due to their unique lithium transport and good performance when lithium metal is used as the electrode.16
As an intermediate possibility, many efforts have been focused on hybrid solid electrolytes (HSEs) which seek to create a synergy and take advantage of the unique individual properties of both inorganic and polymer electrolyte systems.17 Hybrid solid electrolytes are usually composites formed from a polymer matrix, inorganic particles and a lithium salt. Dispersing inorganic particles into a polymer electrolyte is in theory an efficient way to increase its ionic conductivity and further reinforce the soft polymer matrix.18 However, achieving good dispersion of solid particles within a polymer matrix is challenging and particle surface modifiers or surfactants may be needed. This could affect the conduction mechanism between the inorganic and the polymer electrolytes leading to interfacial limitations. For this reason, the expected increase in ionic conductivity is often not achieved, as evident in all the reported HSE literature.
As mentioned before, lithium single-ion conducting polymer electrolytes based on anionic polyelectrolytes or poly(ionic liquid)s have drawn great attention in the last few years.19 Although these anionic polymers are very popular within the polymer electrolyte community, to the best of our knowledge, they have not been investigated in hybrid solid electrolytes. For this reason, we targeted the development of hybrid electrolytes with single-ion lithium conductivity. Furthermore, the ionic charges within the polymer matrix can help in the dispersion of the inorganic particles. For developing the hybrids, we chose LLZO nanoparticles as the inorganic material. LLZO is an attracting filler as it reaches high ionic conductivity (10−4–10−3 S cm−1 at room temperature) and near-unity lithium transference number, presents wide voltage window and is chemically stable towards lithium metal.20 This ceramic has been widely investigated as HSEs in the form of particles, nanowires and even 3D scaffolds.21–24 Typical polymer matrices that have been investigated are poly(ethylene oxide) PEO or poly(vinylene difluoride) PVDF doped with a Li salt (LiClO4, LiTFSI). The reported HSE hybrids in the literature showed in most cases low loading of LLZO (below 25 wt%) and slightly higher ionic conductivity than the sole polymer matrix showing room temperature conductivity values between 10−5 and 10−4 S cm−1.
In this work, we report a new hybrid solid electrolyte (HSIPE) formed by a poly(ethylene glycol) (PEG) type lithium single-ion polymer network, LLZO ceramic garnet type nanoparticles and a propylene carbonate plasticizer. The organic part of the hybrid is based on a recently reported single-ion crosslinked polymer electrolyte formulation (SIPE) which showed a near-unity lithium transference number which allowed room temperature battery operation.25 Flexible free-standing films of the hybrid electrolyte were prepared by a fast UV-photopolymerization process which allows varying its composition easily. The electrochemical, thermal and mechanical properties of the hybrids were investigated using electrochemical impedance spectroscopy (EIS), thermal gravimetric analysis (TGA) and rheology measurements. Understanding of lithium-ion transport within the hybrid polymer electrolytes was elucidated by solid-state nuclear magnetic resonance spectroscopy (NMR). Special attention was paid to investigate the stripping/plating performance of the lithium metal anode with the lithium single-ion HSIPE. For this purpose, galvanostatic tests and electrochemical impedance spectroscopy were performed with lithium symmetrical cells.
Results and discussion
Synthesis and characterization of single-ion hybrid solid electrolytes (HSIPE) by UV photopolymerization
Single-ion hybrid solid electrolytes were prepared by UV-co-polymerization of poly(ethylene glycol) dimethacrylate (PEGDM) and lithium 1-[3-(methacryloyloxy)-propylsulfonyl]-1 (trifluoromethylsulfonyl)imide (LiMTFSI) in the presence of different amounts of inorganic LLZO particles, propylene carbonate (PC) as a plasticizer and using 2-hydroxy-2-methylpropiophene (DAROCUR) as a radical photo-initiator. UV-photopolymerization was selected due to the simplicity and rapidness of the approach. One of the main benefits of this method is that the inorganic particles can be pre-dispersed in the liquid monomer mixture and then polymerized in a rapid way without any visual sedimentation and the need for any additional organic solvent. After the photopolymerization and polymer network formation, self-standing, flexible and white composite membranes could be obtained until an inorganic content of approximately 50 wt% was achieved while keeping constant the plasticizer-to-polymer ratio/formulation which was more successful in our previous work. Once the inorganic content was higher than 50 wt%, the membranes became brittle and difficult to handle. A typical HSIPE-X composition, where X = 5, 9, 26 and 40 wt% LLZO, produces a white flexible membrane as the one shown in the picture of Fig. 1. The different HSIPE compositions investigated in this work are shown in Table 1 in the Experimental section. For comparison, we also prepared some dual-ion HSEs by substituting the LiMTFSI monomer by the LiTFSI salt.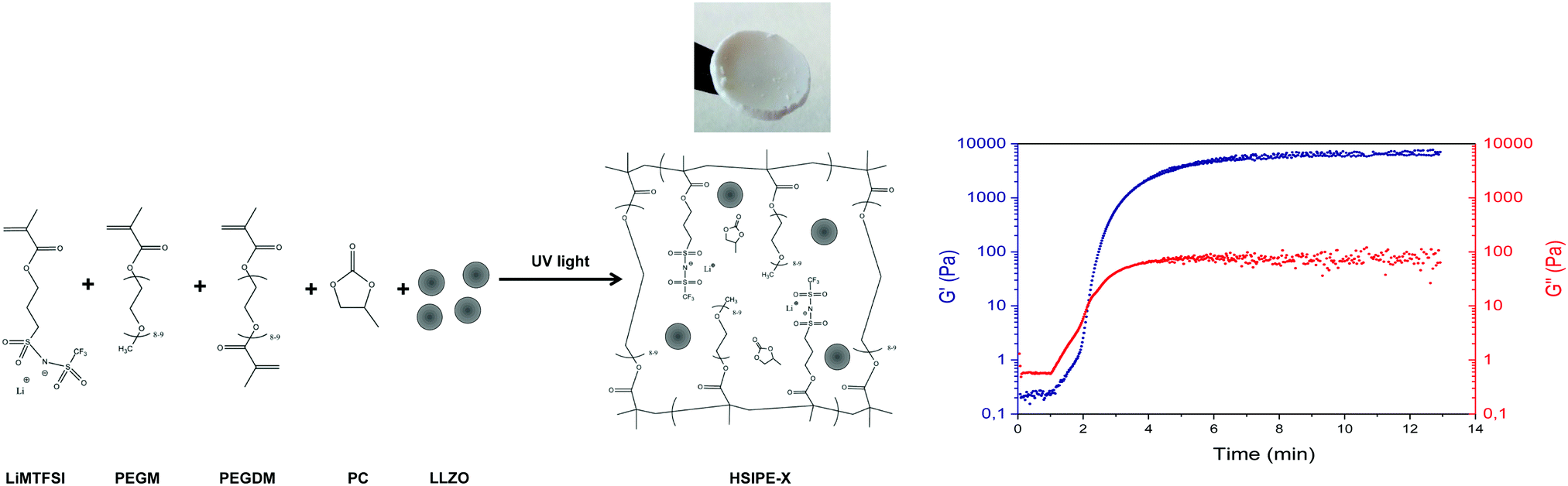 | ||
| Fig. 1 Left side: synthetic route towards lithium single-ion PEG/LLZO crosslinked hybrid solid electrolytes, right side: photo-rheology study showing the (photo)polymerization kinetics. | ||
| PEGM | PEGM | PC | LiMTFSI | LiTFSI | LLZO | |
|---|---|---|---|---|---|---|
| UV initiator 3% w/w of the monomers. | ||||||
| SIPE | 5 | 36 | 50 | 9 | 0 | |
| HPE-26 | 3.7 | 33.3 | 37 | 26 | ||
| HSIPE-5 | 4.7 | 34.2 | 47.6 | 8.5 | 5 | |
| HSIPE-9 | 4.6 | 32.7 | 45.5 | 8.2 | 9 | |
| HSIPE-26 | 3.7 | 26.6 | 37 | 6.7 | 26 | |
| HSIPE-40 | 3 | 21.6 | 30 | 5.4 | 40 | |
| HSIPE-50 | 2.5 | 18 | 25 | 4.5 | 50 | |
| DIPE | 5 | 36.6 | 50.8 | 7.6 | ||
| HDIPE-26 | 3.7 | 27 | 37.5 | 5.8 | 26 | |
In situ rheological measurements were performed during photopolymerization to evaluate the reaction kinetics and determine the gel point of the pristine composition thanks to the rotational rheometer equipped with a UV lamp. Measuring the shear storage G′ and loss G′′ moduli evolution provides accurate information about the formation of a 3D polymer network during irradiation.26,27 The intersection between G′ and G′′ gives the gel point of the crosslinked network, which is the transition between the liquid state of unreacted monomers and the solid state of crosslinked polymers. As shown in Fig. 2, the gel point is reached only after 2 min, which shows the fast polymerization kinetics which should limit the sedimentation of the solid LLZO particles. Nonetheless, mechanical properties were still evolving under irradiation after the gel point. After 15 min, G′ and G′′ finally reached a plateau.
 | ||
| Fig. 2 ESEM images at the cross-section of HSIPE membranes with different LLZO content (wt%): (i) bottom part, (ii) middle part, and (iii) top part; (a) HSIPE-5, (b) HSIPE-26, and (c) HSIPE-50. | ||
To ascertain high conversion of monomers by UV polymerization, the irradiation time was set up to 20 min for the synthesis of pristine and hybrid crosslinked polymer electrolytes. According to the Fourier transform infrared spectroscopy (FTIR) spectra, monomer conversions higher than ≥95% were reached by the disappearance of the 1635 cm−1 band, which is associated with the carbon double bond of methacrylates (Fig. S1, ESI†). This confirms the validity of our synthetic strategy towards HSIPEs.
In order to visualize the dispersion of the LLZO nanoparticles within the polymer matrix, Environmental scanning electron microscopy (ESEM) was carried out on three hybrid electrolytes, having 26, 40 and 50 wt% LLZO as shown in Fig. 3. ESEM allows analysing the hybrid crosslinked networks without the evaporation of the propylene carbonate plasticizer. Cross-section images along the film thickness were taken in order to ascertain that the particles were well distributed in all the film thickness and prove that they did not sediment during UV photopolymerization. ESEM images were taken at three different locations of the HSIPE films: at the bottom, in the middle and at the top part of the cut. As displayed in Fig. 2, hybrid electrolytes with three different compositions were analysed: HSIPE-5 which has 5 wt% LLZO, HSIPE-26 which has 26 wt% of LLZO and HSIPE-50 which has 50 wt%. By looking at the ESEM images shown in Fig. 2, we can see that for LLZO loading up to 26 wt%, particles look very well dispersed from the bottom part to the top one (Fig. 2i-a, i-b, ii-a, ii-b, iii-a and iii-b). In these samples, we can conclude that there is no particle sedimentation in the films for these compositions. On the other hand, some agglomeration of particles is observed in HSIPE-50, especially at the bottom part (Fig. 2i-c, ii-c and iii-c). It seems that dispersing homogeneously LLZO at high content is challenging by just the magnetic stirring used in this work and the fast UV (photo)polymerization is not enough to avoid its sedimentation. ESEM images were taken on the top of the samples to examine the homogeneity of particle dispersion. For compositions HSIPE-26 and HSIPE-40 (Fig. S2, ESI†), no agglomeration was noticed in the ESEM images. However, in HSIPE-50 with 50 wt% of LLZO (Fig. S2c, ESI†), several agglomerates could be seen in the top view.
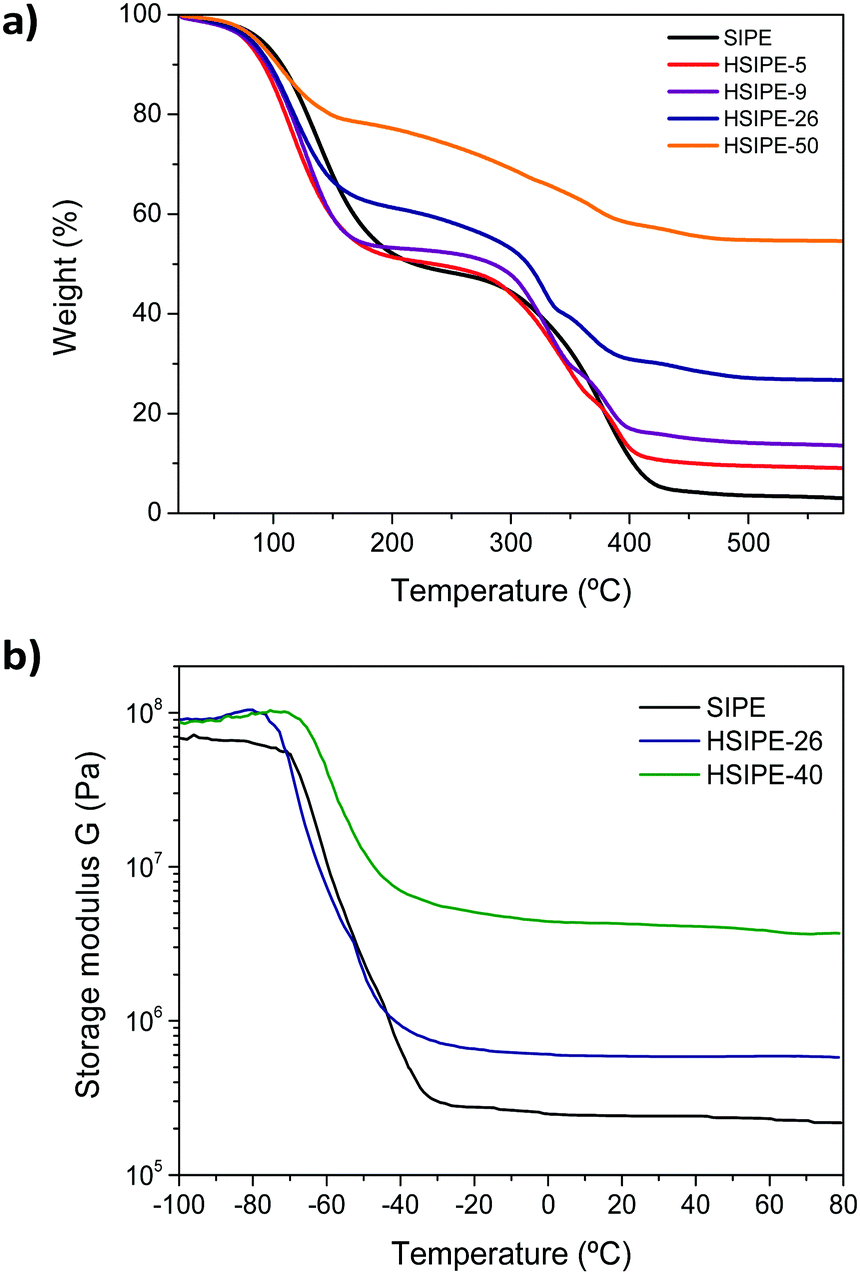 | ||
| Fig. 3 (a) Thermogravimetric analysis of HSIPEs under a nitrogen atmosphere at 10 °C min−1 and (b) DMTA analysis of HSIPEs in the compression mode between −100 °C and 100 °C. | ||
All the HSIPE samples were further characterized from a thermal and mechanical perspective. The thermal stability was assessed by thermal gravimetrical analysis (TGA) under an inert atmosphere (N2) as shown in Fig. 2a. In all cases, the HSIPE materials exhibited a two-step degradation process and curves were slightly shifted at different temperatures, depending on the composition. The initial degradation, in between 150 and 200 °C, was attributed to the initial loss of the propylene carbonate plasticizer. The second degradation, initiated at around 250 °C, was mostly attributed to the degradation of the polymeric matrix. Therefore, the TGA curves of the HSIPEs with higher concentrations of polymer were shifted to higher temperatures. Interestingly, the residue at 500 °C varied in each HSIPE and corresponded quite well with the composition of the inorganic LLZO content in each sample.
Dynamic mechanical thermal analysis (DMTA) (Fig. 3b) was used to investigate the mechanical properties of the HSIPEs. The DMTA spectra show two regimes: (i) from −100 to −70 °C, the material is in the glassy state and (ii) a second plateau in the rubbery state, starting from −40 °C to 80 °C. In between, the glass transition occurs, where the materials are going from the vitreous to the viscous state when the temperature increases. In the glassy state, the storage modulii G′ of pristine and hybrid electrolytes are quite similar (100 MPa). In contrast, the moduli of hybrids dramatically improved compared to the pristine crosslinked polymer despite the viscoelastic state. The storage modulus increases from 73 kPa for the pristine SIPE to 0.58 and 3.7 MPa for hybrids HSIPE-26 and HSIPE-40, respectively, confirming that the HSIPEs have enhanced mechanical properties compared to the pristine polymer electrolyte.
Tan(δ), in other words the ratio between the storage and loss moduli G′/G′′, is also recorded for each material and is depicted in Fig. S3 (ESI†). The maximum of tan(δ) gives us information about the value of glass transition of the HSIPEs. In polymer electrolytes, ionic conductivity is strongly dependent on Tg, as Li ion conduction is closely related to ion hopping mechanisms between polymer chains.28 Thus, it has been widely recognized that low Tg leads to high ionic conductivity for polymer electrolytes although there may be other factors that decouple the ionic conductivity from the Tg value. From DMTA experiments, Tg is determined to be −58.9 °C for pristine SIPE. For HSIPE-26, Tg slightly decreases to −64.75 °C. However, different tendency is observed for the HSIPE-40 composition where Tg increases to −55 °C. All in all, the different HSIPEs show low values of Tg comparable to that of the pristine polymer network, and the presence of LLZO does not seem to affect the local segmental mobility of the polymer chains while still providing a higher modulus material.
Lithium conductivity studies of PEG/LLZO single-ion hybrid solid electrolytes (HSIPE)
Impedance spectroscopy was used to determine the ionic conductivity of the hybrid solid electrolytes. Fig. 4 shows the ionic conductivity of the different hybrid compositions. While increasing the particle loading from 0 to 5, 9, 26 and 40 wt%, the ionic conductivity increases linearly and reaches a maximum for the HSIPE-40 composition. The ionic conductivity shows a highest value of 1.4 × 10−4 S cm−1 at 25 °C which is 3.3-fold higher than the one measured for the pristine SIPE (4.8 × 10−5 S cm−1). This tendency is aligned with the previous studies on hybrid polymer electrolytes with conductive inorganic particles reported in the scientific literature as summarized in Table S4 (ESI†). However, it is worth noting that, in this case, the values of conductivity are higher, probably due to the excellent dispersion of the LLZO particles and the possibility to reach such a high inorganic content (40 wt%) in the HSIPEs. For HSIPE-50, the ionic conductivity value starts to decrease which may be due to the particle agglomeration and sedimentation observed in the FESEM images discussed above. It is noteworthy that the values showed by these membranes are very high compared to the other HSEs shown in the literature, which report optimized ionic conductivity values of around 1.5 × 10−5 S cm−1 (Table S4, ESI†).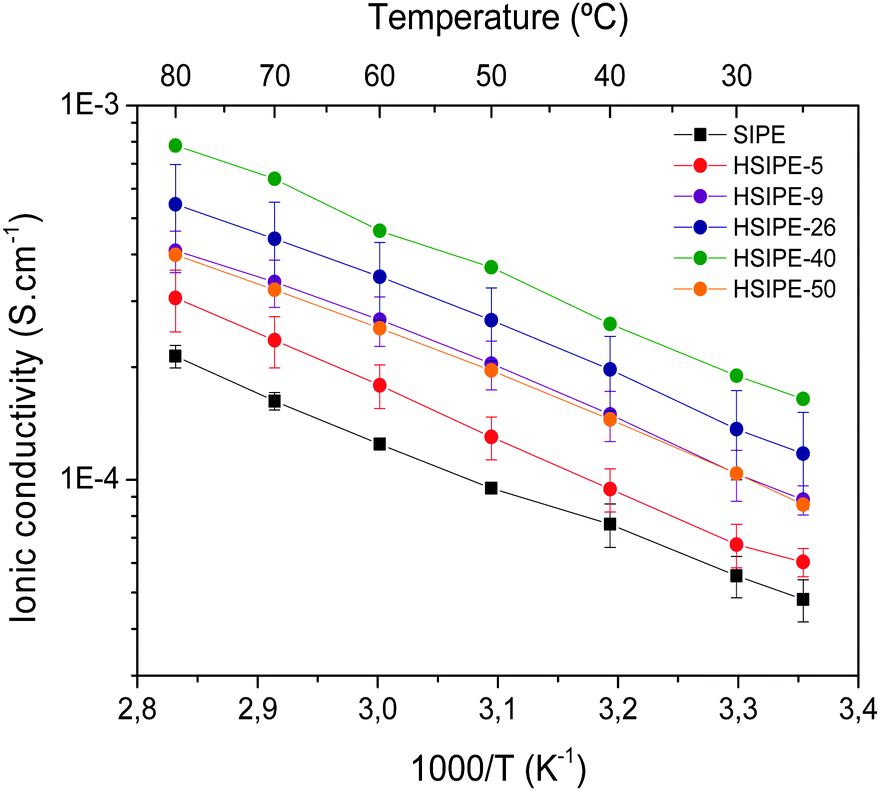 | ||
| Fig. 4 Arrhenius plot for SIPE and HSIPE having different LLZO compositions. | ||
As opposed to previous hybrid or composite electrolytes where a neutral polymer with added salt is typically the polymer electrolyte component, the unique feature of these materials is the combination of a lithium single-ion conducting polymer matrix and LLZO nanoparticles. Both materials separately present a lithium transference number close to unity. To measure the lithium transference number, we collected the EIS spectra for SIPE and HSIPEs before and after polarization as shown in Fig. S5–S7 (ESI†). The red dotted lines represent data fitting from equivalent circuit modelling. The measured tLi+ values are 0.73 for SIPE, 0.63 for HSIPE and 0.57 for HSIPE-26, indicating that tLi+ tends to decrease with the increasing LLZO content which is unexpected. Although we cannot unequivocally explain this observation, it is possible that some anionic impurities within the LLZO particles or the rest of the unreacted anionic monomers are present in the hybrid electrolytes which slightly lowers the tLi+ value.
Solid-state NMR studies of PEG/LLZO single-ion hybrid solid electrolytes (HSIPE)
To further understand the lithium conduction mechanism of the HSIPE, solid-state NMR studies were carried out. Previous solid-state NMR studies focused on the understanding of Li ionic pathways in LLZO hybrid polymer electrolytes, especially for the system LLZO-PEO (LiTFSI) and LLZO-PEO (LiClO4),29,30 and these indicated that ionic transport is greatly influenced by the LLZO content and the presence of a plasticizer. For low LLZO compositions, Li moves preferably in the polymer phase with the ionic conductivity tending to increase due to the suppression of polymer crystallinity. Up to a critical composition, the LLZO particles form a percolated network and the ionic transport mechanism is then altered and the hybrid behaviour is closer to that of a ceramic with Li ions passing only via the inorganic phase.31 Zheng and co-workers also showed that adding a plasticizer (20 wt%) modified Li pathways within this material. It was discovered that in a plasticized hybrid electrolyte, independent of LLZO composition, Li ions favourably move in the polymer phase. To elucidate the composition dependence of ionic conductivity in our materials, solid-state NMR measurements were undertaken for SIPE and HSIPE membranes. To compare these electrolytes with traditional hybrid systems, dual-ion hybrid HSEs, where LiTFSI was added to include free TFSI anions (namely, DIPE and HDIPE), were also synthesized. A non-conductive electrolyte (PEG network without Li source) reinforced by LLZO particles (HPE-26) was also studied to determine if the inorganic conductor by itself without the presence of an additional lithium ion source in the polymer matrix was able to participate in ionic conduction. Table 1 in the Experimental section summarizes the compositions studied.Ionic conductivities were previously measured by EIS to compare with the NMR results (Table S8, ESI†). As demonstrated previously, the ionic conductivity of the pristine single-ion crosslinked polymer is increased by the addition of LLZO particles (SIPE, HSIPE-9 and HSIPE-26). HPE-26, containing LLZO as the unique Li ion source, achieves a lower ionic conductivity than SIPE, up to 10−5 S cm−1. This interesting result implies that Li ions from the LLZO phase can transfer to the polymer matrix and may participate in ionic transport in the HSIPEs. A few studies have previously reported that a possible ionic exchange takes place between LLZO and the PEO matrix32 which appears to be confirmed here. The interface between a Li ion conductive ceramic and polymer has been investigated previously, revealing high interfacial resistance between the two phases.33 This characteristic means slow ionic exchange, leading to a decreased ionic conductivity compared to SIPE. As expected, the DIPE including the free mobile TFSI anions displays higher ionic conduction than SIPE, but somewhat surprisingly, the opposite trend in ionic conductivity behaviour is seen in the case of dual-ion hybrid electrolytes; once 26 wt% LLZO is added to the dual-ion crosslinked polymer matrix (HDIPE-26), the ionic conductivity drops fiercely. These preliminary results stress the importance of using solid NMR to better apprehend the ionic transport in HPEs.
7Li MAS NMR spectra were acquired for the different compositions. First, the SIPE pristine polymer and the hybrid version HPE with 26 wt% LLZO but without an additional lithium monomer or salt are compared (Fig. 5). The difference between these two electrolytes is their Li ion source: in SIPE, LiMTFSI provides Li+ whereas it is solely from LLZO particles for HPE-26. For both electrolytes, one signal is observed with different line width and resonance. For SIPE, one narrow signal is detected at −6.13 ppm. In contrast, one broad signal at −4.01 is distinguished for HPE-26. A narrow signal represents fast Li ion motion34 and so it can be concluded that, in these samples, the narrow line width observed indicates the fast lithium transport in the polymer phase (SIPE) while the the broad one indicates the slow lithium motion in the LLZO phase (HPE-26). These spectra will be used as references to determine Li ion environments in hybrid electrolytes.
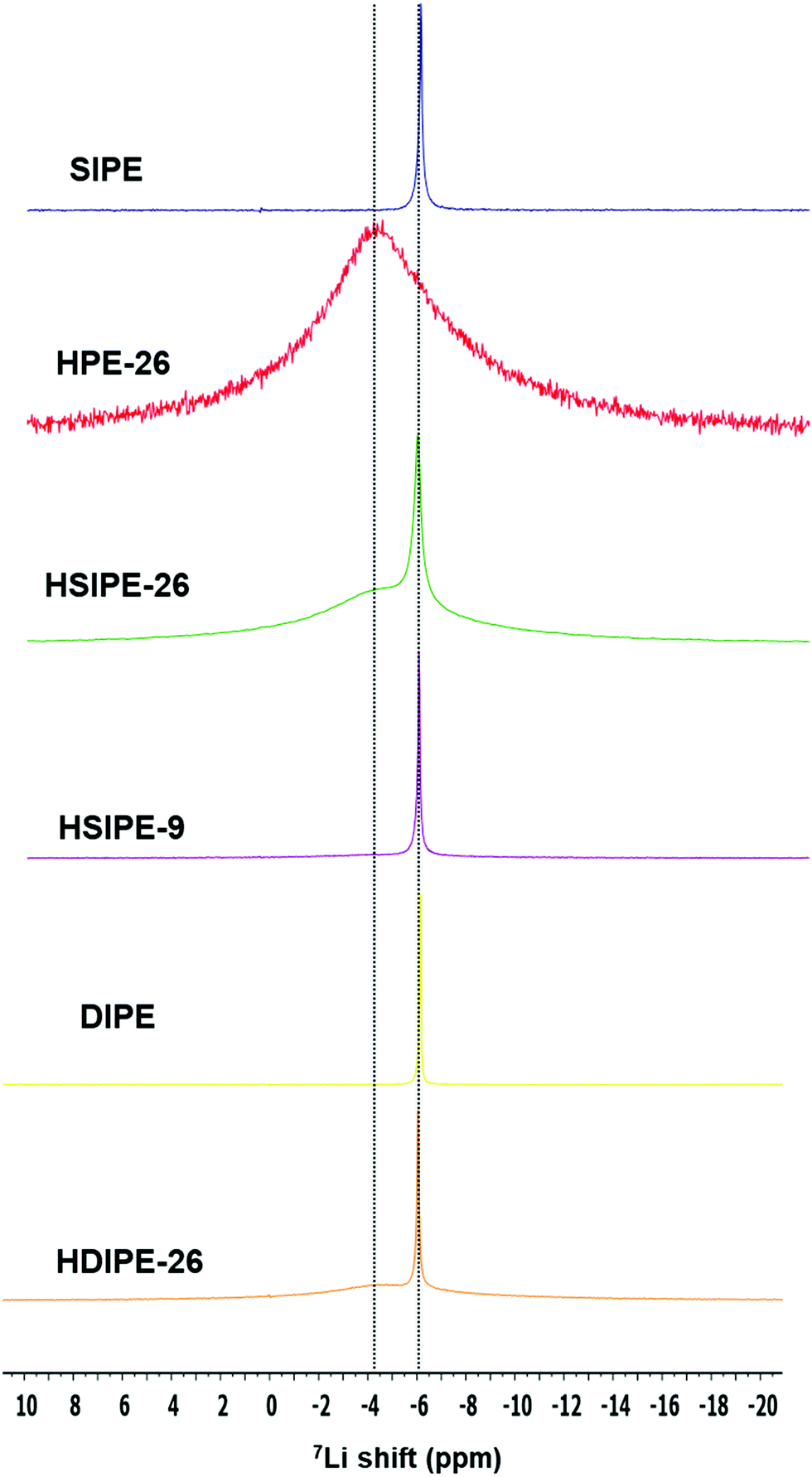 | ||
| Fig. 5 7Li MAS solid NMR on single- and dual-ion crosslinked polymer electrolytes. | ||
HSIPEs are examined to see the effect of LLZO particles on the Li ion environment of the single-ion polymer matrix (HSIPE-26 and HSIPE-9, Fig. 5). HSIPE-26 presents two resonances: a narrow signal at −6.01 ppm and a broad one at −4.89 ppm. The first resonance is assigned to the Li in the polymer phase, the latter to the Li contained in the LLZO particles. These results are in contrast with the ones reported by Zhang and co-workers, who studied the LLZO-TEGDME-PEO(LiClO4) system. After mixing the particles with the plasticized polymer TEGDME-PEO, they could observe two additional signals. They identified them as Li at the LLZO/TEGDME interface and decomposed LLZO diluted in TEGDME. Extra signals are not detected in our study. This discrepancy can be explained by the analysis of different LLZO compositions and their experimental method. Indeed, Li from the interface and decomposed LLZO phase were clearly distinguished up to 40 wt% LLZO. In addition, Zhang and co-workers used 6Li solid NMR which enables higher resolution of Li ion environments within the materials. 7Li resonances are strongly distinctive in terms of chemical shifts once LLZO is added to the SIPE matrix. The signal at −6.13 ppm broadens and shifts downfield. This indicates that the Li ion environment in the polymer phase is strongly modified by the LLZO particles.35 Additionally, Li from the LLZO inorganic phase is also affected as its related Li resonance shifts upfield. These chemical shifts evidence significant structural changes for Li ion environments both in the polymer and LLZO phases. HSIPE-9 only presents one narrow signal at −6.05 ppm. This resonance is attributed to the polymer phase. Signals from LLZO are not discernible for this composition due to the lower concentration of the particles. Nevertheless, the low content of LLZO seems to affect the Li environment in the polymer phase as the peak shifts and slightly broadens.
Solid electrolytes containing LiTFSI salt instead of the monomeric LiMTFSI named DIPE and HDIPEs are also compared to see the impact of free TFSI anions on Li ion environments (Fig. 5). In the case of pristine polymer electrolyte DIPE, only one signal can be observed at −6.15 ppm. When 26 wt% LLZO is added into the dual ion pristine electrolyte (composition HDIPE-26), 7Li resonance displays two Li environments, with a narrow and broad signal observed at −6.05 and −4.47 ppm which are assigned to the Li in the polymer and LLZO, respectively. As observed previously for SIPE compositions, LLZO affects the Li ion environment in the polymer matrix as 7Li resonance shifts downfield.
To deepen our analysis, 19F MAS NMR is carried out on the same compositions to determine if LLZO can also affect the environment of the fluorinated anions. Single-ion compositions are first compared (Fig. 6, compositions SIPE, HSIPE-26 and HSIPE9). In the SIPE composition, a unique signal is present at −85.84 ppm and is assigned to sulfonamide anions anchored covalently to the polymer. In the case of hybrids, an additional “shoulder” is detected at −85.90 and −85.95 ppm for HSIPE-26 and HSIPE-9, respectively. As LLZO does not contain fluorine atoms in its crystallographic structure, these resonances are assigned to two distinct sulfonamide environments in the polymer phase. Once LLZO is added in the SIPE, peaks become narrower and shift downfield. This implies that LLZO can change the sulfonamide environment in the polymer. The “shoulder” signal intensity seems to increase with LLZO content. This additional resonance is assigned to sulphonamide anionic moieties interacting with the LLZO surface. It is worth noting that a few previous 19F solid NMR studies already reported the appearance of a shoulder signal for polymer electrolytes reinforced with inorganic fillers, indicating the strong absorption of fluorinated anions on the surface of the inorganic particles.36,37 The shoulder peak appearing for HSIPEs can be hypothesized as the strong interaction between the LLZO and SIPE matrix.
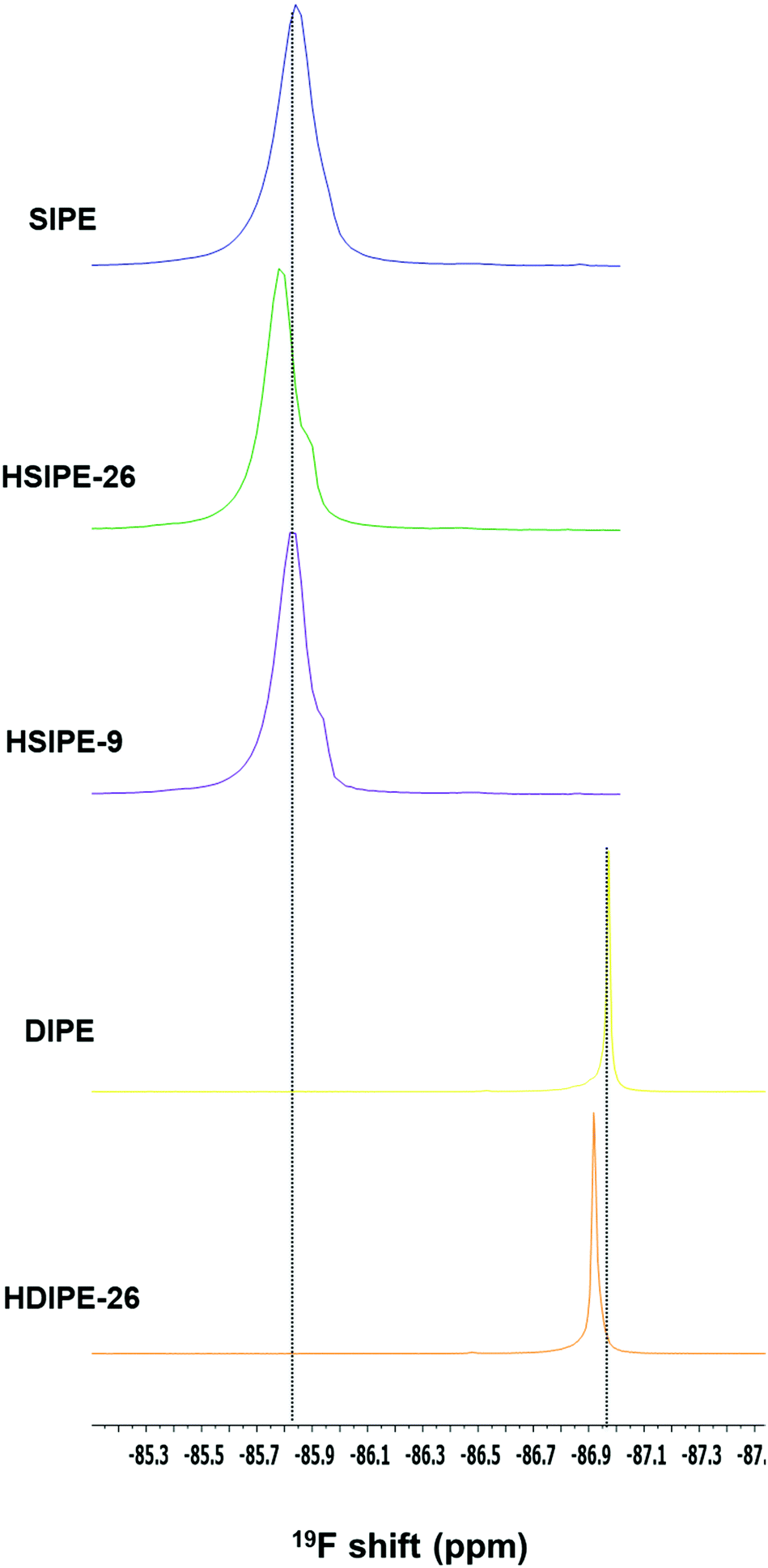 | ||
| Fig. 6 19F MAS solid NMR on single- and dual-ion crosslinked polymer electrolytes. | ||
Dual-ion compositions are also examined by 19F MAS NMR (Fig. 6, compositions DIPE and HDIPE-26). In both cases, one peak is observed, which is assigned to the fluorine environment in the polymer matrix. When the single-ion monomer LiMTFSI is replaced with LiTFSI salt, 19F resonance becomes narrower indicating the higher mobilities of fluorinated anions, and shifts upfield to −86.97 ppm. This result was expected as TFSI− anions are not covalently immobilized into the polymer backbone compared to SIPE.38 Once LLZO is added to the DIPE matrix (HDIPE-26), the peak broadens reflecting a lower mobility of the TFSI− anions which is related to the observed lower ionic conductivity values.
To get more insights into ionic conductivity mechanisms, static 7Li NMR and pulse field gradient (pfg) diffusion NMR are used to determine Li ion mobilities. By peak deconvolution (Fig. 7), the quantification of distinct 7Li species was carried out for HSIPE and HDIPE electrolytes. The narrow component represents 14 ± 2%, 47 ± 8% and 17 ± 2% for compositions HSIPE-26, HSIPE-9 and HDIPE-26, respectively. Note that in the preparation of HSIPE-26, HSIPE-9 and HDIPE-26 samples, the polymer phase consists only 8.2 mol%, 24.1 mol% and 8.5 mol% of the lithium ions which were expected to be mobile (see Table 1). The significantly increased lithium concentrations in the mobile phase suggested the transport of Li ions from the LLZO phase to the polymer phase, for all the three samples. It also suggests that the lithium ions are exchangeable between the inorganic LLZO phase and the polymer phase. However, the HPE-26 sample does not show any narrow component in the 7Li spectra, indicating the absence of mobile Li ions in the polymer phase. Note that HPE-26 is the only sample that does not have any anions. This suggests that the present fluorinated anions is one of the key factors that facilitate Li ion exchange between the inorganic phase and polymer phase.
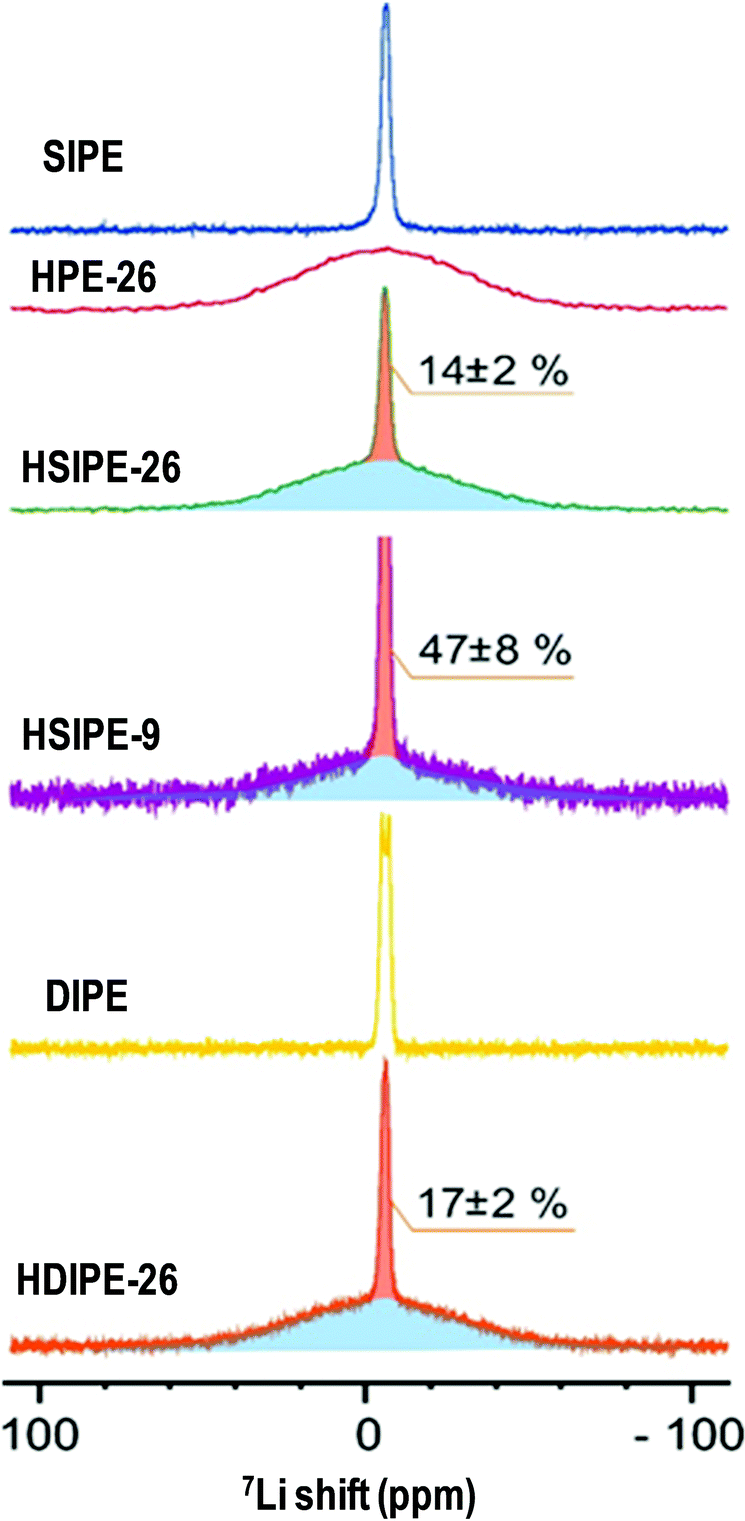 | ||
| Fig. 7 7Li static solid NMR on single- and dual-ion crosslinked polymer electrolytes. | ||
In addition, T1 relaxation times are calculated for pristine and hybrid compositions as shown in Table S9 (ESI†). The narrow and broad components represent the Li in the polymer and LLZO phases, respectively. Mobile Li ions come from the polymer matrix, where the ion transport is faster. T1 relaxations of narrow components increases upon LLZO addition, in both HSIPEs and HDIPEs. This result signifies an increase in local Li ion mobility in the polymer phase (as T1 increases with the increasing mobility if we are on the high temperature side of the minimum). This is in contrast to the trend reported in the work of Zheng and co-workers for the LLZO-PEO(LiTFSI) system;297Li T1 relaxation times became smaller with the increasing LLZO content, which indicates a decrease of Li ion motion in the PEO phase. However, after the addition of a plasticizer (20 wt% TEGDME), a similar trend to that seen here was observed, with Li ion mobility increasing dramatically despite the high content of LLZO. This information emphasizes the beneficial effect of the propylene carbonate plasticizer on Li ion motion in HSIPEs and HDIPEs. Nevertheless, the LLZO phase exhibits very long 7Li T1 relaxation times. Similar values were communicated by Zhang and co-workers for decomposed LLZO.31 They figured that the latter may be affected by its reaction with TEGDME plasticizer. In the present systems, the propylene carbonate may also affect the LLZO phase, as T1 relaxation of bulk LLZO is reported to be close to ∼1 s.
A significant enhancement in the ion diffusion coefficients of Li+ and TFSI− is also observed from pfg NMR, particularly at higher temperatures, as presented in Tables S10 and S11 (ESI†). In the case of HSIPEs (Table S10, ESI†), 7Li diffusion coefficients increase slightly in the whole range of temperature upon LLZO addition to the SIPE matrix. Again, this is a different behaviour to that observed for HDIPEs, in which the 7Li diffusion coefficient is slightly higher at room temperature but decreases at elevated temperatures. These opposite tendencies between HSIPEs and HDIPEs may reveal the different nature of interactions between the LLZO and immobilized/free TFSI− anions. 19F diffusion coefficients were also determined for DIPE and HDIPE-26 (Table S11, ESI†) showing the anion diffusion dropping significantly, especially at low temperatures where it decreases by a factor of 2. These results confirm the presence of physical interactions between the LLZO and LiTFSI salt, which play an important role in the final ionic transport of HDIPEs.
To conclude, Li ion transport in HSIPEs and HDIPEs is strongly distinctive. In HSIPEs, LLZO particles complex with TFSI− moieties anchored in the polymer network as indicated by the observation of two distinct 19F environments in the polymer phase. As a result, Li ions can better dissociate from TFSI− anions. As confirmation, 7Li ion diffusion coefficients are improved with higher LLZO content and are not temperature dependent whereas no such affinity occurs in HDIPEs. However, physical interactions (Lewis) may exist as 7Li and 19F diffusion coefficients are greatly dependent on temperature. For example, fluorine diffusivity is divided by half once 26% LLZO is added to the DIPE matrix, resulting in an enhanced room temperature 7Li mobility in HDIPEs. These distinct interactions between the LLZO and free/immobilized TFSI− moieties can explain the different ionic conductivity trends in HSIPEs and HDIPEs.
Performance of single-ion hybrid electrolytes in lithium symmetric cells
Next, symmetrical lithium cells were assembled to evaluate the electrochemical behaviour of HSIPE solid electrolytes against lithium metal under different current densities. To study the electrochemical performance of hybrid electrolytes, galvanostatic cycling is performed with increased current densities to determine their critical current density (CCD). The CCD is the maximum current density that the electrolyte can withstand without forming dendrites, which is a critical parameter for practical applications.39 For battery electric vehicle (BEV) applications, a critical current density is expected to be superior to 1 mA h cm−2. To assess the CCD, Li symmetrical cells were used to investigate the Li stripping/plating processes under harshest conditions. Use of Li symmetrical cells can decouple the cathode effect and focus especially on the overpotential increasing from the heterogeneous deposition/dissolution of lithium and the evolution of the solid electrolyte interphase (SEI) on the metal anode.40 Observation of voltage oscillations during cycling also gives clues about the morphologies of lithium metal and sheds light on dendrite formation. Besides, the shapes of Li deposits are not only dependent on the electrolyte features (SEI, mechanical properties), but also on the applied current density.41 Galvanostatic cycling was carried out at different current densities and coupled with EIS measurements to monitor the evolution of the overall cell resistance.Cycling performances of both SIPE and HSIPE-26 Li metal symmetric cells are compared in Fig. 8. In the case of the SIPE electrolyte (Fig. 8a-i), two voltage profiles are observed at low and high current densities. At 0.1 mA cm−2, the voltage remains flat, meaning that the Li stripping/plating processes are homogeneous. Between 0.25 and 0.4 mA cm−2, the overpotential starts to increase extensively. This type of voltage profile was reported to correlate to a quasi-homogeneous Li needle morphology.42 A new SEI is formed to accommodate the expansion of the Li metal anode, increasing the overall resistance of the cell. Starting from 0.5 mA cm−2, the voltage response transits to a “neck” shape which is a strong indication of the formation and dissolution of mossy dendritic Li.43 These deposits tend to be detached from the anode surface during cycling, forming a thick layer of dead Li (inactive Li). It can be concluded that the CCD reached for SIPE is 1 mA h cm−2. Nevertheless, a dramatic increase in resistance can be observed during cycling (Fig. 8a-ii). This increase can be explained by the formation of a “bad” SEI, leading gradually to the deterioration of the electrolyte and the lithium.44 PC as an electrolyte solvent was reported to be incompatible for the creation of a stable SEI on Li metal.45 The continuing growth of the SEI by electrolyte decomposition and dead lithium creation is at the origin of the increase of the resistance during cycling.
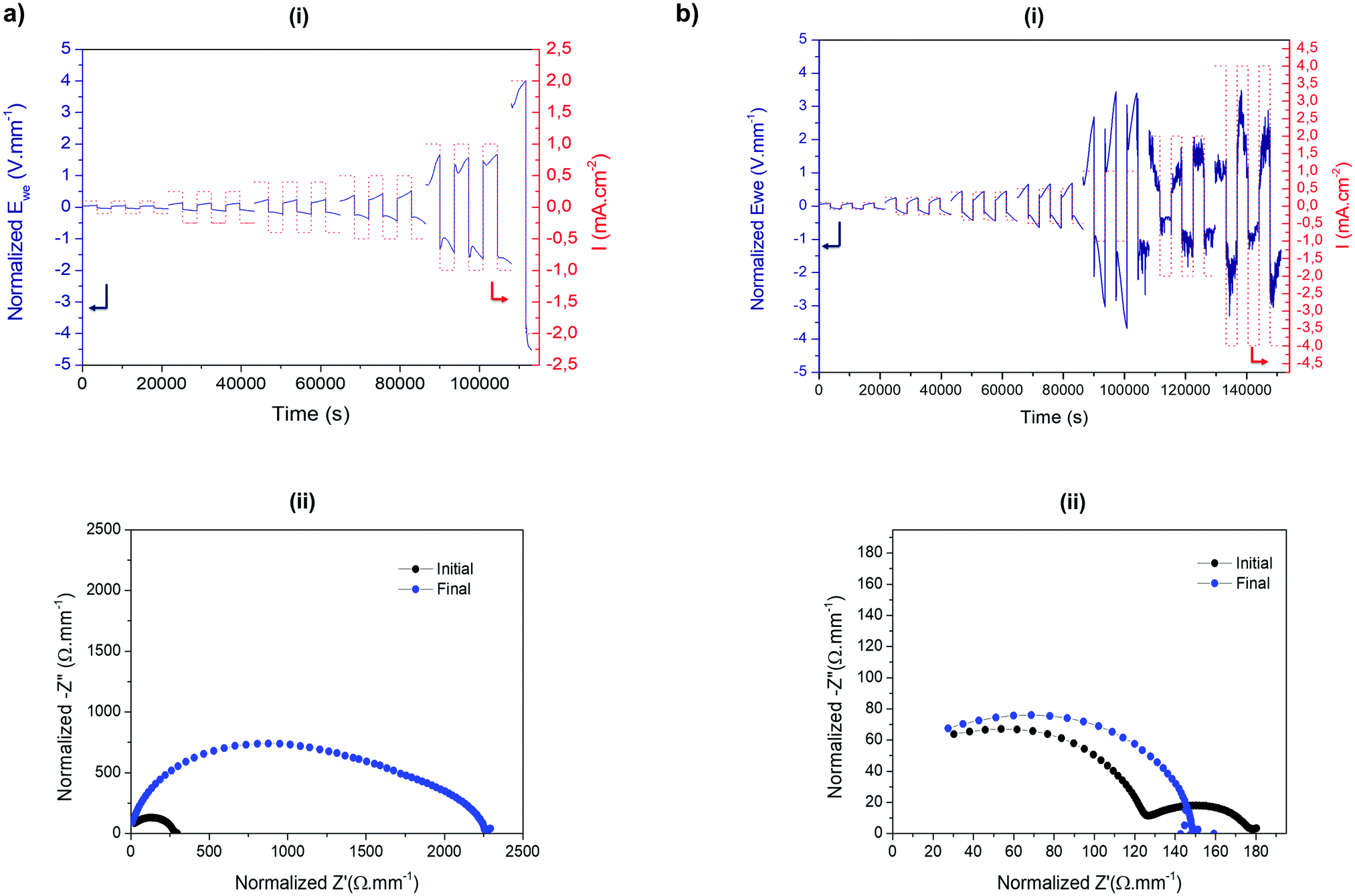 | ||
| Fig. 8 Galvanostatic cycling with increased current densities for (a) Li|SIPE|Li and (b) Li|HSIPE-26|Li. (i) Voltage profiles at different current densities. (ii) EIS spectra before and after galvanostatic cycling. | ||
The galvanostatic cycling of HSIPE-26 is presented in Fig. 8b-i. Three voltage profiles can be seen at low and high current densities. Between 0.1 and 0.25 mA cm−2, Li needle growth takies place. Starting from 0.4 to 1 mA cm−2, mossy like Li is deposited as it indicated by the “neck” shape of the voltage profile. From 2 to 4 mA cm−2, voltage spikes are detected. It was shown in previous studies that the erratic voltage is a sign of dendrite growth.46 Furthermore, the EIS spectrum in Fig. 8b-ii presents a drop in the overall resistance, which is also related to dendrite propagation within the electrolyte.47 It is important to note that these characteristics (voltage spikes, impedance drop) are only observed when the crosslinked polymer electrolyte is reinforced with LLZO particles. Based on these results, it can be concluded that the addition of LLZO, although improving the ionic conductivity, is detrimental for the Li metal stripping/plating processes at high current densities.
Dendrite formation appears to come from the use of LLZO particles, as dendritic growth characteristics are not observable for the cycled SIPE Li battery. To our knowledge, it is the first time that polymer electrolyte reinforced with LLZO particles is reported to form dendrites. LLZO is usually used as an ideal active inorganic filler for polymer hybrid electrolytes. However, most of the studies do not focus on Li stripping and plating processes or use smaller current densities (≤1 mA cm−2), which are less aggressive conditions to form dendrites (Table S4, ESI†). A CCD study similar to our work was carried out by Keller et al.48 with the current density progressively increasing from 0.005 mA cm−2 to 0.1 mA cm−2 but this was too small to investigate fractal dendrite growth. Gupta and co-workers examined the CCD for a PEO-LiTFSI solid electrolyte39 where they applied “practical” current densities, increasing from 0.01 mA cm−2 up to 10 mA cm−2. A resistance drop and erratic voltages were recorded starting at 0.5 mA cm−2 but no short-circuit occurred. Voltage instabilities have also been recorded by Zagórski and co-workers for 30 wt% LLZO-PEO (LiFTSI) at the beginning of a similar cycling protocol (i.e. at 0.1 mA cm−2).33 According to them, this erratic voltage comes from the direct contact of LLZO particles with lithium metal which increases the interfacial resistance. Voltage instabilities observed during the first cycles may also originate from the structural reorganization at the interface Li/electrolyte.
Recently, it has been demonstrated that densified LLZO can form dendrites within itself and is assumed to be a result of a relatively high electronic conductivity.49 This feature can also explain why dendrites form in HSIPEs up to 1 mA cm−2. On the other hand, the CCD for a LLZO single crystal was established to be around 300 μA cm−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 50 and so the CCD obtained here for the hybrid is surprisingly high. In addition, since different Li ion environments are present in HSIPEs, comprising the polymer and LLZO phases and having distinct Li diffusion, this difference in CCD arises by Li stripping/plating more favorably in some areas which can also lead to the formation of dendrites.
50 and so the CCD obtained here for the hybrid is surprisingly high. In addition, since different Li ion environments are present in HSIPEs, comprising the polymer and LLZO phases and having distinct Li diffusion, this difference in CCD arises by Li stripping/plating more favorably in some areas which can also lead to the formation of dendrites.
Experimental
Reactants
Poly(ethylene glycol) methyl ether methacrylate (500 g mol−1) (PEGM), poly(ethylene glycol) dimethacrylate (550 g mol−1) (PEGDM), 2-hydroxy-2-methylpropiophenone (UV initiator, 97% purity) and anhydrous propylene carbonate (PC, 99.8% purity, H2O < 10 ppm) were purchased from Sigma Aldrich. PEGM and PEGDM were dried with molecular sieves before use (4 Å, first dried for 12 h at 300 °C). LiTFSI salt was furnished by TCI. Before use, LiTFSI was dried under a vacuum at 130 °C for 12 h and stored in a glovebox. Lithium 1-[3-(methacryloyloxy)-propylsulfonyl]-1-(trifluoromethylsulfonyl)imide (LiMTFSI) was synthesized according to a previous study.51 LiMTFSI crystallized powder was dried at 90 °C under a vacuum and stored in a glovebox. Al doped Li7−3XAlXLa3Zr2O12 (LLZO) particles were received from MSE (MSE Supplies LLC, Tucson, USA) under an argon atmosphere and were used as received (99.9% purity, 400–600 nm particles size). Li foils (120 μm thickness) were purchased from Rockwood.Equipment and characterization methods
Crosslinked membranes were cured using a Lightningcurve TM LC-L1-V5 UV curing lamp (λ = 365 nm) from Hamamatsu (Sizuoka-ken, JP).The crystallinity of LLZO particles was analyzed with a MinifleX600 X-ray diffractomer from Rigaku (Wroclaw, PL) as shown in Fig. S13 (ESI†). LiMTFSI powder was analyzed by nuclear magnetic resonance (NMR), using deuterated dimethyl sulfoxide (DMSO) with an Advance 500 (500 MHz) spectrometer (Bruker, Billerica, US) as shown in Fig. S12 (ESI†).
Photo-rheology was carried out in order to determine the optimal irradiation time to reach the gel point, using AR-G2 (TA Instrument, Brussels, BE). The liquid monomer mixture was exposed to UV light, and the irradiance was set up at 58 mW cm−2. The liquid was compressed to get membranes with a fixed thickness of 800 μm. The experiment was monitored with a frequency of 1 Hz and an oscillatory shear fixed at 12% strain. The shear storage and loss moduli G′ and G′′ evolutions were recorded while the monomer mixture was exposed to UV light.
Environmental scanning electron microscopy (ESEM-FEI) was used with a QUANTA 500 (ThermoFisher, Waltham, US) to observe LLZO particle dispersion within the hybrid polymer electrolyte. Neat cross-sections of the material were made outside the glovebox by immersing the materials into a liquid N2 bath.
Thermogravimetric analysis (TGA) was performed with a TGA Q500 (TA Instruments, Brussels, BE). Measurements were carried out by heating the sample at a rate of 10 °C min−1 under a nitrogen atmosphere starting at room temperature and heating up to 600 °C.
Dynamical mechanical thermo-analysis (DMTA) was carried out using AR-G2 (TA Instrument, Brussels, BE) with oscillatory compression. Shear storage (G′) and the loss (G′′) moduli were recorded at fixed frequency (1 Hz), in the temperature range from −100 °C to 80 °C.
Solid nuclear magnetic resonance spectroscopy (NMR) was used to better apprehend the ionic transport within hybrid crosslinked electrolytes. Static 7Li NMR, 7Li and 19F magic angle spinning (MAS) NMR were performed using an Advance spectrometer with a 4 mm Bruker HXY triple resonance probe (Bruker, Billerica, USA). The samples were spun at 6 kHz. Recycle delays were 200 s and 10 s for 7Li and 19F, respectively.
Electrochemical impedance spectroscopy (EIS) measurements were carried out with an Autolab 302N potentiostat galvanostat (Metrohm AG, Herisau, CH) with a temperature controller (Microcell HC station). Electrolytes were sandwiched between two stainless steel electrodes for the tests. EIS was monitored using a frequency range of 0.1 Hz to 100 kHz with an amplitude of 10 mV. The ionic conductivity of the electrolytes was determined at different temperatures, between 25 °C and 80 °C. Ionic conductivity measurements were done at least two times on each composition.
A lithium metal/hybrid solid electrolyte/lithium metal cell was assembled and subjected to a 10 mV polarization bias (ΔV) to determine the initial (Ii) and steady-state (Is) currents. EIS was performed by applying a 10 mV perturbation between 100 kHz and 1 Hz under open-circuit conditions to obtain the resistance of the passivation layer before (Ri) and after (Rs) polarization. The lithium transference number tLi+ was calculated using the following equation:
t Li+ = Is(ΔV − IiRi)IIi(ΔV − IsRs)Li symmetrical cells were assembled in an Ar filled glovebox for transference number and galvanostatic cycling investigation, using a VMP3 multichannel potentiostat (Biologic, Seyssinet-Pariset, FR). The Li foil was carefully mechanically scraped to remove the native layer (Li2CO3, Li2O) on the Li surface before making cells. Disks of 11 mm2 were pinched from the prepared Li foils and used as electrodes for the symmetrical cells. The transference numbers were determined thanks to the potentiostatic polarization method, which couples chronoamperometry and EIS. EIS was carried out with an amplitude of 40 mV, before and after cell polarization. The cells were allowed to stabilize at 60 °C thanks to the temperature controlled chamber, for a period of 30 h before the experiment. After 30 h, the solid electrolyte interphase (SEI) between the lithium metal and polymer remained steady and it was possible to launch cell polarization. Galvanostatic tests were executed by increasing the current density, from 0.1, 0.25, 0.5, 1, 2 and to 4 mA cm−2, each step composed of 3 charge/discharge cycles lasting for 2 h (1 h charge and 1 h discharge). EIS was carried out with an amplitude of 40 mV before and after cycling to study the evolution of resistance. Performances of pristine and hybrid electrolytes were examined at 60 °C. It is important to mention that voltage has been recorded every 1s to closely observe the behaviour of Li stripping and plating.
General procedure for the synthesis of the hybrid crosslinked polymer electrolytes
The synthesis of the crosslinked membranes was carried out inside an Ar filled-glovebox with H2O and O2 levels below 10 ppm. As thermal polymerization takes 3 h to obtain fully crosslinked materials, UV curing was preferred in order to limit LLZO to sediment. PEGM, PEGDM, LiMTFSI, PC and UV initiators were magnetically mixed at 450 rpm for 1 h, according to the desired composition (Table 1). Then, appropriate amount of LLZO particles was added and the reactant mixture was left under stirring for 24 h. Different compositions of LLZO particles were studied from 5 to 50 wt% (versus total weight of the electrolyte). After stirring, the mixture was poured into a silicon mold (thickness ∼ 1 mm) and exposed to UV light for 20 min. A similar experimental method was used for the synthesis of dual-ion pristine DIPE and hybrid membrane HDIPE. LiMTFSI was replaced with LiTFSI (equimolar) in the compositions. The distance between the sample and the lamp was set up at 10 cm, the corresponding irradiance was measured to be 58 mW cm−2. The optimal duration of UV exposure was determined before thanks to photo-rheology experiments. After curing, self-standing hybrid crosslinked electrolytes were obtained, with thicknesses between 0.7 and 1 mm.Conclusions
In this article, we show a simple method to prepare single-ion hybrid solid polymer electrolytes (HSIPEs). The hybrid solid electrolyte is formed by a PEG type single-ion polymer network which includes an anionic lithium-single ion monomer, ceramic garnet-type nanoparticles of Li7−3XAlXLa3Zr2O12 (LLZO) and a propylene carbonate plasticizer. Flexible free-standing films of the hybrid electrolyte were prepared by a fast UV-photopolymerization process (<15 min) which allows varying the composition of the HSIPEs easily. This methodology showed excellent dispersion of the LLZO nanoparticles within the gel polymer network with up to 40 wt% ceramic content as shown in the enviromental SEM images which is probably due to the favoured interaction between the anionic polymer backbone and the inorganic material. These hybrid electrolytes show very high ionic conductivity values (1.4 × 10−4 S cm−1 at 25 °C) and unique lithium single-ion conduction as compared to previous hybrid electrolytes. Solid-state and PFG-NMR results show increased lithium mobility and diffusivity with increasing LLZO concentration of up to 26 wt%, with considerably suppressed anion diffusivity.Finally, determination of the critical current density (CCD) has been carried out on pristine and hybrid electrolytes, so as to assess their viable use as solid electrolytes for lithium metal batteries. Lithium dendrite formation appears to come from the LLZO particles, as dendritic growth characteristics are not observable for the cycled polymer SIPE Li cells.
Conflicts of interest
This work was supported by the European Commission's funded Marie Sklodowska-Curie project POLYTE-EID (Project No. 765828). L. P. has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 797295.Notes and references
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Status and challenges in enabling the lithium metal electrode for high-energy and low-cost rechargeable batteries, Nat. Energy, 2018, 3, 16–21, DOI:10.1038/s41560-017-0047-2
.
- C. Yang, K. Fu, Y. Zhang, E. Hitz and L. Hu, Protected lithium–metal anodes in batteries: From liquid to solid, Adv. Mater., 2017, 29, 1701169, DOI:10.1002/adma.201701169
- L. Fan, S. Wei, S. Li, Q. Li and Y. Lu, Recent progress of the solid-state electrolytes for high-energy metal-based batteries, Adv. Energy Mater., 2018, 8, 1702657, DOI:10.1002/aenm.201702657
- J. Schnell, T. Günther, T. Knoche, C. Vieider, L. Köhler, A. Just, M. Keller, S. Passerini and G. Reinhart, All-solid-state lithium-ion and lithium metal batteries – paving the way to large-scale production, J. Power Sources, 2018, 382, 160–175, DOI:10.1016/J.JPOWSOUR.2018.02.062
- Y. Inaguma, C. Liquan, M. Itoh, T. Nakamura, T. Uchida, H. Ikuta and M. Wakihara, High ionic conductivity in lithium lanthanum titanate, Solid State Commun., 1993, 86, 689–693, DOI:10.1016/0038-1098(93)90841-A
- K. Arbi, W. Bucheli, R. Jiménez and J. Sanz, High lithium ion conducting solid electrolytes based on NASICON Li1 + xAlxM2−x(PO4)3 materials (M = Ti, Ge and 0 ≤ x ≤ 0.5), J. Eur. Ceram. Soc., 2015, 35, 1477–1484, DOI:10.1016/J.Jeurceramsoc.2014.11.023
- V. Thangadurai and W. Weppner, Li6ALa2Nb2O12 (A = Ca, Sr, Ba): A new class of fast lithium ion conductors with garnet-like structure, J. Am. Ceram. Soc., 2005, 88, 411–418, DOI:10.1111/j.1551-2916.2005.00060.x
-
(a) K. S. Ngai, S. Ramesh, K. Ramesh and J. C. Juan, A review of polymer electrolytes: fundamental, approaches and applications, Ionics, 2016, 22, 1259–1279, DOI:10.1007/s11581-016-1756-4
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Inorganic solid-state electrolytes for lithium batteries: Mechanisms and properties governing ion conduction, Chem. Rev., 2016, 116, 140–162, DOI:10.1021/acs.chemrev.5b00563
- B. V. Lotsch and J. Maier, Relevance of solid electrolytes for lithium-based batteries: A realistic view, J. Electroceram., 2017, 38, 128–141, DOI:10.1007/s10832-017-0091-0
- L. Porz, T. Swamy, B. W. Sheldon, D. Rettenwander, T. Frömling, H. L. Thaman, S. Berendts, R. Uecker, W. C. Carter and Y. M. Chiang, Mechanism of lithium metal penetration through inorganic solid electrolytes, Adv. Energy Mater., 2017, 7, 1–12, DOI:10.1002/aenm.201701003
- G. Bucci, T. Swamy, Y. M. Chiang and W. C. Carter, Modeling of internal mechanical failure of all-solid-state batteries during electrochemical cycling, and implications for battery design, J. Mater. Chem. A, 2017, 5, 19422–19430, 10.1039/c7ta03199h
-
(a) R. C. Agrawal and G. P. Pandey, Solid polymer eletrolytes: material designing and all solid state battery applications – An overview, J. Phys. D: Appl. Phys., 2008, 41, 223001, DOI:10.1088/0022-3727/41/22/223001
- X. Wang, R. Kerr, F. Chen, N. Goujon, J. M. Pringle, D. Mecerreyes, M. Forsyth and P. C. Howlett, Toward high-energy-density lithium metal batteries: Opportunities and challenges for solid organic electrolytes, Adv. Mater., 2020, 32, 1–21, DOI:10.1002/adma.201905219
- M. Forsyth, L. Porcarelli, X. Wang, N. Goujon and D. Mecerreyes, Innovative electrolytes based on ionic liquids and polymers for next-generation solid-state batteries, Acc. Chem. Res., 2019, 52, 686–694, DOI:10.1021/acs.accounts.8b00566
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, Promising routes to a high Li+ transference number electrolyte for lithium ion batteries, ACS Energy Lett., 2017, 2, 2563–2575, DOI:10.1021/acsenergylett.7b00792
-
(a) M. Keller, A. Varzi and S. Passerini, Hybrid electrolytes for lithium metal batteries, J. Power Sources, 2018, 392, 206–225, DOI:10.1016/j.jpowsour.2018.04.099
- W. Wang and P. Alexandridis, Composite polymer electrolytes: Nanoparticles affect structure and properties, Polymers, 2016, 8, 387, DOI:10.3390/polym8110387
- A. S. Shaplov, R. Marcilla and D. Mecerreyes, Recent advances in innovative polymer electrolytes based on poly(ionic liquid)s, Electrochim. Acta, 2015, 175, 18–34, DOI:10.1016/j.electacta.2015.03.038
- C. K. Chan, T. Yang and J. Mark Weller, Nanostructured Garnet-type Li7La3Zr2O12: Synthesis, properties, and opportunities as electrolytes for Li-ion batteries, Electrochim. Acta, 2017, 253, 268–280, DOI:10.1016/j.electacta.2017.08.130
- X. Zhang, T. Liu, S. Zhang, X. Huang, B. Xu, Y. Lin, B. Xu, L. Li, C. W. Nan and Y. Shen, Synergistic coupling between Li6.75La3Zr175Ta0.25O12 and poly(vinylidene fluoride) induces high ionic conductivity, mechanical strength, and thermal stability of solid composite electrolytes, J. Am. Chem. Soc., 2017, 139, 13779–13785, DOI:10.1021/jacs.7b06364
- T. Yang, J. Zheng, Q. Cheng, Y. Y. Hu and C. K. Chan, Composite polymer electrolytes with Li7La3Zr2O12 Garnet-type nanowires as ceramic fillers: mechanism of conductivity enhancement and role of doping and morphology, ACS Appl. Mater. Interfaces, 2017, 9, 21773–21780, DOI:10.1021/acsami.7b03806
- J. Zhang, X. Zang, H. Wen, T. Dong, J. Chai, Y. Li, B. Chen, J. Zhao, S. Dong, J. Ma, L. Yue, Z. Liu, X. Guo, G. Cui and L. Chen, High-voltage and free-standing poly(propylene carbonate)/Li6.75La3Zr1.75Ta0.25O12 composite solid electrolyte for wide temperature range and flexible solid lithium ion battery, J. Mater. Chem. A, 2017, 5, 4940–4948, 10.1039/c6ta10066j
- M. Falco, L. Castro, J. Nair, F. Bella, F. Barde, G. Meligrana and C. Gerbaldi, UV-cross-linked composite polymer electrolyte for high-rate, ambient temperature lithium batteries, ACS Appl. Energy Mater., 2019, 2(3), 1600–1607 CrossRef CAS
- L. Porcarelli, A. S. Shaplov, F. Bella, J. R. Nair, D. Mecerreyes and C. Gerbaldi, Single-ion conducting polymer electrolytes for lithium metal polymer batteries that operate at ambient temperature, ACS Energy Lett., 2016, 1, 678–682, DOI:10.1021/acsenergylett.6b00216
- K. I. Jung, D. G. Lee, K. W. Bong, S. M. Noh, M. S. Um, W. J. Choi, B. Kim and H. W. Jung, Effects of solvents on rheological and crosslinking properties of photo-polymerized poly(ethylene glycol) hydrogels, Korean J. Chem. Eng., 2017, 34, 1517–1523, DOI:10.1007/s11814-017-0013-5
- J. W. Hwang, S. M. Noh, B. Kim and H. W. Jung, Gelation and crosslinking characteristics of photopolymerized poly(ethylene glycol) hydrogels, J. Appl. Polym. Sci., 2015, 132, 1–6, DOI:10.1002/app.41939
- M. Marcinek, J. Syzdek, M. Marczewski, M. Piszcz, L. Niedzicki, M. Kalita, A. Plewa-Marczewska, A. Bitner, P. Wieczorek, T. Trzeciak, M. Kasprzyk, P. Łężak, Z. Zukowska, A. Zalewska and W. Wieczorek, Electrolytes for Li-ion transport – review, Solid State Ionics, 2015, 276, 107–126, DOI:10.1016/j.ssi.2015.02.006
- J. Zheng and Y. Y. Hu, New insights into the compositional dependence of Li-ion transport in polymer-ceramic composite electrolytes, ACS Appl. Mater. Interfaces, 2018, 10, 4113–4120, DOI:10.1021/acsami.7b17301
- J. Zheng, H. Dang, X. Feng, P.-H. Chien and Y.-Y. Hu, Li-ion transport in a representative ceramic–polymer–plasticizer composite electrolyte: {Li} 7 {La} 3 {Zr} 2 {O} 12 –polyethylene oxide–tetraethylene glycol dimethyl ether, J. Mater. Chem. A, 2017, 5, 18457–18463, 10.1039/C7TA05832B
- L. Chen, Y. Li, S. P. Li, L. Z. Fan, C. W. Nan and J. B. Goodenough, PEO/garnet composite electrolytes for solid-state lithium batteries: From “ceramic-in-polymer” to “polymer-in-ceramic,”, Nano Energy, 2018, 46, 176–184, DOI:10.1016/j.nanoen.2017.12.037
- J. Zagórski, J. M. López Del Amo, M. J. Cordill, F. Aguesse, L. Buannic and A. Llordés, Garnet-polymer composite electrolytes: New insights on local Li-ion dynamics and electrodeposition stability with Li metal anodes, ACS Appl. Energy Mater., 2019, 2, 1734–1746, DOI:10.1021/acsaem.8b01850
- F. Langer, M. S. Palagonia, I. Bardenhagen, J. Glenneberg, F. La Mantia and R. Kun, Impedance spectroscopy analysis of the lithium ion transport through the Li 7 La 3 Zr 2 O 12/P(EO) 20 Li interface, J. Electrochem. Soc., 2017, 164, A2298–A2303, DOI:10.1149/2.0381712jes
-
C. Vinod Chandran and P. Heitjans, Solid-State NMR Studies of Lithium Ion Dynamics Across Materials Classes, 1st edn, Elsevier Ltd, 2016. DOI:10.1016/bs.arnmr.2016.03.001
- M. Forsyth, D. R. MacFarlane, A. Best, J. Adebahr, P. Jacobsson and A. J. Hill, The effect of nano-particle TiO2 fillers on structure and transport in polymer electrolytes, Solid State Ionics, 2002, 147, 203–211, DOI:10.1016/S0167-2738(02)00017-6
- L. van Wüllen and T. K. J. Köster, The interaction between inorganic Li salts (LiTf, LiNTf2) and the surface of alumina particles as studied with solid state nuclear magnetic resonance, Solid State Ionics, 2009, 180, 141–147, DOI:10.1016/j.ssi.2008.12.023
- A. Mejía, S. Devaraj, J. Guzmán, J. M. Lopez Del Amo, N. García, T. Rojo, M. Armand and P. Tiemblo, Scalable plasticized polymer electrolytes reinforced with surface-modified sepiolite fillers – A feasibility study in lithium metal polymer batteries, J. Power Sources, 2016, 306, 772–778, DOI:10.1016/j.jpowsour.2015.12.099
- N. Lago, O. Garcia-Calvo, J. M. del Amo, T. Rojo and M. Armand, All solid state lithium ion batteries with grafted ceramic nanoparticles dispersed in solid polymer electrolytes, ChemSusChem, 2015, 8, 3039–3043, DOI:10.1002/cssc.201500783
- A. Gupta, E. Kazyak, N. Craig, J. Christensen, N. P. Dasgupta and J. Sakamoto, Evaluating the Effects of temperature and pressure on Li/PEO-LiTFSI interfacial stability and kinetics, J. Electrochem. Soc., 2018, 165, A2801–A2806, DOI:10.1149/2.0901811jes
- K. H. Chen, K. N. Wood, E. Kazyak, W. S. Lepage, A. L. Davis, A. J. Sanchez and N. P. Dasgupta, Dead lithium: Mass transport effects on voltage, capacity, and failure of lithium metal anodes, J. Mater. Chem. A, 2017, 5, 11671–11681, 10.1039/c7ta00371d
- L. Frenck, G. K. Sethi, J. A. Maslyn and N. P. Balsara, Factors that control the formation of dendrites and other morphologies on lithium metal anodes, Front. Energy Res., 2019, 7, 115, DOI:10.3389/fenrg.2019.00115
- J. Steiger, G. Richter, M. Wenk, D. Kramer and R. Mönig, Comparison of the growth of lithium filaments and dendrites under different conditions, Electrochem. Commun., 2015, 50, 11–14, DOI:10.1016/j.elecom.2014.11.002
- K. N. Wood, E. Kazyak, A. F. Chadwick, K. H. Chen, J. G. Zhang, K. Thornton and N. P. Dasgupta, Dendrites and pits: Untangling the complex behavior of lithium metal anodes through operando video microscopy, ACS Cent. Sci., 2016, 2, 790–801, DOI:10.1021/acscentsci.6b00260
- K. N. Wood, M. Noked and N. P. Dasgupta, Lithium metal anodes: Toward an improved understanding of coupled morphological, electrochemical, and mechanical behavior, ACS Energy Lett., 2017, 2, 664–672, DOI:10.1021/acsenergylett.6b00650
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J. G. Zhang, High rate and stable cycling of lithium metal anode, Nat. Commun., 2015, 6, 6362, DOI:10.1038/ncomms7362
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Transition of lithium growth mechanisms in liquid electrolytes, Energy Environ. Sci., 2016, 9, 3221–3229, 10.1039/c6ee01674j
- A. Maraschky and R. Akolkar, Mechanism explaining the onset time of dendritic lithium electrodeposition via considerations of the Li+ transport within the solid electrolyte interphase, J. Electrochem. Soc., 2018, 165, D696–D703, DOI:10.1149/2.0601814jes
- M. Keller, G. B. Appetecchi, G.-T. Kim, V. Sharova, M. Schneider, J. Schuhmacher, A. Roters and S. Passerini, Electrochemical performance of a solvent-free hybrid ceramic-polymer electrolyte based on Li7La3Zr2O12 in P(EO)15LiTFSI, J. Power Sources, 2017, 353, 287–297, DOI:10.1016/j.jpowsour.2017.04.014
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, High electronic conductivity as the origin of lithium dendrite formation within solid electrolytes, Nat. Energy, 2019, 4, 187–196, DOI:10.1038/s41560-018-0312-z
- F. Flatscher, M. Philipp, S. Ganschow, H. M. R. Wilkening and D. Rettenwander, The natural critical current density limit for Li 7 La 3 Zr 2 O 12 garnets, J. Mater. Chem. A, 2020, 8, 15782–15788, 10.1039/c9ta14177d
- A. S. Shaplov, P. S. Vlasov, M. Armand, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, O. V. Okatova, G. M. Pavlov, C. Wandrey, I. A. Godovikov and Y. S. Vygodskii, Design and synthesis of new anionic “polymeric ionic liquids” with high charge delocalization, Polym. Chem., 2011, 2, 2609–2618, 10.1039/c1py00282a
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00857a |
| This journal is © The Royal Society of Chemistry 2022 |