
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A caged tris(2-pyridylmethyl)amine ligand equipped with a Ctriazole–H hydrogen bonding cavity†
Gege
Qiu‡
ab,
Donglin
Diao‡
b,
Leo
Chaussy
b,
Sabine
Michaud-Chevallier
b,
A. Jalila
Simaan
b,
Paola
Nava
 b,
Alexandre
Martinez
*b and
Cédric
Colomban
*b
b,
Alexandre
Martinez
*b and
Cédric
Colomban
*b
aCollege of Chemistry and Chemical Engineering, Yantai University, China
bAix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: cedric.colomban@univ-amu.fr; alexandre.martinez@centrale-marseille.fr
First published on 4th May 2022
Abstract
A capped bioinspired ligand built from a tris(2-pyridyl-methyl)amine (TPA) unit and surmounted by a triazole-based intramolecular H-bonding secondary sphere was prepared. The resulting cage provides a well-defined cavity combining the hydrophobic nature with H-bonding properties. Its coordinating properties were explored using Zn(II) and Cu(II) metal ions.
The binding cavities found in metalloproteins govern the selectivity and efficiency of reactions. In their hydrophobic channels, destabilizing (like steric repulsion) and stabilizing (like hydrogen bonding, H-bonding) forces allow for specific enzyme–substrate interactions, substrate positioning and activation/stabilization of highly reactive intermediates.1 H-bonding is particularly important in metalloenzymes involved in dioxygen processing, such as copper-containing oxygenases and oxidases. The postulated oxidative active species in these systems is the highly reactive mononuclear cupric superoxide (CuII–O2˙−).2 Many efforts have been dedicated to the development of artificial Cu ligands able to generate and stabilize such metastable intermediates. Among them, the tris(2-pyridylmethyl)amine (TPA) ligand has been widely used as a scaffold for mimicking the first coordination sphere in the structural and functional models of copper,3 but also iron,4 mono-oxygenases. Interestingly, incorporating intramolecular H-bonding secondary spheres into the TPA ligand was reported as an efficient strategy to stabilize mononuclear hydroperoxo [(L)CuII–OOH]+,5 binuclear peroxodicopper [{(L)CuII}2(O22−)],6 or end-on superoxo [(L)CuII–O2˙−]7 copper–dioxygen intermediates.
On the other hand, synthetic supramolecular chemistry is a powerful tool to build a cage-like second coordination sphere around bioinspired catalysts.8 In particular, the archetypal TPA ligand has been equipped with well-defined cavities by means of its covalent substitution,9,10 or host–guest encapsulation into a H-bonded capsule.11 In this context, TPA-based hemicryptophanes are organic cages built from a bowl-shaped cyclotriveratrylene (CTV) cap, connected to the tripodal ligand via three linkers. We have recently demonstrated that TPA-hemicryptophanes with methylene or phenyl linkers could, respectively, control the helical arrangement of the ligand12 and lead to enhanced oxidation catalysts.13 However, the hydrophobic cavities found in such cages were devoid of H-bonding groups that could allow substrate positioning or intermediate stabilization.
Despite this progress, the preparation of TPA-based complexes combining hydrophobic cavities with intramolecular H-bonding units, at their secondary sphere, is still needed. Designing and discovering new methodologies to prepare such advanced model complexes is in fact crucial to better reproduce the key structural properties of metalloenzymes. Besides their connecting benefits, triazole bridges are particularly interesting due to their H-bonding donor ability. For instance, a triazolo organic cage has recently been reported to be the most efficient chloride-binding receptor to date, by means of Ctriazole–H H-bonding interactions.14 We therefore envisioned that the covalent substitution of the TPA ligand by another C3 symmetrical cap, using triazole spacers, will represent an efficient strategy to construct functionalized cavities.
In this communication, we report the preparation of unprecedented bioinspired complexes displaying a hydrophobic cavity offering three H-bonding triazoles, aiming at reproducing the functionalized hydrophobic channels of metalloenzymes. We design the hemicryptophane Hm-TriA-TPA, where the archetypal TPA ligand is linked to a northern CTV cap via three triazole bridges, resulting in a H-bond donor decorated cavity.
Hm-TriA-TPA was prepared by an eight-step synthetic strategy (Scheme 1). The cage's walls were first prepared (Scheme 1a) before generating the southern TPA and the CTV cap in a final intramolecular macrocyclization closing the structure (Scheme 1b). The aryl propargyl ether derivative 2 was prepared in two steps by alkylation of the starting vanillyl alcohol with propargyl bromide, followed by the protection of the resulting alcohol 1 with THP. 2 was then connected to the pyridine derivative 4 by a triazole link formed in a Cu-catalyzed azide–alkyne cycloaddition reaction (CuAAC). The CuAAC reaction between equimolar amounts of the propargyl 2 and the azide 4 precursors, catalyzed by CuSO4 (10 mol%) in the presence of the sodium ascorbate reducing agent (10 mol%), resulted in the formation of the triazole 5 in 83% yield. Precursor 7 was then prepared in two steps by reduction of 5 into the alcohol 6 followed by its mesylation. The addition of ammonia to 7 in the presence of Cs2CO3, at 90 °C in THF afforded the open TPA derivative 8 in 73% yield. The formation of Hm-TriA-TPA was finally achieved in 29% yield via the intramolecular cyclization of 8 in CH3CN, catalyzed by the Lewis acid scandium triflate Sc(OTf3) under diluted conditions.
 | ||
| Scheme 1 (a) Synthesis of the pyridine-triazole precursor 5. (b) Synthesis of the targeted hemicryptophane Hm-TriA-TPA. | ||
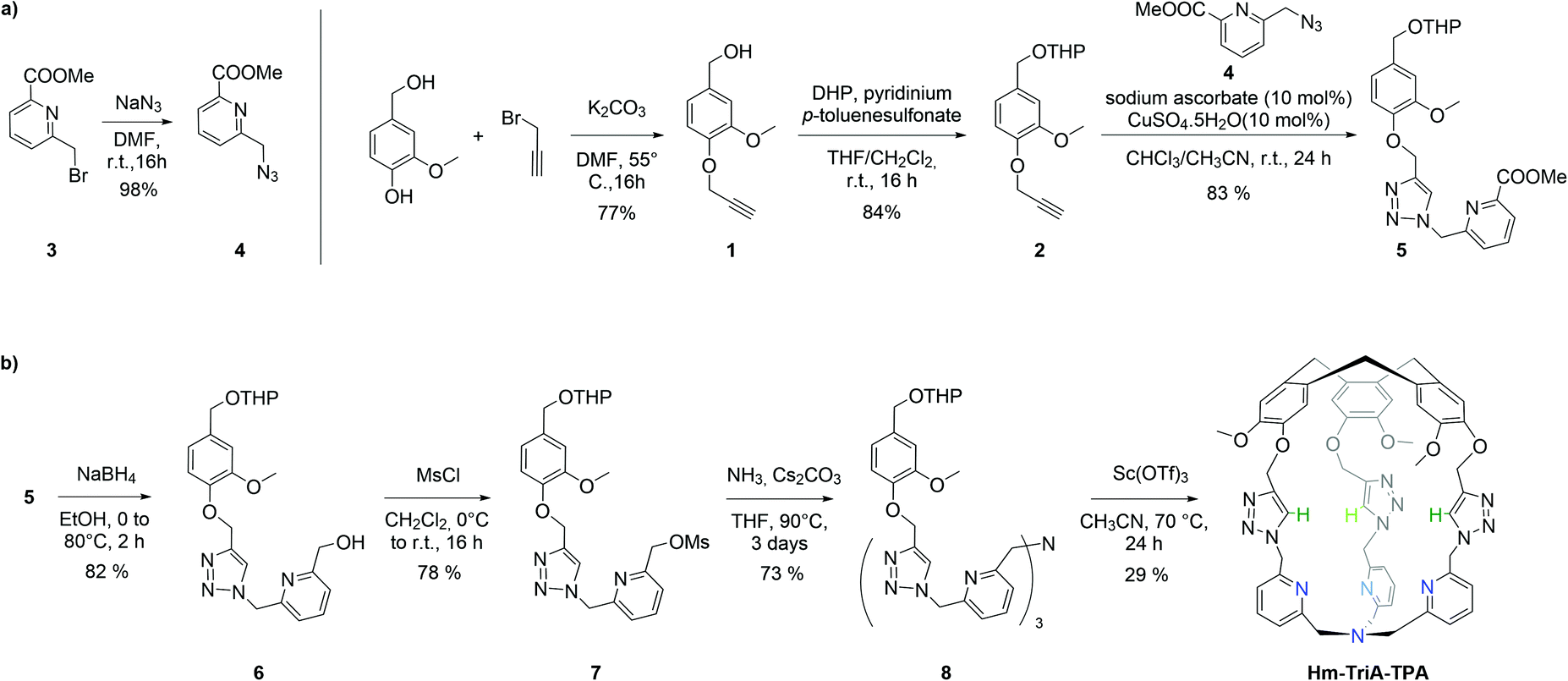
The 1H NMR spectrum of Hm-TriA-TPA shows that it has a C3 symmetrical structure on average in CDCl3 at 298 K (Fig. 1a). Identical, sharp and well-defined signals could be observed for the protons belonging to the northern CTV unit (Hh, Hi, Hj and Hk,k′), the –CH2– links (He,e′ and Hg,g′), the Ctriazole–H bonds (Hf) and the southern TPA (Ha,a′, Hb, Hc and Hd). 2D-NMR experiments (see the ESI†) were used to assign these resonances. Slow diffusion of Et2O into a CH2Cl2 solution of Hm-TriA-TPA afforded single crystals suitable for X-ray diffraction. The XRD structure of Hm-TriA-TPA confirms the endohedral functionalization of the TPA unit by the bowl-shaped CTV via the three triazole bridges having their Ctriazole–H bonds pointing toward the inside of the cavity (Fig. 1b). It should be noted that in the X-ray structure of the cage, a pyridine unit of the TPA resides inside the cavity. This C1 symmetrical conformation observed in the solid state contrasts with the symmetrical 1H NMR spectrum of Hm-TriA-TPA in solution. Fast conformational exchanges between in–out orientation of the pyridines could explain this behavior in solution at 298 K.
 | ||
| Fig. 1 (a) 1H NMR spectra (CDCl3, 400 MHz) of Hm-TriA-TPA along with (b) view of its X-ray crystal structure. Only the hydrogen atoms belonging to the three triazole units have been included for clarity. | ||
We have then investigated the ability of our caged ligand to form metallo-complexes in solution via coordination at its TPA unit.
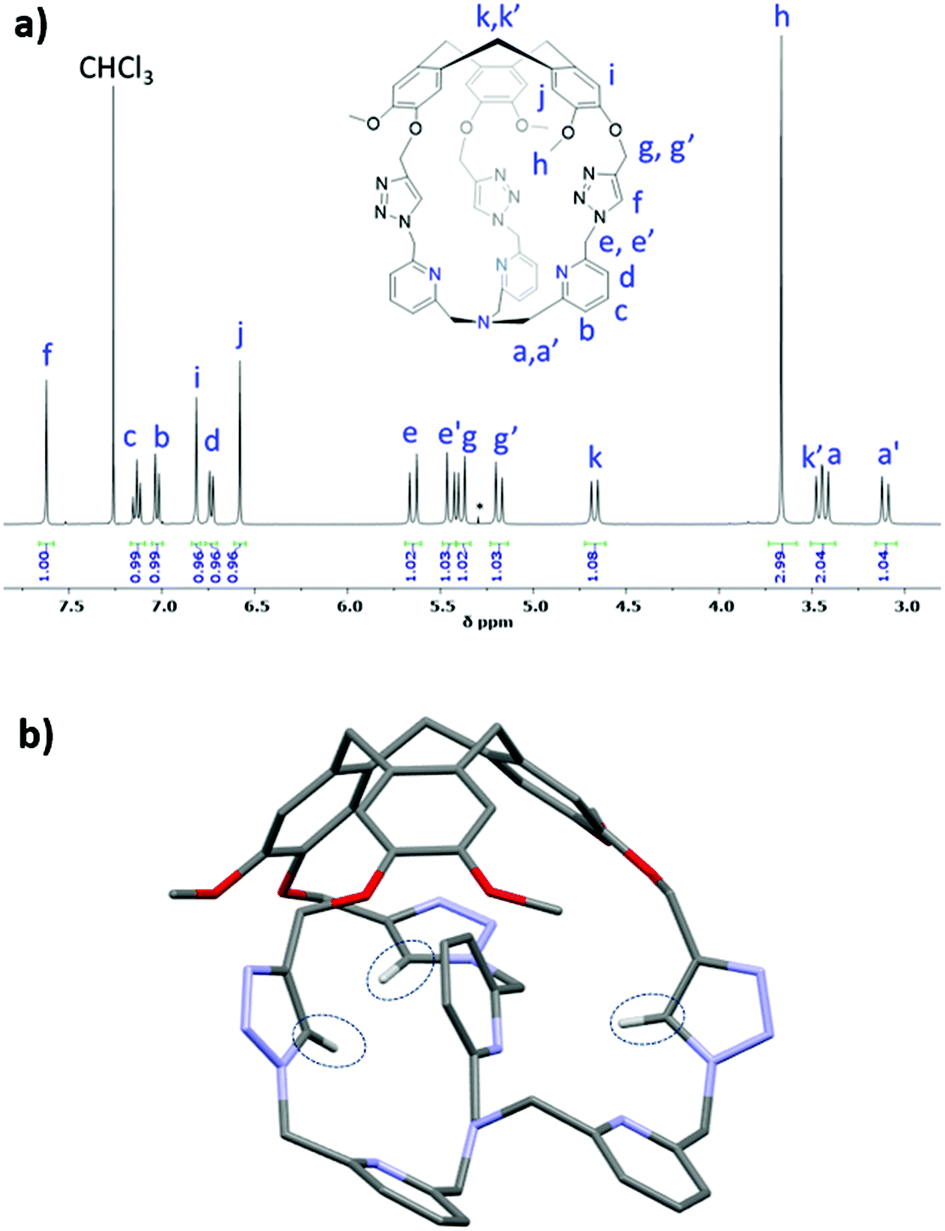
Binding of the air-stable and diamagnetic zinc triflate salt ZnII(OTf)2 was monitored by 1H-NMR in CD3CN at 298 K (Fig. 2 and Fig. S6, ESI†). The 1H-NMR spectra of Hm-TriA-TPA in the presence of 0.5 equiv. of the zinc salt revealed two sets of signals for each proton of the cage, which could be attributed to the presence of ZnII(Hm-TriA-TPA)(OTf)2 and Hm-TriA-TPA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. 2b). Interestingly, in the presence of a stoichiometric amount of the metal salt, the resonances belonging to the free cage fully disappear to the profit of the zinc complex signals, indicating full complexation. Upon metalation, a strong down-field shift occurs for the protons of the TPA's pyridines (Hb, Hc and Hd, Δppm: 0.35–0.7 ppm) that remain equivalent. The triazole bridges appear less affected with a modest upfield-shift observed for the Ctriazole–H bond (Hf, Δppm < 0.03 ppm). Overall, the 1H-NMR analysis of ZnII(Hm-TriA-TPA)(OTf)2 attests to the retention of the C3 symmetry of the caged ligand (on average) with identical and sharp signals for every resonance. Altogether, these observations unambiguously confirm the coordination of the ZnII metal ion at the TPA unit with the retention of the endohedral functionalization of the resulting complex.12 This was further supported by the optimized DFT (Density Functional Theory) structure that clearly reveals a C3 symmetrical caged Zn(II) complex in a trigonal bipyramidal geometry with an apical molecule of acetonitrile (Fig. 2e, Fig. S7, ESI†). Finally, identical spectra were observed upon addition of a second equivalent of ZnII(OTf)2 (Fig. 2d), ruling out the possibility of a second metal-binding event occurring at the triazole crown.15
:1 ratio (Fig. 2b). Interestingly, in the presence of a stoichiometric amount of the metal salt, the resonances belonging to the free cage fully disappear to the profit of the zinc complex signals, indicating full complexation. Upon metalation, a strong down-field shift occurs for the protons of the TPA's pyridines (Hb, Hc and Hd, Δppm: 0.35–0.7 ppm) that remain equivalent. The triazole bridges appear less affected with a modest upfield-shift observed for the Ctriazole–H bond (Hf, Δppm < 0.03 ppm). Overall, the 1H-NMR analysis of ZnII(Hm-TriA-TPA)(OTf)2 attests to the retention of the C3 symmetry of the caged ligand (on average) with identical and sharp signals for every resonance. Altogether, these observations unambiguously confirm the coordination of the ZnII metal ion at the TPA unit with the retention of the endohedral functionalization of the resulting complex.12 This was further supported by the optimized DFT (Density Functional Theory) structure that clearly reveals a C3 symmetrical caged Zn(II) complex in a trigonal bipyramidal geometry with an apical molecule of acetonitrile (Fig. 2e, Fig. S7, ESI†). Finally, identical spectra were observed upon addition of a second equivalent of ZnII(OTf)2 (Fig. 2d), ruling out the possibility of a second metal-binding event occurring at the triazole crown.15
 | ||
| Fig. 2 Monitoring of the 1H NMR spectra (CD3CN, 400 MHz) of Hm-TriA-TPA (a), upon addition of 0.5 equiv. (b), 1.0 equiv. (c), and 2.0 equiv. (d) of Zn(OTf)2 (depicted region: 6.7–8.1 ppm). (e) Views of the DFT-optimized structure (PBE0-D3/def2-TZVP, COSMO, see the ESI† for full computational details) of ZnII(Hm-TriA-TPA)(OTf)2. | ||
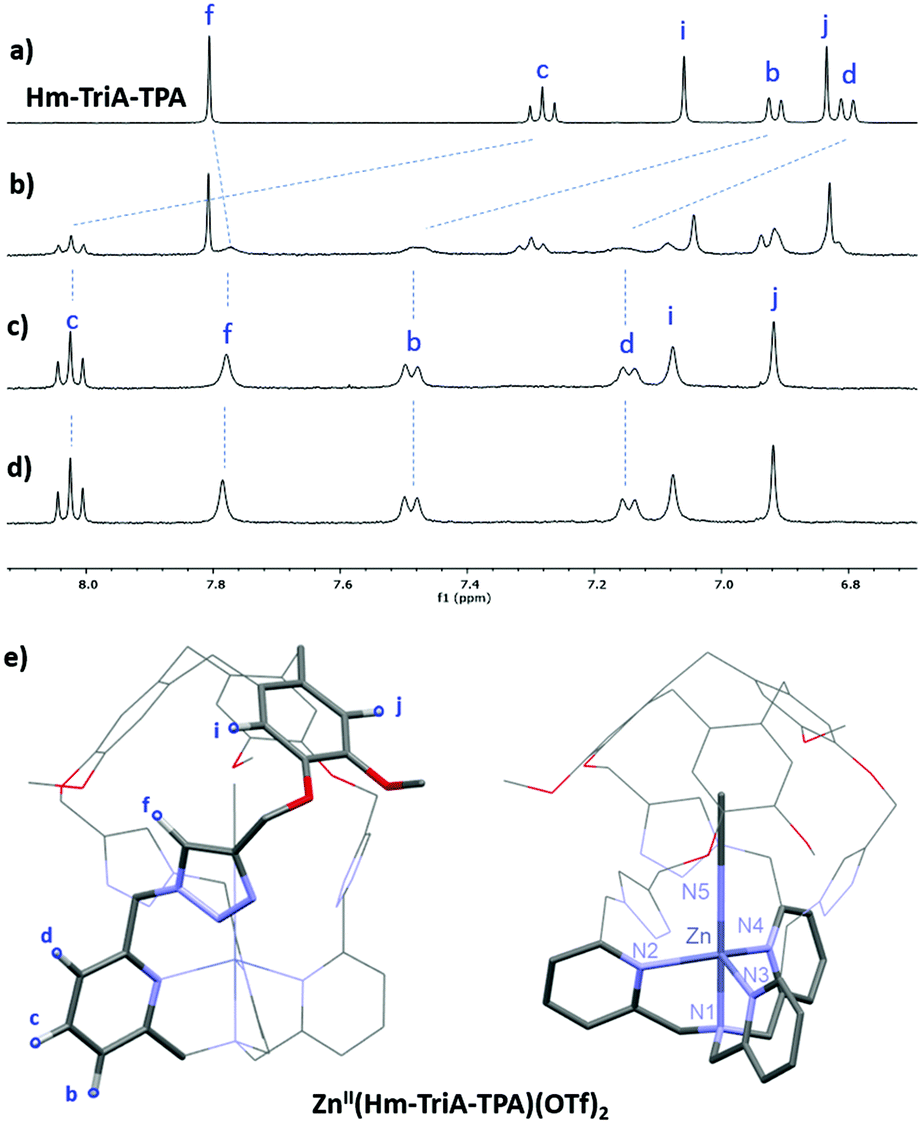
We next investigated the possibility of preparing CuII complexes with our caged ligand. The CuII(Hm-TriA-TPA)(OTf)2 complex was prepared by reacting the ligand with a stoichiometric amount of CuII(OTf)2 in CH3CN, at room temperature (see the ESI†). The formation of the targeted complex, in a 1:1 stoichiometry, was evidenced by High-Resolution Mass Spectrometry analysis (ESI-HRMS, Fig. S8, ESI†). Furthermore, crystallization by slow diffusion of diethyl ether into a CH3CN solution of the complex affords single crystals suitable for X-ray diffraction, allowing for the determination of its solid-state structure. The structure of the CuII(Hm-TriA-TPA)(OTf)2 complex shows a pentacoordinated copper center with a distorted square pyramidal geometry (Fig. 3a). The distorted tetragonal plane comprises the tertiary amine (N1), two pyridines (N2 and N3), and one triazole (N5) that is trans to the tertiary amine, with a similar Cu–N bond length ranging from 1.978 Å to 2.055 Å. The coordination sphere is completed by an apical pyridine (N4) with a Cu–N distance of 2.225 Å. Importantly, a well-defined cavity, described by the northern CTV unit and the tris-triazole crown, could be observed just above the CuII center, confirming its endohedral functionalization. The latter is occupied by a non-bonded guest molecule of acetonitrile (solvent).
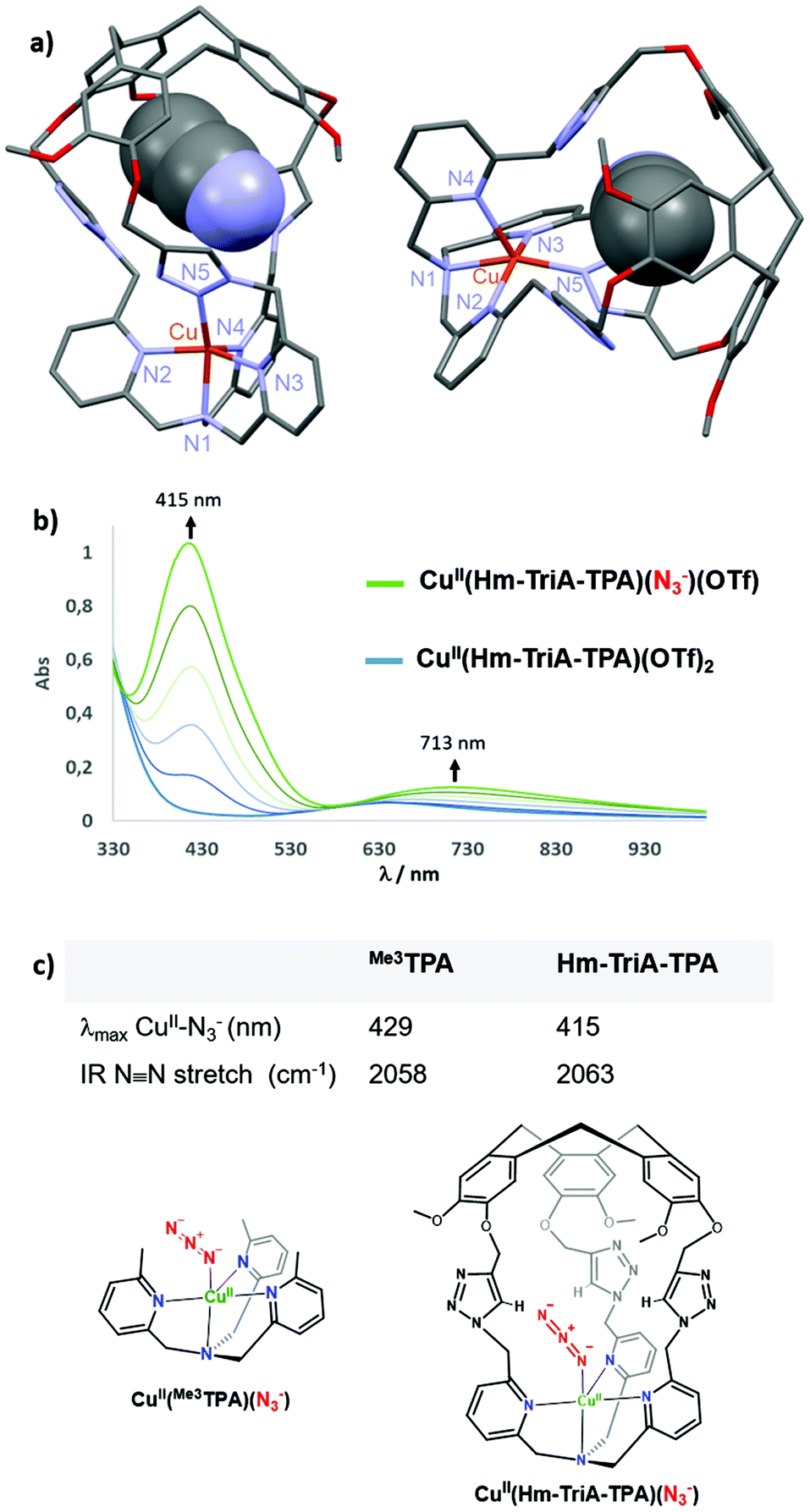 | ||
| Fig. 3 (a) Views of the X-ray crystal structure of CuII(Hm-TriA-TPA)(OTf)2. (b) UV-vis monitoring of the formation of the azido adduct CuII(Hm-TriA-TPA)(N3)(OTf) upon addition of NBu4N3 (0 to 1.0 equiv.) to a 0.5 mM solution of CuII(Hm-TriA-TPA)(OTf)2 in acetone. (c) Spectroscopic features (UV-vis and IR) of the azido complexes stabilized by Me3TPA and Hm-TriA-TPA. | ||
Could the Cu–triazole bond in CuII(Hm-TriA-TPA)(OTf)2 be replaced by anion coordination at the CuII-TPA core?
Anion binding at Cu-TPA derivatives has been reported to result in trigonal–bipyramidal Cu(II) complexes with axial binding of the anion trans to the tertiary amine.16 Among them, azido adducts [(CuII-TPAX)(N3−)]+ have been described as electronic and structural analogues of cupric superoxide intermediates [(CuII-TPAX)(O2˙−)]+. The N3− anion binds to CuII-TPA complexes with very similar bond lengths and angles (CuII–N–N) to those found in [(CuII-TPAX)(O2˙−)]+.7 Azido complexes [(CuII-TPAX)(N3−)]+ have been extensively studied by electronic and vibrational spectroscopies.2,11 They display typical azido → CuII LMCT transitions and ν(N–N) stretching frequency. On this basis, we explored the possibility of binding N3− to our caged Cu(II) complex. UV-vis analysis of CuII(Hm-TriA-TPA)(OTf)2, in acetone, revealed a typical d–d transition centered at 634 nm (0.5 mM, ε = 139 M−1 cm−1, Fig. S9, ESI†). The UV-vis monitored addition of tetrabutylammonium azide NBu4N3 to a 0.5 mM solution of the caged complex, in acetone, results in the appearance of an intense absorption band centered at 415 nm (LMCT, ε = 2068 M−1 cm−1) and a d–d band at 713 nm (ε = 249 M−1 cm−1, Fig. 3b).
The final spectrum was identical to that of the azido adduct prepared using NaN3 and isolated by precipitation (see the ESI†). These UV-vis data are consistent with the formation of the azido complex CuII(Hm-TriA-TPA)(N3)(OTf).2,7,11 In TPA-complexes bearing intramolecular H-bonding donors, the potential presence of stabilizing interactions with the azido moiety has been associated with a blue-shift of the LMCT band, as well as a blue-shift of the ν(N–N) stretching frequency.2,17 The value of the LMCT band observed in the case of Hm-TriA-TPA was therefore compared with that of its corresponding open model ligand Me3TPA devoid of the intramolecular H-bonding group (Fig. 3c). Compared to the open TPA-based complex CuII(Me3TPA)(N3)(OTf) (λmax = 429 nm, ε = 2072 M−1 cm−1, Fig. S10, ESI†), the CuII–N3− LMCT bands in our triazole-containing cage shift to a higher energy (λmax = 415 nm).
To further support the stabilization offered by Hm-TriA-TPA, the two azido complexes have been isolated (see the ESI†) and their antisymmetric N3− IR stretch was compared. Interestingly, CuII(Hm-TriA-TPA)(N3)(OTf) displays a 5 cm−1 blue-shift of the ν(N–N) stretching frequency compared to the “base” ligand Me3TPA (Fig. 3c, Fig. S11 and S12, ESI†). These findings are therefore consistent with a bonding stabilization of the azido adduct within the triazole-containing cavity.
Finally, the electronic influence of our caged ligand was investigated by comparing the CuII/I redox couple of CuII(Hm-TriA-TPA)(OTf)2 with it parent complex CuII(Me3TPA)(OTf)2 (Fig. S13 and S14, ESI†). The caged complex displays a quasi-reversible CuII/I redox couple at −0.112 V vs. Fc (CH3CN; 0.15 M NBu4PF6). This value is slightly shifted to a more negative potential relative to CuII(Me3TPA)(OTf)2 which displays a quasi-reversible CuII/I redox couple at −0.021 V vs. Fc, under identical conditions. This electrochemical shift is consistent with an easier CuI to CuII oxidation in the case of the Hm-TriA-TPA ligand. However, the modest magnitude of the shift (<0.1 V) reveals a moderate influence of the triazole-based cage, compared to its corresponding “base” ligand.
Conclusions
In summary, the preparation of an organic cage where the canonical TPA ligand is surmounted by a H-bonding hydrophobic cavity offering three triazole units is described. We demonstrate that this cage can coordinate zinc(II) and copper(II) metal ions at its TPA unit with an endo-functionalization of the complex, both in the solution and solid states. These are the first examples of bioinspired complexes with a tris-triazole decorated cavity mimicking the functionalized (H-bonding) hydrophobic channels of metalloenzymes. Finally, we found that the azidocopper(II) adduct [(L)CuII–N3−] can be prepared upon addition of N3 to the caged Cu(II) complex. Spectroscopic analysis of this structural analogue of the cupric superoxide intermediates [(L)CuII–O2˙−] suggests the stabilization of the azido adduct within the Ctriazole–H based cavity.We envision that our strategy might find application toward the development of non-enzymatic catalysts able to stabilize reactive intermediates and/or control substrate positioning by their H-bonding hydrophobic cavity. Future work will focus on the use of TPA ligands equipped with our triazole-functionalized cavity to generate, stabilize, and explore the reactivity of end-on superoxocopper(II) complexes upon dioxygen activation.
Author contributions
G. Qiu and D. Diao contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. Wojdyla and T. Borowski, Chem. – Eur. J., 2022, 28, e202104106 CAS.
- M. A. Ehudin, A. W. Schaefer, S. M. Adam, D. A. Quist, D. E. Diaz, J. A. Tang, E. I. Solomon and K. D. Karlin, Chem. Sci., 2019, 10, 2893–2905 RSC.
- S. Y. Quek, S. Debnath, S. Laxmi, M. van Gastel, T. Krämer and J. England, J. Am. Chem. Soc., 2021, 143, 19731–19747 CrossRef CAS PubMed.
- M. Borrell, E. Andris, R. Navrátil, J. Roithová and M. Costas, Nat. Commun., 2019, 10, 901 CrossRef PubMed.
- A. Wada, M. Harata, K. Hasegawa, K. Jitsukawa, H. Masuda, M. Mukai, T. Kitagawa and H. Einaga, Angew. Chem., Int. Ed., 1998, 37, 798–799 CrossRef CAS PubMed.
- E. W. Dahl, H. T. Dong and N. K. Szymczak, Chem. Commun., 2018, 54, 892–895 RSC.
- (a) M. Bhadra, J. Yoon, C. Lee, R. E. Cowley, S. Kim, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2018, 140, 9042–9045 CrossRef CAS PubMed; (b) D. E. Diaz, D. A. Quist, A. E. Herzog, A. W. Schaefer, I. Kipouros, M. Bhadra, E. I. Solomon and K. D. Karlin, Angew. Chem., Int. Ed., 2019, 58, 17572–17576 CrossRef CAS PubMed.
- (a) C. Colomban, V. Martin- Diaconescu, T. Parella, S. Goeb, C. García-Simón, J. Lloret- Fillol, M. Costas and X. Ribas, Inorg. Chem., 2018, 57, 3529–3539S CrossRef CAS PubMed; (b) S. C. Bete, C. Würtele and M. Otte, Chem. Commun., 2019, 55, 4427–4430 RSC; (c) C. Bete and M. Otte, Angew. Chem., Int. Ed., 2021, 60, 18582–18586 CrossRef PubMed.
- C. Bravin, E. Badetti, G. Licini and C. Zonta, Coord. Chem. Rev., 2021, 427, 213558 CrossRef CAS.
- N. Le Poul, B. Colasson, G. Thiabaud, D. Jeanne Dit Fouque, C. Iacobucci, A. Memboeuf, B. Douziech, J. Řezáč, T. Prangé, A. de la Lande, O. Reinaud and Y. Le Mest, Chem. Sci., 2018, 9, 8282–8290 RSC.
- T. Zhang, L. Le Corre, O. Reinaud and B. Colasson, Chem. – Eur. J., 2021, 27, 434–443 CrossRef CAS PubMed.
- G. Qiu, C. Colomban, N. Vanthuyne, M. Giorgi and A. Martinez, Chem. Commun., 2019, 55, 14158–14161 RSC.
- S. A. Ikbal, C. Colomban, D. Zhang, M. Delecluse, T. Brotin, V. Dufaud, J. P. Dutasta, A. B. Sorokin and A. Martinez, Inorg. Chem., 2019, 58, 7220–7228 CrossRef CAS PubMed.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CAS.
- Identical 1H-NMR spectra were obtain after the isolation of the Zn complex by precipitation with Et2O (Fig. S6, ESI†).
- (a) W. T. Eckenhoff and T. Pintauer, Inorg. Chem., 2007, 46, 5844–5846 CrossRef CAS PubMed; (b) C. M. Moore and N. K. Szymczak, Chem. Commun., 2015, 51, 5490–5492 RSC; (c) E. W. Dahl, H. T. Dong and N. K. Szymczak, Chem. Commun., 2018, 54, 892–895 RSC.
- A. Wada, Y. Honda, S. Yamaguchi, S. Nagatomo, T. Kitagawa, K. Jitsukawa and H. Masuda, Inorg. Chem., 2004, 43, 5725–5735 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure and spectral data. CCDC 2153939 and 2154000. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00607c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |