
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging degrader technologies engaging lysosomal pathways
Yu
Ding
 *a,
Dong
Xing
*b,
Yiyan
Fei
*c and
Boxun
Lu
*a
*a,
Dong
Xing
*b,
Yiyan
Fei
*c and
Boxun
Lu
*a
aNeurology Department at Huashan Hospital, State Key Laboratory of Medical Neurobiology and MOE Frontiers Center for Brain Science, School of Life Sciences, Fudan University, Shanghai, China. E-mail: luboxun@fudan.edu.cn; yuding@fudan.edu.cn
bShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai, China. E-mail: dxing@sat.ecnu.edu.cn
cDepartment of Optical Science and Engineering, Shanghai Engineering Research Center of Ultra-Precision Optical Manufacturing, Key Laboratory of Micro and Nano Photonic Structures (Ministry of Education), Fudan University, Shanghai, China. E-mail: fyy@fudan.edu.cn
First published on 11th October 2022
Abstract
Targeted protein degradation (TPD) provides unprecedented opportunities for drug discovery. While the proteolysis-targeting chimera (PROTAC) technology has already entered clinical trials and changed the landscape of small-molecule drugs, new degrader technologies harnessing alternative degradation machineries, especially lysosomal pathways, have emerged and broadened the spectrum of degradable targets. We have recently proposed the concept of autophagy-tethering compounds (ATTECs) that hijack the autophagy protein microtubule-associated protein 1A/1B light chain 3 (LC3) for targeted degradation. Other groups also reported degrader technologies engaging lysosomal pathways through different mechanisms including AUTACs, AUTOTACs, LYTACs and MoDE-As. In this review, we analyse and discuss ATTECs along with other lysosomal-relevant degrader technologies. Finally, we will briefly summarize the current status of these degrader technologies and envision possible future studies.
 Yu Ding | Yu Ding received his BS and PhD degrees in biophysics from Fudan University, China, in 1999 and 2004. From 2004–2022, he worked at Fudan University as a lecturer, associate professor, and professor. From 2009–2011, he moved to the University of Hong Kong as a research associate and research assistant professor. His current research focuses on the structure–function relationship of pathological targets, autophagy-specific proteins, and ATTEC molecules. |
 Dong Xing | Dong Xing received his BS and MS degrees from East China Normal University, China, in 2003 and his PhD from the University of Hong Kong with Professor Dan Yang in 2011. He then joined East China Normal University as an assistant professor. He was promoted to associate professor in 2015. In 2016, he began his postdoctoral research with Professor Guangbin Dong at the University of Texas at Austin and then moved to the University of Chicago with the Dong group. In 2018, he returned to East China Normal University, where his current research is focused on the diversity-oriented synthesis and medicinal chemistry. |
 Yiyan Fei | Yiyan Fei received her PhD degree in optics from the Chinese Academia of Science, China, in 2006. From 2006 to 2012, she was a post-doc at the University of California at Davis, Davis, USA. Since 2012, she has been working in the Department of Optical Science and Engineering, Fudan University, Shanghai, China. Her research focuses on the development of high-throughput biomolecular interaction detection platform and its wide applications. |
 Boxun Lu | Dr Boxun Lu is currently a professor at Fudan University, China. He has been working on Huntington's disease and other neurodegenerative disorders with a focus on degrading the pathogenic proteins for potential therapeutic treatment for these diseases. He proposed the original concept of ATTEC and worked with key collaborators to lead the studies of ATTECs targeting polyQ proteins and lipid droplets. The team is currently expanding the target spectrum of ATTECs and inventing novel protein/organelle manipulating strategies. |
1. Introduction
The traditional inhibitor approach of drug discovery is limited by the target protein's “druggability”: measurable biochemical functions and amenable binding sites, the occupancy of which directly or indirectly influences such functions.1 This limited landscape of druggable targets has been dramatically widened by targeted protein degradation (TPD) strategies, which hijack endogenous degradation pathways to eliminate a target protein rather than merely inhibiting its function.2 With two drug candidates advancing through phase II clinical trials, PROteolysis TArgeting Chimeras (PROTACs) are currently the prevailing TPD approach and have been extensively reviewed in the literature.3 A PROTAC is a synthetic heterobifunctional molecule that links two separate chemical moieties binding, respectively, to a target protein and an E3 ligase or E3 ligase complex.3,4 As such, PROTACs bring the E3 ligase to the target protein in a transient ternary complex that leads to polyubiquitination (polyUb) and subsequent proteasomal degradation of the target protein.5More recently, new TPD strategies have been developed to hijack the lysosomal degradation pathway, the major degradation pathway independent of the proteasome.6 For example, the technologies harnessing autophagy (autophagosome–lysosomal pathway) have been developed, including the autophagy-targeting chimeras (AUTACs) inducing degradation through the selective autophagy pathway,7 the AUTOphagy-TArgeting Chimeras (AUTOTACs) directly engaging SQSTM1/p62,8 and the autophagy-tethering compounds (ATTECs) directly hijacking the macroautophagy pathway to degrade both intracellular proteins and non-protein entities.9,10 The technologies hijacking the endosomal–lysosomal pathway are also emerging, including lysosome targeting chimeras (LYTACs)11,12 and molecular degraders of extracellular proteins through the asialoglycoprotein receptor ASGPR (MoDE-As) degrading membrane or extracellular proteins.13 These novel therapeutic modalities, in addition to PROTACs, provide exciting opportunities to transform small molecule drug discovery beyond the traditional inhibition or antagonism approach. Meanwhile, these nascent technologies require significant additional work to develop their capability and establish them as a platform technology. This review will focus on how these technologies may work and continue to evolve.
2. Degrader technologies engaging autophagy
2.1 Autophagy
Autophagy is a cellular degradation machinery that is highly conserved in all eukaryotes, from yeast to humans.14,15 In mammalian cells, there are three primary types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), all of which deliver cargos into the lysosome for degradation.15 Macroautophagy has the best-characterized and most universal mechanism among these types of autophagy.16 The primary feature distinguishing macroautophagy (autophagy hereafter) from microautophagy and CMA is the formation of double-membrane vesicles named autophagosomes, which engulf different types of cargos such as biomolecules, damaged organelles and protein aggregates, and then deliver them to lysosomes.16Autophagy occurs at a low baseline level constitutively as a quality control machinery and can be further induced under stress conditions, such as nutrient or energy starvation.17 This allows the cells to degrade intracellular materials into metabolites that can be recycled in biosynthetic processes or energy production required for cell survival.17 Because of its critical cellular functions, autophagy plays many essential roles in various physiological and pathophysiological processes. Relevant to targeted degradation technologies, one appealing feature of autophagy is its capability of degrading a wide variety of substrates, including proteins, protein aggregates, DNA/RNA molecules, peroxisomes, ribosomes, lipid droplets, glycogen, damaged mitochondria and microbial pathogens.18,19 This feature provides unprecedented potential for autophagy-hijacking degradation strategies.
Autophagy is a catabolic “self-eating” process that has a series of steps, including initiation of the phagophore, nucleation and expansion of the phagophore, closure and completion of a double-membrane autophagosome that surrounds a portion of the cytoplasm, fusion with lysosomes, and degradation of contents in the autophagosome (Fig. 1).16 Initiation involves the assembly of a protein complex on a small crescent-shaped membrane structure called isolation membrane, which is the origin of autophagosomes (Fig. 1). The initiation induces the formation of the phagophore, which is a protein-bound membrane structure that then nucleates and expands to sequester cargos (Fig. 1). The phagophore then eventually forms the autophagosome after the closure of vesicle membranes (Fig. 1). The autophagosome is a double-membrane structure. Its outer membrane fuses with the lysosome, a low pH single membrane vesicle containing various hydrolases with degradation capability (Fig. 1).20 The inner membrane of the autophagosome is then broken down,21 delivering the cargos to the newly formed autolysosome for degradation (Fig. 1). The breakdown products could be recycled after the release from permeases (proteins that mediate the transport of various molecules across biological membranes) present in the lysosome/autolysosome membrane. All these steps are initiated and controlled by different proteins/protein complexes, which will be discussed in the following text.
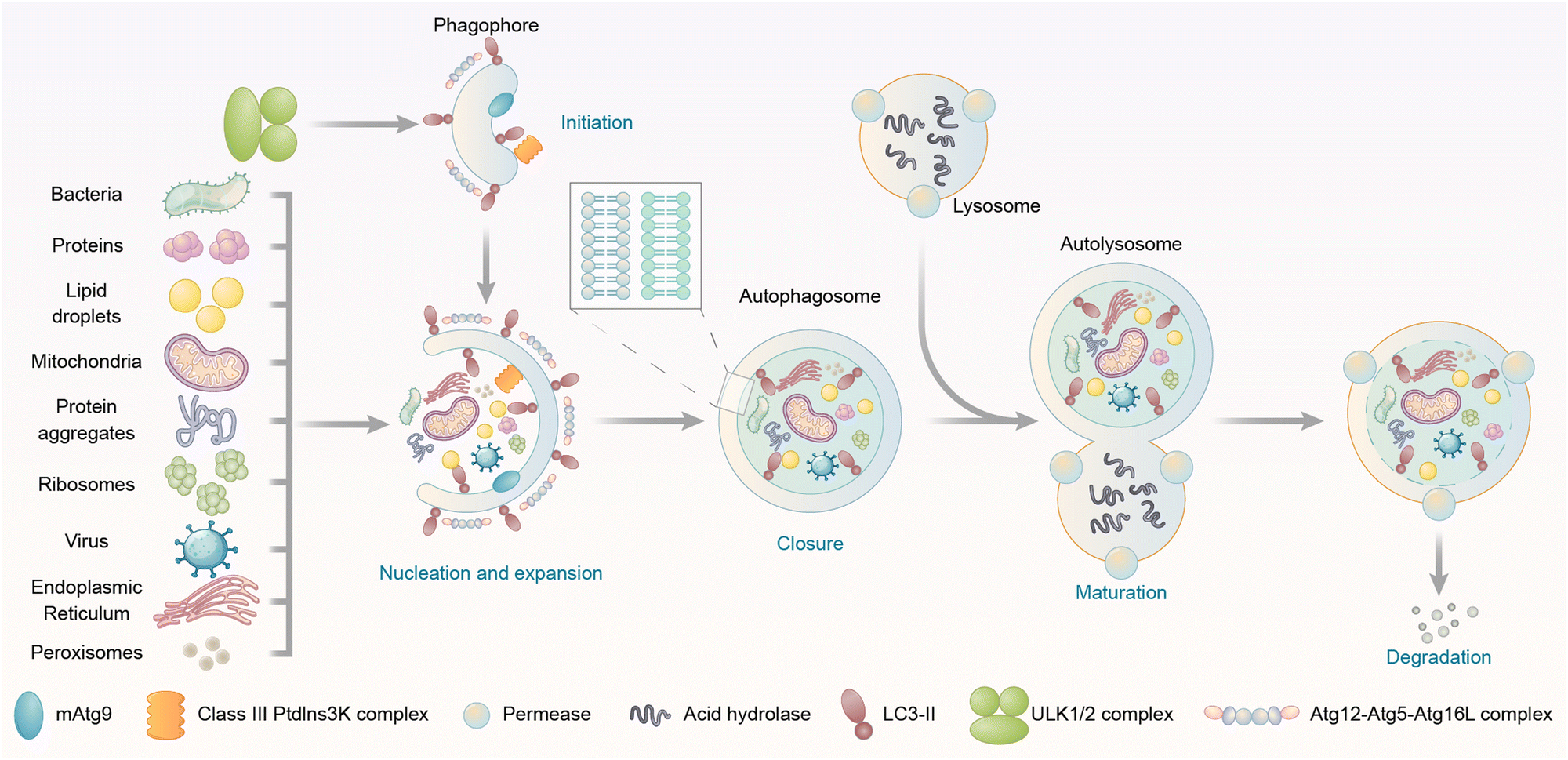 | ||
| Fig. 1 Steps and cargo spectrum of autophagy. Autophagy allows the eukaryotic cells to degrade intracellular materials including proteins, protein aggregates, lipid droplets, DNA/RNA molecules, ribosomes, peroxisomes, glycogen, damaged mitochondria, endoplasmic reticulum, and pathogens such as bacteria and viruses. Autophagy starts with the initiation of the phagophore, followed by the nucleation and expansion of the phagophore, which tethers and engulfs different types of cargos for degradation. The phagophore then undergoes closure and completion of a double-membrane autophagosome, fusion with lysosomes, and degradation of contents in the autophagosome. | ||
2.2 Key autophagosome proteins
The autophagy machinery is orchestrated by a series of biochemical activities involving a set of highly conserved autophagy-related (ATG) proteins hierarchically (Fig. 2). During autophagy, the cargos are engulfed in the autophagosomes, and they may enter the autophagic degradation pathway only during autophagosome formation. Thus, we focus on the molecular mechanism of autophagosome formation and analyse possible proteins that could be utilized as the tethering anchor to enter autophagosomes.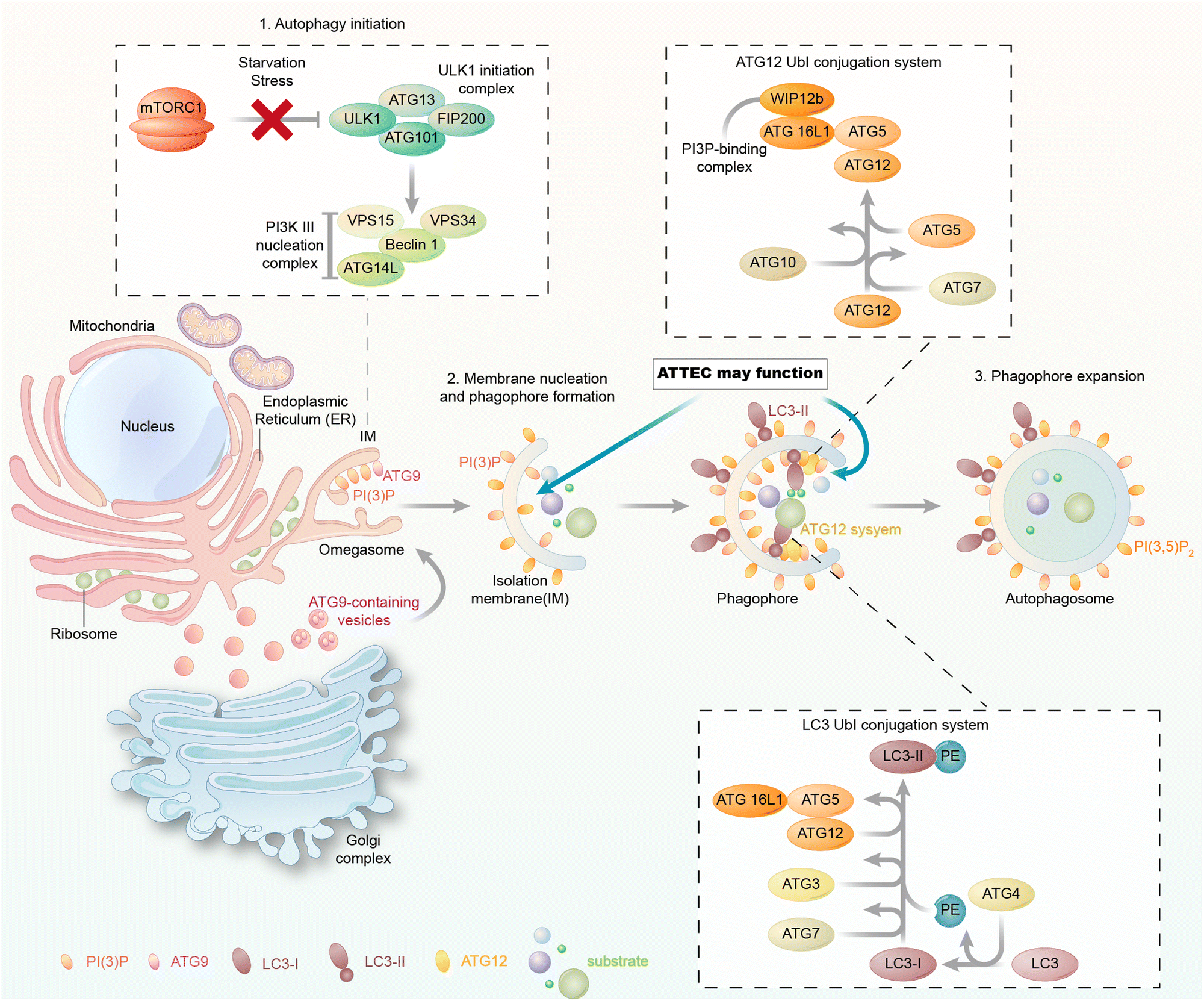 | ||
| Fig. 2 The molecular mechanism of autophagosome formation. The autophagosome formation steps include (1) autophagy initiation, (2) membrane nucleation and phagophore formation and (3) phagophore expansion. Each of these steps involves a group of proteins/protein complexes as indicated in the figure. The membrane-bound autophagy proteins could be potentially used as tethering anchors for targets of interest. Meanwhile, LC3 is involved in all these steps and bound to the phagophore/autophagosome membranes. | ||
In yeast, the autophagosome initiation is mediated by the Atg1:Atg13:Atg17 complex.22 The mammalian counterpart of this complex is the ULK kinase complex, composed of ULK1 or ULK2, FIP200, ATG13, and ATG101.23–25 Inactivation of mTOR by starvation or rapamycin treatment activates ULKs and results in the phosphorylation of ATG13 and FIP200. ATG101 localizes to the phagophore and stabilizes the expression of ATG13.26,27 Subsequently, the ULK complex phosphorylates and activates the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, which is composed of BECN1, VPS34, VPS15, and ATG14.28,29 This leads to the synthesis of phosphatidylinositol-3-phosphate (PtdIns3P, PI(3)P) for the nucleation of the phagophore. The membrane required for phagophore formation is provided by the ATG9 trafficking system, which forms ATG9 vesicles as seeds for membrane formation.30 The following membrane expansion step is mediated by two ubiquitin-like (Ubl) conjugation systems, the ATG12 and LC3 conjugation systems (or the Atg12 and ATG8 conjugation systems in yeast) (Fig. 2).31 ATG12 is conjugated to ATG5 via the action of E1 and E2 enzymes ATG7 and ATG10 and subsequently binds to ATG16L1. The ATG12–ATG5–ATG16L1 complex is assembled and acts like an E3 enzyme in the LC3 conjugation system, which will be discussed below.
2.3 LC3 and its conjugation systems
LC3 is the mammalian homolog of yeast ATG8,32 and it has three different isoforms: LC3A, LC3B, and LC3C.33 All three isoforms were initially characterized as microtubule-associated protein 1A/B light chain 3 (MAP1LC3),33 but were later found to play a conserved role in autophagy.34 Besides LC3, three γ-aminobutyric acid receptor-associated proteins (GABARAPs) are also mammalian homologs of yeast ATG8.35,36 These ATG8 family proteins have been proposed to mediate the expansion and closure of autophagosome membranes,37–43 although their exact functions during autophagosome biogenesis remain to be further elucidated. ATG8 family proteins are possibly dispensable in autophagosome formation, but ATG8-independent autophagosomes may exhibit significant defects.44,45During phagophore expansion, LC3 (and possibly other ATG8 family proteins) is directly conjugated to lipid phosphatidylethanolamine (PE) in the phagophore membrane via the LC3 conjugation system.32,46 Attachment of PE requires the cleavage of LC3 by the protease ATG4, the E1 enzyme ATG7, the E2 enzyme ATG3, and the E3 enzyme ATG12–ATG5–ATG16L1 complex. The PE-conjugated LC3 is called LC3-II. After the completion of the expansion of the phagophore and sequestration of substrates, autophagosome formation starts.
Noticeably, LC3 proteins remain bound to the phagophore and autophagosome membranes throughout the pathway by covalent conjugation with PE and thus are the most widely used markers of autophagosome biogenesis and trafficking.6 Most of the other ATG proteins/protein complexes function catalytically, and many of them do not stay on the phagophore or autophagosome inner membrane. Protein targets tethered to these ATG proteins may enter the autophagy machinery less efficiently due to their detachment from the autophagic membranes. ATG5 may also tether the phagophore or autophagosome membrane,47 but the binding is not covalent, and ATG5 is in a protein complex, which may interfere with compound-binding. Thus, for autophagy-based degrader technologies, LC3 may function better than other ATG proteins as the tethering anchor providing an entrance into the autophagy machinery.
2.4 Autophagy receptors
Besides LC3, the autophagy receptor provides another type of possible docking site for entering the autophagy pathway. The autophagy receptors play a crucial role in selective autophagy, a subtype of macroautophagy. Both selective autophagy and nonselective (also known as bulk) autophagy utilize autophagosomes to deliver cargos to the lysosomes for degradation. Meanwhile, selective autophagy is characterized by high specificity in cargo recognition, whereas nonselective autophagy is thought to engulf cytoplasmic contents randomly and may lack cargo specificity.48 Cargos of selective autophagy include many different types of biomolecules and organelles, such as ubiquitinated proteins, peroxisomes, and damaged mitochondria.49 Selective autophagy relies on autophagy receptors that recognize cargos and tether them to the phagophore.50 The cargo-bound receptors interact with LC3-II anchored in the phagophore membrane during autophagosome formation. The autophagy receptor–LC3 interaction is mediated by 15- to 20-amino-acid-long motifs called the LC3-interacting region (LIR, also called AIM, ATG8-interacting motif) and the LIR docking site (LDS) of LC3. Various autophagy receptors have been identified to recognize different cargo categories, and they have been summarized in review papers.50–52 For example, the ubiquitin-binding protein SQSTM1/p62 targets ubiquitinated protein aggregates and intracellular bacteria to autophagy for degradation by acting as an adaptor protein that interacts with LC3-II.53–55 Besides SQSTM1/p62–LC3 interaction, the cargo recruitment is also dependent on the liquid–liquid phase separation of the protein DAXX.56 NBR1 and OPTN are other receptors that have similar functions to SQSTM1/p62 in targeting ubiquitinated proteins or pathogens to autophagosomes.57,58In 2019, a novel class of LC3 binding autophagy receptors was discovered and named UIM (ubiquitin-interacting motif).59 This class of receptors engages non-canonical UIM-like sequences to ATG8. The discovery of UIM's specific binding to the LC3's UIM-docking site (UDS) expands the available autophagy receptors and adaptors.
Since these autophagy receptors function in recognizing cargos, they are suitable to serve as anchors for the targets to enter the autophagy machinery for degradation. Thus, the autophagy receptor-mediated TPD is worth considering. Meanwhile, since the autophagy receptors function through binding with LC3, tethering to these receptors is indirect and could be less efficient than tethering to LC3 directly. For example, the SQSTM1/p62-mediated TPD is influenced by both the compound–SQSTM1/p62 interaction and the SQSTM1/p62–LC3 interaction, whereas the compound–LC3 interaction only affects the LC3-mediated TPD. Taken together, LC3 is likely a more reliable protein to be engaged in developing novel autophagy-mediated TPD strategies, at least from the biological perspective. We will further analyse its ligandability to discuss the feasibility of engaging LC3 for TPD from a chemical perspective.
2.5 The ligandability of LC3
While the ligandability of E3 ligases targeted in PROTACs has been extensively studied and reviewed,60 the ATG8/LC3 family proteins’ ligandability has not been systematically investigated yet. In the literature, LC3 often stands for LC3B (microtubule associated protein 1 light chain 3β), a major member of the ATG8 family proteins. ATG8 family proteins are evolutionarily conserved, existing in all eukaryotic cells, including fungi, plants and animals. Fig. 3A shows the sequence alignment of Homo sapiens (Human) LC3B to Mus musculus (Mouse) LC3B, Danio rerio (Zebrafish) LC3B, Drosophila melanogaster (Fruit fly) ATG8B, Caenorhabditis elegans LGG1, Arabidopsis thaliana ATG8B (Mouse-ear cress), and Saccharomyces cerevisiae ATG8 (Baker's yeast).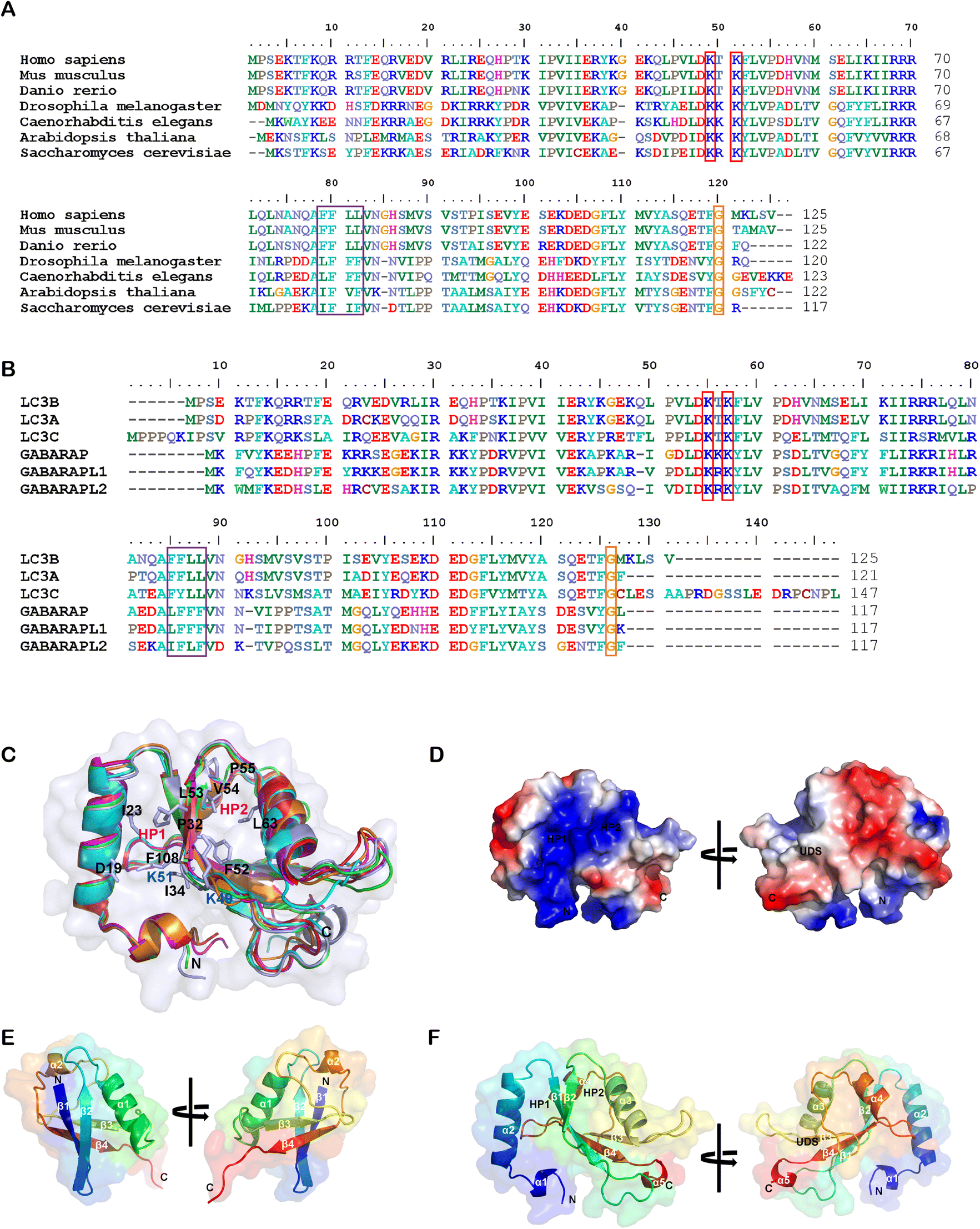 | ||
| Fig. 3 Alignment of the LC3/ATG8 family proteins’ sequences and structures. (A) The sequence alignment of LC3B from Homo sapiens (Uniprot: Q9GZQ8), Mus musculus (Q9CQV6), Danio rerio (Q7ZUD8), Drosophila melanogaster (Q9VEG5), Caenorhabditis elegans (Q09490), Arabidopsis thaliana (Q9XEB5) and Saccharomyces cerevisiae (P38182). Potential acetylation sites are shown in the red box, the UDS core sequence is shown in the purple box, and the glycine site for PE conjugation is shown in the orange box. (B) The sequence alignment of human LC3B (Q9GZQ8), LC3A (Q9H492), LC3C (Q9BXW4), GABARAP (O95166), GABARAPL1 (Q9H0R8) and GABARAPL2 (P60520). (C) Structural alignment of human LC3B (PDB: 6J04), LC3A (3WAL), LC3C (3VVW), GABARAP (1GNU), GABARAPL1 (2R2Q) and GABARAPL2 (4CO7) proteins shown in ribbon diagrams. The N terminus and C terminus are marked as N and C, respectively. The residues that compose HP1 (D19, I23, P32, I34, K51, L53 and F108), HP2 (F52, V54, P55 and L62), and potential acetylation sites (K49 and K51) are also labeled. (D) The electrostatic potential surfaces of human LC3B, the positively charged region is colored in blue and the negatively charged region in red. The regions of hydrophobic pocket 1, hydrophobic pocket 2 and the UIM-docking site are labelled as HP1, HP2 and UDS. (E & F) Comparison of the structure of E. ubiquitin (1CMX) and F. LC3B (6J04). The peptide chain is a rainbow color from N-terminus to C-terminus (blue to red). | ||
Human ATG8 family proteins consist of 6 or 7 orthologs (LC3A has two splicing forms: LC3A-a and LC3A-b): LC3A-a, LC3A-b, LC3B, LC3C, GABARAP, GABARAPL1 and GABARAPL2 (also called Golgi-associated ATPase enhancer of 16 kDa, GATE-16). They have similar but non-overlapped functions. Several review papers have described their roles in autophagy and other biological processes.61–63 Among ATG8 family proteins, LC3B is the most important and well-studied form. Thus we compared all other proteins in the human ATG8 family with LC3B. Despite only moderate similarity at the amino acid sequence level (Fig. 3B), these proteins have similar 3D structures. Fig. 3B shows the amino sequence alignment result of human LC3A-a, LC3C, GABARAP, GABARAPL1 and GABARAPL2 to LC3B, and Fig. 3C shows the structural alignment of these proteins. These proteins consist of 117–147 residues. Even though there are two α helices in all ATG8 family proteins, the properties of these helices are different. The first α helix of the LC3 subfamily is strongly basic (the sizeable blue region in Fig. 3D), whereas the GABARAP subfamily is acidic. The second α helix of LC3 is acidic, whereas the GABARAP is basic and GABARAPL2 is neutral.
ATG8 family proteins exhibit similar structural properties to those of ubiquitin, and thus they are also considered ubiquitin-like proteins (UBLs).62 The overall structure alignment of LC3B with ubiquitin (align to the central helix, α3 of LC3B, and α1 of ubiquitin) is shown in Fig. 3E and 3F. The folding of the central core is similar, including 4 β-sheets and 2 α-helices. The ubiquitin core of the ATG8 family proteins consists of four-stranded central β sheets and 2 α helices, α3 between β2 and β3, and α4 between β3 and β4. The core domain contains a positively charged Arg/Lys-rich region conserved among all ATG8 family proteins to interact with negatively charged residues preceding or within the AIM/LIR motifs. The significant difference between LC3/ATG8 and ubiquitin proteins is that LC3/ATG8 has two additional N terminal α-helices, forming another hydrophobic pocket (HP1) with β-sheets for selective ligand binding, which will also be discussed below.
A series of essential studies by Terje Johansen's group identified the ATG8-interacting motif (AIM) in yeast and the LC3-interacting region (LIR) in mammals.64 The LC3 protein consists of two large hydrophobic pockets (HP1 & HP2), which are critical to the specific interaction with AIM/LIR (Fig. 3D and F). HP1 is slightly larger than HP2 and can accommodate the large side chain of Trp (W), so it is also called the W-site. HP1 is formed by the N terminal α-helix and hydrophobic residue in the core ubiquitin-like domain (Fig. 3C). These residues in the deep pocket provide additional hydrophobic interaction for the binding of AIM/LIR. The hydrophobic pocket 2 (HP2) is smaller and can only accommodate Leu(L)'s more minor side chain and therefore is also called the L-site. Targeting HP1 and HP2 simultaneously will likely provide high binding affinity, which is the primary driving force of AIM/LIR-LC3 interactions. LIRs, including the C terminal of Atg19 and the middle of SQSTM1/p62, are the linear non-structured peptide sequence (WXXL, where X represents any amino acid residue). The LIRs interact with LC3's β2-strand and form anti-parallel intermolecular β-sheets (Fig. 4A). The negatively charged LIR residues also form salt bridges with the positively charged Lys or Arg residue of LC3, further enhancing the binding affinity with AIM/LIR. With more and more AIM/LIR sequences being identified, the general core consensus Θ–X–X–Γ is concluded, in which Θ represents an aromatic (F, W or Y) residue, Γ represents a hydrophobic residue (I, L or V), and X represents any amino acid.
 | ||
| Fig. 4 Structural alignment of LC3B's binding with specific ligands. (A) The structure of the representative LIR (SQSTM1/p62, shown in pink)–LC3 binding interface (PDB: 2ZJD); (B) the structure of the AnkB (green)–LC3 binding interface (5YIS); (C) the structure of the AnkG (green)–LC3 binding interface (5YIQ); (D) the structure of the covalent binding DC-LC3in-A4 (pink)–LC3 binding interface (7ELG); (E) the structure of the novobiocin (cyan)–LC3 binding interface (6TBE); and (F) possible GW5074 (green)–LC3 binding interface by autodocking. | ||
Besides the amino acid sequences, posttranslational modifications (PTMs) also affect the AIM/LIR's interaction with LC3. Phosphorylation and acetylation are the most extensively studied PTMs in LC3.65–67 Tyr phosphorylation in the AIM/LIR region causes a charge shift, thereby negatively regulating the binding and reducing AIM/LIR's binding affinity with LC3. The acetylation in LC3 also alters its binding to AIM/LIR in cells and in vitro. Acetylation of K49 and K51 regulates the shuttling of LC3 between the nuclei and the cells’ cytoplasm, thus influencing LC3's binding to substrates located in specific cellular compartments and affecting the selective autophagy of particular substrates.68,69 Besides the influences on subcellular localization, acetylation may also influence LC3-AIM/LIR binding directly. For example, the acetylation of Lys49 of LC3B could disrupt the interaction between LC3 and AIM/LIR.70
Based on AIM/LIR-LC3 binding knowledge, Luo et al. designed an AIM/LIR based selective degrader that can precisely degrade target proteins.74 They designed a degrader composed of a target-specific binder and an AIM/LIR to tether the protein of interest (POI) to nascent autophagosomes. The system was shown to degrade various fluorescence-tagged proteins and peroxisome organelles. The delivery of plasmids encoding degraders was achieved using a tobacco-based transient expression system and transgenic Arabidopsis expressing engineered receptors, which may be difficult to replicate in mammalian cells. Thus, while this peptide-based degrader study demonstrates the possible ligandability of LC3 for degradation, small-molecule LC3-binders are still desired for drug discovery. Although the binding mechanism of AIM/LIR to LC3 has been extensively investigated, the research on small molecules that can bind to LC3 and modulate LC3 function is still very limited. Several published studies are discussed below.
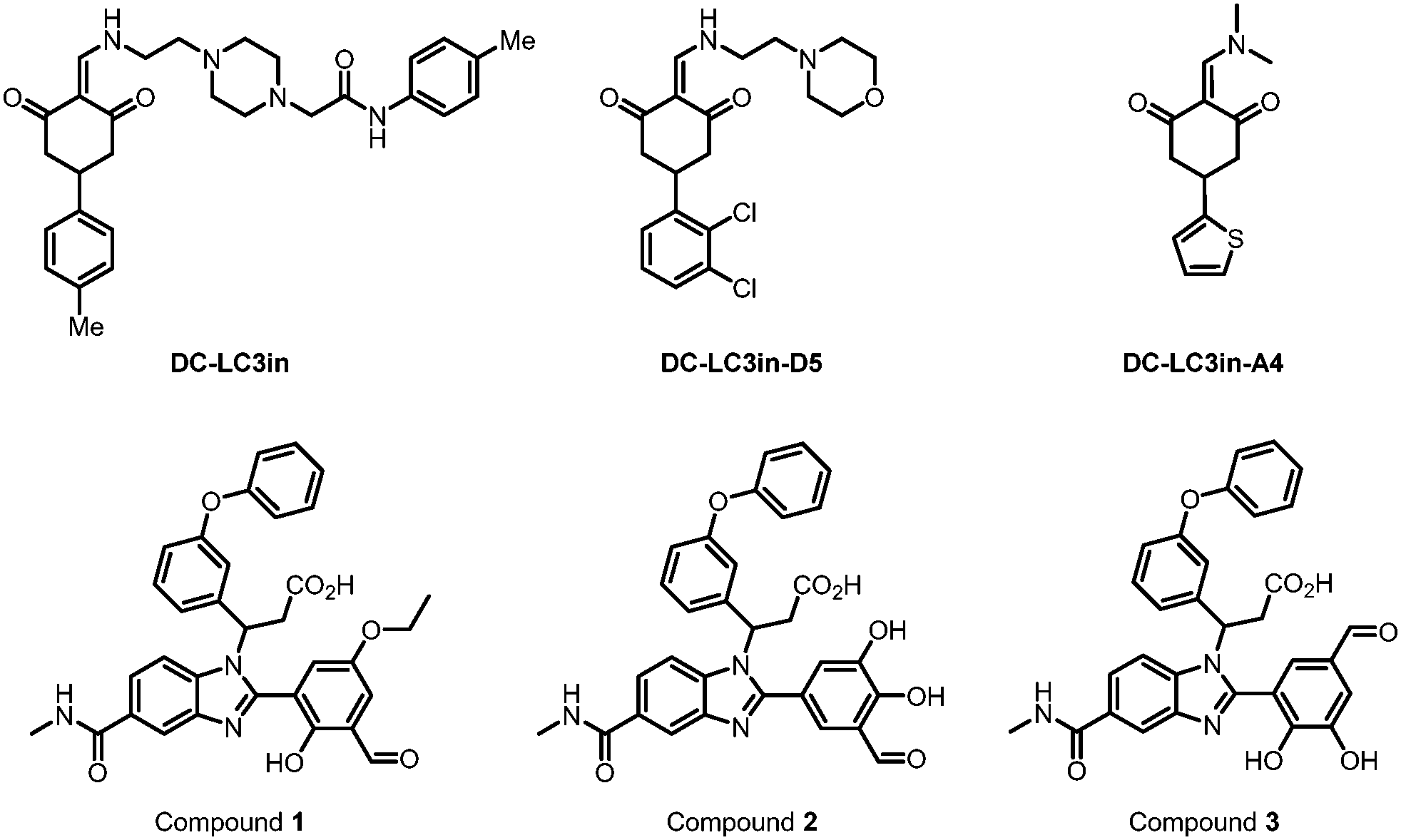 | ||
| Fig. 5 Covalent binders of LC3. DC-LC3in and its derivative DC-LC3in-D5/A4 are efficient covalent modulators of LC3B, while A4 is co-crystallized with LC3B. Compounds 1–3 were found to be reversible covalent binders of LC3 through DEL screening and verified by MS. | ||
Besides forming a covalent bond with Lys49, compound DC-LC3in-A4 could form additional hydrogen bonds with the surrounding Leu53, Lys51 and Arg70 (PDB: 7ELG). A cation–π interaction was also formed between the thiophene ring of DC-LC3in-A4 and ionized Lys30. Meanwhile, another small molecule compound DC-LC3in-D5 exhibited high selectivity in binding with LC3A/LC3B versus GABARAP. This is probably due to the different pocket environments near Lys49 in LC3A/B versus GABARAP, although further chemical biology and mutagenesis studies are needed. The covalent binding of DC-LC3in-D5 to Lys49 also attenuates LC3 lipidation. It inhibits the formation of autophagic structures with low cellular toxicity, providing possible research tools for autophagy research and demonstrating the ligandability of LC3.75
In 2022, Steffek et al. reported 3 small molecular weight compounds (Fig. 5: compounds 1, 2, & 3) that can reversibly form covalent bonds with LC3B. They identified the compounds through DNA-encoded library (DEL) screening.76 The reversible covalent bonds are formed between the compounds’ aldehyde groups and the LC3 protein. Meanwhile, the detailed binding residue (possibly Lys) has not been determined yet. This discovery provides a potential way to develop novel and potent LC3 binder warheads: screening for compounds that can form a potential covalent bond with LC3 with improved specificity and affinity from libraries of compounds with aldehyde or analogy groups. These binders may provide potential warheads for autophagy-dependent degraders, although future functional studies are needed.
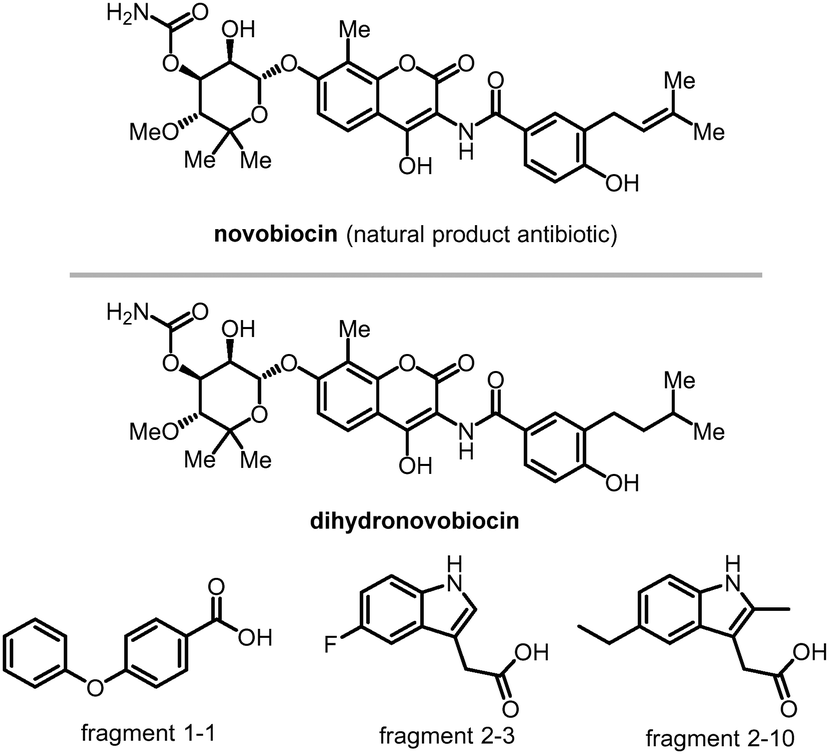 | ||
| Fig. 6 Compounds bind to LC3's HP1 and HP2. Novobiocin and its derivative dihydronovobiocin show good ligandability with LC3A and LC3B adapter proteins. Fragments 1-1, 2-3 and 2-10 are verified to bind with LC3's HP2. | ||
 | ||
| Fig. 7 Hit compounds being identified by glass chip immobilization (with red linker) and analogues that bind with both LC3B and mHTT proteins. | ||
These possible LC3 ligands provide potential possibilities to develop autophagy-dependent degrader technologies. Currently, several TPD technologies based on the autophagy pathway have been reported, including ATTECs, AUTACs, and AUTOTACs (Fig. 8). These technologies are discussed below.
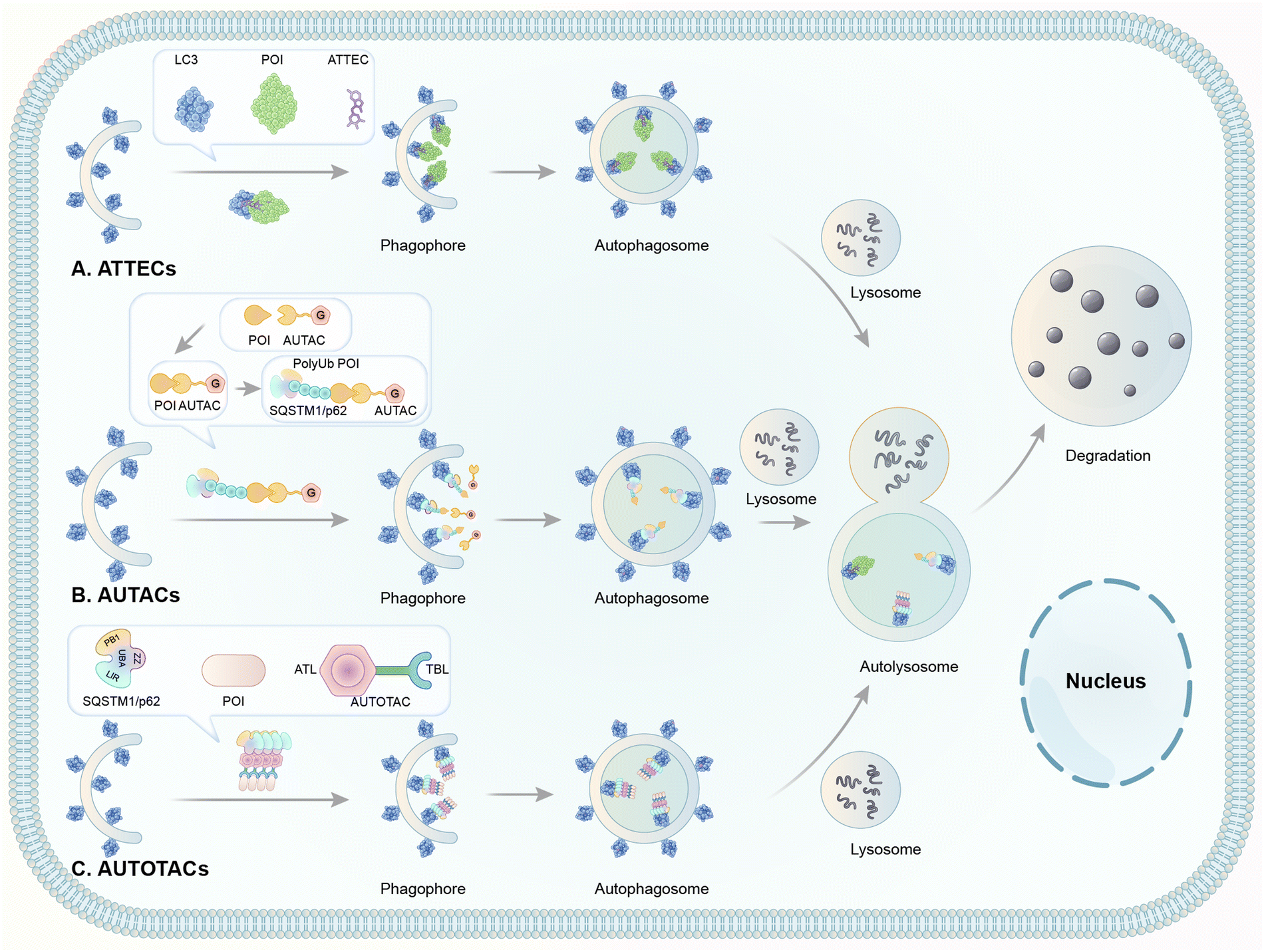 | ||
| Fig. 8 Illustrations of degrader technologies engaging autophagy. (A) ATTECs: ATTECs bind both the POI and LC3, tethering the POI to the phagophores or autophagosomes directly for subsequent autophagic degradation; (B) AUTACs: AUTACs bind to the POI and add a tag mimicking S-guanylation; subsequently, the posttranslational modification triggers the K63-poly-ubiquitination of POI. The POI is then recognized by the autophagy receptor SQSTM1/p62 and recruited to the selective autophagy pathway for degradation; (C) AUTOTACs: AUTOTACs bind to both the POI and SQSTM1/p62, and the complex is then recognized by LC3, tethering the POI to the phagophores for subsequent autophagic degradation. | ||
2.6 ATTECs
Based on the analysis above, we proposed the possibility of developing a novel degrader technology by engaging LC3 and bringing the molecule of interest to the autophagy machinery for degradation. We hypothesized that compounds that interact with both LC3 and the pathogenic biomolecule or organelles might tether the latter to the phagophore and enhance their degradation through autophagy. We named such compounds AuTophagy-TEthering Compounds (ATTECs). Given the broad spectrum of autophagy cargos, ATTECs may greatly expand TPD technologies’ landscape and enable targeted degradation of non-proteinaceous biomolecules or organelles. To demonstrate the feasibility of ATTECs, we have previously performed two proof-of-concept studies using different strategies. Some other groups recently showed that ATTECs could be applied to several oncoprotein targets. These studies are discussed below.Given its potential therapeutic benefits, significant efforts have been made in the field to lower mHTT. At least five strategies have been attempted to achieve this goal (Fig. 9).
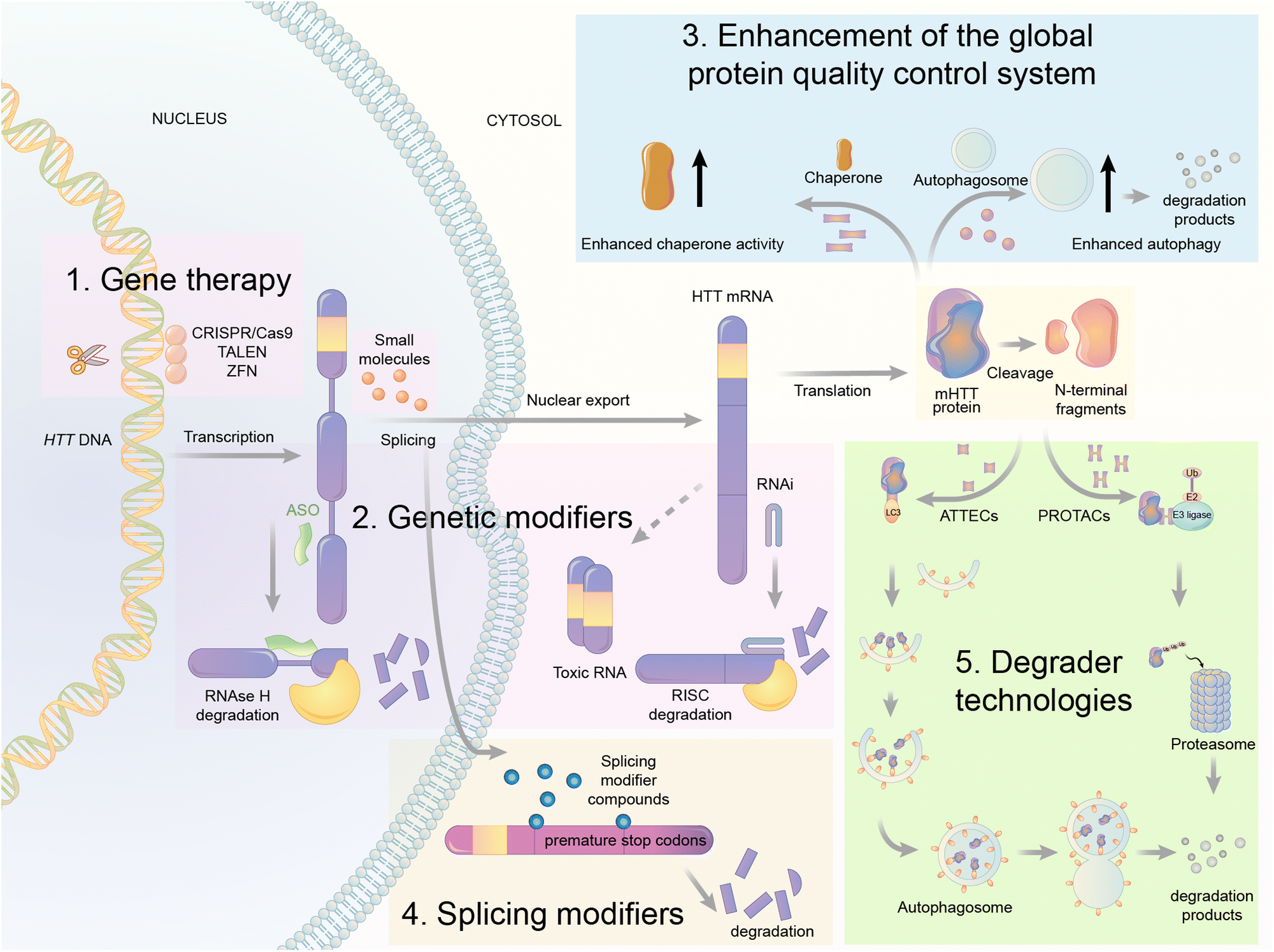 | ||
| Fig. 9 Illustrations of exemplar mHTT lowering strategies. The gene therapy approaches target DNA (CRISPR/Cas9, TALENS, and ZFN) or RNA of HTT via the presented mechanisms. The mHTT protein could also be regulated by genetic modifiers, global protein quality control systems, splicing modifiers, and degrader technologies. These strategies provide opportunities of small molecule drug discoveries for HD treatment. | ||
First is gene therapy. This strategy is currently the best-studied and most advanced in the HD field, with multiple clinical trials attempted already.89,90 This strategy delivers short hairpin RNAs or small interference RNAs (siRNAs) or antisense oligonucleotides (ASOs) or genome-editing reagents (CRISPR/Cas9, TALEN, or ZFP) targeting the RNA or DNA of HTT to lower the level of mHTT.91–94 While this strategy has obtained great success in animal models,92 clinical trials have been unsuccessful [available from: https://en.hdbuzz.net/306]. In addition, it is difficult to deliver gene therapy reagents into the cells, especially for neurological diseases,95 in which the blood–brain barrier (BBB) forms another hurdle. They are also prohibitively expensive.96 Thus, small-molecule-induced mHTT lowering is highly desired.
Second is the use of genetic modifiers, which are upstream genes that regulate mHTT levels. Small-molecule compounds may lower mHTT by inhibiting or activating these modifiers. Screening studies have been performed to identify these genetic modifiers, which showed promising potential in HD animal models.97–102 This strategy also has limitations because targeting these genetic modifiers will induce side effects by altering their physiological functions. This cannot be minimized by medicinal chemistry optimization because mHTT lowering depends on modulating these modifiers’ activity. In addition, the discovered modifiers are non-allele selective, i.e., changing both mHTT and wtHTT levels. This is not ideal because wtHTT likely plays a beneficial role in HD patients.87,103
Third is the enhancement of the global protein quality control system. For example, remodelling the proteostasis network by small-molecule compounds targeting molecular chaperons or inducing stress responses may reduce the misfolding and accumulation of pathogenic proteins, including mHTT.104 Alternatively, enhancing autophagy may also lower mHTT,98,105 because it is known to be degraded by autophagy.106 While these approaches are promising for HD and may have the potential of treating multiple diseases, they induce global changes in the cells that may result in extensive nonspecific or compensatory effects to offset their potential benefits.
Fourth is splicing modifiers. Novartis recently reported a brain penetrant small molecule, branaplam, that can be orally administered (Fig. 10) as an mHTT lowering compound.107 Branaplam promotes the inclusion of a pseudoexon that carries in-frame stop codons in the HTT transcript (Fig. 9), leading to reduced mHTT protein levels in HD patient cells, an HD mouse model, and blood samples from Spinal Muscular Atrophy (SMA) Type I patients dosed branaplam orally for SMA.107
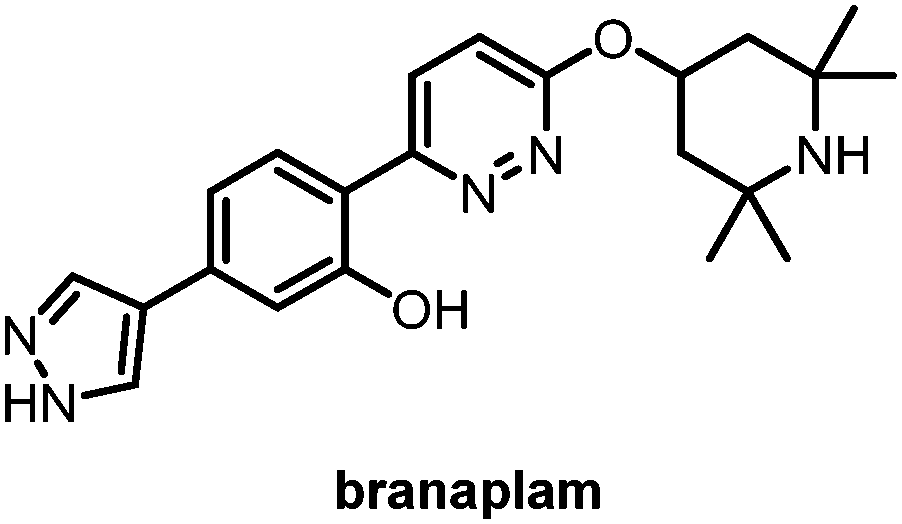 | ||
| Fig. 10 Branaplam, an orally available brain penetrant small molecule that lowers mHTT levels. | ||
The final one is the degrader technology. PROTACs targeting mHTT were designed by linking a probe for general protein aggregates (not mHTT-specific) and a binder of the E3 ligase cIAP108,109 (Fig. 11). Surprisingly, these compounds reduced the soluble HTT protein, including both mHTT and wtHTT, in the human patient fibroblasts,108,109 which have no or little protein aggregates.98 The mechanisms underlying these unexpected biological results remain to be clarified, and they may provide valuable insights into the discovery/designing of other aggregate-targeting degraders.
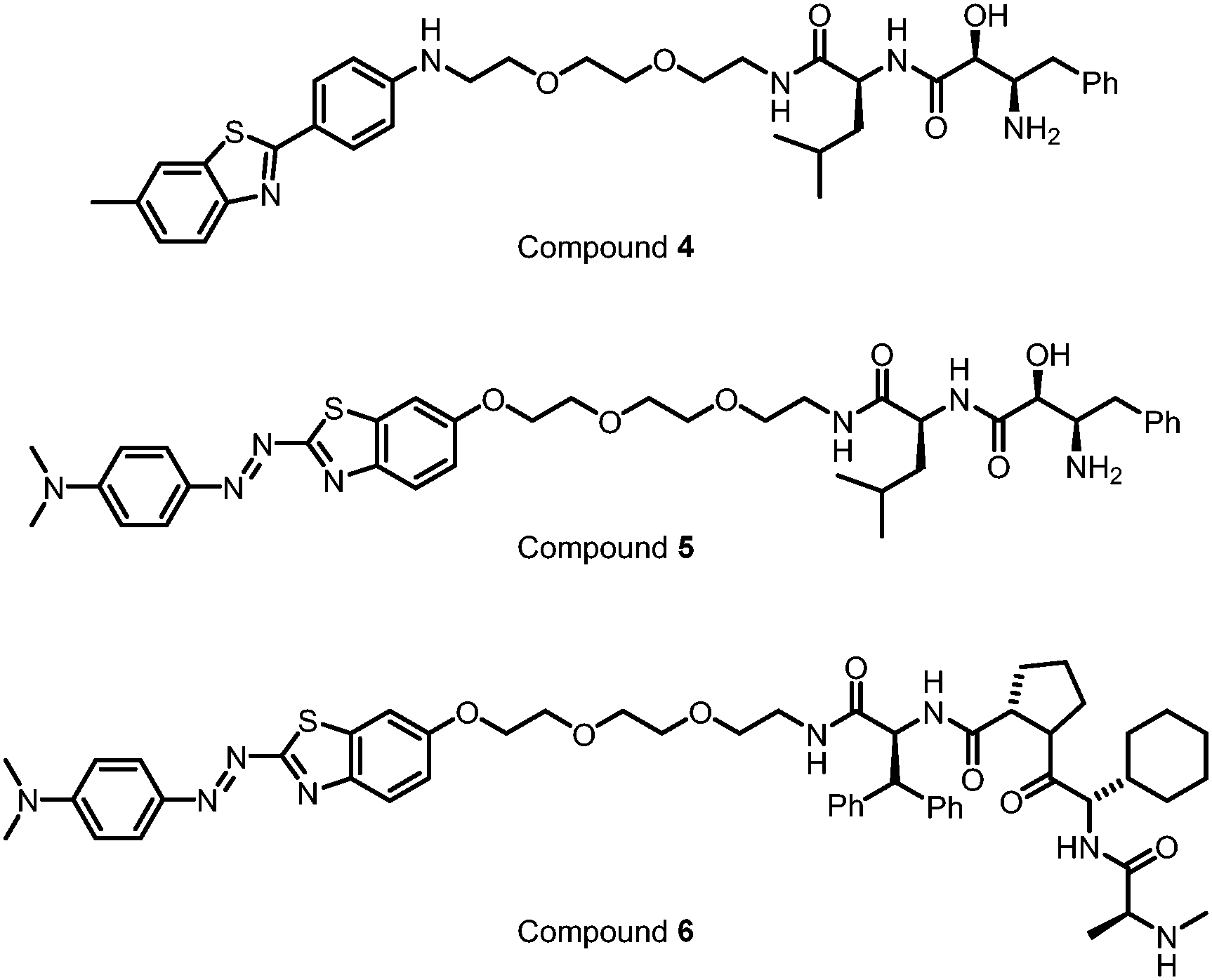 | ||
| Fig. 11 Small molecules designed by linking a general protein aggregate probe with cIAP for the degradation of Huntingtin. | ||
Among the five strategies mentioned above, we believe that the degrader technology may provide an attractive strategy with unique advantages for lowering mHTT. Compared to the gene therapy strategy, the degrader compounds may have advantages in delivery and cost; compared to the genetic modifier strategy, the degrader compounds may target mHTT directly without inducing side-effects by modulating the activity of other targets; compared to the strategies enhancing the global protein quality control system, the degrader compounds may induce much less non-specific effects. Branaplam is promising and has already entered clinical trials. It induces splice-in events of exons enriched for a non-canonical nGA 3′-exonic motif and other splicing changes of different mRNAs,107 and targeting the mHTT protein directly may offer additional specificity and synergistic effects with RNA modulating compounds. Meanwhile, the PROTACs targeting mHTT were not allele-selective.108,109 They were also designed to target protein aggregates rather than mHTT specifically. The mechanism of action also needs further clarification.
Mutant Huntingtin (mHTT) is known to be degraded by autophagy.106 Thus, autophagy-based degrader compounds may also lower mHTT. We have recently reported the discovery of ATTECs that lowered mHTT in an allele-selective manner and rescued HD-relevant phenotypes in mouse models.9 The compounds were discovered using small-molecule-microarray based screens and counter-screens, which identified compounds that directly interact with both LC3 and mHTT but not with wtHTT. Mechanistic studies confirmed that these compounds tether mHTT but not wtHTT to the autophagosomes for degradation without influencing the global autophagy activity per se. In vitro experiments revealed that these compounds distinguish mHTT from wtHTT by their specific interaction with the expanded polyQ stretch directly, possibly by recognizing its unique structural feature different from the short polyQ stretch.83,110–113 Thus, these ATTECs are also capable of degrading other pathogenic polyQ proteins, such as mutant ATXN3,114 which causes spinocerebellar ataxia type III.115 Some of these polyQ·ATTECs can pass the BBB and lower mHTT in the brain tissues in vivo by peripheral injections,9 providing encouraging starting points for drug discovery. The allele-selectivity, the potential to target several different polyQ diseases, and the BBB permeability are attractive properties of the ATTECs compared to the reported mHTT-targeting PROTACs. Meanwhile, maximal degradation (Dmax) of mHTT is lesser with the treatment of polyQ·ATTECs than mHTT PROTACs, possibly because polyQ·ATTECs only recognize some of the mHTT protein molecules exhibiting a unique conformation of the expanded polyQ stretch.
The molecular weights of the discovered polyQ·ATTECs are close to those of molecular glues such as lenalidomide or other immunomodulatory drugs (IMiDs) (Fig. 7).116 As LC3-binding compounds, these polyQ·ATTECs are also potentially useful to be connected to a ligand of the target protein or non-proteinaceous biomolecule. The chimeric molecules generated by this design may enable selective degradation through the ATTEC mechanism. These chimeric ATTECs could be designed directly without tedious screening work. We recently tested this possibility with another proof-of-concept ATTEC study targeting lipid droplets (LDs).10
 | ||
| Fig. 12 Exemplar LD probes. The typical LD probes are shown in the color of their respective emission maximum wavelength. | ||
Based on this design, the assembled chimeric LD·ATTECs can interact with both LC3 and LDs simultaneously, enhancing the engulfment of LDs by autophagosomes and subsequent degradation. The interaction between LD·ATTECs and LDs could be interpreted as hydrophobic forces that lead to better tethering and dissolvement of these compounds into the LDs. Thus, the LD·ATTECs may enhance the tethering of phagophores to LDs and induce the recognition of LDs by autophagosomes, which can envelop small LDs or enter large LDs to engulf portions of them.118,119
For our initial study,10 we selected GW5074 (GW) and 5,7-dihydroxy-4-phenylcoumarin (DP) as the LC3-binding “warheads” in the designed chimeric LD·ATTECs because we identified and validated these two compounds as LC3B-binding compounds that do not influence the overall autophagy functions.9 For the LD-binding moiety, we selected Sudan IV (1-2-methyl-4-[(2-methylphenyl)azo]phenylazo]-2-naphthalenol) or Sudan III (1-[4-(phenylazo)phenyl]azo]-2-naphthalenol) for the synthesis.124–127 We used a simple chemical linker (decane) to connect these two chemical moieties. The four LD·ATTECs synthesized have molecular weights between 700 and 1100 Da (Fig. 13), similar to most PROTACs. In theory, other LC3-binding compounds linked with other LD probes may also degrade LDs, and the molecular weight of LD·ATTECs could be further reduced by using smaller LD probes or LC3-binding compounds. The linkerology design could also be optimized.
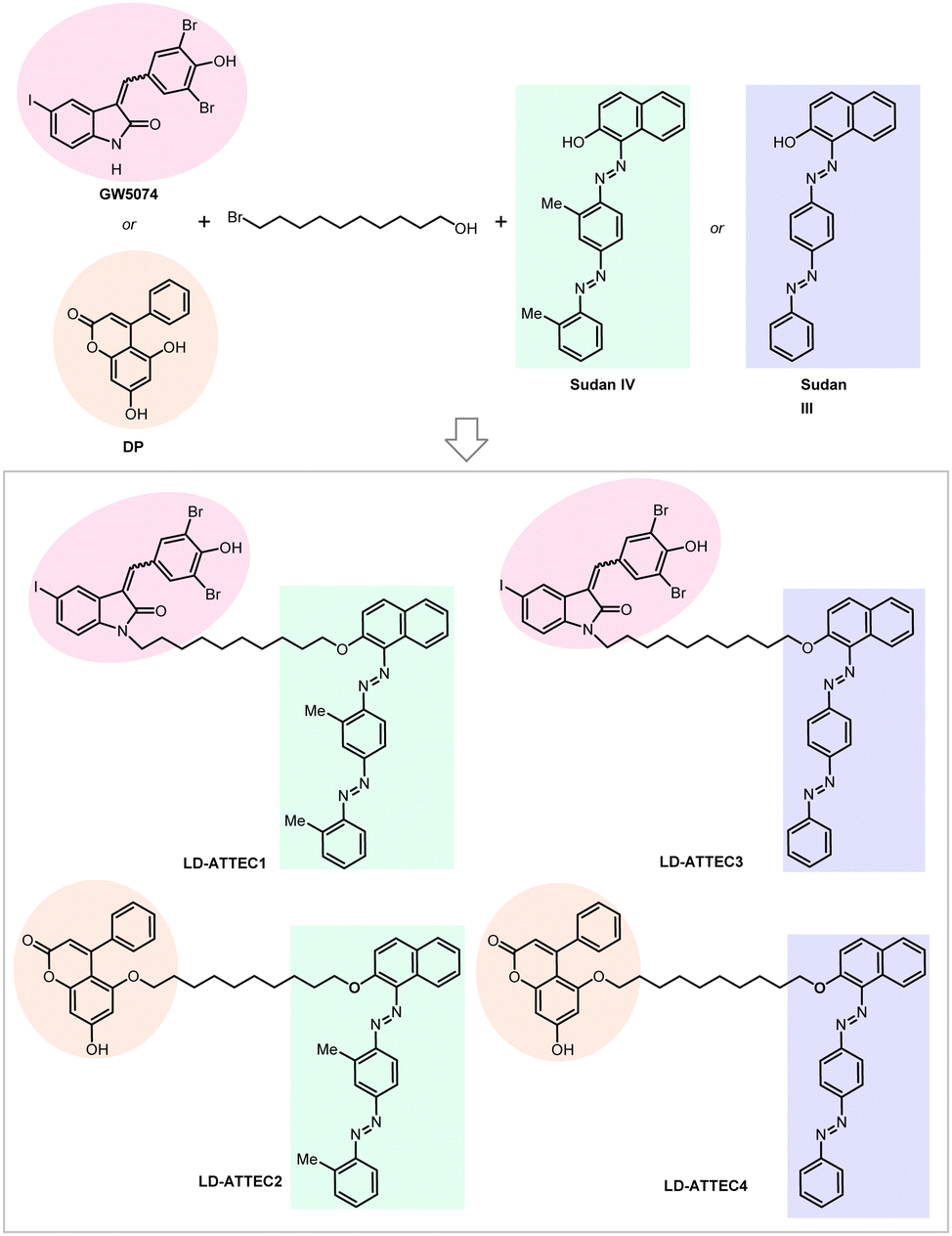 | ||
| Fig. 13 Exemplar LD-ATTECs. | ||
Consistent with the predicted ATTEC mechanism, LD·ATTECs caused near-complete clearance of LDs via autophagy at ∼5 to 15 μM concentrations in several different cellular models, including mouse embryonic fibroblasts (MEFs) and human neuroblastoma cells (SH-SY5Y) with LDs induced by oleic acid, and also in differentiated adipocytes exhibiting endogenous large LDs.10 The observed LD clearance was ameliorated by ATG5 or LC3 knockout, further supporting the target engagement of LC3 and the involvement of autophagy.
Consistent with the cellular data, LD·ATTECs were also effective in two different in vivo mouse models of metabolic disorders,10 including a genetic model (db/db mice, C57BL/6J-Leprdb/Leprdb) with obesity and diabetes,128–130 and a non-alcoholic steatohepatitis (NASH) mouse model generated by a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD, 60 kcal % fat) feeding for 10 weeks.131 LD·ATTECs’ injections for two weeks correlated with a significant reduction in body weight, body fat/lean ratio, liver weight and fat content, and circulating neutral lipids. The liver fat reduction was confirmed using different approaches, including biochemical assays, imaging assays and mass-spectrum lipidomic analyses. In the NASH model, liver fibrosis was significantly attenuated by the LD·ATTEC treatment as well.
Despite the reliance on non-specific hydrophobic interactions for target engagement, the clearance of LDs by LD·ATTECs is relatively selective for LDs, which contain condensates of neutral lipids. Other lipid-containing membranes were not affected, likely because of the polar nature of the membrane lipid composition, which LD·ATTECs do not recognize. The specificity was further demonstrated by lipidomics experiments, which showed that LD·ATTECs only significantly lowered the neutral but not polar lipids. The lowering was also largely irrelevant to the carbon chain length or number of unsaturated bonds. The specificity was further tested at the protein level by proteomics experiments. Significant lowering of the LD marker protein Plin2132 by LD·ATTEC injections was observed, consistent with LD lowering. None of the lipid synthetases or lipases or their cofactors were significantly changed. Autophagic substrates or core pathway genes were not influenced either. Besides the LD marker Plin2, ∼30 other proteins (<1% of the proteome detected) were also changed by injection of each of the LD·ATTECs or the control compound (Sudan III). Interestingly, the protein changes by the two different LD·ATTECs showed substantial overlap, including 8 proteins that Sudan III did not change, and many of these changes likely reflected the consequence of LD lowering. For example, these 8 proteins include Gstm6 and Gstp1, which are down-regulated in livers from mice treated with intravenous lipid emulsions or fed with a high-fat diet,133,134 and were up-regulated in the LD·ATTEC-injected groups possibly due to LD-lowering. Taken together, LD·ATTECs did not directly perturb protein levels on the whole, but indirect and cascading effects on protein levels cannot be entirely ruled out.135
The study also expands the target landscape of degrader technologies to non-proteinaceous targets. ATTECs degrading other targets could be identified or designed similarly to polyQ·ATTECs or LD·ATTECs. Other chimeric ATTEC studies from other independent groups are discussed below.
BRD4 is a well-characterized oncoprotein target by PROTACs presented initially in the milestone studies from three independent groups in 2015.95,139–141 The BRD4 inhibitor JQ1 and the IMiDs interacting with the E3 ligase subunit CRBN were covalently linked to the building up of the PROTAC molecules, which can effectively and selectively degrade BRD4.95,136,142,143 To test whether BRD4 can also be effectively degraded by ATTECs using the LC3-binding warheads, Pei et al. successfully synthesized chimeric compounds by linking the LC3-binding warhead GW5074 from its phenoxyl group to JQ1 with a suitable linker.144 Their data demonstrate that some of these compounds can degrade the BRD4 protein likely through the autophagy pathway. Noticeably, they did a pilot study on the linkerology of the chimeric ATTECs and suggested that the linker length and composition strongly influence the degradation efficacy and potency. This phenomenon is possibly mediated by modifying the coupling between the LC3-ATTEC and the target-ATTEC interactions. Based on the preliminary linkerology study, the authors identified compound 10f (Fig. 14) as the most potent and efficacious compound that can reach a Dmax of 92% with a DC50 of 0.9 μM. They further demonstrated that compound 10f exhibits good anti-proliferative activity in multiple tumor cells. The authors further investigated if the BRD4 degradation by 10f is autophagy-dependent, although the data seemed preliminary. Consistent with the autophagy dependence, the autophagy inhibitor chloroquine largely inhibited the degradation of compound 10f, whereas the autophagy activator rapamycin enhanced the degradation efficiency. Further demonstration of autophagy dependence by genetic approaches is desired to confirm this result. The possible interaction between BRD4 and LC3 after the treatment of 10f in the MDA-MB-231cells was also investigated by the immunofluorescence confocal microscopy analysis, showing colocalization of BRD4 and LC3 puncta. While the cellular colocalization evidence is encouraging, biochemical assays are desired to validate the enhancement of BRD4-LC3 interactions by 10f treatment directly. Finally, the flow cytometry assay showed that 3-methyladenine (3-MA), which inhibits the autophagosome formation, counteracts the apoptosis induced by compound 10f. In contrast, it does not block the apoptosis induced by JQ1, suggesting that the effect of 10f but not its building-block JQ1 is autophagy-dependent. Consistent with our previous studies showing that GW5074 does not influence global autophagy,9 compound 10f does not affect the global autophagy either, as suggested by the lack of changes in the LC3 western blot and mRFP-GFP-LC3 double fluorescence assays.
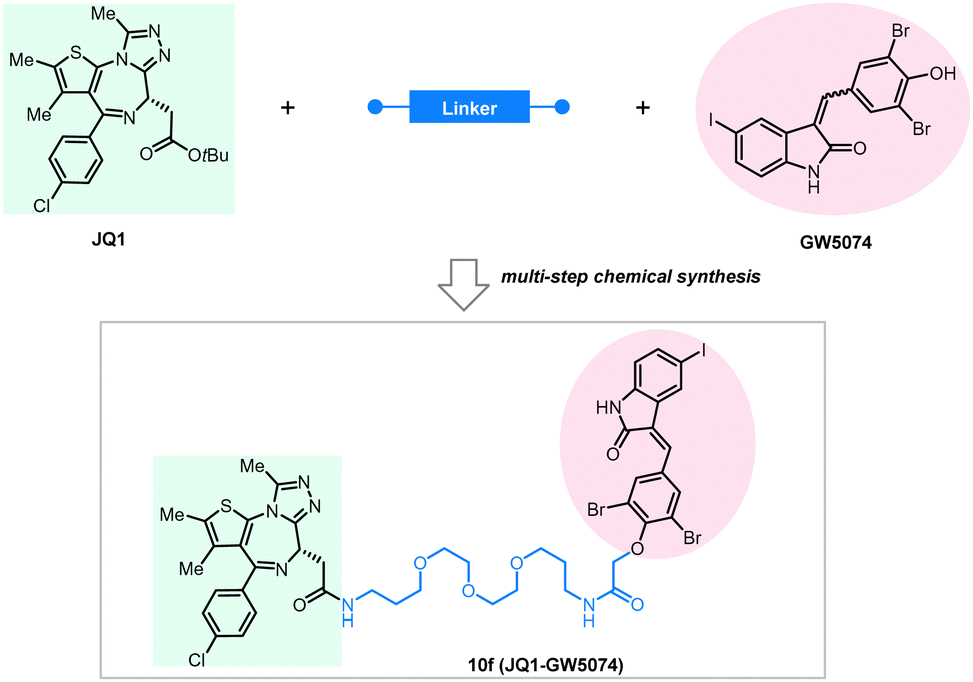 | ||
| Fig. 14 Exemplar BRD4-ATTEC degrader 10f. | ||
It is worth mentioning that the degradation of BRD4 by ATTECs is somewhat unexpected, given that autophagy mainly occurs in the cytoplasm, whereas BRD4 is a nuclear protein. Two potential contributing mechanisms might explain this. First, like other proteins, BRD4 is translated into the cytoplasm by ribosomes and thus has at least a small cytosolic fraction, which could be degraded by ATTECs. This may shift the balance between nuclear and cytoplasmic BRD4, ultimately reducing BRD4 concentrations. Second, Shelley Berger's groups demonstrated that nuclear proteins could be recognized as autophagy substrates and subjected to cytoplasmic autophagosome–lysosome degradation via the autophagy protein LC3.145–147 The detailed mechanisms of BRD4·ATTECs remain to be further clarified.
Taken together, the authors provide promising data demonstrating that BRD4 could be targeted by ATTECs, which may have important therapeutic potentials.
NAMPT is the rate-limiting enzyme that converts nicotinamide to nicotinamide mononucleotide (NMN). It has been reported as a potential oncoprotein and therapeutic target involved in several cancer types, including colon and prostate cancers.148,149 NAMPT is also a potential target for the treatment of inflammatory disorders.150–153 Thus, the development of effective and safe NAMPT inhibition or degradation compounds is desired for both cancer and immune-related disorders. Although several NAMPT inhibitors, including FK866 or CJS828, were developed, they failed in clinical application because of limited efficacy and high dose-dependent toxicity.154–156 The degradation of NAMPT by PROTAC compounds was first verified by Wu et al. in 2021.157 In a very recent study, Dong et al. designed a series of bifunctional compounds connecting a NAMPT inhibitor and an LC3-binding compound using different linkers. They investigated their effects on the degradation of NAMPT in cells.158
They used a novel NAMPT inhibitor MS2 (IC50 = 85 nM) targeting NAMPT, linking it with our previously reported LC3 binding warhead ispinesib at its primary amine terminal with seven alkyl or PEG linkers of different lengths. All the tested compounds showed NAMPT degradation capability in human ovarian cancer A2780 cells after 48 hours of treatment. The linker length is essential for the degradation efficacy, and the compound A3 with an 8-carbon linker (Fig. 15) showed the best degradation efficacy. Shorter or longer linker lengths lead to reduced degradation efficacy. This observation is, in general, consistent with the pilot linkerology study of the BRD4·ATTECs, of which the linker length and structure also have an impact on the degradation efficacy.144 The degradation of NAMPT by A3 reaches 90% at 100 nM, suggesting promising therapeutic potentials. Consistent with this, the authors confirmed that the antitumor potency of A3 is even higher than that of NAMPT inhibitors: A3 showed an excellent anti-tumor cell proliferation effect with <3 nM IC50, whereas the IC50 of the NAMPT inhibitor MS2 was 550 nM in the parallel experiments.
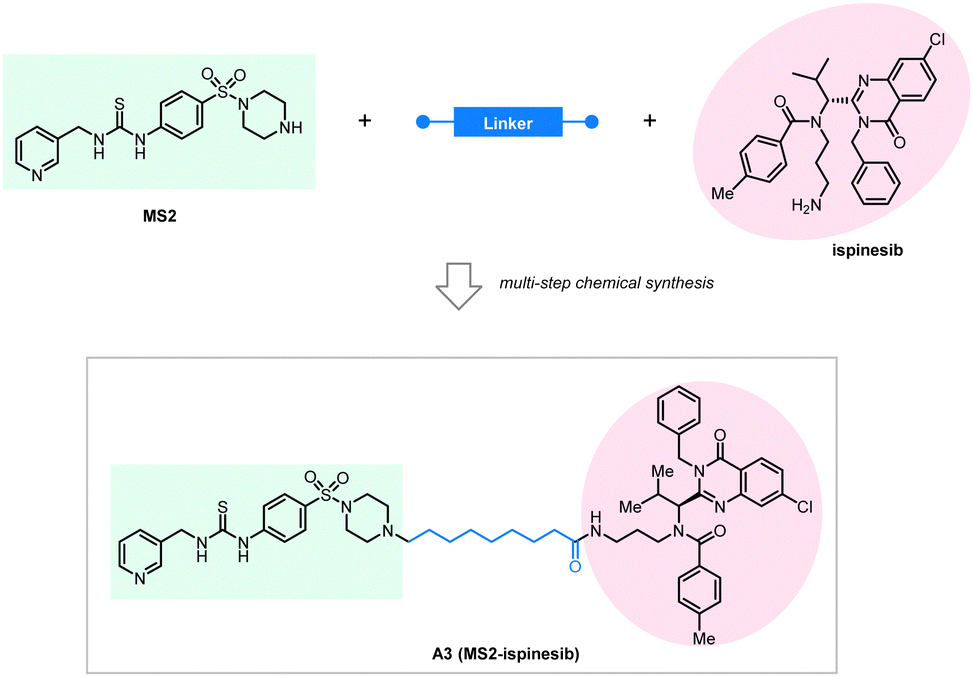 | ||
| Fig. 15 Exemplar NAMPT-ATTEC degrader A3. | ||
It is worth noting that the authors have carried out a thorough mechanistic study of the compounds. To validate the target engagement of LC3, the authors designed a fluorescent probe (P1-8F20) by linking the LC3-targeting compound 8F20 (ispinesib) with P1 (FITC) and tested the binding affinity between P1-8F20 (Fig. 16) and LC3 using the fluorescence polarization (FP) assay, which showed a binding affinity (KD) of 175 nM. P1-8F20 could be engulfed in the autophagosome at the cellular level by confocal microscopy. To further validate the autophagy dependence and the target engagement of NAMPT, they treated A2780 cells with A3 combined with several inhibitor/competitor compounds, including NH4Cl and chloroquine as lysosome inhibitors, wortmannin, 3-methyladenine, and LY294002 as autophagosome inhibitors, and MS2 and FK866 as competitors of NAMPT binding (Fig. 16). All these inhibitors/competitors significantly decreased the efficacy of A3-induced NAMPT degradation, confirming that the degradation of NAMPT is through the autophagy pathway and dependent on compound–NAMPT binding.
 | ||
| Fig. 16 The FITC-ATTEC fluorescent probe and typically used lysosome and autophagosome inhibitors. | ||
The autophagy inhibitors may not be specific enough. As discussed earlier, knockdown or knockout of key autophagy genes is desired to validate autophagy-dependence further. An excellent aspect of the NAMPT study is that the authors also tested the A3's degradation effects in Atg7 knockdown cells, which have damaged autophagy functions. The NAMPT degradation by A3 treatment was also blocked, confirming autophagy dependence.
ATTECs hijack the autophagy pathway by directly tethering the target to the phagophore. While several studies from independent groups illustrate the degradative effects of ATTECs, key questions remain to be addressed in future studies. The structural information of the LC3–ATTEC interaction remains to be resolved. In addition, whether this interaction may interfere with the binding of autophagy receptors and selective autophagy needs to be investigated. Finally, LC3 is involved in secretion159–167 and ER-associated degradation (ERAD).168,169 The potential contribution of those pathways to target-lowering may be worthy of further investigation.
2.7 AUTACs
Hirokazu Arimoto's group developed another degrader technology engaging autophagy: the autophagy-targeting chimera (AUTAC) system.7 Different from ATTECs, AUTACs do not directly utilize LC3 as the docking protein for degradation, and they hijack the selective autophagy by engaging the autophagy receptor SQSTM1/p62 (see Section 2.4 for the discussion on autophagy receptors and selective autophagy). AUTACs also function through poly-ubiquitination, similar to PROTACs. However, instead of tethering the POIs to an E3 ligase and triggering K48-linked polyubiquitination (i.e., ubiquitins conjugate with each other at the lysine 48 position to form a polyubiquitination chain), AUTACs trigger K63-linked polyubiquitination. The engagement of the docking protein (SQSTM1/p62 for AUTACs) is also indirect, unlike PROTACs and ATTECs. The design of AUTAC cleverly utilizes an endogenous process that defends cells against the invading group A Streptococcus (GAS) via antibacterial autophagy (xenophagy).170 Through studying this process, the Arimoto group previously identified S-guanylation (a posttranslational modification of the Cys-cGMP adducts) of the invading GAS by the endogenous nucleotide 8-nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP) as a potential signal to induce K63-linked polyubiquitination,171 which is known to be recognized by SQSTM1/p62 for selective autophagic degradation.172 Based on this, the Arimoto group hypothesized that S-guanylation might function as a standalone tag triggering K63-linked polyubiquitination and subsequent autophagic degradation of the substrate. They validated this hypothesis by inducing the degradation of EGFP fused with a Halo tag (HT) by a chimeric compound with a linker connecting the Cys-S-cGMP and the HT-ligand that covalently interacts with HT. The degradation is dependent on selective autophagy, as illustrated by the lack of compound's effects in Atg5 knockout and SQSTM1/p62 knockout cells. The study then demonstrates S-guanylation by a synthetic guanine derivative (p-fluorobenzyl guanine [FBnG]) also induced autophagic degradation.The authors then established the AUTAC platform to degrade endogenous proteins. An AUTAC molecule contains a degradation tag (guanine derivatives such as FBnG) linked with a small-molecule ligand of the POI. The degradation tag is then attached to the POI through the ligand–POI binding and mimics S-guanylation that destines the substrate protein for selective autophagy by inducing its K63-polyubiquitination (Fig. 8). Based on this principle, the authors designed degraders (Fig. 17) targeting MetAP2, FKBP12, BRD4, and mitochondria, respectively.
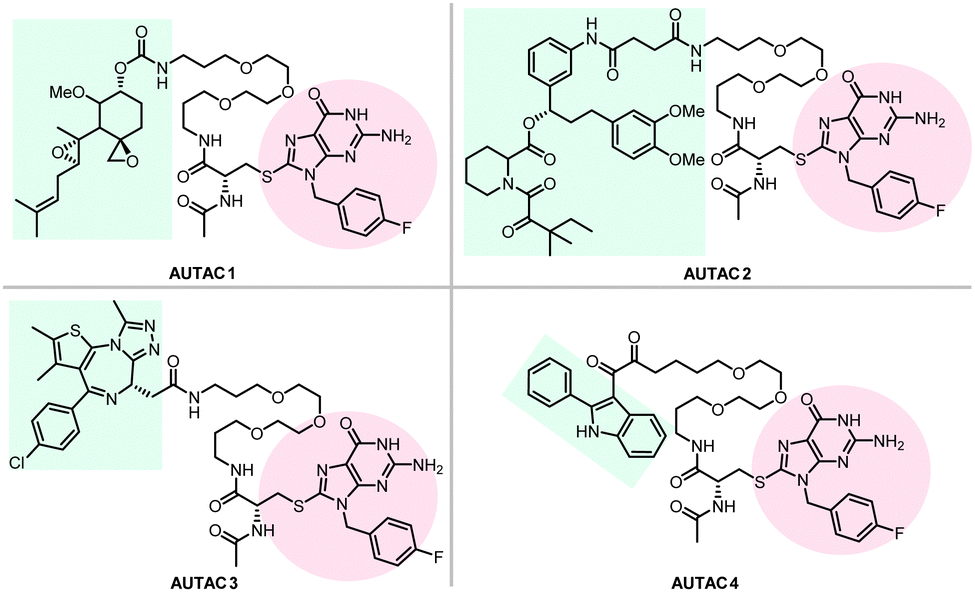 | ||
| Fig. 17 AUTAC degraders targeting MetAP2, FKBP12, BRD4, and mitochondria. | ||
 | ||
| Fig. 18 Thalidomide-based PROTAC by linking an HTL with a thalidomide moiety. | ||
To target endogenous mitochondria without expressing the fusion protein, the authors replaced the HTL moiety with a 2-phenylindole derivative to generate AUTAC4 (Fig. 17). 2-Phenylindole-3-glyoxyamides are ligands of the translocator protein (TSPO) on the OMM and thus may target mitochondria.179,180 A set of experiments similar to the FBnG-HTL ones were performed for AUTAC4 to demonstrate its capability to degrade mitochondria via the predicted mechanism. Encouragingly, AUTAC4 enhanced mitophagy and improved the mitochondrial morphology and membrane potential in Detroit 532 cells, immortalized fibroblasts from a patient with Down syndrome (also called trisomy 21), a genetic disease with mitochondrial dysfunction as a central factor in its etiology.181 These experiments demonstrate the therapeutic potential of AUTAC4 and the AUTAC platform.
The AUTAC platform is elegantly designed and has many advantages. The published study demonstrates that both covalent and noncovalent ligands of POIs may work to assemble AUTACs. It may also trigger the degradation of organelles such as mitochondria, although still through protein targets. Meanwhile, further validation and investigation of the mechanism of action may be necessary. The dose-dependence curves are pretty steep with no hook effects, and the underlying mechanisms may require further exploration. The compound–protein interaction and ternary complex (POI-compound–SQSTM1/p62) formation were assumed but not confirmed by biochemical assays, which are important to support the proposed mechanism. Accumulation of K63-linked polyubiquitin required approximately 8 hours of incubation of AUTAC4.7 This kinetics seems to be slower than typical enzymatic reactions and may provide additional clues into the mechanisms. The PEG linker was used for all AUTACs, and the linkerology study is desired in future AUTAC studies. The possible off-target effects by proteomics studies are also expected. The most crucial issue is probably to clarify the biological process. Unlike other degrader technologies that “tether” the POI to the degradation machinery directly, AUTACs function through mimicking a posttranslational modification, S-guanylation. The subsequent degradation probably involves complicated multiple-step processes that remain to be clarified. In particular, the molecular mechanism mediating the S-guanylation triggered K63-linked ubiquitination of POIs needs to be resolved. The other possible effects of S-guanylation also need further investigation. These studies will clarify which proteins are required for the AUTAC system and provide necessary information to predict whether AUTACs may work in specific cell types. It may also help predict how other signalling pathways may influence the AUTACs’ function and whether AUTACs may cause changes of other cellular functions or the selective autophagy process per se by mimicking S-guanylation. Finally, whether AUTACs are functional in vivo needs to be investigated.
2.8 AUTOTACs
The AUTOphagy-TArgeting Chimera (AUTOTAC) developed by the Yong Tae Kwon group is another degrader technology engaging SQSTM1/p62 (Fig. 8).8 Unlike AUTACs that mimic a posttranslational modification to induce polyubiquitination, AUTOTACs interact with the ZZ domain of SQSTM1/p62 (p62zz) directly and do not require polyubiquitination. p62zz can recognize a degradation signal in proteins – the N-terminal arginine residue (Nt-R).182 Binding of p62zz to Nt-R substrates (Fig. 8) stimulates SQSTM1/p62 aggregation and macroautophagy.183 The p62zz–Nt-R interaction may not be the only mechanism to activate SQSTM1/p62. For example, DAXX drives SQSTM1/p62 liquid phase condensation and activation by inducing SQSTM1/p62 oligomerization.56 In addition, SQSTM1/p62 recognizes K63-linked polyubiquitinated cargos by its ubiquitin-associated (UBA) domain that is separated from p62zz.184 Nonetheless, p62zz may serve as a docking site for degraders, which bind to and ideally are capable of inducing a conformational activation of SQSTM1/p62 as well.Four possible p62zz-binding compounds (Fig. 19A), YOK-2204, YOK-1304, YT-8-8, and YTK-105, were identified either by 3D structure modelling or in vitro pulldown assays.8 Further biochemical and cell biology experiments suggest that these compounds not only activate and target SQSTM1/p62 to autophagic membranes but also facilitate autophagosome biogenesis.8 Meanwhile, the SQSTM1/p62-binding affinity of these compounds (KD values) was not measured. Only ∼10% of SQSTM1/p62 were pulled down by the compounds, suggesting that the affinity was not very high. Importantly, YOK-2204 interacted with SQSTM1/p62 but not NBR1, another autophagic cargo receptor containing a ZZ domain. In addition, it has a much weaker interaction with SQSTM1/p62 carrying the D129A mutant in the ZZ domain.
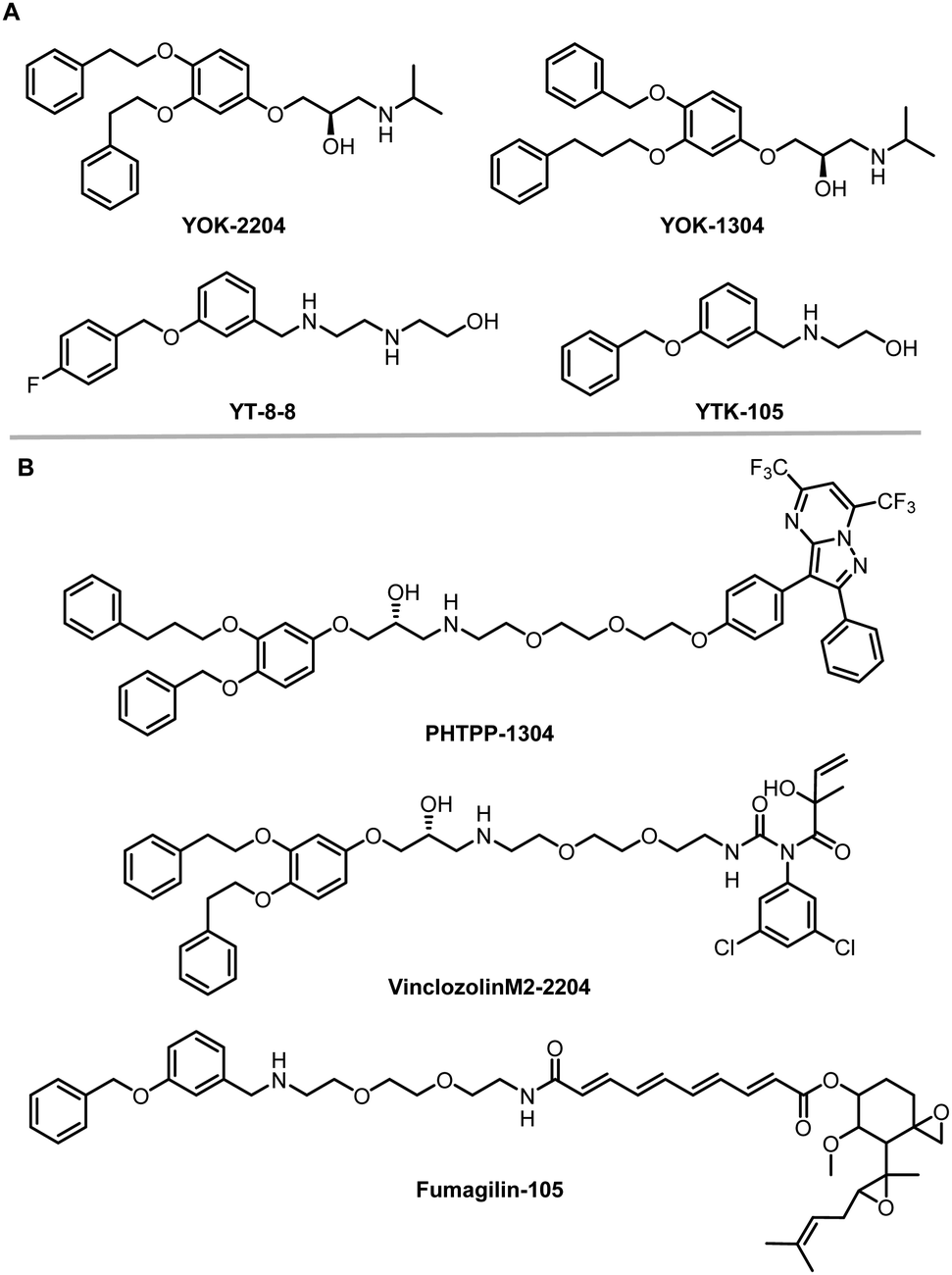 | ||
| Fig. 19 AUTOTAC compounds. (A) p62zz-binding compounds used as AUTOTAC's warheads; (B) AUTOTACs including PHTPP-1304 targeting ERβ, vinclozolinM2-2204 targeting AR and fumagilin-105 targeting MetAP2. | ||
These compounds were considered autophagy-targeting ligands (ATLs) to synthesize AUTOTACs, which are composed of target-binding ligands (TBLs) linked to ATLs via a repeating polyethylene glycol (PEG) moiety. AUTOTACs were designed for different types of targets, including cytosolic soluble proteins and misfolded protein aggregates.
The mechanism of actions of AUTOTACs was further validated by co-localization or knockdown/knockout experiments: VinclozolinM2-2204 induced the formation of AR + LC3+ puncta, and PHTPP-1304 induced SQSTM1/p62 + ERβ+ puncta; degradation of ERβ by PHTPP-1304 was abolished by knockdown of either SQSTM1/p62 or ATG5; AUTOTAC-induced ERβ or MetAP2 puncta formation was present only in wild-type but not SQSTM1/p62−/− or ATG5−/− mouse embryonic fibroblasts. Meanwhile, each AUTOTAC was tested only in some of the mechanistic experiments, and there was no biophysical validation of ternary complex formation for any of the AUTOTACs.
To illustrate whether the mechanism is independent of the classical recognition of polyubiquitinated substrates by the UBA domain of SQSTM1/p62, the authors tested AUTOTACs’ effects under the ubiquitin knockdown or proteasomal inhibition conditions. They observed better efficacy of the compounds, suggesting that AUTOTACs function via the ubiquitination-independent pathways. Meanwhile, ubiquitin is a highly abundant protein with fast turnover, and it also plays a signalling role by mono-ubiquitination.186 Thus, the ubiquitin knockdown experimental results may need further confirmation, such as by using a ubiquitin E1 inhibitor. The proteasomal inhibition experiments may not exclude the possible involvement of UBA's recognition of polyubiquitinated proteins because they are known to be also degraded by autophagy, which is not inhibited by proteasome inhibitors. This pathway is actually the major mechanism of action of AUTACs.
The therapeutic efficacy of these AUTOTACs in cancer signaling was further tested. They inhibited the downstream pathway of their targets with an efficacy that is several fold higher than that of their corresponding TBLs, suggesting that the degraders have more potent effects than simple inhibitors. Consistent with this, they inhibited cancer cell growth and progression more efficiently than their ATL or TBL moieties.
 | ||
| Fig. 20 AUTOTACs targeting misfolded protein aggregates. | ||
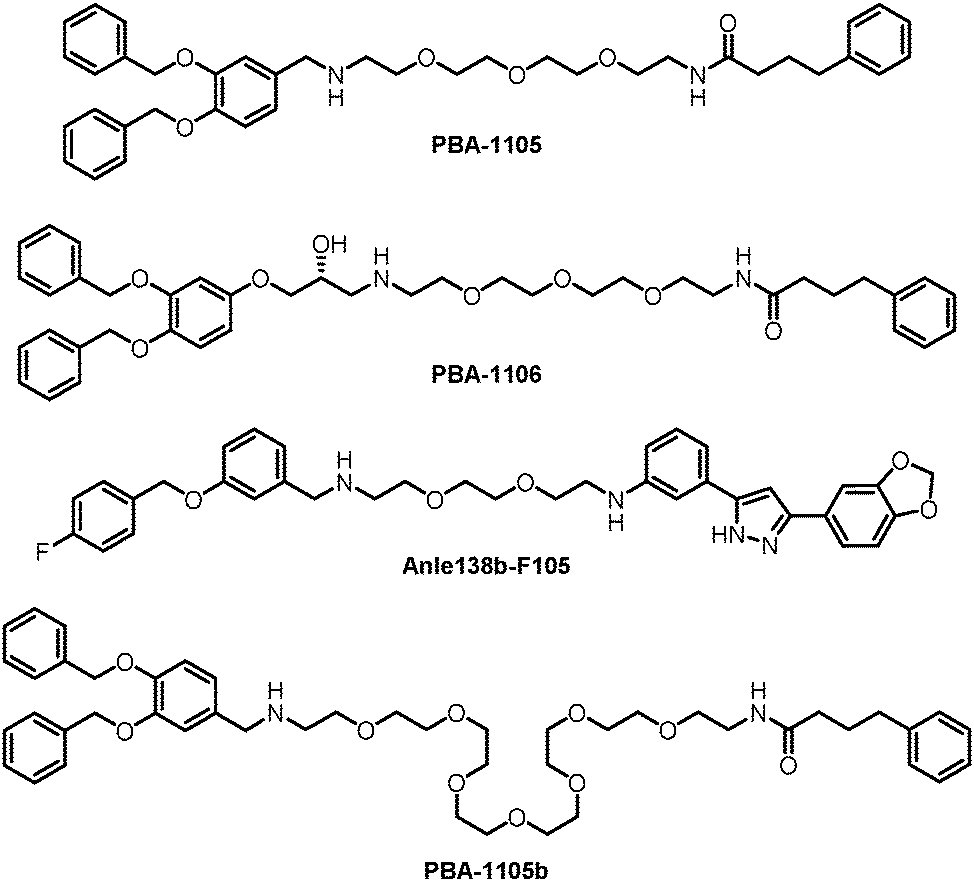
The authors then used another TBL, Anle138b, to assemble AUTOTACs. Anle138b is a phase 1 clinical trial compound that binds oligomers and aggregates of neurodegenerative proteinopathies.188 The effects and mechanisms of the corresponding AUTOTAC Anle138b-F105 (Fig. 20) are highly similar to the ones of PBA-1105.
The authors further tested these AUTOTACs in disease contexts and demonstrated that PBA-1105 and Anle138b-F105 induced the degradation of exogenously expressed mutant aggregation-prone desminL385P, a mutant protein involved in desminopathies, in a concentration- and macroautophagy-dependent manner. The wild-type desmin was not influenced, suggesting specificity on misfolded aggregates.
These AUTOTACs targeting misfolded protein aggregates were then tested for proteins involved in neurodegenerative diseases such as Alzheimer's disease. PBA-1105 and PBA-1106 induced autophagic degradation of the stably expressed aggregation-prone P301L mutant tau protein that forms neurofibrillary tangles because of its prion-like seeding behavior.189 The effects of PBA-1106 are somewhat unexpected because it did not seem to degrade ubiquitin-conjugated protein aggregates in the earlier tests. The ∼1–10 nM DC50 suggested very high efficacy. AUTOTAC-linker length may not be critical for their degradation capability because the degradative efficacy of PBA-1105b (Fig. 20), which carries a drastically longer PEG-based linker than PBA-1105, was found to be similar. Similar to PBA-based degraders, Anle138b-F105 degraded tauP301L at a DC50 of ∼3 nM. The AUTOTACs’ effects were fast and sustained, showing obvious degradation reaching a sustained maximal effect from 3 h onwards and persisting up to at least 8 hours post-washing. Remarkably, PBA-1105 AUTOTAC also showed good efficacy in clearing tauP301L aggregates in vivo in a hTauP301L-BiFC transgenic mouse model when injected intraperitoneally at 20 or 50 mg/kg three times per week for one month. This result is somewhat surprising because it is probably challenging for PBA-1105 to penetrate the BBB, considering its size. Meanwhile, relatively high concentrations and prolonged treatment were applied that possibly overcame the BBB penetration issue.
The mechanisms of tauP301L degradation were confirmed by in vitro pull-down experiments showing compound–tauP301L interaction and colocalization experiments showing autophagic targeting of tauP301L inclusion bodies. The effects were still present in the presence of the phosphatase inhibitor okadaic acid, which induced hyperphosphorylated tau. In addition, co-immunoprecipitation (co-IP) analyses were performed to test the interaction between tauP301L and a mutant SQSTM1/p62 lacking the UBA domain. Treatment of 1 μM Anle138b-F105 seemed to increase interaction, suggesting that UBA or ubiquitin recognition is not required for AUTOTACs. Meanwhile, this result might have complications. Full-length SQSTM1/p62 was not tested for co-IP, whereas SQSTM1/p62 lacking the UBA domain was not tested for degradation.
The AUTOTAC study presents a considerable amount of work illustrating the clearance of three different soluble proteins and misfolded protein aggregates. The AUTOTACs targeting tauP301L were also tested in vivo, showing promising results. Meanwhile, frequent changes in assay conditions and compounds occurred in most mechanistic experiments, making the validation of mechanisms somewhat incomplete for several AUTOTACs. More importantly, the affinities of the compound–SQSTM1/p62 or compound–target interactions were not measured. The ternary complex formation was only supported by colocalization, which could be due to autophagy recognition rather than direct proximity-induction. Finally, the proposed mechanism hijacks the Nt-R recognition by the zz domain of SQSTM1/p62, and thus AUTOTACs may interfere with the degradation of endogenous proteins with Nt-R. AUTOTACs also induce SQSTM1/p62 activation and oligomerization, which may cause additional global effects influencing other substrates of selective autophagy. These potential off-target effects may need further analyses.
Besides autophagy, another lysosomal-dependent pathway, the endosomal–lysosomal pathway initiated by endocytosis, has also been hijacked to develop degrader technologies, which will be discussed in the next section.
3. Degrader technologies engaging the endosomal–lysosomal pathway
3.1 The endosomal–lysosomal pathway
The endosomal–lysosomal pathway is another degradation pathway targeting substrate to lysosomes. Different from autophagy, the endosomal–lysosomal pathway typically internalizes transmembrane or extracellular substrates for degradation. There are several categories of this pathway depending on the cargos and the coat protein of the vesicles. The internalization mechanism of certain extracellular cargos is dependent on ligand–receptor binding, and the corresponding process is referred to as the receptor-mediated endocytosis (RME),190 which is the major pathway hijacked by relevant degrader technologies. RME allows the specificity of extracellular macromolecules and macromolecular complex internalized cells for possible degradation and thus is hijacked for degrader technologies. The pathway involves sequential processing using several membrane-bound intracellular compartments. The ligand–receptor complexes are typically engulfed in clathrin-coated pits upon binding to their transmembrane receptors.191 The pits then form endocytic vesicles that fuse with each other, giving rise to the so-called early endosomes (EEs).The subsequent sorting of these endosomes depends on the nature of their cargos. Metabolic receptors are generally recycled back to the plasma membrane, whereas signalling receptors and their ligands are delivered to multivesicular late endosomes (LEs) and finally degraded by lysosomes (Fig. 21). The latter process is the one that is relevant to degrader technologies. The somewhat vague terminology of the so-called “early” versus “late” endosomes came from the initial studies when cargos were the only marker for endosomes: they were found in tubular–vesicular structures at the cell periphery (early endosomes) in the first 5–10 min of endocytosis and in the vesicles localized in the juxtanuclear region (JNR) also containing lysosomes.192,193 Endocytosed material tends to flow vectorially through the system, proceeding through the early endosomes, the late endosomes, and the lysosomes. Two models explaining how the organelles within the endolysosomal pathway are related to each other have been proposed.194 The “pre-existing compartment model” considers early and late endosomes as stable specialized compartments linked by vesicular traffic. The “maturation model” suggests each organelle along the endocytic pathway as a transient compartment that matures into the following organelle along the pathway. The latter is currently the prevailing view, and comprehensive cellular/molecular mechanisms of endosome maturation have been reported.
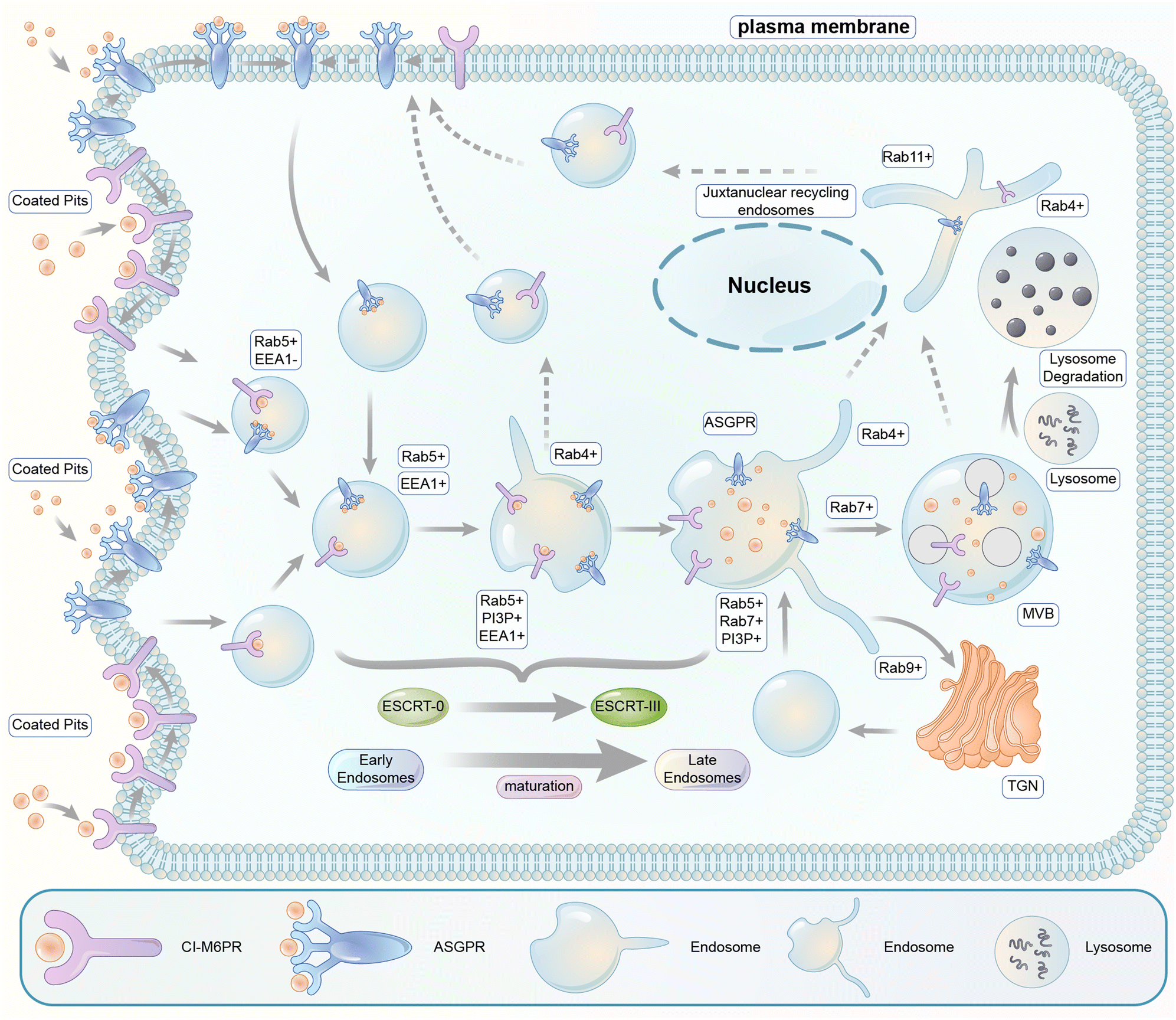 | ||
| Fig. 21 Illustration of the endosomal–lysosomal pathway. Extracellular or transmembrane proteins enter the cell through endocytosis. Following entry through engulfed coated pits, extracellular molecules undergo the processing and sorting by early endosomes. The molecules destined for degradation are passed to specialized late endosomes called multivesicular bodies (MVBs), and the receptors are typically recycled. The fusion of the MVBs with lysosomes finally degraded the targeted biomolecules. | ||
Endocytic vesicles generated from coated pits grow in size via homotypic fusion and are transformed into EEs with the primary function of cargo sorting for recycling or lysosomal degradation: the receptors recycling back into the plasma membrane and the ones directed for degradation in lysosomes are sorted into the tubular part and the vesicular part of EEs, respectively. EEs then undergo gradual composition changes via both the removal of their tubular recycling parts and their fusion with vesicles carrying lysosomal enzymes formed in the trans-Golgi network (TGN).195 Meanwhile, inward invaginations of the EE membrane occur containing the cargos, which become inaccessible to the cytoplasm after the closing and detachment of these invaginations, forming multivesicular bodies (MVBs) containing multiple vesicles. MVBs are considered LEs, which mature and fuse with the lysosomes for cargo degradation.195
Endosomes are not merely passively formed vehicles at the molecular level but a platform with many proteins tightly regulated to perform their functions. First, the ion composition of the endosome lumen is regulated by various ion channels and pumps during endocytosis. The luminal pH is gradually lowered by the incorporation of H+ ATPases and regulated by several types of cation channels/pumps providing maintenance/variation of the concentration of K+, Na+, and Ca2+ ions.196,197 The acidification of the interior space of the endosomes leads to the dissociation of ligand–receptor complexes, allowing the sorting of different ligands and receptors.198 Second, besides the ion composition, endosome sorting and maturation are mediated by the remodelling of its membrane regulated by many proteins. These proteins might be important for endosomal–lysosomal pathway-dependent degrader technologies, and we briefly introduce some of the key pathway proteins in the following text (see Fig. 21).
3.2 Key proteins of endosome sorting and maturation
Several members of the Rab family small GTPases play critical roles in the endosome pathway. Rab proteins are capable of anchoring in the target membrane of specific compartments and involved in the maintenance of membrane structure domains.199A key organizer of the endocytotic pathway is Rab5, a Rab family protein with three isoforms, including Rab5a, b, and c. Knock-down of all Rab5 isoforms below a threshold level leads to sequential disappearance of all endocytotic compartments, including early endosomes, late endosomes and lysosomes.200 Most Rab5 is in the (GTP-bound) active state and associates with endosomal membranes. The Rab5+ endocytic vesicles recruit the phosphoinositol-3-kinase Vps34, leading to the appearance of large domains enriched in phosphatidylinositol-3-monophosphate (PI3P) on the membrane of EEs.201 The emergence of PI3P at the surface of the endosome is recognized by EEA1, the major protein of early endosome tethering protein that interacts with both PI3P and Rab5. This allows tethering of endocytic vesicles to each other.202 Another Rab protein, Rab4, is located in the tubular regions of EEs and functions in recycling endosomes.203 In the endosome recycled through the long pathway via the juxtanuclear recycling endosomes, both Rab 4 and Rab11 are found on the EEs and may play a role in recycling.203 For degradative EEs, Rab5 is replaced by another Rab protein, Rab7, during EE maturation. The Rab5-to-Rab7 conversion provides a major mechanism of the progression from early to late endosomes.204 It is possibly mediated by Mon1-Ccz1, a guanine nucleotide exchange factor (GEF) of Rab7 and also an effector of Rab5.204 In addition, the composition of the vesicular part of the endosomal membrane is also influenced by removing the recycling endosomes and the influx of proteins from the trans-Golgi network (TGN) (Rab9+).205 Besides the change in the membrane Rab protein, the sorting for the lysosomal degradation pathway is also facilitated by the ubiquitination pathway. Typically, the internalized receptors or their transmembrane binding partners recruit ubiquitin ligases to induce their ubiquitination. The ubiquitinated cargos are recognized by the ESCRT (Endosomal Sorting Complex Required for Transport) complexes (0–III), which sort cargos labelled with ubiquitin into invaginations of endosome membranes and then induce their breaking off to form internal vesicles.206 This process leads to the formation of maturing multivesicular endosomes called multivesicular bodies (MVBs), which are considered as LEs.206 Finally, the Rab7 on matured LEs recruits the HOPS complex that initiates vacuole docking by tethering membranes and induces endosome–lysosome fusion.207,208
The cellular and molecular mechanisms of the endosomal–lysosomal pathway discussed above are summarized in Fig. 21, which is an over-simplified model of the pathway. Many other proteins and signalling events are involved, especially the cytoskeleton and intracellular trafficking machinery. Thus, the complexity of the pathway should be appreciated, and many influencing factors need to be considered for degrader technologies hijacking this pathway. Finally, since the substrates/cargos are internalized on the plasma membrane and isolated from the cytosol, the feasible targets for endosomal–lysosomal pathway-dependent degraders are probably extracellular or membrane proteins. In the following sections, we will discuss recently established technologies hijacking this pathway via two different receptors, the cation-independent M6P receptor (CI-M6PR) and the asialoglycoprotein receptor (ASGPR), respectively.
3.3 LYTACs and MoDE-As
Carolyn R. Bertozzi's group first reported the LYTAC (LYsosome TArgeting Chimera) technology that engages CI-M6PR to hijack the endosomal–lysosomal pathway for degradation (Fig. 22).11 CI-M6PR endogenously transports proteins labelled with N-glycans capped with mannose-6-phosphate (M6P) residues to the lysosome, whereas the receptor itself is generally recycled back to the membrane.209 The principle is similar to the endosomal–lysosomal pathway discussed in the previous section (Fig. 21). Based on its endogenous function and wide expression profile, CI-M6PR has been considered a promising docking receptor to deliver recombinant hydrolases to the lysosomes to treat lysosomal storage disorders.210 This inspired the idea of LYTACs, which deliver other proteins to the lysosomes for degradation rather than replenishing depleted hydrolyses.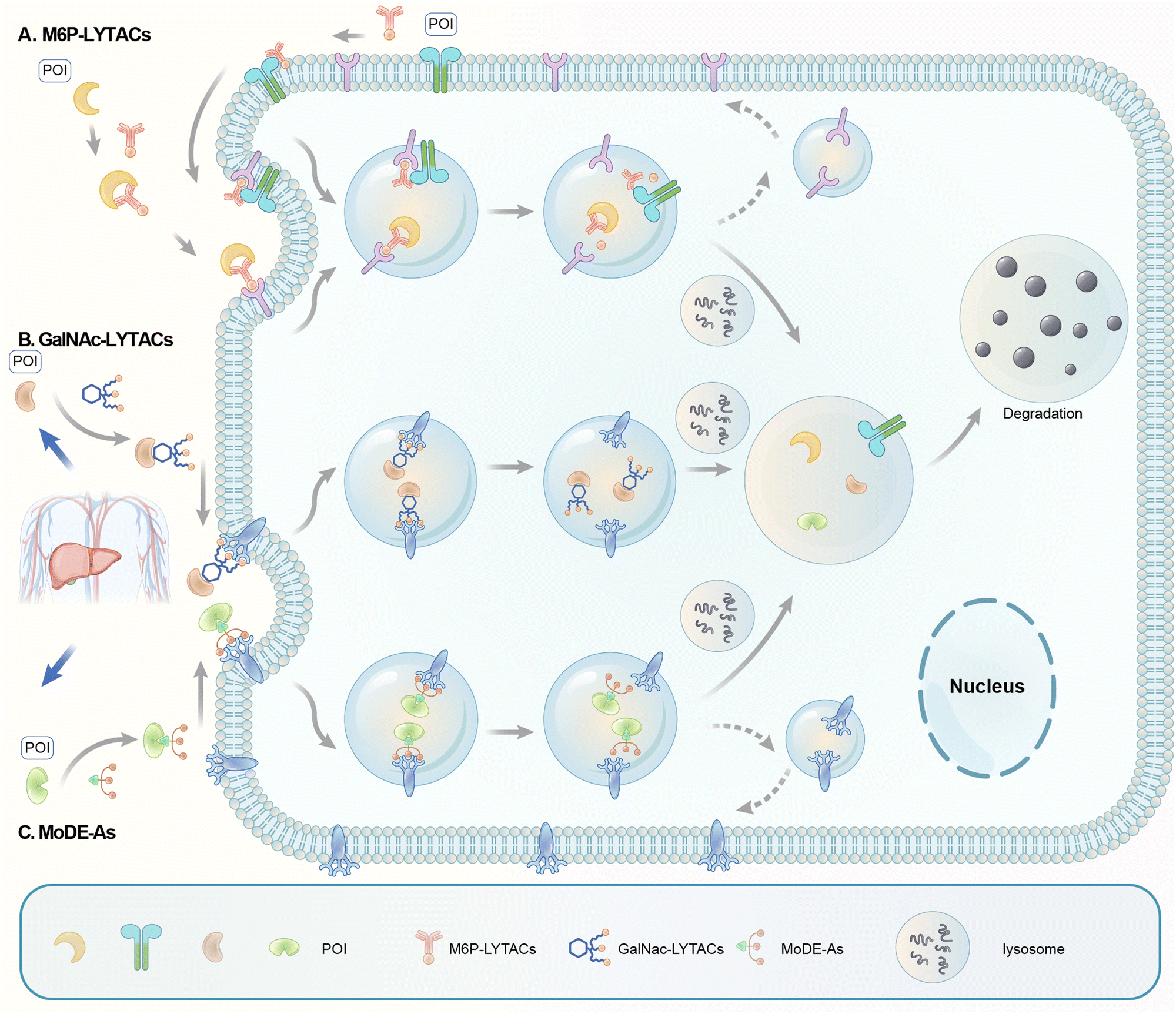 | ||
| Fig. 22 Illustration of degrader technologies engaging the endosomal–lysosomal pathway. (A) M6P-LYTACs: M6P-LYTACs utilize a glycan tag binding with CI-M6PR to mark extracellular or transmembrane POI for intracellular lysosomal degradation following receptor-mediated internalization; (B) GalNAc-LYTACs & (C) MoDE-As utilize a glycan tag binding with the liver-specific ASGPR to mark POI for intracellular lysosomal degradation following receptor-mediated internalization. | ||
To assemble LYTACs, the authors first developed ligands for CI-M6PR by leveraging precedents to enhance lysosomal enzyme replacement therapies and drug delivery platforms. Glycopolypeptides bearing multiple serine-O-mannose-6-phosphonate (M6Pn; Fig. 23) residues were used as a biocompatible phosphatase-inert ligand binding CI-M6PR presented on a modular scaffold. M6Pn glycopolypeptides of various lengths, including short (20 M6Pn) and long (90 M6Pn) variants, were tested. The uptake of extracellular proteins and their lysosomal-targeting were first illustrated using biotinylated M6Pn glycopolypeptides (LYTACs) and extracellular NeutrAvidin-647 (NA-647), an Alexa Fluor-647 (AF647)-labelled protein binding to biotin. NA-647 co-localized with acidic endosomes and lysosomes after only 1 h. In addition, co-incubation with excess exogenous M6P competed with the uptake induced by biotinylated LYTACs and the uptake remained continuous over time, suggesting that surface-receptor recycling was the rate-limiting step for LYTACs’ function.
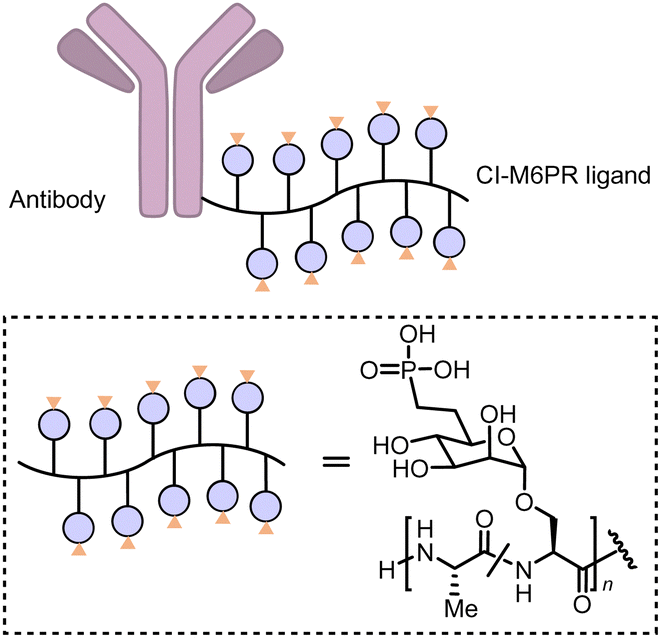 | ||
| Fig. 23 M6P-LYTACs based on the conjugation of the Cl-M6PR ligand poly (M6Pn-co-Ala) with an antibody. | ||
The authors then demonstrated that the LYTAC system could be applied as a research tool to study the CI-M6PR pathway. They performed a CRISPR interference (CRISPRi) pooled genetic screen using this system and identified the exocyst complex components as novel regulators of the pathway by influencing the surface presentation of CI-M6PR. The screening illustrates the value of LYTACs as a research tool to study the molecular pathways regulating cell-surface receptors. Notably, the screening also revealed that CRISPRi of CI-M6PR led to a substantial decrease in NA-647 uptake, providing unbiased confirmation of the engagement of CI-M6PR.
Since NA-647 is an artificial protein, to enable LYTACs to target endogenous extracellular proteins, the authors conjugated antibodies to a poly(M6Pn)-bearing glycopolypeptide to generate LYTACs in the antibody space. As a proof-of-concept, the anti-mouse IgG antibody conjugated with the M6Pn glycopolypeptide, LYTAC Ab-1, efficiently targeted a mouse IgG labelled with Alexa Fluor-488 to the lysosomes. The LYTAC Ab-1 can target not only the mouse IgG but also the mouse IgG's primary target bound to the mouse IgG to the lysosome. This was illustrated by the uptake of mCherry by cells co-incubated with mCherry, Ab-1, and a mouse anti-mCherry antibody, and the uptake of apolipoprotein E4 (ApoE4) by cells co-incubated with ApoE4, Ab-1 and a mouse anti-ApoE4 primary antibody. These extracellular targets were exogenously applied to facilitate the tracking of their uptake and lysosomal localization. The uptake or lysosomal incorporation rather than the “degradation” of the extracellular targets was measured. Demonstration of the degradation of endogenous extracellular targets would be desired in future LYTAC studies.
Besides extracellular targets, LYTACs can also degrade membrane-bound proteins such as transmembrane receptors. The mouse IgG-targeting LYTAC Ab-1 induced >80% degradation of CD71 (transferrin receptor-1) in Jurkat cells when co-incubated with a mouse anti-CD71 antibody. CD71 is a therapeutic cancer target known to cycle between early endosomes and the cell surface rather than being sorted to the lysosome (see Fig. 21 for the recycling versus degradative endosomal pathways).211 The observation suggests that LYTACs can switch the endosomal sorting routes of receptors from the recycling pathway to the lysosomal pathway. The underlying biological mechanism is intriguing because CD71 and CI-M6PR are endogenously sorted to the endosomal recycling pathway. Tethering them together with LYTACs switched CD71 to the lysosomal pathway; this may need further investigation. Noticeably, the degradation of CD71 and the inhibition of its transferrin-transport function were much more substantial than those of the antibody alone, highlighting possible advantages of LYTACs over antibodies alone.
The membrane protein-targeting LYTACs could be further simplified and optimized by directly conjugating M6Pn glycopolypeptide to the antibodies of the membrane protein targets. LYTACs were generated by conjugating M6Pn to cetuximab (ctx), an FDA-approved EGFR-blocking antibody, and an anti-PD-L1 antibody. Treatment of ctx-M6Pn (Ab-2) and anti-PD-L1-M6Pn LYTAC (Ab-3) led to predict the lowering of EGFR and PD-L1, respectively. The maximum degradation was ∼70% for EGFR and ∼80% for PD-L1. Both EGFR and PD-L1 are well-established targets for cancer treatment, and LYTACs may provide an unprecedented platform to tackle these targets. Noticeably, the in vivo clearance of Ab-2 versus ctx alone was tested, which revealed two regimes of in vivo LYTAC clearance, including a rapid initial phase and a slower but more sustained clearance. The in vivo pharmacodynamics of LYTACs on target degradation remains to be tested, possibly after understanding and optimizing these two regimes.
Significant mechanistic and functional work was carried out for the EGFR degrader Ab-2. Degradation of EGFR was observed only by treatment of Ab-2 but not ctx alone, while no changes in the CI-M6PR levels were observed. This observation suggests that EGFR and CI-M6PR may have been sorted into the degradative and the recycling endosomes, respectively. This is consistent with the general principle of the endosomal–lysosomal pathway discussed earlier, in which the substrates are dissociated with the receptor (CI-M6PR) at the acidic pH in the endosomes. The degradative function of Ab-2 is mediated by CI-M6PR, as validated by the CRISPR knock-down and M6P competition experiments. In addition, the lysosome dependence was validated by the lysosomal inhibitor chloroquine. The papain-digested version of Ab-2, Fab-1, which binds to EGFR in a monovalent fashion, induced the degradation of EGFR in a CI-M6PR-dependent manner with a potency similar to that of Ab-2. This excludes the possibility that the degradation was triggered by the crosslinking of EGFR to form dimers. Proteomics analyses were also performed to confirm EGFR lowering. Other co-lowered proteins may represent EGFR interaction partners, co-regulated molecules, or possible off-target effects that need further investigation. From the functional perspective, cells were treated with ctx or Ab-2 and then washed and treated with EGF. EGF activation was further analysed by the rapid phosphorylation of Tyr1068 of EGFR in the HeLa cells or the downstream phosphorylated Akt level in the Hep3B cells. Compared to ctx alone, Ab-2 led to much more potent inhibition of the EGFR activation by EGF, demonstrating possible advantages of LYTACs over inhibitory antibodies. Consistent with this, Ab-2 also inhibited the proliferative effects of EGF stimulation in cancer cell line HepG2 relative to the treatment of ctx alone. The in vivo efficacy of cancer treatment remains to be tested. The dose-dependence of both degradation and potential therapeutic effects also needs characterization.
LYTACs expand the target spectrum of degrader technology to extracellular and membrane proteins. Besides the proof-of-concept study using CI-M6PR, in principle, other endosome-shuttling receptors could also be co-opted for LYTACs, and cell-type-specific ones might provide an avenue for cell-type restricted protein degradation.
Based on this idea, Bertozzi's group developed triantenerrary N-acetylgalactosamine (tri-GalNAc; Fig. 24) conjugated antibodies or peptides as a new type of LYTACs (GalNAc-LYTACs), which engage a liver-specific lysosomal targeting receptor ASGPR (the asialoglycoprotein receptor).12 A back-to-back paper from David A. Spiegel's group reported a small molecule version of the technology called the MoDE-As (Molecular Degraders of Extracellular proteins through the asialoglycoprotein receptor ASGPR). Noticeably, antibodies conjugated with GalNAc were used as negative controls that showed no effects in cells that probably do not express ASGPR, suggesting the possibility of developing cell-type-specific LYTACs. Both GalNAc-LYTACs and MoDE-As utilize tri-GalNAc as the ASGPR-binding moiety. Tri-GalNAc has been established as an ASGPR ligand with low nanomolar affinity and promising efficacy/safety profiles.212–214 The homogeneous nature of the tri-GalNAc ligand enables precise characterization of the ligand to antibody ratios, which is hard to measure for heterogenous M6Pn polymers utilized in the initial LYTAC study.11
 | ||
| Fig. 24 GalNAc-LYTACs based on the conjugation of the ASGPR ligand. | ||
In the GalNAc-LYTAC study,11 Tri-GalNAc conjugated ctx (GalNAc-ctx) caused >70% degradation of both total and surface EGFR in several hepatocellular carcinoma (HCC) cells, but not HeLa cells, consistent with the exclusive expression of ASGPR in hepatocytes.215 A dose-dependence study showed a DC50 of ∼1 nM and maximum degradation achieved at 10 nM or higher concentrations without showing obvious “hook” effects. The downstream pathway kinase phosphorylation confirmed the functional effects. The ASGPR-dependence was confirmed by its knock-down experiments, and the endosomal–lysosomal pathway dependence was confirmed by the lysosomal inhibitor bafilomycin A1 or chloroquine. Meanwhile, the lysosome functions were not impaired by LYTACs, at least not to an extent more severe than the corresponding antibodies alone. ASGPR-directed LYTACs may also degrade other HCC targets such as HER2.
Besides illustrating the possibility of hijacking a cell-type-specific receptor for LYTACs, the study further presents two advances in the LYTAC technology: generating LYTACs using synthetic peptides and in vivo pharmacokinetics studies of site-specific LYTACs. A peptide LYTAC PIP-GalNAc was generated by conjugating tri-GalNAc to a polyspecific integrin-binding peptide (PIP), which was previously engineered to bind to several tumor-associated integrins with high affinity.216 PIP binds to αvβ1, αvβ3, αvβ5, αvβ6, and α5β1 integrins, which are known cancer targets that are elevated in various cancer types.217 PIP-GalNAc led to an apparent lowering of surface αvβ3 and αvβ5 integrins, whereas the PIP treatment alone only caused a slight decrease of αvβ3 and an increase of αvβ5. Cell proliferation assays validated the potential therapeutic effects on PIP-GalNAc. PIP-GalNAc exhibited ASPGR-dependent and wash-out persistent anti-proliferation effects that were much stronger than the PIP treatment alone, suggesting that LYTACs can significantly enhance functional effects compared to the parent binder alone. Noticeably, the effects were tested at 200 nM, and complete dose-dependence may need further investigation. It will also be interesting to measure the total level of integrin to determine if PIP-GalNAc induced degradation or localization changes of integrins. Noticeably, PIP-GalNAc was generated by site-specific conjugation via a single tri-GalNAc-DBCO moiety, illustrating that a single tri-GalNAc ligand conjugated to a proper site might be sufficient to induce efficient degradation. This enables the study of architectural features for LYTAC activity optimization, which was not feasible for non-specific conjugation-based LYTACs. The tri-GalNAc ligand was conjugated to the EGFR antibody ctx or the HER2 antibody pertuzumab (Ptz) at their C-terminus, hinge, or CH1 heavy chain in a site-specific manner. The specific conjugation site influenced both degradation and in vivo pharmacokinetics in a mouse model, but the optimal site may differ for different antibodies.
MoDE-As hijack the same receptor and utilize tri-GalNAc as the ASGPR ligand. Meanwhile, they are within the small molecule modality and thus expand the LYTAC concept to the small molecule space, allowing the small, monodisperse, and nonprotein-based design of the compounds and minimizing the autoimmune responses. Two MoDE-As methods, the D-MoDE-A and the M-MoDE-A (Fig. 25),13 were designed to target the α-DNP antibodies and the migration inhibitory factor (MIF), respectively. Experimental results confirmed that MoDE-As induced degradation of their target likely via the predicted mechanisms. The study tested the formation of a ternary complex between hepatocytes and α-DNP antibody by testing the surface level of the α-DNP antibody after D-MoDE-A treatment. The experiments were carried out below 0 degrees Celsius (on the ice) to inhibit the endocytosis of the cells. The dose-dependence showed strong “hook” effects. While this is not a direct validation of the ASGPR–compound–bait ternary complex, the results provide additional evidence for the predicted mechanism.
 | ||
| Fig. 25 MoDE-As based on tri-GalNAc as the ASGPR ligand. | ||
The ASGPR-dependence was validated by several competition experiments using a set of competitors different from tri-GalNAc alone used in the GalNAc-LYTAC study. Meanwhile, ASGPR knock-down experiments were not performed. The results provide compensatory evidence of ASGPR's involvement. A highlight of the mechanistic experiments of the MoDE-A study is that the endosomal–lysosomal pathway dependence was further validated by several inhibitors, including the global endocytosis inhibitors sodium azide (NaN3) and 2-deoxyglucose (DOG), the clathrin-dependent endocytosis inhibitor sucrose, and the endosome/lysosome acidification inhibitor bafilomycin, chloroquine, and monensin. Meanwhile, the inhibitors of other endocytosis pathways had no effects. The set of inhibitor experiments further suggests the involvement of RME, although genetic knock-down or knockout experiments are desired for further validation. MoDE-As’ effects were further tested in vivo. They were pretty unstable, with a plasma half-life of less than an hour, likely shorter than that of LYTACs. Meanwhile, MoDE-As still accelerated the depletion of their targets. The effects were sustained with daily, twice-daily, or even single-dose injections, likely because MoDE-As induced degradation more rapidly than it is eliminated. On top of this, MoDE-As’ targeted proteins were injected and not replenished in the mice, leading to sustained effects even by a single injection. The MoDE-As was also highly tolerated, possibly due to their rapid clearance, even at relatively high concentrations.
The LYTAC and MoDE-A studies cleverly hijack the endosomal–lysosomal pathway based on previously established delivery technologies and expand the target spectrum of degrader technologies to extracellular and membrane proteins. For extracellular targets, only exogenously added or injection proteins were tested, and application of the technologies on endogenous extracellular proteins is desired. For membrane targets, only LYTACs have been tested, and whether small molecule MoDE-As could be applied to these proteins needs further investigation. Several mechanistic questions are worth further investigation and will be discussed in the next section with the ones of autophagy-based degrader technologies.
4. MoA of degraders engaging lysosomal pathways
4.1 Pathway-dependence
For ATTECs, AUTACs and AUTOTACs, the predicted degradation mechanism is autophagy (Fig. 2 and 8). In addition, AUTACs and AUTOTACs are dependent on SQSTM1/p62, while ATTECs are presumably independent of it. For LYTACs and MoDE-As, the predicted degradation mechanism is the endosomal–lysosomal pathway (Fig. 21 and 22), which includes membrane receptor-mediated internalization (endocytosis), endosome maturation, and lysosomal degradation.There are two major ways to validate the pathway-dependence: pathway inhibitors and knock-down or knockout of key pathway genes. Competition experiments may also provide evidence to validate the pathway, but usually, they are used to validate the engagement of the anchoring proteins, which will be discussed in the next section. The lysosome inhibitors such as bafilomycin A1, chloroquine and NH4Cl, neutralize the lysosomal pH to prevent lysosomal degradation.6,218,219 These inhibitors block both the autophagy and the endosomal–lysosomal pathways, and one or more of them were used in each of the studies discussed above. Noticeably, these inhibitors may also block microautophagy and CMA, which are independent of the LC3 conjugation system and the endocytosis pathway.6 Thus, inhibition of earlier steps of autophagy (macroautophagy) is desired to confirm the autophagy-dependence of ATTECs/AUTACs/AUTOTACs. For example, the PtdIns3K inhibitor 3-methyladenine (3-MA) or SAR405 blocks autophagosome initiation, and they may provide more specific evidence for autophagy-dependence.220,221 Meanwhile, these compounds may also inhibit other kinases and induce opposite effects on autophagy under certain conditions, making data interpretation more complicated.6 In the MoDE-As, the endocytosis inhibitors sodium azide (NaN3), 2-deoxyglucose (DOG) and sucrose have been used, but they have other non-specific effects as well. In general, the chemical inhibitors may provide solid supportive evidence for pathway-dependence.
Meanwhile, given the limitation of the specificity of chemical inhibitors, genetic knockout or knockdown of key pathway genes probably provides better evidence. In the ATTEC/AUTAC/AUTOTAC studies, knockout or knockdown of ATG5 or ATG7 was tested and showed significant blockade of the degradation effects of ATTECs.7–10,158 Such evidence was largely missing in the studies of endosomal–lysosomal-based degrader technologies and might be desired in future studies. For example, sufficient knockdown of Rab5 leads to the disappearance of endocytic compartments, and thus, it could be used to confirm the pathway-dependence of LYTACs/MoDE-As.200
All the evidence for autophagy-dependence in previous reports was obtained in cellular models, and in vivo validation is desired. The best way to achieve this goal is to use genetic models with autophagy deficits by knockout one of the key autophagy genes. Unfortunately, such knockout (for example, Atg3, Atg5, Atg7, Atg9a, Atg12, Atg13 or Ulk1/2 deletion) causes embryonic or neonatal lethality in mice, making it difficult to perform in vivo experiments to validate autophagy dependence.222 Knockout of several other key autophagy genes does not lead to lethality, but they are functionally redundant in autophagy.222 Meanwhile, such experiments could still be performed in conditional knockouts of these genes, although extensive efforts are needed to find the suitable ages and tissues of mice for these experiments. The pathway dependence could also be investigated oppositely: enhancing autophagy or endocytosis. On the other hand, data interpretation might be complicated because enhancing the pathway may have negligible effects in certain scenarios. For example, autophagy enhancement by rapamycin only marginally increased polyQ·ATTECs, possibly because the baseline autophagosome number is probably adequate for polyQ·ATTECs to tether mHTT protein molecules for degradation, at least under the conditions tested.9 This is somewhat consistent with the observation that the total HTT protein contributes to only a tiny fraction (∼ 0.01%) of the whole proteome.9 In contrast, the effects of LD·ATTECs and BRD4·ATTECs were significantly enhanced by the treatment with the autophagy enhancer rapamycin.10,144,223 The exact mechanistic explanation is unclear, but LDs and BRD4 may require a more significant number of autophagosomes for degradation because of LDs’ high level in the OA-induced cells and BRD4's limited accessibility to autophagosomes due to its nuclear localization, respectively.
In control experiments, it is also essential to test the potential involvement of poly-ubiquitination and proteasomes. The AUTOTAC study tested the proteasome inhibitors, which showed enhancement rather than inhibition on the degradation effects of AUTOTACs, possibly because of a compensatory activation of autophagy.8 The poly-ubiquitination of the target proteins was tested in the AUTAC studies, consistent with the proposed mechanism of action. Meanwhile, such experiments have not been performed for other studies, and they are desired to provide additional validation of the proposed mechanisms.
The LYTAC-mediated degradation of membrane receptors may raise another pathway-related question related to the sorting of endosomes. Receptors internalized in the endosomes may be sorted to the recycling endosomes and reused in the cytoplasmic membranes. Thus, enhancing these receptors’ internalization (endocytosis) by LYTACs or their ligands alone may not be sufficient to degrade them, and overturning the recycling program is likely required. LYTACs seemed to be capable of doing this, especially compared to the controls using the ligand alone. Meanwhile, as discussed previously, the LYTAC-engaged receptor CI-M6PR mainly goes to the recycling pathway, and M6P dissociates from CI-M6PR in the endosome. Thus, it is likely that LYTACs continue to function in the endosomes after endocytosis in a CI-M6PR independent manner to influence the sorting of the targeted receptors. There might be interesting novel biology underlying this mechanism, which could be essential to understand how LYTACs may function and whether their functions are dependent on other unknown components in the cells. A CRISPRi screen might be desired to address this issue using CD71 or PD-L1 targeting LYTACs, with an experimental design similar to the NA-647 CRISPRi screen carried out in the original LYTAC study.11
Finally, another important aspect related to pathway dependence is the potential changes in global pathway activities. ATTECs interact with LC3 and thus may influence the global autophagy activity by interacting with LC3 and changing its conformation. Similarly, AUTACs and AUTOTACs may influence the selective autophagy pathway, whereas LYTACs/MoDE-As may influence the endosomal–lysosomal pathway. The global pathway activity has been tested in some studies for ATTECs and MoDE-As, ensuring no significant changes. Meanwhile, certain binders do influence the global activity, and thus, the possible global pathway activity changes are desired to be tested in every study.75
4.2 Target engagement of predicted anchoring proteins
Three major approaches have been used to validate the engagement of the predicted anchoring proteins: genetic deletion/knockdown experiments, competition experiments, and direct biophysical validation of the compound–protein interactions, and label free cellular thermal shift assays (CETSA) may also be applied to validate the engagement.For the first approach, the deletion or knockdown of the anchoring proteins (i.e., LC3, SQSTM1/p62, CI-M6PR, ASGPR) is predicted to ameliorate and block the degraders’ effects. For example, knockout of LC3B in the 293T cells, the major LC3 family protein expressed in these cells, significantly reduced the effects of LD·ATTECs, although the knockout effects for other ATTECs remain to be tested.10 Meanwhile, knockout of the ATG8 family members by genome editing leads to the production of smaller autophagosomes and slower formation of autophagosomes.45 Thus, the ameliorated ATTECs’ effects may also be explained by the deficient autophagosome formation rather than direct engagement of LC3. Knockdown or knockout of SQSTM1/p62 significantly impaired the degradation effects of AUTACs and AUTOTACs;7,8 knockdown of CI-M6PR and ASGPR suppressed the effects of M6Pn-LYTACs and GalNAc-LYTACs, respectively.11,12 These experiments are based on two major assumptions: the deletion or knockdown of the target gene does not cause secondary changes that may mediate the degraders’ effects, and no compensatory mechanisms exist to rescue the degradation effects. Thus, while the genetic deletion/knockdown experiments support justifying the predicted degrader mechanisms, additional mechanistic validations are desired. If the binding sites were resolved for the anchoring proteins, point mutagenesis to abolish the bind sites would give a more convincing proof of the anchoring proteins. No such experiments have been performed for the lysosome-dependent degrader technologies at this point, likely due to a lack of knowledge of the binding sites, which are likely to be revealed in the near future.
For the second approach, the competitor compounds may interact with the anchoring proteins to block their interaction with the degrader, thus reducing the degradation effects. For example, co-incubation of M6P reduced the M6Pn-LYTACs’ effects; treatment of triGalNAc or other ASPGR ligands reduced the effects of GalNAc-LYTACs and MoDE-As.11,12 This approach is also based on the assumption that the binding of the competitor compounds blocks the binding of degraders and that the competitor compounds do not influence other factors required for the degraders’ functions. To achieve more stringent conclusions, competitor compounds could be tested for an independent degrader as a control experiment, i.e., MoDE-As’ competitor compounds could be tested against the effects of M6Pn-LYTACs to ensure that these compounds do not influence the global endocytosis machinery or other key components that might be required for M6Pn-LYTAC-mediated degradation. Besides competition experiments based on competitor compounds, competitor proteins that lack biological activity and compete with the functional protein to sequester the degrader compounds could also be used. For example, the LC3ΔG120 protein, which cannot be lipidated and form autophagosomes, could be used to compete with normal LC3 to sequester ATTECs and prevent them from functioning, leading to reduced efficacy. Similarly, the SQSTM1/p62 protein with the zz domain deleted could also be used to validate the AUTOTAC mechanisms.
The binding of small molecular weight compounds to POI may also be verified by Thermal shift assays (TSAs) because the binding stabilized the POI and increased protein thermal stability. Cellular thermal shift assays (CETSAs) are developed based on TSAs in a more native context in the cultured cells for large-scale screening of the molecular interaction between POI using standard microtiter plate formats.224 This method can be widely used in high-throughput compound discovery and subsequent lead compound optimization. CETSA avoids the tedious steps of purifying target proteins. The traditional CETSA method uses whole cells or cell lysates mixed with candidate compounds, and then aggregated proteins are removed by centrifuge. The soluble target proteins are directly detected by western blot.225 However, the western blot throughput is not satisfactory. Several research groups thus combined high throughput AlphaScreen/AlphaLISA technology and fuse reporter genes to POI (including fragmented Luciferase, GFP, and β-galactosidase, etc.), and CETSA finally developed into a high throughput method.226,227 CETSAs can also be used to confirm cell-based target engagement because of a similar mechanism of action.
These three approaches demonstrate the engagement of anchoring proteins in the cells, providing biological conditions and introducing potential secondary indirect mechanisms that may complicate data interpretation.
Thus, the fourth approach, direct biophysical measurement of compound–target interactions in vitro, may provide critical compensatory validations of the predicted engagement of anchoring proteins. Several potential technologies are briefly introduced as follows. Surface plasmon resonance (SPR) and biolayer interferometry (BLI) are label-free techniques that measure binding kinetics of biomolecular interactions in real-time, providing the affinity of compound–protein interactions.228–232 Oblique-incidence reflectivity difference (OI-RD) can also detect the real-time interaction between compounds and label-free proteins. It is usually performed in a different configuration, in which the small molecules are covalently attached to the microarray chip with the proteins flowing through.150,233–235 This may give better signal to background given the relatively large size of the proteins. Fluorescence polarization (FP) technology measures the molecule rotation of the compounds upon their binding to the target protein and is widely used to study protein–compound interactions in solution.236 The compounds and proteins are not fixed in FP experiments, mimicking intracellular interaction conditions better than the chip-based technologies mentioned above. Meanwhile, the measurement is dependent on the fluorescent labelling of the compounds.
Another assay with both proteins and compounds moving freely in the solution is Isothermal Titration Calorimetry (ITC).237,238 It is also a label-free technique that may have advantages over the label-dependent FP approach. ITC measures the heat released or absorbed upon binding and gives the thermodynamic parameters of the compound–protein binding, including affinity (KA), enthalpy (ΔH), entropy (ΔS), and stoichiometry (n). The enthalpy and entropy measured by ITC reflect different interactions and provide critical determinants of important drug properties. Strong hydrogen bonds and van der Waals interactions between the compound and POI reflect the favorable enthalpic change. In contrast, the unproductive desolvation of polar groups reflects an unfavorable enthalpic change. On the other hand, the desolvation of nonpolar groups reflects favorable entropy.
Compared with ITC, microscale thermophoresis (MST) is based on a similar principle but requires much fewer samples. The principle of MST is to quantitatively detect a temperature-induced change in the fluorescence of a target as a function of the concentration of a non-fluorescent ligand.239,240 Like ITC, MST is also an immobilization-free technology. The disadvantage of MST is that it can only give the KD but no other thermodynamics parameters. Furthermore, the tested proteins need to be labelled using specific fluorescent probes. Finally, pull-down of the target protein by compound-conjugated resins or columns is a direct approach to demonstrate the compound–protein interaction.
Many of these technologies mentioned above, such as SPR, OI-RD, MST, and FP, have been utilized to measure the affinity/kinetics of compound–LC3 interactions in ATTEC studies.9,76,144,158 In addition, pull-down experiments have been applied to validate the compound–SQSTM1/p62 interactions in the AUTOTAC study.8
4.3 Ternary complex formation
Mere interactions between degraders and the anchoring proteins are insufficient to trigger degradation, which requires the formation of transient or stable ternary complexes that trigger the entrance into the degradation machinery. Direct validation of the ternary complex formation in some of the ATTEC and MoDE-A studies might be desired for other degrader technologies.A pull-down assay followed by subsequent western-blot visualizations or ELISA measurements is a relatively straightforward way to test if ternary complexes formed in vitro. In the LD·ATTEC, the ternary complex formation was validated by pull-down followed by ELISA.10 In the polyQ·ATTEC study, the pull-down assays validated the ternary-complex formation by immunoprecipitating either the LC3B or the mHTT protein, showing consistent results. In certain circumstances, the compound may have a high affinity to only one of the proteins, similar to IMiDs with a high affinity only to CRBN. If this is the case, the pull-down assay may only work in one direction if sequential steps are performed: (1) incubating the compound with one protein conjugated to the resin; (2) removing free compound molecules by washing the pull-down solution; and (3) adding the other protein to the resin. If the compound functions as a molecular glue and only interact with one protein, it could be removed during the second step if the resin-bound protein does not interact with it. Noticeably, the polyQ·ATTEC pull-down experiments were performed in this way. The results suggest that the compounds interact with both polyQ proteins and LC3B, consistent with the observed “hook” effects of the compounds.9 The limitation of this method is that it is low-throughput and semi-quantitative nature.
The MoDE-A study validated the ternary complex by measuring the fluorescently labelled target (the α-DNP antibody) on the cell surface by flow cytometry. The assays are more quantitative and exhibited a bell-shaped dose-dependent curve consistent with the predicted prozone effects of ternary complex formation.13 Meanwhile, the assay measures the ternary complex formation with the cell membrane rather than the anchoring protein (ASGPR) directly.
For high-throughput and quantitative assessment of ternary complex formation, HTRF (Homogeneous Time Resolved Fluorescence) and AlphaLISA (amplified luminescence proximity homogeneous assay) are the widely used in TPD studies.241–243 Meanwhile, these technologies have not been used for lysosomal-based degrader technologies yet. In future studies, they may provide valuable tools to screen for or optimize degraders.
It is worth noting that most of the approaches mentioned above are based on antibodies, which may have non-specific binding to proteins. Thus, proper negative controls using other proteins and/or inactive analogs of degraders are desired to validate the conclusion. Antibody-independent biophysical methods such as the ones detecting binary interactions, including SPR, ITC, and MST, could also be used to test ternary complex formation. Since the design is pretty straightforward, we will not expand the relevant discussion here. The poor solubility of some of the degrader compounds may present a hurdle to these biophysical assays. García Jiménez et al. investigated the solubility of 21 commercial PROTAC degraders; 13 of 21 degraders were found to be poor soluble, which may have a negative impact on various stages of the drug discovery process.244 Solubility of the compounds was correlated with lipophilicity descriptors (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), while the ATTEC compounds GW5074 and ispinesib's logP values are 5.31 and 5.56, showing that they are poor soluble in water.245 Thus, novel methods to quantify the formation of ternary complexes with high sensitivity and that require a low concentration of compounds are desired.246 Mass photometry can accurately measure the mass of single molecules in solution without labels, and may provide a promising method to quantify the formation of ternary complex, especially for compounds with poor solubility.247–249
P), while the ATTEC compounds GW5074 and ispinesib's logP values are 5.31 and 5.56, showing that they are poor soluble in water.245 Thus, novel methods to quantify the formation of ternary complexes with high sensitivity and that require a low concentration of compounds are desired.246 Mass photometry can accurately measure the mass of single molecules in solution without labels, and may provide a promising method to quantify the formation of ternary complex, especially for compounds with poor solubility.247–249
4.4 Intracellular recycling of degraders
PROTACs have an advantage over traditional inhibitors by utilizing an event-driven MoA with a catalytic nature. In theory, the compounds could be reused after poly-ubiquitination and degradation.250 This may explain why the effective concentrations of PROTACs are usually lower than their affinity (KD) to the E3 ligase subunits.251 This principle may also apply to autophagy–lysosomal based degraders because the major function of autophagy is to recycle metabolites in the cells.The lysosomes not only function as degradation machinery but also as cellular storage compartments.252 Numerous transporters on the lysosomal membranes export metabolites from the lysosomal lumen to the cytosol to reuse them, which could be generated by the breakdown of large biomolecules or organelles in the lysosomes.252 Thus, small molecule degraders hijacking the autophagy pathways, including ATTECs, AUTACs, and AUTOTACs, may enter lysosomes and then get released. The degraders may then be exported to the cytosol for their reuse, at least in theory. Thus, ATTECs, AUTACs, and AUTOTACs likely function in a reusable and catalytic manner as well, similar to the proposed properties of PROTACs. This may also explain the observed phenomenon that the effective concentrations of the published ATTECs, such as polyQ·ATTECs, BRAD4·ATTECs, and NAMPT·ATTECs are lower than their KD of LC3-binding.9,158
On the other hand, the LYTACs are larger molecules and likely degraded in the lysosomes, preventing their reuse. The MoDE-As are small molecules that could be released into the cytosol after entering the lysosomes, but they need to function extracellularly to interact with ASGPR. Thus, recycling MoDE-As as degraders is unlikely, although still possible by exocytosis that may further release MoDE-As to the extracellular space. Recycling-related issues for LYTACs and MoDE-As, as indicated by their pharmacokinetics studies, are that they are pretty unstable and have a relatively short half-life in vivo.11–13,253 This could be contributed by the internalization and degradation of these degraders via the endosomal–lysosomal pathway, which is required for their functioning. Thus, the space for optimizing their stability might be limited, although site-specific conjugation of triGalNAc does have some effects.12 Meanwhile, the effects of certain LYTACs were wash-out persistent, and the underlying mechanisms may desire further study to optimize in vivo dosing.
The evidence of the recycling of the lysosomal-dependent degraders is currently missing and technically challenging. Craig Crews’ group has illustrated the catalytic activity of PROTACs based on the modelling of in vitro poly-ubiquitination assay results,254 which is inapplicable for ATTECs, AUTOTACs, LYTACs and MoDE-As, which require complete cellular pathways that involves multiple organelles for possible recycling of degraders. Meanwhile, AUTACs function via poly-ubiquitination and thus could be tested for potential catalytic activity using in vitro assays, although the molecular mechanism and components of the S-guanylation induced poly-ubiquitination are needed to be resolved before carrying out such experiments. Investigations of the catalytic activities in the cells are mainly missing for all degraders. These could be potentially performed by subcellular tracking the compounds in different cellular compartments at other time points, ideally at the single-cell level.
4.5 Lessons from PROTACs
The MoA of PROTACs is certainly more established compared to the lysosome-based degraders, partially due to the fact that many more groups have been on PROTACs. Meanwhile, many mechanistic studies on PROTACs provide valuable information that may benefit the study of lysosome-based degraders. The evidence of UPS-proteasomal pathway dependence is largely based on inhibitor or genetic knockout experiments.137,255,256 Similar experiments were performed for autophagy-based degraders by applying autophagy inhibitors or knockout of key autophagy genes.7–10 Such experiments were desired for endosome–lysosome-based degraders by knocking out key pathway genes such as Rab7. An additional pathway demonstration of PROTACs is the enhanced poly-ubiquitination of the target protein,257 which has not been tested for most lysosome-based degraders including ATTECs, AUTOTACs, LYTACs, and MoDE-As. Poly-ubiquitination changes are desired to be tested in future studies as control experiments, considering that these lysosome-based degraders are expected to function independently of the UPS pathway. The target engagement of PROTACs is usually validated by in vitro biophysical binding experiments, knockout experiments, and/or competition experiments.255,258 All these experiments have been performed in one or more studies of different lysosome-base degraders as discussed above, although this experimental set is typically incomplete in each single study possibly due to lack of tools or resources. This could be further improved in future studies. The largest piece of missing evidence of the MoA of lysosome degraders compared to the one of PROTACS is the structural information of the ternary complexes. As discussed in section 6, a number of ternary complexes of PROTACs and relevant molecular glues have been resolved at the atomic level,258,259 providing definitive evidence demonstrating the PROTAC MoA and valuable information for compound optimization. Such evidence is missing in all the lysosome-based degraders, including ATTECs, AUTACs, AUTOTACs, LYTACs, and MoDE-As. We and several other groups are making such efforts and have obtained some progresses. Meanwhile, compounds with better affinity and solubility are desired to facilitate such efforts.The mechanistic insights of the degraders’ functions may provide key information for the optimization of the degraders and dosing strategies and the selection of new targets for these technologies. We will discuss the target selection strategies as follows.
5. The target selection strategies
Lysosomes are capable of degrading many types of substrates, including proteins, protein aggregates, damaged organelles, etc.260,261 Thus, the theoretical target spectrum of lysosome-engaging degradation technology is broad, including essentially all large biomolecules and organelles. Meanwhile, several factors need to be considered in the target selection for each technology. They may significantly impact the success rate of identifying degraders with high efficacy and potency. These factors are discussed as follows:5.1 Biological/clinical evidence
Degrader technologies need to be applied to pathogenic biomolecules/organelles to provide possible therapeutic benefits. In contrast, degradation of targets with key physiological functions may have harmful effects. Human genetic studies may reveal the genetic mutations that cause specific diseases, but mutated gene products (proteins) may cause the disease via either the loss-of-function (LOF) or the gain-of-function mechanisms. For the LOF mechanism, degrader technologies including lysosome-engaging ones may not provide any therapeutic benefits because restoration of the protein function rather than ablation of the protein is required to treat such diseases. Thus, biological/clinical evidence demonstrating a gain-of-function mechanism is required for the lysosome-engaging target selection. In some cases, disease-relevant proteins are identified using pathological and biochemical experiments, which typically reveal correlative but not causal relationships between the proteins and the diseases. Additional biological/clinical evidence is also needed to establish the causal connection. Ideally, experimental validation that the lowering of the target provides therapeutic benefits in patients or at least in animal models is desired for target selection of degrader technologies. This could be potentially tested using gene therapy approaches.5.2 Subcellular localization
The desired target subcellular localization of autophagy-based degraders (ATTECs, AUTACs and AUTOTACs) and the one for endosome–lysosome-based degraders (LYTACs and MoDE-As) are different. Based on the current knowledge, autophagy mainly occurs in the cytoplasm, where autophagosomes are formed.262 Thus, compared to extracellular targets or targets in other subcellular localizations, cytoplasmic targets are more likely to be degraded by autophagy-based degraders, and they should be prioritized during target selection. Meanwhile, autophagy could also degrade non-cytosolic proteins, at least under certain conditions. The nuclear lamina and the nuclear protein SIRT1 could be degraded by autophagy in response to tumorigenic stress and in senescence, respectively.145–147,263 The process involves interaction between the target and LC3 in the nuclei, and thus ATTECs may strengthen this interaction and enhance the autophagic degradation of the nuclear constituents. In fact, ATTECs are capable of degrading the nuclear protein BRD4.144 It is important to note that the nuclear autophagy may not involve SQSTM1/p62, and thus the SQSTM1/p62-engaging degrader including AUTACs or AUTOTACs may have limited effects on nuclear protein, consistent with the observation that AUTAC3, the BRD4-targeting AUTAC, has little effect on the nuclear protein BRD4.7 The other potential mechanism that may allow ATTECs to degrade nuclear proteins is the balance between cytoplasmic and nuclear distribution of the POI. The nuclear proteins are translated in the cytoplasm, where ribosomes are located.264 These nuclear proteins usually have the nuclear localization sequence(s) (NLS) that is recognized by the importin family proteins, which facilitate the nuclear pore complex.265 Thus, there is always a fraction of the nuclear protein remaining in the cytoplasm, which ATTECs for autophagic degradation could capture. This may shift the cytoplasmic-nuclear balance of the target protein and eventually reduce the POI in the nucleus. A similar scenario may also apply to membrane proteins and secreted proteins, which have a cytoplasmic fraction as well. In summary, while the cytoplasmic targets should be prioritized for autophagy-based degraders, targets in other subcellular localizations could also be considered.For endosome–lysosome-based degraders (LYTACs and MoDE-As), only extracellular or membrane proteins could be targeted. In fact, MoDE-As have only been tested for extracellular targets,13 whereas LYTACs have been shown to be able to degrade certain membrane receptors.11,12 In theory, all extracellular proteins could be targeted by LYTACs or MoDE-As, but some membrane receptors may not be suitable targets for them. EGFR is a suitable target because it tends to be sorted to the lysosomes rather than recycling endosomes after its endocytosis.266 Meanwhile, many other targets that tend to enter the recycling endosomes might not be degraded by LYTACs or MoDE-As efficiently, and desired properties of membrane receptor targets may need further investigation. The proportion of receptors or other transmembrane targets entering the recycling endosomes probably has a strong effect on degradation efficiency, and is worth considering for target selection of LYTACs and MoDE-As.
5.3 Proteasomal degradation and endogenous half-life
This aspect mainly influences the target selection for autophagy-based degraders. In theory, autophagy-based degraders can induce the autophagic degradation of proteins that are initially degraded by the proteasome because tethering the POI to LC3 or SQSTM1/p62 can induce its engulfment by the autophagosomes for subsequent lysosomal degradation. Meanwhile, there is actually an opportunity cost to doing so because POIs entering the autophagosomes prevent their degradation by the Ubiquitin/Proteasome System (UPS). Thus, if the target is efficiently degraded by the UPS, the degradation effects of autophagy-based degraders are likely weak or even in the opposite direction due to reduced degradation by UPS. This scenario may also apply in the presence of PROTACs. Thus, combined treatment of both PROTACs and ATTECs/AUTACs/AUTOTACs of the same target may not be an excellent strategy to increase the degradation efficiency. The above analysis is based on the assumption that the autophagy-based degraders cannot distinguish the poly-ubiquitinated target from the non-ubiquitinated target.A related factor is the half-life of the POI, which is influenced by its endogenous degradation pathway. Generally speaking, the proteins degraded via the UPS tend to have short half-lives, whereas the long-lived proteins (half-life >18 hours) are usually autophagy substrates.267 Based on this principle, measuring the degradation rate of long-lived proteins is a classical assay for the measurement of the autophagic flux.267,268 This suggests that autophagic degradation may take a longer time. Consistent with this, autophagosome formation, engulfment of cargos, and autophagosome-lysosome fusion may take hours.21 Thus, if the target has a very short endogenous half-life, it may not be an efficient target for autophagy-based degraders. Interestingly, the pulse-chase proteomic analyses revealed that most proteins are long-lived, with a median half-life of 46 hours.269,270 Thus, most proteins are suitable for targeted degradation by autophagy-based degraders from this perspective.
For ATTECs, we have also developed a mathematical model for the degradation effects taking into account both the degradation rate of autophagy and the formation of ternary complexes.271 The nonspecific protein degradation rate of autophagy under basal conditions is about 1.5% h−1 of total cellular protein,272 which is mostly due to the limited number of autophagosomes in each cell (about 13), even though the lifetime of autophagosome is short (approximately 20 minutes).21,273 The degradation of proteins directly sequestered by LC3 is likely accelerated, as demonstrated by SQSTM1/p62 with a half-life of ∼6 hours.274 Thus, degradation of the proteins sequestered by LC3 through ATTEC may be similarly accelerated. Based on this, we performed kinetic simulation of the protein target of ATTECs with binding affinities changing from nM to μM, and we found that the time needed for ATTECs to reach the degradation steady-state levels varied from 30 hours to 120 hours for low-expression proteins like mHTT.271 This is consistent with experimental data and a much longer than the degradation time of 1–6 hours for PROTACs, which takes advantage of proteasomes with high abundance (1% of total soluble protein) and a fast degradation rate for each proteasome (2.5 substrates per min) inside the cell.275,276
5.4 Autophagy activity and lysosome functions
All the lysosome-based degraders’ function relies on the autophagy or endosomal–lysosomal pathways. Certain disease cells have impaired autophagy activity and/or lysosome functions, and whether degraders may still work in these cells needs a careful assessment.Lysosome storage disorders (LSDs) refer to a group of rare diseases caused by lysosomal defects.277 LSDs are caused by the LOF mutations of certain specific lysosome proteins, including enzymes or membrane proteins.277,278 Thus, most LSDs seem to be outside the scope of lysosome-engaging degrader technologies including ATTECs, AUTACs, AUTOTACs, LYTACs, and MoDE-As, due to their LOF nature and lysosomal deficits.
Defective autophagy and/or lysosome functions have been reported in many neurodegenerative disorders. Autophagosomes accumulate in the brain of AD patients and mouse models likely due to deficits in their retrograde transport, maturation and lysosomal clearance.279–284 A very recent study also revealed that the autolysosome acidification is impaired in the AD mouse model, forming unique flower-like perikaryal rosettes termed PANTHOS (poisonous anthos (flower)).285 A hallmark of Parkinson's disease is the formation of intracellular α-synuclein inclusions, known as Lewy bodies.286 α-Synuclein aggregates may cause a deficiency in autophagy initiation in neurons.287 Consistent with this, α-synuclein overexpression in cell lines inhibits autophagosome biogenesis and fusion with lysosomes.287–289 Huntington's disease (HD) cells also may have deficient autophagy functions compared to the wild-type. While the studies are not completely consistent, the widely accepted evidence showed increased presence of endocytic and autophagic compartments and reduced cargo/mHTT recognition.290–297 The autophagy deficits in neurodegenerative disorders do not necessarily prevent ATTECs/AUTACs/AUTOTACs from being a possible therapeutic strategy. In fact, both mHTT and Tau, two targets of neurodegenerative disorders, have been successfully degraded by ATTECs or AUTOTACs.8,9 mHTT comprises only a tiny fraction (∼0.01%) of the total cellular proteins based on proteomics data,9 and the baseline autophagy could be sufficient to degrade mHTT.9 Meanwhile, AUTOTACs enhance the autophagic flux while tethering the target to SQSTM1/p62, and this may overcome possible impairment of autophagy in certain neurodegenerative disorders.
The autophagy activity is also altered in cancer,298 and this may impact the functions of autophagy-based degraders targeting oncoproteins. Autophagy plays a complex and highly context-dependent role in tumorigenesis, which may proceed with a temporary inhibition of autophagy followed by restoration or enhancement of autophagy.299 In the initial stage, defects in autophagy may facilitate the acquisition of malignant features by healthy cells.299 Once malignancy is established and the tumor grows, autophagy may be activated in the nutrient-deprived environment caused by the rapid proliferation of cancer cells.299 This response may be essential to support the survival, proliferation, and growth of these cells in an adverse microenvironment. Thus, autophagy-based degraders might have higher efficiency and potency in cancer cells at later stages after the establishment of malignancy, when the autophagy activity is restored and enhanced.
Taken together, several factors need to be considered during target selection for lysosome-engaging degrader technologies. Biological evidence is undoubtedly required to justify the potential benefits of degrading the target. Cytosolic long-lived targets in autophagy proficient cells are generally the best targets for autophagy-based degraders, whereas extracellular targets and some of the transmembrane proteins are suitable targets for LYTACs and MoDE-As. Once the target is determined, the primary task is to identify or design the corresponding degrader compounds, and the possible strategies are discussed as follows.
6. The strategies to identify or design degraders engaging lysosomal pathways
There are potentially 2 different strategies to identify or design degraders engaging lysosomal pathways. One is the smaller molecular glue strategy, the other is the bifunctional chimeric compound strategy. Since the two strategies are somewhat different and have pros and cons, we will discuss them separately.6.1 Molecular glue
In TPD, molecular glues are typically small chemical molecules that act on the interface between the POI and the protein in the degradation machinery. Unlike chimeric compounds, they are usually smaller (typically <500 Da) and do not have clear separable components such as the linker and the chemical moieties binding to each protein. Molecular glues normally interact with at least one of the two proteins they work on and may interact with both of them. The key is that they enhance protein–protein interaction by directly engaging the interaction surface. The widely used CRBN-binding IMiDs used for PROTACs were initially identified as molecular glues.116,300,301Molecular glues are believed to be better drug-like because of their small sizes and previous success examples. Although it has been 30 years since the first discovery of molecular glues (under a broader definition of compounds inducing neo-protein–protein associations), the molecular glues successfully used in the clinic or clinical trials are still primarily discovered in serendipity.116,245,302 Among them, molecular glue degraders are the major class that researchers are most interested in. At least for E3-based molecular glues, there are already many precedents in the literature. Since the discovery of thalidomide, there have been over 10 different small molecular glues with similar structural motifs; 5 typical thalidomide-derived IMiDs (thalidomide, lenalidomide, pomalidomide, CC-885 and CC-90009) are listed in Fig. 26.303,304 They induce neo associations between distinct E3 ligase enzymes, their corresponding adaptors, and target proteins. For example, thalidomide binds to CRBN, the substrate receptor of the E3 ubiquitin ligase.
 | ||
| Fig. 26 Thalidomide-derived IMiDs as molecular glues to bind with CRBN and neosubstrates. | ||
It has been reported that a series of aryl sulfonamides function as molecular glues to promote the E3 ubiquitin ligase CUL4-DCAF15 (DDB1 CUL4 Associated Factor 15) and RBM39 (splicing factor RNA binding motif protein 39) interaction, therefore to induce the degradation of RBM39. Several aryl sulfonamides, such as indisulam, CQS, and tasisulam, have been evaluated in clinical trials as antitumor candidates (Fig. 27), sharing the same mechanism of action.
 | ||
| Fig. 27 Aryl sulfonamide derivatives as molecular glues to promote RBM39–DCAF15 interaction to trigger RBM39 degradation. | ||
Among the discussed autophagy degraders, ATTECs and AUTOTACs have the potential to be molecular glue degraders based on their mechanisms of action. ATTECs may function as molecular glues binding with both the POI and LC3B, forming a stable ternary complex, whereas AUTOTACs may function as molecular glues binding with both the POI and SQSTM1/p62. The polyQ·ATTECs are examples of possible autophagy-based molecular glues,9 while there is still no demonstration of AUTOTAC molecular glues. Since AUTACs depend on mimicking the S-guanylation by a separate chemical moiety, developing AUTAC molecular glues may be difficult.
For endosomal–lysosomal pathway-based degraders, although LYTAC and MoDE-A for membrane proteins and extracellular proteins can theoretically be designed and developed as molecular glues, the available endosomal–lysosomal degraders all adopt the bifunctional molecule mode similar to PROTAC. Also, since the existing LYTAC and MoDE-A molecules are all relatively large. It is difficult to screen small molecular weight compounds that bind to CI-M6PR or ASGPR with high affinity, and molecular glue degraders should bind to both POI and oligosaccharide receptors. It may be challenging to obtain lead compounds with sufficiently low molecular weight using existing technical methods.
Our previously published polyQ·ATTECs are molecular glue-like molecules. Based on the discovery of these ATTECs, we propose an approach to screen for possible molecular glues by in vitro biophysical assays, followed by phenotypic characterization. The key to identifying molecular glues is discovering compounds that may directly enhance ternary complex formation. This could be potentially achieved by in vitro screening, which is much more cost-effective than cellular phenotypic screening. It may also facilitate biophysical validation at later stages as well. One possible strategy to screen for ATTECs is similar to the one presented in the polyQ·ATTECs study. It utilizes a novel high-throughput screening platform, as illustrated in Fig. 28, which is based on the small molecule microarray (SMM) (Fig. 28A) and oblique-incidence reflectivity difference (OI-RD) microscopy (Fig. 28B) technologies.
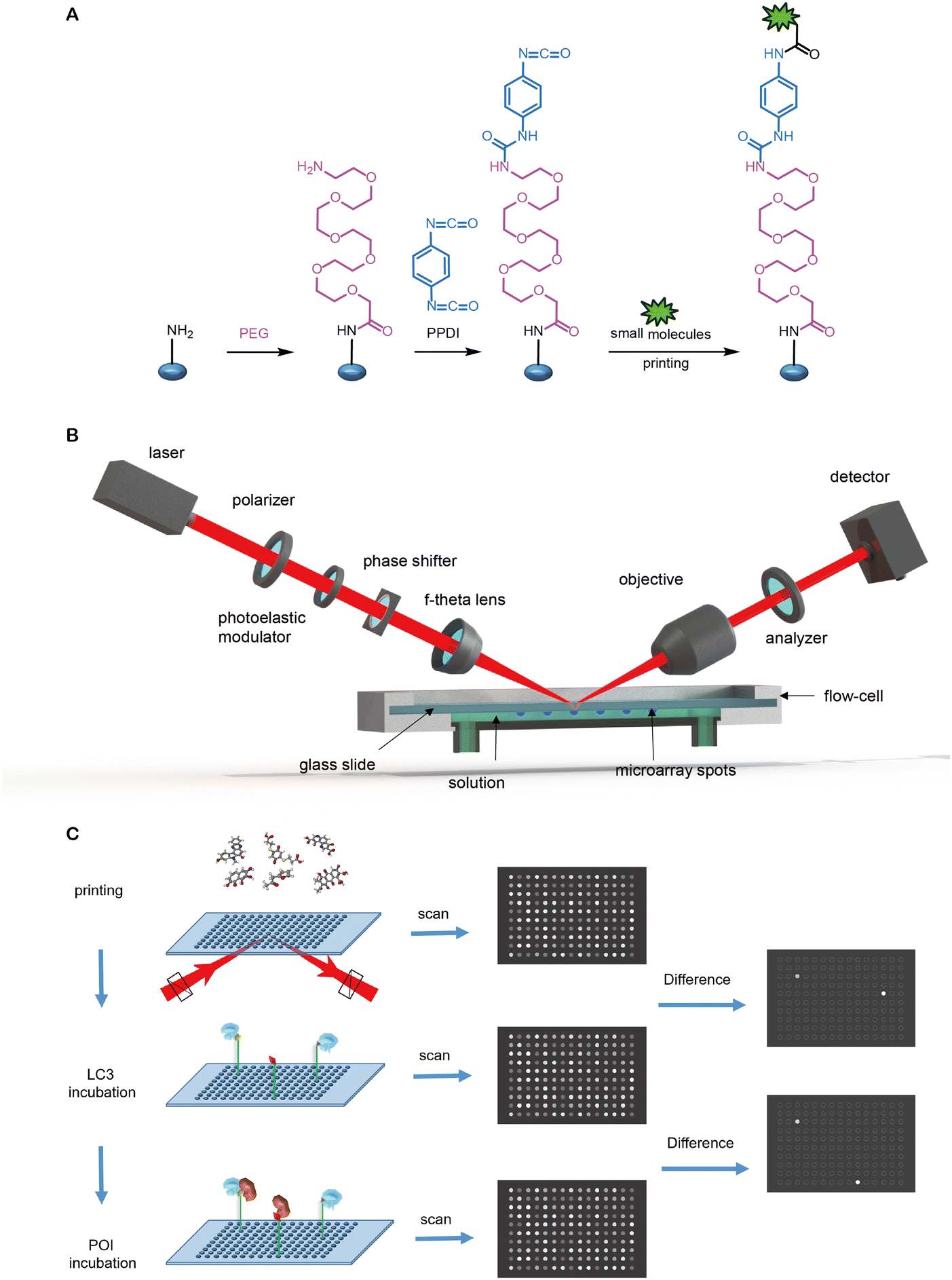 | ||
| Fig. 28 A high-throughput molecular glue ATTEC discovery platform. (A) Immobilizing thousands of small molecules on isocyanate functionalized glass slides to generate a small molecule microarray (SMM); (B) scheme of oblique-incidence reflectivity difference (OI-RD) microscope; and (C) the screening procedure to discover molecular glue ATTECs. | ||
SMMs are microarrays with thousands of small molecules immobilized on one solid support in a regular array format and provide a high throughput screening substrate for drug screening. To immobilize as many as compounds on glass slides, amine coated glass slides are functionalized as isocyanate functionalized glass slides, onto which thousands of compounds are printed and immobilized through covalent bonds between nucleophilic residues in compounds and isocyanate groups on glass slides (Fig. 28A).235 Tens of SMMs can be prepared simultaneously and stored at −20 °C until further use.
SMMs are then scanned using an OI-RD microscope which can detect the surface mass density of biomolecules deposited on a solid substrate (Fig. 28B), enabling OI-RD to probe biomolecular interactions without the need to label any molecule.305 By housing the transparent glass slides in a flow cell that provides a liquid-flowing environment, each operation, including washing, blocking, and reaction can be monitored in situ by taking SMM images or monitoring real-time signal changes. The screening platform, based on SMM and OI-RD microscope, is able to screen for ATTECs in situ in a label-free high-throughput mode, at least in theory.
To screen for compounds that bind to a particular target protein, they are flown over an SMM housed in the flow cell and OI-RD images of the SMM before the reaction and after the reaction with the target protein are scanned sequentially. The bright spots appeared in the difference image between the OI-RD image after the reaction with the protein and the OI-RD image before the reaction with the protein stand for those compounds which could bind to the target protein (Fig. 28C). Noticeably, after flowing through each protein, a wash with PBS is needed to wash away the non-specific bound proteins. In addition, control proteins are needed to exclude “sticky” compounds that may interact with many proteins non-specifically. Since ATTECs are compounds which bind to two target proteins simultaneously; one screening strategy is flowing through the recombinant purified LC3 and POI proteins subsequently over the same SMM (Fig. 28C); the other feasible strategy is flowing through the recombinant purified LC3 and POI proteins separately over different SMMs. ATTECs can be obtained by identifying compounds binding to both LC3 and POI in either screening strategy.
Our previous identification of polyQ·ATTECs demonstrates this feasibility at least for one example. Meanwhile, the polyQ protein may have specific conformation properties that facilitate the screening. For example, the expanded polyQ may have conformational polymorphisms that adapt the binding of molecular glues on the chip.294,306 We need to screen for more targets and test the degradation effects of the identified compounds to validate this approach entirely.
Compared with traditional inhibitors, the ATTECs’ binding sites of the proteins are not limited to the active centre. Thus the positive hit rate of screening could be much higher than traditional inhibitor screens because the whole surface of POI is much larger than the active center. In addition, because these binding sites may not reside in the active center, the cells treated with these compounds may have a smaller selection pressure, decreasing the possibility of drug resistance via mutagenesis of the ATTEC-engaged proteins. If the identified compounds also bind to the active site of POI, it may have dual activity of both inhibition and degradation to enhance their efficacy, similar to some PROTACs.307
Besides the SMM chip and OIRD-based platform mentioned above, other biophysical technologies that detect compound–protein interactions could also potentially be applied to identify and validate molecular glues. Meanwhile, it is worth noting that most of these biophysical assays, especially the high-throughput compatible ones, are designed for binary rather than ternary binding detection. Thus, significant exploration and assay development are needed to use them for molecular glue screening.
Other molecular glue discovery strategies, including data mining, phenotypic screening and structural-based optimization, could also be used to identify molecular glues. Previously, most of the molecular glue degraders are discovered by serendipity. By correlating compound toxicity and the ubiquitin ligase expression in hundreds of cancer cell lines, Slabicki et al. discovered that the CDK12 kinase inhibitor CR8 is a cyclin K molecular glue degrader bridging the interaction of CDK12 and DDB1 (CRL4 adaptor). With the help of CR8, CDK12-bound cyclin K can be ubiquitinated by CRL4 and degraded by the proteasome.308
Mayor-Ruiz et al. developed a scalable glue degrader discovery strategy based on chemical screening coupled with a multi-omics target deconvolution method.309 They identified molecular glue degraders that induce ubiquitination and degradation of cyclin K by inducing the interaction of CDK12–cyclin K with a CRL4B ligase complex. This screening strategy can be applied to larger chemical libraries to discover novel degraders with novel mechanisms.
Through phenotype-based high-throughput small-molecule screening, Lv et al. found that HQ461 acts as a molecular glue degrader by binding to the CDK12's kinase domain, thus creating a novel CDK12 surface that can recruit DDB1, the subunit of the DDB1-CUL4-RBX1 E3 ubiquitin ligase. The binding of HQ461 converts CDK12 into a substrate-specific receptor for DDB1-CUL4-RBX1, triggering the polyubiquitination and subsequent degradation of CDK12's binding partner cyclin K.310
Through phenotypic screens of cell viability in HeLa cells, Li et al. identified that 17-β-estradiol and its related steroid hormones induce apoptosis by directly binding to phosphodiesterase 3A and SLFN12.311 Further mechanistic and structural studies showed that 17-β-estradiol, anagrelide, nauclefine, and DNMDP are molecular glue degraders binding with PDE3A and SLFN12, occupying the same binding pocket in PDE3A and forming a heterotetrameric complex with a butterfly-like shape, thus initiating apoptosis.312 These strategies could potentially be modified and applied to discover molecular glues based on autophagy-lysosome pathways like ATTEC or AUTOTAC molecules.
For molecular glues, the more significant challenges probably lie in the later optimization stage. Although molecular glues are more druglike and obey the Lipinski's Rule of Five when compared with bifunctional chimeric molecules, the complexity of their pharmacology complicates SAR elucidation due to a less clear knowledge of the binding mode and MoA.313,314 In addition, although the thalidomide family IMiD are good examples of molecular glues, the molecular surfaces buried when binding with POI are much smaller than those of PROTACs.259 This may cause specificity problems, resulting in higher off-target effects or toxic side effects. Therefore, it is necessary to perform proteomics analyses to elucidate the potential off-target effects of glue degraders. Meanwhile, their property-based optimization as drug candidates is facilitated by their physiochemistry compared to heterobifunctional molecules.
High-resolution crystal structures may provide direct evidence of the existence of the ternary complex and the atomic resolution structure information that can guide the SAR studies and the further optimization process. However, the co-crystallization of the binary or ternary complex is still tricky. For CRBN based molecular glue and PROTAC, the first binary complex structure was resolved in 2014 by Fischer et al.315 The structure of the DDB1–CRBN complex bound to thalidomide, lenalidomide, or pomalidomide showed that relatively small IMiD's binding could modulate E3 ubiquitin ligase and substrate selectivity. In 2016, a ternary complex of CK1α-lenalidomide-CRBN was solved (Fig. 29A); the most exciting feature is that CRBN and lenalidomide jointly provide the binding interface for CK1α's β-hairpin-loop.316 The CK1α's β-hairpin residue Gly40 is crucial for the ternary complex formation, although the lenalidomide–CK1α binding interface only provides a 96.8 Å2 buried surface; the total binding surface is more than 1000 Å2 (CRBN–CK1α, 718.1 Å2 and CRBN–lenalidomide, 236.6 Å2; total, 1051.5 Å2), and the CRBN–CK1α interface induced by lenalidomide provided the most significant binding interface, which is crucial for the stable ternary complex formation.
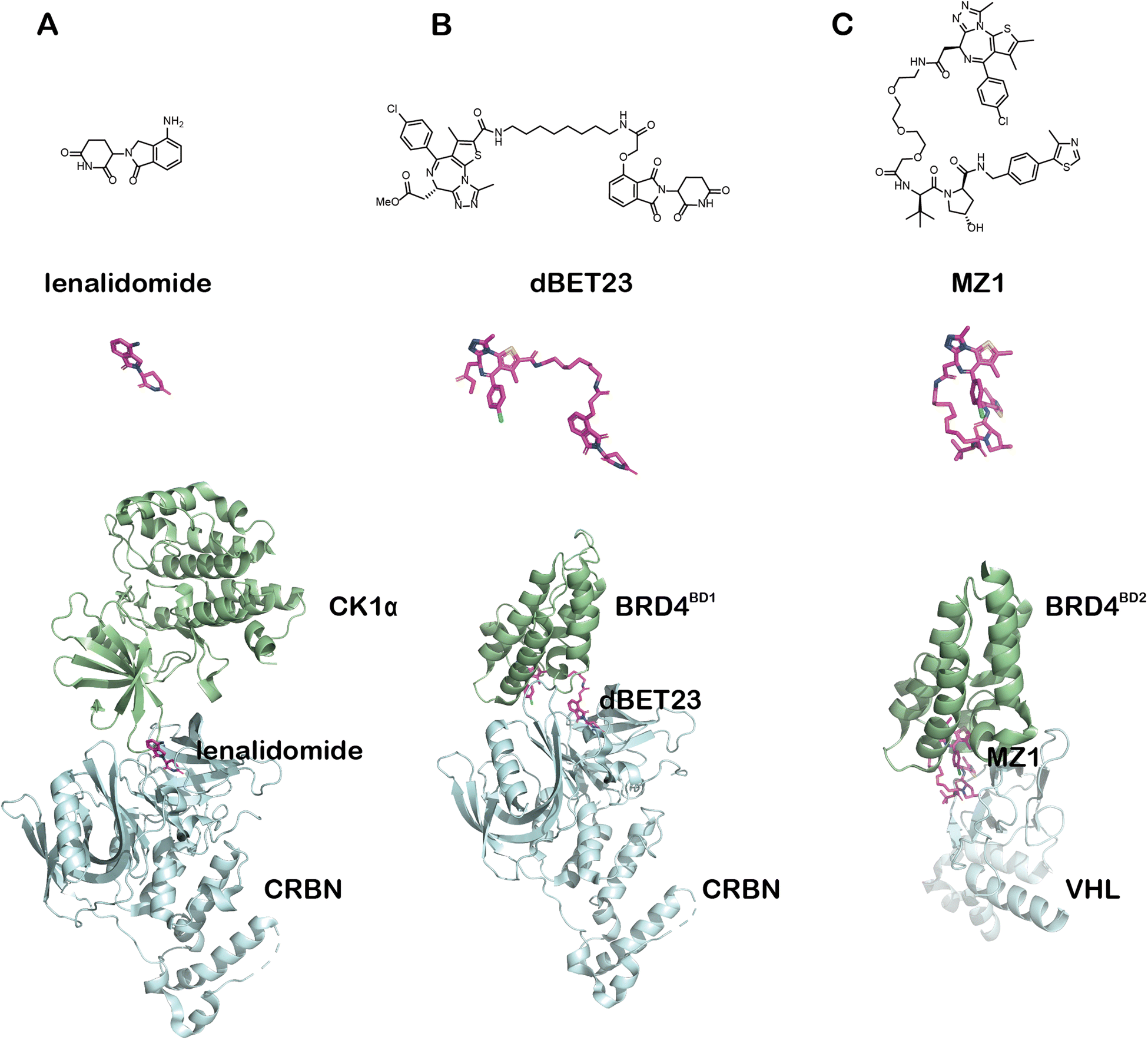 | ||
| Fig. 29 Comparison of 3 representative ternary complex molecular glues or PROTAC crystal structures. Upper: structural formula of the compound; middle: the compound's actual 3D structure in the crystal; lower: the crystal structure of the ternary complex (POI CK1α, BRD4BD1 or BRD4BD2: green, compound lenalidomide, dBET23 or MZ1: magenta, and E3 ligase CRBN or VHL: cyan). (A) The ternary CK1α–lenalidomide–CRBN molecule glue complex (PDB: 5FQD); (B) the ternary BRD4BD1–dBET23–CRBN PROTAC complex (6BN7); and (C) the ternary BRD4BD2–MZ1–VHL PROTAC complex (5T35). | ||
For all the published ATTECs, the low solubility in water and relatively low binding affinity to LC3 may hinder the purification and crystallization of the ternary complex. The problem may be solved by crystallizing with optimized compounds with higher water solubility and higher binding affinity to LC3. The ternary complex structures were missing in all the other autophagy or endosomal–lysosomal pathway-based degraders, possibly due to similar reasons.
6.2 Bifunctional chimeric degraders
The autophagy–lysosomal based ATTEC, AUTAC and AUTOTAC degraders and endosome–lysosomal based LYTAC and MoDE-A degraders introduced in this paper all have successful samples of bifunctional chimeric degraders. Bifunctional chimeric degraders have broad-spectrum and extensibility. Once suitable warheads are identified, these degraders can be designed relatively easily.As discussed above, LD·ATTECs and oncoprotein-targeting ATTECs, including BRD4·ATTECs and NAMPT·ATTECs, utilized the bifunctional chimeric strategy similar to the design of PROTACs: connecting the LD- or POI-binding warhead with an LC3-binding warhead via a chemical linker. These chimeric compounds may then tether the target macromolecules, including lipid droplets, NAMPT, or BRD4 to LC3 and degrade them through the autophagy pathway.
The published AUTACs and AUTOTACs are also bifunctional chimeric degraders, The AUTAC warheads targeting POI (MetAP2, FKBP12, BRD4 and mitochondria specific OMM transporter protein) are connected to the warhead mimicking S-guanylation via a linker. The induced K63-linked polyubiquitination process thus hijacked the autophagy receptor SQSTM1/p62 and specifically degraded the targeted POI or organelle. AUTOTACs are designed by connecting the warheads targeting POI (ERβ, AR, MetAP2 and misfolded protein aggregates) and SQSTM1/p62's ZZ domain, respectively, via a linker.
Compared with PROTACs, bifunctional chimeric ATTECs and AUTOTACs may have a potential advantage as they do not require specific lysine residues of POI that are available for polyubiquitination. When designing PROTACs, it is necessary to consider whether the target protein has specific lysine residues exposed to the solvent that is accessible to the E2 ligases to be poly-ubiquitinylated. The chimeric compounds need to be designed to avoid the potential competition of the poly-ubiquitination sites either. This limitation was recently demonstrated by Lv et al.317 They developed a PROTAC that can simultaneously degrade BCL-xL and BCL-2. During the optimization process, they found that the protonated state, flexibility, and orientation of the substrates’ lysine (K20, K87 of BCL-xL, and K17 of BCL-2) and nearby residues have a strong influence on the interaction between the E2 and the POI, leading to different efficiency of subsequent proteasomal degradation. Since ATTECs’ and AUTOTACs’ MoA requires only tethering but not enzymatic catalysis, this factor need not be considered.
The stable ternary complex formation is important for good bifunctional degraders.232,251,318 Structural analysis can validate this, including NMR, X-ray crystallography, or single-particle cryo-EM. The formation of the ternary complex may change the microenvironment near the binding interface and thus can be detected by the nuclear Overhasuer effect spectroscopy (NOESY) or 1H-15N HSQC spectra of ATTEC treated/untreated samples.319,320
X-Ray-based crystallography or single-particle cryo-EM may provide the most definitive evidence of ternary complex formation and reveal the molecular details of the binding mechanism. Although currently there are no ternary complex structures available for all the published degraders targeting the lysosomal pathway, the available PROTAC ternary structures may provide some clues for the analyses of chimeric lysosomal degraders.
For PROTACs, the formation of the ternary complex is essential for the effective degradation of POI (Fig. 29B and C).138 The first PROTAC ternary complex structure of VHL-MZ1-BRD4BD2 was resolved in 2017 (Fig. 29C).259 Interestingly, ITC results showed that the PROTAC molecule MZ1 causes a positive synergistic effect for the binding with VHL and BRD4BD2, and the MZ1's binding forms a novel protein–protein interaction interface. The linker's length and rigidity affect the formation of the ternary complex and thus change the effectiveness and selectivity of POI's degradation. The structures of the ternary complexes show that the linker is particularly important to bifunctional chimeric molecules. The linker is not just a simple linking element connecting the two warheads in the traditional sense. It provides a suitable distance and orientation between the POI and the E3 ligase and helps the stable ternary complex formation to facilitate POI's poly-ubiquitination by the E3 ligase. In addition to the binding of the warheads, the linker may contribute to the binding force from at least two aspects. First, the linker itself may provide interaction forces. Taking MZ1 as an example. The PEG linker of MZ1 in the ternary complex is not exposed to the hydrophilic solvent at all, and this provides van der Waals interactions with BRD4BD2's loop and hydrogen bonds with BRD4's His437. PROTACs based on thalidomide also showed a similar scenario (Fig. 29B); the PROTAC dBET23's linker interacts with BRD4BD1's residues.321 Given the experience from PROTACs, it is reasonable to rationalize that installing certain types of functional groups on the linker of ATTECs may make crucial contributions in promoting the compounds’ interaction with the POI's or LC3's nearby surface. Second, the linkers with a suitable length may induce novel POI-E3 protein–protein interaction and thus build significant hydrophobic surface burial that stabilizes the ternary complex. For example, the total surface area buried in VHL-MZ1-BRD4BD2 is at least 2600 Å2,259 even larger than the ones in the molecular glue ternary complexes: 1830 Å2 in CRBN–lenalidomide–CK1α,316 and 2390 Å2 in CRBN-CC-885-GSPT1.303
Bifunctional chimeric lysosomal degrader molecules with optimized linkers may likely form stable ternary complexes by burying larger hydrophobic surface areas similar to PROTACs so that relatively stable ternary complexes could be formed to facilitate effective engulfment of the POI into autophagosomes for degradation. In addition, compared with PROTACs, which require precise orientation of the interface formed by the E3 and POI to trigger effective poly-ubiquitination, lysosomal degraders except AUTACs do not need to ubiquitinate the POIs. They thus may not require such stringent restrictions on the spatial orientation of the complex. Once a suitable linker is introduced, it will likely provide a new interface that can promote the POI/LC3 surface interaction. In silico, protein–protein docking may provide models to guide the design.322 The structures of binary or ternary complexes will provide important atomic resolution information for the development of a lysosomal based degradation platform. If the structures cannot be solved, biophysical methods amended by in silico structural analyses may also provide valuable insights for optimizing chimeric degraders targeting the lysosomal pathway.
Compared with molecular glues, bifunctional chimeric lysosomal degraders may have larger molecular weight and less ideal druglikeness. Meanwhile, they could be invented by rationale design that does not require tedious screen efforts. In addition, the optimization of linkers may provide better efficacy and specificity.
6.3 Antibodies or large molecules
One of the main advantages of biological large molecules or antibodies as degraders is their target specificity. The affinity (usually ∼ nM or even ∼ pM KD) of these molecules binding to POI is typically much higher than the ordinary small molecular weight compounds to their target, and thus they can bind to the POI more specifically and has significantly lower off-target effects, providing better safety profile during the late stage of development. Developing antibody-based degradation technologies can fully use the well-developed antibodies as the ligand, which may significantly reduce the workload of screening target-specific binding molecules. In addition, the antibody–drug conjugates (ADCs) are also bifunctional molecules based on antibodies and toxic small molecular weight compounds. Since ADC-related technologies have been verified in many clinical research, expanding them to the design of biomacromolecule-based degraders may significantly speed up the development process.Meanwhile, biological macromolecules, especially antibodies, also face other hurdles during development. For example, they have difficulties in entering the cells to tackle intracellular targets. In addition, the stability of biomacromolecules, the transportation, the preservation, and the challenging delivery are critical factors to consider during development.
7. Potential advantages and limitations of degraders engaging lysosomal pathways
The degrader technology field has been booming in recent years. The PROTACs have already entered the clinic.323 The lysosome-based degrader technologies are emerging and greatly expanded the target spectrum. For example, ATTECs may degrade LDs and neutral lipids; LYTACs/MoDE-As may degrade extracellular targets; AUTACs may degrade mitochondria; and AUTOTACs may degrade protein aggregates. These targets are probably intactable by PROTACs. Meanwhile, other aspects such as the target half-life and subcellular localization may limit the target spectrum of these lysosome-based technologies.Autophagy is highly conserved in eukaryotic cells and present in essentially all mammalian cell types. Thus, unlike degrader technologies dependent on specific E3 ligase subunits that may not be expressed in all cell types, degrader technology engaging autophagy are potentially active in all cell types. This could be both an advantage and a disadvantage. Dependence on specific E3 ligase subunits may limit the degraders’ application to particular cell types and cause cancer cell resistance following chronic treatment.324 Targeting the autophagy pathway may not have this limitation. On the other hand, some disease targets might be pathogenic in certain cell types and beneficial in others and thus require cell-type-specific treatment. This could be challenging for autophagy pathway degraders because autophagosomes are present in most cell types. Meanwhile, the ATG8 family proteins exhibit a certain degree of cell type-specific expression. For example, LC3C is low-expressed in most tissues but only highly expressed in the lung and ovary.80 The mRNA of GABARAPL1 or GABARAPL2 is mainly expressed in the central nervous system, especially in the brain. GABARAPL1 is specifically expressed in the pons or diencephalon, important for regulating somatomotor or endocrine function. Also, GABARAP is highly expressed in the endocrine glands.78,79,81 These may allow the design of relatively cell-type-specific degraders engaging autophagy. For LYTACs and MoDE-As, since ASGPR has good liver-targeted tissue specificity, it may be easy to develop liver-targeted endosomal–lysosomal degraders, but this could be challenging to achieve for other organs.
Similar to other degrader technologies, the mathematical modelling of lysosomal pathway degradation effects is very limited. The Spiegel group has established a comprehensive mathematical model for the ternary complex formation without considering degradation.325 Riching et al. performed quantitative live-cell degradation kinetics and mechanistic profiling of PROTACs, but with little mathematical modelling.257 We have completed a very preliminary mathematical modelling of the ternary complex formation, the degradation kinetics, and the steady-state dose-dependent curves of ATTECs, but the model is somewhat over-simplified and lacks experimental validation.271 For AUTACs, AUTOTACs, LYTACs and MoDE-As, there's still no reported mathematical model of the degradation. More detailed modelling with experimental validation is desired to predict the degradation effects and guide the optimization of degraders engaging the lysosomal pathways.
Optimization of degraders engaging lysosomal pathways is also limited by the current knowledge of the structure–activity relationship. For bifunctional chimeric ATTECs, the preliminary linkerology studies of the BRD4·ATTECs and NAMPT·ATTECs suggest that a medium-length carbon chain might give the best efficacy and potency. Different LC3 binders with suitable linking terminals have been used to link with LD, BRD4, or NAMPT for different types of chimeric ATTECs; for example, GW5704 has either been linked with LD probes at its amide NH group to form LD·ATTECs or linked with JQ1 from its phenoxyl group to form BRD4·ATTECs. With ispinesib as the LC3 binder, a NAMPT inhibitor MS2 has also been used to link it at its primary amine terminal with suitable linkers to form NAMPT·ATTECs. The medicinal chemistry optimization of ATTECs is currently limited by a lack of structural information on the LC3–ATTEC complex. The AUTOTACs faced a similar problem during optimization of the AUTOTAC-SQSTM1/p62 complex. In addition, new LC3-binding warheads could be developed based on recently reported LC3-binding chemicals with structure information (Fig. 4).75–77 Meanwhile, their potential influence on LC3 functions and global autophagy need to be tested, and proof-of-concept degradation studies using them as warheads are desired.
On the other hand, for heterobifunctional lysosome-based degradation technologies, optimization of degraders may have advantages over PROTACs, given that the mechanism of action such as ATTECs/AUTOTACs/LYTACs/MoDE-As does not involve the subsequent ubiquitination process induced by PROTACs. Since the PROTAC's enzymatic ubiquitination process requires relatively high spatial relative positions and distances of substrates and ubiquitin molecules, developing degradation technologies based on lysosome degradation technology may be less complicated. Finally, a common issue of heterobifunctional lysosome-based degraders, as for PROTACs, is the technical challenges of determining free drug levels due to their physiochemistry properties that may lead to high off-target or non-specific binding such as plasma protein binding, which may hamper the downstream in vivo translational studies.
8. Conclusions
In summary, lysosome-based degraders including ATTECs/AUTACs/AUTOTACs/LYTACs/MoDE-As provide unprecedented technologies of harnessing autophagy or endosomal–lysosomal pathways for the selective degradation of pathogenic proteins and non-proteinaceous targets, fundamentally expanding the target spectrum of degrader technologies. These degraders may have different formats, including molecular glues and bifunctional chimeric compounds. The MoA needs to be further analysed to exclude other possible mechanisms. Biologically and clinically validated, long-lived cytosolic targets in autophagy proficient cells are ideal targets for autophagy-based degraders, whereas extracellular and some of the transmembrane targets could be tackled by endosome–lysosome-based degraders. Structural information, more selective warheads with higher affinities, additional SAR information, and more accurate modelling supported by experimental data are desired to develop these technologies.Author contributions
B. L. organized the draft, designed the figures and wrote the cell biology related sections; Y. D. wrote the biochemical and biophysical-related sections and prepared the figures; Y. D. and D. X. prepared the chemistry related figures and wrote relevant sections; Y. F. wrote the screening relevant sections; Y. D. and B. L. put together the manuscript; all authors read and revised the manuscript.Conflicts of interest
Y. D., Y. F., and B. L. are shareholders of ATTEC-related biotech companies and patent inventors of ATTEC-related patents.Acknowledgements
We’d like to thank Drs Huaixiang Hao and Nan Ji for critical reading of the earlier versions of the manuscript. This work was supported by grants 2021YFA0805200 from the National Key Research and Development Program of China, 81925012, 82050008, 92049301 and 82030106, from the National Natural Science Foundation of China, 20JC1410900 and 20ZR1403700, from the Shanghai Science and Technology Commission, NAF/R1/191045 (Advanced Newton Fellowship) from the Academy of Medical Science (UK), Xplorer Prize, Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01), ZJ Lab, and Shanghai Center for Brain Science and Brain-Inspired Technology.Notes and references
- Y. Ding, Y. Fei and B. Lu, Trends Pharmacol. Sci., 2020, 41, 464–474 CrossRef CAS PubMed.
- B. Dale, M. Cheng, K. S. Park, H. Kaniskan, Y. Xiong and J. Jin, Nat. Rev. Cancer, 2021, 21, 638–654 CrossRef CAS PubMed.
- S. M. Qi, J. Dong, Z. Y. Xu, X. D. Cheng, W. D. Zhang and J. J. Qin, Front. Pharmacol., 2021, 12, 692574 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- Y. Zou, D. Ma and Y. Wang, Cell Biochem. Funct., 2019, 37, 21–30 CrossRef CAS PubMed.
- D. J. Klionsky, K. Abdelmohsen, A. Abe, M. J. Abedin, H. Abeliovich, A. Acevedo Arozena, H. Adachi, C. M. Adams, P. D. Adams, K. Adeli, P. J. Adhihetty, S. G. Adler, G. Agam, R. Agarwal, M. K. Aghi, M. Agnello, P. Agostinis, P. V. Aguilar, J. Aguirre-Ghiso, E. M. Airoldi, S. Ait-Si-Ali, T. Akematsu, E. T. Akporiaye, M. Al-Rubeai, G. M. Albaiceta, C. Albanese, D. Albani, M. L. Albert, J. Aldudo and H. Algul, et al. , Autophagy, 2016, 12, 1–222 CrossRef PubMed.
- D. Takahashi, J. Moriyama, T. Nakamura, E. Miki, E. Takahashi, A. Sato, T. Akaike, K. Itto-Nakama and H. Arimoto, Mol. Cell, 2019, 76(797–810), e710 Search PubMed.
- C. H. Ji, H. Y. Kim, M. J. Lee, A. J. Heo, D. Y. Park, S. Lim, S. Shin, S. Ganipisetti, W. S. Yang, C. A. Jung, K. Y. Kim, E. H. Jeong, S. H. Park, S. Bin Kim, S. J. Lee, J. E. Na, J. I. Kang, H. M. Chi, H. T. Kim, Y. K. Kim, B. Y. Kim and Y. T. Kwon, Nat. Commun., 2022, 13, 904 CrossRef CAS PubMed.
- Z. Li, C. Wang, Z. Wang, C. Zhu, J. Li, T. Sha, L. Ma, C. Gao, Y. Yang, Y. Sun, J. Wang, X. Sun, C. Lu, M. Difiglia, Y. Mei, C. Ding, S. Luo, Y. Dang, Y. Ding, Y. Fei and B. Lu, Nature, 2019, 575, 203–209 CrossRef CAS PubMed.
- Y. Fu, N. Chen, Z. Wang, S. Luo, Y. Ding and B. Lu, Cell Res., 2021, 31(9), 965–979 CrossRef CAS PubMed.
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Nature, 2020, 584, 291–297 CrossRef CAS PubMed.
- G. Ahn, S. M. Banik, C. L. Miller, N. M. Riley, J. R. Cochran and C. R. Bertozzi, Nat. Chem. Biol., 2021, 17, 937–946 CrossRef CAS PubMed.
- D. F. Caianiello, M. Zhang, J. D. Ray, R. A. Howell, J. C. Swartzel, E. M. J. Branham, E. Chirkin, V. R. Sabbasani, A. Z. Gong, D. M. McDonald, V. Muthusamy and D. A. Spiegel, Nat. Chem. Biol., 2021, 17, 947–953 CrossRef CAS PubMed.
- J. S. King, Autophagy, 2012, 8, 1159–1162 CrossRef CAS PubMed.
- K. R. Parzych and D. J. Klionsky, Antioxid. Redox Signaling, 2014, 20, 460–473 CrossRef CAS PubMed.
- Y. Yang and D. J. Klionsky, Cell Death Differ., 2020, 27, 858–871 CrossRef PubMed.
- T. Yorimitsu and D. J. Klionsky, Cell Death Differ., 2005, 12(2), 1542–1552 CrossRef CAS PubMed.
- T. Ichimiya, T. Yamakawa, T. Hirano, Y. Yokoyama, Y. Hayashi, D. Hirayama, K. Wagatsuma, T. Itoi and H. Nakase, Int. J. Mol. Sci., 2020, 21(23), 8974 CrossRef CAS PubMed.
- O. B. Kotoulas, S. A. Kalamidas and D. J. Kondomerkos, Pathol., Res. Pract., 2006, 202, 631–638 CrossRef CAS PubMed.
- L. Yu, Y. Chen and S. A. Tooze, Autophagy, 2018, 14, 207–215 CrossRef CAS PubMed.
- K. Tsuboyama, I. Koyama-Honda, Y. Sakamaki, M. Koike, H. Morishita and N. Mizushima, Science, 2016, 354, 1036–1041 CrossRef CAS PubMed.
- H. Cheong, U. Nair, J. Geng and D. J. Klionsky, Mol. Biol. Cell, 2008, 19, 668–681 CrossRef CAS PubMed.
- I. G. Ganley, H. Lam du, J. Wang, X. Ding, S. Chen and X. Jiang, J. Biol. Chem., 2009, 284, 12297–12305 CrossRef CAS PubMed.
- N. Hosokawa, T. Hara, T. Kaizuka, C. Kishi, A. Takamura, Y. Miura, S. Iemura, T. Natsume, K. Takehana, N. Yamada, J. L. Guan, N. Oshiro and N. Mizushima, Mol. Biol. Cell, 2009, 20, 1981–1991 CrossRef CAS PubMed.
- C. H. Jung, C. B. Jun, S. H. Ro, Y. M. Kim, N. M. Otto, J. Cao, M. Kundu and D. H. Kim, Mol. Biol. Cell, 2009, 20, 1992–2003 CrossRef CAS PubMed.
- N. Hosokawa, T. Sasaki, S. Iemura, T. Natsume, T. Hara and N. Mizushima, Autophagy, 2009, 5, 973–979 CrossRef CAS PubMed.
- C. A. Mercer, A. Kaliappan and P. B. Dennis, Autophagy, 2009, 5, 649–662 CrossRef CAS PubMed.
- R. C. Russell, Y. Tian, H. Yuan, H. W. Park, Y. Y. Chang, J. Kim, H. Kim, T. P. Neufeld, A. Dillin and K. L. Guan, Nat. Cell Biol., 2013, 15, 741–750 CrossRef CAS PubMed.
- J. M. Park, C. H. Jung, M. Seo, N. M. Otto, D. Grunwald, K. H. Kim, B. Moriarity, Y. M. Kim, C. Starker, R. S. Nho, D. Voytas and D. H. Kim, Autophagy, 2016, 12, 547–564 CrossRef CAS PubMed.
- J. Sawa-Makarska, V. Baumann, N. Coudevylle, S. von Bülow, V. Nogellova, C. Abert, M. Schuschnig, M. Graef, G. Hummer and S. Martens, Science, 2020, 369(6508), eaaz7714 CrossRef CAS PubMed.
- H. Nakatogawa, Essays Biochem., 2013, 55, 39–50 CrossRef CAS PubMed.
- Y. Kabeya, N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi and T. Yoshimori, EMBO J., 2000, 19, 5720–5728 CrossRef CAS PubMed.
- S. S. Mann and J. A. Hammarback, J. Biol. Chem., 1994, 269, 11492–11497 CrossRef CAS PubMed.
- I. Tanida, T. Ueno and E. Kominami, Methods Mol. Biol., 2008, 445, 77–88 CrossRef CAS PubMed.
- M. R. Slobodkin and Z. Elazar, Essays Biochem., 2013, 55, 51–64 CrossRef CAS PubMed.
- P. Wild, D. G. McEwan and I. Dikic, J. Cell Sci., 2014, 127, 3–9 CAS.
- H. Nakatogawa, Y. Ichimura and Y. Ohsumi, Cell, 2007, 130, 165–178 CrossRef CAS PubMed.
- N. Fujita, M. Hayashi-Nishino, H. Fukumoto, H. Omori, A. Yamamoto, T. Noda and T. Yoshimori, Mol. Biol. Cell, 2008, 19, 4651–4659 CrossRef CAS PubMed.
- H. Weidberg, E. Shvets, T. Shpilka, F. Shimron, V. Shinder and Z. Elazar, EMBO J., 2010, 29, 1792–1802 CrossRef CAS PubMed.
- E. Itakura, C. Kishi-Itakura, I. Koyama-Honda and N. Mizushima, J. Cell Sci., 2012, 125, 1488–1499 CAS.
- M. Manil-Ségalen, C. Lefebvre, C. Jenzer, M. Trichet, C. Boulogne, B. Satiat-Jeunemaitre and R. Legouis, Dev. Cell, 2014, 28, 43–55 CrossRef PubMed.
- J. Sawa-Makarska, C. Abert, J. Romanov, B. Zens, I. Ibiricu and S. Martens, Nat. Cell Biol., 2014, 16, 425–433 CrossRef CAS PubMed.
- A. Stolz, A. Ernst and I. Dikic, Nat. Cell Biol., 2014, 16, 495–501 CrossRef CAS PubMed.
- K. Juenemann and E. A. Reits, Int. J. Cell Biol., 2012, 2012, 189794 Search PubMed.
- T. N. Nguyen, B. S. Padman, J. Usher, V. Oorschot, G. Ramm and M. Lazarou, J. Cell Biol., 2016, 215, 857–874 CrossRef CAS PubMed.
- T. Kirisako, M. Baba, N. Ishihara, K. Miyazawa, M. Ohsumi, T. Yoshimori, T. Noda and Y. Ohsumi, J. Cell Biol., 1999, 147, 435–446 CrossRef CAS PubMed.
- J. Romanov, M. Walczak, I. Ibiricu, S. Schüchner, E. Ogris, C. Kraft and S. Martens, EMBO J., 2012, 31, 4304–4317 CrossRef CAS PubMed.
- D. Gatica, V. Lahiri and D. J. Klionsky, Nat. Cell Biol., 2018, 20, 233–242 CrossRef CAS PubMed.
- H. Weidberg, E. Shvets and Z. Elazar, Annu. Rev. Biochem., 2011, 80, 125–156 CrossRef CAS PubMed.
- T. Johansen and T. Lamark, J. Mol. Biol., 2020, 432, 80–103 CrossRef CAS PubMed.
- A. Gubas and I. Dikic, FEBS J., 2022, 289(1), 75–89 CrossRef CAS PubMed.
- Z. Deng, K. Purtell, V. Lachance, M. S. Wold, S. Chen and Z. Yue, Trends Cell Biol., 2017, 27, 491–504 CrossRef CAS PubMed.
- Y. T. Zheng, S. Shahnazari, A. Brech, T. Lamark, T. Johansen and J. H. Brumell, J. Immunol., 2009, 183, 5909–5916 CrossRef CAS PubMed.
- G. Bjørkøy, T. Lamark, A. Brech, H. Outzen, M. Perander, A. Overvatn, H. Stenmark and T. Johansen, J. Cell Biol., 2005, 171, 603–614 CrossRef PubMed.
- R. K. Vadlamudi, I. Joung, J. L. Strominger and J. Shin, J. Biol. Chem., 1996, 271, 20235–20237 CrossRef CAS PubMed.
- Y. Yang, T. L. Willis, R. W. Button, C. J. Strang, Y. Fu, X. Wen, P. R. C. Grayson, T. Evans, R. J. Sipthorpe, S. L. Roberts, B. Hu, J. Zhang, B. Lu and S. Luo, Nat. Commun., 2019, 10, 3759 CrossRef PubMed.
- V. Kirkin, T. Lamark, Y. S. Sou, G. Bjørkøy, J. L. Nunn, J. A. Bruun, E. Shvets, D. G. McEwan, T. H. Clausen, P. Wild, I. Bilusic, J. P. Theurillat, A. Øvervatn, T. Ishii, Z. Elazar, M. Komatsu, I. Dikic and T. Johansen, Mol. Cell, 2009, 33, 505–516 CrossRef CAS PubMed.
- P. Wild, H. Farhan, D. G. McEwan, S. Wagner, V. V. Rogov, N. R. Brady, B. Richter, J. Korac, O. Waidmann, C. Choudhary, V. Dötsch, D. Bumann and I. Dikic, Science, 2011, 333, 228–233 CrossRef CAS PubMed.
- R. S. Marshall, Z. Hua, S. Mali, F. McLoughlin and R. D. Vierstra, Cell, 2019, 177, 766–781.e724 CrossRef CAS PubMed.
- B. P. Belcher, C. C. Ward and D. K. Nomura, Biochemistry, 2021 DOI:10.1021/acs.biochem.1c00464.
- M. B. Schaaf, T. G. Keulers, M. A. Vooijs and K. M. Rouschop, FASEB J., 2016, 30, 3961–3978 CrossRef CAS PubMed.
- V. Kirkin and V. V. Rogov, Mol. Cell, 2019, 76, 268–285 CrossRef CAS PubMed.
- B. L. Heckmann, E. Boada-Romero, L. D. Cunha, J. Magne and D. R. Green, J. Mol. Biol., 2017, 429, 3561–3576 CrossRef CAS PubMed.
- S. Pankiv, T. H. Clausen, T. Lamark, A. Brech, J. A. Bruun, H. Outzen, A. Overvatn, G. Bjorkoy and T. Johansen, J. Biol. Chem., 2007, 282, 24131–24145 CrossRef CAS PubMed.
- Y. Thielmann, O. H. Weiergräber, P. Ma, M. Schwarten, J. Mohrlüder and D. Willbold, Proteins, 2009, 77, 637–646 CrossRef CAS PubMed.
- L. Shang, S. Chen, F. Du, S. Li, L. Zhao and X. Wang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4788–4793 CrossRef CAS PubMed.
- S. J. Cherra, 3rd, S. M. Kulich, G. Uechi, M. Balasubramani, J. Mountzouris, B. W. Day and C. T. Chu, J. Biol. Chem., 2010, 190, 533–539 Search PubMed.
- R. Huang, Y. Xu, W. Wan, X. Shou, J. Qian, Z. You, B. Liu, C. Chang, T. Zhou, J. Lippincott-Schwartz and W. Liu, Mol. Cell, 2015, 57, 456–466 CrossRef CAS PubMed.
- A. Bánréti, M. Sass and Y. Graba, Autophagy, 2013, 9, 819–829 CrossRef PubMed.
- T. Song, H. Su, W. Yin, L. Wang and R. Huang, FEBS Lett., 2019, 593, 414–422 CrossRef CAS PubMed.
- R. S. Marshall, F. Li, D. C. Gemperline, A. J. Book and R. D. Vierstra, Mol. Cell, 2015, 58, 1053–1066 CrossRef CAS PubMed.
- W. C. Tseng, P. M. Jenkins, M. Tanaka, R. Mooney and V. Bennett, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1214–1219 CrossRef CAS PubMed.
- J. Li, R. Zhu, K. Chen, H. Zheng, H. Zhao, C. Yuan, H. Zhang, C. Wang and M. Zhang, Nat. Chem. Biol., 2018, 14, 778–787 CrossRef CAS PubMed.
- N. Luo, D. Shang, Z. Tang, X. Huang, L.-Z. Tao, L. Liu, C. Gao, Y. Qian, Q. Xie and F. Li, bioRxiv, 2021 DOI:10.1101/2021.06.11.448008,2021.2006.2011.448008.
- S. Fan, L. Yue, W. Wan, Y. Zhang, B. Zhang, C. Otomo, Q. Li, T. Lin, J. Hu, P. Xu, M. Zhu, H. Tao, Z. Chen, L. Li, H. Ding, Z. Yao, J. Lu, Y. Wen, N. Zhang, M. Tan, K. Chen, Y. Xie, T. Otomo, B. Zhou, H. Jiang, Y. Dang and C. Luo, Angew. Chem., Int. Ed., 2021, 60(50), 26105–26114 CrossRef CAS PubMed.
- M. Steffek, E. Helgason, N. Popovych, L. Rougé, J. M. Bruning, K. S. Li, D. J. Burdick, J. Cai, T. Crawford, J. Xue, W. Decurtins, C. Fang, F. Grubers, M. J. Holliday, A. Langley, A. Petersen, A. L. Satz, A. Song, D. Stoffler, Q. Strebel, J. Y. K. Tom, N. Skelton, S. T. Staben, M. Wichert, M. M. Mulvihill and E. C. Dueber, Biochemistry, 2022 DOI:10.1021/acs.biochem.1c00682.
- M. Hartmann, J. Huber, J. S. Kramer, J. Heering, L. Pietsch, H. Stark, D. Odadzic, I. Bischoff, R. Fürst, M. Schröder, M. Akutsu, A. Chaikuad, V. Dötsch, S. Knapp, R. M. Biondi, V. V. Rogov and E. Proschak, J. Med. Chem., 2021, 64, 3720–3746 CrossRef CAS PubMed.
- F. Tolle, P. Y. Risold, V. Mansuy-Schlick, E. Rossi, M. Boyer-Guittaut, A. Fraichard and M. Jouvenot, Brain Res., 2008, 1210, 103–115 CrossRef CAS PubMed.
- Y. Sagiv, A. Legesse-Miller, A. Porat and Z. Elazar, EMBO J., 2000, 19, 1494–1504 CrossRef CAS PubMed.
- H. He, Y. Dang, F. Dai, Z. Guo, J. Wu, X. She, Y. Pei, Y. Chen, W. Ling, C. Wu, S. Zhao, J. O. Liu and L. Yu, J. Biol. Chem., 2003, 278, 29278–29287 CrossRef CAS PubMed.
- Y. K. Lee and J. A. Lee, BMB Rep., 2016, 49, 424–430 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- X. Feng, S. Luo and B. Lu, Trends Biochem. Sci., 2018, 43, 424–435 CrossRef CAS PubMed.
- H. S. McLoughlin, L. R. Moore and H. L. Paulson, Neurobiol. Dis., 2020, 134, 104635 CrossRef CAS PubMed.
- D. C. Rubinsztein and J. Carmichael, Expert Rev. Mol. Med., 2003, 5, 1–21 CrossRef PubMed.
- M. P. Duyao, A. B. Auerbach, A. Ryan, F. Persichetti, G. T. Barnes, S. M. McNeil, P. Ge, J. P. Vonsattel, J. F. Gusella and A. L. Joyner, et al. , Science, 1995, 269, 407–410 CrossRef CAS PubMed.
- A. Reiner, I. Dragatsis, S. Zeitlin and D. Goldowitz, Mol. Neurobiol., 2003, 28, 259–276 CrossRef CAS PubMed.
- J. M. Van Raamsdonk, J. Pearson, D. A. Rogers, N. Bissada, A. W. Vogl, M. R. Hayden and B. R. Leavitt, Hum. Mol. Genet., 2005, 14, 1379–1392 CrossRef CAS PubMed.
- A. M. Travessa, F. B. Rodrigues, T. A. Mestre and J. J. Ferreira, J. Huntington's Dis., 2017, 6, 157–163 CAS.
- S. J. Tabrizi, B. R. Leavitt, G. B. Landwehrmeyer, E. J. Wild, C. Saft, R. A. Barker, N. F. Blair, D. Craufurd, J. Priller, H. Rickards, A. Rosser, H. B. Kordasiewicz, C. Czech, E. E. Swayze, D. A. Norris, T. Baumann, I. Gerlach, S. A. Schobel, E. Paz, A. V. Smith, C. F. Bennett and R. M. Lane, N. Engl. J. Med., 2019, 380, 2307–2316 CrossRef CAS PubMed.
- C. F. Bennett and E. E. Swayze, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 259–293 CrossRef CAS PubMed.
- H. B. Kordasiewicz, L. M. Stanek, E. V. Wancewicz, C. Mazur, M. M. McAlonis, K. A. Pytel, J. W. Artates, A. Weiss, S. H. Cheng, L. S. Shihabuddin, G. Hung, C. F. Bennett and D. W. Cleveland, Neuron, 2012, 74, 1031–1044 CrossRef CAS PubMed.
- E. J. Wild and S. J. Tabrizi, Lancet Neurol., 2017, 16, 837–847 CrossRef CAS PubMed.
- S. J. Tabrizi, R. Ghosh and B. R. Leavitt, Neuron, 2019, 101, 801–819 CrossRef CAS PubMed.
- M. Zengerle, K. H. Chan and A. Ciulli, ACS Chem. Biol., 2015, 10, 1770–1777 CrossRef CAS PubMed.
- Nat. Med., 2021, 27, 1311 Search PubMed.
- B. Lu, I. Al-Ramahi, A. Valencia, Q. Wang, F. Berenshteyn, H. Yang, T. Gallego-Flores, S. Ichcho, A. Lacoste, M. Hild, M. Difiglia, J. Botas and J. Palacino, Nat. Neurosci., 2013, 16, 562–570 CrossRef CAS PubMed.
- M. Yu, Y. Fu, Y. Liang, H. Song, Y. Yao, P. Wu, Y. Yao, Y. Pan, X. Wen, L. Ma, S. Hexige, Y. Ding, S. Luo and B. Lu, Cell Res., 2017, 27, 1441–1465 CrossRef CAS PubMed.
- H. Song, H. Li, S. Guo, Y. Pan, Y. Fu, Z. Zhou, Z. Li, X. Wen, X. Sun, B. He, H. Gu, Q. Zhao, C. Wang, P. An, S. Luo, Y. Hu, X. Xie and B. Lu, Brain, 2018, 141, 1782–1798 CrossRef PubMed.
- I. Al-Ramahi, B. Lu, S. Di Paola, K. Pang, M. de Haro, I. Peluso, T. Gallego-Flores, N. T. Malik, K. Erikson, B. A. Bleiberg, M. Avalos, G. Fan, L. E. Rivers, A. M. Laitman, J. R. Diaz-García, M. Hild, J. Palacino, Z. Liu, D. L. Medina and J. Botas, Cell Syst., 2018, 7, 28–40.e24 CrossRef CAS PubMed.
- T. S. Onur, A. Laitman, H. Zhao, R. Keyho, H. Kim, J. Wang, M. Mair, H. Wang, L. Li, A. Perez, M. de Haro, Y. W. Wan, G. Allen, B. Lu, I. Al-Ramahi, Z. Liu and J. Botas, eLife, 2021, 10, e64564 CrossRef CAS PubMed.
- M. Jimenez-Sanchez, W. Lam, M. Hannus, B. Sönnichsen, S. Imarisio, A. Fleming, A. Tarditi, F. Menzies, T. E. Dami, C. Xu, E. Gonzalez-Couto, G. Lazzeroni, F. Heitz, D. Diamanti, L. Massai, V. P. Satagopam, G. Marconi, C. Caramelli, A. Nencini, M. Andreini, G. L. Sardone, N. P. Caradonna, V. Porcari, C. Scali, R. Schneider, G. Pollio, C. J. O'Kane, A. Caricasole and D. C. Rubinsztein, Nat. Chem. Biol., 2015, 11, 347–354 CrossRef CAS PubMed.
- L. R. Gauthier, B. C. Charrin, M. Borrell-Pagès, J. P. Dompierre, H. Rangone, F. P. Cordelières, J. De Mey, M. E. MacDonald, V. Lessmann, S. Humbert and F. Saudou, Cell, 2004, 118, 127–138 CrossRef CAS PubMed.
- A. J. Sala, L. C. Bott and R. I. Morimoto, J. Cell Biol., 2017, 216, 1231–1241 CrossRef CAS PubMed.
- A. Djajadikerta, S. Keshri, M. Pavel, R. Prestil, L. Ryan and D. C. Rubinsztein, J. Mol. Biol., 2020, 432, 2799–2821 CrossRef CAS PubMed.
- S. Sarkar, J. E. Davies, Z. Huang, A. Tunnacliffe and D. C. Rubinsztein, J. Biol. Chem., 2007, 282, 5641–5652 CrossRef CAS PubMed.
- C. G. Keller, Y. Shin, A. M. Monteys, N. Renaud, M. Beibel, N. Teider, T. Peters, T. Faller, S. St-Cyr, J. Knehr, G. Roma, A. Reyes, M. Hild, D. Lukashev, D. Theil, N. Dales, J. H. Cha, B. Borowsky, R. Dolmetsch, B. L. Davidson and R. Sivasankaran, Nat. Commun., 2022, 13, 1150 CrossRef CAS PubMed.
- S. Tomoshige, S. Nomura, K. Ohgane, Y. Hashimoto and M. Ishikawa, Bioorg. Med. Chem. Lett., 2018, 28, 707–710 CrossRef CAS PubMed.
- S. Tomoshige, S. Nomura, K. Ohgane, Y. Hashimoto and M. Ishikawa, Angew. Chem., Int. Ed., 2017, 56, 11530–11533 CrossRef CAS PubMed.
- X. Sun, Y. Fu, Y. Pan and B. Lu, Autophagy, 2017, 13, 2111–2112 CrossRef CAS PubMed.
- Y. Fu, X. Sun and B. Lu, Autophagy, 2018, 14, 169–170 CrossRef CAS PubMed.
- J. Miller, M. Arrasate, E. Brooks, C. P. Libeu, J. Legleiter, D. Hatters, J. Curtis, K. Cheung, P. Krishnan, S. Mitra, K. Widjaja, B. A. Shaby, G. P. Lotz, Y. Newhouse, E. J. Mitchell, A. Osmand, M. Gray, V. Thulasiramin, F. Saudou, M. Segal, X. W. Yang, E. Masliah, L. M. Thompson, P. J. Muchowski, K. H. Weisgraber and S. Finkbeiner, Nat. Chem. Biol., 2011, 7, 925–934 CrossRef CAS PubMed.
- C. Peters-Libeu, J. Miller, E. Rutenber, Y. Newhouse, P. Krishnan, K. Cheung, D. Hatters, E. Brooks, K. Widjaja, T. Tran, S. Mitra, M. Arrasate, L. A. Mosquera, D. Taylor, K. H. Weisgraber and S. Finkbeiner, J. Mol. Biol., 2012, 421, 587–600 CrossRef CAS PubMed.
- G. M. Burslem and C. M. Crews, Cell, 2020, 181, 102–114 CrossRef CAS PubMed.
- R. A. M. Buijsen, L. J. A. Toonen, S. L. Gardiner and W. M. C. van Roon-Mom, Neurotherapeutics, 2019, 16, 263–286 CrossRef CAS PubMed.
- G. Lu, R. E. Middleton, H. Sun, M. Naniong, C. J. Ott, C. S. Mitsiades, K. K. Wong, J. E. Bradner and W. G. Kaelin, Jr., Science, 2014, 343, 305–309 CrossRef CAS PubMed.
- G. Onal, O. Kutlu, D. Gozuacik and S. Dokmeci Emre, Lipids Health Dis., 2017, 16, 128 CrossRef PubMed.
- R. Singh, S. Kaushik, Y. Wang, Y. Xiang, I. Novak, M. Komatsu, K. Tanaka, A. M. Cuervo and M. J. Czaja, Nature, 2009, 458, 1131–1135 CrossRef CAS PubMed.
- H. Dong and M. J. Czaja, Trends Endocrinol. Metab., 2011, 22, 234–240 CrossRef CAS PubMed.
- K. L. Donnelly, C. I. Smith, S. J. Schwarzenberg, J. Jessurun, M. D. Boldt and E. J. Parks, J. Clin. Invest., 2005, 115, 1343–1351 CrossRef CAS PubMed.
- L. Liu, K. Zhang, H. Sandoval, S. Yamamoto, M. Jaiswal, E. Sanz, Z. Li, J. Hui, B. H. Graham, A. Quintana and H. J. Bellen, Cell, 2015, 160, 177–190 CrossRef CAS PubMed.
- S. Sharma, J. V. Adrogue, L. Golfman, I. Uray, J. Lemm, K. Youker, G. P. Noon, O. H. Frazier and H. Taegtmeyer, FASEB J., 2004, 18, 1692–1700 CrossRef CAS PubMed.
- A. Mehlem, C. E. Hagberg, L. Muhl, U. Eriksson and A. Falkevall, Nat. Protoc., 2013, 8, 1149–1154 CrossRef PubMed.
- C. V. de Castro Ghizoni, F. R. Gasparin, A. S. Junior, F. O. Carreno, R. P. Constantin, A. Bracht, E. L. Ishii Iwamoto and J. Constantin, Mol. Cell. Biochem., 2013, 373, 265–277 CrossRef PubMed.
- C. de Koning, M. Beekhuijzen, M. Tobor-Kaplon, S. de Vries-Buitenweg, D. Schoutsen, N. Leeijen, B. van de Waart and H. Emmen, Birth Defects Res., Part B, 2015, 104, 253–272 CrossRef CAS PubMed.
- I. S. Sobolevskaia, O. D. Miadelets and V. N. Grushin, Morfologiia, 2012, 142, 78–82 CAS.
- T. Yuan, Y. Zhong, Y. Wang, T. Zhang, R. Lu, M. Zhou, Y. Lu, K. Yan, Y. Chen, Z. Hu, J. Liang, J. Fan and Y. Cheng, Lipids Health Dis., 2019, 18, 69 CrossRef PubMed.
- H. Chen, O. Charlat, L. A. Tartaglia, E. A. Woolf, X. Weng, S. J. Ellis, N. D. Lakey, J. Culpepper, K. J. Moore, R. E. Breitbart, G. M. Duyk, R. I. Tepper and J. P. Morgenstern, Cell, 1996, 84, 491–495 CrossRef CAS PubMed.
- D. A. Gapp, E. H. Leiter, D. L. Coleman and R. W. Schwizer, Diabetologia, 1983, 25, 439–443 CrossRef CAS PubMed.
- K. P. Hummel, M. M. Dickie and D. L. Coleman, Science, 1966, 153, 1127–1128 CrossRef CAS PubMed.
- L. Recena Aydos, L. Aparecida do Amaral, R. Serafim de Souza, A. C. Jacobowski, E. Freitas Dos Santos and M. L. Rodrigues Macedo, Nutrients, 2019, 11(12), 3067 CrossRef PubMed.
- T. H. Tsai, E. Chen, L. Li, P. Saha, H. J. Lee, L. S. Huang, G. S. Shelness, L. Chan and B. H. Chang, Autophagy, 2017, 13, 1130–1144 CrossRef CAS PubMed.
- C. Ferrucci-Da Silva, L. Zhan, J. Shen, B. Kong, M. J. Campbell, N. Memon, T. Hegyi, L. Lu and G. L. Guo, Acta Pharm. Sin. B, 2020, 10, 153–158 CrossRef PubMed.
- I. A. Kirpich, L. N. Gobejishvili, M. Bon Homme, S. Waigel, M. Cave, G. Arteel, S. S. Barve, C. J. McClain and I. V. Deaciuc, J. Nutr. Biochem., 2011, 22, 38–45 CrossRef CAS PubMed.
- E. De Vita, D. Lucy and E. W. Tate, Cell Res., 2021, 31, 945–946 CrossRef CAS PubMed.
- Z. Hu and C. M. Crews, ChemBioChem, 2022, 23, e202100270 CAS.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- S. He, G. Dong, J. Cheng, Y. Wu and C. Sheng, Med. Res. Rev., 2022, 42, 1280–1342 CrossRef PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755–763 CrossRef CAS PubMed.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- C. Y. Yang, C. Qin, L. Bai and S. Wang, Drug Discovery Today: Technol., 2019, 31, 43–51 CrossRef PubMed.
- F. Spriano, A. Stathis and F. Bertoni, Pharmacol. Ther., 2020, 215, 107631 CrossRef CAS PubMed.
- J.-P. Pei, X. Pan, A. Wang, W. Shuai, F. Bu, P. Tang, S. Zhang, Y. Zhang, G. Wang and L. Ouyang, Chem. Commun., 2021, 57, 13194–13197 RSC.
- C. Xu, L. Wang, P. Fozouni, G. Evjen, V. Chandra, J. Jiang, C. Lu, M. Nicastri, C. Bretz, J. D. Winkler, R. Amaravadi, B. A. Garcia, P. D. Adams, M. Ott, W. Tong, T. Johansen, Z. Dou and S. L. Berger, Nat. Cell Biol., 2020, 22, 1170–1179 CrossRef CAS PubMed.
- Z. Dou, A. Ivanov, P. D. Adams and S. L. Berger, Autophagy, 2016, 12, 1416–1417 CrossRef CAS PubMed.
- Z. Dou, C. Xu, G. Donahue, T. Shimi, J. A. Pan, J. Zhu, A. Ivanov, B. C. Capell, A. M. Drake, P. P. Shah, J. M. Catanzaro, M. D. Ricketts, T. Lamark, S. A. Adam, R. Marmorstein, W. X. Zong, T. Johansen, R. D. Goldman, P. D. Adams and S. L. Berger, Nature, 2015, 527, 105–109 CrossRef CAS PubMed.
- A. Lucena-Cacace, D. Otero-Albiol, M. P. Jiménez-García, S. Muñoz-Galvan and A. Carnero, Clin. Cancer Res., 2018, 24, 1202–1215 CrossRef CAS PubMed.
- V. Audrito, EBioMedicine, 2020, 62, 103109 CrossRef CAS PubMed.
- V. Audrito, V. G. Messana and S. Deaglio, Front Oncol., 2020, 10, 358 CrossRef PubMed.
- V. K. Pulla, D. S. Sriram, V. Soni, S. Viswanadha, D. Sriram and P. Yogeeswari, Chem. Biol. Drug Des., 2015, 86, 881–894 CrossRef CAS PubMed.
- C. Travelli, G. Colombo, S. Mola, A. A. Genazzani and C. Porta, Pharmacol. Res., 2018, 135, 25–36 CrossRef CAS PubMed.
- S. Imai, Curr. Pharm. Des., 2009, 15, 20–28 CrossRef CAS PubMed.
- K. Holen, L. B. Saltz, E. Hollywood, K. Burk and A. R. Hanauske, Invest. New Drugs, 2008, 26, 45–51 CrossRef CAS PubMed.
- A. von Heideman, A. Berglund, R. Larsson and P. Nygren, Cancer Chemother. Pharmacol., 2010, 65, 1165–1172 CrossRef PubMed.
- L. Korotchkina, D. Kazyulkin, P. G. Komarov, A. Polinsky, E. L. Andrianova, S. Joshi, M. Gupta, S. Vujcic, E. Kononov, I. Toshkov, Y. Tian, P. Krasnov, M. V. Chernov, J. Veith, M. P. Antoch, S. Middlemiss, K. Somers, R. B. Lock, M. D. Norris, M. J. Henderson, M. Haber, O. B. Chernova and A. V. Gudkov, Leukemia, 2020, 34, 1828–1839 CrossRef CAS PubMed.
- Y. Wu, C. Pu, Y. Fu, G. Dong, M. Huang and C. Sheng, Acta Pharm. Sin. B, 2022, 12, 2859–2868 CrossRef CAS PubMed.
- G. Dong, Y. Wu, J. Cheng, L. Chen, R. Liu, Y. Ding, S. Wu, J. Ma and C. Sheng, J. Med. Chem., 2022, 65(11), 7619–7628 CrossRef CAS PubMed.
- Y. Gao, Y. Liu, L. Hong, Z. Yang, X. Cai, X. Chen, Y. Fu, Y. Lin, W. Wen, S. Li, X. Liu, H. Huang, A. Vogt, P. Liu, X. M. Yin and M. Li, Cell Death Dis., 2016, 7, e2330 CrossRef CAS PubMed.
- T. Torisu, K. Torisu, I. H. Lee, J. Liu, D. Malide, C. A. Combs, X. S. Wu Rovira, II, M. M. Fergusson, R. Weigert, P. S. Connelly, M. P. Daniels, M. Komatsu, L. Cao and T. Finkel, Nat. Med., 2013, 19, 1281–1287 CrossRef CAS PubMed.
- K. Ishibashi, T. Uemura, S. Waguri and M. Fukuda, Mol. Biol. Cell, 2012, 23, 3193–3202 CrossRef CAS PubMed.
- J. D. Dickinson, Y. Alevy, N. P. Malvin, K. K. Patel, S. P. Gunsten, M. J. Holtzman, T. S. Stappenbeck and S. L. Brody, Autophagy, 2016, 12, 397–409 CrossRef CAS PubMed.
- S. Bel, M. Pendse, Y. Wang, Y. Li, K. A. Ruhn, B. Hassell, T. Leal, S. E. Winter, R. J. Xavier and L. V. Hooper, Science, 2017, 357, 1047–1052 CrossRef CAS PubMed.
- N. Dupont, S. Jiang, M. Pilli, W. Ornatowski, D. Bhattacharya and V. Deretic, EMBO J., 2011, 30, 4701–4711 CrossRef CAS PubMed.
- T. Kimura, J. Jia, S. Kumar, S. W. Choi, Y. Gu, M. Mudd, N. Dupont, S. Jiang, R. Peters, F. Farzam, A. Jain, K. A. Lidke, C. M. Adams, T. Johansen and V. Deretic, EMBO J., 2017, 36, 42–60 CrossRef CAS PubMed.
- J. Thorburn, H. Horita, J. Redzic, K. Hansen, A. E. Frankel and A. Thorburn, Cell Death Differ., 2009, 16, 175–183 CrossRef CAS PubMed.
- J. Shi, Y. Zhao, K. Wang, X. Shi, Y. Wang, H. Huang, Y. Zhuang, T. Cai, F. Wang and F. Shao, Nature, 2015, 526, 660–665 CrossRef CAS PubMed.
- T. Calì, C. Galli, S. Olivari and M. Molinari, Biochem. Biophys. Res. Commun., 2008, 371, 405–410 CrossRef PubMed.
- F. Reggiori, I. Monastyrska, M. H. Verheije, T. Calì, M. Ulasli, S. Bianchi, R. Bernasconi, C. A. de Haan and M. Molinari, Cell Host Microbe, 2010, 7, 500–508 CrossRef CAS PubMed.
- I. Nakagawa, A. Amano, N. Mizushima, A. Yamamoto, H. Yamaguchi, T. Kamimoto, A. Nara, J. Funao, M. Nakata, K. Tsuda, S. Hamada and T. Yoshimori, Science, 2004, 306, 1037–1040 CrossRef CAS PubMed.
- T. Sawa, M. H. Zaki, T. Okamoto, T. Akuta, Y. Tokutomi, S. Kim-Mitsuyama, H. Ihara, A. Kobayashi, M. Yamamoto, S. Fujii, H. Arimoto and T. Akaike, Nat. Chem. Biol., 2007, 3, 727–735 CrossRef CAS PubMed.
- M. L. Seibenhener, J. R. Babu, T. Geetha, H. C. Wong, N. R. Krishna and M. W. Wooten, Mol. Cell. Biol., 2004, 24, 8055–8068 CrossRef CAS PubMed.
- H. Shimizu, S. Yamagishi, H. Chiba and M. Ghazizadeh, Adv. Clin. Exp. Med., 2016, 25, 117–128 CrossRef PubMed.
- M. Lin, X. Zhang, B. Jia and S. Guan, J. Neuro-Oncol., 2018, 136, 243–254 CrossRef CAS PubMed.
- N. Sin, L. Meng, M. Q. Wang, J. J. Wen, W. G. Bornmann and C. M. Crews, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6099–6103 CrossRef CAS PubMed.
- J. Friedman and I. Weissman, Cell, 1991, 66, 799–806 CrossRef CAS PubMed.
- J. Liu, J. D. Farmer, Jr., W. S. Lane, J. Friedman, I. Weissman and S. L. Schreiber, Cell, 1991, 66, 807–815 CrossRef CAS PubMed.
- X. Zhang, V. M. Crowley, T. G. Wucherpfennig, M. M. Dix and B. F. Cravatt, Nat. Chem. Biol., 2019, 15, 737–746 CrossRef CAS PubMed.
- R. Rupprecht, V. Papadopoulos, G. Rammes, T. C. Baghai, J. Fan, N. Akula, G. Groyer, D. Adams and M. Schumacher, Nat. Rev. Drug Discovery, 2010, 9, 971–988 CrossRef CAS PubMed.
- L. Xie, J. Yui, A. Hatori, T. Yamasaki, K. Kumata, H. Wakizaka, Y. Yoshida, M. Fujinaga, K. Kawamura and M. R. Zhang, J. Hepatol., 2012, 57, 1076–1082 CrossRef CAS PubMed.
- D. Valenti, N. Braidy, D. De Rasmo, A. Signorile, L. Rossi, A. G. Atanasov, M. Volpicella, A. Henrion-Caude, S. M. Nabavi and R. A. Vacca, Free Radical Biol. Med., 2018, 114, 69–83 CrossRef CAS PubMed.
- H. Cha-Molstad, J. E. Yu, Z. Feng, S. H. Lee, J. G. Kim, P. Yang, B. Han, K. W. Sung, Y. D. Yoo, J. Hwang, T. McGuire, S. M. Shim, H. D. Song, S. Ganipisetti, N. Wang, J. M. Jang, M. J. Lee, S. J. Kim, K. H. Lee, J. T. Hong, A. Ciechanover, I. Mook-Jung, K. P. Kim, X. Q. Xie, Y. T. Kwon and B. Y. Kim, Nat. Commun., 2017, 8, 102 CrossRef PubMed.
- Y. Zhang, S. R. Mun, J. F. Linares, J. Ahn, C. G. Towers, C. H. Ji, B. E. Fitzwalter, M. R. Holden, W. Mi, X. Shi, J. Moscat, A. Thorburn, M. T. Diaz-Meco, Y. T. Kwon and T. G. Kutateladze, Nat. Commun., 2018, 9, 4373 CrossRef PubMed.
- N. Myeku and M. E. Figueiredo-Pereira, J. Biol. Chem., 2011, 286, 22426–22440 CrossRef CAS PubMed.
- K. M. Sakamoto, Pediatr. Res., 2010, 67, 505–508 CrossRef CAS PubMed.
- S. Sigismund, S. Polo and P. P. Di Fiore, Curr. Top. Microbiol. Immunol., 2004, 286, 149–185 CAS.
- L. Cortez and V. Sim, Prion, 2014, 8, 197–202 CrossRef CAS PubMed.
- S. Hashimoto, Y. Matsuba, N. Kamano, N. Mihira, N. Sahara, J. Takano, S. I. Muramatsu, T. C. Saido and T. Saito, Nat. Commun., 2019, 10, 2394 CrossRef PubMed.
- K. H. Strang, C. L. Croft, Z. A. Sorrentino, P. Chakrabarty, T. E. Golde and B. I. Giasson, J. Biol. Chem., 2018, 293, 4579 CrossRef CAS PubMed.
- E. S. Kornilova, Biochemistry, 2014, 79, 865–878 CAS.
- S. A. Mousavi, L. Malerød, T. Berg and R. Kjeken, Biochem. J., 2004, 377, 1–16 CrossRef CAS PubMed.
- J. Gruenberg, G. Griffiths and K. E. Howell, J. Cell Biol., 1989, 108, 1301–1316 CrossRef CAS PubMed.
- N. Ali and W. H. Evans, Biochem. J., 1990, 271, 193–199 CrossRef CAS PubMed.
- C. S. Pillay, E. Elliott and C. Dennison, Biochem. J., 2002, 363, 417–429 CrossRef CAS PubMed.
- C. C. Scott, F. Vacca and J. Gruenberg, Semin. Cell Dev. Biol., 2014, 31, 2–10 CrossRef CAS PubMed.
- C. C. Cain, D. M. Sipe and R. F. Murphy, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 544–548 CrossRef CAS PubMed.
- M. Forgac, L. Cantley, B. Wiedenmann, L. Altstiel and D. Branton, Semin. Cell Dev. Biol., 1983, 80, 1300–1303 CAS.
- I. Mellman, J. Exp. Biol., 1992, 172, 39–45 CrossRef CAS PubMed.
- J. Somsel Rodman and A. Wandinger-Ness, J. Cell Sci., 2000, 113(2), 183–192 CrossRef PubMed.
- A. Zeigerer, J. Gilleron, R. L. Bogorad, G. Marsico, H. Nonaka, S. Seifert, H. Epstein-Barash, S. Kuchimanchi, C. G. Peng, V. M. Ruda, P. Del Conte-Zerial, J. G. Hengstler, Y. Kalaidzidis, V. Koteliansky and M. Zerial, Nature, 2012, 485, 465–470 CrossRef CAS PubMed.
- H. W. Shin, M. Hayashi, S. Christoforidis, S. Lacas-Gervais, S. Hoepfner, M. R. Wenk, J. Modregger, S. Uttenweiler-Joseph, M. Wilm, A. Nystuen, W. N. Frankel, M. Solimena, P. De Camilli and M. Zerial, J. Cell Biol., 2005, 170, 607–618 CrossRef CAS PubMed.
- D. C. Lawe, A. Chawla, E. Merithew, J. Dumas, W. Carrington, K. Fogarty, L. Lifshitz, R. Tuft, D. Lambright and S. Corvera, J. Biol. Chem., 2002, 277, 8611–8617 CrossRef CAS PubMed.
- B. Sönnichsen, S. De Renzis, E. Nielsen, J. Rietdorf and M. Zerial, J. Cell Biol., 2000, 149, 901–914 CrossRef PubMed.
- L. Langemeyer, A. C. Borchers, E. Herrmann, N. Füllbrunn, Y. Han, A. Perz, K. Auffarth, D. Kümmel and C. Ungermann, eLife, 2020, 9, e56090 CrossRef PubMed.
- P. Barbero, L. Bittova and S. R. Pfeffer, J. Cell Biol., 2002, 156, 511–518 CrossRef CAS PubMed.
- C. Raiborg and H. Stenmark, Nature, 2009, 458, 445–452 CrossRef CAS PubMed.
- H. J. Balderhaar and C. Ungermann, J. Cell Sci., 2013, 126, 1307–1316 CrossRef CAS PubMed.
- C. M. Hickey and W. Wickner, Mol. Biol. Cell, 2010, 21, 2297–2305 CrossRef CAS PubMed.
- P. Ghosh, N. M. Dahms and S. Kornfeld, Nat. Rev. Mol. Cell Biol., 2003, 4, 202–212 CrossRef CAS PubMed.
- M. Gary-Bobo, P. Nirdé, A. Jeanjean, A. Morère and M. Garcia, Curr. Med. Chem., 2007, 14, 2945–2953 CrossRef CAS PubMed.
- Y. Shen, X. Li, D. Dong, B. Zhang, Y. Xue and P. Shang, Am. J. Cancer Res., 2018, 8, 916–931 CAS.
- Y. C. Lee, R. R. Townsend, M. R. Hardy, J. Lönngren, J. Arnarp, M. Haraldsson and H. Lönn, J. Biol. Chem., 1983, 258, 199–202 CrossRef CAS PubMed.
- J. K. Nair, J. L. Willoughby, A. Chan, K. Charisse, M. R. Alam, Q. Wang, M. Hoekstra, P. Kandasamy, A. V. Kel'in, S. Milstein, N. Taneja, J. O'Shea, S. Shaikh, L. Zhang, R. J. van der Sluis, M. E. Jung, A. Akinc, R. Hutabarat, S. Kuchimanchi, K. Fitzgerald, T. Zimmermann, T. J. van Berkel, M. A. Maier, K. G. Rajeev and M. Manoharan, J. Am. Chem. Soc., 2014, 136, 16958–16961 CrossRef CAS PubMed.
- X. Huang, J. C. Leroux and B. Castagner, Bioconjugate Chem., 2017, 28, 283–295 CrossRef CAS PubMed.
- T. S. Zimmermann, V. Karsten, A. Chan, J. Chiesa, M. Boyce, B. R. Bettencourt, R. Hutabarat, S. Nochur, A. Vaishnaw and J. Gollob, Mol. Ther., 2017, 25, 71–78 CrossRef CAS PubMed.
- J. H. Shi, W. Z. Guo, Y. Jin, H. P. Zhang, C. Pang, J. Li, P. D. Line and S. J. Zhang, Cancer Med., 2019, 8, 1269–1278 CrossRef CAS PubMed.
- J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9–22 CrossRef CAS PubMed.
- P. O. Seglen, B. Grinde and A. E. Solheim, Eur. J. Biochem., 1979, 95, 215–225 CrossRef CAS PubMed.
- T. Yoshimori, A. Yamamoto, Y. Moriyama, M. Futai and Y. Tashiro, J. Biol. Chem., 1991, 266, 17707–17712 CrossRef CAS PubMed.
- P. O. Seglen and P. B. Gordon, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 1889–1892 CrossRef CAS PubMed.
- B. Pasquier, Autophagy, 2015, 11, 725–726 CrossRef CAS PubMed.
- A. Kuma, M. Komatsu and N. Mizushima, Autophagy, 2017, 13, 1619–1628 CrossRef CAS PubMed.
- S. Sarkar and D. C. Rubinsztein, Autophagy, 2006, 2, 132–134 CrossRef CAS PubMed.
- M. J. Henderson, M. A. Holbert, A. Simeonov and L. A. Kallal, SLAS Discovery, 2020, 25, 137–147 CrossRef PubMed.
- D. Martinez Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef PubMed.
- M. L. Dart, T. Machleidt, E. Jost, M. K. Schwinn, M. B. Robers, C. Shi, T. A. Kirkland, M. P. Killoran, J. M. Wilkinson, J. R. Hartnett, K. Zimmerman and K. V. Wood, ACS Med. Chem. Lett., 2018, 9, 546–551 CrossRef CAS PubMed.
- J. Shaw, I. Dale, P. Hemsley, L. Leach, N. Dekki, J. P. Orme, V. Talbot, A. J. Narvaez, M. Bista, D. Martinez Molina, M. Dabrowski, M. J. Main and D. Gianni, SLAS Discovery, 2019, 24, 121–132 CrossRef CAS PubMed.
- R. Karlsson and A. Fält, J. Immunol. Methods, 1997, 200, 121–133 CrossRef CAS PubMed.
- B. Douzi, Methods Mol. Biol., 2017, 1615, 257–275 CrossRef PubMed.
- J. Concepcion, K. Witte, C. Wartchow, S. Choo, D. Yao, H. Persson, J. Wei, P. Li, B. Heidecker, W. Ma, R. Varma, L. S. Zhao, D. Perillat, G. Carricato, M. Recknor, K. Du, H. Ho, T. Ellis, J. Gamez, M. Howes, J. Phi-Wilson, S. Lockard, R. Zuk and H. Tan, Comb. Chem. High Throughput Screening, 2009, 12, 791–800 CrossRef CAS PubMed.
- A. Sultana and J. E. Lee, Curr. Protoc. Protein Sci., 2015, 79, 19.25.11–19.25.26 Search PubMed.
- M. J. Roy, S. Winkler, S. J. Hughes, C. Whitworth, M. Galant, W. Farnaby, K. Rumpel and A. Ciulli, ACS Chem. Biol., 2019, 14, 361–368 CrossRef CAS PubMed.
- C. Zhu, B. Ge, R. Chen, X. Zhu, L. Mi, J. Ma, X. Wang, F. Zheng and Y. Fei, Sensors, 2018, 18(2), 524 CrossRef PubMed.
- Y. Fei, J. P. Landry, Y. Sun, X. Zhu, X. Wang, J. Luo, C. Y. Wu and K. S. Lam, J. Biomed. Opt., 2010, 15, 016018 CrossRef PubMed.
- C. Zhu, X. Zhu, J. P. Landry, Z. Cui, Q. Li, Y. Dang, L. Mi, F. Zheng and Y. Fei, Sensors, 2016, 16(3), 378 CrossRef PubMed.
- X. Huang and A. Aulabaugh, Methods Mol. Biol., 2016, 1439, 115–130 CrossRef CAS PubMed.
- M. M. Pierce, C. S. Raman and B. T. Nall, Methods, 1999, 19, 213–221 CrossRef CAS PubMed.
- G. Bruylants, J. Wouters and C. Michaux, Curr. Med. Chem., 2005, 12, 2011–2020 CrossRef CAS PubMed.
- M. Jerabek-Willemsen, C. J. Wienken, D. Braun, P. Baaske and S. Duhr, Assay Drug Dev. Technol., 2011, 9, 342–353 CrossRef CAS PubMed.
- C. J. Wienken, P. Baaske, U. Rothbauer, D. Braun and S. Duhr, Nat. Commun., 2010, 1, 100 CrossRef PubMed.
- F. Degorce, A. Card, S. Soh, E. Trinquet, G. P. Knapik and B. Xie, Curr. Chem. Genomics, 2009, 3, 22–32 CrossRef CAS PubMed.
- A. K. Hagan and T. Zuchner, Anal. Bioanal. Chem., 2011, 400, 2847–2864 CrossRef CAS PubMed.
- M. Bielefeld-Sevigny, Assay Drug Dev. Technol., 2009, 7, 90–92 CrossRef CAS PubMed.
- D. García Jiménez, M. Rossi Sebastiano, M. Vallaro, V. Mileo, D. Pizzirani, E. Moretti, G. Ermondi and G. Caron, J. Med. Chem., 2022 DOI:10.1021/acs.jmedchem.2c00201.
- G. Dong, Y. Ding, S. He and C. Sheng, J. Med. Chem., 2021, 64, 10606–10620 CrossRef CAS PubMed.
- F. Soltermann, W. B. Struwe and P. Kukura, Phys. Chem. Chem. Phys., 2021, 23, 16488–16500 RSC.
- G. Young, N. Hundt, D. Cole, A. Fineberg, J. Andrecka, A. Tyler, A. Olerinyova, A. Ansari, E. G. Marklund, M. P. Collier, S. A. Chandler, O. Tkachenko, J. Allen, M. Crispin, N. Billington, Y. Takagi, J. R. Sellers, C. Eichmann, P. Selenko, L. Frey, R. Riek, M. R. Galpin, W. B. Struwe, J. L. P. Benesch and P. Kukura, Science, 2018, 360, 423–427 CrossRef CAS PubMed.
- D. Wu and G. Piszczek, Eur. Biophys. J., 2021, 50, 403–409 CrossRef CAS PubMed.
- E. D. B. Foley, M. S. Kushwah, G. Young and P. Kukura, Nat. Methods, 2021, 18, 1247–1252 CrossRef CAS PubMed.
- M. Pettersson and C. M. Crews, Drug Discovery Today: Technol., 2019, 31, 15–27 CrossRef PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2018, 25, 78–87.e75 CrossRef CAS PubMed.
- S. Rudnik and M. Damme, FEBS J., 2021, 288, 4168–4182 CrossRef CAS PubMed.
- E. D. Carstea, J. A. Morris, K. G. Coleman, S. K. Loftus, D. Zhang, C. Cummings, J. Gu, M. A. Rosenfeld, W. J. Pavan, D. B. Krizman, J. Nagle, M. H. Polymeropoulos, S. L. Sturley, Y. A. Ioannou, M. E. Higgins, M. Comly, A. Cooney, A. Brown, C. R. Kaneski, E. J. Blanchette-Mackie, N. K. Dwyer, E. B. Neufeld, T. Y. Chang, L. Liscum, J. F. Strauss, 3rd, K. Ohno, M. Zeigler, R. Carmi, J. Sokol and D. Markie, et al. , Science, 1997, 277, 228–231 CrossRef CAS PubMed.
- D. P. Bondeson, A. Mares, I. E. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed.
- R. P. Nowak and L. H. Jones, SLAS discovery, 2021, 26, 474–483 CrossRef CAS PubMed.
- L. Zhao, J. Zhao, K. Zhong, A. Tong and D. Jia, Signal Transduction Targeted Ther., 2022, 7, 113 CrossRef CAS PubMed.
- K. M. Riching, S. Mahan, C. R. Corona, M. McDougall, J. D. Vasta, M. B. Robers, M. Urh and D. L. Daniels, ACS Chem. Biol., 2018, 13, 2758–2770 CrossRef CAS PubMed.
- R. Casement, A. Bond, C. Craigon and A. Ciulli, Methods Mol. Biol., 2021, 2365, 79–113 CrossRef PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K. H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
- H. Xu and D. Ren, Annu. Rev. Physiol., 2015, 77, 57–80 CrossRef CAS PubMed.
- O. Florey and M. Overholtzer, Trends Cell Biol., 2012, 22, 374–380 CrossRef CAS PubMed.
- T. J. Melia, A. H. Lystad and A. Simonsen, J. Cell Biol., 2020, 219(6), e202002085 CrossRef PubMed.
- L. Wang, C. Xu, T. Johansen, S. L. Berger and Z. Dou, Autophagy, 2021, 17, 593–595 CrossRef CAS PubMed.
- Y. Yoneda, J. Biochem., 1997, 121, 811–817 CrossRef CAS PubMed.
- J. Lu, T. Wu, B. Zhang, S. Liu, W. Song, J. Qiao and H. Ruan, Cell Commun. Signaling, 2021, 19, 60 CrossRef CAS PubMed.
- H. A. Alwan, E. J. van Zoelen and J. E. van Leeuwen, J. Biol. Chem., 2003, 278, 35781–35790 CrossRef CAS PubMed.
- J. Zhang, J. Wang, S. Ng, Q. Lin and H. M. Shen, Autophagy, 2014, 10, 901–912 CrossRef CAS PubMed.
- D. J. Klionsky, A. M. Cuervo and P. O. Seglen, Autophagy, 2007, 3, 181–206 CrossRef CAS PubMed.
- T. Mathieson, H. Franken, J. Kosinski, N. Kurzawa, N. Zinn, G. Sweetman, D. Poeckel, V. S. Ratnu, M. Schramm, I. Becher, M. Steidel, K. M. Noh, G. Bergamini, M. Beck, M. Bantscheff and M. M. Savitski, Nat. Commun., 2018, 9, 689 CrossRef PubMed.
- B. Schwanhäusser, D. Busse, N. Li, G. Dittmar, J. Schuchhardt, J. Wolf, W. Chen and M. Selbach, Nature, 2011, 473, 337–342 CrossRef PubMed.
- H. Zhang, P. An, Y. Fei and B. Lu, Neurosci. Bull., 2021, 37, 255–260 CrossRef CAS PubMed.
- C. M. Schworer, K. A. Shiffer and G. E. Mortimore, J. Biol. Chem., 1981, 256, 7652–7658 CrossRef CAS PubMed.
- A. du Toit, J. S. Hofmeyr, T. J. Gniadek and B. Loos, Autophagy, 2018, 14, 1060–1071 CAS.
- G. Bjorkoy, T. Lamark, A. Brech, H. Outzen, M. Perander, A. Overvatn, H. Stenmark and T. Johansen, J. Cell Biol., 2005, 171, 603–614 CrossRef PubMed.
- K. Tanaka and A. Ichihara, Biochem. Biophys. Res. Commun., 1989, 159, 1309–1315 CrossRef CAS PubMed.
- M. F. Princiotta, D. Finzi, S. B. Qian, J. Gibbs, S. Schuchmann, F. Buttgereit, J. R. Bennink and J. W. Yewdell, Immunity, 2003, 18, 343–354 CrossRef CAS PubMed.
- F. M. Platt, A. d'Azzo, B. L. Davidson, E. F. Neufeld and C. J. Tifft, Nat. Rev. Dis. Primers, 2018, 4, 27 CrossRef PubMed.
- M. L. Schultz, L. Tecedor, M. Chang and B. L. Davidson, Trends Neurosci., 2011, 34, 401–410 CrossRef CAS PubMed.
- A. M. Cataldo, C. M. Peterhoff, J. C. Troncoso, T. Gomez-Isla, B. T. Hyman and R. A. Nixon, Am. J. Pathol., 2000, 157, 277–286 CrossRef CAS PubMed.
- R. A. Nixon, J. Wegiel, A. Kumar, W. H. Yu, C. Peterhoff, A. Cataldo and A. M. Cuervo, J. Neuropathol. Exp. Neurol., 2005, 64, 113–122 CrossRef PubMed.
- W. H. Yu, A. M. Cuervo, A. Kumar, C. M. Peterhoff, S. D. Schmidt, J. H. Lee, P. S. Mohan, M. Mercken, M. R. Farmery, L. O. Tjernberg, Y. Jiang, K. Duff, Y. Uchiyama, J. Näslund, P. M. Mathews, A. M. Cataldo and R. A. Nixon, J. Cell Biol., 2005, 171, 87–98 CrossRef CAS PubMed.
- B. Boland, A. Kumar, S. Lee, F. M. Platt, J. Wegiel, W. H. Yu and R. A. Nixon, J. Neurosci., 2008, 28, 6926–6937 CrossRef CAS PubMed.
- Y. J. Lee, S. B. Han, S. Y. Nam, K. W. Oh and J. T. Hong, Arch. Pharmacal Res., 2010, 33, 1539–1556 CrossRef CAS PubMed.
- P. Tammineni and Q. Cai, Autophagy, 2017, 13, 982–984 CrossRef CAS PubMed.
- J. H. Lee, D. S. Yang, C. N. Goulbourne, E. Im, P. Stavrides, A. Pensalfini, H. Chan, C. Bouchet-Marquis, C. Bleiwas, M. J. Berg, C. Huo, J. Peddy, M. Pawlik, E. Levy, M. Rao, M. Staufenbiel and R. A. Nixon, Nat. Neurosci., 2022, 25, 688–701 CrossRef CAS PubMed.
- L. Stefanis, Cold Spring Harbor Perspect. Med., 2012, 2, a009399 Search PubMed.
- A. R. Winslow, C. W. Chen, S. Corrochano, A. Acevedo-Arozena, D. E. Gordon, A. A. Peden, M. Lichtenberg, F. M. Menzies, B. Ravikumar, S. Imarisio, S. Brown, C. J. O'Kane and D. C. Rubinsztein, J. Cell Biol., 2010, 190, 1023–1037 CrossRef CAS PubMed.
- S. A. Tanik, C. E. Schultheiss, L. A. Volpicelli-Daley, K. R. Brunden and V. M. Lee, J. Biol. Chem., 2013, 288, 15194–15210 CrossRef CAS PubMed.
- L. A. Volpicelli-Daley, K. C. Luk and V. M. Lee, Nat. Protoc., 2014, 9, 2135–2146 CrossRef CAS PubMed.
- S. W. Davies, M. Turmaine, B. A. Cozens, M. DiFiglia, A. H. Sharp, C. A. Ross, E. Scherzinger, E. E. Wanker, L. Mangiarini and G. P. Bates, Cell, 1997, 90, 537–548 CrossRef CAS PubMed.
- E. Sapp, C. Schwarz, K. Chase, P. G. Bhide, A. B. Young, J. Penney, J. P. Vonsattel, N. Aronin and M. DiFiglia, Ann. Neurol., 1997, 42, 604–612 CrossRef CAS PubMed.
- K. B. Kegel, M. Kim, E. Sapp, C. McIntyre, J. G. Castaño, N. Aronin and M. DiFiglia, J. Neurosci., 2000, 20, 7268–7278 CrossRef CAS PubMed.
- E. Nagata, A. Sawa, C. A. Ross and S. H. Snyder, NeuroReport, 2004, 15, 1325–1328 CrossRef PubMed.
- Y. Fu, P. Wu, Y. Pan, X. Sun, H. Yang, M. Difiglia and B. Lu, Nat. Chem. Biol., 2017, 13, 1152–1154 CrossRef CAS PubMed.
- Y. N. Rui, Z. Xu, B. Patel, Z. Chen, D. Chen, A. Tito, G. David, Y. Sun, E. F. Stimming, H. J. Bellen, A. M. Cuervo and S. Zhang, Nat. Cell Biol., 2015, 17, 262–275 CrossRef CAS PubMed.
- J. Ochaba, T. Lukacsovich, G. Csikos, S. Zheng, J. Margulis, L. Salazar, K. Mao, A. L. Lau, S. Y. Yeung, S. Humbert, F. Saudou, D. J. Klionsky, S. Finkbeiner, S. O. Zeitlin, J. L. Marsh, D. E. Housman, L. M. Thompson and J. S. Steffan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16889–16894 CrossRef CAS PubMed.
- M. Martinez-Vicente, Z. Talloczy, E. Wong, G. Tang, H. Koga, S. Kaushik, R. de Vries, E. Arias, S. Harris, D. Sulzer and A. M. Cuervo, Nat. Neurosci., 2010, 13, 567–576 CrossRef CAS PubMed.
- A. K. Verma, P. S. Bharti, S. Rafat, D. Bhatt, Y. Goyal, K. K. Pandey, S. Ranjan, S. A. Almatroodi, M. A. Alsahli, A. H. Rahmani, A. Almatroudi and K. Dev, Oxid. Med. Cell. Longevity, 2021, 2021, 8832541 Search PubMed.
- L. Galluzzi, F. Pietrocola, J. M. Bravo-San Pedro, R. K. Amaravadi, E. H. Baehrecke, F. Cecconi, P. Codogno, J. Debnath, D. A. Gewirtz, V. Karantza, A. Kimmelman, S. Kumar, B. Levine, M. C. Maiuri, S. J. Martin, J. Penninger, M. Piacentini, D. C. Rubinsztein, H. U. Simon, A. Simonsen, A. M. Thorburn, G. Velasco, K. M. Ryan and G. Kroemer, EMBO J., 2015, 34, 856–880 CrossRef CAS PubMed.
- T. Ito, H. Ando, T. Suzuki, T. Ogura, K. Hotta, Y. Imamura, Y. Yamaguchi and H. Handa, Science, 2010, 327, 1345–1350 CrossRef CAS PubMed.
- J. Krönke, N. D. Udeshi, A. Narla, P. Grauman, S. N. Hurst, M. McConkey, T. Svinkina, D. Heckl, E. Comer, X. Li, C. Ciarlo, E. Hartman, N. Munshi, M. Schenone, S. L. Schreiber, S. A. Carr and B. L. Ebert, Science, 2014, 343, 301–305 CrossRef PubMed.
- S. L. Schreiber, Cell, 2021, 184, 3–9 CrossRef CAS PubMed.
- M. E. Matyskiela, G. Lu, T. Ito, B. Pagarigan, C. C. Lu, K. Miller, W. Fang, N. Y. Wang, D. Nguyen, J. Houston, G. Carmel, T. Tran, M. Riley, L. Nosaka, G. C. Lander, S. Gaidarova, S. Xu, A. L. Ruchelman, H. Handa, J. Carmichael, T. O. Daniel, B. E. Cathers, A. Lopez-Girona and P. P. Chamberlain, Nature, 2016, 535, 252–257 CrossRef CAS PubMed.
- C. Surka, L. Jin, N. Mbong, C. C. Lu, I. S. Jang, E. Rychak, D. Mendy, T. Clayton, E. Tindall, C. Hsu, C. Fontanillo, E. Tran, A. Contreras, S. W. K. Ng, M. Matyskiela, K. Wang, P. Chamberlain, B. Cathers, J. Carmichael, J. Hansen, J. C. Y. Wang, M. D. Minden, J. Fan, D. W. Pierce, M. Pourdehnad, M. Rolfe, A. Lopez-Girona, J. E. Dick and G. Lu, Blood, 2021, 137, 661–677 CrossRef CAS PubMed.
- Y. Y. Fei, J. P. Landry, Y. S. Sun, X. D. Zhu, J. T. Luo, X. B. Wang and K. S. Lam, Rev. Sci. Instrum., 2008, 79, 013708 CrossRef CAS PubMed.
- S. Yu, Y. Liang, J. Palacino, M. Difiglia and B. Lu, Trends Pharmacol. Sci., 2014, 35, 53–62 CrossRef CAS PubMed.
- X. Sun, H. Gao, Y. Yang, M. He, Y. Wu, Y. Song, Y. Tong and Y. Rao, Signal Transduction Targeted Ther., 2019, 4, 64 CrossRef PubMed.
- M. Słabicki, Z. Kozicka, G. Petzold, Y. D. Li, M. Manojkumar, R. D. Bunker, K. A. Donovan, Q. L. Sievers, J. Koeppel, D. Suchyta, A. S. Sperling, E. C. Fink, J. A. Gasser, L. R. Wang, S. M. Corsello, R. S. Sellar, M. Jan, D. Gillingham, C. Scholl, S. Fröhling, T. R. Golub, E. S. Fischer, N. H. Thomä and B. L. Ebert, Nature, 2020, 585, 293–297 CrossRef PubMed.
- C. Mayor-Ruiz, S. Bauer, M. Brand, Z. Kozicka, M. Siklos, H. Imrichova, I. H. Kaltheuner, E. Hahn, K. Seiler, A. Koren, G. Petzold, M. Fellner, C. Bock, A. C. Müller, J. Zuber, M. Geyer, N. H. Thomä, S. Kubicek and G. E. Winter, Nat. Chem. Biol., 2020, 16, 1199–1207 CrossRef CAS PubMed.
- L. Lv, P. Chen, L. Cao, Y. Li, Z. Zeng, Y. Cui, Q. Wu, J. Li, J. H. Wang, M. Q. Dong, X. Qi and T. Han, eLife, 2020, 9, e59994 CrossRef CAS PubMed.
- D. Li, J. Chen, Y. Ai, X. Gu, L. Li, D. Che, Z. Jiang, L. Li, S. Chen, H. Huang, J. Wang, T. Cai, Y. Cao, X. Qi and X. Wang, Mol. Cell, 2019, 75, 1103–1116.e1109 CrossRef CAS PubMed.
- J. Chen, N. Liu, Y. Huang, Y. Wang, Y. Sun, Q. Wu, D. Li, S. Gao, H. W. Wang, N. Huang, X. Qi and X. Wang, Nat. Commun., 2021, 12, 6204 CrossRef PubMed.
- M. Kostic and L. H. Jones, Trends Pharmacol. Sci., 2020, 41, 305–317 CrossRef CAS PubMed.
- S. B. Alabi and C. M. Crews, J. Biol. Chem., 2021, 296, 100647 CrossRef CAS PubMed.
- E. S. Fischer, K. Böhm, J. R. Lydeard, H. Yang, M. B. Stadler, S. Cavadini, J. Nagel, F. Serluca, V. Acker, G. M. Lingaraju, R. B. Tichkule, M. Schebesta, W. C. Forrester, M. Schirle, U. Hassiepen, J. Ottl, M. Hild, R. E. Beckwith, J. W. Harper, J. L. Jenkins and N. H. Thomä, Nature, 2014, 512, 49–53 CrossRef CAS PubMed.
- G. Petzold, E. S. Fischer and N. H. Thomä, Nature, 2016, 532, 127–130 CrossRef CAS PubMed.
- D. Lv, P. Pal, X. Liu, Y. Jia, D. Thummuri, P. Zhang, W. Hu, J. Pei, Q. Zhang, S. Zhou, S. Khan, X. Zhang, N. Hua, Q. Yang, S. Arango, W. Zhang, D. Nayak, S. K. Olsen, S. T. Weintraub, R. Hromas, M. Konopleva, Y. Yuan, G. Zheng and D. Zhou, Nat. Commun., 2021, 12, 6896 CrossRef CAS PubMed.
- S. Imaide, K. M. Riching, N. Makukhin, V. Vetma, C. Whitworth, S. J. Hughes, N. Trainor, S. D. Mahan, N. Murphy, A. D. Cowan, K. H. Chan, C. Craigon, A. Testa, C. Maniaci, M. Urh, D. L. Daniels and A. Ciulli, Nat. Chem. Biol., 2021, 17, 1157–1167 CrossRef CAS PubMed.
- O. Cala, F. Guillière and I. Krimm, Anal. Bioanal. Chem., 2014, 406, 943–956 CrossRef CAS PubMed.
- W. Becker, K. C. Bhattiprolu, N. Gubensäk and K. Zangger, Chem. Phys. Chem., 2018, 19, 895–906 CrossRef CAS PubMed.
- R. P. Nowak, S. L. DeAngelo, D. Buckley, Z. He, K. A. Donovan, J. An, N. Safaee, M. P. Jedrychowski, C. M. Ponthier, M. Ishoey, T. Zhang, J. D. Mancias, N. S. Gray, J. E. Bradner and E. S. Fischer, Nat. Chem. Biol., 2018, 14, 706–714 CrossRef CAS PubMed.
- A. Sircar, S. Chaudhury, K. P. Kilambi, M. Berrondo and J. J. Gray, Proteins, 2010, 78, 3115–3123 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2019, 18, 237–239 Search PubMed.
- L. Zhang, B. Riley-Gillis, P. Vijay and Y. Shen, Mol. Cancer Ther., 2019, 18, 1302–1311 CrossRef CAS PubMed.
- E. F. Douglass, Jr., C. J. Miller, G. Sparer, H. Shapiro and D. A. Spiegel, J. Am. Chem. Soc., 2013, 135, 6092–6099 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |