
Bacteria-responsive biopolymer-coated nanoparticles for biofilm penetration and eradication†
Yingying
Wang
 a and
Anita
Shukla
*b
a and
Anita
Shukla
*b
aDepartment of Chemistry, Brown University, Providence, Rhode Island 02912, USA
bSchool of Engineering, Center for Biomedical Engineering, Brown University, Providence, Rhode Island 02912, USA. E-mail: anita_shukla@brown.edu
First published on 13th April 2022
Abstract
Biofilm infections are common and can be extremely difficult to treat. Nanoparticles that respond to multiple bacterial stimuli have the potential to successfully prevent and eradicate biofilms. Here, we developed a hyaluronic acid and chitosan coated, antibiotic loaded gelatin nanoparticle, which can undergo hyaluronidase- and gelatinase-mediated degradation regulated by chitosan protonation and swelling in the acidic biofilm microenvironment. We examined the antibiofilm properties of these nanoparticles using a Gram-negative biofilm forming pathogenic bacteria, Vibrio vulnificus. Non-drug loaded responsive nanoparticle formulations exhibited excellent biofilm penetration and retention in preformed V. vulnificus biofilms. Drug loaded formulations were found to exhibit excellent biofilm eradication efficacy, eliminating the biofilm matrix and effectively causing bacterial cell death, which was not observed for treatment with free drug at equivalent concentrations. Overall, these multi-stimuli-responsive nanoparticles have the potential to provide effective and efficient antibiofilm treatment.
1 Introduction
Biofilm-associated bacterial infections are a significant cause of patient morbidity and mortality worldwide. It is estimated that there are ∼17 million new biofilm-associated infections annually in the United States alone causing nearly 550![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths.1 Bacterial biofilms are complex, three-dimensional bacterial communities that are embedded in a self-produced matrix of extracellular polymeric substances (EPS), which includes proteins, polysaccharides, extracellular DNA, and lipids.2 These bacterial clusters are often associated with surfaces, from necrotic tissue to implants.3 Biofilms are involved in various conditions including dental caries, urinary tract infections, burn wound infections, and diabetic foot ulcers.4 Currently, the most common approach to treating acute infections is oral or intravenous administration of United States Food and Drug Administration (FDA) approved antibiotics.5 However, the recalcitrance of biofilms toward antibiotics impairs the therapeutic efficacy of this traditional approach, leading to chronic infections, and the need for higher antibiotic doses and more invasive approaches (e.g., debridement, removal of biofilm-associated devices).6,7 In fact, high antibiotic doses (∼10 to 1000 times greater than concentrations effective against planktonic bacteria) and prolonged treatment times are usually required for the treatment of biofilm-associated bacterial infections, which can exacerbate toxicity and resistance.6,8–10
000 deaths.1 Bacterial biofilms are complex, three-dimensional bacterial communities that are embedded in a self-produced matrix of extracellular polymeric substances (EPS), which includes proteins, polysaccharides, extracellular DNA, and lipids.2 These bacterial clusters are often associated with surfaces, from necrotic tissue to implants.3 Biofilms are involved in various conditions including dental caries, urinary tract infections, burn wound infections, and diabetic foot ulcers.4 Currently, the most common approach to treating acute infections is oral or intravenous administration of United States Food and Drug Administration (FDA) approved antibiotics.5 However, the recalcitrance of biofilms toward antibiotics impairs the therapeutic efficacy of this traditional approach, leading to chronic infections, and the need for higher antibiotic doses and more invasive approaches (e.g., debridement, removal of biofilm-associated devices).6,7 In fact, high antibiotic doses (∼10 to 1000 times greater than concentrations effective against planktonic bacteria) and prolonged treatment times are usually required for the treatment of biofilm-associated bacterial infections, which can exacerbate toxicity and resistance.6,8–10
With recent advances in nanotechnology, nanomaterials have shown promise as new antimicrobials and delivery systems to combat biofilm-associated bacterial infection.11 Numerous nanomaterials have been reported as promising antibiotic alternatives, such as metal-based nanoparticles (NPs) (e.g., silver,12,13 gold,14 and palladium NPs15), cationic polymer NPs,16–18 and quantum dots.19 In addition to their direct use as antimicrobial agents, NPs have great utility as antibacterial drug delivery systems. Specifically, responsive NPs that target infection and biofilm features show great potential to prevent and eradicate biofilms. The unique bacterial microenvironment is characterized by several features that can be used as stimuli for responsive drug delivery systems. For example, bacterial infection sites and biofilms often exhibit lowered pH due to the acidic byproducts of bacterial metabolism.20,21 These sites are also characterized by overexpression of various proteases.22–24 For example, bacterial gelatinases are known to contribute to biofilm formation and virulence through the degradation of a broad range of host substrates.25 Hyaluronidases are another class of enzymes commonly produced by biofilm bacteria, which also serve as virulence factors that are involved in the invasion and penetration of host tissues.26 Although antimicrobial NPs have been designed to respond to these biofilm microenvironmental features,27–30 most reported NPs respond only to a specific stimuli, limiting the broad antibiofilm potential of these materials. Smart NP drug delivery systems that are responsive to multiple bacterial stimuli may greatly enhance the antibiofilm properties of these materials.
In this work, we developed a gelatin NP (GNP) drug delivery system that responds to the lowered pH of the biofilm microenvironment as well as the presence of gelatinases and hyaluronidases to combat bacterial biofilms. GNPs have been widely used in drug delivery since the 1980s,31 with more recent applications ranging from cancer treatment, protein and vaccine delivery, gene delivery, ocular drug delivery, and pulmonary drug delivery to nutraceutical delivery.32,33 Various GNP surface modification strategies have been used to incorporate targeting agents, increase NP stability, and control drug loading and release.34–40 Here, we used layer-by-layer (LbL) self-assembly to adsorb a bilayer of the polycation, chitosan (CS), and the polyanion, hyaluronic acid (HA), on the surface of antibiotic loaded GNPs, with each layer serving a specific mechanistic purpose (Fig. 1). We hypothesized that at biofilm infection sites, bacterial hyaluronidases will degrade the HA layer exposing the underlying CS. The positive surface charge of the CS coated GNPs in the acidic biofilm environment will enable the NPs to more readily attach to biofilm bacteria and the surrounding EPS, allowing enhanced penetration, retention, and possible disruption of the EPS.41,42 The presence of nano- to micro-scale water channels, which are abundant in biofilms, may also support the initial penetration of these NPs into the biofilm.4 The acidic biofilm microenvironment will also trigger swelling of the CS providing bacterial gelatinases greater access to the GNP core.43,44 Gelatinase-triggered degradation of the GNP will increase release of the NP loaded antibiotics, leading to efficient bacteria death. Overall, we hypothesized that these biopolymer coated NPs would lead to effective biofilm eradication.
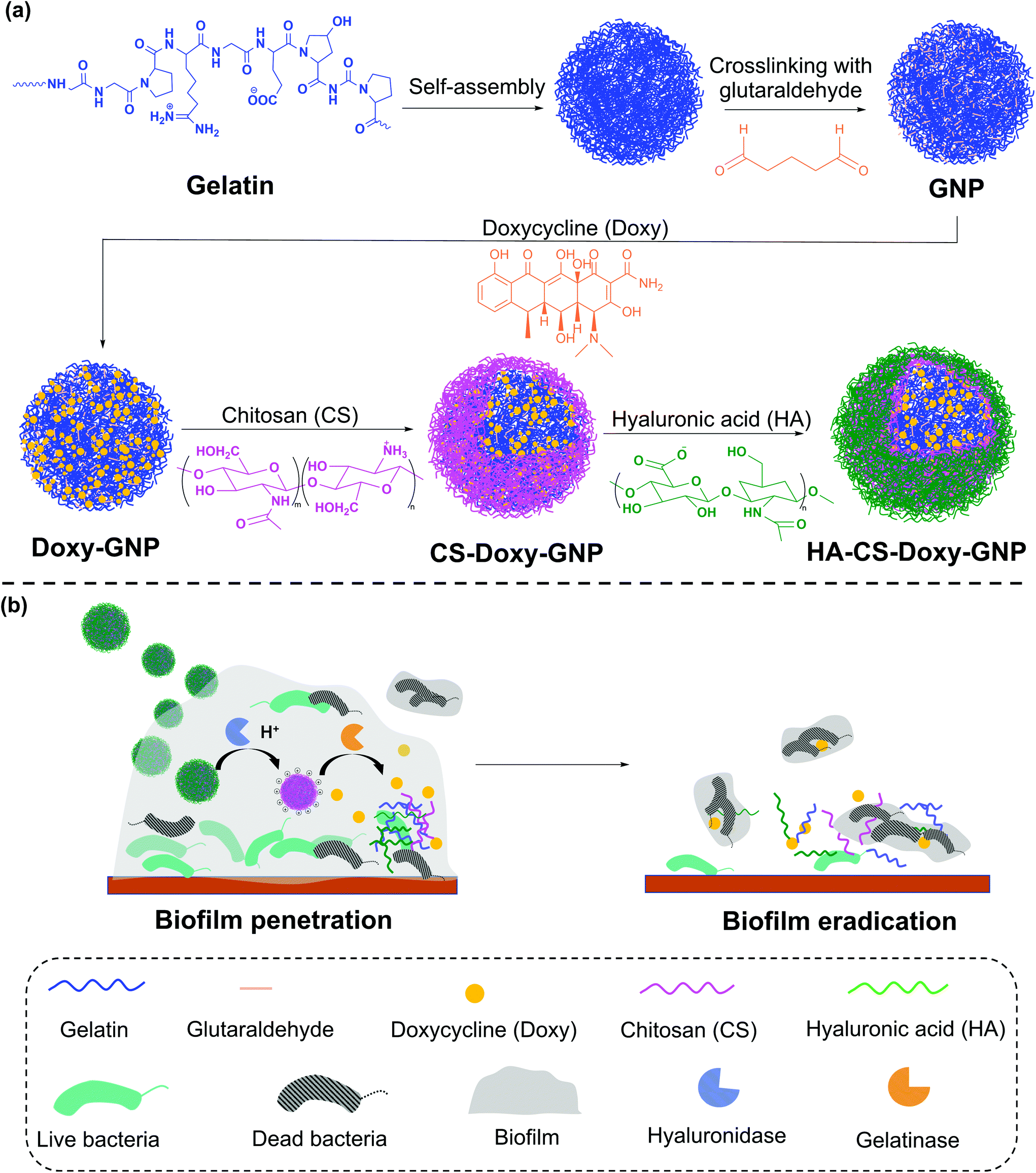 | ||
| Fig. 1 Schematic of synthesis and antibiofilm mechanism of bacteria-responsive HA and CS biopolymer-coated GNPs. (a) Following a two-step desolvation driven self-assembly of GNPs, GNP crosslinking with glutaraldehyde, and Doxy loading, a bilayer of CS and HA is formed on the GNP core via electrostatic interactions, leading to HA-CS-Doxy-GNPs. (b) Bacteria in biofilms secrete hyaluronidases and acidic metabolites, which promotes the degradation of the outermost HA layer, exposing the underlying CS layer. The increasing positive charge of the CS layer in the acidic biofilm microenvironment enhances NP interaction with the bacteria and EPS components, enabling penetration and retention in the biofilm and potential disruption of the EPS. Swelling of the CS at these conditions, increases access of bacterial gelatinases to the GNP core, causing degradation of the core and increased Doxy release to promote bacterial cell death. | ||
We synthesized these biopolymer coated GNPs and characterized their activity against a pathogenic, Gram-negative bacteria, Vibrio vulnificus. V. vulnificus causes severe wound infections and sepsis with a fatality rate of ∼20%.45V. vulnificus also forms biofilms and secretes common enzymes found in many biofilm microenvironments.46 Doxycycline (Doxy), an FDA-approved tetracycline antibiotic, was loaded in the GNPs due to its common use in treatment of V. vulnificus infections.47 The physicochemical properties and drug release characteristics of Doxy loaded GNPs, including non-coated (Doxy-GNPs), CS coated (CS-Doxy-GNPs), and the complete CS and HA coated GNPs (HA-CS-Doxy-GNPs) were evaluated. We confirmed pH and enzyme-responsive drug release from these formulations, and observed promising biofilm penetration ability, antibiofilm properties, and EPS disruption by the HA and CS coated responsive NPs. The results of this work, support the use of multi-stimuli-responsive NPs to combat bacterial biofilms.
2 Experimental section
2.1 Materials
Gelatin (Type B, Bloom number of ∼225 g), glutaraldehyde (50% (w/w) in water), doxycycline hydrochloride, CS (≥75% deacetylated), sucrose, type IV collagenase (i.e., gelatinase) from Clostridium histolyticum, hyaluronidase from bovine testes, 10× Dulbecco's phosphate buffered saline (PBS, pH 7.4), acetone, ethanol (200 proof, anhydrous, 99.5%), sodium chloride (NaCl), glucose, and vancomycin were obtained from Sigma-Aldrich (St Louis, MO). HA sodium salt (average molecular weight ∼36 kDa) was obtained from Lifecore Biomedical (Chaska, MN). Silicon wafers were purchased from WaferPro (Santa Clara, CA). V. vulnificus (ATCC 27562), human umbilical vein endothelial cells (HUVEC), and mouse embryonic fibroblasts (NIH 3T3) were obtained from American Type Culture Collection (ATCC, Manassas, VA). Tryptic soy broth (TSB), FilmTracer SYPRO Ruby biofilm matrix stain, and LIVE/DEAD BacLight Bacterial Viability Kit were purchased from Thermo Fisher Scientific (Waltham, MA). Lennox broth (LB) and crystal violet were purchased from Millipore Sigma (St Louis, MO). Bacto agar was obtained from BD Biosciences (San Jose, CA). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco-BRL (Grand Island, NY). Penicillin–streptomycin was obtained from Caisson Laboratories (Smithfield, UT). Endothelial Cell Growth Medium-2 (EGM-2) BulletKit was purchased from Lonza (Basel, Switzerland). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies (Tokyo, Japan). Bovine red blood cells (BRBCs) (10% in 1× PBS) and single donor human red blood cells (HRBCs) were purchased from Innovative Research (Novi, MI). Frozen porcine skin tissue was obtained from a butcher shop (Providence, RI). All chemicals were of analytical reagent quality or high-performance liquid chromatography grade. Ultrapure water (18.2 MΩ cm Milli-Q, Millipore Sigma, Billerica, MA) was utilized in all experiments requiring water. Room temperature (RT) refers to ∼23 °C.2.2 Synthesis of NPs
GNPs were prepared using two-step desolvation, as previously reported.28,34 Briefly, 1.25 g gelatin was dissolved in 25 mL of water at 50 °C while stirring (360 rpm). In the first desolvation step, 25 mL of acetone was added into the solution dropwise while stirring (360 rpm). After the addition of acetone was complete, stirring was halted. The gel-like gelatin fractions precipitated after 15 min and the opaque supernatant containing the lower molecular weight gelatin was discarded. The sediment was re-suspended by adding 25 mL of water at 50 °C while stirring (360 rpm), and pH was adjusted to 11. Next, 75 mL of acetone was added at a flow rate of approximately 1 mL min−1 to form the GNPs under continuous stirring (600 rpm), inducing the second desolvation step and particle formation. Finally, 150 μL of 25% glutaraldehyde was added dropwise to crosslink the particles. This solution was left to stir for 18 h at RT. The GNPs obtained were collected and washed three times with water. All NP collection and wash steps were carried out via centrifugation at RT (16000g for 20 min).
Doxy loaded GNPs (Doxy-GNPs) were synthesized by mixing varying ratios of lyophilized GNPs with Doxy solution (15 mg mL−1). After swelling and loading of Doxy into GNPs for 24 h at RT, the NPs were washed three times with water at RT to remove unloaded free Doxy. To form the (CS/HA) bilayer coating on Doxy-GNPs, first the CS layer was adsorbed by gradual addition of 10 mL of 1 mg mL−1 Doxy-GNP suspension in water (pH 6) to 10 mL of 1 mg mL−1 aqueous CS (pH 6) under stirring (500 rpm). After stirring for 1 h, the CS-Doxy-GNPs were collected and washed three times with water, followed by a final resuspension in 10 mL of water (pH 6). The HA layer was fabricated by adding 10 mL of 1 mg mL−1 aqueous HA (pH 6) to the CS-Doxy-GNP suspension dropwise under stirring (500 rpm) for 1 h. The resulting HA-CS-Doxy-GNPs were collected and washed three times with water. All washes were collected for quantification of Doxy loss during bilayer assembly.
2.3 Characterization of NPs
2.4 Effects of NPs on planktonic bacteria
The minimum inhibitory concentrations (MICs) of NPs against V. vulnificus were determined using microdilution assays as previously reported.50,51 Briefly, free Doxy and Doxy loaded NPs were serially diluted 2-fold in LB in 96-well plates to obtain Doxy concentrations ranging from 0.03 to 4.0 μg mL−1. V. vulnificus in LB media in its logarithmic growth phase was added to each well at a final concentration of 1 × 105 colony forming units (CFU) per mL. Positive controls (PC) of bacteria cultured in LB only and negative controls (NC) of LB without bacteria were included. After 16–18 h of shaking (100 rpm) at 37 °C, the optical density at 600 nm (OD600) was measured using a plate reader. The normalized bacteria density (%) was calculated as follows: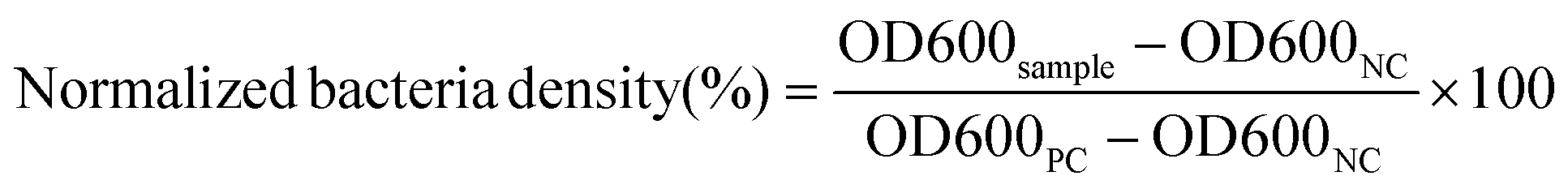
2.5 Effects of NPs on biofilm bacteria
:500 (v/v) in LB media supplemented with 2% (w/v) NaCl (LBS) at pH 5.5, and OD600 was monitored over time. Once in its logarithmic growth phase (OD600 = 0.1, which is ∼1.5 × 107 CFU mL−1), biofilms were formed by static incubation of bacteria at 37 °C for 48 h either in chamber slides or multi-well plates (8-well chamber slides and 96-well plates utilized a 200 μL culture volume per well, while 48-well plates utilized a 300 μL culture volume per well). Every 24 h, the biofilms were gently washed three times with 1× PBS and fresh LBS was added.
Further methods utilized in characterization of gelatinase and hyaluronidase production by V. vulnificus are included in ESI.†
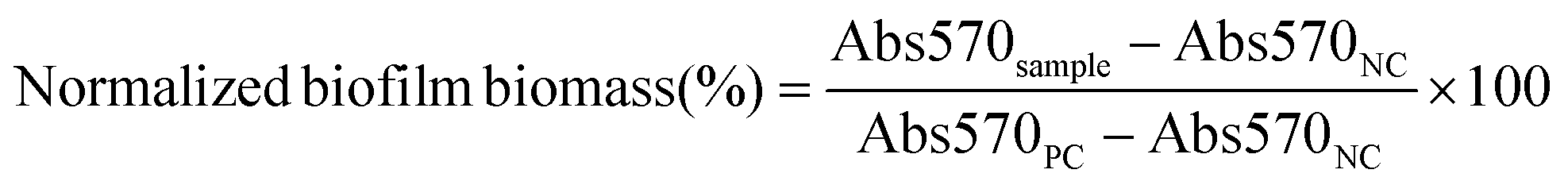
To assess morphology, biofilms were formed on silicon wafer surfaces (0.5 cm × 0.5 cm) in 24-well plates. After treatment, the biofilms were gently washed three times in 1× PBS and fixed with 2.5% (w/v) glutaraldehyde at 4 °C for 4 h. After the biofilms were washed with 1× PBS, samples were dehydrated gradually through a series of incubations with 50%, 60%, 70%, 90% and 99.5% ethanol (10 min each). The dehydrated samples were lyophilized, sputter coated with gold-palladium, and imaged via SEM as described for NP characterization.
For LIVE/DEAD viability staining, biofilms were formed on 8-well chambered cover glass. After treatment, staining was conducted according to the manufacturer protocol. Briefly, a working fluorescent stain solution was prepared by adding 3 μL of SYTO 9 and 3 μL of propidium iodide to 1 mL of water. Treated biofilms were incubated with 200 μL of this working solution for 30 min at RT, followed by rinsing three times with water to remove excess stain. The stained bacteria were imaged with CLSM with an Apo LWD 25×/1.10 W water immersion objective.
Colony enumeration was also conducted by first dispersing biofilms formed in 48-well plates by sonication following treatment. Each sample was then serially diluted in LB, and 10 μL of the dilution was plated onto LB agar plates. Agar plates were imaged and CFU were counted following incubation for 24 h at 37 °C.
Biofilm biomass was assessed upon incubation of 48 h aged biofilms in 96-well plates with serial dilutions of the various treatment groups at Doxy concentrations ranging from 0.78 to 50 μg mL−1 for 24 h. PCs of biofilms cultured in LBS at pH 5.5 and NCs of LBS at pH 5.5 without bacteria were included. Following a 24 h incubation, the biofilm biomass was quantified using crystal violet staining as described earlier.
2.6 Ex vivo porcine skin infection model
Frozen porcine skin tissue was thawed and washed with 1× PBS. A biopsy punch (8 mm) was used to create tissue sections from freshly thawed tissue. A 1.5 mm deep well was created in the center of each of these skin samples using a smaller 3 mm biopsy punch. The tissue pieces were sterilised by immersing in 70% ethanol for 2 h and drying for 30 min in a CellGard™ Energy Saver class II, type A2 biosafety cabinet (NuAire, Plymouth, MN). The sterile tissue was then placed on soft TSB agar plates containing 0.5% (w/v) agar with 100 μg mL−1 vancomycin. Note that vancomycin was included to prevent contamination by non-V. vulnificus bacteria. The skin was then infected by adding 10 μL of V. vulnificus suspension (OD600 = 0.1) in the 3 mm diameter well in each sample. The inoculated skin pieces were incubated for 48 h at 37 °C in a humidified chamber. The tissue samples were transferred to a new agar plate every 24 h. At 48 h, 15 μL of Doxy or HA-CS-Doxy-GNPs at a Doxy concentration of 10, 50, or 100 μg mL−1 was added to the infected skin samples and incubated for an additional 24 h. Controls of tissue inoculated with bacteria only and tissue without bacteria treated with LBS at pH 5.5 were included. After incubation, skin samples were washed with 1× PBS and homogenized using a gentle MACS™ dissociator (Miltenyi Biotec, Waltham, MA). The homogenized samples were serially diluted 10-fold (101 to 106) and each dilution plated onto LB agar plates. The plates were incubated at 37 °C for 24 h followed by CFU enumeration.2.7 Assessing NP cytocompatibility
The cytotoxicity of each NP formulation was evaluated for HUVEC and NIH 3T3 using a CCK-8 viability assay. Briefly, HUVEC and NIH 3T3 cells were seeded at a density of ∼5000 cells per cm2 in EGM-2 and DMEM supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin, respectively, and incubated at 37 °C with 5% CO2. After 24 h, cells were treated with free Doxy, Doxy-GNPs, CS-Doxy-GNPs, or HA-CS-Doxy-GNPs at a Doxy concentration of 50 μg mL−1 in their respective medias. Cells treated with media only and wells containing no cells or NPs were included as PCs and NCs, respectively. Following a 24 h incubation, the media was removed, cells were rinsed with 1× PBS three times and 10 μL of CCK-8 solution was added to each well. After incubation for 4 h at 37 °C with 5% CO2, the absorbance was read at 450 nm (Abs450) using a plate reader. Normalized cell viability was calculated as follows: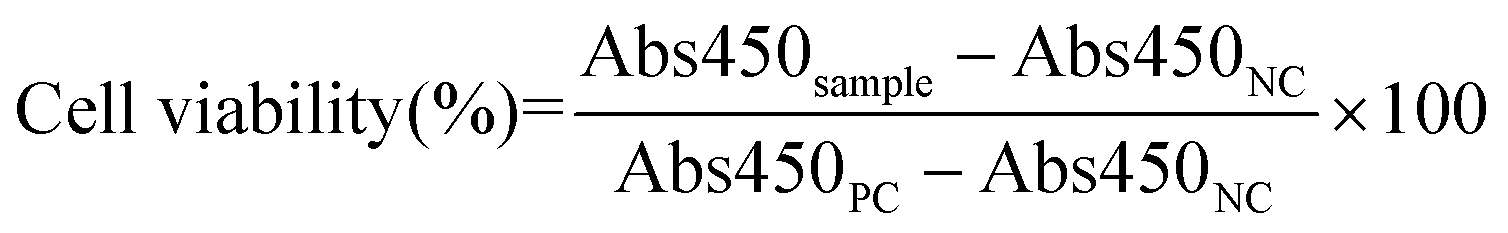
RBC hemolysis upon exposure to the NP formulations was also assessed as previously reported52,53 and described briefly in ESI.†
2.8 Statistical analysis
Data are presented as mean ± standard deviation of three biological replicates at minimum. Statistical analysis was performed with GraphPad Prism™ using either one- or two-way analysis of variance (ANOVA; α = 0.05) with Tukey's post hoc analysis.3 Results and discussion
3.1 Justification for NP design
In this work, we developed a NP drug delivery system to combat bacterial biofilms, using V. vulnificus as a pathogenic, biofilm forming bacteria to investigate efficacy. These NPs were functionalized with a bacteria-responsive LbL biopolymer coating consisting of an inner layer of CS and outer layer of HA. The gelatin core was chosen as the antibiotic depot that can degrade in the presence of bacterial gelatinases, while the CS layer was chosen as a pH-responsive layer to regulate this gelatinase induced degradation and encourage electrostatic interactions with the biofilm. Finally, the HA layer was chosen as a bacterial hyaluronidase-responsive layer to regulate CS swelling in the acidic biofilm microenvironment. Prior to synthesizing these NPs, we examined V. vulnificus biofilm formation and enzyme production to provide support for the proposed NP design.We first confirmed V. vulnificus biofilm formation using crystal violet staining. Although V. vulnificus formed biofilms in various culture conditions, the most robust biofilm formation was observed in LBS at pH 5.5 (Fig. S2a†). SEM images of the V. vulnificus biofilm showed an abundant EPS, absent from SEM images of planktonic V. vulnificus (Fig. S2b and c†). Next we examined the production of gelatinases and hyaluronidases by the specific strain of V. vulnificus used in this study. It has previously been seen that these enzymes are among the virulence factors produced by V. vulnificus.46 We found that liquid gelatin at 50 °C remained liquid following incubation with V. vulnificus and cooling to below 25 °C, where it normally forms a gel (Fig. S3a†). These results confirmed gelatinase production by the V. vulnificus. Hyaluronidase production was also confirmed using a hyaluronic acid agar plate assay as previously reported,54 indicated by a clear region surrounding the V. vulnificus (Fig. S3b†). Having confirmed robust V. vulnificus biofilm formation at acidic pH, where CS is protonated and can swell, and V. vulnificus production of gelatinases and hyaluronidases, we proceeded to fabricate the proposed responsive NPs.
3.2 Characterizing NP physicochemical properties
Gelatin is a naturally derived, biocompatible, biodegradable, and relatively inexpensive macromolecule that has been widely used in drug delivery.34,55–58 The GNP core was prepared using a two-step desolvation approach, followed by Doxy loading. As these are the first reported Doxy loaded GNP formulations, we first examined several Doxy to GNP ratios for fabrication. A 0.2 (w/w) ratio was found to lead to an optimal Doxy encapsulation efficiency and drug loading of ∼77% and ∼12%, respectively (Fig. S4a†). These Doxy-GNPs were then coated with responsive biopolymer layers via adsorption of CS followed by HA. The LbL coating approach applied here has previously been used to develop functionalized nanoparticles primarily for applications in cancer.59–61 LbL has also been applied to NP coatings for antibacterial applications, mainly aimed at eliminating planktonic bacteria or preventing biofilm formation,62,63 with a limited investigation of biofilm eradication.64 In this work, we combined the LbL coating of bacteria-responsive polymers with a bacteria-responsive NP core and investigated both biofilm prevention and eradication effects. After coating with CS and HA, the Doxy encapsulation efficiency and drug loading decreased to ∼26% and ∼6%, respectively (Fig. S4b†). Similar decreases in drug loading are typically observed for NPs functionalized with LbL coatings for other applications.65,66Following NP fabrication, we investigated the size, charge, and morphology of GNPs, Doxy-GNPs, CS-Doxy-GNPs, and HA-CS-Doxy-GNPs. DLS analysis showed no significant increase in the hydrodynamic diameter of ∼215 nm for the GNP core before and after Doxy loading (Fig. 2a). The average hydrodynamic diameter was observed to increase with the addition of the polymer coating layers from ∼215 nm for Doxy-GNPs to ∼240 nm and ∼290 nm for CS-Doxy-GNPs and HA-CS-Doxy-GNPs, respectively, indicating successful biopolymer adsorption. The ζ-potential of the GNP formulations was also monitored to further confirm biopolymer coating (Fig. 2a). As with the size, the GNP core exhibited no significant difference in ζ-potential before and after Doxy loading (∼−20 mV). However, after CS coating, the mean ζ-potential of the NPs increased dramatically to ∼32 mV. At the assembly conditions used, the amino groups of the cationic CS can both interact with the carboxylate on the surface of the Doxy-GNPs and cause charge reversal of the Doxy-GNPs once adsorbed, leading to the positive ζ-potential of CS-Doxy-GNPs. Upon coating with HA, which can interact with CS via electrostatic interactions due to its carboxyl groups, a surface charge reversal was observed with a new ζ-potential of ∼−25 mV. Together, the size increase and surface charge reversal during LbL assembly confirmed the successful fabrication of the GNP formulations. Environmental SEM showed the generally spherical morphology of the NPs with uniform sizes (Fig. 2b). The polydispersity index (PDI) obtained via DLS also confirmed this observation, indicating PDIs of 0.20 ± 0.01, 0.26 ± 0.02, 0.13 ± 0.02, and 0.26 ± 0.01 for GNPs, Doxy-GNPs, CS-Doxy-GNPs, and HA-CS-Doxy-GNPs, respectively.
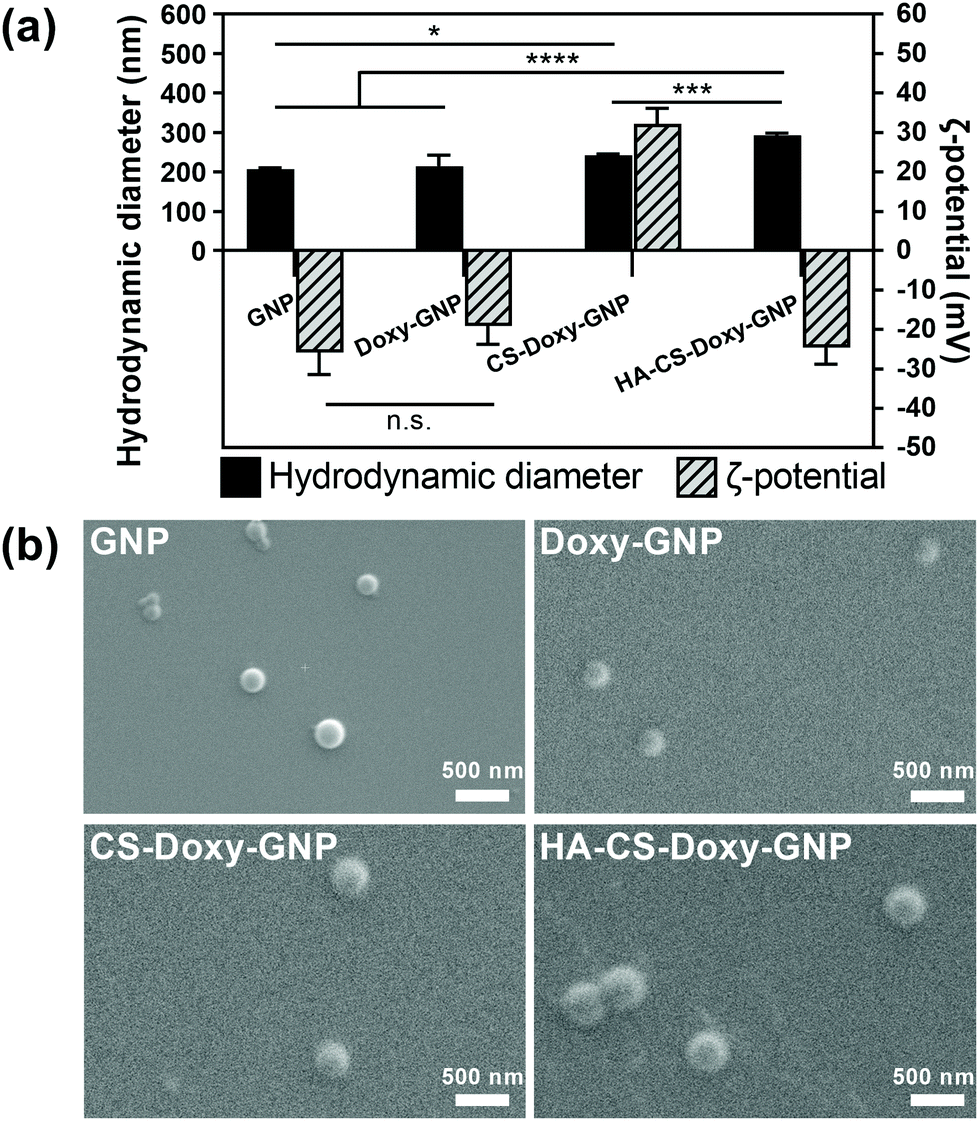 | ||
| Fig. 2 Size, charge, and morphology of responsive NP formulations. (a) Hydrodynamic diameter and ζ-potential of GNPs determined by DLS. Results are shown as mean ± standard deviation. Statistical significance (*p < 0.05, **p < 0.001, and ****p < 0.0001) between groups was determined using two-way ANOVA with Tukey's post hoc analysis; n = 3. (b) SEM images of NPs. Scale bar: 500 nm. SEM images are representative of at least three independent experiments per NP formulation. | ||
3.3 Assessing responsive NP drug release
With the NP formulations successfully synthesized, we proceeded to quantify Doxy release from Doxy-GNPs, CS-Doxy-GNPs, and HA-CS-Doxy-GNPs. Release was examined over 48 h at 37 °C in various incubation conditions including 1× PBS at pH 7.4, 1× PBS at pH 5, gelatinases in 1× PBS at pH 5, hyaluronidases in 1× PBS at pH 5, and V. vulnificus conditioned culture medium at pH 5.5 (Fig. 3). As shown in (Fig. 3a), Doxy-GNPs exhibited a rapid release at all conditions, releasing more than 40% of the encapsulated Doxy within 4 h. Following 4 h, additional release was only observed in gelatinases or conditioned medium resulting in greater than 80% cumulative Doxy release by 48 h (Fig. 3d). The addition of the CS layer in the CS-Doxy-GNPs reduced the burst release observed from Doxy-GNPs at 4 h from ∼40% down to ∼10% (Fig. 3b). Over time, low pH conditions resulted in greater drug release from CS-Doxy-GNPs compared to other release conditions, likely due to CS protonation and swelling. As shown in Fig. 3e, after 48 h CS-Doxy-GNP in the 1× PBS and hyaluronidase incubation conditions (both at pH 5) led to ∼45% cumulative Doxy release compared to ∼30% release in 1× PBS at pH 7.4. Incubation in gelatinases or conditioned medium also increased release as expected, resulting in ∼60% cumulative Doxy release over 48 h. The addition of the HA layer further slowed drug release. Less than 20% of the encapsulated Doxy was released from HA-CS-Doxy-GNPs at all conditions over the first ∼12 h (Fig. 3c). Following this period, only incubation in hyaluronidases or conditioned medium influenced release (∼40% release by 48 h) (Fig. 3f). Together these results demonstrated that the biopolymer coated GNPs exhibited pH, hyaluronidase, and gelatinase responsive drug release behavior.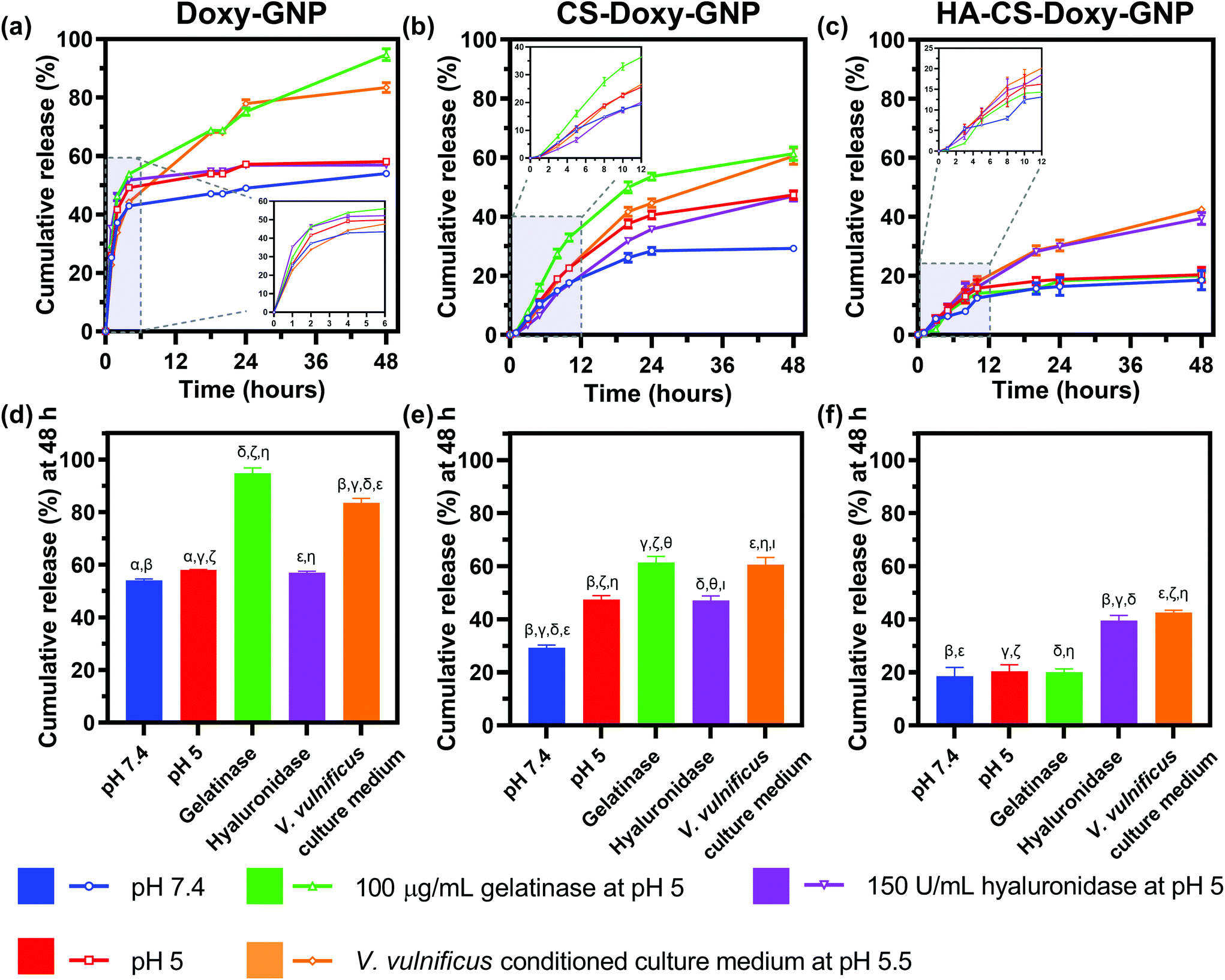 | ||
| Fig. 3 Drug release from all responsive NP formulations. Normalized cumulative Doxy release from (a) Doxy-GNPs, (b) CS-Doxy-GNPs, and (c) HA-CS-Doxy-GNPs in 1× PBS at pH 7.4, 1× PBS at pH 5, gelatinases in 1× PBS at pH 5, hyaluronidases in 1× PBS at pH 5, and V. vulnificus conditioned culture medium at pH 5.5 at 37 °C over 48 h. Insets show the early release time points. Cumulative drug release at 48 h from (d) Doxy-GNPs, (e) CS-Doxy-GNPs, and (f) HA-CS-Doxy-GNPs in the varying release conditions. Results are shown as mean ± standard deviation. Statistical significance between conditions is indicated by matching Greek letters using one-way ANOVA with Tukey's post hoc analysis (α indicates p < 0.05 while β, γ, δ, ε, ζ, η, θ, and ι indicate p < 0.0001); n = 3. | ||
3.4 Assessing the biofilm penetration ability of responsive NPs
The biofilm EPS plays a critical role in antimicrobial resistance of biofilms. The net negative charge of the EPS can sequester positively charged antimicrobial agents and/or repel negatively charged antimicrobial agents. Therefore, it is important to study the biofilm penetration ability for any new therapeutic or antimicrobial drug delivery system. We monitored the penetration of NPs formulated without Doxy encapsulation into V. vulnificus biofilms using CLSM (Fig. 4). Biofilm EPS was observed through staining with SYPRO Ruby matrix stain, while GNPs, CS-GNPs, and HA-CS-GNPs were visualized via their autofluorescence. There are two groups that produce autofluorescence in the GNP core, namely the bonds of the Schiff base (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and carbon–carbon double bonds (CC) formed during glutaraldehyde-mediated crosslinking of the gelatin.67 Fig. S5† shows the fluorescence spectra upon 480 nm excitation of the GNPs and the gelatin, CS, and HA biopolymers alone, confirming autofluorescence of the GNPs. In Fig. 4 the biofilm matrix appears red and the NPs are green. In the PBS treatment control we observed no green signal as expected. For all of the NP formulations, no penetration was observed at the early 1 h timepoint. However, by 3 h some level of NP interaction with the biofilm was observed for all formulations. For GNPs, by 24 h no green was seen in the biofilm, likely indicating degradation of the NPs via V. vulnificus gelatinases at these conditions. CS-GNPs were still observed at 24 h in the biofilms, indicating some protection against early degradation enabled by the biopolymer coating. Finally, for HA-CS-GNP treated biofilms, an intense NP green was observed throughout the entire biofilm structure at 24 h, indicating that these NPs were able to penetrate and remain stable for longer within preformed V. vulnificus biofilms than the GNP core and single biopolymer layer coated, CS-GNP formulation.
N) and carbon–carbon double bonds (CC) formed during glutaraldehyde-mediated crosslinking of the gelatin.67 Fig. S5† shows the fluorescence spectra upon 480 nm excitation of the GNPs and the gelatin, CS, and HA biopolymers alone, confirming autofluorescence of the GNPs. In Fig. 4 the biofilm matrix appears red and the NPs are green. In the PBS treatment control we observed no green signal as expected. For all of the NP formulations, no penetration was observed at the early 1 h timepoint. However, by 3 h some level of NP interaction with the biofilm was observed for all formulations. For GNPs, by 24 h no green was seen in the biofilm, likely indicating degradation of the NPs via V. vulnificus gelatinases at these conditions. CS-GNPs were still observed at 24 h in the biofilms, indicating some protection against early degradation enabled by the biopolymer coating. Finally, for HA-CS-GNP treated biofilms, an intense NP green was observed throughout the entire biofilm structure at 24 h, indicating that these NPs were able to penetrate and remain stable for longer within preformed V. vulnificus biofilms than the GNP core and single biopolymer layer coated, CS-GNP formulation.
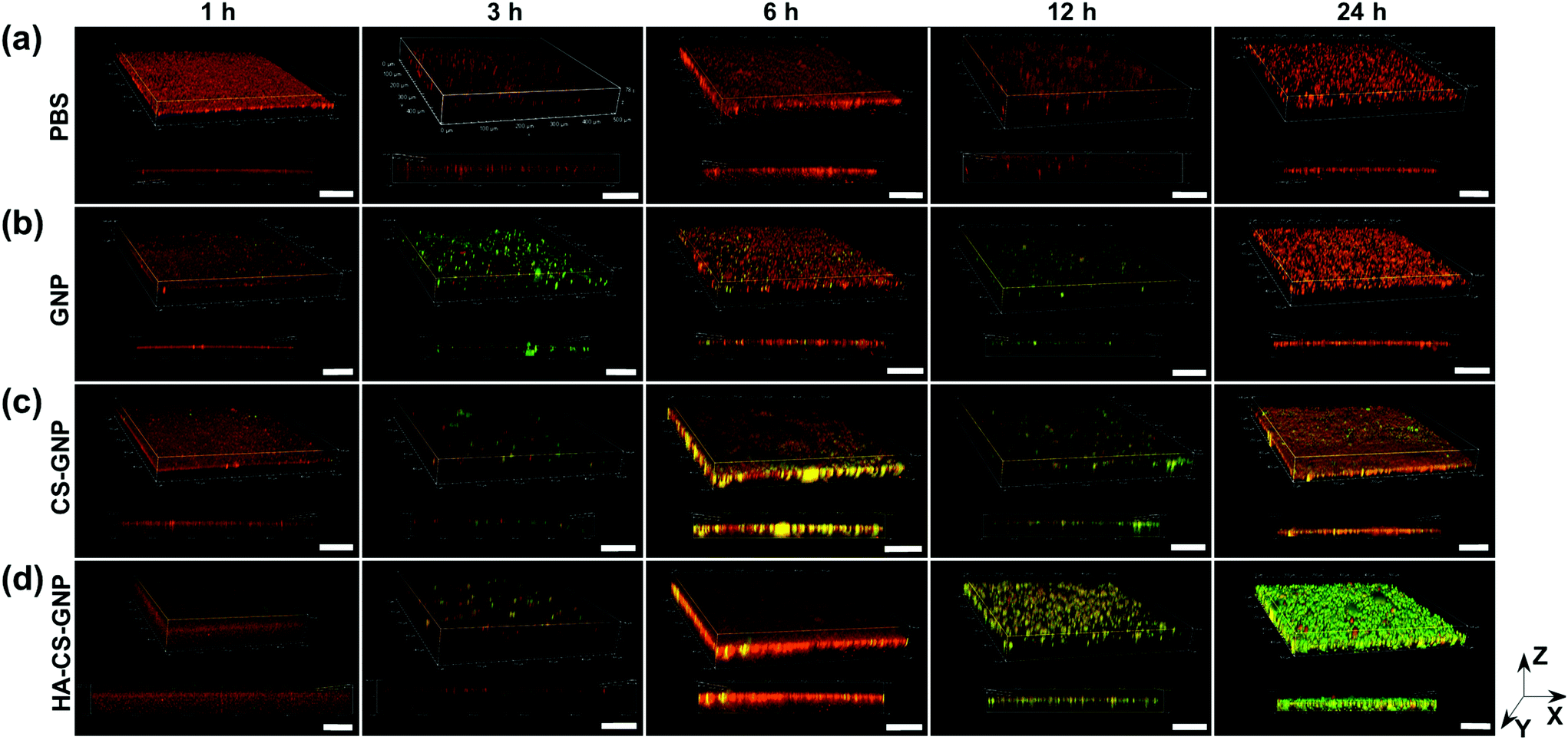 | ||
| Fig. 4 Biofilm penetration ability of NP formulations against 48 h aged, preformed V. vulnificus biofilms. CLSM images of V. vulnificus biofilms after treatment with (a) 1× PBS, (b) GNPs (1 mg mL−1), (c) CS-GNPs (1 mg mL−1), or (d) HA-CS-GNPs (1 mg mL−1) over 24 h. Both three-dimensional and cross-sectional images are shown. The biofilm EPS was stained with SYPRO Ruby (red) and the NPs exhibited autofluorescence (green). Scale bar: 100 μm. CLSM images are representative of at least three independent biofilms. | ||
3.5 Investigating the antibiofilm properties of responsive NPs
After observing excellent biofilm penetration by the HA-CS-GNPs, we proceeded to investigate the antibiofilm efficacy of the Doxy loaded formulations. As a comparison, we initially examined the antibacterial properties of these NPs against planktonic V. vulnificus bacteria. Using microdilution assays, we determined the MIC of the NP formulations and free Doxy against V. vulnificus (note, MIC is the lowest drug concentration that prevents the growth of V. vulnificus). Fig. 5a shows the normalized V. vulnificus density over a range of concentrations for free Doxy and Doxy loaded NPs. The MICs of all treatments against V. vulnificus were similar, with free Doxy exhibiting a slightly lower MIC of ∼0.25 μg mL−1 compared with Doxy loaded NP formulations, which all exhibited MICs of ∼0.5 μg mL−1 Doxy. We noted greater inhibition of bacterial growth at concentrations below the MIC for free Doxy compared with specific NP formulations. For example, at an equivalent concentration of 0.06 μg mL−1 Doxy, treatment with free Doxy led to a significantly lower normalized bacteria density compared with all of the NP formulations examined. This result is likely due to the increased availability of this free Doxy to the planktonic bacteria compared to Doxy loaded in the NP formulations, which is not completely released during the timescale of this experiment.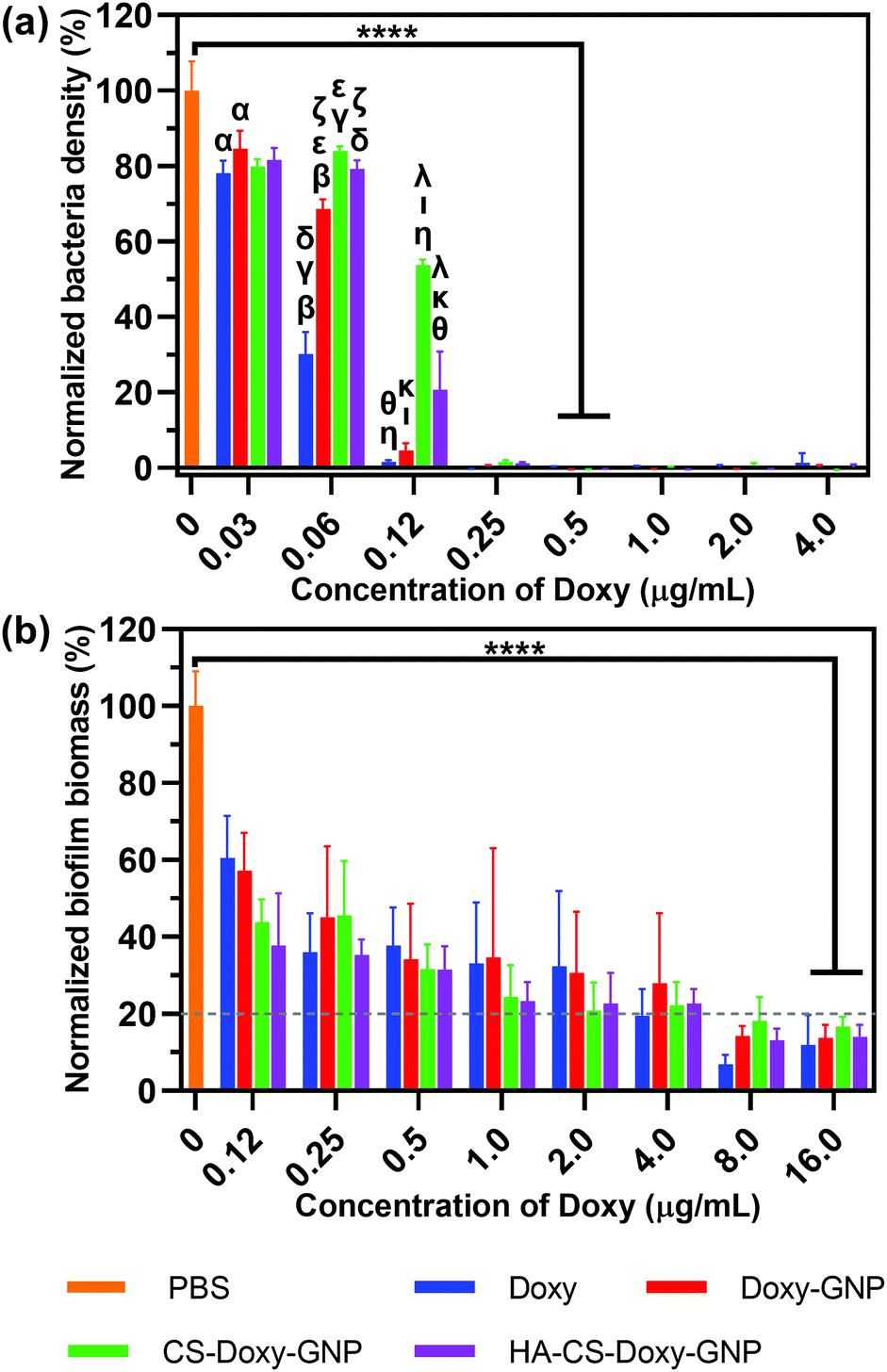 | ||
| Fig. 5 Antibacterial efficacy and inhibition of biofilm formation upon NP treatment. (a) Normalized bacteria density for planktonic V. vulnificus and (b) normalized biofilm biomass were determined after treatment with varying concentrations of free Doxy or Doxy loaded NPs at equivalent Doxy concentrations. Results are shown as mean ± standard deviation. Statistical significance between different NP formulations is indicated by matching Greek letters using one-way ANOVA with Tukey's post hoc analysis (α indicates p < 0.05 while β, γ, δ, ε, ζ, η, θ, ι, κ, and λ indicate p < 0.0001). Statistical significance (****p < 0.0001) between the no treatment control and the MIC or MBIC80 was determined using one-way ANOVA with Tukey's post hoc analysis; n = 3. | ||
Both biofilm prevention and eradication are critical for effective treatment of biofilm-associated infections.6 We first evaluated the ability of the free Doxy and Doxy loaded NP formulations to inhibit the formation of biofilms in a static V. vulnificus culture using crystal violet staining. Fig. 5b shows the decrease in normalized biofilm biomass with increasing concentrations of free Doxy and Doxy loaded NPs. An MBIC80 was determined over the concentration range examined as the minimum biofilm inhibitory concentration (MBIC) at which biofilm formation is inhibited by ≥80% compared to an untreated control. As with activity against planktonic V. vulnificus, there was no significant difference in the MBIC80 (∼16 μg mL−1) of free Doxy and all Doxy loaded NP formulations. The greater Doxy concentration required for biofilm inhibition compared to the MIC against planktonic V. vulnificus was expected given the static conditions utilized in these biofilm cultures.
Next, we investigated the ability of free Doxy and the Doxy loaded NP formulations to eradicate preformed, 48 h aged V. vulnificus biofilms. The eradication of existing biofilms is arguably one of the most difficult tasks in combating biofilms. In fact, typical methods for removing or dispersing these biofilms at an accessible site include physical debridement and application of harsh chemicals such as bleach.68–70 NP therapeutics offer an interesting possibility of combating these biofilms using biocompatible biomaterials and relatively benign administration routes. The morphology of the preformed biofilms after treatment with a PBS control, free Doxy (50 μg mL−1), and Doxy loaded NPs at an equivalent Doxy concentration was assessed via scanning electron microscopy. As shown in Fig. 6a, compared with the PBS treated biofilms, free Doxy and all of the Doxy loaded NPs exhibited some disruption of the biofilm structure. Most notably, treatment with HA-CS-Doxy-GNPs led to a drastic disruption of the biofilm structure, with a lack of EPS and predominantly isolated single cells remaining. The enhanced biofilm penetration ability of the HA-CS-GNP carrier (Fig. 4) likely enables the superior biofilm disruption exhibited by the HA-CS-Doxy-GNPs.
 | ||
| Fig. 6 Biofilm eradication ability of NP formulations against 48 h aged, preformed V. vulnificus biofilms. (a) SEM images and (b) CLSM images of LIVE/DEAD staining of V. vulnificus biofilms after treatment with PBS, 50 μg mL−1 Doxy, and Doxy loaded NP formulations at an equivalent Doxy concentration for 24 h (live bacteria appear green; membrane damaged bacteria appear red). Scale bar in (a): 50 μm and (b): 100 μm. SEM and CLSM images are representative of at least three independent biofilms. | ||
Viability of the bacteria following treatment of the V. vulnificus biofilms was also assessed using LIVE/DEAD viability staining (Fig. 6b). Here, SYTO 9 permeates and labels all bacterial cells (green), while propidium iodide (red) only enters damaged bacterial membranes. As expected, the PBS treated control biofilm stained completely green, indicating the presence of live bacteria. Free Doxy treatment led to a mix of live and membrane damaged bacteria, with more live bacteria than what was observed when biofilms were treated with the biopolymer-coated NP formulations. As with the SEM imaging, the greatest differences between the PBS control treatment and Doxy treatment were for biofilms treated with HA-CS-Doxy-GNPs. For these biofilms, all bacteria remaining following treatment exhibited membrane damage and limited to no green signal was observed. These results further suggest that HA-CS-Doxy-GNPs can effectively eliminate V. vulnificus biofilms.
We also quantified the MBEC90, which was defined as the minimal biofilm eradication concentration (MBEC) that reduced the initial biofilm biomass by ≥90% after treatment. The MBEC90 of free Doxy, Doxy-GNPs, CS-Doxy-GNPs, and HA-CS-Doxy-GNPs, determined by examining the biomass with crystal violet staining, was found to be comparable for all treatments at ∼50 μg mL−1 (Fig. S6a†). These results contrasted what was observed for SEM and LIVE/DEAD staining where the HA-CS-Doxy-GNPs exhibited the greatest biofilm eradication. We hypothesize that this discrepancy is a result of the nature of crystal violet staining, which is unable to distinguish between components on the stained structure (potentially including NP components, dead bacteria, etc.) Colony enumeration showed an ∼2 log reduction (99%) in CFU for the free Doxy and Doxy loaded NP treatments compared to the PBS control, with the HA-CS-Doxy-GNP treatment leading to the lowest mean CFU value (Fig. S6b and c†).
3.6 Assessing the antimicrobial effects of responsive NPs in an ex vivo porcine skin infection model
As an early step towards translation, we investigated the impact of treatment with free Doxy and HA-CS-Doxy-GNPs in an ex vivo porcine skin V. vulnificus infection model (Fig. 7a). After forming the porcine skin puncture and inoculation with V. vulnificus, Doxy and HA-CS-Doxy-GNPs treatments were applied at varying Doxy concentrations and compared with a PBS treatment control (Fig. S7†and Fig. 7b). Compared to the PBS control, we found an ∼84% and ∼92% reduction in CFU upon treatment with free Doxy and HA-CS-Doxy-GNPs at Doxy concentrations of 50 and 100 μg mL−1, respectively. Although no significant difference between CFU for treatment with free Doxy and HA-CS-Doxy-GNPs was noted in this experimental model, the significant reduction in V. vulnificus bacteria in this complex tissue environment is promising for future exploration and elucidation of differences in the treatment with free drug versus NP formulations. We hypothesize that large differences will exist in the EPS structure in this tissue-biofilm microenvironment upon NP treatment.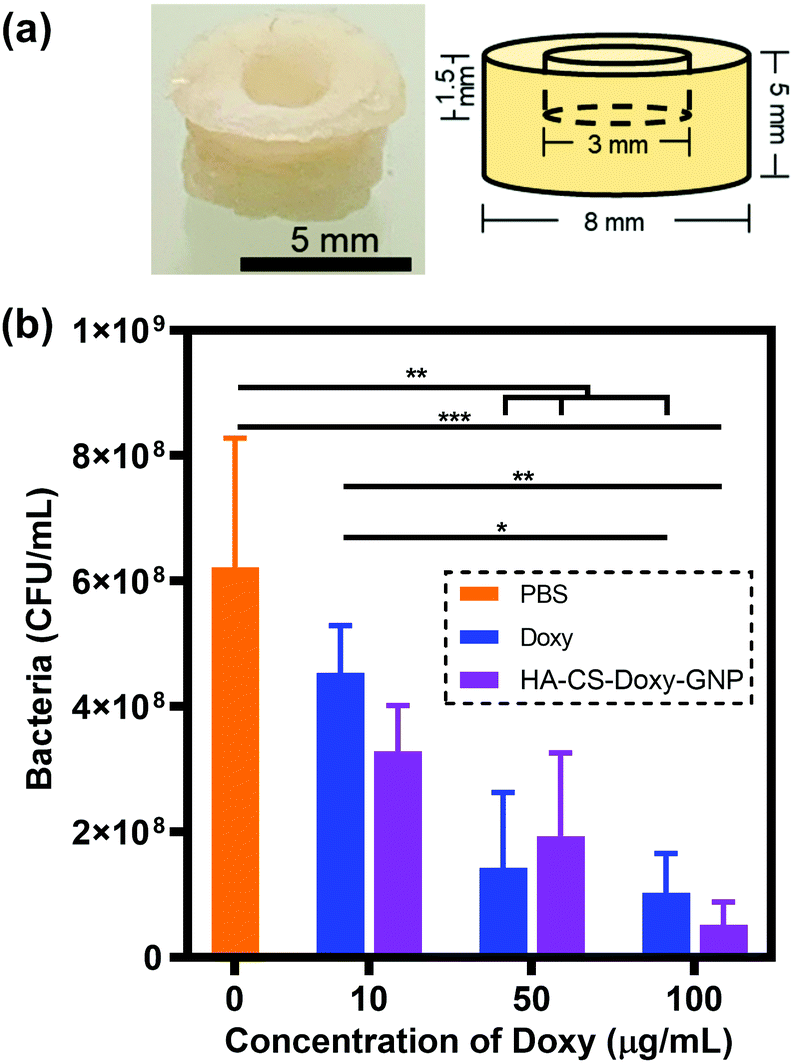 | ||
| Fig. 7 Antibacterial efficacy of NPs in an ex vivo porcine skin infection model. (a) Digital photo of a porcine skin sample with puncture. (b) CFU per mL from free Doxy and HA-CS-Doxy-GNP treated biofilms. Results are shown as mean ± standard deviation. Statistical significance (*p < 0.05, **p < 0.01, and ***p < 0.001) between groups was determined using two-way ANOVA with Tukey's post hoc analysis; n = 3. | ||
3.7 Examining the cytocompatibility of responsive NPs
Along with ensuring the antibacterial and antibiofilm efficacy of the NPs developed in this work, toxicity towards mammalian cells is also critical to evaluate. We investigated the toxicity profiles of Doxy and Doxy loaded NPs with 50 μg mL−1 Doxy on HUVEC and NIH 3T3 cells. Using a CCK-8 cell viability assay, we found that free Doxy exhibited a viability of ∼75% and ∼54% for NIH 3T3 cells and HUVEC compared with untreated controls, respectively (Fig. 8). Viability of these cells was significantly improved to ∼80% when incubated with Doxy loaded NPs at the same equivalent Doxy concentration, which is a likely result of the lack of a Doxy burst release from these formulations (Fig. 3). We also investigated the hemolytic activity upon incubation of free Doxy or Doxy loaded and non-loaded NPs with both HRBCs and BRBCs at various concentrations, including concentrations above and below the experimentally determined MIC, MBIC80, and MBEC90 values (Fig. S8†). Minimal to no hemolysis was observed for both RBC types with the greatest percent hemolysis (∼5% and ∼15% for HRBCs and BRBCs, respectively) observed for non-drug loaded GNP and CS-GNP formulations at an extreme concentration of 2000 μg mL−1. Percent hemolysis of BRBCs and HRBCs observed was extremely low (<0.5%) for the HA-CS-Doxy-GNPs even at concentrations well above the MBEC90, indicating the excellent hemocompatibility of these NPs. Together these results lend support for future use of these NPs in antibiofilm therapies without any significant concern for host toxicity.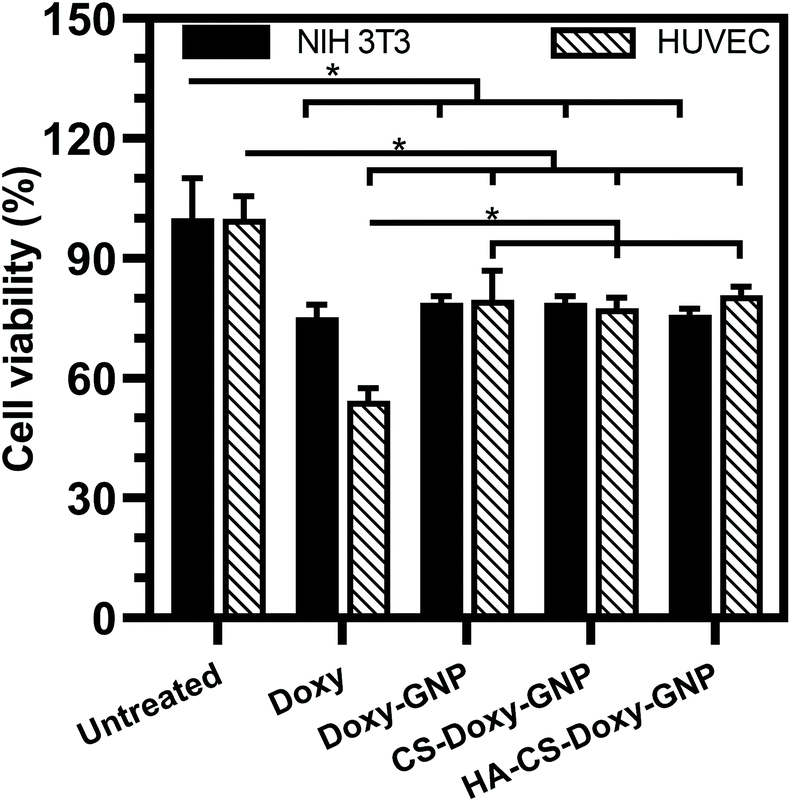 | ||
| Fig. 8 Cytocompatibility of NPs. Normalized HUVEC and NIH 3T3 cell viability upon exposure to media incubated with 50 μg mL−1 free Doxy and Doxy loaded NP formulations at an equivalent Doxy concentration for 24 h. Results are shown as mean ± standard deviation. Statistical significance (*p < 0.05) between groups was determined using two-way ANOVA with Tukey's post hoc analysis; n = 3. | ||
4 Conclusions
In this work we developed a new bacteria-responsive NP formulation and characterized its antibiofilm efficacy. These NPs were designed to exhibit both pH and bacterial enzyme-responsive drug release and interaction with the bacterial biofilm structure. The Doxy loaded GNP core was responsive to bacterial gelatinases, while the HA and CS LbL coating of the NPs enabled both a pH-regulated and bacterial hyaluronidase-controlled response. This is the first report of a hyaluoranidase, gelatinase, and pH-responsive animicrobial NP. Indeed we observed that HA-CS-Doxy-GNPs exhibited both pH and enzyme-responsive drug release behavior. We found that HA-CS-Doxy-GNPs also exhibited excellent V. vulnificus biofilm penetration and eradication ability compared with free Doxy, with SEM confirming a significant reduction in EPS and viability staining indicating significant membrane damaged following HA-CS-Doxy-GNP treatment of preformed biofilms. These HA-CS-Doxy-GNPs also reduced bacterial burden in an ex vivo porcine V. vulnificus infection model, providing support for future translation. Additionally, these NPs were found to remain generally biocompatible with fibroblasts, endothelial cells, and RBCs. The multi-stimuli-responsive NP platform has the potential to exhibit similar antibiofilm properties against a broad range of biofilm forming bacteria including other Gram-negative as well as Gram-positive bacteria, as these enzyme triggers and reduced pH are hallmarks of many bacterial species. Additionally, this responsive drug delivery system could also be used to deliver multiple drugs including those aimed specifically at biofilms (e.g., antibiofilm peptides) or signaling molecules for infection detection (e.g., fluorescent dyes) for the effective broad-spectrum treatment and detection of infections.Author contributions
Yinging Wang: Conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, visualization; Anita Shukla: Conceptualization, methodology, resources, writing – review and editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the Office of Naval Research (N000141712120) is gratefully acknowledged. We thank Professor Robert Hurt at Brown for use of his DLS. Finally, we thank Anthony McCormick from the Brown University Electron Microscopy Core Facility for his assistance with SEM.References
- R. J. Worthington, J. J. Richards and C. Melander, Org. Biomol. Chem., 2012, 10, 7457–7474 RSC.
- R. Joseph, A. Naugolny, M. Feldman, I. M. Herzog, M. Fridman and Y. Cohen, J. Am. Chem. Soc., 2016, 138, 754–757 CrossRef CAS PubMed.
- Y. Dhar and Y. Han, Eng. Regener., 2020, 1, 64–75 Search PubMed.
- Y. Liu, L. Shi, L. Su, H. C. van der Mei, P. C. Jutte, Y. Ren and H. J. Busscher, Chem. Soc. Rev., 2019, 48, 428–446 RSC.
- C. Deusenbery, Y. Wang and A. Shukla, ACS Infect. Dis., 2021, 7, 695–720 CrossRef CAS PubMed.
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2, 114–122 CrossRef CAS PubMed.
- D. Lebeaux, J.-M. Ghigo and C. Beloin, Microbiol. Mol. Biol. Rev., 2014, 78, 510–543 CrossRef PubMed.
- T.-F. Mah, B. Pitts, B. Pellock, G. C. Walker, P. S. Stewart and G. A. O'Toole, Nature, 2003, 426, 306–310 CrossRef CAS PubMed.
- P. Bowler, C. Murphy and R. Wolcott, Antimicrob. Resist. Infect. Control, 2020, 9, 1–5 CrossRef PubMed.
- T. F. C. Mah and G. A. O'Toole, Trends Microbiol., 2001, 9, 34–39 CrossRef CAS PubMed.
- S. Fulaz, S. Vitale, L. Quinn and E. Casey, Trends Microbiol., 2019, 27, 915–926 CrossRef CAS PubMed.
- Y. Dong, H. Zhu, Y. Shen, W. Zhang and L. Zhang, PLoS One, 2019, 14, e0222322 CrossRef CAS PubMed.
- X. Dai, Q. Guo, Y. Zhao, P. Zhang, T. Zhang, X. Zhang and C. Li, ACS Appl. Mater. Interfaces, 2016, 8, 25798–25807 CrossRef CAS PubMed.
- X. Yang, J. Yang, L. Wang, B. Ran, Y. Jia, L. Zhang, G. Yang, H. Shao and X. Jiang, ACS Nano, 2017, 11, 5737–5745 CrossRef CAS PubMed.
- G. Fang, W. Li, X. Shen, J. M. Perez-Aguilar, Y. Chong, X. Gao, Z. Chai, C. Chen, C. Ge and R. Zhou, Nat. Commun., 2018, 9, 129 CrossRef PubMed.
- A. Gupta, R. F. Landis, C.-H. Li, M. Schnurr, R. Das, Y.-W. Lee, M. Yazdani, Y. Liu, A. Kozlova and V. M. Rotello, J. Am. Chem. Soc., 2018, 140, 12137–12143 CrossRef CAS PubMed.
- L. Liu, K. Xu, H. Wang, P. K. J. Tan, W. Fan, S. S. Venkatraman, L. Li and Y.-Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS PubMed.
- A. Ivanova, K. Ivanova, J. Hoyo, T. Heinze, S. Sanchez-Gomez and T. Tzanov, ACS Appl. Mater. Interfaces, 2018, 10, 3314–3323 CrossRef CAS PubMed.
- C. M. Courtney, S. M. Goodman, J. A. McDaniel, N. E. Madinger, A. Chatterjee and P. Nagpal, Nat. Mater., 2016, 15, 529–534 CrossRef CAS PubMed.
- S. Fulaz, D. Hiebner, C. H. N. Barros, H. Devlin, S. Vitale, L. Quinn and E. Casey, ACS Appl. Mater. Interfaces, 2019, 11, 32679–32688 CrossRef CAS PubMed.
- A. M. Scharnow, A. E. Solinski and W. M. Wuest, MedChemComm, 2019, 10, 1057–1067 RSC.
- G.-B. Qi, D. Zhang, F.-H. Liu, Z.-Y. Qiao and H. Wang, Adv. Mater., 2017, 29, 1703461 CrossRef PubMed.
- L.-L. Li, H.-L. Ma, G.-B. Qi, D. Zhang, F. Yu, Z. Hu and H. Wang, Adv. Mater., 2016, 28, 254–262 CrossRef CAS PubMed.
- X. Wang, J. Wu, P. Li, L. Wang, J. Zhou, G. Zhang, X. Li, B. Hu and X. Xing, ACS Appl. Mater. Interfaces, 2018, 10, 34905–34915 CrossRef CAS.
- L. R. Thurlow, V. C. Thomas, S. Narayanan, S. Olson, S. D. Fleming and L. E. Hancock, Infect. Immun., 2010, 78, 4936–4943 CrossRef CAS PubMed.
- W. L. Hynes and S. L. Walton, FEMS Microbiol. Lett., 2000, 183, 201–207 CrossRef CAS.
- C. Wang, W. Zhao, B. Cao, Z. Wang, Q. Zhou, S. Lu, L. Lu, M. Zhan and X. Hu, Chem. Mater., 2020, 32, 7725–7738 CrossRef CAS.
- J. Xu, R. Danehy, H. Cai, Z. Ao, M. Pu, A. Nusawardhana, D. Rowe-Magnus and F. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 14640–14646 CrossRef CAS PubMed.
- Y. Liu, A. Lin, J. Liu, X. Chen, X. Zhu, Y. Gong, G. Yuan, L. Chen and J. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 26590–26606 CrossRef CAS PubMed.
- Q. Deng, P. Sun, L. Zhang, Z. Liu, H. Wang, J. Ren, X. Qu, Q. Deng, P. Sun, L. Zhang, Z. Liu, H. Wang, J. Ren and X. Qu, Adv. Funct. Mater., 2019, 29, 1903018 CrossRef.
- R. C. Oppenheim, J. J. Marty and P. Speiser, U.S. Patent4107288, 1978 Search PubMed.
- A. O. Elzoghby, J. Controlled Release, 2013, 172, 1075–1091 CrossRef CAS PubMed.
- K. Vaghasiya, E. Ray, R. Singh, K. Jadhav, A. Sharma, R. Khan, O. P. Katare and R. K. Verma, Mater. Sci. Eng., C, 2021, 123, 112027 CrossRef PubMed.
- A. Lin, Y. Liu, X. Zhu, X. Chen, J. Liu, Y. Zhou, X. Qin and J. Liu, ACS Nano, 2019, 13, 13965–13984 CrossRef CAS PubMed.
- L.-L. Li, J.-H. Xu, G.-B. Qi, X. Zhao, F. Yu and H. Wang, ACS Nano, 2014, 8, 4975–4983 CrossRef CAS PubMed.
- J. Su, R. Zhang, Y. Lian, Z. Kamal, Z. Cheng, Y. Qiu and M. Qiu, Pharmaceutics, 2019, 11, 93 CrossRef CAS PubMed.
- S. Kirar, N. S. Thakur, J. K. Laha and U. C. Banerjee, ACS Appl. Bio Mater., 2019, 2, 4202–4212 CrossRef CAS PubMed.
- S. Balthasar, K. Michaelis, N. Dinauer, H. v. Briesen, J. Kreuter and K. Langer, Biomaterials, 2005, 26, 2723–2732 CrossRef CAS PubMed.
- T. G. Shutava, S. S. Balkundi, P. Vangala, J. J. Steffan, R. L. Bigelow, J. A. Cardelli, D. P. O'Neal and Y. M. Lvov, ACS Nano, 2009, 3, 1877–1885 CrossRef CAS PubMed.
- X.-h. Tian, F. Wei, T.-x. Wang, D. Wang, J. Wang, X.-n. Lin, P. Wang and L. Ren, Mater. Lett., 2012, 68, 94–96 CrossRef CAS.
- Z. V. Feng, I. L. Gunsolus, T. A. Qiu, K. R. Hurley, L. H. Nyberg, H. Frew, K. P. Johnson, A. M. Vartanian, L. M. Jacob, S. E. Lohse, M. D. Torelli, R. J. Hamers, C. J. Murphy and C. L. Haynes, Chem. Sci., 2015, 6, 5186–5196 RSC.
- R. M. Pinto, F. A. Soares, S. Reis, C. Nunes and P. Van Dijck, Front. Microbiol., 2020, 11, 952 CrossRef PubMed.
- W. Wang, X. Hao, S. Chen, Z. Yang, C. Wang, R. Yan, X. Zhang, H. Liu, Q. Shao and Z. Guo, Polymer, 2018, 158, 223–230 CrossRef CAS.
- S.-M. Jung, G. H. Yoon, H. C. Lee, M. H. Jung, S. I. Yu, S. J. Yeon, S. K. Min, Y. S. Kwon, J. H. Hwang and H. S. Shin, Sci. Rep., 2015, 5, 18089 CrossRef CAS PubMed.
- C. Baker-Austin and J. D. Oliver, Environ. Microbiol., 2018, 20, 423–430 CrossRef PubMed.
- J. D. Oliver, J. E. Wear, M. B. Thomas, M. Warner and K. Linder, Diagn. Microbiol. Infect. Dis., 1986, 5, 99–111 CrossRef CAS PubMed.
- S. A. Trinh, H. E. Gavin and K. J. F. Satchell, Antimicrob. Agents Chemother., 2017, 61, e01106–e01117 CrossRef CAS PubMed.
- Y. Sun, H. Qin, Z. Yan, C. Zhao, J. Ren and X. Qu, Adv. Funct. Mater., 2019, 29, 1808222 CrossRef.
- H. Ji, K. Dong, Z. Yan, C. Ding, Z. Chen, J. Ren and X. Qu, Small, 2016, 12, 6200–6206 CrossRef CAS PubMed.
- T. Gwisai, N. R. Hollingsworth, S. Cowles, N. Tharmalingam, E. Mylonakis, B. B. Fuchs and A. Shukla, Biomed. Mater., 2017, 12, 045010 CrossRef PubMed.
- D. Alkekhia and A. Shukla, J. Biomed. Mater. Res., Part A, 2019, 107, 1324–1339 CrossRef CAS PubMed.
- N. Vera-González, C. M. Bailey-Hytholt, L. Langlois, F. de Camargo Ribeiro, E. L. de Souza Santos, J. C. Junqueira and A. Shukla, J. Biomed. Mater. Res., Part A, 2020, 108, 2263–2276 CrossRef PubMed.
- H. Liu, S. Shukla, N. Vera-González, N. Tharmalingam, E. Mylonakis, B. B. Fuchs and A. Shukla, Front. Cell. Infect. Microbiol., 2019, 9, 37 CrossRef CAS PubMed.
- R. F. Smith and N. P. Willett, Appl. Microbiol., 1968, 16, 1434–1436 CrossRef CAS PubMed.
- R. Yasmin, M. Shah, S. A. Khan and R. Ali, Nanotechnol. Rev., 2017, 6, 191–207 CAS.
- X. Song, K. Gan, S. Qin, L. Chen, X. Liu, T. Chen and H. Liu, Sci. Rep., 2019, 9(1), 1–11 CAS.
- S. Kirar, N. S. Thakur, J. K. Laha, J. Bhaumik and U. C. Banerjee, ACS Biomater. Sci. Eng., 2018, 4, 473–482 CrossRef CAS PubMed.
- J. H. Xu, F. P. Gao, X. F. Liu, Q. Zeng, S. S. Guo, Z. Y. Tang, X. Z. Zhao and H. Wang, Chem. Commun., 2013, 49, 4462–4464 RSC.
- D. Alkekhia, P. T. Hammond and A. Shukla, Annu. Rev. Biomed. Eng., 2020, 22, 1–24 CrossRef CAS PubMed.
- S. M. Kong, D. F. Costa, A. Jagielska, K. J. Van Vliet and P. T. Hammond, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2104826118 CrossRef CAS PubMed.
- C. H. Kapadia, S. A. Ioele and E. S. Day, J. Biomed. Mater. Res., Part A, 2020, 108, 601–613 CrossRef CAS PubMed.
- X. Zhu and X. J. Loh, Biomater. Sci., 2015, 3, 1505–1518 RSC.
- S. Duan, S. Duan, X. Zhao, Z. Su, C. Wang and Y. Lin, ACS Appl. Bio Mater., 2020, 3, 3673–3680 CrossRef CAS PubMed.
- A. Ivanova, K. Ivanova, A. Tied, T. Heinze and T. Tzanov, Adv. Funct. Mater., 2020, 30, 2001284 CrossRef CAS.
- W. Feng, X. Zhou, C. He, K. Qiu, W. Nie, L. Chen, H. Wang, X. Mo and Y. Zhang, J. Mater. Chem. B, 2013, 1, 5886–5898 RSC.
- T. Ramasamy, Z. S. Haidar, T. H. Tran, J. Y. Choi, J. H. Jeong, B. S. Shin, H. G. Choi, C. S. Yong and J. O. Kim, Acta Biomater., 2014, 10, 5116–5127 CrossRef CAS PubMed.
- B. Cai, L. Rao, X. Ji, L.-L. Bu, Z. He, D. Wan, Y. Yang, W. Liu, S. Guo and X.-Z. Zhao, J. Biomed. Mater. Res., Part A, 2016, 104, 2854–2860 CrossRef CAS PubMed.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740–755 CrossRef CAS PubMed.
- S. Galié, C. García-Gutiérrez, E. M. Miguélez, C. J. Villar and F. Lombó, Front. Microbiol., 2018, 9, 898 CrossRef PubMed.
- A. D. Verderosa, M. Totsika and K. E. Fairfull-Smith, Front. Chem., 2019, 7, 824 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00361a |
| This journal is © The Royal Society of Chemistry 2022 |