
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The transition metal-catalysed hydroboration reaction†
Stephen J.
Geier
 *,
Christopher M.
Vogels
,
Jennifer A.
Melanson
and
Stephen A.
Westcott
*,
Christopher M.
Vogels
,
Jennifer A.
Melanson
and
Stephen A.
Westcott
Department of Chemistry and Biochemistry, Mount Allison University, Sackville, NB E4L 1G8, Canada. E-mail: sgeier@mta.ca
First published on 7th October 2022
Abstract
The use of transition metals to catalyse the addition of hydridoboranes to unsaturated organic molecules was initially realised several decades ago. Although this area of chemistry received considerable attention at the time, interest in this reaction and its use in organic synthesis waned for several years. Like a phoenix rising from the ashes, this amazing catalytic reaction has grown to include the use of earth-abundant metal catalysts and a much wider range of organic substrates. Indeed, it is now commonly utilised as a diagnostic tool to assess the reactivity and catalytic ability of newly generated transition metal and main group complexes. As this field is progressing so rapidly, this review highlights some important advances up to the end of 2021 and into early 2022. Excluded from this review are ‘hydroboration’ reactions using diboron sources.
 Stephen J. Geier | Steve Geier is a native of Sackville, New Brunswick. He completed his BSc degree at Mount Allison University and went on to pursue his PhD degree at the University of Windsor under the supervision of Dr Douglas W. Stephan. His PhD work focused on “Frustrated Lewis Pair” chemistry. He accompanied Dr Stephan in his move to the University of Toronto, completing his PhD in 2010. He then spent 2 years as an NSERC Postdoctoral Fellow in the lab of Dr Jeffrey R. Long at the University of California, Berkeley where he helped develop and test new metal–organic frameworks for gas separations. In January 2013, he returned to Steve Westcott's group at Mount Allison University as a Research Associate and has been there ever since. During his time as a Research Associate, he completed a MLIS (Masters of Library and Information Studies) degree from the University of Alberta. His career interests include research dissemination and, of course, all things boron. |
 Christopher M. Vogels | Christopher M. Vogels received his BSc from Mount Allison University in 1993 and his MS from New Mexico State University in 1996 under the supervision of Michael Johnson. He has been working with the Wild Toads since 1997 and his current research interests include the synthesis and reactivity of E–B compounds (where E = S, Se, Ge and P). |
 Jennifer A. Melanson | Jennifer completed her BSc degree at Mount Allison University as a Wild Toad in 2010 and her PhD degree in 2015 at Dalhousie University under the supervision of Dr Alison Thompson. She was then recruited by Dr Victor A. Snieckus to become a postdoctoral fellow at Queen's University and remained there until late 2018. Jennifer has since returned to work with the Wild Toads as a research associate. Her current research focus is on the modification of natural products for medicinal and industrial use. |
 Stephen A. Westcott | Steve Westcott was born in the sixties somewhere around Tecumseh and received his PhD from the University of Waterloo under the joint supervision of Drs Todd B. Marder (now at Universität Würzburg) and R. Tom Baker (now at the University of Ottawa) working on metal-catalyzed hydroborations. He was an NSERC PDF, where he spent one year at Emory University in Atlanta with Dr Lanny Liebeskind, and more than one year working with Dr Maurice Brookhart at the University of North Carolina at Chapel Hill, NC. He was at Mount Allison University from August 1995 through April 2022 and was a Canada Research Chair in Boron Chemistry in the later years of his employment there. He was also a Fellow of the Chemical Institute of Canada and the Royal Society of Chemistry (UK). His research interests included boron chemistry, catalysis, and the synthesis and development of biologically active small molecules. His research group was called the ‘Wild Toads’ (and it is best not to ask why). With his group, Westcott had published 172 papers with 172 different undergraduate authors as of November 2021 and helped send 125 students to graduate school in chemistry and biochemistry. Steve sadly passed away in April of 2022. |
1 Introduction
The hydroboration reaction is the addition of a boron–hydrogen bond to an unsaturated organic group and was first reported by H. C. Brown in 1956.1 The pioneering work of Brown on organoborane chemistry resulted in his receipt of the Nobel Prize in 1979. The importance of the hydroboration reaction owes largely to the incredible synthetic versatility of organoboranes. Indeed, organoboranes can be readily transformed into several other functionalities with relative ease and in high yields (Scheme 1). While early efforts focused on converting the resulting organoboranes to the corresponding alcohol, the development of other transformations, in particular the carbon–carbon bond formation arising from the Suzuki–Miyaura cross-coupling reaction, has made the preparation of organoboranes even more important in organic synthesis.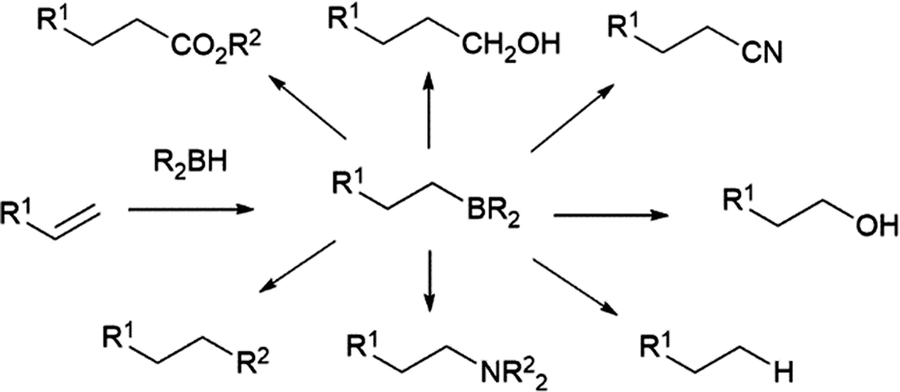 | ||
| Scheme 1 Derivatization of organoboranes. | ||
Addition of boron hydride reagents to unsaturated organic functionalities usually occurs in a cis-fashion. Borane, BH3, which exists as a dimer or Lewis base adduct in solution, is found to react rapidly with unhindered alkenes, adding a B–H bond across the carbon–carbon double bonds of three alkenes, generating a trialkyl borane. Reactions with hindered substrates occur more slowly and, in the case of unsymmetrical alkenes, the boryl group (BR2) ends up on the less-substituted carbon atom. Though this regioselectivity is largely a steric effect, it is also electronically favoured, as the more electronegative hydrogen atom is added to the more substituted carbon of the alkene. One illustrative example of a hydroboration with a BH3-Lewis base adduct is the preparation of a very commonly used hydroborating agent, 9-borabicyclo[3.3.1]nonane (9-BBN, Scheme 2).2–7
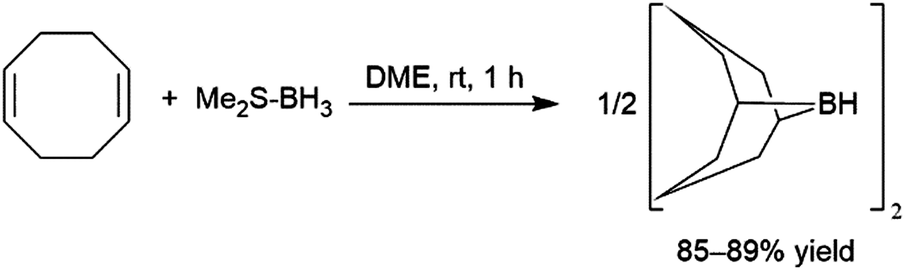 | ||
| Scheme 2 Preparation of 9-borabicyclo[3.3.1]nonane dimer.2–7 | ||
While borane and many dialkyl boranes often react rapidly with alkenes at temperatures as low as −80 °C, other boranes, particularly bulky polyhedral boranes and more electron-rich boranes, react much more sluggishly. The reaction of polyhedral boranes with acetylene is believed to proceed via an initial hydroboration step, however the elevated reaction temperatures required for this addition resulted in several products arising from competing rearrangement reactions. The first reported transition metal-catalysed hydroboration investigated the addition of 2-butyne with B5H9, using the bimetallic alkyne-bridged dimer dimethylacetylene dicobalthexacarbonyl. The catalytic reaction was desirable in this case because in the absence of catalyst, the elevated temperatures required for the reaction resulted in cage fragmentation and other undesired products. Although turnover numbers for the catalyst were quite low, the transition metal-catalysed hydroboration reaction takes place at 75 °C in the presence of 10 mol% catalyst and afforded the desired product in 22% yield.8 Subsequent thermolysis of this product gave higher yields of corresponding carborane products than reactions involving the direct thermolysis of B5H9 and dimethylacetylene (Scheme 3).
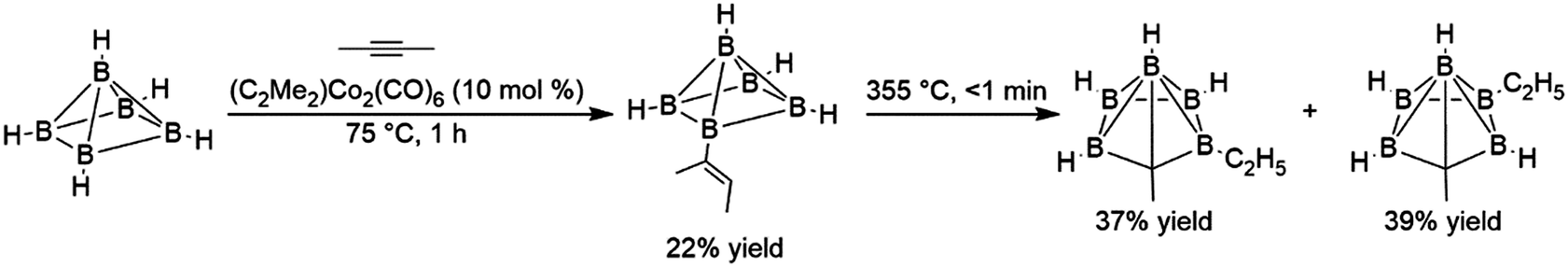 | ||
| Scheme 3 Hydroboration of dimethylacetylene using a polyhedral borane (bridging hydrides between each equatorial boron atom are excluded for clarity).8 | ||
Intramolecular hydroboration of the alkenyl borane product was previously proposed as the first step of the thermolysis reaction, whereupon subsequent rearrangement gave predominantly two alkyl-substituted CB5H9 carborane products. The same reaction was subsequently examined using different alkynes and with Ir(CO)(Cl)(PPh3)2 as the catalyst precursor.9 While moderately successful, hydroborations using the iridium catalyst were ultimately limited by the complex's poor solubility and eventual deactivation. The authors concluded that while the iridium catalyst was more active for terminal alkynes, the cobalt catalyst was more active for internal alkynes and gave products with the opposite regioselectivity in reactions using propyne (Scheme 4), implying that the catalytic cycles for the two systems were significantly different. Alkyne insertions of related carboranes catalysed by analogous alkyne-bridged dinuclear cobalt carbonyl complexes were also reported.10 Olefin insertion into a rhodium-bound carborane has been observed, and catalytic turnover was noted in the presence of excess carborane.11 Subsequent work improved activity for catalytic hydroborations of alkynes and alkenes involving polyhedral boranes and carboranes.12–25 Hydroborations of alkynes and alkenes with borazine and related compounds have been studied in detail.26–30 Due to prohibitive costs, reactions involving these boranes are generally not synthetically useful and thus will not be comprehensively covered in this review.
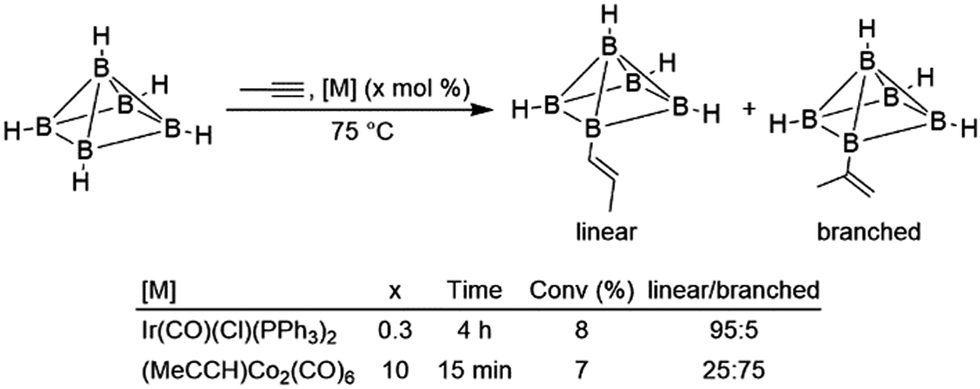 | ||
| Scheme 4 Hydroboration of propyne using a polyhedral borane (bridging hydrides between each equatorial boron atom are excluded for clarity).9 | ||
Another borane that is much less reactive than most dialkyl borane reagents is catecholborane (HBcat; cat = 1,2-O2C6H4), which is significantly more electron-rich than dialkyl boranes due to π-dative interactions between the neighbouring oxygen atoms and the boron center. First prepared in 1975 from the reaction of catechol with THF–BH3, catecholborane requires elevated temperatures (70 °C and up) to react with alkynes and alkenes.31 This borane is known to decompose at elevated temperatures or in the presence of nucleophiles,32 to give a variety of boron-containing species, including a diboron species containing a bridging catecholato group (B2cat3), Lewis base adducts of BH3, and salts of the tetracoordinate anion Bcat2−.
The first publication to describe the transition metal-catalysed hydroboration of alkenes and alkynes with catecholborane came from Männig and Nöth in 1985.33 With low loadings of Wilkinson's catalyst, RhCl(PPh3)3, excellent yields and selectivities were observed in hydroborations at room temperature. Perhaps most interestingly, the authors found that the RhCl(PPh3)3-catalysed hydroboration of 5-hexen-2-one offered complementary chemoselectivity to the uncatalysed reaction, with the catalysed hydroboration occurring selectively at the less-reactive alkene moiety. The catalytic protocol offered high yields and selectivities using mild reaction conditions. Since then, many studies of catalysed hydroboration reactions employing catecholborane have been reported. Other boron reagents have also been developed for catalytic hydroborations, with the majority of them having heteroatom-substituted (N, O, S) ancillary groups.34–39 One example is pinacolborane (HBpin; pin = 1,2-O2C2Me4),40 which is a close relative of HBcat that yields more stable hydroboration products and, in some cases, offers complementary selectivity to catecholborane. Organoboron products derived from pinacolborane are of singular interest in organic synthesis as most are relatively stable to air, water, and chromatography, and therefore isolation of these valuable compounds is considerably easier than their reactive catecholato equivalents. Following the initial disclosure by Männig and Nöth, a number of reports emerged seeking to expand the substrate scope and elucidate the mechanism of these reactions. Particular attention has been paid to hydroborations yielding products with selectivities that are complementary to those observed in uncatalysed hydroborations. There have been many advances in transition metal-catalysed hydroborations in the nearly four decades that have passed since the Männig and Nöth publication, including the development of increasingly active and selective catalysts, enantioselective catalysts, and new hydroboration reagents. Recent efforts have also expanded the scope to immobilized (and reusable) catalysts as well as the use of more affordable earth-abundant transition metals.
This review will cover the development of the homogeneous transition metal-catalysed hydroboration reaction up to early 2022 using mono-boron hydrides, as well as the use of the resulting products in organic synthesis. A number of reviews have been published on catalytic hydroborations, however they are now dated, or only cover a subset of catalytic hydroborations.41–62 This field of chemistry has had a resurgence in recent years and we hope to represent some of the major advances in this area.
2 Mechanisms
2.1 General
As previously mentioned, uncatalysed hydroboration reactions proceed in a concerted syn-fashion with boron adding to the less substituted carbon, while the more electronegative hydrogen atom adds to the more substituted carbon atom, which can better stabilize a partial carbocation transition state. The reaction is believed to proceed via an initial coordination of the alkene unit to the Lewis-acidic empty p-type orbital of the boron atom, which is followed by a four-centered transition state (Scheme 5), resulting in syn-addition. The same selectivity trends are observed in additions of boranes to carbonyls and imines, with the more electropositive boron atom adding to the more electronegative heteroatom. | ||
| Scheme 5 Typical hydroboration of an alkene without a catalyst. | ||
2.2 Oxidative addition
The generally accepted catalytic cycle for the rhodium-catalysed hydroboration varies only slightly from that proposed by Männig and Nöth in their landmark report, and a mechanism previously proposed for rhodium-catalysed hydroboration of olefins with polyhedral boranes (Scheme 6). The catalytic hydroboration reaction proceeds through the rhodium(III) compound RhCl(H)(Bcat)(PPh3)2, which was prepared independently,63 generated from the stoichiometric reaction of catecholborane with RhCl(PPh3)3 (the tri-isopropylphosphine analog of this compound was later characterized crystallographically64). Olefin coordination occurs next, followed by insertion of the alkene into the rhodium hydrogen bond. Finally, reductive elimination liberates the alkyl borane and regenerates the active catalyst. Interestingly, some reports have suggested that insertion of the alkene into the rhodium–boron bond may be favoured in certain circumstances. Among the clearest evidence for the feasibility of this pathway is the fact that vinylboronate esters are often observed in hydroborations of monosubstituted aryl alkenes.65 These products presumably arise from insertion of the alkene into the rhodium–boron bond, followed by β-hydride elimination. As hydrogen is a byproduct of this reaction, hydrogenation products are also frequently observed. Stoichiometric insertion reactions of ketones, imines, and alkenes into a rhodium–boron bond have been performed.66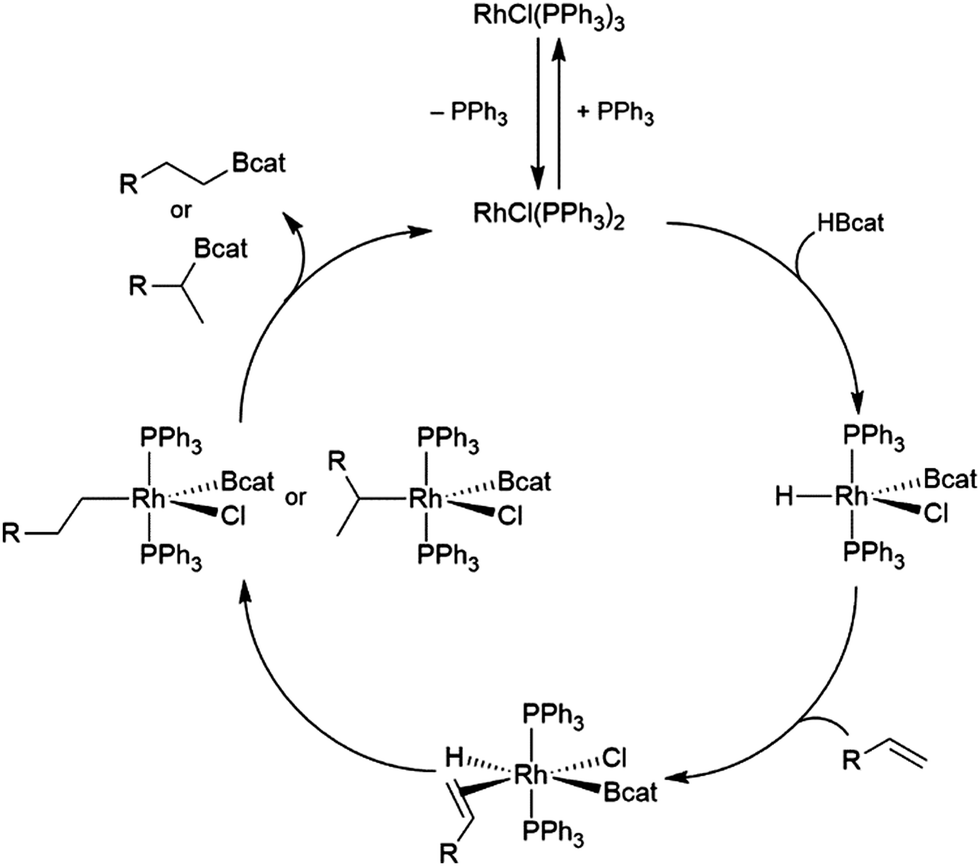 | ||
| Scheme 6 Proposed catalytic cycle for the rhodium-catalysed hydroboration of alkenes. | ||
A computational study later suggested that insertion of the alkene into the rhodium–hydride bond is more facile, however this leads to a more difficult reductive elimination step; insertion into the rhodium–boron bond is more thermodynamically uphill, but leads to a more favourable reductive elimination step.67 Several theoretical studies have examined the potential mechanism of the rhodium-catalysed hydroboration. Among the most notable is the aforementioned study, which found that the most stable conformation has the hydride and the boryl group trans-disposed to one another in the octahedral rhodium(III) intermediate, with both groups cis to the coordinated alkene (Scheme 6, bottom).67 The study concluded that associative and dissociative mechanisms for olefin coordination, followed by hydride or boryl migration, are all viable. Earlier studies focused exclusively on either the associative68 or dissociative69 pathways. A study on related NHC-RhCl hydroboration catalysis suggested that with those catalysts the formation of the terminal hydroboration product proceeds through boryl migration as steric effects disfavour hydride migration.70
While regioselectivity trends for simple alkyl-substituted alkenes are normally similar for catalysed and uncatalysed hydroboration reactions, differences have been observed in some cases with aryl alkenes. For uncatalysed hydroboration reactions of monosubstituted terminal aryl alkenes, the boryl group traditionally adds to the terminal carbon. Initial results from the analogous catalytic hydroboration reactions were conflicting, as in many cases, the rhodium catalysed hydroboration of aryl alkenes using HBcat generally yielded the unexpected branched products, where the boryl group has added to the internal carbon. This selectivity is rationalized by insertion of the alkene into the rhodium–hydride bond to yield a relatively stabilized benzylic rhodium intermediate, whereupon a subsequent reductive elimination step would generate the branched product.
Studies were conducted utilizing DBcat in the catalysed hydroboration of 1-decene or styrene with Wilkinson's catalyst.71 The authors found that while the deuterium label appeared cleanly on the carbon α to the alcohol in the hydroboration and subsequent oxidation of styrene, the label was scrambled between the 1- and 2-positions of 1-decanol. This result implies that the reversibility of individual steps in the catalytic cycle can be substrate dependent. Likewise, a deuterium labelling study with a rhodium-catalysed hydroboration of styrene using DBpin demonstrated deuterium scrambling between the benzylic and terminal carbons, suggesting that reversibility within the catalytic cycle can also be borane- and/or catalyst-dependent.72
2.3 trans-Hydroboration of alkynes
Alkynes have been reported to undergo trans-hydroboration using rhodium or iridium catalysts to give (Z)-alkenyl boronates. In the initial report of this reactivity, it was suggested that the reaction proceeds through initial oxidative addition of the terminal C–H bond.73 Hydride migration from the metal then leads to a vinylidene complex, which oxidatively adds the B–H bond of the borane (Scheme 7). A stereospecific 1,2-migration of the boryl group leads to the metal-alkyl-hydride complex, which can then reductively eliminate the trans-hydroborated product. The key to promoting the trans-hydroboration product is the presence of triethylamine, which hinders the competitive oxidative addition of a B–H bond (instead of the alkynyl C–H bond).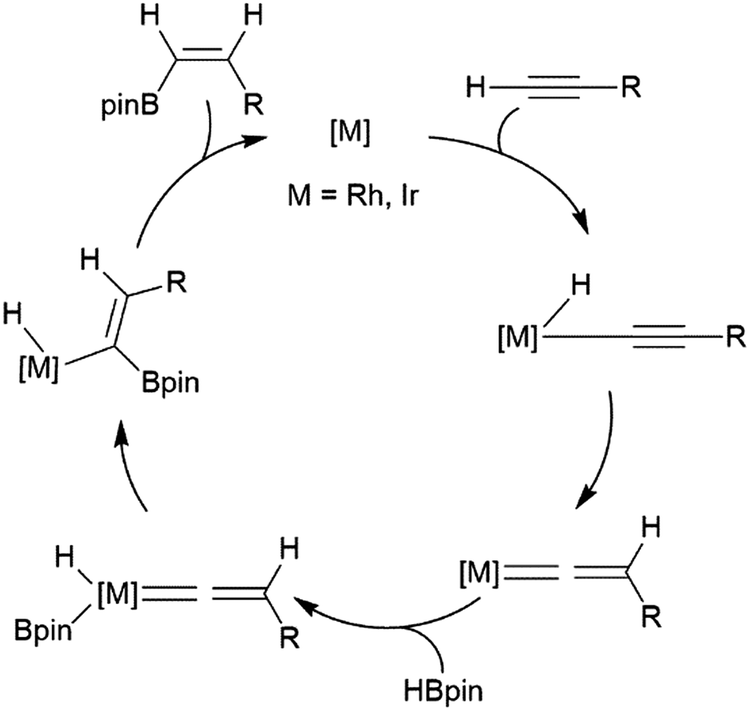 | ||
| Scheme 7 Proposed mechanism for the rhodium-catalysed trans-hydroboration of terminal alkynes.73 | ||
Similarly, a ruthenium complex has been shown to be capable of trans-hydroboration of internal alkynes. This is proposed to proceed through a metallacyclopropene intermediate (Scheme 8).74,75 Steric bulk is thought to play a key role in promoting the rotation about the carbon–carbon bond, as the same reactions using RuCp (Cp = cyclopentadienyl) catalyst precursors showed greatly reduced stereoselectivity.
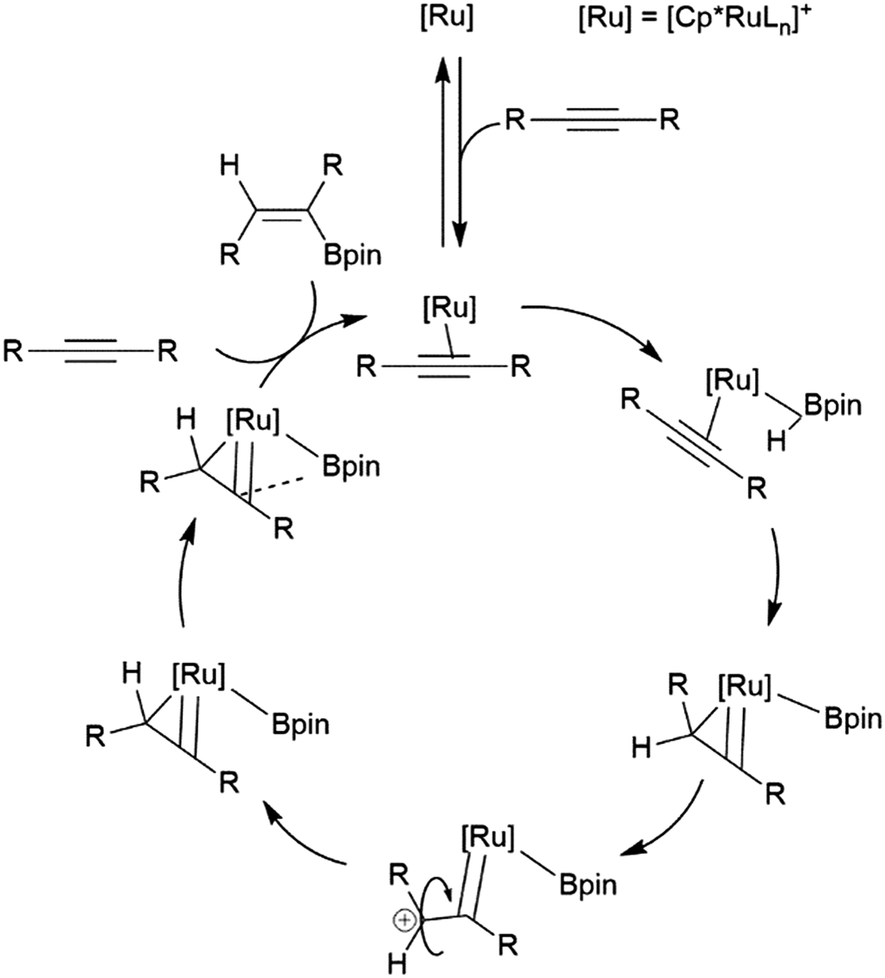 | ||
| Scheme 8 Proposed mechanism for the ruthenium-catalysed trans-hydroboration of internal alkynes.73,74 | ||
2.4 σ-Bond metathesis
While many transition metal-catalysed hydroboration reactions utilize late transition metals, there are also hydroboration reactions using early transition metal catalysts. Noteworthy reports of catalysis by trivalent lanthanides,76–78 niobium,79 titanium,78,80–82 zirconium78,83–87 and, more recently, scandium88 have been reported. Although some reactions are found to be plagued by the metal-catalysed formation of BH3,79,80,89 which can then act as a catalyst for the hydroboration of the substrate with the original borane, or add to the substrate itself, two other mechanisms are implicated in these reactions. Most suggested mechanisms involved the initial formation of a transition metal hydride by reaction of HBcat with the catalyst precursor. Subsequent insertion of an alkene or alkyne results in a transition metal-alkyl or transition metal-alkenyl species, respectively. Metathesis with HBcat yields the organoborane product and regenerates the transition metal hydride (Scheme 9).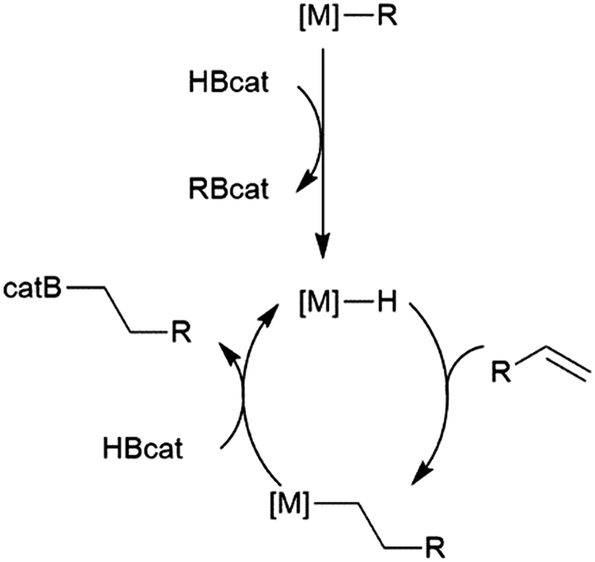 | ||
| Scheme 9 Proposed general mechanism for hydroboration via σ-bond metathesis. | ||
Hartwig and co-workers found that reactions catalysed by Ti(II) precursors proceed through alternate pathways that differ slightly for alkenes and alkynes.82 Cp2Ti(CO)2 was found to be an active catalyst for the hydroboration of alkynes. The cycle begins with displacement of one CO ligand for the alkyne substrate, followed by displacement of the second CO by HBcat (Scheme 10). Elimination of the product from the titanium alkyne-borane/β-borylvinyl-hydride resonance hybrid and coordination of CO restarts the catalytic cycle.
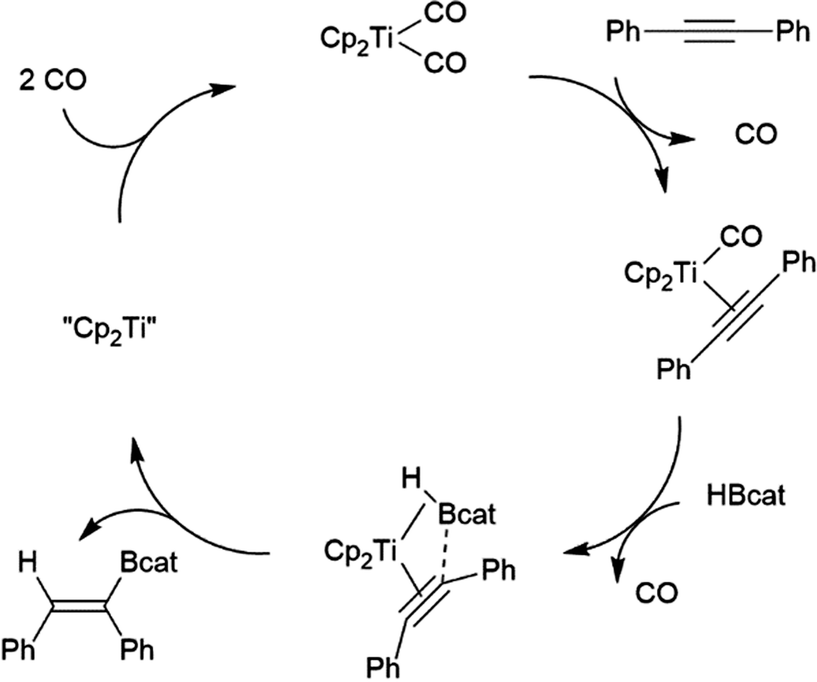 | ||
| Scheme 10 Proposed mechanism for Ti(II)-catalysed hydroboration of alkynes.82 | ||
Hydroboration of alkenes catalysed by dimethyltitanocene was found to proceed through the titanium(II) bis(catecholborane) complex (Scheme 11).82 The reaction proceeds through the substitution of one equivalent of HBcat for the alkene substrate. Elimination of the product from the resulting titanium alkene-borane/β-borylalkyl-hydride resonance hybrid and coordination of HBcat regenerates the active catalyst.
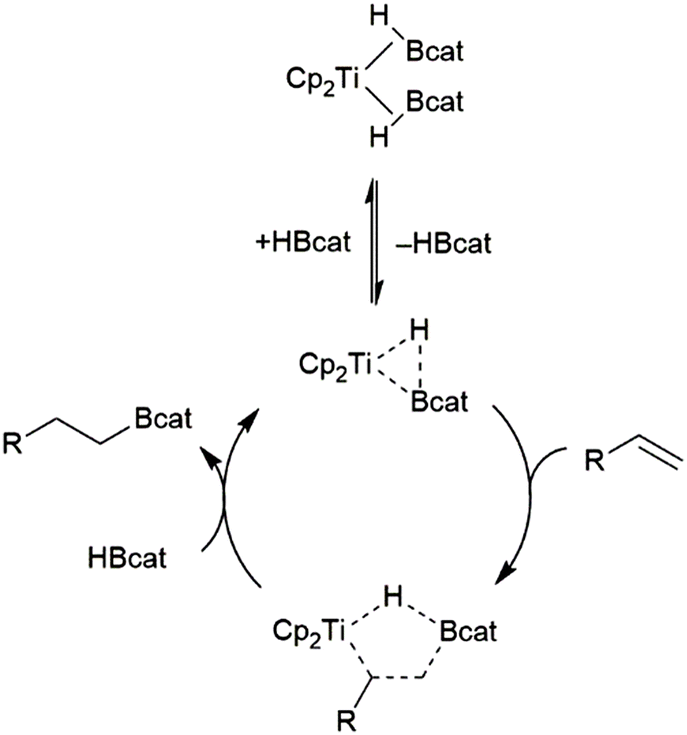 | ||
| Scheme 11 Proposed mechanism for Ti(II)-catalysed hydroboration of alkenes.82 | ||
A computational study of nickel-catalysed hydroboration of CO2 revealed that the transformation can be broken down into three separate hydride transfer steps (Scheme 12).90 The first involves insertion of CO2 into the metal–hydride bond in the catalyst precursor. Metathesis with HBcat results in the formation of HCO2Bcat and regeneration of the nickel hydride. Insertion of HCO2Bcat into the nickel hydride bond generates Ni(O2CH2Bcat). Reaction with another equivalent of catecholborane produces CH2O, catBOBcat, and the nickel hydride catalyst precursor. Insertion of formaldehyde into the nickel-hydride bond yields a nickel methoxide complex, which reacts with a third equivalent of catecholborane, releasing CH3OBcat and the nickel hydride catalyst precursor. The reaction mixture can be quenched with water to complete the transformation of CO2 to methanol. Further experimental study of the system revealed that bulky groups on the phosphorus atom of the pincer ligand results in faster reactions. The use of 9-BBN as the boron source results in a similar reaction, but at a slower rate, while using HBpin resulted in the formation of HCO2Bpin which did not react further. The use of BH3 (from H3B–SMe2) results in the formation of a catalytically inactive nickel(II) borohydride complex.91,92 The catalytic cycle proposed for the copper-catalysed reduction of CO2 with pinacolborane closely resembles the first hydride transfer in the nickel-catalysed reaction. As is the case with the nickel catalysed reduction with HBpin, the pinacolated borylformate in this case does not react further (Scheme 13).93
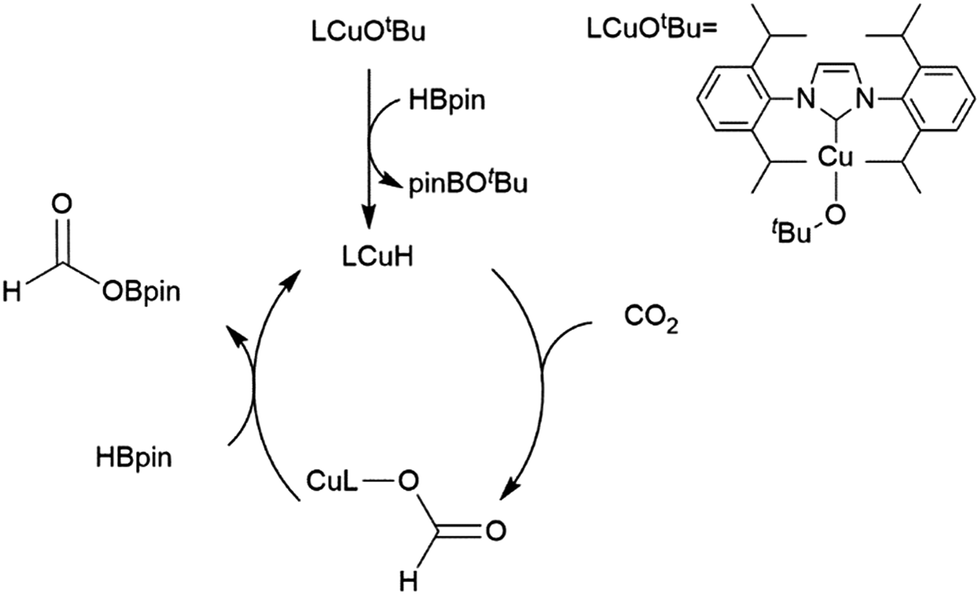 | ||
| Scheme 12 Proposed mechanism for the copper-catalysed hydroboration of CO2.93 | ||
 | ||
| Scheme 13 Hydroboration of CO2 catalysed by a nickel hydride species.90 | ||
3 Stereochemistry
The potential for hydroborations to create chiral centers was seen from the seminal paper by Männig and Nöth.33 In the following years, chiral boranes and catalysts have been examined for their potential use in enantioselective hydroboration reactions. Appropriate ligand choice has the potential to provide asymmetric induction with only a catalytic amount of the chiral reagent needed.3.1 Chiral boranes
Hydroboration reactions using chiral boranes have been performed with rhodium catalysts and aryl alkenes as substrates. The hydroboration of aryl alkenes using these boranes were examined, catalysed by chiral and achiral catalysts.94 The most promising result is a 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 enantiomeric ratio obtained using a rhodium/dppf catalyst system and a pseudoephedrine-derived borane. However, the reaction only showed 82% selectivity for the α-alcohol upon oxidation (Scheme 14). The relatively poor selectivity, along with the cost of using a stoichiometric amount of chiral reagent limits the utility of this approach. The use of some chiral catalysts with these chiral boranes showed no improvement in enantioselectivity.
:12 enantiomeric ratio obtained using a rhodium/dppf catalyst system and a pseudoephedrine-derived borane. However, the reaction only showed 82% selectivity for the α-alcohol upon oxidation (Scheme 14). The relatively poor selectivity, along with the cost of using a stoichiometric amount of chiral reagent limits the utility of this approach. The use of some chiral catalysts with these chiral boranes showed no improvement in enantioselectivity.
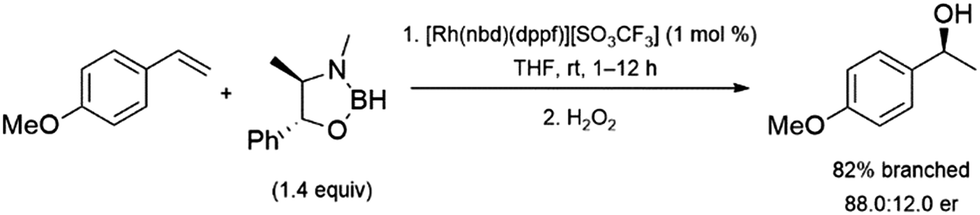 | ||
| Scheme 14 Rhodium-catalysed hydroboration of 4-methoxystyrene with a chiral borane.94 | ||
3.2 Catecholborane-monodentate ligands
Monodentate ligands have not been studied to the same extent as their bidentate counterparts in the catalytic hydroboration reaction. In 2005, a series of α-chiral monophosphine ligands were prepared and used with a cationic rhodium(I) precursor for the catalytic hydroboration of styrene with HBcat.95 Conversions and yields were generally good, however, only relatively modest enantioselectivity was achieved (Scheme 15).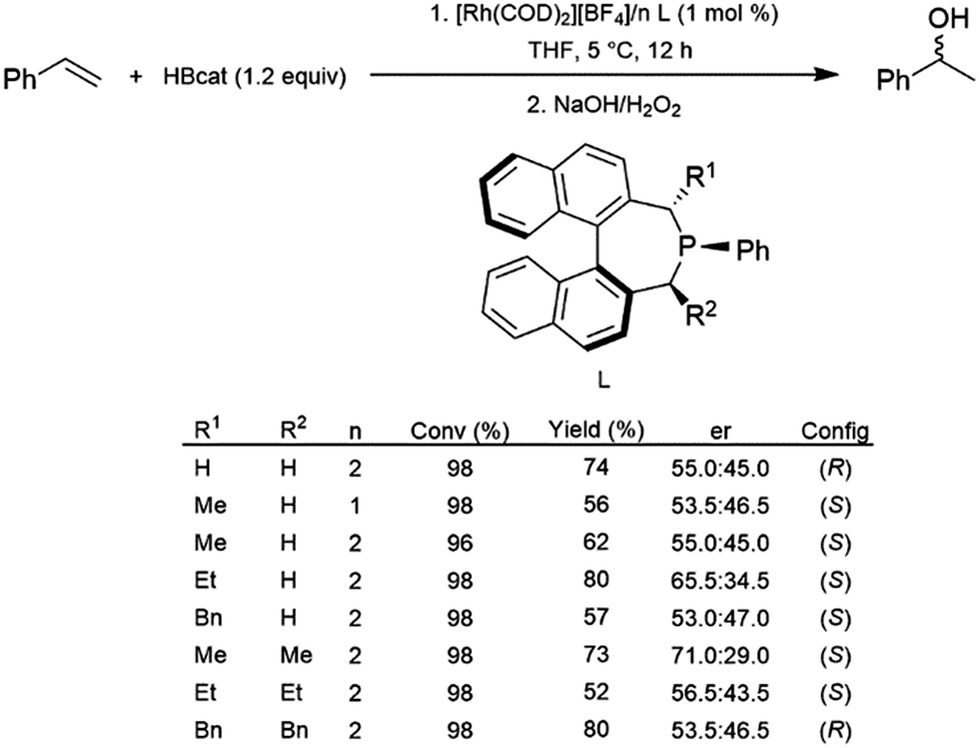 | ||
| Scheme 15 Rhodium-catalysed asymmetric hydroboration of styrene using a chiral phosphine ligand.95 | ||
3.3 Catecholborane-bidentate ligands
Bidentate ligands have been quite popular in catalytic hydroboration reactions. The stronger binding and greater tunability when compared to monodentate ligands make them the ligands of choice in the enantioselective hydroboration of many substrates. Several transition metal starting materials contain labile ligands that can be readily substituted by a bidentate ligand. Thus, the catalyst precursor is often prepared in situ, prior to addition of the substrate and borane.The branched product of styrene hydroboration, which can be transformed into a secondary alcohol upon oxidation, is chiral, offering the opportunity for asymmetric induction. This potential was realized quite early, as initial reports of styrene hydroboration96–99 included a cationic rhodium/binap catalyst system that was found to offer enantiomeric ratios of up to 98:2 at −78 °C (Scheme 16).96
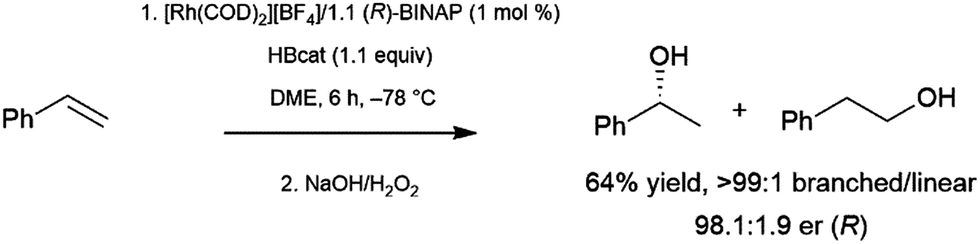 | ||
| Scheme 16 Asymmetric hydroboration of styrene catalysed by a cationic rhodium(I)/BINAP catalyst.96 | ||
Other ligands found to offer highly enantioselective catalyst systems in combination with a rhodium(I) catalyst precursor include QUINAP derivatives,100–102 ferrocene-based diphosphine,103 and P,N ligands (Fig. 1).104,105 A dendrimer version of JOSIPHOS also gave an active catalyst on combination with [Rh(COD)2][BF4], with similar enantioselectivity to the heterogeneous analog, but poorer regioselectivity.106 Heterogeneous analogs of BINAP107,108 and QUINAP107,109 have been successfully prepared and achieved good selectivities in rhodium-catalysed hydroboration reactions. As with their homogeneous analogs, QUINAP outperformed BINAP significantly in asymmetric induction. Both systems could be reused 4 times without appreciable loss in catalytic activity or selectivity. In terms of enantioselectivity, this methodology outperformed previously reported systems derived from zeolite-supported N,N- and P,N-ligands.110,111
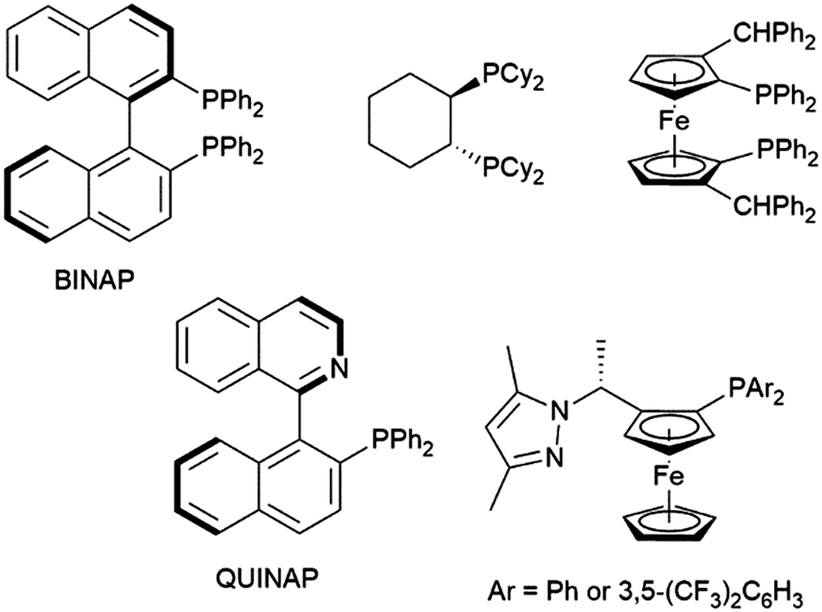 | ||
| Fig. 1 Commonly used chiral ligands in asymmetric hydroboration reactions. | ||
A variety of substituted aryl alkenes were explored in the initial study of the cationic Rh-BINAP catalyst system, finding that electron-rich aryl alkenes underwent hydroboration with greater enantioselectivities than their electron-poor counterparts. The same study also found that asymmetric induction was poorer for α- and β-substituted aryl alkenes, which is likely at least partly due to the sluggish nature of these reactions which necessitates higher reaction temperatures than monosubstituted aryl alkenes.99
3.4 Pinacolborane-monodentate ligands
Pinacolborane was introduced as a hydroboration reagent in 1992.40 While some alkenes and alkynes can undergo hydroboration with pinacolborane in the absence of a catalyst, it tends to be less reactive than HBcat and thus could offer some advantages in terms of selectivity. However, hydroboration reactions of aryl alkenes with HBpin tend to proceed with lower selectivity for the branched isomer than their catecholborane counterparts86 and can give significant amounts of vinylboronate esters from dehydrogenative borylation.112TADDOL-derived phosphite and phosphoramidite ligands have been successfully utilized with rhodium catalysts in the asymmetric hydroboration of aryl alkenes with HBpin.113,114 Up to 98:2 er's are achieved along with high yields, however the ligands providing the highest enantioselectivities of the branched product offer only modest regioselectivity, with significant amount of the linear product also being generated (Scheme 17). Conversion and selectivity for the branched isomer was good in the hydroboration of ortho-substituted aryl alkenes, however enantioselectivity was poor.115
 | ||
| Scheme 17 Rhodium-catalysed asymmetric hydroboration using a chiral TADDOL-derived phosphoramidite ligand.113 | ||
3.5 Pinacolborane-bidentate ligands
While hydroboration reactions using an iridium complex derived from [IrCl(COD)]2 and dppb gave >99% selectivity for the linear isomer, the branched isomer was obtained with 96% selectivity using a cationic complex derived from [Rh(COD]2[BF4] and dppb.116 The same study found that asymmetric hydroboration reactions of aryl alkenes using a cationic rhodium/JOSIPHOS system gave er's from 88:12 to 94:6 and selectivity for the branched isomer of 72–95% (Scheme 18).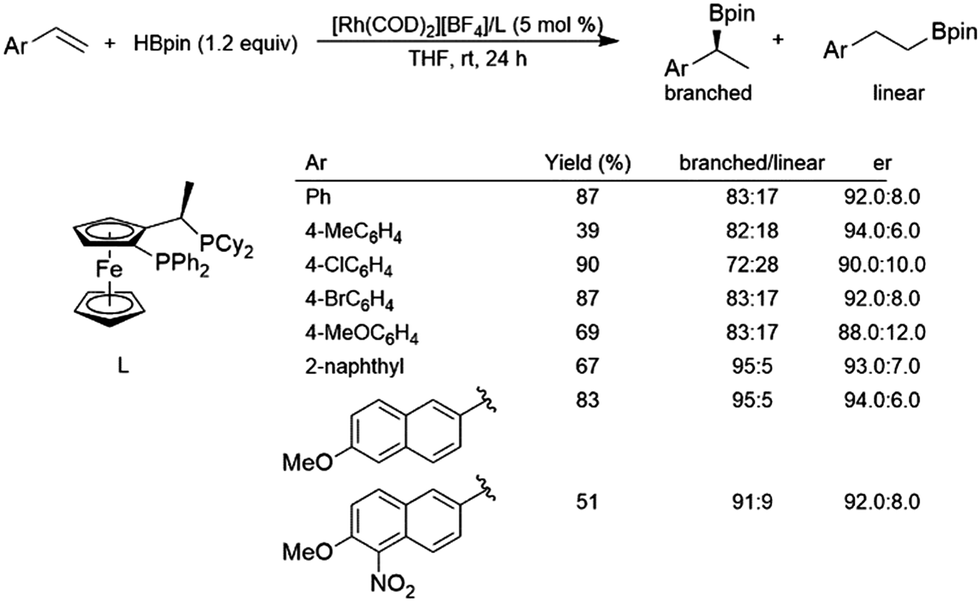 | ||
| Scheme 18 Asymmetric hydroboration of styrene derivatives using a rhodium(I)/JOSIPHOS catalyst system.116 | ||
A study using a Rh-(R)-QUINAP system found that while catalysed hydroboration reactions of aryl alkenes provided similar good yields, regioselectivities, and enantioselectivity, the use of a Rh-(R)-BINAP system showed poorer and inverse enantioselectivity, along with lower yields and regioselectivities.117 A supramolecular catalyst comprised of a zinc complex bearing pendant TADDOL-derived phosphites and [Rh(nbd)2][BF4] provided excellent regioselectivities and yields in the hydroboration of meta-substituted aryl alkenes.119
In recently developed chemistry, copper(I) diphosphine catalysts have been reported that give highly regio- and enantioselective hydroboration of aryl alkenes with HBpin.120 A computational study found that bidentate phosphines accelerate the reaction by destabilizing intermediates involving aryl alkene coordination to a copper(I)–hydride complex and the ensuing copper(I) alkyl complex.121 A later publication detailed a more reactive catalyst system, capable of the asymmetric hydroboration of electron-rich and β-substituted aryl alkenes (Scheme 19).118
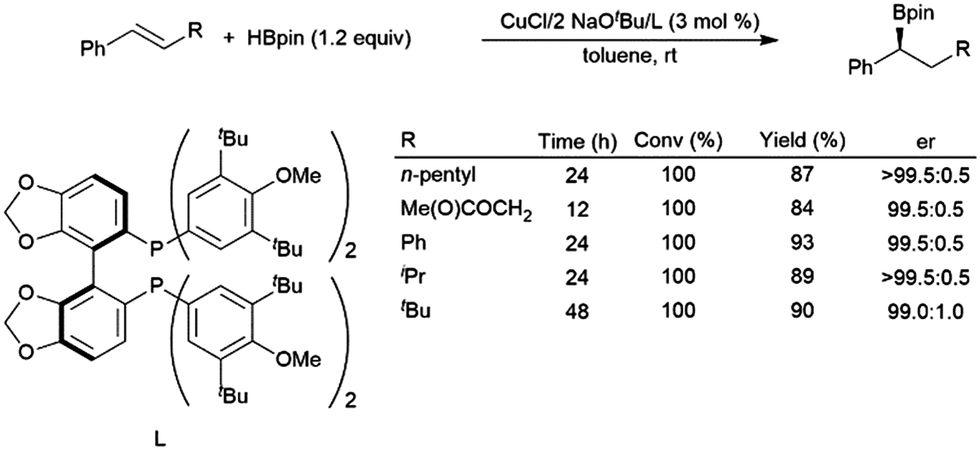 | ||
| Scheme 19 Asymmetric hydroboration of trans-substituted aryl alkenes using a chiral bidentate phosphine ligand.118 | ||
4 Scope
4.1 Aliphatic alkenes
Catalytic hydroboration reactions of simple alkyl-substituted alkenes have traditionally received relatively little attention, mainly because these reactions generally result in the same achiral linear products accessible through uncatalysed hydroboration reactions. Several systems have shown the capability of carrying out the hydroboration reaction to mono-substituted aliphatic alkenes, with selectivities favouring the terminal hydroboration product. Given the utility of pinacol boronate esters, reactions using HBpin have received considerable recent attention, particularly focusing on the use of first row transition metals for this transformation.122,123 Where catalytic hydroboration reactions of mono-substituted alkenes initially drew attention, many focused on alkenes bearing other reducible or directing functionalities. For example, the addition of HBcat to terminal alkenes using RhCl(PPh3)3 can be carried out in the presence of ketone and nitrile groups (Scheme 20).33 In a similar fashion, the chemoselective addition of HBcat to the terminal alkene in 3-vinyl cyclohexene proceeded with excellent conversion at room temperature.33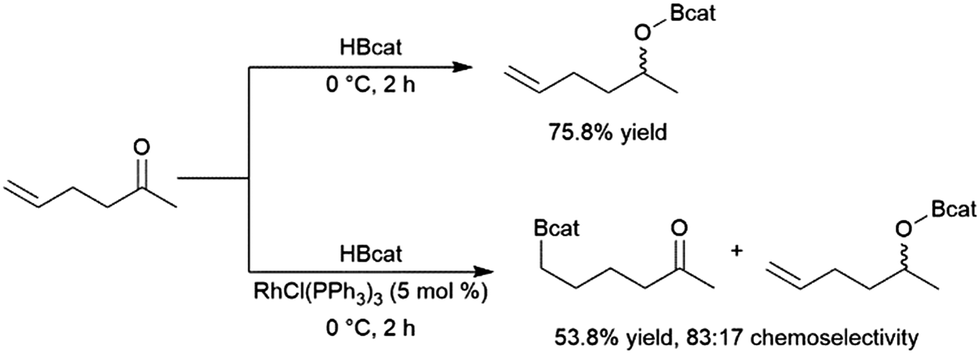 | ||
| Scheme 20 Hydroboration of 5-hexen-2-one by HBcat in the absence of a catalyst (top) and using Wilkinson's catalyst (bottom).33 | ||
Catalytic hydroboration reactions of ethylene are not common in the literature but have been investigated, using ruthenium catalysts and HBpin or HBcat;125,126 and the rhodium catalysed hydroboration using N-alkylborazines.30 In the case of the ruthenium catalysed hydroboration reactions, the vinylboronate ester product is also observed depending on the solvent used. Bromoethylene undergoes hydroboration with HBcat at room temperature in the presence of Wilkinson's catalyst, whereupon the branched product was isolated exclusively.127
A number of allylic sulfones have been subjected to catalytic hydroboration with HBcat using Wilkinson's catalyst.124 The authors noted selectivity that strongly favoured the branched isomer (∼90:10), regardless of the sulfone substituent. Although the product distribution in the uncatalysed reaction with BH3·THF also favoured the branched product, selectivity was greatly improved in the catalytic reaction suggesting complexation of the sulfone group to the rhodium center may serve to direct the hydroboration reaction (Scheme 21).
 | ||
| Scheme 21 Rhodium-catalysed hydroboration of allylic sulfones.124 | ||
The synthesis of diborylalkanes from terminal aliphatic alkenes has been reported using a zirconium(IV) alkoxide catalyst.128 The reaction proceeds through initial dehydrogenative borylation, followed by hydroboration of the boryl alkene. Catalytic hydroboration of allyl benzene with HBcat has been investigated using several rhodium catalysts.129 While the linear hydroboration product is predominant with the three catalyst precursors utilized, significant amounts of benzylic and branched products were also observed (Scheme 22). The benzylic (1-boryl-1-aryl) product is formed as a result of alkene isomerization at the metal center towards the thermodynamically favourable aryl alkene, which subsequently undergoes hydroboration favouring the branched isomer (Scheme 22, α). A more recent publication highlights a cobalt catalyst capable for this transformation, showing isomerization of chains up to 12 carbon atoms long to selectively form the α-arylboronate ester.130 A nickel(II) catalyst has been found to provide excellent selectivity for the terminal hydroboration product with pinacolborane.131
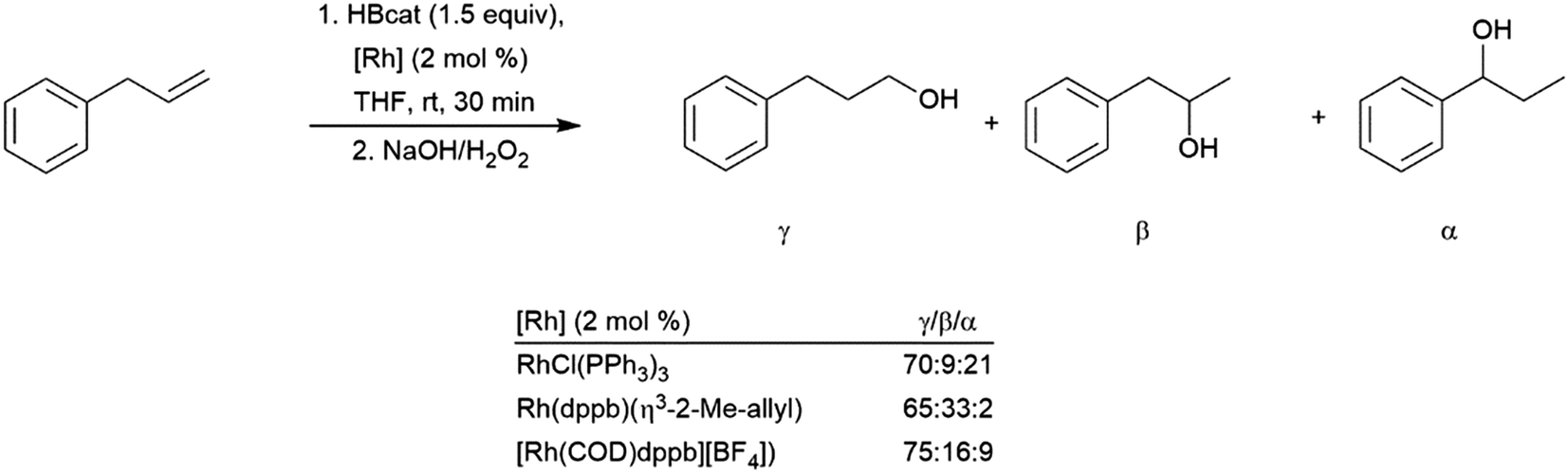 | ||
| Scheme 22 Catalytic hydroboration of allyl benzene.129 | ||
Teskey and co-workers have reported a photoswitchable cobalt(I)–hydride system which effects the hydroboration of terminal alkenes under visible light irradiation, while selectively effecting isomerization in the dark (only in the presence of pinacolborane and without subsequent hydroboration).132 Iridium-catalysed hydroboration of alkenes bearing pendant amides have been studied. It was found that [Ir(COD)(PCy3)(py)][PF6] catalysed the hydroboration of monosubstituted β,γ-unsaturated amides with a regioselectivity slightly favouring the branched isomer.133 Under identical conditions, 1-hexene gave almost exclusively the linear product suggesting that the amide group was directing the addition of the borane in these reactions.
Enamines can undergo highly selective hydroboration reactions that are catalyst and borane dependent. For instance, 9-vinylcarbazole was efficiently hydroborated by HBcat using Rh(acac)(dppb) to selectively generate the branched product, whereas using the same catalyst with HBpin selectively gave the terminal borylated product.134 The hydroboration of 1-vinyl-2-pyrrolidinone catalysed by Rh(acac)(dppb) proceeded efficiently with high selectivity for the linear hydroboration product (Scheme 23). Uncatalysed reactions with HBcat were complicated by partial reduction of the carbonyl group, and catalysed reactions gave significant amounts of dehydrogenative borylation.
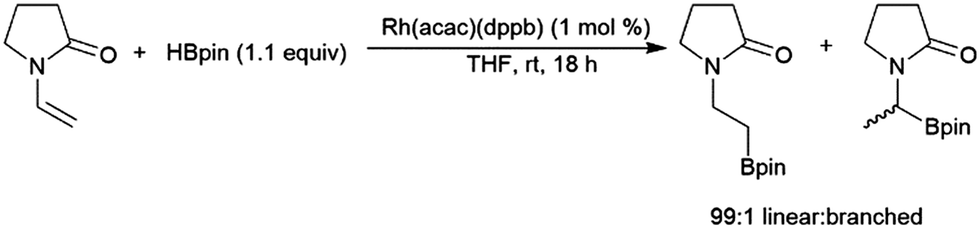 | ||
| Scheme 23 Rhodium-catalysed hydroboration of 1-vinyl-2-pyrrolidinone.134 | ||
A recent study has shown that judicious selection of rhodium catalyst and reaction conditions can provide the linear or branched isomers resulting from the hydroboration of vinylphosphine. This builds on previous work showing relatively low activity and selectivity for rhodium-catalysed hydroborations of the same substrate. Silyl enols can also undergo rhodium-catalysed asymmetric hydroboration in good yield and enantioselectivity.136 Terminal alkenes can also be selectively hydroborated under appropriate conditions in the presence of internal alkenes,33,137,138 1,1-disubstituted alkenes,138 ketone,33,138 and cyano33,139 groups. Pendant alkene groups have been hydroborated to functionalize a polymer.140
4.2 1,1-Disubstituted aliphatic alkenes
As is the case with most monosubstituted aliphatic alkenes, the terminal alkylboronate ester tends to be preferentially generated in catalytic hydroboration reactions of 1,1-disubstituted aliphatic alkenes. This regioselectivity is often even higher than with the monosubstituted aliphatic alkenes due to increased steric crowding, which also results in more sluggish reactions. Although the terminal carbon atom bearing the boron group in the product is not chiral, in the case of unsymmetrical 1,1-disubstituted alkenes a chiral center is created at the internal carbon. As such, a number of studies have looked at asymmetric hydroboration of 1,1-disubstituted alkenes.In the early days of transition metal-catalysed hydroboration reactions, allylic alcohol,77,129,135,141–149 and amine144,150,151 derivatives drew extensive attention as substrates as it was noted that these reactions produced predominantly syn isomers while uncatalysed variants gave the anti diol products on oxidation (Scheme 24).
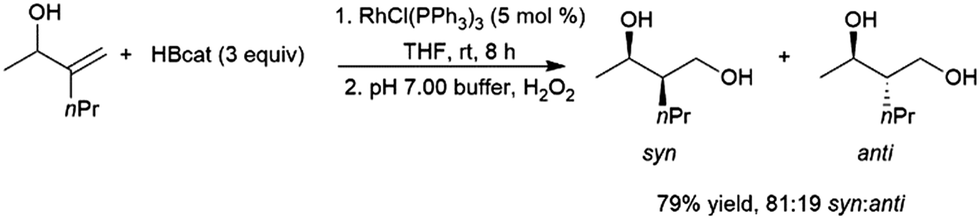 | ||
| Scheme 24 Rhodium-catalysed hydroboration of an allylic alcohol.135 | ||
In a similar fashion, 1-methylidine-4-cyclohexane derivatives undergo hydroboration/oxidation giving good diastereoselectivity in favour of the syn isomers, using RhCl(PPh3)3 or [RhCl(COD)]2/4 PPh3 as catalyst precursors (Scheme 25).152,153
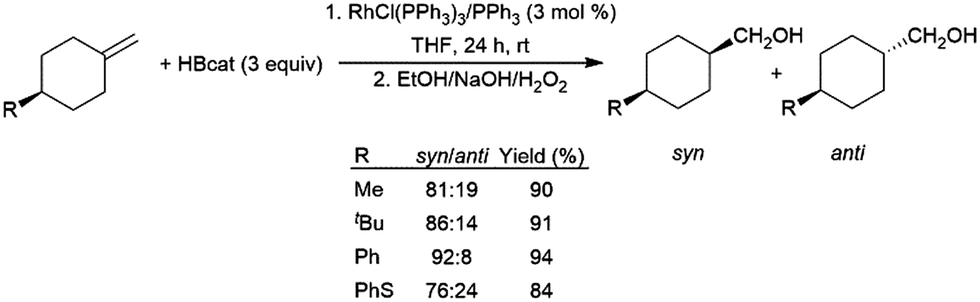 | ||
| Scheme 25 Rhodium-catalysed hydroboration of 1-methylidine-4-cyclohexane derivatives.152 | ||
Chiral catalysts have been employed to induce chirality in hydroborations of 2,3,3-trimethylbut-1-ene, albeit with relatively modest enantioselectivity. Optimal conditions employed 2 equivalents of (R,R)-DIOP as a ligand with 1 mol% [RhCl(COD)]2 at −5 °C in THF, achieving 84.5:15.5 er favouring the R-isomer (Scheme 26).154
 | ||
| Scheme 26 Asymmetric hydroboration of 2,3,3-trimethylbut-1-ene using a rhodium(I) phosphine catalyst.154 | ||
1,1-Disubstituted aliphatic alkenes have been selectively reduced using hydridoboronate esters in the presence of a trisubstituted alkene,135,137,155–157 even in cases containing ester and amide functionalities.147,158 Interestingly, RhCl(PPh3)3-catalysed hydroboration of 2,2-disubstituted-1-methylidene cyclopropanes gave ring-opened borylated products bearing a terminal monosubstituted alkene.159 Similarly, vinylcyclopropanes underwent ring-opening upon iron-catalysed hydroboration to give homoallylic organoboronic esters. As with other substrates, much recent attention has focused on the use of iron and cobalt as catalysts for the hydroboration of 1,1-disubstituted aliphatic alkenes.137,157,160,161 A recent report highlights the ability of a polystyrene-supported copper catalyst is effective for the efficient hydroboration of a 1,1-disubstituted alkene.162
4.3 1,2-Disubstituted aliphatic alkenes
Regioselectivity is frequently problematic in the catalysed hydroboration of 1,2-disubstituted aliphatic alkenes. In addition to normal regioselectivity issues present in other substrates, competitive isomerization can lead to the formation of linear products.77,86,122,135,141,160,163–169 Some recent work has shown that these linear products of isomerization and subsequent hydroboration can be produced quite selectively.130,166,168–175 A zirconium(IV) catalyst has been shown to enable the isomerization, dehydrogenative borylation, and hydroboration of internal alkynes, generating 1,1-diborylalkanes from internal alkenes.128 Frequently, to avoid isomerization, cyclic substrates were used. In particular, norbornene and derivatives have been investigated extensively as substrates in catalytic hydroborations.33,35,76,77,86,97–100,125,146,154–157,163,176–189 Transition metal-catalysed hydroborations are selective for giving the exo-hydroboration products. Several chiral ligands have also been used for the asymmetric hydroboration of these substrates, however many chiral rhodium complexes offered only modest levels of enantioselectivity.97–100,154,155,163,179 Systems containing copper,180 cobalt,157 or rhodium154,179 catalyst precursors have all shown promise in asymmetric hydroborations of these substrates (Scheme 27).180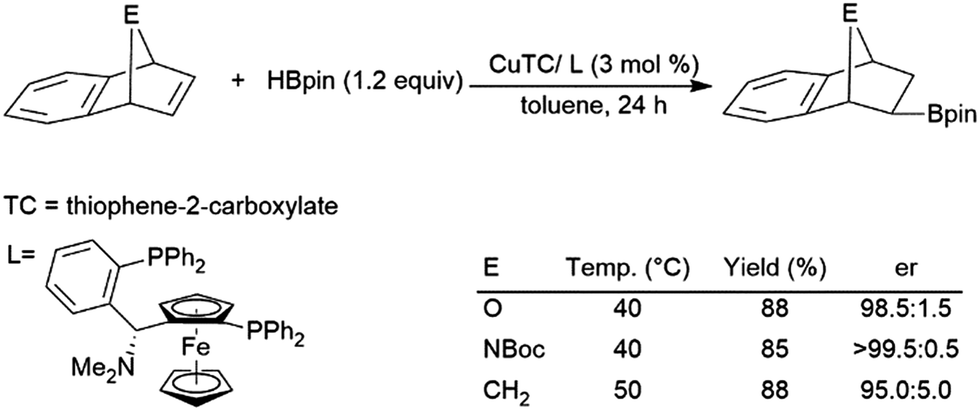 | ||
| Scheme 27 Copper-catalysed hydroboration of norbornene derivatives.180 | ||
Norbornene has been a frequently examined substrate in catalytic asymmetric hydroboration reactions. In an initial study, BINAP was found to provide the best enantioselectivity for this reaction (Scheme 28).98 It was subsequently discovered that bis-aminophosphine ligands could also provide reasonable yields and enantioselectivities for the hydroboration of norbornene.184
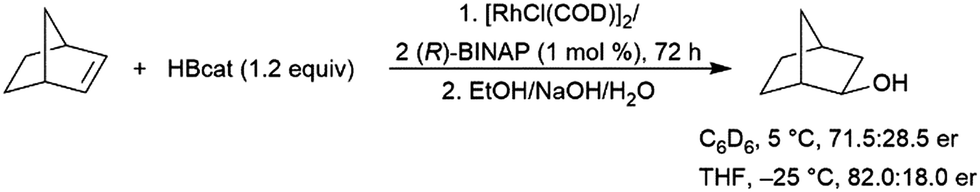 | ||
| Scheme 28 Asymmetric hydroboration of norbornene.98 | ||
Closely related meso-bicyclic hydrazines can also undergo asymmetric hydroboration, with er's up to 93:7. The resulting alcohols are of great interest and can be reduced to diaminocyclopentane species (Scheme 29).176 Interestingly, enantioselectivity for the iridium catalysed reaction is opposite to that observed with the rhodium analogs.190
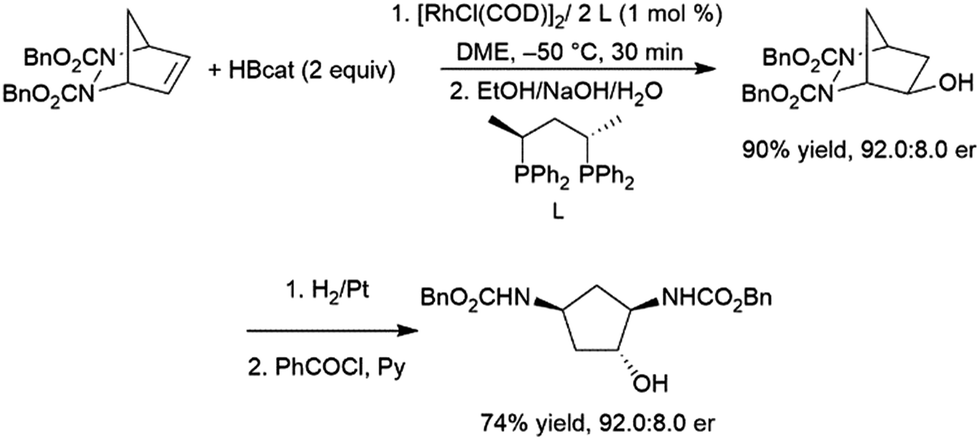 | ||
| Scheme 29 Asymmetric hydroboration of meso-bicyclic hydrazines.176 | ||
Allylic alcohol derivatives have been substrates of interest due to the reversed regioselectivity observed in catalytic hydroborations versus uncatalysed hydroborations. Electronic effects cause hydroboration selective for the boron atom adding to the carbon α to the alcohol in uncatalysed hydroborations while steric effects dominate the catalytic process resulting in selectivity favouring the trans-1,3-disubstituted product (Scheme 30).142,155,191,192 An extra carbon atom between the alkoxide group and the alkene double bond results in generally poorer selectivity,191,193 frequently favouring the cis-1,3-disubstituted product.
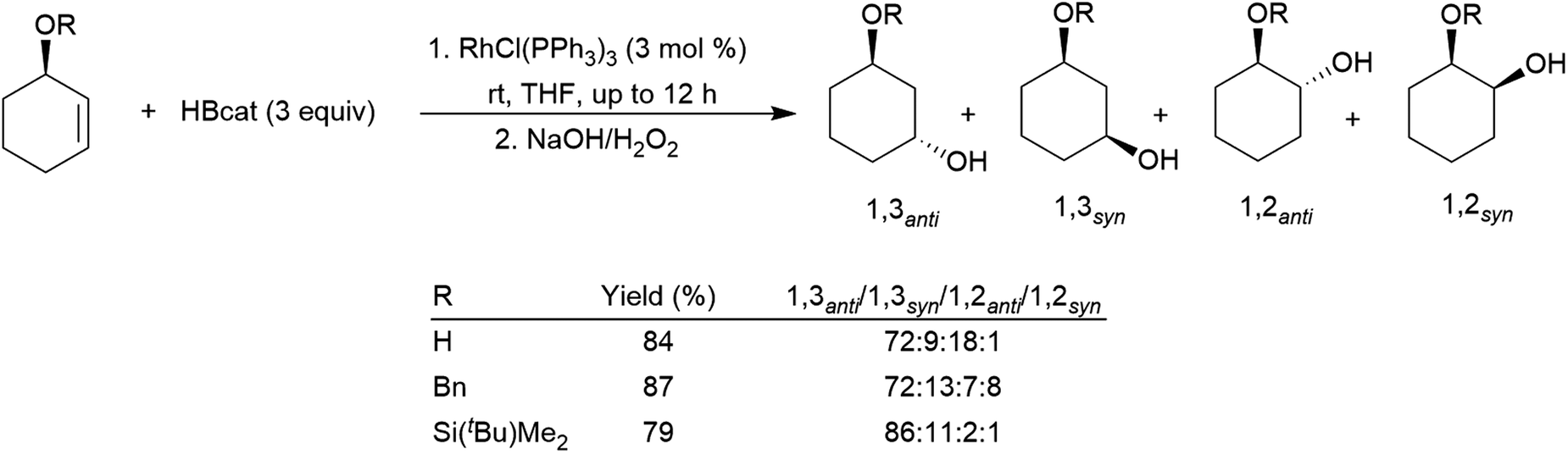 | ||
| Scheme 30 Rhodium-catalysed hydroborations of cyclic allylic alcohol derivatives.142 | ||
1,2-Disubstituted194 alkenes bearing pendant amide groups have also been examined extensively as substrates in metal-catalysed hydroboration reactions.133,135,195,196 β,γ-Alkenyl amides generally show strong regioselectivity favouring the boryl group in the β-position, though Zhang, Zu, and co-workers have reported an iridium-catalysed protocol which strongly favours the γ-boryl hydroboration product.197 Judicious ligand selection also allows for excellent enantioselectivity in these reactions.195,196 Similarly, γ,δ-alkenyl amides underwent highly selective asymmetric hydroboration incorporating the boryl group in the γ-position.198 Cyclic alkenes bearing exocyclic amide groups have similarly shown potential for highly regio- and enantioselective metal-catalysed hydroborations.133,135,199 Similarly, 2,4,6-trichlorobenzoate substituents have been utilized as directing groups in the asymmetric copper-catalysed hydroboration of internal alkenes.200
Z-Alkenes bearing a halogen substituent undergo hydroboration with catecholborane catalysed by RhCl(PPh3)3, with the boryl moiety being regioselectively added to the carbon bearing the halogen atom.127 Cyclopropenes undergo catalytic hydroboration reactions with selectivities favouring addition of the boryl group trans to the larger group. Methyl esters serve as directing groups, with the boryl group adding on the same face as the methyl ester (Scheme 31).194,201
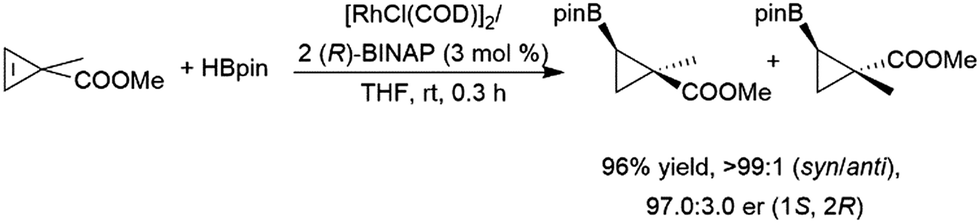 | ||
| Scheme 31 Rhodium-catalysed asymmetric hydroboration of a cyclopropene derivative.194,201 | ||
A platinum-coordinated 4-(3-cyclohexen-1-yl)pyridine was submitted to RhCl(PPh3)3-catalysed hydroboration using catecholborane.202 Regioselectivity was, perhaps not surprisingly, quite poor as there is little steric or electronic differentiation between the two carbon atoms.
4.4 Trisubstituted alkenes
Among the most challenging substrates for catalytic hydroborations are trisubstituted alkenes. Many attempts at catalytic hydroboration reactions are plagued by low conversions185 and competitive substrate isomerization.77,129,160,166,167 Isomerization can be highly selective, resulting in the formation of a terminal alkene, which subsequently undergoes hydroboration, forming alternative hydroboration products (Scheme 32).173 A zirconium(IV) system was used to generate the corresponding 1,1-diborylalkane product through isomerization and subsequent dehydrogenative borylation.128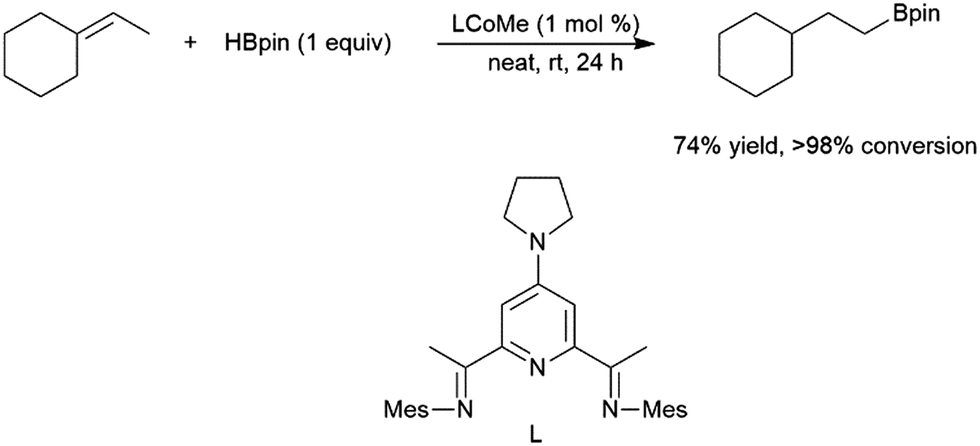 | ||
| Scheme 32 Cobalt-catalysed hydroboration of a trisubstituted alkene.173 | ||
Trisubstituted allylic amides undergo catalytic hydroborations using a variety of rhodium-based catalysts and pinacolborane.195,203,204 The amide directing group, combined with steric effects, results in hydroboration selective for the boryl group β to the amide (Scheme 33).203
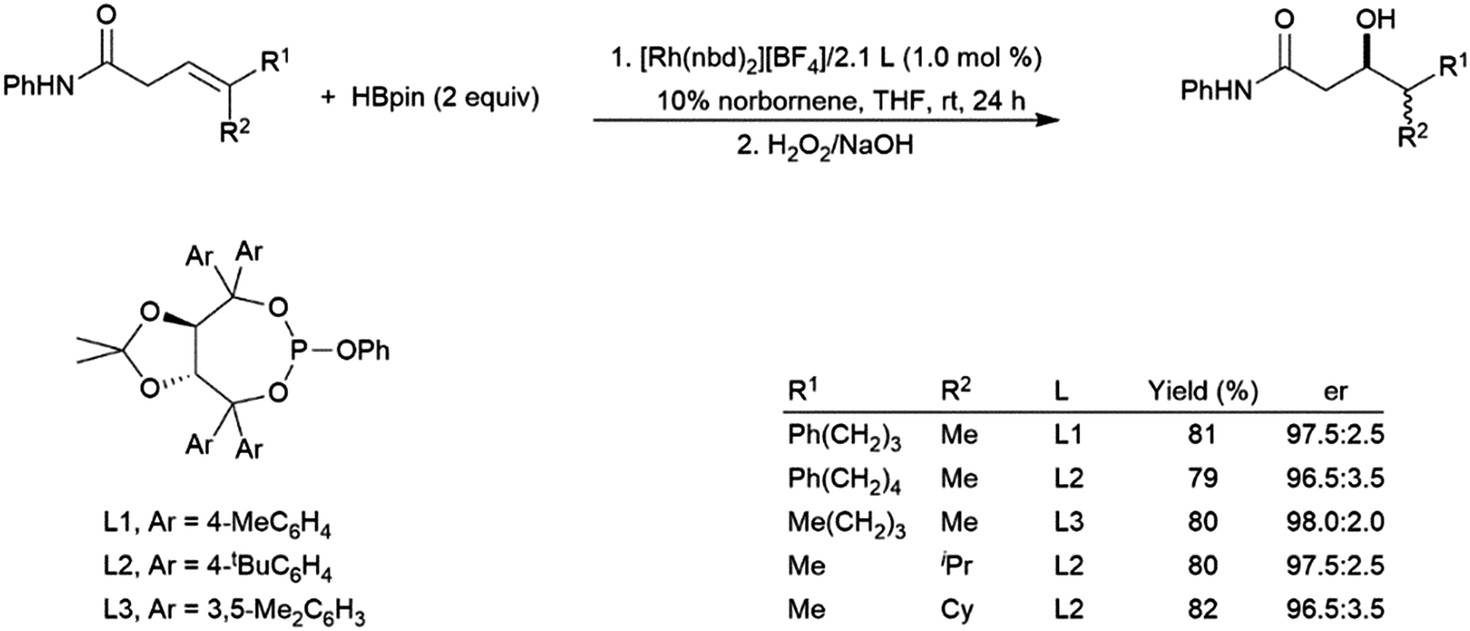 | ||
| Scheme 33 Asymmetric amide-directed hydroboration of trisubstituted alkenes.203 | ||
The utilization of oxime205 and phosphonate206,207 directing groups has allowed for the asymmetric hydroboration of trisubstituted alkenes with the boryl group selectively adding to the disubstituted carbon, resulting in the formation of chiral tertiary boronic esters (Scheme 34). Substrates with an aryl group206 instead of an alkyl group207gem- to the directing group provided complimentary enantioselectivity.
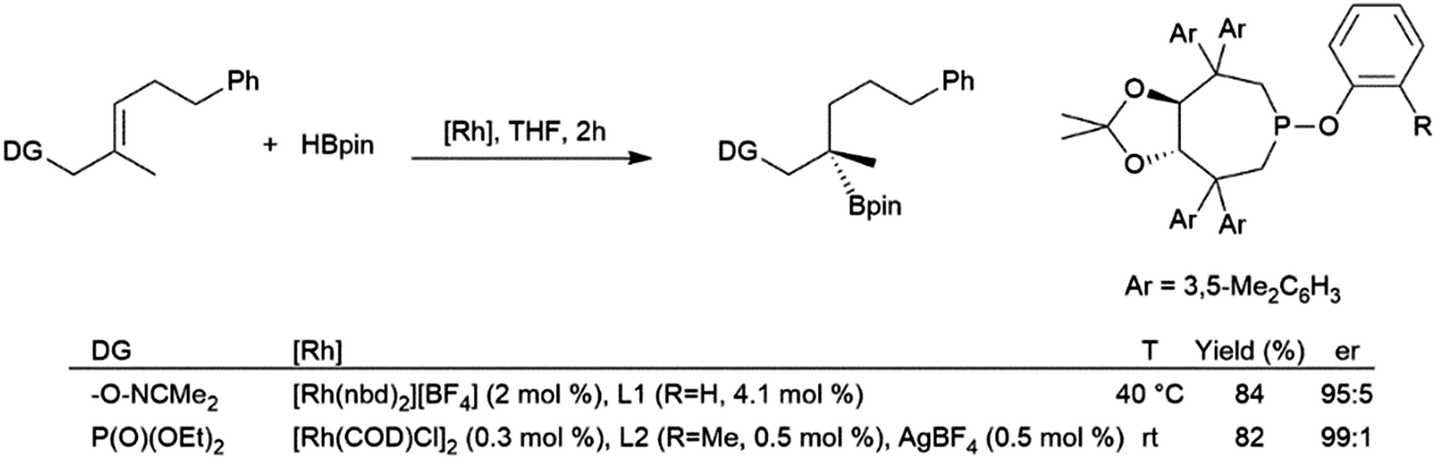 | ||
| Scheme 34 Oxime-205 and phosphonate-206,207 directed asymmetric hydroboration of trisubstituted alkenes. | ||
A recent publication cites the in situ formation of HBpin and a cobalt-catalysed hydroboration of a 1,2,2-trisubstituted aryl alkene as a key step in the synthesis of 1,1,1-tris(boronates) from the reaction of monosubstituted aryl alkenes with B2pin2 (Scheme 35).208 Cobalt(II) hydrides appended to bipy-type functionalities in metal–organic frameworks (MOFs) proved to be effective in the hydroboration of 2-methyl-2-butene, in addition to other mono- and di-substituted alkenes, as well as ketones and aldehydes, albeit with relatively long reaction times in all cases.209
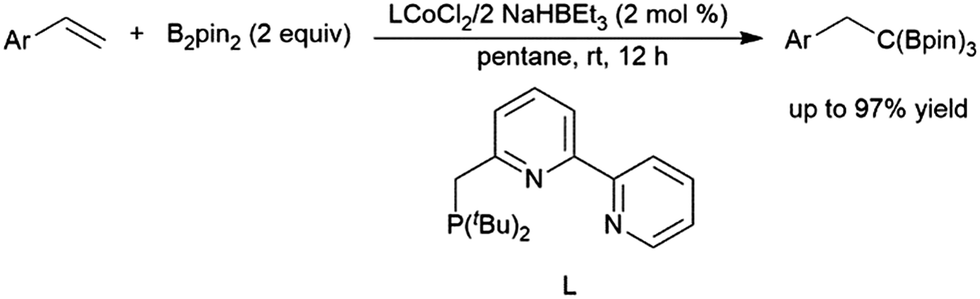 | ||
| Scheme 35 Cobalt-catalysed synthesis of tris(boronates).208 | ||
4.5 Tetrasubstituted alkenes
There are several examples of hydroboration reactions of 2,3-dimethylbut-2-ene. To the best of our knowledge, the most selective hydroboration of this substrate was the first one reported in 1992 by Baker, Marder, and co-workers (Scheme 36).129 While several of the catalysts examined were inactive for the reaction, two rhodium bisphosphine η3-2-methylallyl catalyst precursors provided nearly quantitative yield of the hydroboration product at room temperature, albeit with extended reaction times. More recent attempts to hydroborate the same substrate have been plagued by alkene isomerization.160,166,173 | ||
| Scheme 36 Rhodium-catalysed hydroboration of 2,3-dimethylbut-2-ene.129 | ||
4.6 Dienes
Dienes have also been found to present problems in terms of selectivity. Hydroboration of the terminal alkene occurs in hydroborations using dialkylboranes, leaving the other π-bond unaltered. Good selectivity for 1,2-hydroboration is observed using Pd(0), Pd(II), and Rh(I) catalyst precursors (Scheme 37).210 In a similar fashion, an Ir(I) system was found to catalyse the 3,4-hydroboration of 2-substituted 1,3-dienes, giving the terminal hydroboration product.211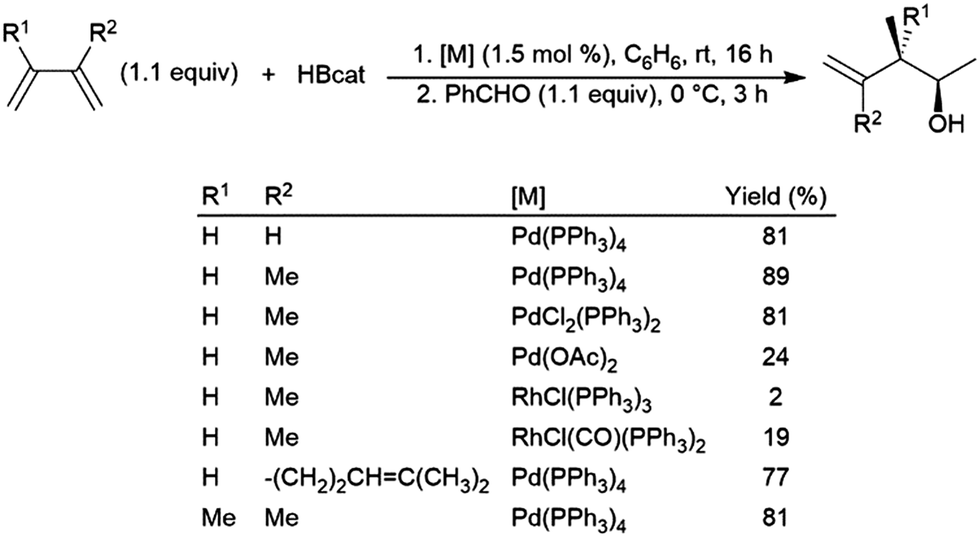 | ||
| Scheme 37 Catalytic hydroboration of dienes.210 | ||
Another report has also highlighted the 1,2-hydroboration of the terminal double bond of dienes using a CuCl/diphosphine catalyst system.118 Using 2.5 equivalents of catecholborane and a variety of Rh(I) catalyst precursors the bis-hydroboration of 1-phenylbutadiene was achieved in moderate yields and with facial selectivity (Scheme 38).212
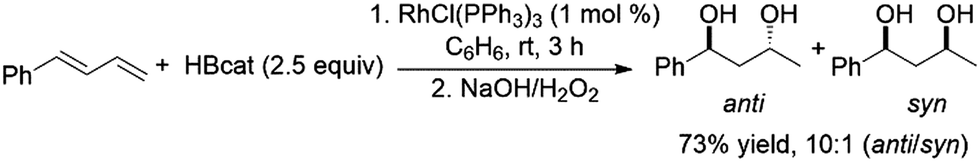 | ||
| Scheme 38 Rhodium-catalysed bis-hydroboration of dienes.212 | ||
Chemo- and regioselectivity are often issues in Ni(II) and Co(II) diphosphine-catalysed hydroborations using catecholborane, giving significant amounts of products resulting from differing regioselectivity in 1,2-hydroborations and also products of 1,4-hydroboration.214–216 Improved selectivity for 1,4-hydroboration is observed using Ni(0)217–221 and Fe(II)213,222 catalyst precursors (Scheme 39).213
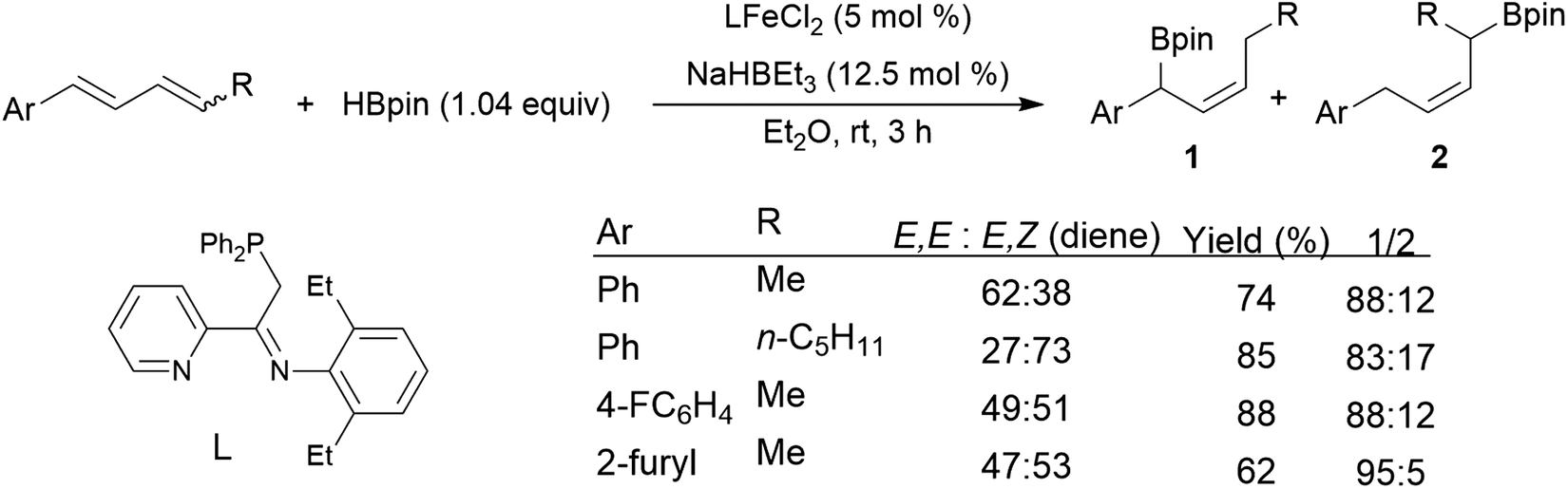 | ||
| Scheme 39 Iron-catalysed hydroboration of 1,4-di-substituted 1,3-dienes.213 | ||
Rajanbabu and co-workers reported divergent selectivity depending on ligand choice in cobalt(I) catalytic hydroboration of dienes (Scheme 40).223 The best achiral system for 1,4-hydroboration of 2-substituted 1,3-dienes came from the in situ generation of a cobalt(I) dppp complex. Chiral systems derived from bulky 2-oxazolyl-phenyldi(aryl)phosphine ligands (with chirality built into the oxazolyl fragment) were found to provide good regio- and enantioselectivities (up to 94:6 er) in this transformation. A computational study from Liu, Jiang, and Chen found that the dppp ligand donates strongly and provides steric bulk, while the narrower bite angle of dppe alongside the reduced bulk and electron donating ability of (2-oxazolyl)-phenyldiphenylphosphine alter the reaction pathway to favour 1,4-addition.224 A CuCl/diphosphine-based catalyst system was found to be quite selective for the 1,4-hydroboration of 1,3-cyclohexadiene and the 1,2-hydroboration of 1-phenylbutadiene (Scheme 41).225
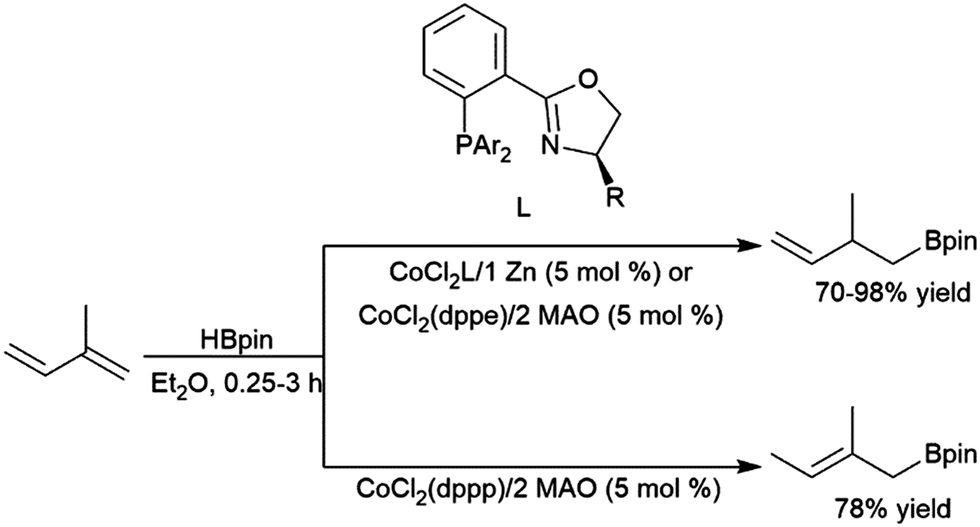 | ||
| Scheme 40 Cobalt-catalysed hydroboration of dienes.223 | ||
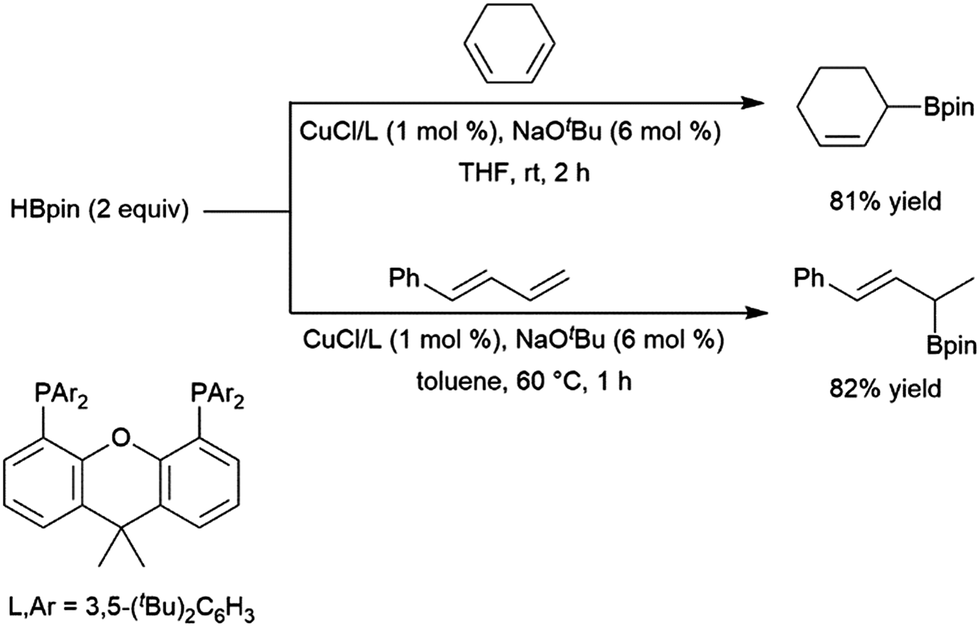 | ||
| Scheme 41 Copper-catalysed hydroboration of dienes.225 | ||
4.7 Enynes
Enynes have been shown to exhibit a wide variety of reactivity in catalytic hydroboration reactions. In many cases, hydroboration occurs selectively at the more reactive terminal alkyne functionality. Regioselectivity for the cis-hydroboration of the terminal alkyne in conjugated enynes has been found to be excellent with a catalyst system based on Schwartz's reagent, triethylamine, and pinacolborane (Scheme 42).226,227 Selectivity for the linear borylated product is reduced somewhat using a NiCl2/dppe catalyst system.228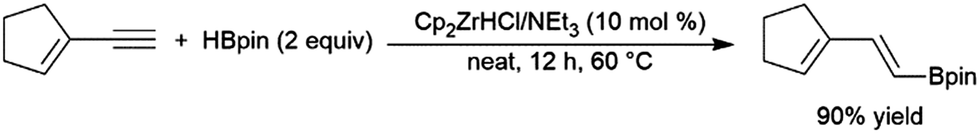 | ||
| Scheme 42 Zirconium-catalysed hydroboration of terminal alkynes.236,237 | ||
trans-Hydroboration reactions have also been demonstrated using catecholborane and a palladium(0)/1,4-azoborine-based phosphine ligand system.229,230 As previously discussed for simple terminal alkynes, trans-hydroboration of a terminal alkyne functionality of an enyne is also possible using a rhodium(I) tri-isopropyl phosphine/triethylamine catalyst system with pinacolborane.73 This reaction has seen some practical applications with enynes in synthetic chemistry (Scheme 43).231,232
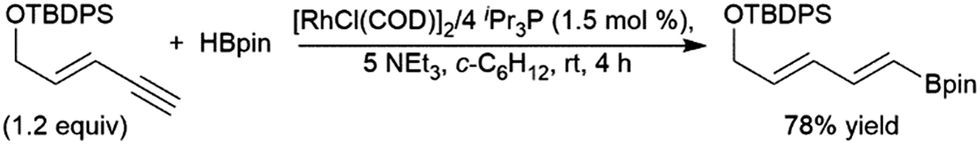 | ||
| Scheme 43 Selective trans-hydroboration of the alkyne functionality of an enyne.231 | ||
In reactivity analogous to that observed with conjugated dienes, 1,4-hydroborations of enynes generatesing allenes with the boryl group tending to add to the end of what was the alkynyl moiety.233 This can be achieved using Pd(0)210,234–236 and, more recently, enantioselective reactions using Cu(II) catalyst precursors (Scheme 44).237,238
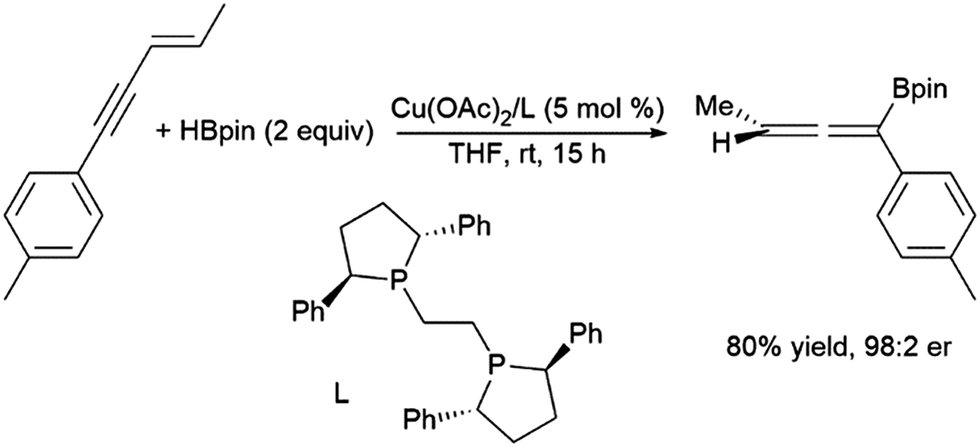 | ||
| Scheme 44 Copper-catalysed hydroboration of enynes.246 | ||
1,6-Enynes can undergo hydroboration resulting in ring-closing carbon–carbon bond formation, first reported in 2006, using a rhodium(I) catalyst precursor (Scheme 45).239 The authors propose that the reaction must proceed via η4-coordination of the enyne to the cationic rhodium(III) intermediate after the initial B–H oxidative addition, followed by either insertion of the alkyne into the Rh–B bond or insertion of the alkene into the Rh–H bond followed by insertion of the other unsaturated functionality into the resulting Rh–C bond forming the 5-membered ring. Reductive elimination yields the cyclic product.
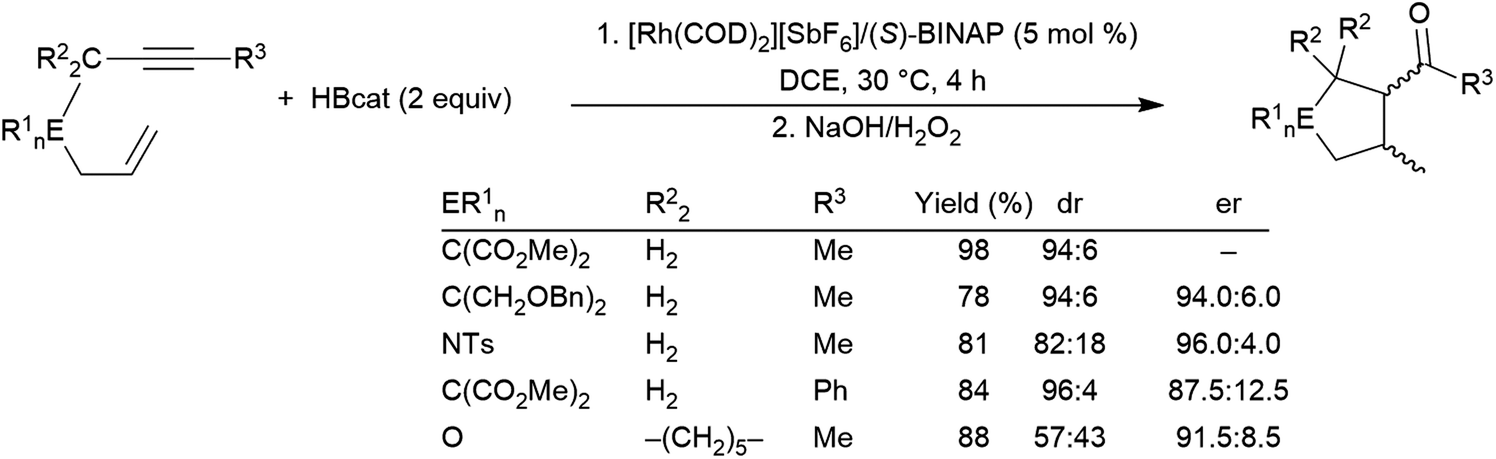 | ||
| Scheme 45 Rhodium-catalysed cyclization of 1,6-enynes via hydroboration.239 | ||
More recent work has shown the ability of the simple cobalt(II) catalyst precursor Co(acac)2 in combination with chiral phosphine ligands to promote the reductive cyclization of 1,6-240,241 and 1,7-enynes.242 Depending on the nature of the substrate, vinyl boronate ester or alkyl boronate esters can be generated. A similar cyclization of 1,6-enynes was also reported, using a bulky cobalt(II) catalyst precursor in combination with NaHBEt3.243 This protocol offered complementary regioselectivity to that found with the Co(acac)2/(R,R)-Quinox-P combination with some substrates (Scheme 46). Similar reactivity has since been observed with Ni(II)244 and Fe(II)245 catalysts. 1,5-Ynones have been cyclized with Cu(I) catalysts and trimethylamine-borane as the boron source.246
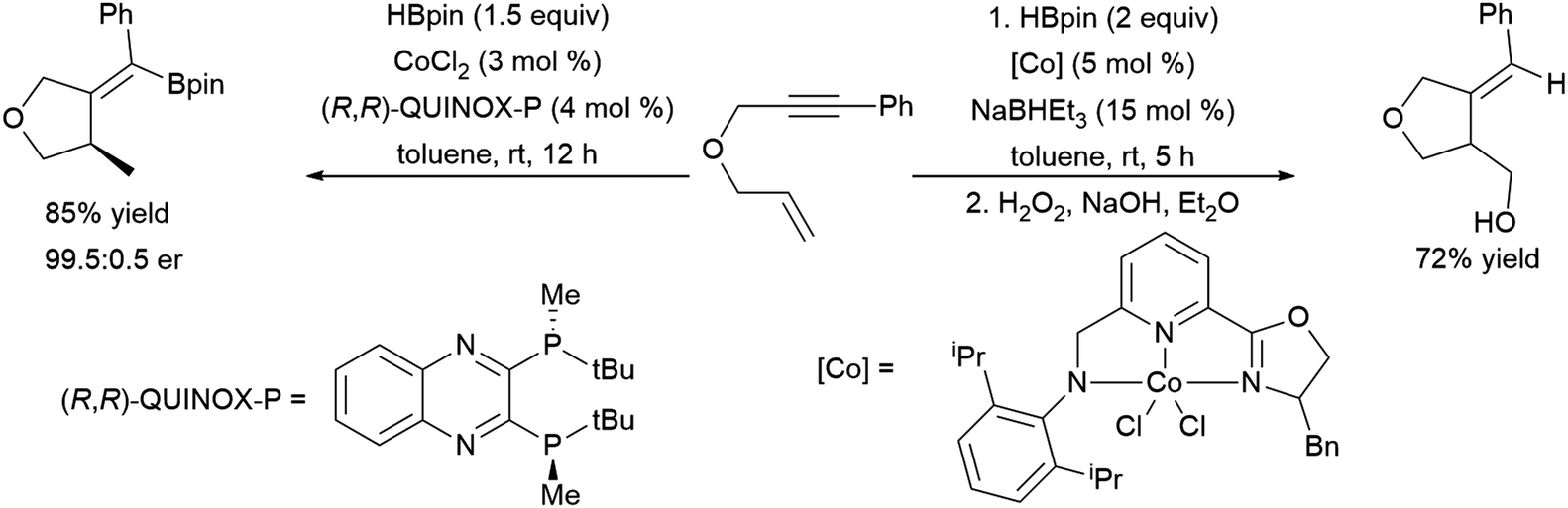 | ||
| Scheme 46 Cobalt-catalysed cyclization of enyne derivatives via hydroboration.241,243 | ||
4.8 Allenes
While less extensively studied than many other substrates, allenes have proven to be interesting substrates in transition metal-catalysed hydroborations as the utilization of different ligands under identical conditions can offer products with different regioselectivities.202,203 This is particularly true of Pt(0) catalyst precursors (Scheme 47).247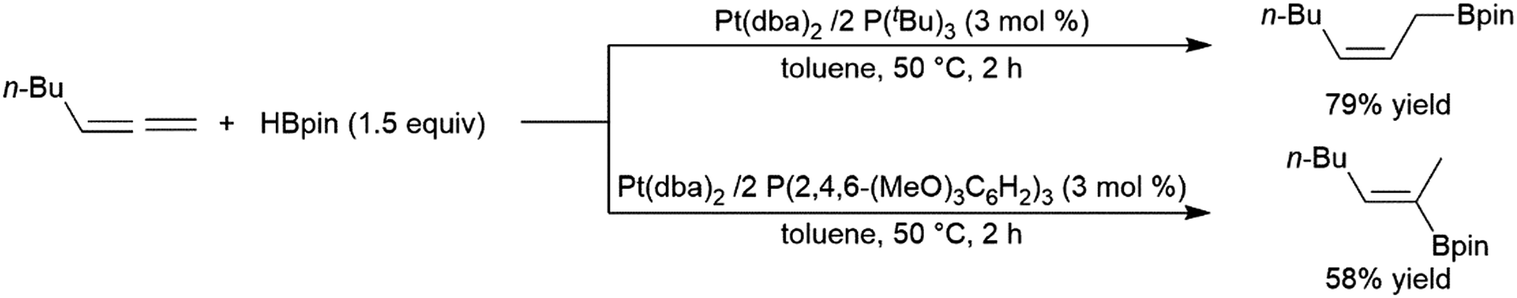 | ||
| Scheme 47 Platinum-catalysed hydroboration of allenes.247 | ||
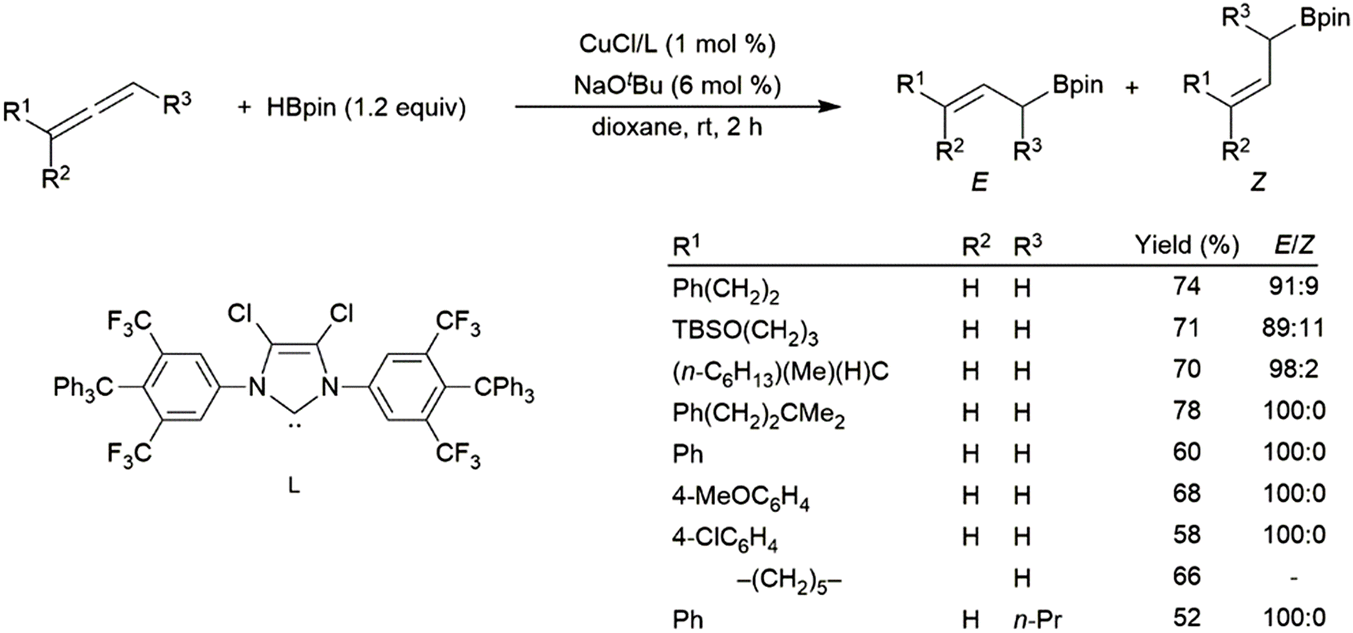
Rhodium(I)-based catalysts were found to be less effective in the reaction, providing low yields and little regioselectivity.247 Recently, copper(I) catalysts have proven to be effective in the hydroboration of allenes, with strong selectivity for the linear hydroboration product. The remaining double bond exists as a mixture of E- and Z-isomers, generally favouring the E-isomer (Scheme 48).225 More recent work has unearthed a cobalt-based protocol which yields primarily the Z-isomer on the terminal end of the allene,248 a metal-dependant regio-divergent protocol that selectively installs the boron moiety on the central carbon of the allene using either a nickel- or cobalt-catalyst,249 and a chromium-based protocol which yields boronated (E,Z)-skipped dienes through tandem dimerization and hydroboration.250
 | ||
| Scheme 48 Copper(I)-catalysed hydroboration of allenes.225 | ||
4.9 Monosubstituted aryl alkenes
From the very early days of transition metal-catalysed hydroborations, styrene and derivatives have been of great interest. In the absence of a catalyst, the reaction is highly selective for the linear hydroboration product. In 1989, it was reported that some cationic rhodium(I) phosphine complexes were able to catalyse hydroborations using catecholborane that are highly selective for the branched borylated product.96 Over the next few years contradicting reports of the selectivity of hydroborations using Wilkinson's catalyst (RhCl(PPh3)3) and catecholborane were published.96,99,251 Soon thereafter, these contrasting results were explained by the partial oxidation of the rhodium(I) catalyst in some cases.129,141 Selectivity for either the branched or linear products is achievable, depending on catalyst selection (Scheme 49).129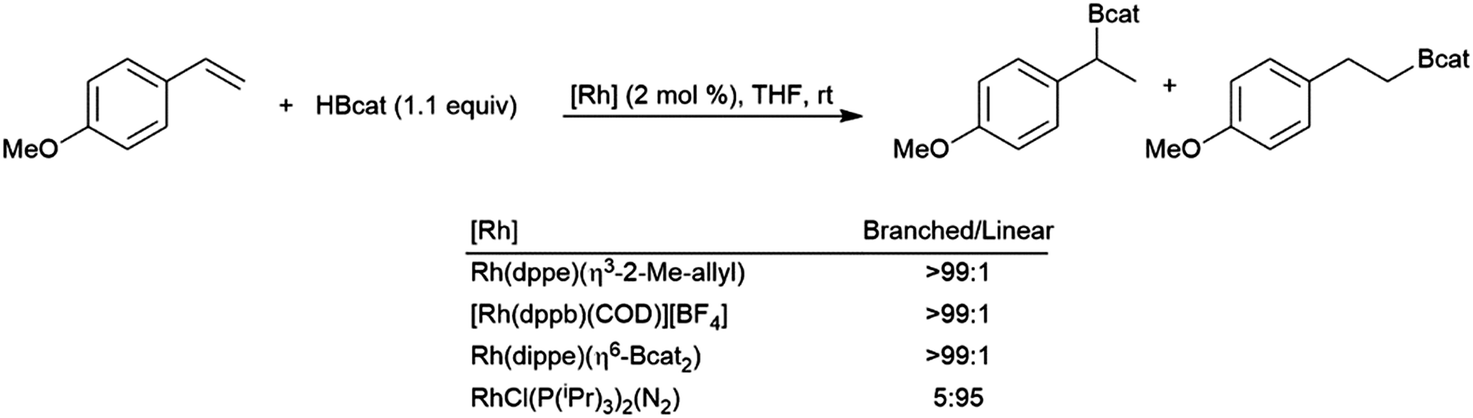 | ||
| Scheme 49 Rhodium-catalysed hydroboration of 4-methoxystyrene.129 | ||
The frequently observed selectivity for the branched isomer is rationalized by the preferential formation of a benzylic intermediate following insertion of the alkene into the rhodium–hydrogen bond. Further evidence for this assertion comes from the results of the hydroborations of ortho-substituted aryl alkenes. While regioselectivity in the rhodium-catalysed hydroboration of 2,4-dimethylstyrene is quite high at 95–97%, this number drops significantly in the hydroboration of 2,4,6-trimethylstyrene to 63% due to the steric constraint that destabilizes the planar benzylic intermediate in this substrate.252 Rhodium-BABAR-phos complexes were found to be remarkably stable, however regioselectivity in the catalysed hydroboration of aryl alkenes was relatively poor.253,254
Iridium-catalysed reactions tend to produce the linear isomer.35,116,117,156,185,255–258 It has been suggested that this may be due to olefin insertion first into the M–B bond, as opposed to initial insertion into the M–H bond, as is the case with many rhodium catalysts. The readily available ruthenium-based catalyst [Ru(p-cymene)Cl2]2 was also found to provide terminal-selective hydroboration of vinyl arenes.259
In recent years, first row transition metals have been utilized in this reaction. Many of these initially provided poor selectivity or selectivity for the linear isomer. Metals utilized included cobalt,123,260–264 iron,265–267 and manganese.268,269 More recently, earth-abundant catalysts providing branched selectivity in the hydroboration of vinyl arenes have been developed, including nickel,270,271 cobalt,131,272 and iron.273,274 These developments are covered in a 2019 review.275 This has been extended to heterogeneous catalysis using a cobalt(II) coordination polymer.276
Catecholborane could be added to a platinum complex of 2-vinylpyridine using RhCl(PPh3)3 as a catalyst, but the regioselectivity observed under these reaction conditions was poor.202 Each hydroboration isomer accounted for 45% of the product mixture, while dehydrogenative borylation and hydrogenation products were also observed.
Heterogeneous catalyst systems based on rhodium(I) catalyst precursors in combination with silica bearing diphenylphosphine groups showed good regioselectivity for the branched isomer in the hydroboration of aryl alkenes with catecholborane, particularly when Rh(acac)(COE)2 was used as the rhodium(I) source.277 Only a slight decrease in activity was observed over 3 runs. The synthesis of diborylalkanes from terminal aryl alkenes has been effected in a zirconium(IV)-catalysed dehydrogenative borylation-hydroboration sequence.128
4.10 1,1-Disubstituted aryl alkenes
Like their aliphatic counterparts, hydroborations of 1,1-disubstituted aryl alkenes are more sluggish than their monosubstituted analogs. An early study discovered that selectivity in the rhodium-catalysed hydroboration of 2-phenylpropene with catecholborane could be altered by the choice of catalyst.65 The combination of [MCl(COE)2]2 (M = Rh, Ir) with one equivalent of PPh3 per M was found to give >98% selectivity for the linear product while using Rh(acac)(dppb) as the catalyst results in 95% selectivity for the tertiary product (Scheme 50).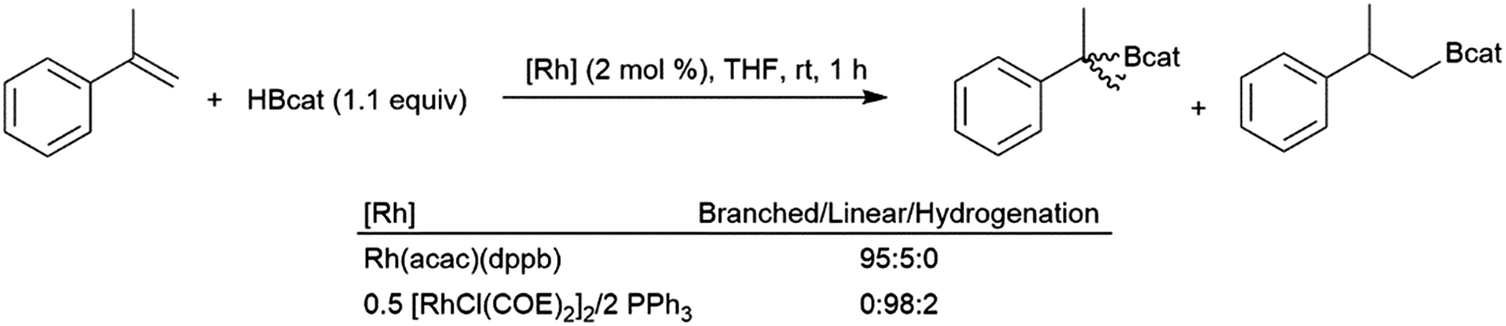 | ||
| Scheme 50 Rhodium-catalysed hydroboration of 2-phenylpropene.65 | ||
Selective formation of the tertiary product is relatively rare and unique to some rhodium-based systems.99,129,154,258,278,279 Most hydroboration catalysts based on rhodium,65,97–100,129,154,156,163,167,280,281 iridium,281–283 cobalt,157,160–162 lanthanum,284 samarium,77 nickel,181 iron,137,168,285 and copper286 selectively provide the linear product. Chiral cobalt161,287,288 and iron catalyst285 precursors gave good activity and enantioselectivity in this transformation. Asymmetric cobalt-catalysed hydroborations of 1-aryl-1-silyl alkenes have drawn interest as the second part of a hydrosilylation/hydroboration sequence, starting with the hydrosilylation of a terminal alkyne.289–292 Substrates that are bulkier99 or more electron-poor278 than 2-phenylpropene also showed increased selectivity for the linear hydroboration product in rhodium-catalysed reactions. The copper catalysed hydroboration of a 1-phenyl-1-borylalkene gave the tertiary diborylated product (Scheme 51).293
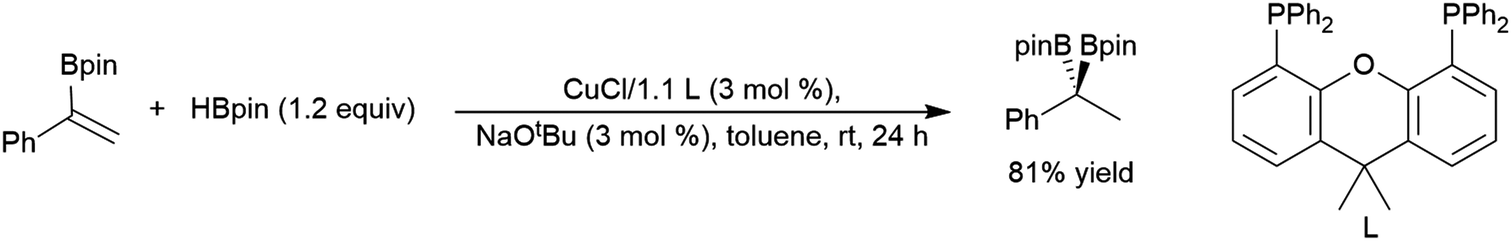 | ||
| Scheme 51 Catalytic hydroboration of a 1-phenyl-1-borylalkene.293 | ||
Recent work has demonstrated the ability of amide and oxime ether groups to direct asymmetric hydroborations using HBpin and chiral Rh catalysts. Interestingly, while a phosphonate group provided regioselectivity for the quaternary isomer when the substrate's aromatic group is unsubstituted or had meta- or para-substituents, the use of ortho-substituted aryl groups led to an inversion in regioselectivity and a significant loss of asymmetric induction (Scheme 52).206 Oxime ether directing groups were found to promote differing regioselectivity in catalytic asymmetric hydroborations of the same substrate, depending on the ligand utilized.294
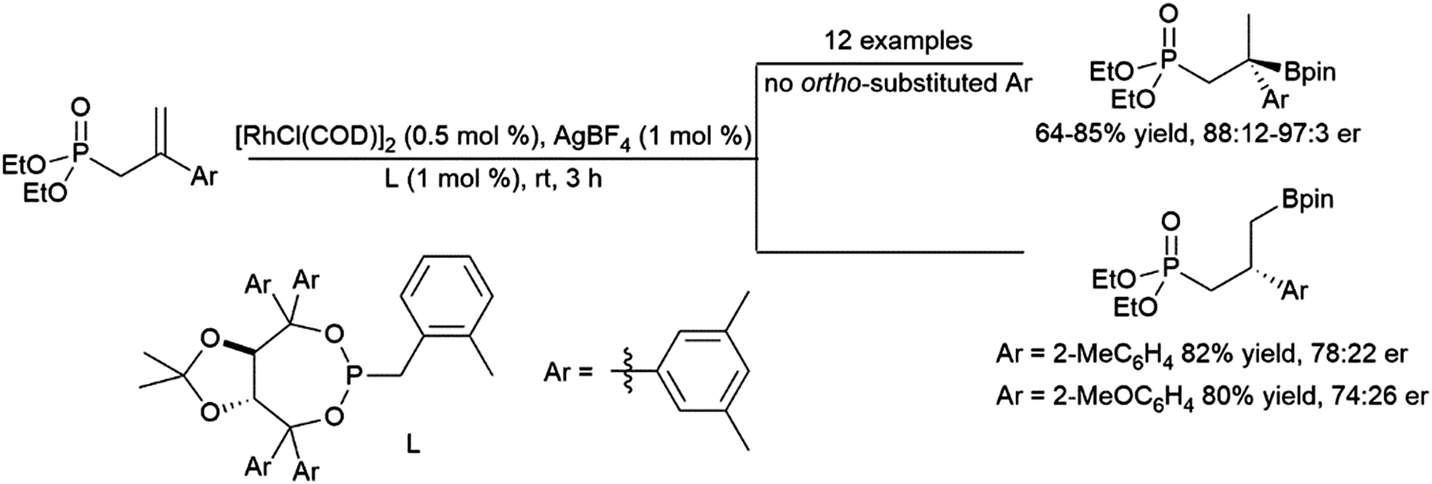 | ||
| Scheme 52 Phosphonate-directed rhodium-catalysed hydroborations of 1,1-disubstituted aryl alkenes.206 | ||
4.11 1,2-Disubstituted aryl alkenes
Rhodium-catalysed hydroborations of trans-3-phenyl-2-propene, cis-3-phenyl-2-propene, indene, and other 1-aryl-2-alkyl alkenes generally proceed with the boryl group adding α to the aromatic ring. As with monosubstituted aromatic alkenes, this selectivity is rationalized through the formation of a rhodium-benzyl intermediate.Some 1,2-disubstituted aryl alkenes, including indene and dihydronaphthalene and their derivatives are commonly used substrates in asymmetric hydroboration reactions. Rhodium catalysts with QUINAP-type ligands are among the most effective ligands for these transformations, achieving excellent er's at room temperature (Scheme 53).100,101,108,252,295–301
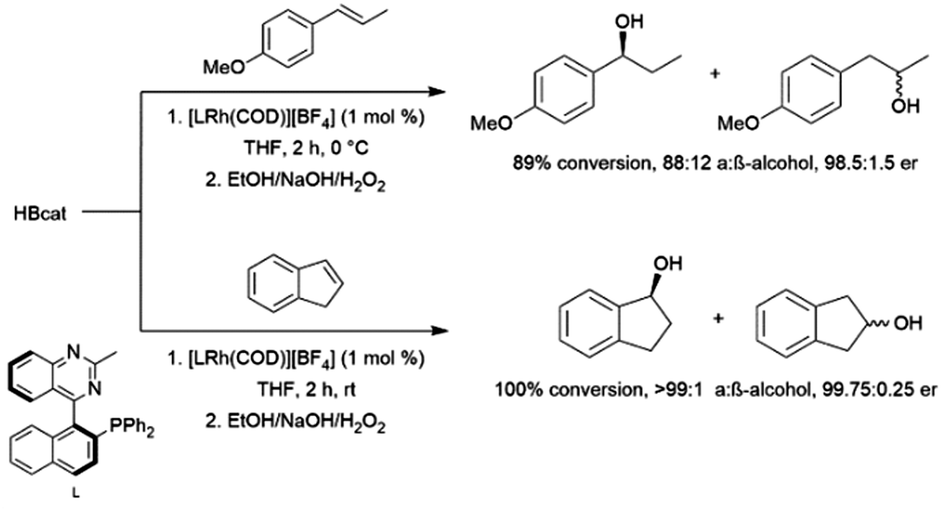 | ||
| Scheme 53 Rhodium-catalysed hydroboration of trans-1-methoxy-(1-propenyl)benzene (top) and indene (bottom).295 | ||
Recent work has demonstrated the potential of cobalt catalysts with amide ligands in the asymmetric hydroboration of 1,2-disubstituted aryl alkenes, with enantiomeric ratios up to >99:1 (Scheme 54).264 The reaction is thought to proceed through a benzylic intermediate similar to that found in the case of many rhodium catalysts.
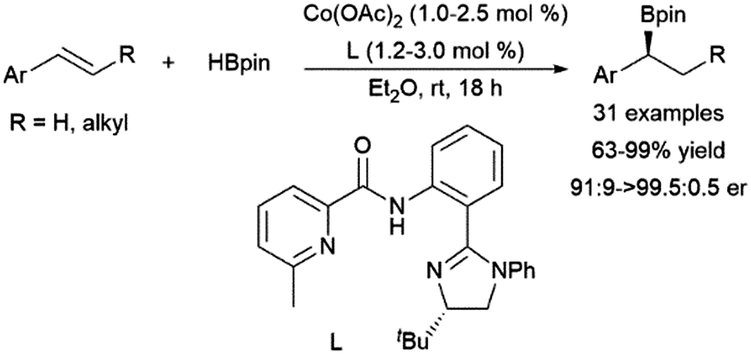 | ||
| Scheme 54 Cobalt-catalysed α-selective hydroborations of trans-1,2-disubstituted aryl alkenes.264 | ||
Later work by Takacs showed that the use of amide,302 or phosphonate303 directing groups in the substrate, in rhodium-catalysed hydroborations with bulky chiral TADDOL-based ligands led to highly regio- and enantioselective hydroborations of 1,2-disubstituted aryl alkenes. A zirconium(IV) catalyst has demonstrated the isomerization, dehydrogenative borylation, and hydroboration of trans-β-methylstyrene, generating the 1,1-diborylalkane.128
4.12 Terminal alkynes
A variety of catalysts have found use in the cis-hydroboration of terminal alkynes304 including nickel,181,214,305 palladium,306–308 rhodium,182,183,309–313 ruthenium,314 iron,267,315–317 copper,318–320 titanium,81,82,321 scandium,322 silver,323 gold,324 iridium,38,39 and molybdenum.325,326 However, the most popular catalyst system for this transformation has been the combination of pinacolborane with a catalytic amount of Schwartz's reagent (Scheme 55, top85).83–85,327–336 While rates and selectivities are frequently acceptable in the absence of triethylamine, the additive was found to improve the results in some cases.226,337–346 As discussed in the enyne section, the system provides generally good yields and excellent selectivity for cis-hydroboration of the terminal alkyne functionality, with the boryl group being added to the terminal carbon. In competition experiments, using a gold(I) catalyst, terminal alkynes were hydroborated preferentially over alkenes.324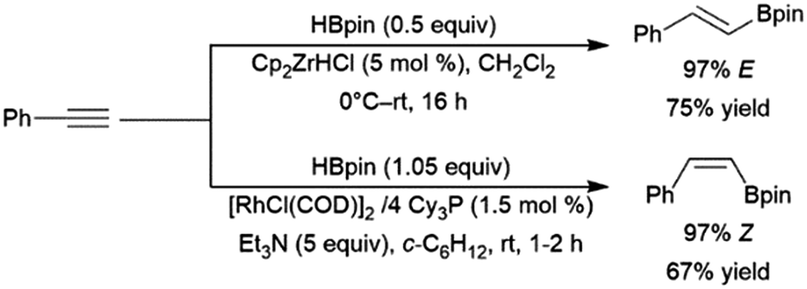
In many cases, the hydroboration of terminal alkynes catalysed by rhodium(I) complexes was found to produce a mixture of products containing the trans-hydroboration product.33,129,304,309,314,347 Building on these results, a protocol was developed for trans-selective hydroboration using rhodium348 or iridium catalysts (Scheme 55, bottom).73 Since its development, this protocol has seen some use in synthetic chemistry.73,349–354 Ruthenium,268,309,355,356 copper,357 cobalt,358,359 and iron360,361 catalysts have since also been shown to be capable of the same transformation. It should be noted that in many cases, the reaction proceeds through initial migration of the alkynyl proton (see Section 2.3 trans-Hydroboration of alkynes).314,362
Dehydrogenative borylation (replacing the terminal hydrogen atom with a boryl group) is another possible outcome for transition metal-catalysed reactions of boranes with terminal alkynes, this has been achieved in high selectivity with a copper(I) catalyst system.363 See Section 4.13 Internal alkynes for reactions involving the dehydrogenative borylation of terminal alkynes followed by hydroboration.
Copper(I)-based hydroboration catalyst systems have found use in multi-step catalytic reactions, including a sequential hydroboration-hydrocupration-palladium catalysed cross-coupling reaction, resulting in the dihydrofunctionalization of terminal alkynes (Scheme 56).364
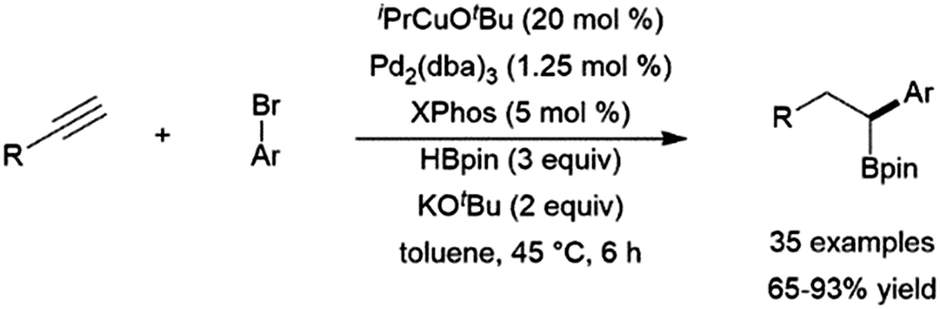 | ||
| Scheme 56 One pot hydroboration-hydrocupration-cross-coupling reaction.364 | ||
Gade and co-workers found that cobalt(II) complexes could catalyse α-selective hydroborations of terminal alkynes (Scheme 57).365 The use of similar N,N,N-pincer cobalt(II) complexes and HBpin to achieve this Markovnikov-type selectivity was later mechanistically explored by Chen and co-workers, whereby they suggest that the reaction proceeds by alkyne coordination with a cobalt hydride intermediate, followed by alkyne insertion and σ-bond metathesis with HBpin to provide the α-selective product.366
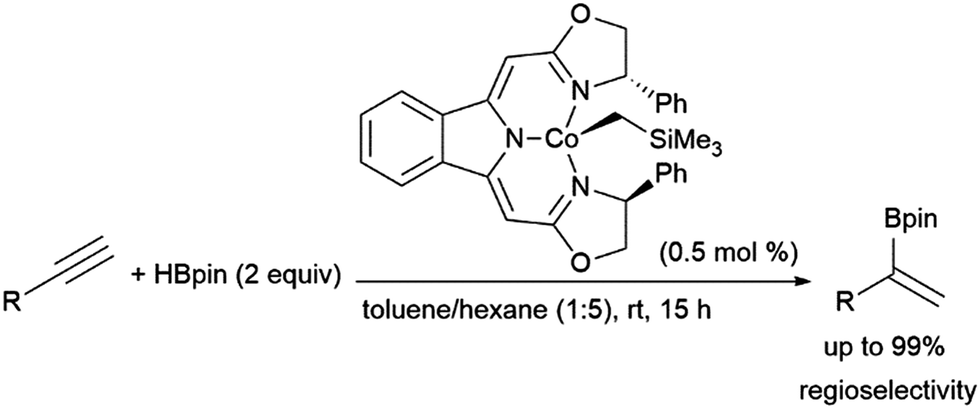 | ||
| Scheme 57 Cobalt-catalysed α-selective hydroboration of terminal alkynes.365 | ||
As hydroborations of 1,2-disubstituted alkenes are well established, in some cases terminal alkynes can be hydroborated twice to achieve geminal substitution on the terminal carbon. Copper,293 cobalt,367 platinum,368 and rhodium313,369 catalysts have been shown to demonstrate this reactivity (Scheme 58).293,370
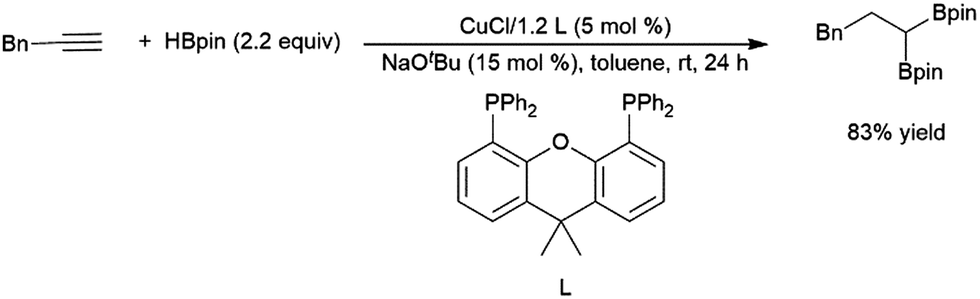 | ||
| Scheme 58 Copper-catalysed bis-hydroboration of a terminal alkyne.293 | ||
Recently, onium salt-stabilized nanocatalysts of Ru, Ir, Rh, and Pt were effective in the hydroboration of phenylacetylene in supercritical carbon dioxide. These reactions were quite selective for cis-hydroboration, though conversions were often not quantitative and reusing the catalyst led to a decrease in activity.371 Separate studies have looked specifically at ruthenium catalysts in ionic liquids (with or without supercritical CO2),372 in supercritical CO2,373 or in polyethylene glycol,374 finding good selectivity through multiple catalytic cycles.
4.13 Internal alkynes
A variety of catalysts have been used for the hydroboration of internal alkynes. In addition to rhodium catalysts,305,322,375 this reactivity has also been demonstrated using titanium,81,82 zirconium,85 nickel,214,305 palladium,305,376 copper,320,341,377–381 ruthenium,382,383 manganese,384 molybdenum,325 cobalt,385,386 iron,137,315,316,387,388 platinum,368 and iridium39,322 catalysts. A recent report has highlighted the ability of cobalt centers appended to a MOF to effect the transformation (albeit quite slowly).389Alkynes with alkyl and α-ester substituents undergo copper(I)-catalysed hydroborations with pinacolborane adding in a cis-fashion with the boryl group preferentially adding to the carbon bearing the ester group.377,378 Catalytic hydroborations of thioalkynes generally proceed with the boryl group adding to the carbon bearing the alkyl group.305,376 Conversely, a more recent report has shown the inverse regioselectivity can be strongly favoured using a copper(I) catalyst and pinacolborane (Scheme 59).320
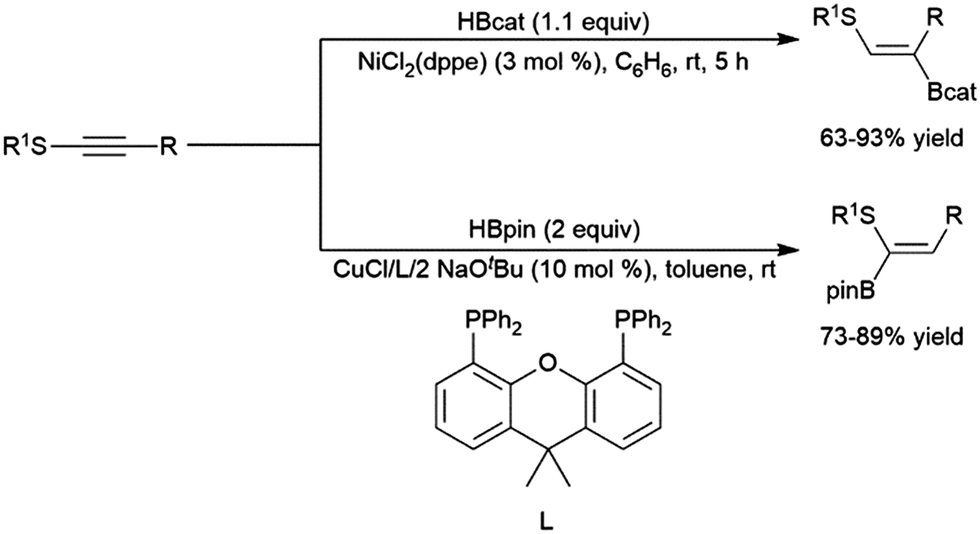 | ||
| Scheme 59 Catalytic hydroborations of thioalkynes using nickel304,372 (top) and copper320 (bottom) catalyst systems. | ||
A ruthenium system was found to catalyse the preferential trans-hydroboration of internal alkynes, including macrocyclic examples (Scheme 60).74 While providing some functional group tolerance, problems with the ruthenium-catalysed trans-hydroboration protocol were encountered with conjugated enynes, terminal alkynes and small cyclic substrates, which were either non-reactive, showed poor selectivity, or underwent polymerization. A recent report has demonstrated the trans-selective hydroboration of ynamides using a carbene–BH3 adduct and ZnEt2 as a catalyst.390
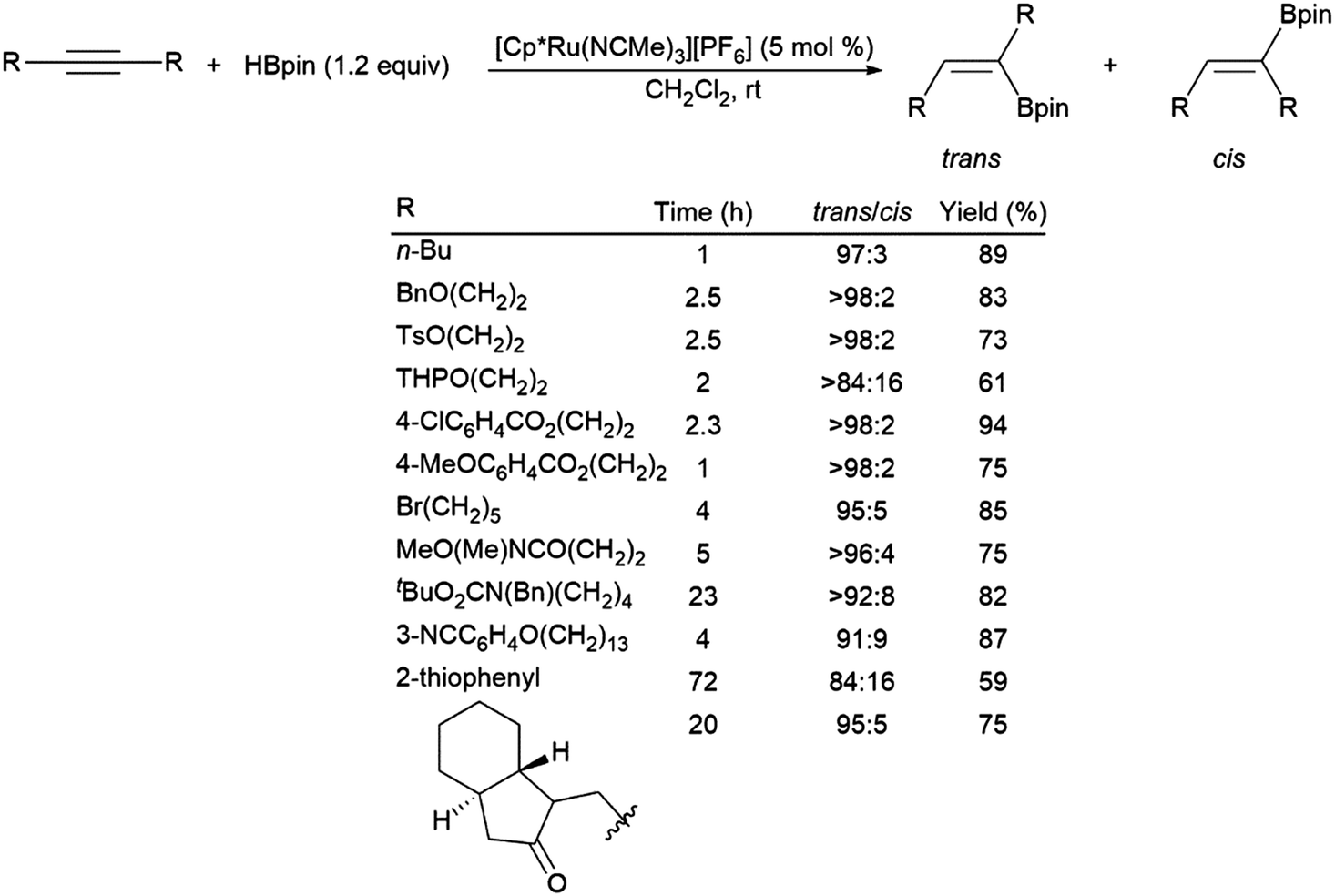 | ||
| Scheme 60 Ruthenium-catalysed trans-hydroborations of internal alkynes.74 | ||
Simple unsymmetrical internal alkynes undergo hydroboration with the boryl group being preferentially added to the carbon bearing the smaller substituent, but selectivities are often relatively poor. Alkynes with aryl and alkyl substituents are catalytically hydroborated with the boryl group typically adding preferentially to the carbon bearing the aryl group. The most regioselective and general protocols for unsymmetrical alkynes use pinacolborane and copper(I) catalysts.378,379 Additionally, one of these systems even functions in the presence of air (Scheme 61).379
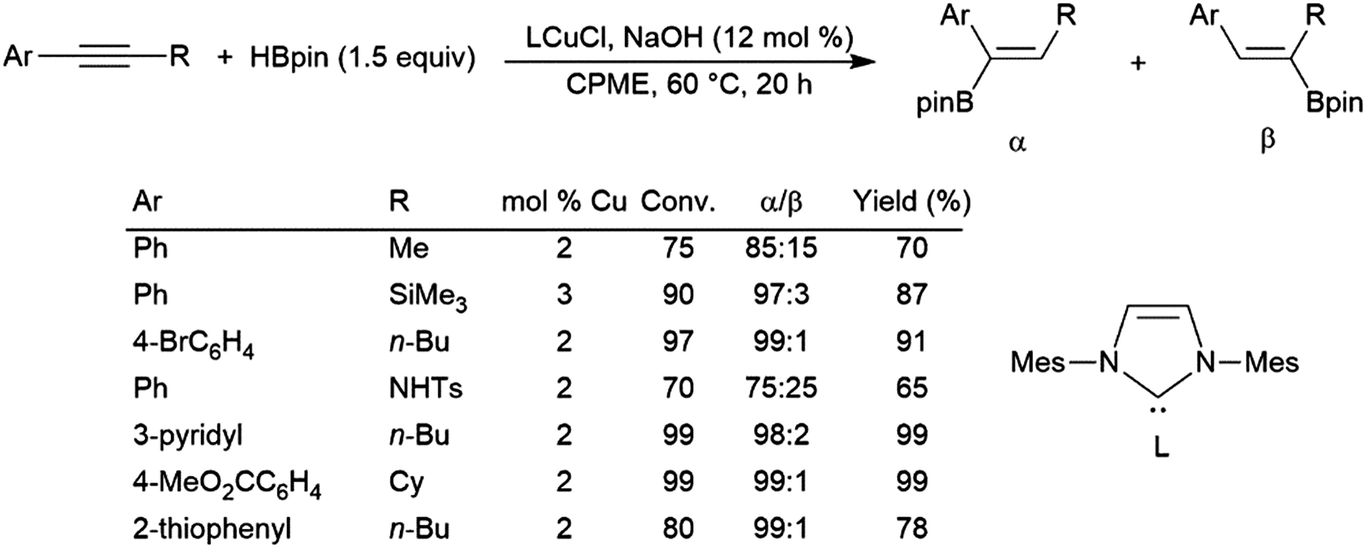 | ||
| Scheme 61 Copper-catalysed α-selective hydroboration of internal aryl alkynes.379 | ||
Bis-hydroboration of diphenylacetylene with catecholborane and rhodium catalysts has also been examined, although selectivity is relatively low, with a maximum of 68% selectivity for the diol following oxidative workup.375 Shi and co-workers have reported an elegant intramolecular hydroboration catalysed by gold(I) complexes in the synthesis of cyclic amine boranes (Scheme 62).391 This methodology would later be modified to incorporate the catalyst in a porous organic polymer392 and to include the analogous intramolecular reactions with terminal alkynes.393
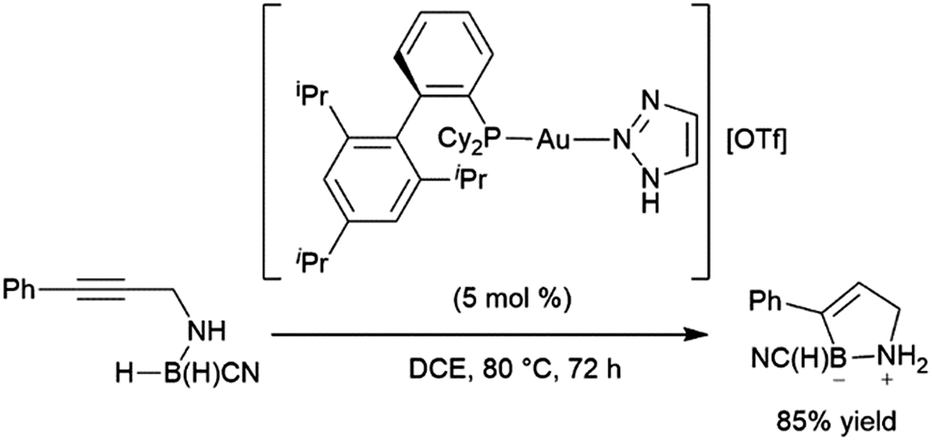 | ||
| Scheme 62 Intramolecular gold-catalysed hydroboration of internal alkynes.391 | ||
Engle and co-workers demonstrated a stepwise reaction involving dehydrogenative hydroboration of terminal alkynes with HBdan (HBdan = 1,8-diaminonaphthylatoborane), followed by hydroboration with pinacolborane to generate the 1,1-bis-boryl alkene product.394 1,2-Bis-boryl alkene products are also accessible: Weber and co-workers reacted a bench-stable manganese(I) alkyl complex and HBpin with terminal alkynes to provide trans-1,2-bis-boryl alkene products,269 and Lai and Ozerov produced the cis-1,2-bis-boryl products using a Ir/CO/tBuNC system.397 A protocol by Ingleson and co-workers employing pinacolborane and a zinc-hydride catalyst was shown to perform dehydrogenative borylation and subsequent bis-hydroboration of terminal alkynes, generating 1,1,1-triborylalkanes.398 Notably, the final step of the reaction demonstrates that the hydroboration of the bis-borylalkene requires the addition of catalytic THF–BH3 to proceed. Iridium-catalysed hydroboration of a 2-borylalkyne serves as part of the preparation of triboryl alkenes from terminal alkynes (Scheme 63).395 Initial dehydrogenative borylation of terminal alkyl or aryl alkynes generates the boryl alkyne, which undergoes hydroboration and subsequent dehydrogenative borylation to give the triboryl alkene. The synthesis of multiboronate esters by catalytic hydroboration has been recently included in a review.370
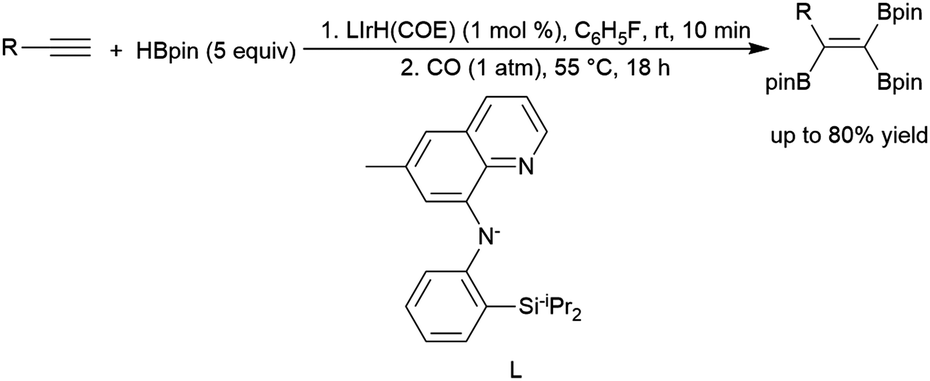 | ||
| Scheme 63 Iridium-catalysed preparation of triborylalkenes.395 | ||
Another multi-step, one-pot reaction proceeds via alkyne hydroboration followed by asymmetric hydrogenation, generating chiral alkylboronate esters.399 One-pot hydrosilylation and asymmetric hydroboration has also been achieved, producing chiral gem-(borylsilyl)alkanes.400 A ruthenium-catalysed geminal hydroboration of silyl alkynes was reported by Chung, Wu, Sun, and co-workers (Scheme 64).396 Silyl migration via a ruthenium carbene intermediate is proposed to allow this transformation. Hydrosilylation of alkynylboranes was found to provide the same products, supporting the proposed migration from a α-boryl-α-silyl intermediate.
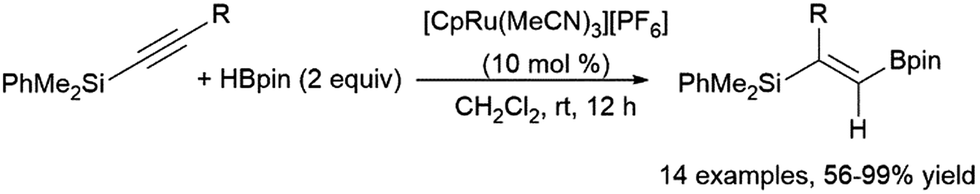 | ||
| Scheme 64 Ruthenium-catalysed gem-hydroboration of a silylalkyne.396 | ||
4.14 Aldehydes
In the early days of catalytic hydroborations, reactions employing simple aldehydes did not spark much interest. This is likely because the regioselectivity is fixed, with the boryl group adding to the oxygen atom, and the transformation is readily achieved using conventional reagents in the absence of catalysts. Despite this, there is a recent increase of reports on the reaction. A number of catalysts promote the addition of pinacolborane to simple aryl or alkyl aldehydes, including a titanocene(II) catalyst,401 titanium(IV),402 hafnium,403 ruthenium,404,405 silver,406 rhenium,407 manganese,408 nickel,409–411 zinc,412,413 palladium,414 iron,415–420 and copper.421,422 Using [Ru(p-cymene)Cl2]2, aldehydes could undergo hydroboration selectively in the presence of ketone or ester functionalities.405 A titanium(IV) amide was also found to be highly selective for hydroboration of aldehydes over their corresponding methyl ketones.402 Notably, while both copper(I) and copper(II) catalysts have been reported, the copper(II) catalyst exhibits greater chemoselectivity for aldehydes over ketones.422 A report has highlighted the potential of zirconium amide compounds as homogeneous and heterogeneous catalysts appended to mesoporous silica nanoparticles.423 Catalytic reactions involving catecholborane addition to aldehydes are relatively rare, likely due to the relatively rapid uncatalysed reaction, noted in a publication examining rhodium-catalysed addition of catecholborane to carbonyls.424Reusable options for heterogeneous catalysis include iron(II)425 and cobalt(II)426 coordination polymers, Fe2O3 nanoparticles,427 along with Co(II)428 or Ti(IV)429 sites in MOFs.
4.15 Ketones
As ketones are a more challenging substrate in the absence of a catalyst, the potential for catalysis and asymmetric catalysis with catechol- or pinacolborane is attractive. In many cases for the hydroboration of carbonyls, the catalyst functions mainly as a Lewis acid, activating the carbonyl carbon for nucleophilic attack by the hydride of the borane reagent. A number of different transition metals have been used to catalyse this reaction,430 including rhodium,424,431 titanium,401,402,432–438 hafnium,403 molybdenum,325 silver,406 cobalt,169,439–442 ruthenium,404 palladium,414,443 zinc,412,413,444–450 iron,415,418–420,439,440,451,452 nickel,409,410 manganese,408,440,453 copper,421,422 and yttrium.454,455 Chiral diols combined with titanium(IV) complexes provide up to 99:1 enantiomeric ratios in the hydroboration of simple ketones with catecholborane.432,434,435,438 The most effective zinc-catalysed hydroboration with catecholborane involves the use of bulky iminooxazoline, which provided er's of at least 92.5:7.5 for most ketones examined.447 Hydroborations with pinacolborane can be catalysed by chiral iron and cobalt catalysts, providing excellent enantioselectivity (Scheme 65).439,456 Heterogeneous and homogeneous zirconium amide catalysts were also effective for the catalytic hydroboration of ketones.423 Other heterogeneous systems are also capable of catalysing the hydroboration of ketones including Co(II) sites in MOFs428 and coordination polymers,426 iron(II) sites in MOFs457,458 and coordination polymers,425,459 Fe2O3 nanoparticles,427 a manganese coordination polymer,463 and Ti(IV) sites in a MOF.429
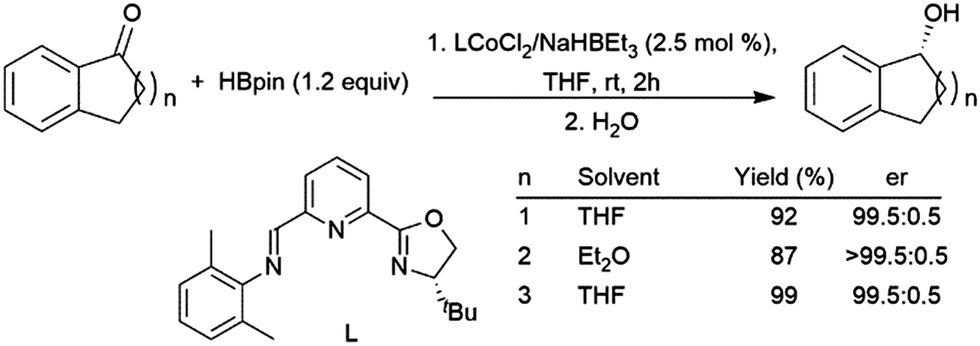 | ||
| Scheme 65 Cobalt-catalysed asymmetric hydroboration of cyclic ketones.430 | ||
Using an excess of catecholborane, to protect the alcohol, β-hydroxyketones can be hydroborated in a diastereoselective fashion using Wilkinson's catalyst RhCl(PPh3)3, producing cis-1,3-diols (Scheme 66).431
 | ||
| Scheme 66 Diastereoselective rhodium-catalysed hydroboration of a β-hydroxyketone.431 | ||
Chen and co-workers have reported reduction of the α-ketone functionality in α-ketoamides, catalysed by chiral oxido-vanadium(V) complexes.464 Fascinatingly, enantioselectivity was found to be nearly reversed depending on the use of pinacolborane or catecholborane. The same systems were also used in the hydroboration of β-ketoamides with pinacolborane to achieve moderate to high enantioselectivity.465
4.16 α,β-Unsaturated carbonyls
Catalytic hydroborations of α,β-unsaturated ketones, esters, and amides can occur in a 1,4-fashion.460 Quenching with an aqueous buffer solution gives the reduced ketone (Scheme 67). | ||
| Scheme 67 Rhodium-catalysed 1,4-hydroboration of an α,β-unsaturated ester.460 | ||
Addition of pinacolborane to cyclohexen-2-one catalysed by a titanocene(II) complex occurs selectively at the ketone, leaving the carbon–carbon double bond intact.401 Several other catalyst systems have been shown to hydroborate the carbonyl groups while maintaining the carbon–carbon double bond.417 Protocols for asymmetric hydroboration of the ketone groups of α,β-unsaturated ketones have been developed using a copper(I)/(R)-DBTM-SEGPHOS system,466 a manganese(II) pincer catalyst precursor,467 a cobalt(II)/chiral 8-oxazoline iminoquinoline system,468 and a nickel(0)/chiral bidentate oxazoline-based ligand system (Scheme 68).461
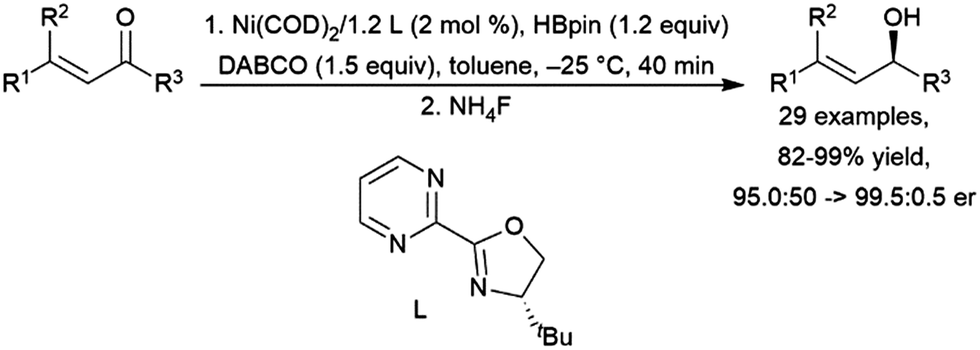 | ||
| Scheme 68 Nickel-catalysed hydroboration of the ketone group of an α,β-unsaturated ketone.461 | ||
Hydroboration of substituted cyclohexenone substrates by catecholborane, catalysed by a chiral rhodium complex, has been shown to produce moderate yields of the product of alkene hydroboration with low enantioselectivity.184 Optimization of a cationic rhodium phosphine catalyst precursor gave good yields and high enantioselectivity for the alkene reduction product of α,β-unsaturated amides.462 Selection of Z- or E-alkene and the appropriate chiral ligand allowed for the synthesis of all diastereomers of a product with two vicinal chiral centers created by the hydroboration reaction (Scheme 69).
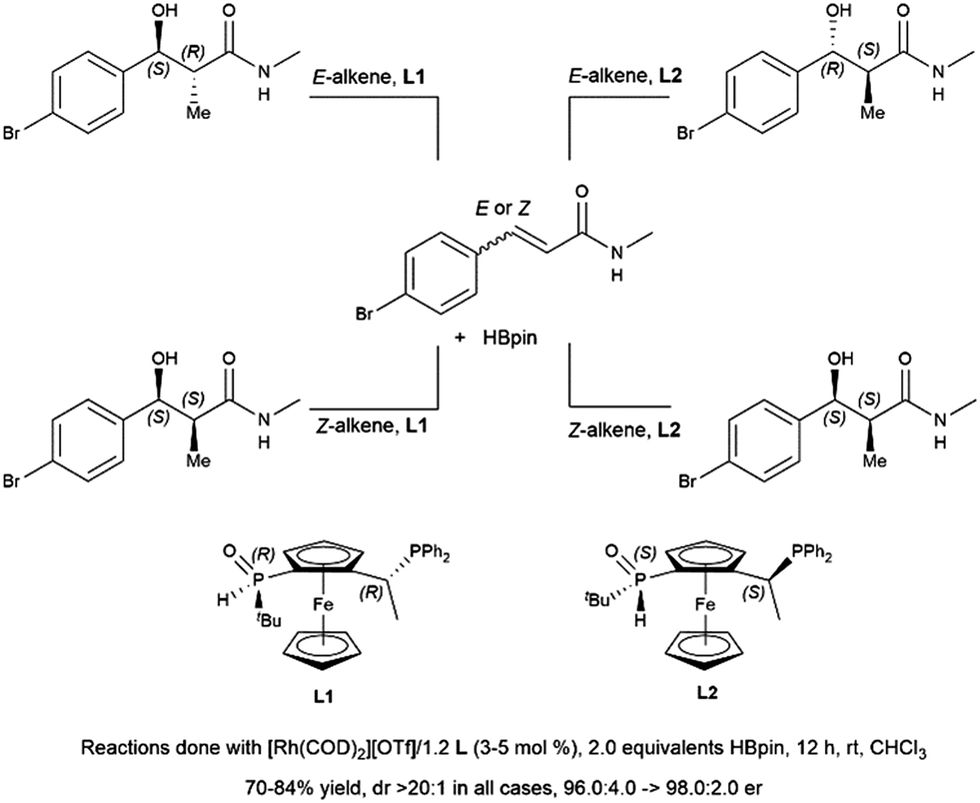 | ||
| Scheme 69 Rhodium-catalysed asymmetric hydroboration of E- and Z-alkene isomers of α,β-unsaturated amides.462 | ||
Teskey and co-workers recently reported the photo-controlled hydroboration of unsaturated carbonyls, providing differing regioselectivity depending on whether the reaction was conducted in presence or absence of light (Scheme 70).469
 | ||
| Scheme 70 Photoswitchable cobalt-catalysed hydroboration of α,β-unsaturated ketones.469 | ||
Copper(I) catalysts have been employed in the hydroboration of unsaturated carbonyls. For example, the addition of diethylborane to an α,β-unsaturated ketone was carried out as part of a one pot hydroboration/aldol cascade reaction (Scheme 71).470 The use of borane instead of silane resulted in improved selectivity for the syn-product when acyclic enones are used and for anti-products in cyclic enones. A similar procedure has been employed involving the 1,4-hydroboration of unsaturated esters and addition to ketimines.473 Achiral systems were able to provide good diastereoselectivity, and excellent enantioselectivity was obtained with a chiral ligand.
 | ||
| Scheme 71 One-pot hydroboration-aldol cascade reaction.470 | ||
A similar protocol was developed for the asymmetric hydroboration of coumarin derivatives using chiral copper(I) catalysts (Scheme 72).471 Upon quenching with water, the reduced ketones are obtained in high er's (over 95:5).
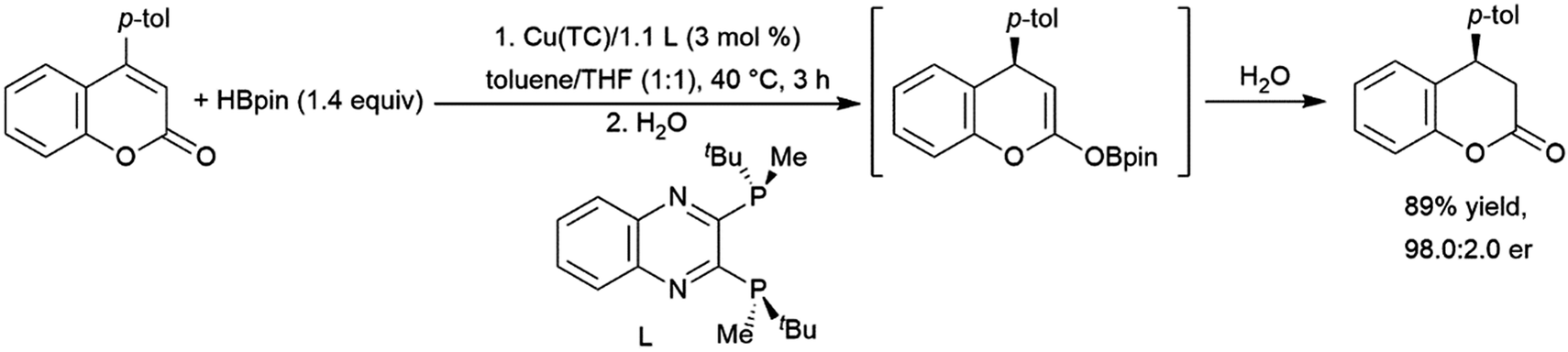 | ||
| Scheme 72 Asymmetric hydroboration of coumarin derivatives.471 | ||
The aldol cycloreduction of certain enone–ketone substrates with pinacolborane catalysed by a rhodium(I) complex results in carbon–carbon bond formation, closing a 5- or 6-membered ring (Scheme 73).474 While with some substrates the same reaction can be accomplished with catecholborane without the need for a catalyst, the use of pinacolborane and a rhodium catalyst increased the yield in the case of a phenyl-substituted substrate. The near-racemic mixture implies the ring closing step occurs via a boron-enolate intermediate as opposed to a rhodium-enolate.
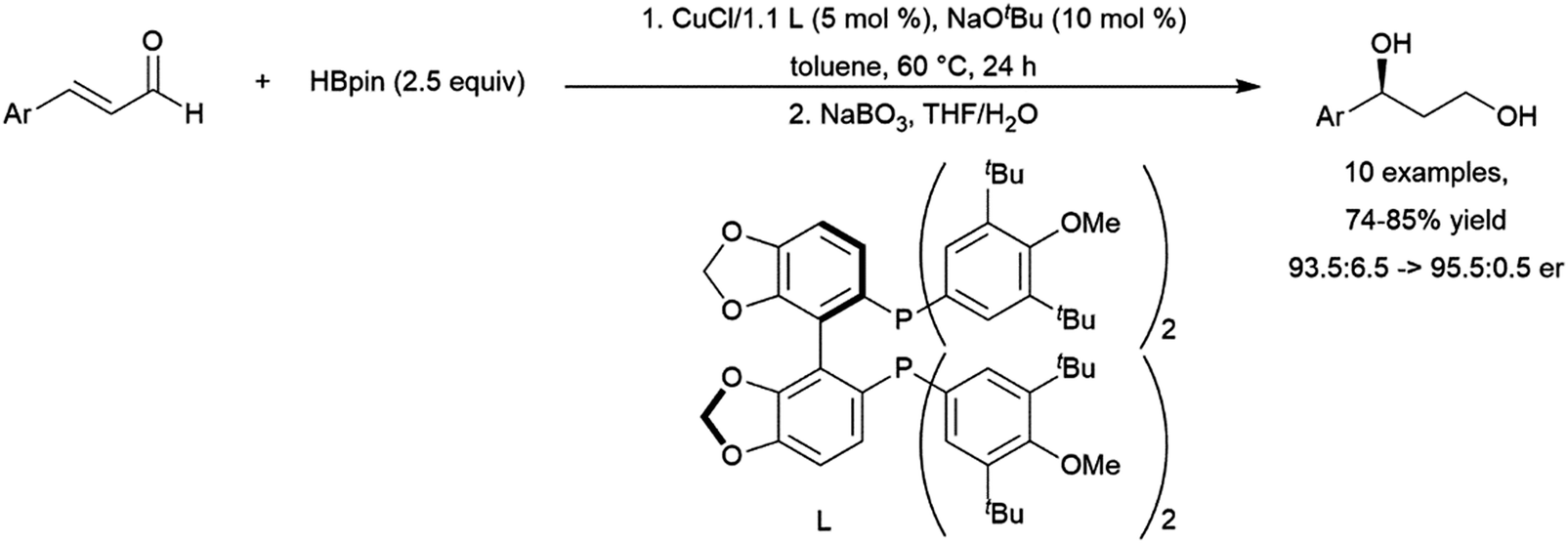 | ||
| Scheme 73 Bis-hydroboration of an α,β-unsaturated aldehyde.472 | ||
Yun and co-workers have reported the synthesis of chiral β-hydroxy pinacolboronates by asymmetric hydroboration of α,β-unsaturated aldehydes.472 The reaction proceeds through initial 1,2-reduction of the aldehyde, followed by the asymmetric hydroboration of the aryl alkene product (Scheme 74).
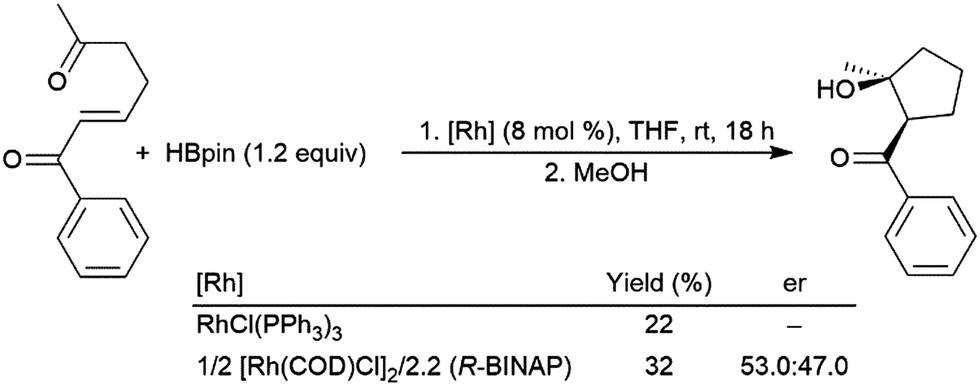 | ||
| Scheme 74 Rhodium-catalysed aldol cycloreduction of an enone-ketone.474 | ||
Gade and co-workers have developed asymmetric hydroboration of N,N-disubstituted α-amino ketones and chloroalkyl ketones. Use of basic conditions with α-, β-, γ-chloroketones, and chloro keto ethers caused the hydroboration products to cyclize and produce chiral heterocyclic compounds in good yields (Scheme 75).475 A manganese analogue of this iron catalyst precursor was also found to be effective for the asymmetric hydroboration of ketones.467,477
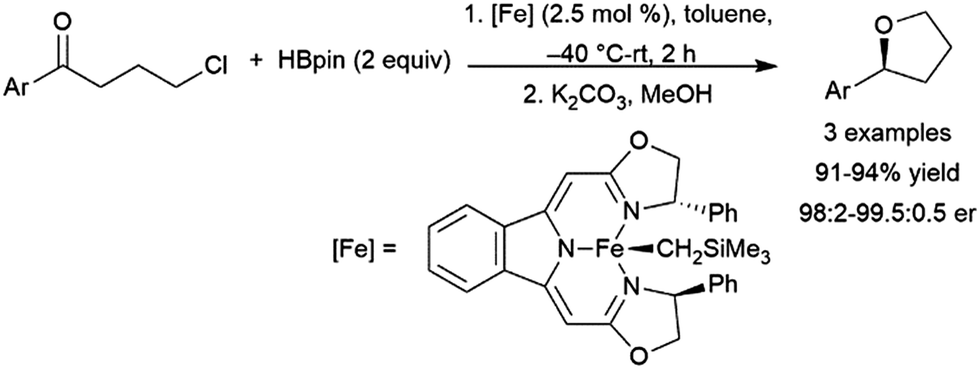 | ||
| Scheme 75 Generation of chiral heterocyclic compounds via hydroboration.475 | ||
4.17 Esters, amides, acyl silanes, carboxylic acids, and carbonates
Ethyl acetate has been shown to undergo catalytic hydroboration with 2 equivalents of catecholborane using molybdenum catalysts, resulting in the formation of EtOBcat and presumably, the loss of MeOBcat.325,326 In a similar fashion, manganese476,478,479 and ruthenium480,481 catalysts were able to reduce carboxylic acids to generate the equivalent alkylboronate and O(Bpin)2. One of the manganese systems was also found to reduce carbonates, resulting in the loss of MeOBpin and the formation of alkylboronic acid esters (Scheme 76).476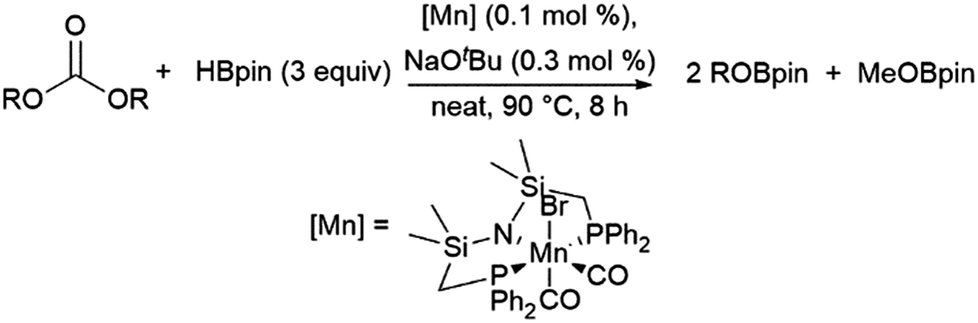 | ||
| Scheme 76 Manganese-catalysed hydroboration of carbonates.476 | ||
The same catalyst system was also found to be effective for the reduction of carboxylic acids, generating the corresponding boron-protected alcohol, O(Bpin)2, and hydrogen (Scheme 77).476 Similarly, a vanadium(III) catalyst was able to promote the deoxygenation of esters and carboxamides.482 The deoxygenation of amides can be achieved using yttrium,483 zirconium,484 and manganese485 catalysts, and a zinc catalyst was also found to selectively reduce an isocyanate group to a N-boryl formamide, a N-,O-bis(boryl)hemiaminal, or a N-borylmethylamine using 1, 2, or 3 equivalents of HBpin respectively.486 Deoxygenative reduction of amides through use of hydroboration, as well as other methods, has been recently reviewed.487
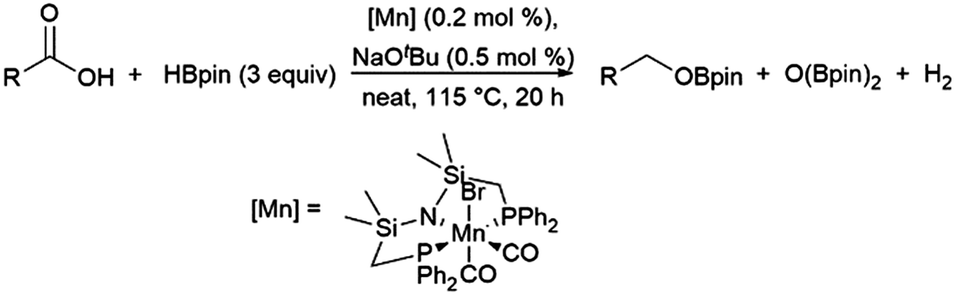 | ||
| Scheme 77 Manganese-catalysed hydroboration of carboxylic acids.476 | ||
Riant, Leyssens, and co-workers have demonstrated the asymmetric reduction of acylsilanes to generate the corresponding chiral α-hydroxysilanes using a copper(II)-phosphine catalyst and pinacolborane.488
A nickel(II) iminophosphinite pincer complex has been utilized in the catalytic reduction of primary and secondary amides to their corresponding boryl amines.410 Hydroboration of esters was complicated by intramolecular boryl group transfer from the initially reduced carbonyl group to the alkoxy group, generating aldehydes in addition to boryl ethers. The hydroboration of N-methylsuccinimide proceeded cleanly with the addition of pinacolborane to just one carbonyl group.
4.18 Thioketones
There is one paper describing the rhodium-catalysed hydroboration of thiocarbonyls, forming the expected thioboronate esters and generating trace amounts of borane degradation products and thiols.489 Nonetheless, yields were improved when compared to the uncatalysed hydroboration of the same substrate with 9-BBN.4.19 Nitriles
Uncatalysed addition of HBpin to nitriles proceeds slowly, adding one equivalent of borane.490 The monohydroboration can also be effected more efficiently using 9-BBN and a ruthenium catalyst.491 It has been demonstrated that several molybdenum imido complexes could catalyse the addition of two equivalents of catecholborane to a variety of nitriles (Scheme 78)325,326 and that a molybdenum nitride could even convert N2 to ammonia.492 Titanium(IV) amidophosphine-borane complexes were found to add two equivalents of pinacolborane to nitriles.321 Under their reaction conditions, ketones, alkenes, and alkynes underwent hydroboration selectively over nitriles. More recent publications highlight the ability of ruthenium,493,494 osmium,495 manganese,479,485,496 nickel,497 cobalt,123,498,499 silver,500 a MOF-based cobalt system,389 iron,501 and an iron–indium502 catalyst system to catalyse nitrile bis-hydroboration. In one case the diborylamines were shown to subsequently react with aldehydes in situ, providing an elegant route from nitriles to aldimines.498 The iron–indium system was also found to catalyse the addition of one equivalent each of HBpin and of silane to acetonitrile, forming a borylsilylamine.502 This chemistry has been recently reviewed.506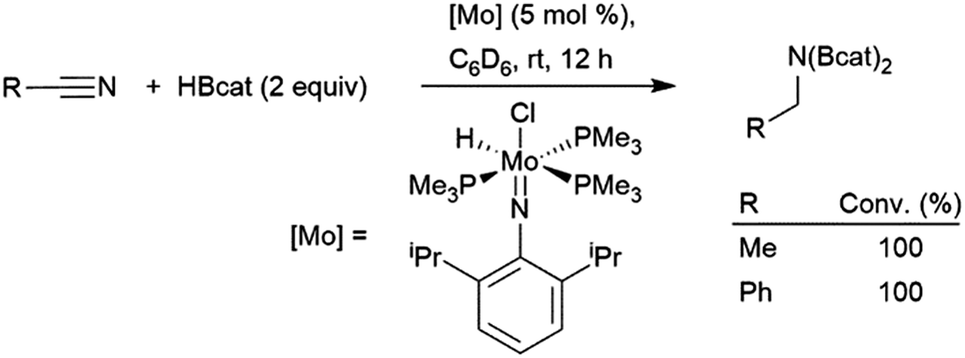 | ||
| Scheme 78 Molybdenum-catalysed bis-hydroboration of nitriles.325,326 | ||
4.20 Imines
The catalytic hydroboration of aldimines and 2-methyl-2-thiazoline using gold(I) precursors has been reported.503 In the hydroboration of aldimines, gold species were found to be more active than RhCl(PPh3)3. For 2-methyl-2-thiazoline, selectivity for the N-borylamine hydroboration product was much higher with gold than with rhodium and was complementary to the uncatalysed hydroboration, which generates only the adduct in a stoichiometric reaction or the ring-opened product with excess catecholborane (Scheme 79).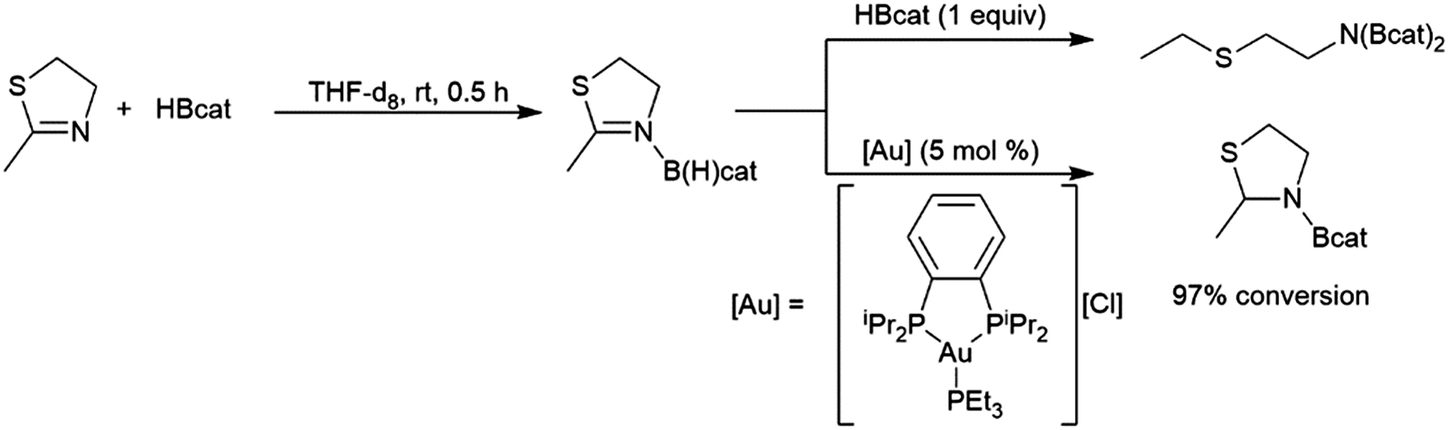 | ||
| Scheme 79 Hydroboration of 2-methyl-2-thiazoline.503 | ||
A series of allyl imines react with two equivalents of HBcat in the presence of various rhodium catalysts forming a mixture of products that favour the terminal hydroboration of the alkene and the imine (Scheme 80).504 The same study also reported the hydroboration of an aldimine derived from 2-thiophenecarboxaldehyde. A ruthenium(II) dimer was found to catalyse the hydroboration of aldimines.404
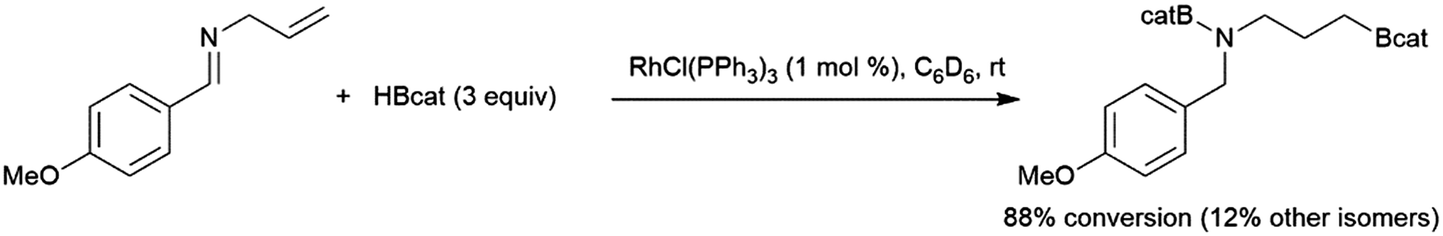 | ||
| Scheme 80 Rhodium-catalysed hydroboration of an allyl imine.504 | ||
Rhodium-catalysed addition of 4-tert-butylcatecholborane (HBcat′) to ketimines gives substrate-dependent mixtures of products arising from multiple hydroborations and β–hydride eliminations (Scheme 81).505
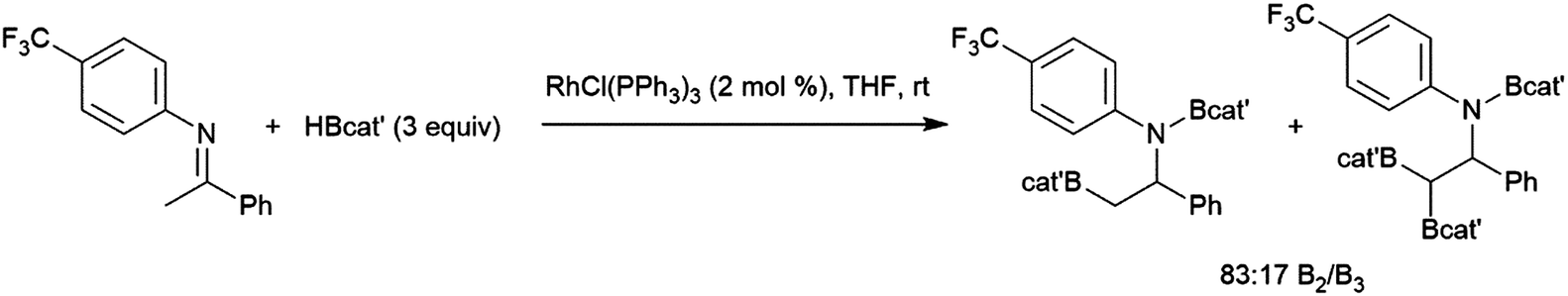 | ||
| Scheme 81 Rhodium-catalysed hydroboration/borylation of an imine.505 | ||
Many more reports have since emerged, often accompanying studies of hydroborations of carbonyl groups. These include catalysis using nickel,409,411 rhenium,407 a cobalt coordination polymer,426 rhodium,424 ruthenium,494,507 iron,501 and zinc.449 Most tend to focus on benzylic aldimines, with only isolated examples of the catalysed hydroboration of imines derived from acetophenone424 and benzophenone.409 More recently, Gade and co-workers presented a broadly applicable method for the asymmetric hydroboration of N-alkyl imines using an iron catalyst (See Scheme 102 for an example).508 Eisen, Tamm, and co-workers have demonstrated the mono-hydroboration of carbodiimides using a hafnium(IV) catalyst.403
4.21 Pyridines
Pyridines can undergo hydroboration with pinacolborane catalysed by rhodium(I) complexes.509 Careful selection of the phosphine ligand provides the 1,2- or 1,4-addition product in good conversion, although the generation of 1,4-addition product was plagued by the formation of unidentified byproducts (Scheme 82). Since this initial report several publications have emerged detailing pyridine hydroboration. Selective 1,2-hydroborations of pyridines and related compounds have been catalysed by zinc,511,512 iron,513 and nickel,514 though selectivity and activity can suffer with ortho-substituted pyridines. A report has also emerged detailing organolanthanide-catalysed 1,2-hydroborations of pyridine derivatives.515 Selective 1,4-hydroborations of pyridines have also been catalysed by ruthenium,516 nickel,517 zinc,449 and trivalent zirconium or hafnium-based MOFs.518 A heterobimetallic iron–copper protocol was found to have enhanced activity and selectivity for the 1,4-hydroboration product when compared to the analogous copper-only reactions.519,520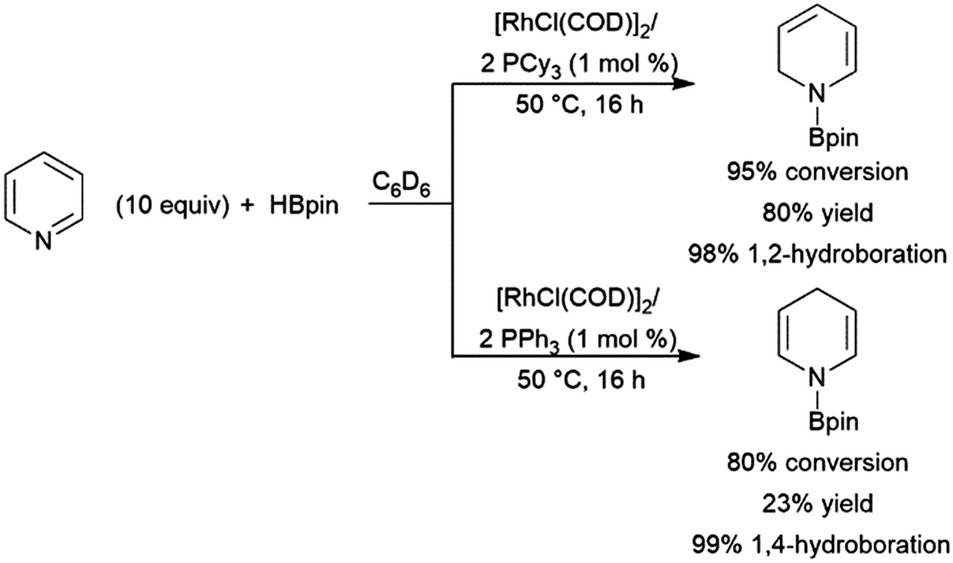 | ||
| Scheme 82 Rhodium-catalysed hydroboration of pyridine.509 | ||
4.22 Carbon dioxide
Recent interest in chemistry involving the reduction of carbon dioxide has led researchers to look into the hydroboration reaction.521 The first transition metal-catalysed catalytic hydroboration of carbon dioxide was reported in 2010. A nickel(II) pincer complex catalyses the reaction of CO2 with 3 equivalents of catecholborane (Scheme 83).510 The reaction is performed under 1 atm of CO2 and in the presence of 500 equivalents of catecholborane, relative to the catalyst. An exceptional turnover frequency of 495 per hour was observed. Since then, several closely related Ni-POCOP systems were also found to be effective in this transformation.522–526 A related nickel system containing a P–Si–P pincer ligand catalysed the reduction of CO2 with HBpin, although it yielded only 13% of the desired MeOBpin product.527 Another P–Si–P pincer ligand system was used with nickel to produce H2C(OBpin)2 in good selectivity.174 Hazari and co-workers found that selectivity could be altered through the choice of nickel or palladium-based catalyst, ligand, borane, and reaction conditions.528 Guan and co-workers also reported a palladium-POCOP catalyst system capable of the efficient and selective reduction of CO2 to MeOBcat and O(Bcat)2.529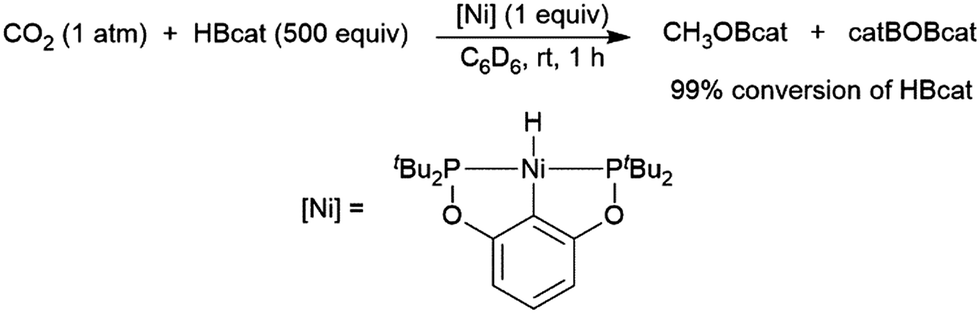 | ||
| Scheme 83 Nickel-catalysed hydroboration of CO2.510 | ||
A copper(I) catalyst system capable of reducing carbon dioxide with HBpin to generate HCO2Bpin, which could be protonated or used in situ as a formylation reagent for primary amines and some secondary amines (Scheme 84) was reported.93 A heterogeneous analogue of this system was shown to be active for the same reaction.530 The catalyst for this system is a MOF which incorporated cationic copper(I) centers and can be readily reused, albeit with slightly diminishing performance. More recently, copper(I) systems based on tripodal phosphine ligands have been employed in the catalytic hydroboration of CO2 using 9-BBN giving mixtures of CH2(O-BBN)2, MeO-BBN, and HCO2BBN in varying selectivity.531,532 A heterobimetallic copper/gold system was also found capable of reducing CO2 using 9-BBN, with good selectivity for the boryl formate derivative, but relatively low turnover numbers.533
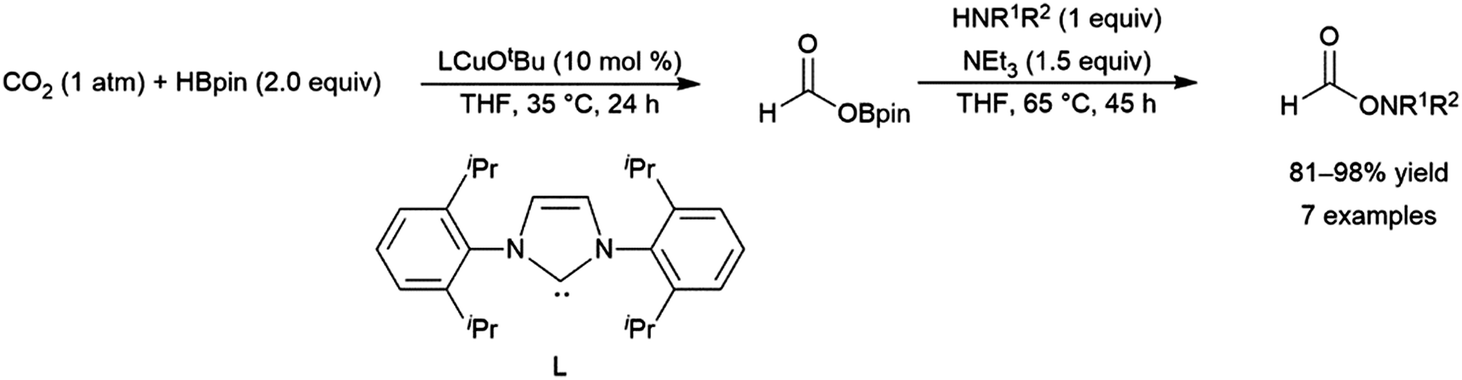 | ||
| Scheme 84 One pot hydroboration–amination reaction of CO2.93 | ||
Ruthenium complexes have also shown catalytic activity in the reduction of carbon dioxide. The first example came in 2012,534 and is a somewhat unique case in that it is proposed to proceed via initial insertion of CO2 into a ruthenium–phosphorus bond and subsequent hydroboration of the pendant carbon–oxygen bond by HBpin. Reaction with a second equivalent of HBpin affords pinBOBpin and CH3OBpin. The reaction was found to be quite sluggish (0.1 turnovers per hour), however 9 turnovers can be achieved in 96 hours at 50 °C. A zinc hydridotriphenylborate catalyst supported by a macrocyclic polyamine has been demonstrated to have similar reactivity, generating a 1:1 mixture of pinBOBpin and CH3OBpin through the hydroboration of CO2 over 16 hours with 10 mol% catalyst.449
The catalytic hydroboration of CO2 using other ruthenium complexes has also been reported.535 While pinacolborane was generally completely consumed during the reaction, mixtures of boron-containing products were consistently observed (Scheme 85). It was later confirmed that formaldehyde reacts with borylformate to give pinBOCH2OCOH.536 Complete consumption of the borylformate product and a larger conversion to pinBOCH2OCOH was observed upon changing the PCy3 ligands to P(c-C5H9)3 ligands.537 Formaldehyde could be trapped as its corresponding imine in up to 74% yield through the addition of 2,6-diisopropylaniline. A Density Functional Theory (DFT) study of this transformation suggested a ruthenium dihydride carbonyl complex (generated through deoxygenation of CO2) as the catalytically active complex responsible for the formation of the C2 species.538
 | ||
| Scheme 85 Ruthenium-catalysed hydroboration of CO2.535 | ||
A cationic ruthenium pincer complex in tandem with an alkali metal salt was found to be capable of the transformation of CO2 to HCO2Bpin in selectivity as high as 76%.539 The complex was found to be inactive in the absence of the salt, which is proposed to coordinate to HBpin, activating it for addition to a pendant CO bond in a ruthenium formate intermediate. It was later reported that zinc hydride catalysts were also capable of the selective generation of HCO2Bpin from CO2.540 In contrast, the use of HBcat generated MeOBcat, while the use of 9-BBN produced a mixture of H2C(BBN)2 and MeOBBN.
In an interesting case of ligand-based catalysis, Song and co-workers have demonstrated the ability of ruthenium and zinc diazafluorenyl ligands to add pinacolborane and catecholborane to carbon dioxide.541 A computational study suggested that the reaction proceeds through a hydride-shuttle mechanism, in which the metal is essentially a spectator while reactivity takes place at the diazafluorenyl C–H bond.542
Palladium catalysts have also shown promise in this transformation. In an elegant study examining the carboxylation of alkynes, a palladium(II) pincer complex was found to be an incredibly active catalyst for the selective reduction of CO2 to HCO2Bpin, with turnover numbers of up to 63500 being observed.543 A molybdenum system was able to catalyse the reduction of CO2 to H3COBpin and O(Bpin)2 using HBpin.544 Upon quenching with water, a 58% yield of methanol was obtained. Several Mn(I) catalysts have also been shown to selectively generate methoxyboranes upon hydroboration of CO2476,545 and the reaction mechanism for this has been elucidated using DFT calculations.546
Using Ni and Pd catalysts, Hazari and co-workers studied the effect of varying experimental conditions on the nature of products obtained from the hydroboration of CO2 with various boranes.528 The authors were able to selectively form products of two (HCO2BR2), four (H2C(OBR2)2), and six (MeOBR2) electron reductions of CO2 using the same catalyst. Lewis acid additives were found to promote greater reduction of CO2. Likewise, utilization of catecholborane was found to promote further reduction of CO2, possibly due to its stronger Lewis acidity (compared to HBpin) and reduced steric bulk (compared to 9-BBN).
Bis-hydroboration of CO2 using HBpin, HBcat, or 9-BBN, and an iron catalyst has been reported.420,547 Most interestingly, they found that the bis-boryl species R2B–O–CH2–O–BR2 are excellent methylene transfer reagents, allowing for the formation of new C–N, C–O, C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and CN bonds in two-step, one pot protocols. In one such case, L-erythrulose was prepared from CO2 in a chemo-enzymatic cascade.548 A cobalt catalyst derived from Co(acac)3 and Na(HBEt3) was found to reduce CO2 with a variety of boranes, giving mixtures of products.549 With Me2S–BH3 for example, (MeOBO)3 is selectively generated.
C, and CN bonds in two-step, one pot protocols. In one such case, L-erythrulose was prepared from CO2 in a chemo-enzymatic cascade.548 A cobalt catalyst derived from Co(acac)3 and Na(HBEt3) was found to reduce CO2 with a variety of boranes, giving mixtures of products.549 With Me2S–BH3 for example, (MeOBO)3 is selectively generated.
The nature of products from the reduction of CO2 using a copper catalyst system and HBpin could be altered through the nature of a heterobimetallic Cu/M catalyst system.550 While some catalyst systems generated HCO2Bpin, others gave primarily HOBpin or O(Bpin)2. The authors propose that while copper reduces CO2 to HCO2Bpin, the other metal can catalyse further reduction, resulting in the loss of a carbonyl oxygen as O(Bpin)2.
Iridium CNP pincer complexes were able to selectively generate HCO2Bpin and MeOBcat (and O(Bcat)2) through the hydroboration of CO2 with the respective boranes.551 Rates were enhanced with the addition of water, prompting the authors to suggest that water plays a role in activating the catalyst.
4.23 Sulfoxides and nitro compounds
In chemistry closely related to the reduction of CO2, which can involve the loss of an oxygen atom as (RO)2BOB(OR)2, sulfoxides can be deoxygenated through transition metal-catalysed hydroboration. The reaction was first reported using rhodium(I) catalysts and catecholborane.552 Good conversion could also be achieved in the absence of a catalyst, but an excess of borane was required. Subsequent studies showed that rhenium553,554 and molybdenum554–557 catalysts could also provide the corresponding thioethers in good yields. One advantage to molybdenum and rhenium catalysts is that they are less active as catalysts for the hydroboration of alkenes or alkynes. This allows the selective deoxygenation of sulfoxides with these functional groups (Scheme 86). In a similar fashion, the nitro group in 4-nitroanisole could be reduced to the diboryl amine using an excess of catecholborane and a rhodium(I) catalyst,558 to the monoboryl amine using an excess of HBpin and a cobalt(II) catalyst,559 or to the amine using HBpin and chromium(0) species.560 | ||
| Scheme 86 Rhenium-catalysed deoxygenation of sulfoxides.553,554 | ||
4.24 Other
Partial hydroboration of the strained hydrocarbon quadricyclane has been reported to give norbornenol as part of a mixture of products, with the dominant product being norbornadiene, an isomer of quadricyclane (Scheme 87, top).561 In a similar fashion, biphenylene can undergo ring-opening reduction using a variety of reducing agents including pinacolborane, formally cleaving a σ-bond to form a borylated 2,2′-biphenyl species (Scheme 87, bottom).562 Ring-opening reductions can be performed on cyclopropanes.565 | ||
| Scheme 87 Hydroboration of strained carbon–carbon bonds in quadricyclane (top)561 and biphenylene (bottom).562 | ||
Ring-opening reactions of epoxides have been demonstrated with iron(II)566 and nickel(II)567 catalyst precursors. While the iron system is believed to break the C–O bond in a ligand-assisted reaction, the nickel system isomerizes the epoxide to the corresponding aldehyde, which then undergoes hydroboration. An yttrium complex was found to catalyse the cleavage of Si–O bonds via a hydroboration, which proceeds through a pinacolborane adduct of an yttrium alkoxide complex.568 A ruthenium catalyst has been shown to cleave C–O bonds in ethers by hydroboration, under relatively harsh conditions (Scheme 88).563
 | ||
| Scheme 88 Ruthenium-catalysed cleavage of ethers.563 | ||
Electron-poor aromatic rings were shown to undergo 1,4-hydroboration using a carbene–borane adduct and an iridium(III) catalyst in the presence of ethyldiisopropylamine and light (Scheme 89).564 A number of other boranes (including pinacolborane) and borane-Lewis base adducts were ineffective in this transformation.
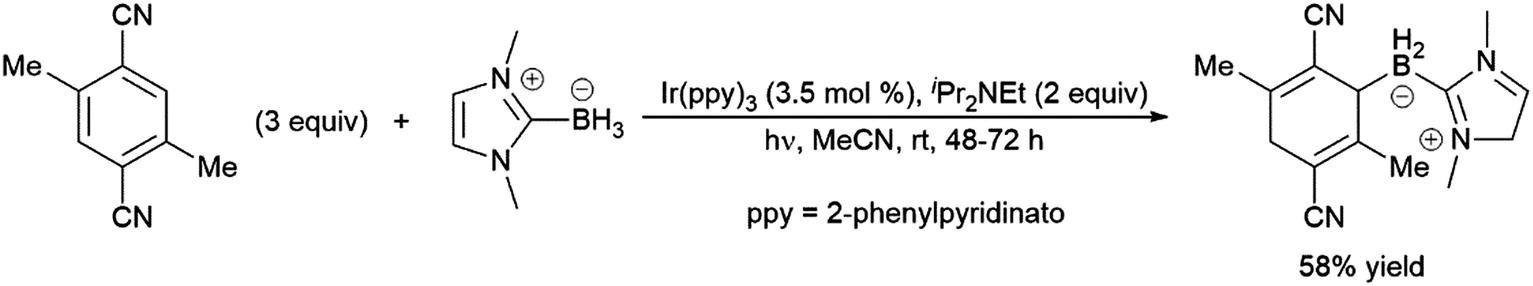 | ||
| Scheme 89 Hydroboration of an electron-poor aromatic ring using a carbene–borane adduct and an iridium catalyst.564 | ||
Phosphaalkenes were found to initially form adducts with BH3; however, when excess Me2S–BH3 is added, the adduct undergoes hydroboration (Scheme 90, bottom). Reaction of catecholborane with phosphaalkenes catalysed by RhCl(PPh3)3 gives the hydroboration product (Scheme 90, top).569 With a 1,2-disubstituted phosphaalkene, significant amounts of the hydrolyzed reduction product is also observed.
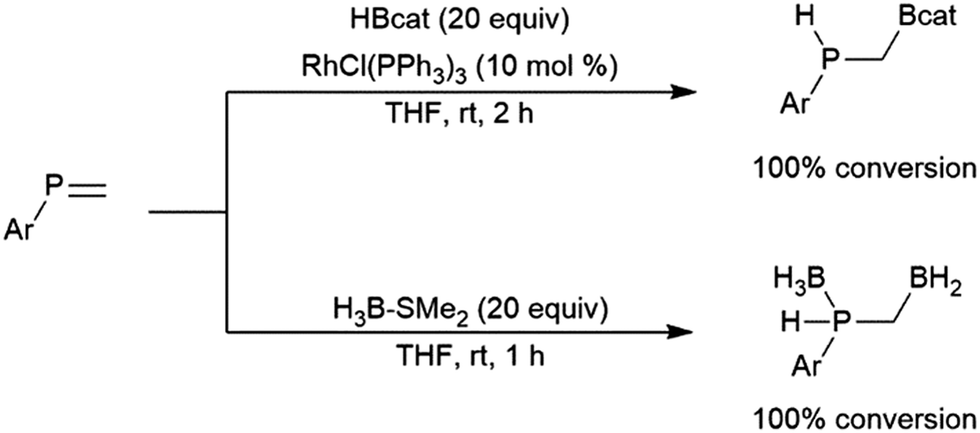 | ||
| Scheme 90 Hydroboration of phosphaalkenes.569 | ||
5 Applications to synthesis
One of the major advantages of the hydroboration reaction is the synthetic versatility of the organoborane product. Though traditionally these products are converted to their corresponding alcohols via oxidative workup, the boryl group can be utilized to install a variety of functional groups. In a one-pot fashion, hydroboration products of dienes210,218 and enynes210,234 could be added to aldehydes, giving homoallylic and homoacetylenic alcohols respectively (Scheme 91).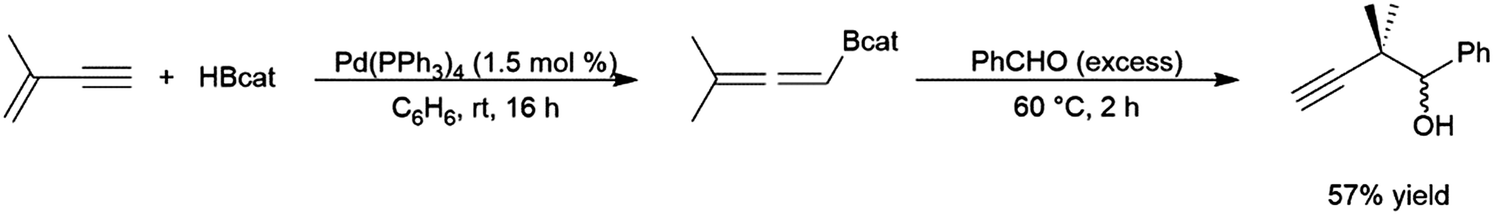 | ||
| Scheme 91 Hydroboration as a step in the synthesis of a homoacetylenic alcohol.210 | ||
Merlic and co-workers prepared a series of bis-vinylboronate species that could undergo oxidative cyclization using palladium(II) catalysts (Scheme 92).341
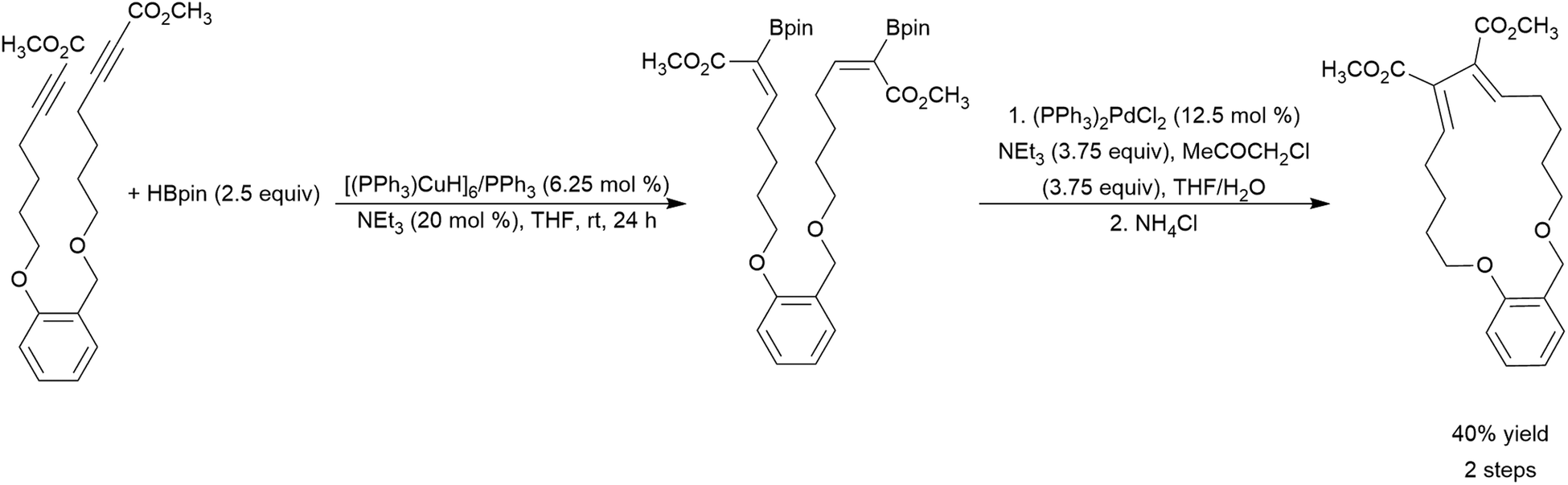 | ||
| Scheme 92 Hydroboration-oxidative cyclization of a di-yne.341 | ||
A reductive coupling reaction involving propargylic alcohols, catalysed by a ruthenium complex has been reported (Scheme 93).570 The reaction proceeds through initial formation of a ruthenium-allylidene, which undergoes hydroboration with HBpin. The authors hypothesize that trace oxygen generates a radical while eliminating the boryl group. Deprotonation and rearrangement of the radical leads to a ruthenium-alkyne radical, which couples with a second such molecule. The ensuing species is protonated, cleaving the ruthenium–carbon bonds.
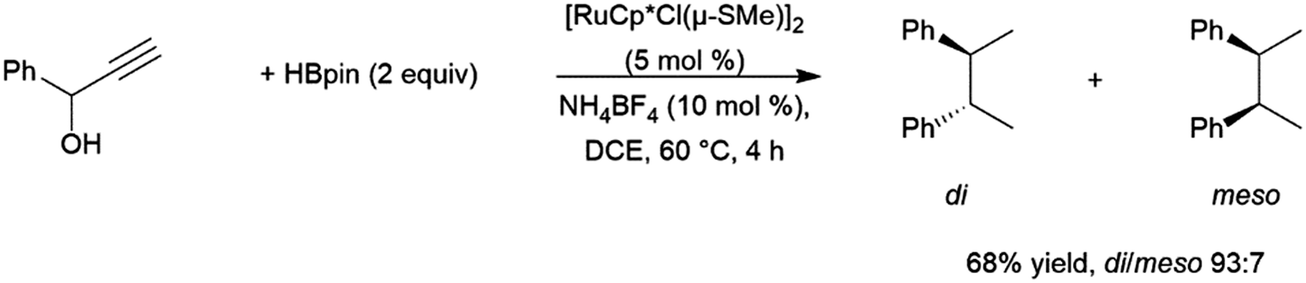 | ||
| Scheme 93 Hydroboration-homocoupling of a propargylic alcohol.570 | ||
5.1 Reactions of boronate esters with organometallic reagents
 | ||
| Scheme 94 A hydroboration-homologation-oxidation reaction sequence.571 | ||
Oxidation of the α-chloroboronate ester with basic hydrogen peroxide provided the corresponding alcohol. An alternative one carbon homologating agent, TMSCHN2, has also been successfully used to homologate organoboronate esters.575 The same reaction has been reported in the reverse order, a one-pot methylenation–hydroboration reaction where a carbonyl group is converted to a terminal alkene using TMSCHN2 under Rh-catalysis, then catecholborane is added to hydroborate the terminal alkene.576
Multiple homologations occur when LiCH2Br or LiCH2I are employed in the reaction,577 thus LiCH2Cl is the reagent of choice for the homologation reaction. Multiple homologation reactions using chiral lithium reagents have been employed to elegantly generate extended chain asymmetric boronate esters and alcohols.574 The study showed that the stepwise addition of chiral lithium salts to a chiral biphenylboronate could generate a tailor-made extended chain boronate ester, and the boronate ester could then be readily converted to alcohols or protected alcohols (Scheme 95).
 | ||
| Scheme 95 Multiple homologation reaction sequence, starting with the product of a chiral hydroboration.574 | ||
A related carbon–carbon bond forming reaction was used to prepare amino acid derivatives.578 In this case the boronate ester must first be converted to a more reactive 9-BBN derivative, prior to homologation and chiral protonation giving the chiral imine ester compound.
 | ||
| Scheme 96 Zinc transmetallation-alkylation reaction sequence of a boronate ester.149 | ||
Alkylmagnesium species are more nucleophilic and thus potentially more synthetically useful than organozinc compounds, thus there has been an effort to prepare them from organoboronate esters. A method developed for the exchange of magnesium for trialkyl boranes580 was later modified to include pinacolboronate esters.156 These organomagnesium salts were then used in palladium and copper catalysed cross-coupling reactions, 1,2-addition reactions, and alkylations (Scheme 97).
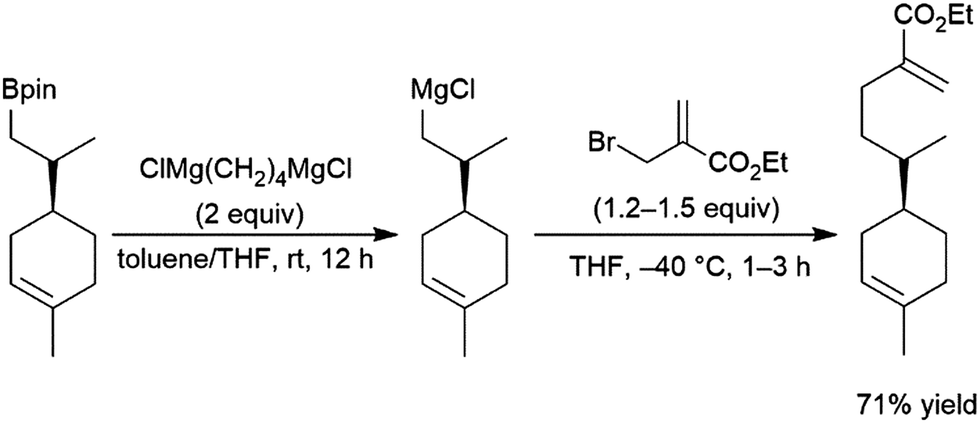 | ||
| Scheme 97 Magnesium transmetallation-alkylation reaction sequence.156 | ||
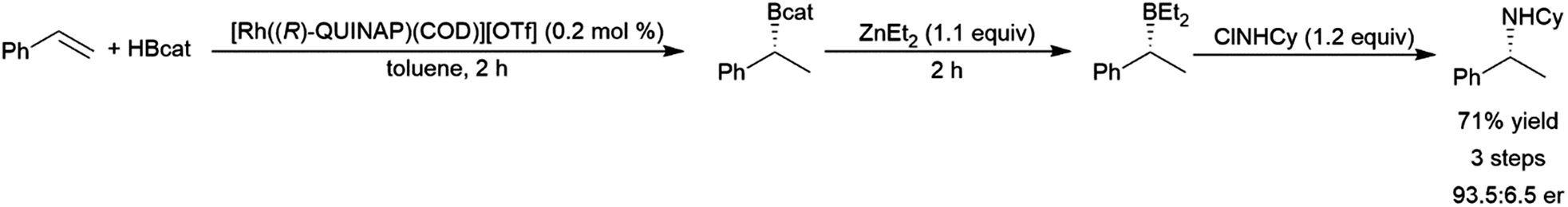 | ||
| Scheme 98 Hydroboration–amination reaction sequence.130 | ||
An alternative approach to the same reaction has been published, where the boronate ester is first converted to the RBCl2 derivative through reaction with BCl3, prior to addition of benzyl azide to generate the amine (Scheme 99).583 A very similar intramolecular amination was reported, where an organic azide containing a pendant boronate ester group reacts following the conversion of the boronate ester to a difluoroborane.584
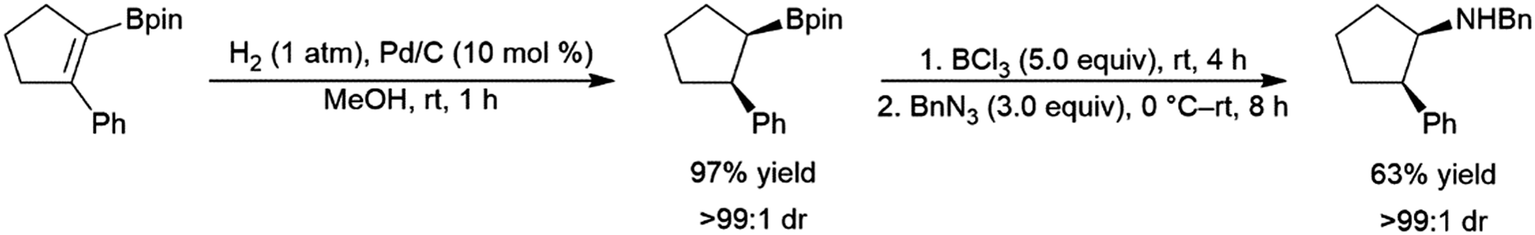 | ||
| Scheme 99 Hydrogenation–amination of a vinyl boronate ester.583 | ||
Using rhodium-catalysed hydroboration, alkylboronate esters were prepared and were found to be suitable radical precursors for addition to methyl acrylate. In addition to a protected cyclohexenol derivative (Scheme 100), (S)-(−)-limonene and norbornene were suitable starting materials for the multistep synthesis.155
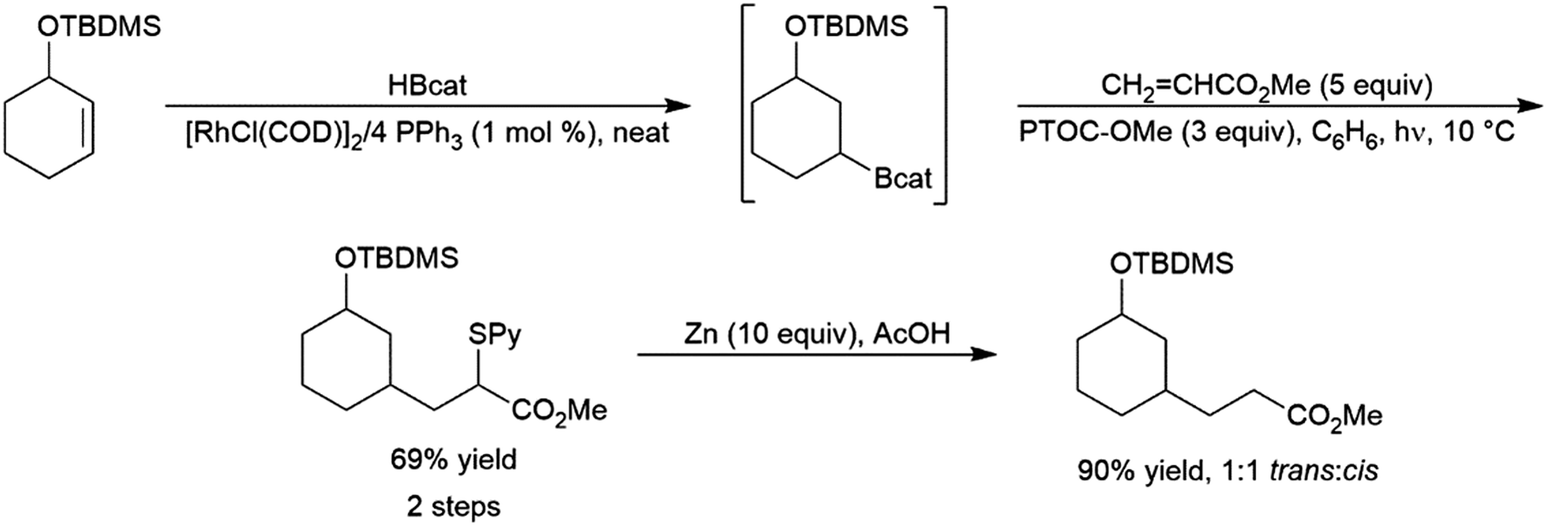 | ||
| Scheme 100 Addition of methyl acrylate to an alkylboronate ester (PTOC-OMe = N-methoxycarbonyloxypyridine-2-thione).155 | ||
5.2 Synthesis of natural products and medicinal compounds
The asymmetric hydroboration of aryl alkenes has been exploited in the synthesis of Ibuprofen™ and Naproxen™, two popular anti-inflammatory drugs. An asymmetric hydroboration, followed by homologation gave Ibuprofen™ in 74% overall yield, with 96.5:3.5 er (Scheme 101).571 A similar protocol gave Naproxen™ (Scheme 101, right) in 66% yield with 94:6 er.116 An attempt was made to use a stereoselective rhodium catalysed hydroboration in the preparation of (+)-macbecin I, a compound with antibiotic and antitumor properties, however the predominant isomer was not the desired product.585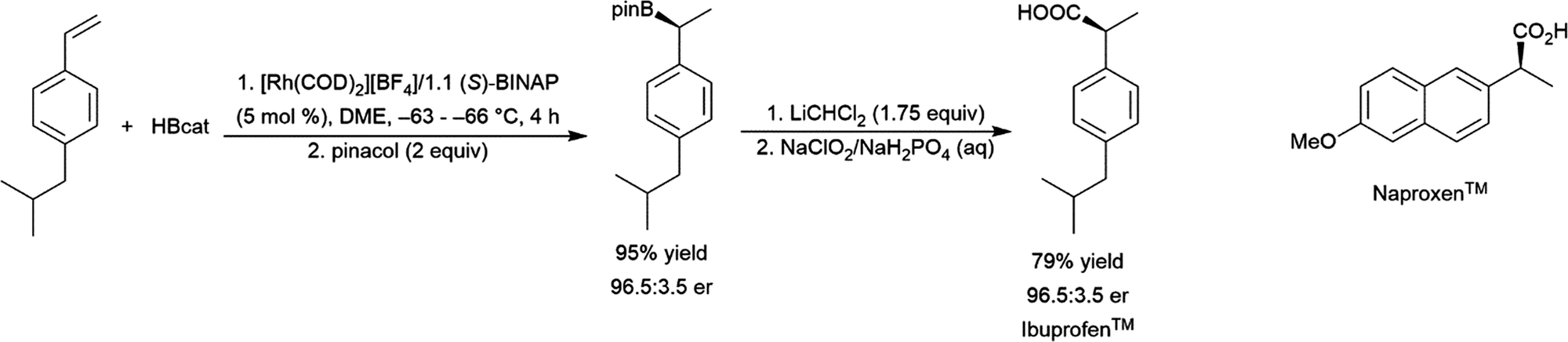 | ||
| Scheme 101 Preparation of Ibuprofen™ from its corresponding styrene.571 | ||
In a similar fashion, the asymmetric hydroboration of imines was used in the synthesis of (R)-Fendiline and (R)-Tecalcet, compounds showing promise in the treatment of coronary heart disease and hyperparathyroidism respectively (Scheme 102).508
 | ||
| Scheme 102 Preparation of (R)-Fendiline via asymmetric hydroboration of an imine.508 | ||
 | ||
| Scheme 103 Preparation of sertraline, including kinetic resolution of the starting alkene through asymmetric hydroboration.582 | ||
 | ||
| Scheme 104 Preparation of (+)-catalponol through hydroboration.586 | ||
:6 diastereoselectivity.
 | ||
| Scheme 105 Preparation of a synthon of Lonomycin A, the hydroboration–oxidation of an alkene.147 | ||
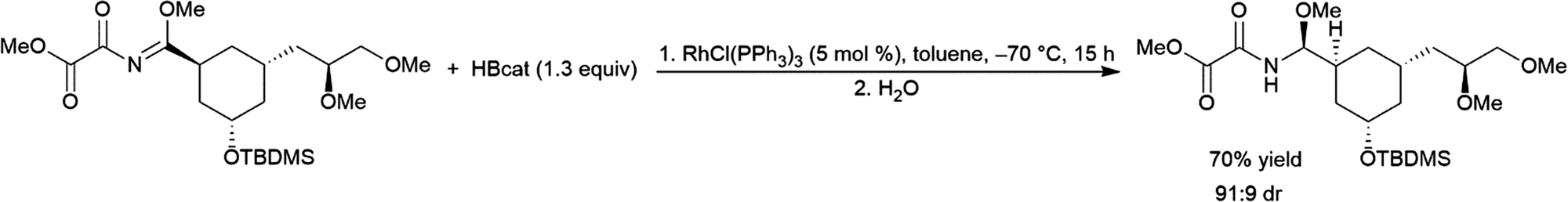 | ||
| Scheme 106 The hydroboration of an N-acyl imidate included in the synthesis of pederin.587 | ||
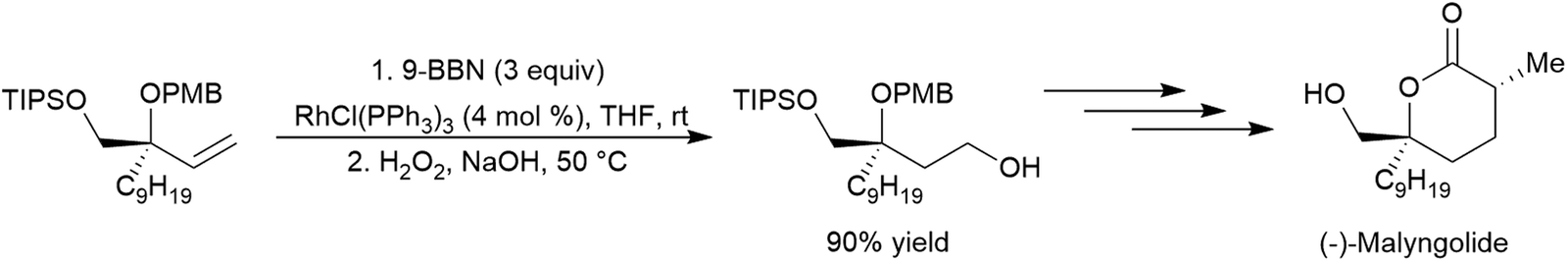 | ||
| Scheme 107 Catalytic hydroboration of an alkene as part of the synthesis of (−)-malyngolide.588 | ||
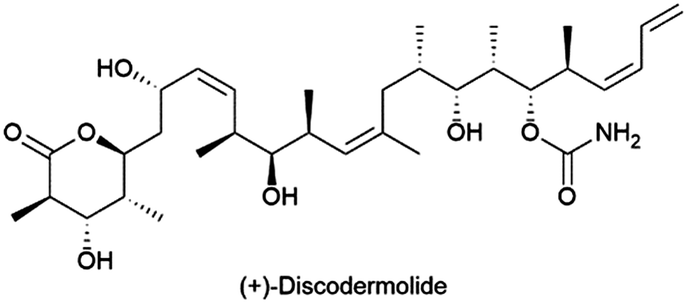 | ||
| Fig. 2 (+)-Discodermolide. | ||
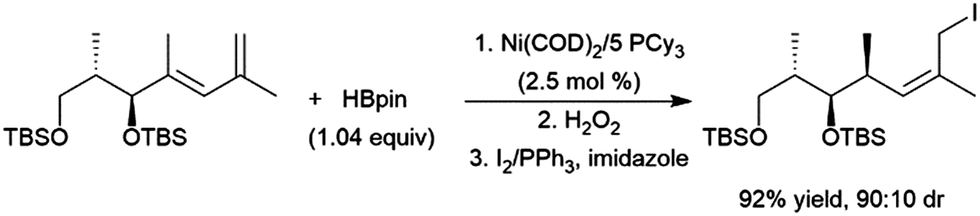 | ||
| Scheme 108 Hydroboration of a diene as a step in the preparation of an intermediate in the preparation of (+)-discodermolide.221 | ||
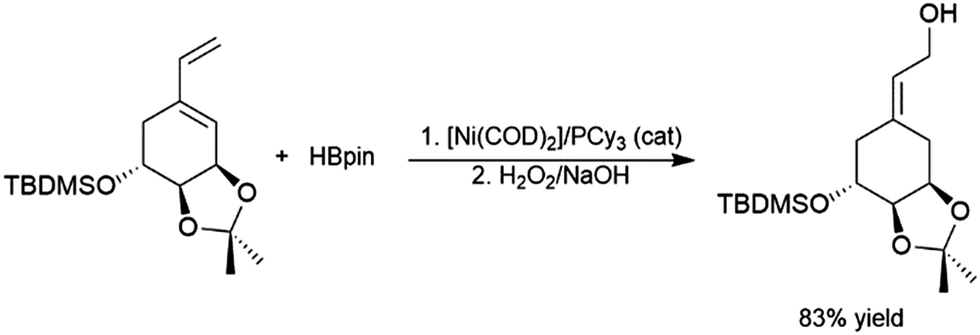 | ||
| Scheme 109 Nickel-catalysed hydroboration-oxidation in the preparation of an intermediate en route to vitamin D3.590 | ||
6 Comparison with other methods
6.1 Other hydroborations
The uncatalysed hydroboration reaction has been mentioned extensively in this chapter and is obviously closely related to the transition metal-catalysed variant. The biggest downside to the uncatalysed version is that more reactive boranes and/or harsher reaction conditions are required in general, and the selectivity can be somewhat limited. Catalysis by transition metals can offer complementary and often superior selectivity, and also asymmetric induction through the use of chiral catalysts. Catalysis can allow for more stable boranes to be used, facilitating isolation (for example, pinacolborane-derived products are often stable to column chromatography).Other catalysts have also been used in catalytic hydroborations, including lanthanides,76,77,515,591 magnesium592–596 and, more recently, frustrated Lewis pairs.597–602 While these catalysts are effective and often quite selective, the lack of participation from d-orbitals makes selectivity generally similar to uncatalysed hydroborations.
Recent reviews have covered alkali and alkaline earth-catalysed hydroborations,603 organolanthanide- and organoactinide-catalysed hydroborations,604 and aluminum-catalysed hydroborations.605
6.2 Reactions of B2(OR)4
Recent years have seen a rapid expansion in the exploration of the chemistry of B2(OR)4 reagents, in particular B2pin2. Among the most relevant developments include transition metal-catalysed diboration and boryl addition reactions. This area has recently been reviewed.606Diborations catalysed by zero-valent platinum complexes tend to proceed via a catalytic cycle involving oxidative addition of the B–B bond in a manner very similar to rhodium-catalysed hydroborations.607–609 Copper-catalysed diborations are also common and the metathesis pathway is similar to that proposed for copper-catalysed hydroborations.610–612 The most obvious difference between diboration and hydroboration is the functionalization at two sites; in 1,2-diborations of simple monosubstituted alkenes, a chiral center is always generated. A comparison of asymmetric cationic rhodium-catalysed generation of 1,2-diboryl alkenes from 1,2-diboration of alkenes and from dihydroboration of alkynes found far superior selectivity for the dihydroboration reaction.375 Although diboration reactions are somewhat new in comparison to hydroborations, they have been studied quite extensively and good asymmetric inductions have been achieved in some cases. For a more detailed description and comparison of the syntheses of multi(boronate) esters from diboration of hydroboration please see recent reviews from Xia, Wu and co-workers,370 and Salvado and Fernandez.613
Boryl addition reactions can result in a formal hydroboration. These reactions involve the addition of an electrophilic boryl group followed by quenching with a proton source, which is often methanol. Thus, selectivity in these reactions can often be complementary to that observed in hydroborations. For example, the boryl addition to α,β-unsaturated carbonyls can give the β-boryl carbonyl (Scheme 110).614
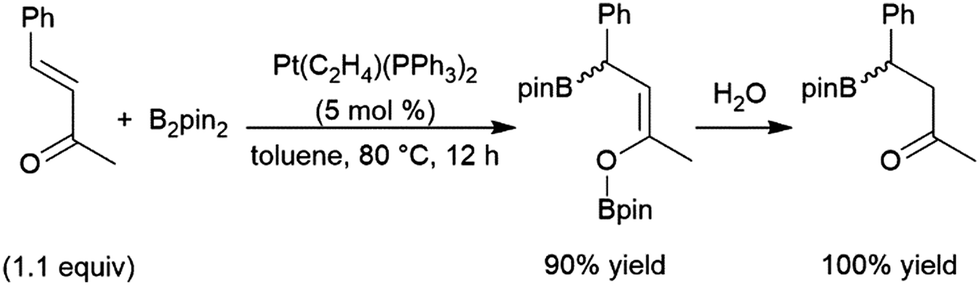 | ||
| Scheme 110 Platinum-catalysed diboration-protonation of an α,β-unsaturated ketone.614 | ||
6.3 Hydrosilylation
Silanes are like boranes in a number of ways, perhaps not surprising given their diagonal relationship on the periodic table. Like boranes, silanes possess an E–H bond that is polarized towards the hydrogen atom. Similar catalyst systems to those used in hydroborations are frequently used in hydrosilylations.615–617 As is the case with hydroborations, side reactions can occur, including dehydrogenative silylation and hydrogenation. Complete regioselectivity and good asymmetric induction have been elusive.6187 Challenges and future directions
The main challenges of hydroboration reactivity include the water sensitivity of reagents and products, competitive reactions that reduce the yield of the desired products (e.g., dehydrogenative borylation),619 and competing catalytic pathways achieved by other boranes formed in situ.89,620 Development of methodology which reduces or prevents the occurrence of these challenges would be a beneficial development to this field. Future directions will include continued development of earth-abundant catalysts for reactions with boron reagents, including B2(OR)4 species as outlined in the comparison with other methods section.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for the Natural Sciences and Engineering Research Council of Canada for funding. This article is dedicated to the many talented scientists we have had the privilege of working with in the Wild Toads Research Laboratory.Notes and references
- H. C. Brown, Hydroboration, W. A. Benjamin, New York, NY, 1962 Search PubMed.
- J. A. Soderquist and A. Negron, Org. Synth., 1992, 70, 169–176 CrossRef CAS.
- E. F. Knights and H. C. Brown, J. Am. Chem. Soc., 1968, 90, 5281–5283 CrossRef CAS.
- E. F. Knights and H. C. Brown, J. Am. Chem. Soc., 1968, 90, 5280–5281 CrossRef CAS.
- H. C. Brown, E. F. Knights and C. G. Scouten, J. Am. Chem. Soc., 1974, 96, 7765–7770 CrossRef CAS.
- H. C. Brown, A. K. Mandal and S. U. Kulkarni, J. Org. Chem., 1977, 42, 1392–1398 CrossRef CAS.
- J. A. Soderquist and H. C. Brown, J. Org. Chem., 1981, 46, 4599–4600 CrossRef CAS.
- R. Wilczynski and L. G. Sneddon, J. Am. Chem. Soc., 1980, 102, 2857–2858 CrossRef CAS.
- R. Wilczynski and L. G. Sneddon, Inorg. Chem., 1981, 20, 3955–3962 CrossRef CAS.
- R. Wilczynski and L. G. Sneddon, Inorg. Chem., 1982, 21, 506–514 CrossRef CAS.
- J. D. Hewes, C. W. Kreimendahl, T. B. Marder and M. F. Hawthorne, J. Am. Chem. Soc., 1984, 106, 5757–5759 CrossRef CAS.
- L. G. Sneddon, Pure Appl. Chem., 1987, 59, 837–846 CrossRef CAS.
- R. L. Ernest, W. Quintana, R. Rosen, P. J. Carroll and L. G. Sneddon, Organometallics, 1987, 6, 80–88 CrossRef CAS.
- M. G. L. Mirabelli and L. G. Sneddon, J. Am. Chem. Soc., 1988, 110, 449–453 CrossRef CAS.
- M. G. L. Mirabelli, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 1989, 111, 592–597 CrossRef CAS.
- K. Mazighi, P. J. Carroll and L. G. Sneddon, Inorg. Chem., 1993, 32, 1963–1969 CrossRef CAS.
- M. J. Pender, T. Wideman, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 1998, 120, 9108–9109 CrossRef CAS.
- D. E. Kadlecek, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2000, 122, 10868–10877 CrossRef CAS.
- M. J. Pender, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2001, 123, 12222–12231 CrossRef CAS PubMed.
- U. Kusari, Y. Li, M. G. Bradley and L. G. Sneddon, J. Am. Chem. Soc., 2004, 126, 8662–8663 CrossRef CAS PubMed.
- U. Kusari, P. J. Carroll and L. G. Sneddon, Inorg. Chem., 2008, 47, 9203–9215 CrossRef CAS PubMed.
- S. Chatterjee, P. J. Carroll and L. G. Sneddon, Inorg. Chem., 2010, 49, 3095–3097 CrossRef CAS PubMed.
- S. Chatterjee, P. J. Carroll and L. G. Sneddon, Inorg. Chem., 2013, 52, 9119–9130 CrossRef CAS PubMed.
- R. Cheng, Z. Qiu and Z. Xie, Chem. – Eur. J., 2020, 26, 7212–7218 CrossRef CAS PubMed.
- K. Cao, T.-T. Xu, J. Wu, C.-Y. Zhang, X.-Y. Wen and J. Yang, Inorg. Chem., 2021, 60, 1080–1085 CrossRef CAS PubMed.
- A. T. Lynch and L. G. Sneddon, J. Am. Chem. Soc., 1989, 111, 6201–6209 CrossRef.
- A. T. Lynch and L. G. Sneddon, J. Am. Chem. Soc., 1987, 109, 5867–5868 CrossRef CAS.
- P. J. Fazen and L. G. Sneddon, Organometallics, 1994, 13, 2867–2877 CrossRef CAS.
- J.-K. Jeon, Q. D. Nghiem, D.-P. Kim and J. Lee, J. Organomet. Chem., 2004, 689, 2311–2318 CrossRef CAS.
- Y. Yamamoto, K. Miyamoto, J. Umeda, Y. Nakatani, T. Yamamoto and N. Miyaura, J. Organomet. Chem., 2006, 691, 4909–4917 CrossRef CAS.
- H. C. Brown and S. K. Gupta, J. Am. Chem. Soc., 1975, 97, 5249–5255 CrossRef CAS.
- S. A. Westcott, H. P. Blom, T. B. Marder, R. T. Baker and J. C. Calabrese, Inorg. Chem., 1993, 32, 2175–2182 CrossRef CAS.
- D. Männig and H. Nöth, Angew. Chem., Int. Ed. Engl., 1985, 24, 878–879 CrossRef.
- A. Lang, H. Noeth and M. Thomann-Albach, Chem. Ber./Recl., 1997, 130, 363–369 CrossRef CAS.
- C. B. Fritschi, S. M. Wernitz, C. M. Vogels, M. P. Shaver, A. Decken, A. Bell and S. A. Westcott, Eur. J. Inorg. Chem., 2008, 779–785 CrossRef CAS.
- S. W. Hadebe and R. S. Robinson, Eur. J. Org. Chem., 2006, 4898–4904 CrossRef CAS.
- D. H. Motry, A. G. Brazil and M. R. Smith, J. Am. Chem. Soc., 1997, 119, 2743–2744 CrossRef CAS.
- H. Yoshida, M. Kimura, H. Tanaka, Y. Murashige, I. Kageyuki and I. Osaka, Chem. Commun., 2019, 55, 5420–5422 RSC.
- N. Iwadate and M. Suginome, Org. Lett., 2009, 11, 1899–1902 CrossRef CAS PubMed.
- C. E. Tucker, J. Davidson and P. Knochel, J. Org. Chem., 1992, 57, 3482–3485 CrossRef CAS.
- C. M. Vogels and S. A. Westcott, Curr. Org. Chem., 2005, 9, 687–699 CrossRef CAS.
- A.-M. Carroll, T. P. O’Sullivan and P. J. Guiry, Adv. Synth. Catal., 2005, 347, 609–631 CrossRef CAS.
- T. Hayashi, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer-Verlag, Berlin Heidelberg, 1999, ch. 9, pp. 351–364 Search PubMed.
- S. Huang, Y. Xie, S. Wu, M. Jia, J. Wang, W. Xu and H. Fang, Curr. Org. Synth., 2013, 10, 683–696 CrossRef CAS.
- G. C. Fu, in Transition Metals for Organic Synthesis, ed. M. Beller and C. Bohm, Wiley-VCH Verlag GmbH, Weinheim, 2008, ch. 1.5, pp. 193–198 Search PubMed.
- C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695–4712 CrossRef CAS.
- M. Zaidlewicz, in Kirk-Othmer Encyclopedia of Chemical Technology, ed. Kirk-Othmer, John Wiley & Sons, Ltd, New York, 2004, vol. 13, pp. 631–684 Search PubMed.
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS.
- M. Zaidlewicz, A. Wolan and M. Budny, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, 2nd edn, 2014, ch. 24, vol. 8, pp. 877–963 Search PubMed.
- P. J. Guiry, A. G. Coyne and A. M. Carroll, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, vol. 10, pp. 839–869 Search PubMed.
- I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957–5026 CrossRef CAS.
- H. Wadepohl, Angew. Chem., Int. Ed. Engl., 1997, 36, 2441–2444 CrossRef CAS.
- T. Fujihara, K. Semba, J. Terao and Y. Tsuji, Catal. Sci. Technol., 2014, 4, 1699–1709 RSC.
- E. Fernández and A. M. Segarra, in Iridium Complexes in Organic Synthesis, ed. L. A. Oro and C. Claver, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009, pp. 173–194 Search PubMed.
- C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyas and E. Fernandez, Org. Biomol. Chem., 2010, 8, 2667–2682 RSC.
- A. L. Reznichenko and K. C. Hultzsch, Struct. Bonding, 2010, 137, 1–48 CrossRef CAS.
- A. G. Coyne and P. J. Guiry, in Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, ch. 3, pp. 65–86 Search PubMed.
- S. A. Westcott, P. Nguyen, H. P. Blom, N. J. Taylor, T. B. Marder, R. T. Baker and J. C. Calabrese, Spec. Publ. – R. Soc. Chem., 1994, 143, 68–71 CAS.
- D. Hong, G. Rong, X. Hou and L. Dai, Youji Huaxue, 1994, 14, 131–141 CAS.
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC.
- N. Arai and T. Ohkuma, in Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, ch. 7, pp. 159–181 Search PubMed.
- S. A. Westcott and R. T. Baker, Modern Reduction Methods, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, ch. 12, pp. 297–319 Search PubMed.
- H. Kono, K. Ito and Y. Nagai, Chem. Lett., 1975, 1095–1096 CrossRef CAS.
- S. A. Westcott, N. J. Taylor, T. B. Marder, R. T. Baker, N. J. Jones and J. C. Calabrese, J. Chem. Soc., Chem. Commun., 1991, 304–305 RSC.
- S. A. Westcott, T. B. Marder and R. T. Baker, Organometallics, 1993, 12, 975–979 CrossRef CAS.
- S. I. Kallane, T. Braun, B. Braun and S. Mebs, Dalton Trans., 2014, 43, 6786–6801 RSC.
- C. Widauer, H. Gruetzmacher and T. Ziegler, Organometallics, 2000, 19, 2097–2107 CrossRef CAS.
- D. G. Musaev, A. M. Mebel and K. Morokuma, J. Am. Chem. Soc., 1994, 116, 10693–10702 CrossRef CAS.
- A. E. Dorigo and P. v R. Schleyer, Angew. Chem., Int. Ed. Engl., 1995, 34, 115–118 CrossRef CAS.
- H. Shao, Y. Wang, C. W. Bielawski and P. Liu, ACS Catal., 2020, 10, 3820–3827 CrossRef CAS.
- D. A. Evans and G. C. Fu, J. Org. Chem., 1990, 55, 2280–2282 CrossRef CAS.
- D. R. Edwards, Y. B. Hleba, C. J. Lata, L. A. Calhoun and C. M. Crudden, Angew. Chem., Int. Ed., 2007, 46, 7799–7802 CrossRef CAS PubMed.
- T. Ohmura, Y. Yamamoto and N. Miyaura, J. Am. Chem. Soc., 2000, 122, 4990–4991 CrossRef CAS.
- B. Sundararaju and A. Fuerstner, Angew. Chem., Int. Ed., 2013, 52, 14050–14054 CrossRef CAS PubMed.
- L.-J. Song, T. Wang, X. Zhang, L. W. Chung and Y.-D. Wu, ACS Catal., 2017, 7, 1361–1368 CrossRef CAS.
- K. N. Harrison and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 9220–9221 CrossRef CAS.
- D. A. Evans, A. R. Muci and R. Stuermer, J. Org. Chem., 1993, 58, 5307–5309 CrossRef CAS.
- E. A. Bijpost, R. Duchateau and J. H. Teuben, J. Mol. Catal. A: Chem., 1995, 95, 121–128 CrossRef CAS.
- K. Burgess and M. Jaspars, Tetrahedron Lett., 1993, 34, 6813–6816 CrossRef CAS.
- K. Burgess and W. A. van der Donk, Organometallics, 1994, 13, 3616–3620 CrossRef CAS.
- X. He and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 1696–1702 CrossRef CAS.
- J. F. Hartwig and C. N. Muhoro, Organometallics, 2000, 19, 30–38 CrossRef CAS.
- R. W. Hoffmann, J. Kruger and D. Bruckner, New J. Chem., 2001, 25, 102–107 RSC.
- R. W. Hoffmann and D. Bruckner, New J. Chem., 2001, 25, 369–373 RSC.
- S. Pereira and M. Srebnik, Organometallics, 1995, 14, 3127–3128 CrossRef CAS.
- S. Pereira and M. Srebnik, Tetrahedron Lett., 1996, 37, 3283–3286 CrossRef CAS.
- S. Pereira and M. Srebnik, J. Am. Chem. Soc., 1996, 118, 909–910 CrossRef CAS.
- S. Mandal, P. K. Verma and K. Geetharani, Chem. Commun., 2018, 54, 13690–13693 RSC.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed.
- F. Huang, C. Zhang, J. Jiang, Z.-X. Wang and H. Guan, Inorg. Chem., 2011, 50, 3816–3825 CrossRef CAS PubMed.
- S. Chakraborty, J. Zhang, Y. J. Patel, J. A. Krause and H. Guan, Inorg. Chem., 2013, 52, 37–47 CrossRef CAS PubMed.
- S. Chakraborty, Y. J. Patel, J. A. Krause and H. Guan, Polyhedron, 2012, 32, 30–34 CrossRef CAS.
- R. Shintani and K. Nozaki, Organometallics, 2013, 32, 2459–2462 CrossRef CAS.
- J. M. Brown and G. C. Lloyd-Jones, Tetrahedron: Asymmetry, 1990, 1, 869–872 CrossRef CAS.
- P. Kasak, K. Mereiter and M. Widhalm, Tetrahedron: Asymmetry, 2005, 16, 3416–3426 CrossRef CAS.
- T. Hayashi, Y. Matsumoto and Y. Ito, J. Am. Chem. Soc., 1989, 111, 3426–3428 CrossRef CAS.
- M. Sato, N. Miyaura and A. Suzuki, Tetrahedron Lett., 1990, 31, 231–234 CrossRef CAS.
- K. Burgess and M. J. Ohlmeyer, J. Org. Chem., 1988, 53, 5178–5179 CrossRef CAS.
- T. Hayashi, Y. Matsumoto and Y. Ito, Tetrahedron: Asymmetry, 1991, 2, 601–612 CrossRef CAS.
- J. M. Brown, D. I. Hulmes and T. P. Layzell, J. Chem. Soc., Chem. Commun., 1993, 1673–1674 RSC.
- J. M. Valk, G. A. Whitlock, T. P. Layzell and J. M. Brown, Tetrahedron: Asymmetry, 1995, 6, 2593–2596 CrossRef CAS.
- A. Korostylev, I. Gridnev and J. M. Brown, J. Organomet. Chem., 2003, 680, 329–334 CrossRef CAS.
- J. Kang, J. H. Lee, J. B. Kim and G. J. Kim, Chirality, 2000, 12, 378–382 CrossRef CAS PubMed.
- A. Togni, C. Breutel, A. Schnyder, F. Spindler, H. Landert and A. Tijani, J. Am. Chem. Soc., 1994, 116, 4062–4066 CrossRef CAS.
- A. Schnyder, L. Hintermann and A. Togni, Angew. Chem., Int. Ed. Engl., 1995, 34, 931–933 CrossRef CAS.
- C. Kollner and A. Togni, Can. J. Chem., 2001, 79, 1762–1774 CrossRef CAS.
- A. M. Segarra, R. Guerrero, C. Claver and E. Fernandez, Chem. Commun., 2001, 1808–1809 RSC.
- A. M. Segarra, R. Guerrero, C. Claver and E. Fernandez, Chem. – Eur. J., 2003, 9, 191–200 CrossRef CAS PubMed.
- A. M. Segarra, F. Guirado, C. Claver and E. Fernandez, Tetrahedron: Asymmetry, 2003, 14, 1611–1615 CrossRef CAS.
- A. Carmona, A. Corma, M. Iglesias, J. A. San and F. Sanchez, J. Organomet. Chem., 1995, 492, 11–21 CrossRef CAS.
- A. Corma, M. I. De Dios, M. Iglesias and F. Sanchez, An. Quim., 1995, 91, 277–283 CAS.
- M. Murata, S. Watanabe and Y. Masuda, Tetrahedron Lett., 1999, 40, 2585–2588 CrossRef CAS.
- S. A. Moteki, D. Wu, K. L. Chandra, D. S. Reddy and J. M. Takacs, Org. Lett., 2006, 8, 3097–3100 CrossRef CAS PubMed.
- H. Shao, S. Chakrabarty, X. Qi, J. M. Takacs and P. Liu, J. Am. Chem. Soc., 2021, 143, 4801–4808 CrossRef CAS PubMed.
- S. A. Moteki and J. M. Takacs, Angew. Chem., Int. Ed., 2008, 47, 894–897 CrossRef CAS PubMed.
- C. M. Crudden, Y. B. Hleba and A. C. Chen, J. Am. Chem. Soc., 2004, 126, 9200–9201 CrossRef CAS PubMed.
- A. M. Segarra, E. Daura-Oller, C. Claver, J. M. Poblet, C. Bo and E. Fernandez, Chem. – Eur. J., 2004, 10, 6456–6467 CrossRef CAS PubMed.
- D. Noh, S. K. Yoon, J. Won, J. Y. Lee and J. Yun, Chem. – Asian J., 2011, 6, 1967–1969 CrossRef CAS PubMed.
- S. A. Moteki, K. Toyama, Z. Liu, J. Ma, A. E. Holmes and J. M. Takacs, Chem. Commun., 2012, 48, 263–265 RSC.
- D. Noh, H. Chea, J. Ju and J. Yun, Angew. Chem., Int. Ed., 2009, 48, 6937 CrossRef CAS PubMed.
- J. Won, D. Noh, J. Yun and J. Y. Lee, J. Phys. Chem. A, 2010, 114, 12112–12115 CrossRef CAS PubMed.
- M. Pang, C. Wu, X. Zhuang, F. Zhang, M. Su, Q. Tong, C.-H. Tung and W. Wang, Organometallics, 2018, 37, 1462–1467 CrossRef CAS.
- A. D. Ibrahim, S. W. Entsminger and A. R. Fout, ACS Catal., 2017, 7, 3730–3734 CrossRef CAS.
- X. Hou, D. Hong, G. Rong, Y. Guo and L. Dai, Tetrahedron Lett., 1993, 34, 8513–8516 CrossRef CAS.
- V. Montiel-Palma, M. Lumbierres, B. Donnadieu, S. Sabo-Etienne and B. Chaudret, J. Am. Chem. Soc., 2002, 124, 5624–5625 CrossRef CAS PubMed.
- A. Caballero and S. Sabo-Etienne, Organometallics, 2007, 26, 1191–1195 CrossRef CAS.
- S. Elgendy, G. Patel, V. V. Kakkar, G. Claeson, D. Green, E. Skordalakes, J. A. Baban and J. Deadman, Tetrahedron Lett., 1994, 35, 2435–2436 CrossRef CAS.
- X. Wang, X. Cui, S. Li, Y. Wang, C. Xia, H. Jiao and L. Wu, Angew. Chem., Int. Ed., 2020, 59, 13608–13612 CrossRef CAS PubMed.
- K. Burgess, d D. van, A. Wilfred, S. A. Westcott, T. B. Marder, R. T. Baker and J. C. Calabrese, J. Am. Chem. Soc., 1992, 114, 9350–9359 CrossRef CAS.
- M. L. Scheuermann, E. J. Johnson and P. J. Chirik, Org. Lett., 2015, 17, 2716–2719 CrossRef CAS PubMed.
- G. Vijaykumar, M. Bhunia and S. K. Mandal, Dalton Trans., 2019, 48, 5779–5784 RSC.
- E. Bergamaschi, F. Beltran and C. J. Teskey, Chem. – Eur. J., 2020, 26, 5180–5184 CrossRef CAS PubMed.
- D. A. Evans and G. C. Fu, J. Am. Chem. Soc., 1991, 113, 4042–4043 CrossRef CAS.
- M. J. Geier, C. M. Vogels, A. Decken and S. A. Westcott, J. Organomet. Chem., 2009, 694, 3154–3159 CrossRef CAS.
- D. A. Evans, G. C. Fu and A. H. Hoveyda, J. Am. Chem. Soc., 1992, 114, 6671–6679 CrossRef CAS.
- W. Dong, X. Xu, H. Ma, Y. Lei, Z. Lin and W. Zhao, J. Am. Chem. Soc., 2021, 143, 10902–10909 CrossRef CAS PubMed.
- M. D. Greenhalgh and S. P. Thomas, Chem. Commun., 2013, 49, 11230–11232 RSC.
- L. Zhang, Z. Zuo, X. Leng and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 2696–2700 CrossRef CAS PubMed.
- G. A. Molander and T. Ito, Org. Lett., 2001, 3, 393–396 CrossRef CAS PubMed.
- C. B. Wagner and A. Studer, Eur. J. Org. Chem., 2010, 5782–5786 CrossRef CAS.
- D. A. Evans, G. C. Fu and B. A. Anderson, J. Am. Chem. Soc., 1992, 114, 6679–6685 CrossRef CAS.
- D. A. Evans, G. C. Fu and A. H. Hoveyda, J. Am. Chem. Soc., 1988, 110, 6917–6918 CrossRef CAS.
- K. Burgess and M. J. Ohlmeyer, Tetrahedron Lett., 1989, 30, 395–398 CrossRef CAS.
- K. Burgess and M. J. Ohlmeyer, Tetrahedron Lett., 1989, 30, 5861–5864 CrossRef CAS.
- K. Burgess, J. Cassidy and M. J. Ohlmeyer, J. Org. Chem., 1991, 56, 1020–1027 CrossRef CAS.
- K. Burgess and M. Jaspars, Organometallics, 1993, 12, 4197–4200 CrossRef CAS.
- D. A. Evans and G. S. Sheppard, J. Org. Chem., 1990, 55, 5192–5194 CrossRef CAS.
- D. A. Evans and J. R. Gage, J. Org. Chem., 1992, 57, 1958–1961 CrossRef CAS.
- E. Hupe, M. I. Calaza and P. Knochel, Chem. – Eur. J., 2003, 9, 2789–2796 CrossRef CAS PubMed.
- K. Burgess and M. J. Ohlmeyer, Tetrahedron Lett., 1989, 30, 5857–5860 CrossRef CAS.
- K. Burgess and M. J. Ohlmeyer, J. Org. Chem., 1991, 56, 1027–1036 CrossRef CAS.
- X.-L. Hou, Q.-C. Xie and L.-X. Dai, J. Chem. Res., Synop., 1997, 436 RSC.
- X.-L. Hou, Q.-C. Xie and L.-X. Dai, J. Chem. Res., 1997, 2665 Search PubMed.
- K. Burgess, W. A. van der Donk and M. J. Ohlmeyer, Tetrahedron: Asymmetry, 1991, 2, 613–621 CrossRef CAS.
- P. Renaud, C. Ollivier and V. Weber, J. Org. Chem., 2003, 68, 5769–5772 CrossRef CAS PubMed.
- M. A. Reichle and B. Breit, Angew. Chem., Int. Ed., 2012, 51, 5730–5734 CrossRef CAS PubMed.
- L. Zhang, Z. Zuo, X. Wan and Z. Huang, J. Am. Chem. Soc., 2014, 136, 15501–15504 CrossRef CAS PubMed.
- S. M. Smith, G. L. Hoang, R. Pal, M. O. B. Khaled, L. S. W. Pelter, X. C. Zeng and J. M. Takacs, Chem. Commun., 2012, 48, 12180–12182 RSC.
- S. Simaan, A. F. G. Goldberg, S. Rosset and I. Marek, Chem. – Eur. J., 2010, 16, 774–778 CrossRef CAS PubMed.
- W. N. Palmer, T. Diao, I. Pappas and P. J. Chirik, ACS Catal., 2015, 5, 622–626 CrossRef CAS.
- J. Chen, T. Xi, X. Ren, B. Cheng, J. Guo and Z. Lu, Org. Chem. Front., 2014, 1, 1306–1309 RSC.
- T. Iwai, T. Harada, H. Shimada, K. Asano and M. Sawamura, ACS Catal., 2017, 7, 1681–1692 CrossRef CAS.
- M. P. Doyle, L. J. Westrum, M. N. Protopopova, M. Y. Eismont and M. B. Jarstfer, Mendeleev Commun., 1993, 81–82 CrossRef CAS.
- S. W. Hadebe and R. S. Robinson, Tetrahedron Lett., 2006, 47, 5419 CrossRef CAS.
- J. Zheng, J.-B. Sortais and C. Darcel, ChemCatChem, 2014, 6, 763–766 CrossRef CAS.
- J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107–19110 CrossRef CAS PubMed.
- C. J. Lata and C. M. Crudden, J. Am. Chem. Soc., 2010, 132, 131–137 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, Org. Lett., 2013, 15, 2680–2683 CrossRef CAS PubMed.
- A. J. Ruddy, O. L. Sydora, B. L. Small, M. Stradiotto and L. Turculet, Chem. – Eur. J., 2014, 20, 13918–13922 CrossRef CAS PubMed.
- N. M. Weliange, D. S. McGuinness, M. G. Gardiner and J. Patel, Dalton Trans., 2016, 45, 10842–10849 RSC.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed.
- C. M. Macaulay, S. J. Gustafson, J. T. Fuller, D.-H. Kwon, T. Ogawa, M. J. Ferguson, R. McDonald, M. D. Lumsden, S. M. Bischof, O. L. Sydora, D. H. Ess, M. Stradiotto and L. Turculet, ACS Catal., 2018, 8, 9907–9925 CrossRef CAS.
- T. Ogawa, A. J. Ruddy, O. L. Sydora, M. Stradiotto and L. Turculet, Organometallics, 2017, 36, 417–423 CrossRef CAS.
- L. J. Murphy, H. Hollenhorst, R. McDonald, M. Ferguson, M. D. Lumsden and L. Turculet, Organometallics, 2017, 36, 3709–3720 CrossRef CAS.
- A. J. Ruddy, O. L. Sydora, B. L. Small, M. Stradiotto and L. Turculet, Chemistry, 2016, 22, 12589 CrossRef CAS PubMed.
- A. P. Luna, M. A. Ceschi, M. Bonin, L. Micouin, H. P. Husson, S. Gougeon, G. Estenne-Bouhtou, B. Marabout, M. Sevrin and P. George, J. Org. Chem., 2002, 67, 3522–3524 CrossRef CAS PubMed.
- A. Alexakis, D. Polet, C. Bournaud, M. Bonin and L. Micouin, Tetrahedron: Asymmetry, 2005, 16, 3672–3675 CrossRef CAS.
- T. Bunlaksananusorn, A. P. Luna, M. Bonin, L. Micouin and P. Knochel, Synlett, 2003, 2240–2242 CAS.
- K. Burgess, M. J. Ohlmeyer and K. H. Whitmire, Organometallics, 1992, 11, 3588–3600 CrossRef CAS.
- H. Lee, B. Y. Lee and J. Yun, Org. Lett., 2015, 17, 764–766 CrossRef CAS PubMed.
- G. W. Kabalka, C. Narayana and N. K. Reddy, Synth. Commun., 1994, 24, 1019–1023 CrossRef CAS.
- J. J. J. Juliette, D. Rutherford, I. T. Horvath and J. A. Gladysz, J. Am. Chem. Soc., 1999, 121, 2696–2704 CrossRef CAS.
- J. J. J. Juliette, I. T. Horvath and J. A. Gladysz, Angew. Chem., Int. Ed. Engl., 1997, 36, 1610–1612 CrossRef CAS.
- J.-M. Brunel and G. Buono, Tetrahedron Lett., 1999, 40, 3561–3564 CrossRef CAS.
- Y. Yamamoto, R. Fujikawa, T. Umemoto and N. Miyaura, Tetrahedron, 2004, 60, 10695–10700 CrossRef CAS.
- N. M. Hunter, C. M. Vogels, A. Decken, A. Bell and S. A. Westcott, Inorg. Chim. Acta, 2011, 365, 408–413 CrossRef CAS.
- A. Arase, M. Hoshi, A. Mijin and K. Nishi, Synth. Commun., 1995, 25, 1957–1962 CrossRef CAS.
- B. Nguyen and J. M. Brown, Adv. Synth. Catal., 2009, 351, 1333–1343 CrossRef CAS.
- K. M. J. Brands and A. S. Kende, Tetrahedron Lett., 1992, 33, 5887–5890 CrossRef CAS.
- L. A. Perez, M. Bonin, L. Micouin and H.-P. Husson, J. Am. Chem. Soc., 2002, 124, 12098–12099 CrossRef PubMed.
- C. E. Garrett and G. C. Fu, J. Org. Chem., 1998, 63, 1370–1371 CrossRef CAS.
- L. Bunch, T. Liljefors, J. R. Greenwood, K. Frydenvang, H. Braeuner-Osborne, P. Krogsgaard-Larsen and U. Madsen, J. Org. Chem., 2003, 68, 1489–1495 CrossRef CAS PubMed.
- M. Toganoh, Y. Matsuo and E. Nakamura, J. Organomet. Chem., 2003, 683, 295–300 CrossRef CAS.
- A. Edwards, M. Rubina and M. Rubin, Chem. – Eur. J., 2018, 24, 1394–1403 CrossRef CAS PubMed.
- S. M. Smith and J. M. Takacs, Org. Lett., 2010, 12, 4612–4615 CrossRef CAS PubMed.
- S. M. Smith, M. Uteuliyev and J. M. Takacs, Chem. Commun., 2011, 47, 7812–7814 RSC.
- H. Zhao, Q. Gao, Y. Zhang, P. Zhang and S. Xu, Org. Lett., 2020, 22, 2861–2866 CrossRef CAS PubMed.
- G. L. Hoang and J. M. Takacs, Chem. Sci., 2017, 8, 4511–4516 RSC.
- G. L. Hoang, Z.-D. Yang, S. M. Smith, R. Pal, J. L. Miska, D. E. Pérez, L. S. W. Pelter, X. C. Zeng and J. M. Takacs, Org. Lett., 2015, 17, 940–943 CrossRef CAS PubMed.
- Y. Xi and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 6703–6706 CrossRef CAS PubMed.
- M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2003, 125, 7198–7199 CrossRef CAS PubMed.
- M. P. Shaver, C. M. Vogels, A. I. Wallbank, T. L. Hennigar, K. Biradha, M. J. Zaworotko and S. A. Westcott, Can. J. Chem., 2000, 78, 568–576 CrossRef CAS.
- S. M. Smith and J. M. Takacs, J. Am. Chem. Soc., 2010, 132, 1740–1741 CrossRef CAS PubMed.
- Y. Cai, D. Tan, Q. Zhang, W. Lv, Q. Li and H. Wang, Chin. Chem. Lett., 2021, 32, 417–420 CrossRef CAS.
- V. M. Shoba, N. C. Thacker, A. J. Bochat and J. M. Takacs, Angew. Chem., Int. Ed., 2016, 55, 1465–1469 CrossRef CAS PubMed.
- S. Chakrabarty and J. M. Takacs, ACS Catal., 2018, 8, 10530–10536 CrossRef CAS PubMed.
- S. Chakrabarty and J. M. Takacs, J. Am. Chem. Soc., 2017, 139, 6066–6069 CrossRef CAS PubMed.
- L. Zhang and Z. Huang, J. Am. Chem. Soc., 2015, 137, 15600–15603 CrossRef CAS PubMed.
- T. Zhang, K. Manna and W. Lin, J. Am. Chem. Soc., 2016, 138, 3241–3249 CrossRef CAS PubMed.
- M. Satoh, Y. Nomoto, N. Miyaura and A. Suzuki, Tetrahedron Lett., 1989, 30, 3789–3792 CrossRef CAS.
- D. Fiorito and C. Mazet, ACS Catal., 2018, 8, 9382–9387 CrossRef CAS.
- Y. Matsumoto and T. Hayashi, Tetrahedron Lett., 1991, 32, 3387–3390 CrossRef CAS.
- Y. Cao, Y. Zhang, L. Zhang, D. Zhang, X. Leng and Z. Huang, Org. Chem. Front., 2014, 1, 1101–1106 RSC.
- M. Zaidlewicz and J. Meller, Main Group Met. Chem., 2000, 23, 765–772 CAS.
- M. Zaidlewicz and J. Meller, Tetrahedron Lett., 1997, 38, 7279–7282 CrossRef CAS.
- M. Zaidlewicz, J. Binkul, M. Giminska, M. Krzeminski and J. Meller, Spec. Publ. – R. Soc. Chem., 2000, 253, 415–421 CAS.
- R. J. Ely, J. P. Morken, P. M. Tadross and B. M. Stoltz, Org. Synth., 2011, 88, 342–352 CrossRef CAS.
- R. J. Ely and J. P. Morken, J. Am. Chem. Soc., 2010, 132, 2534–2535 CrossRef CAS PubMed.
- L. A. Brozek, M. J. Ardolino and J. P. Morken, J. Am. Chem. Soc., 2011, 133, 16778–16781 CrossRef CAS PubMed.
- R. J. Ely, Z. Yu and J. P. Morken, Tetrahedron Lett., 2015, 56, 3402–3405 CrossRef CAS PubMed.
- Z. Yu, R. J. Ely and J. P. Morken, Angew. Chem., Int. Ed., 2014, 53, 9632–9636 CrossRef CAS PubMed.
- J. Y. Wu, B. Moreau and T. Ritter, J. Am. Chem. Soc., 2009, 131, 12915–12917 CrossRef CAS PubMed.
- K. Duvvuri, K. R. Dewese, M. M. Parsutkar, S. M. Jing, M. M. Mehta, J. C. Gallucci and T. V. Rajanbabu, J. Am. Chem. Soc., 2019, 141, 7365–7375 CrossRef CAS PubMed.
- Y. Liu, Z. Jiang and J. Chen, Org. Biomol. Chem., 2020, 18, 3747–3753 RSC.
- K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem. – Eur. J., 2013, 19, 7125–7132 CrossRef CAS PubMed.
- D. J. Winternheimer and C. A. Merlic, Org. Lett., 2010, 12, 2508–2510 CrossRef CAS PubMed.
- B. François, L. Eberlin, F. Berrée, A. Whiting and B. Carboni, J. Org. Chem., 2020, 85, 5173–5182 CrossRef PubMed.
- M. Zaidlewicz and J. Meller, Collect. Czech. Chem. Commun., 1999, 64, 1049–1056 CrossRef CAS.
- Y. Yang, J. Jiang, H. Yu and J. Shi, Chemistry, 2018, 24, 178–186 CrossRef CAS PubMed.
- S. Xu, Y. Zhang, B. Li and S.-Y. Liu, J. Am. Chem. Soc., 2016, 138, 14566–14569 CrossRef CAS PubMed.
- O. Robles and F. E. McDonald, Org. Lett., 2009, 11, 5498–5501 CrossRef CAS PubMed.
- H. N. Lim and K. A. Parker, Org. Lett., 2013, 15, 398–401 CrossRef CAS PubMed.
- S. Manna, K. K. Das, D. Aich and S. Panda, Adv. Synth. Catal., 2021, 363, 2444–2463 CrossRef CAS.
- K. Fuji, M. Sakurai, T. Kinoshita, T. Tada, A. Kuroda and T. Kawabata, Chem. Pharm. Bull., 1997, 45, 1524–1526 CrossRef CAS.
- T. Hayashi, Yuki Gosei Kagaku Kyokaishi, 1994, 52, 900–911 CrossRef CAS.
- Y. Matsumoto, M. Naito and T. Hayashi, Organometallics, 1992, 11, 2732–2734 CrossRef CAS.
- Y. Huang, J. del Pozo, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2018, 140, 2643–2655 CrossRef CAS PubMed.
- D.-W. Gao, Y. Xiao, M. Liu, Z. Liu, M. K. Karunananda, J. S. Chen and K. M. Engle, ACS Catal., 2018, 8, 3650–3654 CrossRef CAS PubMed.
- R. E. Kinder and R. A. Widenhoefer, Org. Lett., 2006, 8, 1967–1969 CrossRef CAS PubMed.
- C. Wang and S. Ge, J. Am. Chem. Soc., 2018, 140, 10687–10690 CrossRef CAS PubMed.
- S. Yu, C. Wu and S. Ge, J. Am. Chem. Soc., 2017, 139, 6526–6529 CrossRef CAS PubMed.
- C. Wu, J. Liao and S. Ge, Angew. Chem., Int. Ed., 2019, 58, 8882–8886 CrossRef CAS PubMed.
- T. Xi and Z. Lu, ACS Catal., 2017, 7, 1181–1185 CrossRef CAS.
- N. Cabrera-Lobera, M. T. Quiros, E. Bunuel and D. J. Cardenas, Catal. Sci. Technol., 2019, 9, 1021–1029 RSC.
- N. Cabrera-Lobera, P. Rodriguez-Salamanca, J. C. Nieto-Carmona, E. Bunuel and D. J. Cardenas, Chem. – Eur. J., 2018, 24, 784–788 CrossRef CAS PubMed.
- J.-M. Yang, Z.-Q. Li, M.-L. Li, Q. He, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 3784–3789 CrossRef CAS PubMed.
- Y. Yamamoto, R. Fujikawa, A. Yamada and N. Miyaura, Chem. Lett., 1999, 1069–1070 CrossRef CAS.
- C. Li, Z. Yang, L. Wang, Y. Guo, Z. Huang and S. Ma, Angew. Chem., Int. Ed., 2020, 59, 6278–6283 CrossRef CAS PubMed.
- Y. Yang, J.-H. Zeng and Z.-P. Zhan, Org. Chem. Front., 2021, 8, 2537–2542 RSC.
- Y. Zhao and S. Ge, Angew. Chem., Int. Ed., 2021, 60, 2149–2154 CrossRef CAS PubMed.
- J. Zhang, B. Lou, G. Guo and L. Dai, J. Org. Chem., 1991, 56, 1670–1672 CrossRef CAS.
- H. Doucet, E. Fernandez, T. P. Layzell and J. M. Brown, Chem. – Eur. J., 1999, 5, 1320–1330 CrossRef CAS.
- J. Liedtke, H. Ruegger, S. Loss and H. Grutzmacher, Angew. Chem., Int. Ed., 2000, 39, 2478–2481 CrossRef CAS PubMed.
- H. Grutzmacher, J. Liedtke, G. Frasca, F. Lang and N. Pe, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 1771–1774 CrossRef CAS.
- J. A. Melanson, C. M. Vogels, A. Decken and S. A. Westcott, Inorg. Chem. Commun., 2010, 13, 1396–1398 CrossRef CAS.
- S. A. Westcott, T. B. Marder, R. T. Baker and J. C. Calabrese, Can. J. Chem., 1993, 71, 930–936 CrossRef CAS.
- G. M. Lee, C. M. Vogels, A. Decken and S. A. Westcott, Eur. J. Inorg. Chem., 2011, 2433–2438 CrossRef CAS.
- C. M. Vogels, A. Decken and S. A. Westcott, Can. J. Chem., 2006, 84, 146–153 CrossRef CAS.
- S. Kisan, V. Krishnakumar and C. Gunanathan, ACS Catal., 2017, 7, 5950–5954 CrossRef CAS.
- T. F. C. Cruz, L. F. Veiros and P. T. Gomes, Inorg. Chem., 2018, 57, 14671–14685 CrossRef CAS PubMed.
- J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595–600 CrossRef CAS PubMed.
- R. Agahi, A. J. Challinor, N. B. Carter and S. P. Thomas, Org. Lett., 2019, 21, 993–997 CrossRef CAS PubMed.
- J. Peng, J. H. Docherty, A. P. Dominey and S. P. Thomas, Chem. Commun., 2017, 53, 4726–4729 RSC.
- X. Chen, Z. Cheng and Z. Lu, ACS Catal., 2019, 9, 4025–4029 CrossRef CAS.
- T. F. C. Cruz, L. C. J. Pereira, J. C. Waerenborgh, L. F. Veiros and P. T. Gomes, Catal. Sci. Technol., 2019, 9, 3347–3360 RSC.
- M. Espinal-Viguri, C. R. Woof and R. L. Webster, Chem. – Eur. J., 2016, 22, 11605–11608 CrossRef CAS PubMed.
- A. Singh, S. Shafiei-Haghighi, C. R. Smith, D. K. Unruh and M. Findlater, Asian J. Org. Chem., 2020, 9, 416–420 CrossRef CAS.
- J. R. Carney, B. R. Dillon, L. Campbell and S. P. Thomas, Angew. Chem., Int. Ed., 2018, 57, 10620–10624 CrossRef CAS PubMed.
- S. Weber, D. Zobernig, B. Stöger, L. F. Veiros and K. Kirchner, Angew. Chem., Int. Ed., 2021, 60, 24488–24492 CrossRef CAS PubMed.
- F. Ulm, Y. Cornaton, J.-P. Djukic, M. J. Chetcuti and V. Ritleng, Chem. – Eur. J., 2020, 26, 8916–8925 CrossRef CAS PubMed.
- E. E. Touney, R. Van Hoveln, C. T. Buttke, M. D. Freidberg, I. A. Guzei and J. M. Schomaker, Organometallics, 2016, 35, 3436–3439 CrossRef CAS.
- S. R. Tamang, D. Bedi, S. Shafiei-Haghighi, C. R. Smith, C. Crawford and M. Findlater, Org. Lett., 2018, 20, 6695–6700 CrossRef CAS PubMed.
- X. Chen, Z. Cheng and Z. Lu, Org. Lett., 2017, 19, 969–971 CrossRef CAS PubMed.
- G. Zhang, J. Wu, M. Wang, H. Zeng, J. Cheng, M. C. Neary and S. Zheng, Eur. J. Org. Chem., 2017, 5814–5818 CrossRef CAS.
- W. Fan, L. Li and G. Zhang, J. Org. Chem., 2019, 84, 5987–5996 CrossRef CAS PubMed.
- G. Zhang, J. Wu, S. Li, S. Cass and S. Zheng, Org. Lett., 2018, 20, 7893–7897 CrossRef CAS PubMed.
- M. J. Geier, S. J. Geier, C. M. Vogels, F. Beland and S. A. Westcott, Synlett, 2009, 477–481 CAS.
- K. Burgess and W. A. van der Donk, Inorg. Chim. Acta, 1994, 220, 93–98 CrossRef CAS.
- A. Black, J. M. Brown and C. Pichon, Chem. Commun., 2005, 5284–5286 RSC.
- C. A. G. Carter, R. T. Baker, W. Tumas and S. P. Nolan, Chem. Commun., 2000, 347–348 RSC.
- J. Cipot, C. M. Vogels, R. McDonald, S. A. Westcott and M. Stradiotto, Organometallics, 2006, 25, 5965–5968 CrossRef CAS.
- C. Mazet and D. Gerard, Chem. Commun., 2011, 47, 298–300 RSC.
- M. Magre, M. Biosca, O. Pamies and M. Dieguez, ChemCatChem, 2015, 7, 114–120 CrossRef CAS.
- Y. Horino, T. Livinghouse and M. Stan, Synlett, 2004, 2639–2641 CAS.
- J. Chen, T. Xi and Z. Lu, Org. Lett., 2014, 16, 6452–6455 CrossRef CAS PubMed.
- W. J. Jang, S. M. Song, J. H. Moon, J. Y. Lee and J. Yun, J. Am. Chem. Soc., 2017, 139, 13660–13663 CrossRef CAS PubMed.
- H. Zhang and Z. Lu, ACS Catal., 2016, 6, 6596–6600 CrossRef CAS.
- J. Chen, T. Xi and Z. Lu, Org. Chem. Front., 2018, 5, 247–253 RSC.
- J. Guo and Z. Lu, Angew. Chem., Int. Ed., 2016, 55, 10835–10838 CrossRef CAS PubMed.
- H. Wen, X. Wan and Z. Huang, Angew. Chem., Int. Ed., 2018, 57, 6319–6323 CrossRef CAS PubMed.
- Z. Zuo, J. Yang and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 10839–10843 CrossRef CAS PubMed.
- Z. Cheng, J. Guo, Y. Sun, Y. Zheng, Z. Zhou and Z. Lu, Angew. Chem., Int. Ed., 2021, 60, 22454–22460 CrossRef CAS PubMed.
- S. Lee, D. Li and J. Yun, Chem. – Asian J., 2014, 9, 2440–2443 CrossRef CAS PubMed.
- A. J. Bochat, V. M. Shoba and J. M. Takacs, Angew. Chem., Int. Ed., 2019, 58, 9434–9438 CrossRef CAS PubMed.
- D. J. Connolly, P. M. Lacey, M. McCarthy, C. P. Saunders, A.-M. Carroll, R. Goddard and P. J. Guiry, J. Org. Chem., 2004, 69, 6572–6589 CrossRef CAS PubMed.
- A. C. Maxwell, S. P. Flanagan, R. Goddard and P. J. Guiry, Tetrahedron: Asymmetry, 2010, 21, 1458–1473 CrossRef CAS.
- W. J. Fleming, H. Mueller-Bunz and P. J. Guiry, Eur. J. Org. Chem., 2010, 5996–6004 CrossRef CAS.
- M. McCarthy, P. J. Guiry and M. W. Hooper, Chem. Commun., 2000, 1333–1334 RSC.
- M. McCarthy and P. J. Guiry, Polyhedron, 2000, 19, 541–543 CrossRef CAS.
- E. Fernandez, K. Maeda, M. W. Hooper and J. M. Brown, Chem. – Eur. J., 2000, 6, 1840–1846 CrossRef CAS PubMed.
- E. Fernandez, M. W. Hooper, F. I. Knight and J. M. Brown, Chem. Commun., 1997, 173–174 RSC.
- G. L. Hoang, S. Zhang and J. M. Takacs, Chem. Commun., 2018, 54, 4838–4841 RSC.
- S. Chakrabarty, H. Palencia, M. D. Morton, R. O. Carr and J. M. Takacs, Chem. Sci., 2019, 10, 4854–4861 RSC.
- S. Rej, A. Das and T. K. Panda, Adv. Synth. Catal., 2021, 363, 4818–4840 CrossRef CAS.
- I. D. Gridnev, N. Miyaura and A. Suzuki, Organometallics, 1993, 12, 589–592 CrossRef CAS.
- D.-Y. Yang and X. Haung, Synth. Commun., 1996, 26, 4617–4621 CrossRef CAS.
- D.-Y. Yang and X. Huang, J. Chem. Res., Synop., 1997, 62–63 RSC.
- Y. Yu and H. Xian, Synth. Commun., 1997, 27, 345–349 CrossRef CAS.
- T. Lee, C. Baik, I. Jung, K. H. Song, S. Kim, D. Kim, S. O. Kang and J. Ko, Organometallics, 2004, 23, 4569–4575 CrossRef CAS.
- A. Fukuda, Y. Kobayashi, T. Kimachi and Y. Takemoto, Tetrahedron, 2003, 59, 9305–9313 CrossRef CAS.
- A. Plikhta, A. Poethig, E. Herdtweck and B. Rieger, Inorg. Chem., 2015, 54, 9517–9528 CrossRef CAS PubMed.
- K. Wang and R. W. Bates, Synthesis, 2017, 2749–2752 CAS.
- C. N. von Hahmann, M. Talavera, C. Xu and T. Braun, Chem. – Eur. J., 2018, 24, 11131–11138 CrossRef CAS PubMed.
- M. Murata, S. Watanabe and Y. Masuda, J. Chem. Res., Synop., 2002, 142–143 CrossRef CAS.
- M. Haberberger and S. Enthaler, Chem. – Asian J., 2013, 8, 50–54 CrossRef CAS PubMed.
- K.-N. T. Tseng, J. W. Kampf and N. K. Szymczak, ACS Catal., 2015, 5, 411–415 CrossRef CAS.
- K. Nakajima, T. Kato and Y. Nishibayashi, Org. Lett., 2017, 19, 4323–4326 CrossRef CAS PubMed.
- T. Bai, Y. Yang and C. Han, Tetrahedron Lett., 2017, 58, 1523–1527 CrossRef CAS.
- E. A. Romero, R. Jazzar and G. Bertrand, J. Organomet. Chem., 2017, 829, 11–13 CrossRef CAS.
- G. Zhu, W. Kong, H. Feng and Z. Qian, J. Org. Chem., 2014, 79, 1786–1795 CrossRef CAS PubMed.
- J. Bhattacharjee, A. Harinath, K. Bano and T. K. Panda, ACS Omega, 2020, 5, 1595–1606 CrossRef CAS PubMed.
- E. Lu, Q. Zhou, Y. Li, J. Chu, Y. Chen, X. Leng and J. Sun, Chem. Commun., 2012, 48, 3403–3405 RSC.
- R. Mamidala, V. K. Pandey and A. Rit, Chem. Commun., 2019, 55, 989–992 RSC.
- A. Leyva, X. Zhang and A. Corma, Chem. Commun., 2009, 4947–4949 RSC.
- A. Y. Khalimon, P. Farha, L. G. Kuzmina and G. I. Nikonov, Chem. Commun., 2012, 48, 455–457 RSC.
- A. Y. Khalimon, P. M. Farha and G. I. Nikonov, Dalton Trans., 2015, 44, 18945–18956 RSC.
- N. G. Bandur, D. Brückner, R. W. Hoffmann and U. Koert, Org. Lett., 2006, 8, 3829–3831 CrossRef CAS PubMed.
- B. D. Stevens, C. J. Bungard and S. G. Nelson, J. Org. Chem., 2006, 71, 6397–6402 CrossRef CAS PubMed.
- A. Furstner and J. Ackerstaff, Chem. Commun., 2008, 2870–2872 RSC.
- R. G. Iafe, J. L. Kuo, D. G. Hochstatter, T. Saga, J. W. Turner and C. A. Merlic, Org. Lett., 2013, 15, 582–585 CrossRef CAS PubMed.
- D. G. Batt, G. C. Houghton, W. F. Daneker and P. K. Jadhav, J. Org. Chem., 2000, 65, 8100–8104 CrossRef CAS PubMed.
- M. Sindelar, T. A. Lutz, M. Petrera and K. T. Wanner, J. Med. Chem., 2013, 56, 1323–1340 CrossRef CAS PubMed.
- H. P. Shunatona, N. Früh, Y.-M. Wang, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 7724–7727 CrossRef CAS PubMed.
- H.-S. Shin, Y.-G. Jung, H.-K. Cho, Y.-G. Park and C.-G. Cho, Org. Lett., 2014, 16, 5718–5720 CrossRef CAS PubMed.
- H.-K. Cho, H.-Y. Lim and C.-G. Cho, Org. Lett., 2013, 15, 5806–5809 CrossRef CAS PubMed.
- G. Pattison, G. Piraux and H. W. Lam, J. Am. Chem. Soc., 2010, 132, 14373–14375 CrossRef CAS PubMed.
- Y. D. Wang, G. Kimball, A. S. Prashad and Y. Wang, Tetrahedron Lett., 2005, 46, 8777–8780 CrossRef CAS.
- L. Lercher, J. F. McGouran, B. M. Kessler, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2013, 52, 10553–10558 CrossRef CAS PubMed.
- H. M. Lee, C. Nieto-Oberhuber and M. D. Shair, J. Am. Chem. Soc., 2008, 130, 16864–16866 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Chem. Commun., 2013, 49, 2747–2749 RSC.
- R. G. Iafe, D. G. Chan, J. L. Kuo, B. A. Boon, D. J. Faizi, T. Saga, J. W. Turner and C. A. Merlic, Org. Lett., 2012, 14, 4282–4285 CrossRef CAS PubMed.
- H. Burghart-Stoll and R. Brückner, Org. Lett., 2011, 13, 2730–2733 CrossRef CAS PubMed.
- D. G. Chan, D. J. Winternheimer and C. A. Merlic, Org. Lett., 2011, 13, 2778–2781 CrossRef CAS PubMed.
- C. G. Frost, S. D. Penrose and R. Gleave, Org. Biomol. Chem., 2008, 6, 4340–4347 RSC.
- K. Hishikawa, H. Nakagawa, T. Furuta, K. Fukuhara, H. Tsumoto, T. Suzuki and N. Miyata, Bioorg. Med. Chem. Lett., 2010, 20, 302–305 CrossRef CAS PubMed.
- J. Li, C. E. Kendig and E. E. Nesterov, J. Am. Chem. Soc., 2007, 129, 15911–15918 CrossRef CAS PubMed.
- D. M. Khramov, E. L. Rosen, J. A. V. Er, P. D. Vu, V. M. Lynch and C. W. Bielawski, Tetrahedron, 2008, 64, 6853–6862 CrossRef CAS.
- Y. Lyu, N. Toriumi and N. Iwasawa, Org. Lett., 2021, 23, 9262–9266 CrossRef CAS PubMed.
- N.-H. Chang, X.-C. Chen, H. Nonobe, Y. Okuda, H. Mori, K. Nakajima and Y. Nishihara, Org. Lett., 2013, 15, 3558–3561 CrossRef CAS PubMed.
- J. Cid, J. J. Carbo and E. Fernandez, Chem. – Eur. J., 2012, 18, 1512–1521 CrossRef CAS PubMed.
- D. Gao and G. A. O’Doherty, Org. Lett., 2010, 12, 3752–3755 CrossRef CAS PubMed.
- S. Essig, S. Bretzke, R. Müller and D. Menche, J. Am. Chem. Soc., 2012, 134, 19362–19365 CrossRef CAS PubMed.
- H. Shimizu, T. Igarashi, T. Miura and M. Murakami, Angew. Chem., Int. Ed., 2011, 50, 11465–11469 CrossRef CAS PubMed.
- J. Carreras, A. Caballero and P. J. Pérez, Chem. – Asian J., 2019, 14, 329–343 CrossRef CAS PubMed.
- S. Murakami, R. Matsubara and M. Hayashi, J. Organomet. Chem., 2021, 933, 121653 CrossRef CAS.
- K. Pathak, S. Gayen, S. Saha, C. Nandi, S. Mishra and S. Ghosh, Chem. – Eur. J., 2022, 28, e202104393 CrossRef CAS PubMed.
- W. J. Jang, W. L. Lee, J. H. Moon, J. Y. Lee and J. Yun, Org. Lett., 2016, 18, 1390–1393 CrossRef CAS PubMed.
- J. V. Obligacion, J. M. Neely, A. N. Yazdani, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2015, 137, 5855–5858 CrossRef CAS PubMed.
- R. Matveeva, C. K. Blasius, H. Wadepohl and L. H. Gade, Dalton Trans., 2021, 50, 6802–6810 RSC.
- N. Gorgas, L. G. Alves, B. Stoeger, A. M. Martins, L. F. Veiros and K. Kirchner, J. Am. Chem. Soc., 2017, 139, 8130–8133 CrossRef CAS PubMed.
- N. Gorgas, B. Stoeger, L. F. Veiros and K. Kirchner, ACS Catal., 2018, 8, 7973–7982 CrossRef CAS.
- C. Gunanathan, M. Hoelscher, F. Pan and W. Leitner, J. Am. Chem. Soc., 2012, 134, 14349–14352 CrossRef CAS PubMed.
- E. A. Romero, R. Jazzar and G. Bertrand, Chem. Sci., 2017, 8, 165–168 RSC.
- M. K. Armstrong and G. Lalic, J. Am. Chem. Soc., 2019, 141, 6173–6179 CrossRef CAS PubMed.
- C. K. Blasius, V. Vasilenko, R. Matveeva, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2020, 59, 23010–23014 CrossRef CAS PubMed.
- J. Chen, X. Shen and Z. Lu, Angew. Chem., Int. Ed., 2021, 60, 690–694 CrossRef CAS PubMed.
- Z. Zuo and Z. Huang, Org. Chem. Front., 2016, 3, 434–438 RSC.
- L. C. Moraes, R. C. Figueiredo, J. P. Espinós, F. Vattier, A. Franconetti, C. Jaime, B. Lacroix, J. Rojo, P. Lara and S. Conejero, Nanoscale, 2020, 12, 6821–6831 RSC.
- K. Endo, M. Hirokami and T. Shibata, Synlett, 2009, 1331–1335 CrossRef CAS.
- X. Wang, Y. Wang, W. Huang, C. Xia and L. Wu, ACS Catal., 2021, 11, 1–18 CrossRef CAS.
- V. Liautard, O. Pascu, C. Aymonier and M. Pucheault, Catal. Today, 2015, 255, 60–65 CrossRef CAS.
- J. Szyling, A. Franczyk, K. Stefanowska, H. Maciejewski and J. Walkowiak, ACS Sustainable Chem. Eng., 2018, 6, 10980–10988 CrossRef CAS.
- J. Szyling, A. Franczyk, K. Stefanowska, M. Klarek, H. Maciejewski and J. Walkowiak, ChemCatChem, 2018, 10, 531–539 CrossRef CAS.
- J. Szyling, A. Franczyk, K. Stefanowska and J. Walkowiak, Adv. Synth. Catal., 2018, 360, 2966–2974 CrossRef CAS.
- J. Ramírez, A. M. Segarra and E. Fernández, Tetrahedron: Asymmetry, 2005, 16, 1289–1294 CrossRef.
- I. D. Gridnev, N. Miyaura and A. Suzuki, J. Org. Chem., 1993, 58, 5351–5354 CrossRef CAS.
- B. H. Lipshutz, Ž. V. Bošković and D. H. Aue, Angew. Chem., Int. Ed., 2008, 47, 10183–10186 CrossRef CAS PubMed.
- K. Semba, T. Fujihara, J. Terao and Y. Tsuji, Chem. – Eur. J., 2012, 18, 4179–4184 CrossRef CAS PubMed.
- Y. D. Bidal, F. Lazreg and C. S. J. Cazin, ACS Catal., 2014, 4, 1564–1569 CrossRef CAS.
- J. Doroszuk, M. Musiejuk, Ł. Ponikiewski and D. Witt, Eur. J. Org. Chem., 2018, 6333–6337 CrossRef CAS.
- J. W. Hall, D. M. L. Unson, P. Brunel, L. R. Collins, M. K. Cybulski, M. F. Mahon and M. K. Whittlesey, Organometallics, 2018, 37, 3102–3110 CrossRef CAS.
- B. Sundararaju and A. Fuerstner, Angew. Chem., Int. Ed., 2013, 52, 14050–14054 CrossRef CAS PubMed.
- S. Gupta, S. Su, Y. Zhang, P. Liu, D. J. Wink and D. Lee, J. Am. Chem. Soc., 2021, 143, 7490–7500 CrossRef CAS PubMed.
- A. Brzozowska, V. Zubar, R.-C. Ganardi and M. Rueping, Org. Lett., 2020, 22, 3765–3769 CrossRef CAS PubMed.
- L. Ferrand, Y. Lyu, A. Rivera-Hernandez, B. J. Fallon, M. Amatore, C. Aubert and M. Petit, Synthesis, 2017, 3895–3904 CAS.
- M. J. González, F. Bauer and B. Breit, Org. Lett., 2021, 23, 8199–8203 CrossRef PubMed.
- D. Bedi, A. Brar and M. Findlater, Green Chem., 2020, 22, 1125–1128 RSC.
- F. Rami, F. Bächtle and B. Plietker, Catal. Sci. Technol., 2020, 10, 1492–1497 RSC.
- X. Feng, P. Ji, Z. Li, T. Drake, P. Oliveres, E. Y. Chen, Y. Song, C. Wang and W. Lin, ACS Catal., 2019, 9, 3327–3337 CrossRef CAS.
- K. Wang, Z. Zhuang, H. Ti, P. Wu, X. Zhao and H. Wang, Chin. Chem. Lett., 2020, 31, 1564–1567 CrossRef CAS.
- Q. Wang, S. E. Motika, N. G. Akhmedov, J. L. Petersen and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 5418–5422 CrossRef CAS PubMed.
- R. Cai, X. Ye, Q. Sun, Q. He, Y. He, S. Ma and X. Shi, ACS Catal., 2017, 7, 1087–1092 CrossRef CAS PubMed.
- S. E. Motika, Q. Wang, N. G. Akhmedov, L. Wojtas and X. Shi, Angew. Chem., Int. Ed., 2016, 55, 11582–11586 CrossRef CAS PubMed.
- Y. Gao, Z.-Q. Wu and K. M. Engle, Org. Lett., 2020, 22, 5235–5239 CrossRef CAS PubMed.
- C.-I. Lee, W.-C. Shih, J. Zhou, J. H. Reibenspies and O. V. Ozerov, Angew. Chem., Int. Ed., 2015, 54, 14003–14007 CrossRef CAS PubMed.
- Q. Feng, H. Wu, X. Li, L. Song, L. W. Chung, Y.-D. Wu and J. Sun, J. Am. Chem. Soc., 2020, 142, 13867–13877 CrossRef CAS PubMed.
- Q. Lai and O. V. Ozerov, J. Organomet. Chem., 2021, 931, 121614 CrossRef CAS.
- M. Uzelac, K. Yuan and M. J. Ingleson, Organometallics, 2020, 39, 1332–1338 CrossRef CAS.
- J. Guo, B. Cheng, X. Shen and Z. Lu, J. Am. Chem. Soc., 2017, 139, 15316–15319 CrossRef CAS PubMed.
- Y. e You and S. Ge, Angew. Chem., Int. Ed., 2021, 60, 20684–20688 CrossRef CAS PubMed.
- A. A. Oluyadi, S. Ma and C. N. Muhoro, Organometallics, 2013, 32, 70–78 CrossRef CAS.
- A. Harinath, J. Bhattcharjee, K. R. Gorantla, B. S. Mallik and T. K. Panda, Eur. J. Org. Chem., 2018, 3180–3192 CrossRef CAS.
- M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, Eur. J. Org. Chem., 2020, 3153–3160 CrossRef CAS.
- L. Koren-Selfridge, H. N. Londino, J. K. Vellucci, B. J. Simmons, C. P. Casey and T. B. Clark, Organometallics, 2009, 28, 2085–2090 CrossRef CAS.
- A. Kaithal, B. Chatterjee and C. Gunanathan, Org. Lett., 2015, 17, 4790–4793 CrossRef CAS PubMed.
- S. A. Orr, J. A. Kelly, A. J. Boutland and V. L. Blair, Chem. – Eur. J., 2020, 26, 4947–4951 CrossRef CAS PubMed.
- R. Arevalo, C. M. Vogels, G. A. MacNeil, L. Riera, J. Perez and S. A. Westcott, Dalton Trans., 2017, 46, 7750–7757 RSC.
- S. Vijjamarri, T. M. O’Denius, B. Yao, A. Kubátová and G. Du, Organometallics, 2020, 39, 3375–3383 CrossRef CAS.
- A. E. King, S. C. E. Stieber, N. J. Henson, S. A. Kozimor, B. L. Scott, N. C. Smythe, A. D. Sutton and J. C. Gordon, Eur. J. Inorg. Chem., 2016, 1635–1640 CrossRef CAS.
- K. A. Gudun, M. Segizbayev, A. Adamov, P. N. Plessow, K. A. Lyssenko, M. P. Balanay and A. Y. Khalimon, Dalton Trans., 2019, 48, 1732–1746 RSC.
- I. Hossain and J. A. R. Schmidt, Eur. J. Inorg. Chem., 2020, 1877–1884 CrossRef CAS.
- M. J. C. Dawkins, E. Middleton, C. E. Kefalidis, D. Dange, M. M. Juckel, L. Maron and C. Jones, Chem. Commun., 2016, 52, 10490–10492 RSC.
- G. S. Kumar, A. Harinath, R. Narvariya and T. K. Panda, Eur. J. Inorg. Chem., 2020, 467–474 CrossRef CAS.
- N. Sarkar, M. Mahato and S. Nembenna, Eur. J. Inorg. Chem., 2020, 2295–2301 CrossRef CAS.
- S. R. Tamang and M. Findlater, J. Org. Chem., 2017, 82, 12857–12862 CrossRef CAS PubMed.
- U. K. Das, C. S. Higman, B. Gabidullin, J. E. Hein and R. T. Baker, ACS Catal., 2018, 8, 1076–1081 CrossRef CAS.
- J. Liu, A. Zhang, H. Song, Q. Tong, C.-H. Tung and W. Wang, Chin. Chem. Lett., 2018, 29, 949–953 CrossRef CAS.
- X. Qi, T. Zheng, J. Zhou, Y. Dong, X. Zuo, X. Li, H. Sun, O. Fuhr and D. Fenske, Organometallics, 2019, 38, 268–277 CrossRef CAS.
- A. Baishya, S. Baruah and K. Geetharani, Dalton Trans., 2018, 47, 9231–9236 RSC.
- S. Lau, C. B. Provis-Evans, A. P. James and R. L. Webster, Dalton Trans., 2021, 50, 10696–10700 RSC.
- S. Bagherzadeh and N. P. Mankad, Chem. Commun., 2016, 52, 3844–3846 RSC.
- H. Zeng, J. Wu, S. Li, C. Hui, A. Ta, S.-Y. Cheng, S. Zheng and G. Zhang, Org. Lett., 2019, 21, 401–406 CrossRef CAS PubMed.
- N. Eedugurala, Z. Wang, U. Chaudhary, N. Nelson, K. Kandel, T. Kobayashi, I. I. Slowing, M. Pruski and A. D. Sadow, ACS Catal., 2015, 5, 7399–7414 CrossRef CAS.
- R. J. Newland, J. M. Lynam and S. M. Mansell, Chem. Commun., 2018, 54, 5482–5485 RSC.
- G. Zhang, J. Cheng, K. Davis, M. G. Bonifacio and C. Zajaczkowski, Green Chem., 2019, 21, 1114–1121 RSC.
- J. Wu, H. Zeng, J. Cheng, S. Zheng, J. A. Golen, D. R. Manke and G. Zhang, J. Org. Chem., 2018, 83, 9442–9448 CrossRef CAS PubMed.
- M. L. Shegavi, A. Baishya, K. Geetharani and S. K. Bose, Org. Chem. Front., 2018, 5, 3520–3525 RSC.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Nat. Commun., 2016, 7, 12610 CrossRef CAS PubMed.
- Z. Huang, D. Liu, J. Camacho-Bunquin, G. Zhang, D. Yang, J. M. Lopez-Encarnacion, Y. Xu, M. S. Ferrandon, J. Niklas, O. G. Poluektov, J. Jellinek, A. Lei, E. E. Bunel and M. Delferro, Organometallics, 2017, 36, 3921–3930 CrossRef CAS.
- R. Wang and S. Park, ChemCatChem, 2021, 13, 1898–1919 CrossRef CAS.
- D. A. Evans and A. H. Hoveyda, J. Org. Chem., 1990, 55, 5190–5192 CrossRef CAS.
- F. Almqvist, L. Torstensson, A. Gudmundsson and T. Frejd, Angew. Chem., Int. Ed. Engl., 1997, 36, 376–377 CrossRef CAS.
- I. Sarvary, F. Almqvist and T. Frejd, Chem. – Eur. J., 2001, 7, 2158–2166 CrossRef CAS PubMed.
- G. Giffels, C. Dreisbach, U. Kragl, M. Weigerding, H. Waldmann and C. Wandrey, Angew. Chem., Int. Ed. Engl., 1995, 34, 2005–2006 CrossRef CAS.
- A. Friberg, I. Sarvary, O. F. Wendt and T. Frejd, Tetrahedron: Asymmetry, 2008, 19, 1765–1777 CrossRef CAS.
- C. R. Sarko, I. C. Guch and M. DiMare, J. Org. Chem., 1994, 59, 705–706 CrossRef CAS.
- C. W. Lindsley and M. DiMare, Tetrahedron Lett., 1994, 35, 5141–5144 CrossRef CAS.
- S. Phothongkam and B.-J. Uang, Asian J. Org. Chem., 2015, 4, 794–799 CrossRef CAS.
- J. Guo, J. Chen and Z. Lu, Chem. Commun., 2015, 51, 5725–5727 RSC.
- J. Wenz, V. Vasilenko, A. Kochan, H. Wadepohl and L. H. Gade, Eur. J. Inorg. Chem., 2017, 5545–5556 CrossRef CAS.
- Y.-N. Liu, H.-F. Su, Y.-W. Li, Q.-Y. Liu, Z. Jaglicic, W.-G. Wang, C.-H. Tung and D. Sun, Inorg. Chem., 2019, 58, 4574–4582 CrossRef CAS PubMed.
- E. Bergamaschi, D. Lunic, L. A. McLean, M. Hohenadel, Y.-K. Chen and C. J. Teskey, Angew. Chem., Int. Ed., 2022, 61, e202114482 CrossRef CAS PubMed.
- S. Mahato, P. Rawal, A. K. Devadkar, M. Joshi, A. Roy Choudhury, B. Biswas, P. Gupta and T. K. Panda, Org. Biomol. Chem., 2022, 20, 1103–1111 RSC.
- P. A. Lummis, M. R. Momeni, M. W. Lui, R. McDonald, M. J. Ferguson, M. Miskolzie, A. Brown and E. Rivard, Angew. Chem., Int. Ed., 2014, 53, 9347–9351 CrossRef CAS PubMed.
- S. G. Roh, J. U. Yoon and J. H. Jeong, Polyhedron, 2004, 23, 2063–2067 CrossRef CAS.
- S.-G. Roh, Y.-C. Park, D.-K. Park, T.-J. Kim and J. H. Jeong, Polyhedron, 2001, 20, 1961–1965 CrossRef CAS.
- M. Locatelli and P. G. Cozzi, Angew. Chem., Int. Ed., 2003, 42, 4928–4930 CrossRef CAS PubMed.
- M. Bandini, P. G. Cozzi, M. de Angelis and A. Umani-Ronchi, Tetrahedron Lett., 2000, 41, 1601–1605 CrossRef CAS.
- D. Mukherjee, A.-K. Wiegand, T. P. Spaniol and J. Okuda, Dalton Trans., 2017, 46, 6183–6186 RSC.
- R. K. Sahoo, M. Mahato, A. Jana and S. Nembenna, J. Org. Chem., 2020, 85, 11200–11210 CrossRef CAS PubMed.
- S. Khoo, J. Cao, F. Ng and C.-W. So, Inorg. Chem., 2018, 57, 12452–12455 CrossRef CAS PubMed.
- T. M. Maier, M. Gawron, P. Coburger, M. Bodensteiner, R. Wolf, N. P. van Leest, B. de Bruin, S. Demeshko and F. Meyer, Inorg. Chem., 2020, 59, 16035–16052 CrossRef CAS PubMed.
- M. R. Elsby, M. Son, C. Oh, J. Martin, M.-H. Baik and R. T. Baker, ACS Catal., 2021, 11, 9043–9051 CrossRef CAS.
- S. Anga, J. Acharya and V. Chandrasekhar, J. Org. Chem., 2021, 86, 2224–2234 CrossRef CAS PubMed.
- K. Lou, F. Zu, J. Yi and C. Cui, Organometallics, 2021, 40, 4092–4097 CrossRef CAS.
- W. Liu, J. Guo, S. Xing and Z. Lu, Org. Lett., 2020, 22, 2532–2536 CrossRef CAS PubMed.
- R. Newar, N. Akhtar, N. Antil, A. Kumar, S. Shukla, W. Begum and K. Manna, Angew. Chem., Int. Ed., 2021, 60, 10964–10970 CrossRef CAS PubMed.
- N. Antil, N. Akhtar, R. Newar, W. Begum, A. Kumar, M. Chauhan and K. Manna, ACS Catal., 2021, 11, 10450–10459 CrossRef CAS.
- L. Li, E. Liu, J. Cheng and G. Zhang, Dalton Trans., 2018, 47, 9579–9584 RSC.
- D. A. Evans and G. C. Fu, J. Org. Chem., 1990, 55, 5678–5680 CrossRef CAS.
- F. Chen, Y. Zhang, L. Yu and S. Zhu, Angew. Chem., Int. Ed., 2017, 56, 2022–2025 CrossRef CAS PubMed.
- T.-T. Gao, W.-W. Zhang, X. Sun, H.-X. Lu and B.-J. Li, J. Am. Chem. Soc., 2019, 141, 4670–4677 CrossRef CAS PubMed.
- G. Zhang, H. Zeng, S. Li, J. Johnson, Z. Mo, M. C. Neary and S. Zheng, Dalton Trans., 2020, 49, 2610–2615 RSC.
- R. Agarwal, Y. Liao, D.-J. Lin, Z.-X. Yang, C.-F. Lai and C.-T. Chen, Org. Chem. Front., 2020, 7, 2505–2510 RSC.
- C.-T. Chen, N. C. Maity, R. Agarwal, C.-F. Lai, Y. Liao and W.-R. Yu, J. Org. Chem., 2020, 85, 6408–6419 CrossRef CAS PubMed.
- E. W. H. Ng, K.-H. Low and P. Chiu, J. Am. Chem. Soc., 2018, 140, 3537–3541 CrossRef CAS PubMed.
- V. Vasilenko, C. K. Blasius, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2017, 56, 8393–8397 CrossRef CAS PubMed.
- X. Ren and Z. Lu, Org. Lett., 2021, 23, 8370–8374 CrossRef CAS PubMed.
- F. Beltran, E. Bergamaschi, I. Funes-Ardoiz and C. J. Teskey, Angew. Chem., Int. Ed., 2020, 59, 21176–21182 CrossRef CAS PubMed.
- B. H. Lipshutz and P. Papa, Angew. Chem., Int. Ed., 2002, 41, 4580–4582 CrossRef CAS PubMed.
- H. Kim and J. Yun, Adv. Synth. Catal., 2010, 352, 1881–1885 CrossRef CAS.
- W. J. Jang, S. M. Song, Y. Park and J. Yun, J. Org. Chem., 2019, 84, 4429–4434 CrossRef CAS PubMed.
- Y. Du, L.-W. Xu, Y. Shimizu, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2008, 130, 16146–16147 CrossRef CAS PubMed.
- R. R. Huddleston, D. F. Cauble and M. J. Krische, J. Org. Chem., 2003, 68, 11–14 CrossRef CAS PubMed.
- C. K. Blasius, V. Vasilenko and L. H. Gade, Angew. Chem., Int. Ed., 2018, 57, 10231–10235 CrossRef CAS PubMed.
- C. Erken, T. Weyhermuller, C. Werle, W. Leitner, C. Erken, A. Kaithal, S. Sen, M. Holscher and W. Leitner, Nat. Commun., 2018, 9, 4521 CrossRef PubMed.
- V. Vasilenko, C. K. Blasius and L. H. Gade, J. Am. Chem. Soc., 2018, 140, 9244–9254 CrossRef CAS PubMed.
- M. K. Barman, K. Das and B. Maji, J. Org. Chem., 2019, 84, 1570–1579 CrossRef CAS PubMed.
- R. Thenarukandiyil, V. Satheesh, L. J. W. Shimon and G. de Ruiter, Chem. – Asian J., 2021, 16, 999–1006 CrossRef CAS PubMed.
- S. Kisan, V. Krishnakumar and C. Gunanathan, ACS Catal., 2018, 8, 4772–4776 CrossRef CAS.
- Q. Wang, L. Li, P. Li, X. Yue, Z. Yang, M. Pu and M. Lei, New J. Chem., 2019, 43, 11493–11496 RSC.
- G. Zhang, J. Wu, S. Zheng, M. C. Neary, J. Mao, M. Flores, R. J. Trovitch and P. A. Dub, J. Am. Chem. Soc., 2019, 141, 15230–15239 CrossRef CAS PubMed.
- P. Ye, Y. Shao, X. Ye, F. Zhang, R. Li, J. Sun, B. Xu and J. Chen, Org. Lett., 2020, 22, 1306–1310 CrossRef CAS PubMed.
- B. Han, J. Zhang, H. Jiao and L. Wu, Chin. J. Catal., 2021, 42, 2059–2067 CrossRef CAS.
- P. Ghosh and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2021, 60, 16035–16043 CrossRef CAS PubMed.
- R. K. Sahoo, N. Sarkar and S. Nembenna, Angew. Chem., Int. Ed., 2021, 60, 11991–12000 CrossRef CAS PubMed.
- A. Y. Khalimon, Dalton Trans., 2021, 50, 17455–17466 RSC.
- A. Nagy, L. Collard, K. Indukuri, T. Leyssens and O. Riant, Chem. – Eur. J., 2019, 25, 8705–8708 CAS.
- C. A. G. Carter, C. M. Vogels, D. J. Harrison, M. K. J. Gagnon, D. W. Norman, R. F. Langler, R. T. Baker and S. A. Westcott, Organometallics, 2001, 20, 2130–2132 CrossRef CAS.
- M. Yalpani, R. Köster and R. Boese, Chem. Ber., 1993, 126, 285–288 CrossRef CAS.
- T. Kitano, T. Komuro and H. Tobita, Organometallics, 2019, 38, 1417–1420 CrossRef CAS.
- T. Itabashi, K. Arashiba, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, Organometallics, 2021, 41, 366–373 CrossRef.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed.
- A. Kaithal, B. Chatterjee and C. Gunanathan, J. Org. Chem., 2016, 81, 11153–11161 CrossRef CAS PubMed.
- J. C. Babón, M. A. Esteruelas, I. Fernández, A. M. López and E. Oñate, Organometallics, 2020, 39, 3864–3872 CrossRef.
- T. T. Nguyen, J.-H. Kim, S. Kim, C. Oh, M. Flores, T. L. Groy, M.-H. Baik and R. J. Trovitch, Chem. Commun., 2020, 56, 3959–3962 RSC.
- G. Nakamura, Y. Nakajima, K. Matsumoto, V. Srinivas and S. Shimada, Catal. Sci. Technol., 2017, 7, 3196–3199 RSC.
- K. A. Gudun, A. Slamova, D. Hayrapetyan and A. Y. Khalimon, Chem. – Eur. J., 2020, 26, 4963–4968 CrossRef CAS PubMed.
- C. Li, S. Song, Y. Li, C. Xu, Q. Luo, Y. Guo and X. Wang, Nat. Commun., 2021, 12, 3813 CrossRef CAS PubMed.
- V. K. Pandey, C. S. Tiwari and A. Rit, Org. Lett., 2021, 23, 1681–1686 CrossRef CAS PubMed.
- A. R. Bazkiaei, M. Wiseman and M. Findlater, RSC Adv., 2021, 11, 15284–15289 RSC.
- M. Ito, M. Itazaki and H. Nakazawa, Inorg. Chem., 2017, 56, 13709–13714 CrossRef CAS PubMed.
- R. T. Baker, J. C. Calabrese and S. A. Westcott, J. Organomet. Chem., 1995, 498, 109–117 CrossRef CAS.
- C. M. Vogels, P. E. O’Connor, T. E. Phillips, K. J. Watson, M. P. Shaver, P. G. Hayes and S. A. Westcott, Can. J. Chem., 2001, 79, 1898–1905 CrossRef CAS.
- T. M. Cameron, R. T. Baker and S. A. Westcott, Chem. Commun., 1998, 2395–2396 RSC.
- M. Itazaki and H. Nakazawa, Molecules, 2018, 23, 2769 CrossRef PubMed.
- S. Pradhan, S. Thiyagarajan and C. Gunanathan, Org. Biomol. Chem., 2021, 19, 7147–7151 RSC.
- C. K. Blasius, N. F. Heinrich, V. Vasilenko and L. H. Gade, Angew. Chem., Int. Ed., 2020, 59, 15974–15977 CrossRef CAS PubMed.
- K. Oshima, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2012, 134, 3699–3702 CrossRef CAS PubMed.
- S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS PubMed.
- J. L. Lortie, T. Dudding, B. M. Gabidullin and G. I. Nikonov, ACS Catal., 2017, 7, 8454–8459 CrossRef CAS.
- X. Wang, Y. Zhang, D. Yuan and Y. Yao, Org. Lett., 2020, 22, 5695–5700 CrossRef CAS PubMed.
- F. Zhang, H. Song, X. Zhuang, C.-H. Tung and W. Wang, J. Am. Chem. Soc., 2017, 139, 17775–17778 CrossRef CAS PubMed.
- J. Liu, J.-Y. Chen, M. Jia, B. Ming, J. Jia, R.-Z. Liao, C.-H. Tung and W. Wang, ACS Catal., 2019, 9, 3849–3857 CrossRef CAS.
- A. S. Dudnik, V. L. Weidner, A. Motta, M. Delferro and T. J. Marks, Nat. Chem., 2014, 6, 1100–1107 CrossRef CAS PubMed.
- A. Kaithal, B. Chatterjee and C. Gunanathan, Org. Lett., 2016, 18, 3402–3405 CrossRef CAS PubMed.
- S. R. Tamang, A. Singh, D. K. Unruh and M. Findlater, ACS Catal., 2018, 8, 6186–6191 CrossRef CAS.
- P. Ji, X. Feng, S. S. Veroneau, Y. Song and W. Lin, J. Am. Chem. Soc., 2017, 139, 15600–15603 CrossRef CAS PubMed.
- H.-C. Yu, S. M. Islam and N. P. Mankad, ACS Catal., 2020, 10, 3670–3675 CrossRef CAS.
- J. Mi, S. Huo, L. Meng and X. Li, Mol. Catal., 2021, 511, 111722 CrossRef CAS.
- S. Kostera, M. Peruzzini and L. Gonsalvi, Catalysts, 2021, 11, 58 CrossRef CAS.
- T. Liu, W. Meng, Q. Q. Ma, J. Zhang, H. Li, S. Li, Q. Zhao and X. Chen, Dalton Trans., 2017, 46, 4504–4509 RSC.
- J. Zhang, J. Chang, T. Liu, B. Cao, Y. Ding and X. Chen, Catalysts, 2018, 8, 508 CrossRef.
- N. P. N. Wellala, H. T. Dong, J. A. Krause and H. Guan, Organometallics, 2018, 37, 4031–4039 CrossRef CAS.
- N. Ma, C. Tu, Q. Xu, W. Guo, J. Zhang and G. Zhang, Dalton Trans., 2021, 50, 2903–2914 RSC.
- N. Ma, Q. Xu, C. Tu, W. Guo and G. Zhang, New J. Chem., 2021, 45, 11275–11283 RSC.
- H.-W. Suh, L. M. Guard and N. Hazari, Polyhedron, 2014, 84, 37–43 CrossRef CAS.
- M. R. Espinosa, D. J. Charboneau, A. Garcia de Oliveira and N. Hazari, ACS Catal., 2019, 9, 301–314 CrossRef CAS.
- Q.-Q. Ma, T. Liu, S. Li, J. Zhang, X. Chen and H. Guan, Chem. Commun., 2016, 52, 14262–14265 RSC.
- A. Burgun, R. S. Crees, M. L. Cole, C. J. Doonan and C. J. Sumby, Chem. Commun., 2014, 50, 11760–11763 RSC.
- A. Aloisi, J.-C. Berthet, C. Genre, P. Thuery and T. Cantat, Dalton Trans., 2016, 45, 14774–14788 RSC.
- E. S. Smirnova, F. Acuna-Pares, E. C. Escudero-Adan, C. Jelsch and J. Lloret-Fillol, Eur. J. Inorg. Chem., 2018, 2612–2620 CrossRef CAS.
- A. Hicken, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2017, 56, 15127–15130 CrossRef CAS PubMed.
- M. J. Sgro and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 11343–11345 CrossRef CAS PubMed.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS PubMed.
- S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2013, 52, 10253–10255 CrossRef CAS PubMed.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2014, 136, 4419–4425 CrossRef CAS PubMed.
- F. Huang, Q. Wang, J. Guo, M. Wen and Z.-X. Wang, Dalton Trans., 2018, 47, 4804–4819 RSC.
- C. K. Ng, J. Wu, T. S. A. Hor and H.-K. Luo, Chem. Commun., 2016, 52, 11842–11845 RSC.
- X. Wang, K. Chang and X. Xu, Dalton Trans., 2020, 49, 7324–7327 RSC.
- T. Janes, K. M. Osten, A. Pantaleo, E. Yan, Y. Yang and D. Song, Chem. Commun., 2016, 52, 4148–4151 RSC.
- L. Li, H. Zhu, L. Liu, D. Song and M. Lei, Inorg. Chem., 2018, 57, 3054–3060 CrossRef CAS PubMed.
- H.-W. Suh, L. M. Guard and N. Hazari, Chem. Sci., 2014, 5, 3859–3872 RSC.
- R. Pal, T. L. Groy and R. J. Trovitch, Inorg. Chem., 2015, 54, 7506–7515 CrossRef CAS PubMed.
- S. Kostera, M. Peruzzini, K. Kirchner and L. Gonsalvi, ChemCatChem, 2020, 12, 4625–4631 CrossRef CAS.
- X. Wan, M. Li and R.-Z. Liao, Chem. – Asian J., 2021, 16, 2529–2537 CrossRef CAS PubMed.
- G. Jin, C. G. Werncke, Y. Escudie, S. Sabo-Etienne and S. Bontemps, J. Am. Chem. Soc., 2015, 137, 9563–9566 CrossRef CAS PubMed.
- S. Desmons, K. Grayson-Steel, N. Nuñez-Dallos, L. Vendier, J. Hurtado, P. Clapés, R. Fauré, C. Dumon and S. Bontemps, J. Am. Chem. Soc., 2021, 143, 16274–16283 CrossRef CAS PubMed.
- S. R. Tamang and M. Findlater, Dalton Trans., 2018, 47, 8199–8203 RSC.
- S. Bagherzadeh and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 10898–10901 CrossRef CAS PubMed.
- P. Sanchez, M. Hernandez-Juarez, N. Rendon, J. Lopez-Serrano, E. Alvarez, M. Paneque and A. Suarez, Dalton Trans., 2018, 47, 16766–16776 RSC.
- D. J. Harrison, N. C. Tam, C. M. Vogels, R. F. Langler, R. T. Baker, A. Decken and S. A. Westcott, Tetrahedron Lett., 2004, 45, 8493–8496 CrossRef CAS.
- A. C. Fernandes, J. A. Fernandes, C. C. Romao, L. F. Veiros and M. J. Calhorda, Organometallics, 2010, 29, 5517–5525 CrossRef CAS.
- I. Cabrita, S. C. A. Sousa and A. C. Fernandes, Tetrahedron Lett., 2010, 51, 6132–6135 CrossRef CAS.
- A. C. Fernandes and C. C. Romão, Tetrahedron Lett., 2007, 48, 9176–9179 CrossRef CAS.
- M. J. Calhorda and P. J. Costa, Dalton Trans., 2009, 8155–8161 RSC.
- L. Huang and H. Wei, New J. Chem., 2014, 38, 5421–5428 RSC.
- C. M. Vogels, A. Decken and S. A. Westcott, Tetrahedron Lett., 2006, 47, 2419–2422 CrossRef CAS.
- K. A. Gudun, R. Zakarina, M. Segizbayev, D. Hayrapetyan, A. Slamova and A. Y. Khalimon, Adv. Synth. Catal., 2022, 364, 601–611 CrossRef CAS.
- L. Zhao, C. Hu, X. Cong, G. Deng, L. L. Liu, M. Luo and X. Zeng, J. Am. Chem. Soc., 2021, 143, 1618–1629 CrossRef CAS PubMed.
- D. Chen, K. Gillman, P. Feng and T. C. Morrill, Main Group Met. Chem., 1994, 17, 413–416 CAS.
- T. Matsuda and H. Kirikae, Organometallics, 2011, 30, 3923–3925 CrossRef CAS.
- A. Kaithal, D. Kalsi, V. Krishnakumar, S. Pattanaik, A. Bordet, W. Leitner and C. Gunanathan, ACS Catal., 2020, 10, 14390–14397 CrossRef CAS.
- W. Dai, S. J. Geib and D. P. Curran, J. Am. Chem. Soc., 2020, 142, 6261–6267 CrossRef PubMed.
- Y. Wang, J. Bai, Y. Yang, W. Zhao, Y. Liang, D. Wang, Y. Zhao and Z. Shi, Chem. Sci., 2021, 12, 3599–3607 RSC.
- H. Song, K. Ye, P. Geng, X. Han, R. Liao, C.-H. Tung and W. Wang, ACS Catal., 2017, 7, 7709–7717 CrossRef CAS.
- A. N. Desnoyer, J. Geng, M. W. Drover, B. O. Patrick and J. A. Love, Chem. – Eur. J., 2017, 23, 11509–11512 CrossRef CAS PubMed.
- K. Aoyagi, K. Matsumoto, S. Shimada, K. Sato and Y. Nakajima, Organometallics, 2019, 38, 210–212 CrossRef CAS.
- M. Yoshifuji, H. Takahashi and K. Toyota, Heteroat. Chem., 1999, 10, 187–196 CrossRef CAS.
- G. Onodera, Y. Nishibayashi and S. Uemura, Organometallics, 2006, 25, 35–37 CrossRef CAS.
- A. C. Chen, L. Ren and C. M. Crudden, Chem. Commun., 1999, 611–612 RSC.
- D. S. Matteson, Tetrahedron, 1998, 54, 10555–10607 CrossRef CAS.
- D. S. Matteson and D. Majumdar, J. Organomet. Chem., 1980, 184, C41–C43 CrossRef CAS.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183–188 CrossRef CAS PubMed.
- J.-P. Goddard, T. Le Gall and C. Mioskowski, Org. Lett., 2000, 2, 1455–1456 CrossRef CAS PubMed.
- H. Lebel and C. Ladjel, J. Organomet. Chem., 2005, 690, 5198–5205 CrossRef CAS.
- L. Ren and C. M. Crudden, Chem. Commun., 2000, 721–722 RSC.
- M. J. O’Donnell, J. T. Cooper and M. M. Mader, J. Am. Chem. Soc., 2003, 125, 2370–2371 CrossRef PubMed.
- E. Hupe, M. I. Calaza and P. Knochel, Tetrahedron Lett., 2001, 42, 8829–8831 CrossRef CAS.
- K. Kondo and S.-I. Murahashi, Tetrahedron Lett., 1979, 20, 1237–1240 CrossRef.
- F. I. Knight, J. M. Brown, D. Lazzari, A. Ricci and A. J. Blacker, Tetrahedron, 1997, 53, 11411–11424 CrossRef CAS.
- K. Maeda and J. M. Brown, Chem. Commun., 2002, 310–311 RSC.
- E. Hupe, I. Marek and P. Knochel, Org. Lett., 2002, 4, 2861–2863 CrossRef CAS PubMed.
- D. S. Matteson and G. Y. Kim, Org. Lett., 2002, 4, 2153–2155 CrossRef CAS PubMed.
- J. S. Panek, F. Xu and A. C. Rondón, J. Am. Chem. Soc., 1998, 120, 4113–4122 CrossRef CAS.
- T. Suzuki, Ismiyarto, Y. Ishizaka, D.-Y. Zhou, K. Asano and H. Sasai, Org. Lett., 2015, 17, 5176–5179 CrossRef CAS PubMed.
- P. Kocienski, K. Jarowicki and S. Marczak, Synthesis, 1991, 1191–1200 CrossRef CAS.
- B. M. Trost, W. Tang and J. L. Schulte, Org. Lett., 2000, 2, 4013–4015 CrossRef CAS PubMed.
- K. A. Scheidt, A. Tasaka, T. D. Bannister, M. D. Wendt and W. R. Roush, Angew. Chem., Int. Ed., 1999, 38, 1652–1655 CrossRef CAS PubMed.
- K. Ibe, H. Aoki, H. Takagi, K. Ken-mochi, Y.-S. Hasegawa, N. Hayashi and S. Okamoto, Tetrahedron Lett., 2015, 56, 2315–2318 CrossRef CAS.
- Z. Hou, Y. Zhang and Y. Wakatsuki, Kidorui, 1997, 30, 40–41 CAS.
- C. Weetman, M. S. Hill and M. F. Mahon, Chem. Commun., 2015, 51, 14477–14480 RSC.
- D. Mukherjee, A. Ellern and A. D. Sadow, Chem. Sci., 2014, 5, 959–964 RSC.
- M. Arrowsmith, M. S. Hill and G. Kociok-Koehn, Chem. – Eur. J., 2013, 19, 2776–2783 CrossRef CAS PubMed.
- M. Arrowsmith, T. J. Hadlington, M. S. Hill and G. Kociok-Koehn, Chem. Commun., 2012, 48, 4567–4569 RSC.
- M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Kohn and C. Weetman, Organometallics, 2011, 30, 5556–5559 CrossRef CAS.
- R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Legare, M.-A. Courtemanche, K. S. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513–2520 CrossRef CAS.
- F.-G. Fontaine, M.-A. Courtemanche and M.-A. Legare, Chem. – Eur. J., 2014, 20, 2990–2996 CrossRef CAS PubMed.
- M.-A. Courtemanche, J. Larouche, M.-A. Legare, W. Bi, L. Maron and F.-G. Fontaine, Organometallics, 2013, 32, 6804–6811 CrossRef CAS.
- T. Wang and D. W. Stephan, Chem. – Eur. J., 2014, 20, 3036–3039 CrossRef CAS PubMed.
- C. Chauvier, A. Tlili, C. Das Neves Gomes, P. Thuery and T. Cantat, Chem. Sci., 2015, 6, 2938–2942 RSC.
- C. Das Neves Gomes, E. Blondiaux, P. Thuery and T. Cantat, Chem. – Eur. J., 2014, 20, 7098–7106 CrossRef CAS PubMed.
- M. Magre, M. Szewczyk and M. Rueping, Chem. Rev., 2022, 122, 8261–8312 CrossRef CAS PubMed.
- H. Liu and M. S. Eisen, Synthesis, 2020, 629–644 CAS.
- A. Das, S. Rej and T. K. Panda, Dalton Trans., 2022, 51, 3027–3040 RSC.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- T. Ishiyama, N. Matsuda, M. Murata, F. Ozawa, A. Suzuki and N. Miyaura, Organometallics, 1996, 15, 713–720 CrossRef CAS.
- H. Braunschweig and A. Damme, Chem. Commun., 2013, 49, 5216–5218 RSC.
- C. N. Iverson and M. R. Smith, Organometallics, 1996, 15, 5155–5165 CrossRef CAS.
- D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036–11037 CrossRef CAS PubMed.
- V. Lillo, M. R. Fructos, J. Ramirez, A. A. C. Braga, F. Maseras, D.-R. M. Mar, P. J. Perez and E. Fernandez, Chem. – Eur. J., 2007, 13, 2614–2621 CrossRef CAS PubMed.
- O. Salvadó and E. Fernández, Molecules, 2020, 25, 1758 CrossRef PubMed.
- Y. G. Lawson, M. J. Gerald Lesley, N. C. Norman, C. R. Rice and T. B. Marder, Chem. Commun., 1997, 2051–2052 RSC.
- Y. Mutoh, K. Yamamoto, Y. Mohara and S. Saito, Chem. Rec., 2021, 21, 3429–3441 CrossRef CAS PubMed.
- J. Guo, Z. Cheng, J. Chen, X. Chen and Z. Lu, Acc. Chem. Res., 2021, 54, 2701–2716 CrossRef CAS PubMed.
- P. Schlichter and C. Werlé, Synthesis, 2022, 517–534 CAS.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- S. J. Geier and S. A. Westcott, Rev. Inorg. Chem., 2015, 35, 69–79 CAS.
- A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2020, 10, 13479–13486 CrossRef CAS.
Footnote |
| † Dedicated to the memory of Professor Stephen A. Westcott, a great mentor and friend. |
| This journal is © The Royal Society of Chemistry 2022 |