
Bacteria-responsive biopolymer-coated nanoparticles for biofilm penetration and eradication†
 a
and
Anita
Shukla
*b
a
and
Anita
Shukla
*b
Abstract
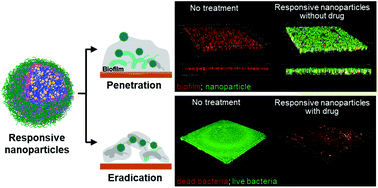
Biofilm infections are common and can be extremely difficult to treat. Nanoparticles that respond to multiple bacterial stimuli have the potential to successfully prevent and eradicate biofilms. Here, we developed a hyaluronic acid and chitosan coated, antibiotic loaded gelatin nanoparticle, which can undergo hyaluronidase- and gelatinase-mediated degradation regulated by chitosan protonation and swelling in the acidic biofilm microenvironment. We examined the antibiofilm properties of these nanoparticles using a Gram-negative biofilm forming pathogenic bacteria, Vibrio vulnificus. Non-drug loaded responsive nanoparticle formulations exhibited excellent biofilm penetration and retention in preformed V. vulnificus biofilms. Drug loaded formulations were found to exhibit excellent biofilm eradication efficacy, eliminating the biofilm matrix and effectively causing bacterial cell death, which was not observed for treatment with free drug at equivalent concentrations. Overall, these multi-stimuli-responsive nanoparticles have the potential to provide effective and efficient antibiofilm treatment.
- This article is part of the themed collection: Biomaterials Science Emerging Investigator Series
 Please wait while we load your content...
Please wait while we load your content...

