
Charged droplet-driven fast formation of nickel–iron (oxy)hydroxides with rich oxygen defects for boosting overall water splitting†
Jianing
Dong
,
Yanjie
Wang
,
Qiaorong
Jiang
,
Zi-Ang
Nan
,
Feng Ru
Fan
 * and
Zhong-Qun
Tian
* and
Zhong-Qun
Tian
State Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), Tan Kah Kee Innovation Laboratory, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: frfan@xmu.edu.cn
First published on 17th August 2021
Abstract
NiFe (oxy)hydroxides (NiFeOOH) are considered as one of the most efficient electrocatalysts for water splitting. Although several strategies recently have been developed to fabricate NiFeOOH electrodes, such as electrodeposition, hydrothermal reaction, corrosion engineering, etc., it has been a challenge to directly tune the energy level, conductivity and surface properties (such as molecular adsorption) of NiFeOOH with less time-consuming and convenient synthesis to overcome the relatively sluggish reaction kinetics. Herein, we, for the first time, utilize charged droplets to synthesize NiFeOOH with abundant oxygen vacancies (Ov) via the convenient electrospraying (ESI) approach without necessitating additional post-treatments. Specifically, the optimized NiFeOOH delivers much lower overpotentials of 145 mV, 215 mV and 360 mV for the hydrogen evolution reaction (HER), the oxygen evolution reaction (OER) and overall water splitting in 1 M KOH at 10 mA cm−2 with long-term durability over 180 h. The good performing electrocatalyst is much improved compared with the benchmark Pt/C/NF in the high current density region, RuO2/NF and other NiFe-based electrocatalysts. Simultaneously, the unique reaction environment of the droplet (confinement, superacid and desolvation) was further explored to understand the synthesis reaction acceleration process and related oxygen defect formation mechanisms. Also, we used density functional theory (DFT) to investigate the change of free energy of the reaction pathway caused by oxygen vacancies and understand the reaction mechanism of electrocatalysts prepared by different treatment methods. The calculation results show that the shortened Fe–O bonds and oxygen defects of NiFeOOH synergistically improve the interaction between the metal cations and the intermediate species and further accelerate the overall reaction kinetics. This work demonstrates that the charged microdroplet chemistry offers a novel means to expedite the design of powerful electrocatalysts.
1. Introduction
Water splitting utilizing electricity converted from solar, wind and other renewable energy sources proposes a strategic approach to solve the energy crisis. In principle, the electrolysis of water consists of two half reactions: the HER on the cathode and the OER on the anode.1 To overcome the activation barrier, especially for the four-electron transfer sluggish OER, various electrocatalysts including transition-metal sulfides,2 oxides,3 phosphides4 and (oxy)hydroxides5 have been exploited to replace the widely used but expensive Pt and RuO2. Among them, transition-metal (oxy)hydroxides, especially NiFe-based (oxy)hydroxides, are considered to be one of the most promising candidates due to their abundant reserves and excellent OER activity. Recently, “corrosion engineering” has been reported to fabricate effective OER electrodes by immersing Ni foam in an iron-containing salt solution, and vice versa.6 Although electrocatalysts produced from “corrosion engineering” demonstrate good OER performance, the entire preparation process takes a long time (at least 12 hours)6a or requires high temperature (e.g. 100 °C)6c to promote the process, which is not favorable for wide use. Moreover, because of the few active sites, slow charge transfer and weak adsorption of dissociated hydrogen atoms (Hads), NiFe-based electrocatalysts not only require a large overpotential in the OER to achieve high current density, but also exhibit poor performance in the HER.7 Therefore, it is of great importance to develop a novel and less time-consuming and convenient method for preparing NiFe-based electrodes with considerable activity.It is well known that the formation of oxygen vacancies in NiFe-based (oxy)hydroxides is a paramount approach to reduce the energy gap between the metal 3d and O-2p-band centers, and thus increase the covalency of the metal–oxygen bond of NiFe-based electrocatalysts. The electrocatalyst with rich oxygen vacancies can promote structural self-reconstruction and the surface adsorption/desorption of reactants and further accelerate the overall water splitting efficiency of long-term operation.8 Various methods of manipulating oxygen vacancies have been explored, including thermal treatment,9 reduction processing,10 plasma treatment,11 laser radiation,12 and ball-milling,13 which can be summarized as top-down approaches. Although these approaches notably improve the performance of electrocatalysts, it is still hard to achieve large-scale applications because they require complex operating procedures and expensive equipment. In contrast, bottom-up approaches based on the non-equilibrium stoichiometric reaction (e.g. more metal cations and fewer non-metal species in a confined system,14 cation doping15 and anion doping16) are mostly applied to hydrothermal reactions that require high temperature and high pressure assistance. As far as we know, there has not been any report based on sub-stoichiometric chemistry synthesis at room temperature. Therefore, it would be highly desired to find some new ways to combine the synthesis efficiency with defect construction to synthesize NiFe-based electrocatalysts to achieve spectacular overall water splitting.
In the past two decades, the confinement reaction in microdroplets produced by microfluidic systems,17 Leidenfrost suspension droplets,18 different kinds of spray droplets19 and thin films on surfaces,20 has been reported. The rate of many organic reactions in these “droplet-reactors”, especially in charged droplets, is strikingly expedited compared with those in bulk water, which can be attributed to the extreme pH of the interface, electrical double layer, concentration enrichment and evaporation, and fast diffusion and mixing.21 Hence, the accelerated reactions at the aqueous interface (including the gas–liquid interface,22 the liquid–liquid interface23 and the solid–liquid interface24) offer a forward-looking perspective that will help us to form new cognitions in chemical reactions and material synthesis. Moreover, the microdroplet is an exquisite confined reactor. Once the droplet is charged, the molecules in the droplet will have an oriented distribution, thereby forming a confined reaction area under the action of an electric field.25 Therefore, the charged droplets could precisely control the ratio of reactants to achieve unique chemical reactions under non-equilibrium conditions. This droplet reactor is favorable for the formation of oxygen vacancies based on the unbalanced ratio of metal salt to oxygen content under confinement and charged conditions.14a
Herein, inspired by the accelerated reaction in charged droplets, we report the reaction between Ni foams (NF) and charged droplets via electrospraying ionization to accelerate the spontaneous formation of NiFe (oxy)hydroxides with abundant oxygen vacancies (named as E-NiFeOOH) at a confined and charged solid–liquid interface. E-NiFeOOH is significantly superior to the benchmark Pt/C/NF, RuO2/NF and other NiFe-based electrocatalysts. The electrocatalytic mechanism was investigated by DFT calculations. Our work not only provides a feasible and convenient synthesis method for the construction of catalyst defects, but also helps us to further understand the chemical reaction at the confined and charged solid–liquid interface.
2. Results and discussion
2.1 Charged droplet accelerated corrosion engineering and the formation of oxygen defects
In a general metal corrosion engineering process, there are several factors that can influence the foam etching and the growth rate of electrocatalysts: solution concentration; the reaction atmosphere, temperature and time; diffusion rates of cations, anions and surrounding gases. These factors can effectively change the structure, composition, and geometry of the corrosion layer, which has a remarkable impact on the performance of the electrocatalyst.26 However, the current metal corrosion engineering methods reported are all performed in the bulk phase. All these reactions in bulk solution actually have similar chemical reaction states. Taking the Ni foam corrosion process in FeCl3 aqueous as an example (shown in Fig. S1a,† the electrocatalyst named as I-NiFeOOH), the overall system shows a strong ion solvation state, uniform gas distribution under equilibrium conditions and a setting pH circumstance. The whole reaction process can be represented by eqn (1)–(4) as follows:| Fe3+ + e− = Fe2+ | (1) |
| Ni − 2e− = Ni2+ | (2) |
| O2 + 2H2O + 4e− = 4OH− | (3) |
| xFe3+ + (1 − x)Ni + yOH− = NiFeOOH | (4) |
Such a reaction environment leads to extremely slow reaction kinetics for electrocatalyst synthesis, and it is difficult to regulate the structure of the electrocatalyst, if there are no extra supporting factors (heating, stirring or additive agent). In contrast, the charged droplet reactor displays a rather different scenario compared with the bulk phase, including increasing reagent concentration by continuous and rapid solvent evaporation, with extreme pH at the interface, electrical double layer at the interface and so on.27 These factors will naturally affect the chemical process of the corrosion reaction. In this work, several factors (extreme pH, concentration enrichment and desolvation structure) have been discussed to illustrate the acceleration of reaction and the formation of oxygen defects, and the corresponding reaction schemes are shown in Fig. S1b.†
Fig. 1a illustrates the fabrication process of NiFeOOH with oxygen vacancies via electrospray. Briefly, a high positive voltage was applied on the spray needle to polarize the droplets with positive charges. The positively charged droplets will produce abundant H3O+ at the droplet interface. According to previous reports, this H3O+ layer on the surface of the droplet can exhibit a super-acidic behavior,27b which can provide super-acidic conditions. Compared to the reaction in the bulk phase, the reaction mediated by positively charged droplets can achieve rapid foam etching without adding an additional acidic solution in a green way. Fig. 1b and c show the morphology of different methods after 20 min of reaction. The reaction mediated by positively charged droplets shows faster foam corrosion and electrocatalyst growth than that in the bulk phase. To further prove the pH effect by the polarity of droplets, we polarize the droplets with negative charges, which have been reported to present alkaline properties, to perform the corrosion reaction. As shown in Fig. S2,† compared with the positively charged droplets and bulk conditions, the corrosion reaction efficiency of nickel foam mediated by negatively charged droplets is rather low. We speculate that the high negative electric field leads to the transformation of Fe3+ into Fe2+ because of its high redox potential (0.77 V), and this can lead to weak oxidation of nickel foam. And the potential chlorine evolution also reduces the etching reaction.28 Anyway, this result shows that charged droplets with different polarities could display different pH effects, which may offer a unique way to provide an acid or alkali environment without other cations or anions involved for some pH and ion sensitive material synthesis, such as synthesis of nanocrystals.29 And in this work, positively charged droplets are beneficial for corrosion engineering. In addition, the non-equilibrium stoichiometric reaction in confined spaces has been reported to be a valid means to synthesize oxygen vacancies.14 In the confined charged droplets, the tiny droplets have low oxygen content and the oxygen adsorption corrosion reaction has been largely reduced due to the lower pH surroundings in the positively charged droplets. This circumstance causes a non-equilibrium stoichiometric reaction between oxygen species-OH− (low) and partially solvated Fe3+ (high) to construct oxygen defects. Therefore, NiFeOOH with abundant oxygen vacancies can be reasonably prepared at the confined and charged interface. Moreover, as we know, electrospraying is a desolvation process. Therefore, the Fe3+ and Cl− are partially solvated, and these partially solvated reactants in charged droplets can have thermodynamic advantages. They can overcome the small solvation energy and the activation energy to form products.30 For instance, partially solvated Cl− can exhibit stronger corrosion characteristics than in bulk aqueous solution. And partially solvated Fe3+ can more easily approach Ni2+ to form NiFeOOH. This solvation strategy has also been reported for the rapid formation of ion pairs to construct the SEI layer, thus promoting the stability of lithium batteries.31 Therefore, the electrocatalyst via this spontaneous desolvation process shows better geometry in a shorter time and can realize better electrocatalytic performances. Importantly, under the electrical field at droplets, the reactants are enriched on the surface of droplets, causing a concentration effect on the reaction rate. This characteristic can further intensify the above process in terms of both thermodynamics and kinetics. According to the above analysis, the synergistic effect of multiple factors makes the charged droplets show faster corrosion reaction and electrocatalyst growth with more oxygen vacancies than the bulk solution. Based on the above discussions, as shown in Fig. 1d, the potential energy barrier for the formation of E-NiFeOOH is greatly reduced, thereby accelerating the entire reaction process.
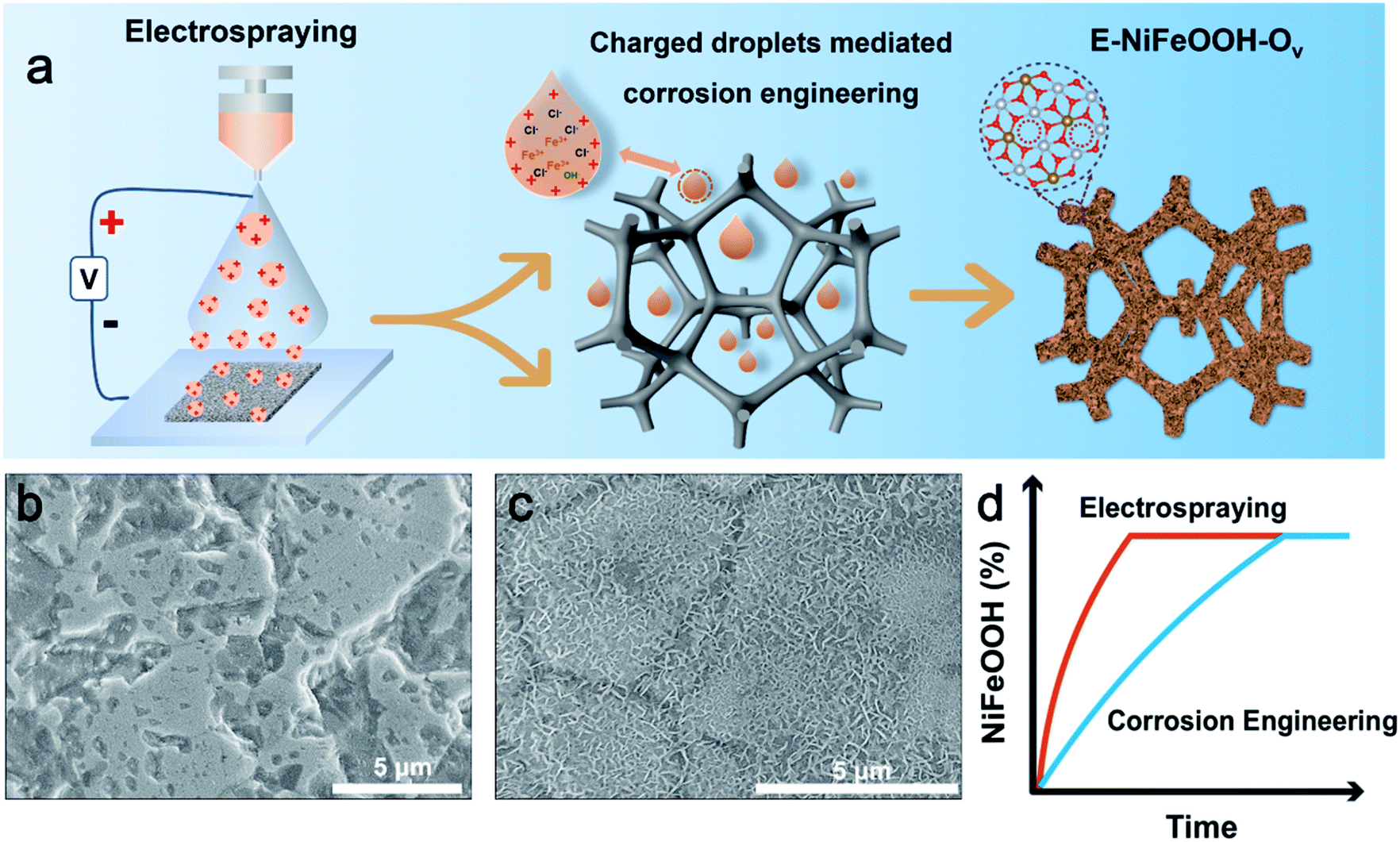 | ||
| Fig. 1 (a) Schematic illustration of the reaction process of E-NiFeOOH composites via electrospraying. (b) SEM images of I-NiFeOOH-20 min. (c) SEM images of E-NiFeOOH-20 min. (d) The content (%) of NiFeOOH changes over time in different systems: electrospraying and corrosion engineering. | ||
2.2 Morphology and structural characterization of electrocatalysts
The morphology and composition of the electrodes before and after the ESI processes were examined by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The nickel foam displays a flat and regular structure as shown in Fig. 2a. After the charged droplets were sprayed onto the foam surface, a typical flake structure is gradually formed as the reaction time increases (Fig. 2b, c and S3†). When the spraying time is extended to 60 minutes, the small spherical objects will adhere to the surface, which are FeOOH produced by the hydrolysis of iron salt. SEM results validate that the charged droplets can accelerate the formation of NiFeOOH, and as the reaction time increases, Ni on the surface will be depleted and the excess Fe3+ will form FeOOH. In contrast, the morphology of I-NiFeOOH-20 min produced by traditional corrosion engineering can be observed in Fig. 1b, and only a rough surface is formed. The color of I-NiFeOOH is much lighter than that of E-NiFeOOH, which implies a sluggish reaction kinetics in bulk water compared with the charged droplets (Fig. S4†). TEM characterization further reveals the structural features, as shown in Fig. 2d. E-NiFeOOH exhibits an ultra-thin layer structure, which is beneficial for charge and mass transfer during the water splitting reaction. AFM characterization is further employed to investigate the thickness of the electrocatalyst. As shown in Fig. S5,† the height of the as-prepared ultra-thin nanosheets was measured and the thickness of the nanosheets is determined to be ca. 3.5–4 nm by AFM, corresponding to the bilayer and three-layer nanosheet morphologies. Furthermore, Fig. 2e displays the high-resolution transmission electron microscopy (HRTEM) image and corresponding selected-area electron diffraction (SAED) pattern of E-NiFeOOH. The characteristics of 0.25 nm and 0.33 nm can be attributed to the (012) lattice plane of NiFe layered double hydroxide (LDH) and the (310) d-spacing plane of FeOOH.6a The SAED pattern (the inset of Fig. 2e) shows well-defined diffraction rings, which can be indexed to the (012) plane of NiFe LDH and the (310) plane of FeOOH. Especially, the observed hierarchical porous structure of E-NiFeOOH can expose more active sites on the surface to demonstrate better electrocatalytic performance. In addition, the elemental mapping images (Fig. 2f) verify the coexistence and distribution of Ni, Fe and O, suggesting the sufficient redox reaction at the liquid–solid interface. | ||
| Fig. 2 SEM images of the Ni foam (a) and E-NiFeOOH with different reaction times: (b) 30 min and (c) 40 min. (d) TEM image of E-NiFeOOH. (e) HRTEM image and SAED pattern of E-NiFeOOH. (f) EDX map of E-NiFeOOH containing O, Ni and Fe elements. | ||
X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) were used to study the structure, chemical composition and valence state of different samples. To avoid the interference of the strong diffraction peak of nickel, a low diffraction angle and a low scan rate were employed to perform XRD measurements. As shown in Fig. S5a,† there are no obvious diffraction peaks of NiFe LDH and α-FeOOH. Therefore, we believe that E-NiFeOOH is a poorly crystalline or amorphous phase. As shown in Fig. S6b,† O 2p, Fe 2p and Ni 2p with different peak intensities can be found in the broad XPS spectra of both I-NiFeOOH and E-NiFeOOH samples. Fig. 3a presents the high-resolution spectrum of Fe 2p.32 The fitted peaks at 711.3 eV and 725.1 eV represent Fe3+ 2p3/2 and Fe3+ 2p1/2. Similarly, the binding energies of 709.3 eV and 722.4 eV are attributed to Fe2+ 2p3/2 and Fe2+ 2p1/2, which confirms that Fe3+ was reduced to Fe2+ by Ni. Nevertheless, the Fe 2p spectrum of I-NiFeOOH is quite different. The intensity of peaks is extremely lower than that of E-NiFeOOH within the vertical axis of the same intensity. Interestingly, the fitting peak of Fe3+ 2p3/2 (711.3 eV) of E-NiFeOOH shifts to lower energy compared with that of I-NiFeOOH (712.0 eV), since the electron transfer from NiFeLDH to FeOOH may increase the Fermi level of FeOOH, thereby optimizing the interaction with the intermediate state of the reaction.33 Furthermore, the detailed Ni 2p spectrum of E-NiFeOOH is presented in Fig. 3b, and the two peaks located at 855.3 eV and 873.0 eV are attributed to the Ni2+ 2p3/2 and Ni2+ 2p1/2,34 along with two typically satellite peaks. In contrast, there is no obvious peak of metallic Ni foams (the peaks of Ni0 are located at 852.6 eV and 869.9 eV).35 Therefore, the elemental ratio of Ni to Fe in NiFeOOH can be evaluated by XPS measurements, and is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in E-NiFeOOH and 1:9 in I-NiFeOOH. Moreover, when the Fe content rises above 25%, the high content of iron in NiOOH can greatly improve the OER performance, possibly due to the presence of FeOOH phase nucleation.36 In the high-resolution spectrum of the O region (Fig. 3c), the peaks at 529.3 eV, 531.2 eV and 532.6 eV can be attributed to the lattice oxygen–metal bond, oxygen vacancies in the low oxygen coordination and adsorbed oxygen contents on the surface, respectively.37 It is well known that the oxygen vacancies (Ov) formed can be evaluated by determining the peak area ratio between oxygen vacancies and lattice oxygen. As can be seen from Fig. 3c, the Ov density in E-NiFeOOH is more than twice as high as that in I-NiFeOOH. The calculation results of the Ov density are shown in Table S1.† In addition, the electron paramagnetic resonance (EPR) spectrum in Fig. 3d shows that the major peaks are located at 1.93, 1.96 and 2.006 of the g values. This result indicates the presence of unpaired electrons of Fe3+ and oxygen vacancies.38,39 Compared with the EPR spectrum of I-NiFeOOH, the Fe3+ and oxygen defect peak intensity of E-NiFeOOH is much higher. This indicates that more oxygen-rich vacancies are formed in E-NiFeOOH, which is consistent with the XPS results.
:3 in E-NiFeOOH and 1:9 in I-NiFeOOH. Moreover, when the Fe content rises above 25%, the high content of iron in NiOOH can greatly improve the OER performance, possibly due to the presence of FeOOH phase nucleation.36 In the high-resolution spectrum of the O region (Fig. 3c), the peaks at 529.3 eV, 531.2 eV and 532.6 eV can be attributed to the lattice oxygen–metal bond, oxygen vacancies in the low oxygen coordination and adsorbed oxygen contents on the surface, respectively.37 It is well known that the oxygen vacancies (Ov) formed can be evaluated by determining the peak area ratio between oxygen vacancies and lattice oxygen. As can be seen from Fig. 3c, the Ov density in E-NiFeOOH is more than twice as high as that in I-NiFeOOH. The calculation results of the Ov density are shown in Table S1.† In addition, the electron paramagnetic resonance (EPR) spectrum in Fig. 3d shows that the major peaks are located at 1.93, 1.96 and 2.006 of the g values. This result indicates the presence of unpaired electrons of Fe3+ and oxygen vacancies.38,39 Compared with the EPR spectrum of I-NiFeOOH, the Fe3+ and oxygen defect peak intensity of E-NiFeOOH is much higher. This indicates that more oxygen-rich vacancies are formed in E-NiFeOOH, which is consistent with the XPS results.
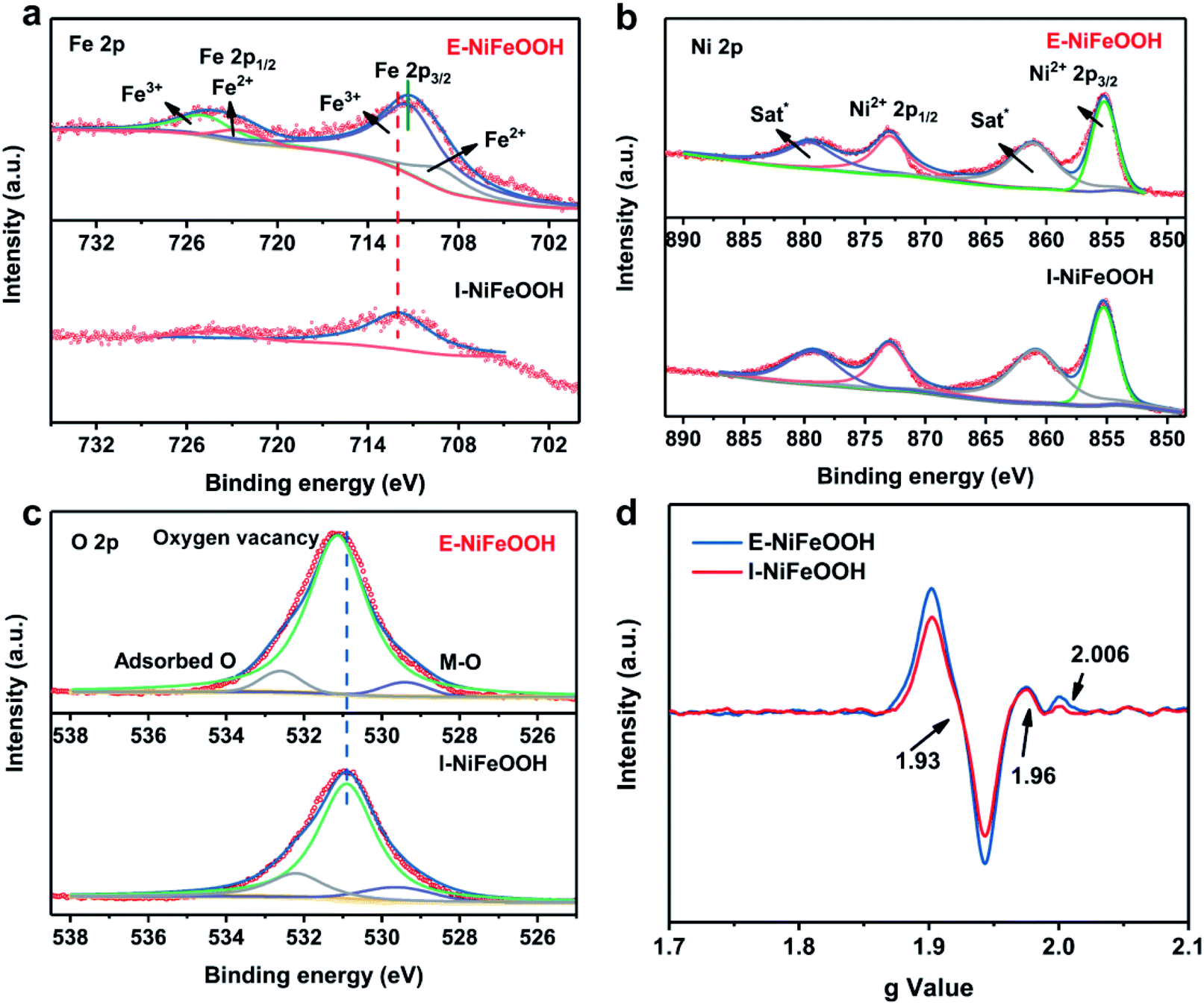 | ||
| Fig. 3 (a–c) High-resolution XPS spectra of Fe 2p, Ni 2p and O 2p. (d) EPR spectra of I-NiFeOOH and E-NiFeOOH. | ||
2.3 HER, OER and overall water splitting electrocatalytic activity
To explore the influence of material composition and structure on the catalytic performance of water splitting, we evaluated the HER activity of Ni foam, I-NiFeOOH, E-NiFeOOH and Pt/C/NF in 1 M KOH in an ambient environment (as shown in Fig. S7a and S8a†). The reaction of the HER in the alkaline medium generally follows the Volmer–Heyrovsky pathway, in which H2O needs to be debonded to form metal-Hads. As presented in Fig. S7a,† the electrodes of E-NiFeOOH-x min exhibit lower overpotentials at 10 mA cm−2 compared with that of the pristine Ni foam (211 mV). The minimum overpotential value of E-NiFeOOH is 145 mV (E-NiFeOOH-30 min), which is inferior to that of excellent Pt/C/NF (84 mV). Interestingly, although E-NiFeOOH exhibits worse performance than Pt/C/NF in the low-current region, the HER electrocatalytic performance is still significantly higher than that of I-NiFeOOH (Fig. S8a†) and other NiFe-based HER electrocatalysts (Fig. S7d and Table S2†), which may overcome the main bottleneck of overall water splitting in large-scale applications of NiFe LDH. Moreover, under the overpotential of 300 mV, E-NiFeOOH-30 min delivered a higher current density of about 155 mA cm−2, while the current density of Pt/C/NF is about 137 mA cm−2 (Fig. S8c†). To evaluate the intrinsic relationship between the reaction kinetics process and the catalytic performance, we calculated the Tafel slopes of different electrocatalysts. As shown in Fig. S7b,† the Tafel slope of E-NiFeOOH-30 min is 115.1 mV dec−1 near to the value of 120 mV dec−1 (theoretical value of the Volmer step). This result suggests that the discharge step (Volmer step) is the rate-limiting step due to the weak adsorption of Fe–Hads with low electronegativity, which is consistent with the previous literature reports.37,39 In contrast, I-NiFeOOH demonstrates more sluggish reaction kinetics and a larger Tafel slope (Fig. S8b†). This indicates that the oxygen defects in E-NiFeOOH significantly accelerate charge transfer from the electrodes to the active sites, thereby boosting the HER process.40 In addition, we explore the practical application potential through cyclic measurements and stability testing. After 1500 CV cycles and maintaining 15 mA cm−2 under −1.1 V (vs. Hg/HgO) for 10 hours, E-NiFeOOH exhibited very negligible changes, which highlights its robust stability (Fig. S7c†).Unlike the HER procedure, the OER is an inherently complicated 4e− transfer process with a sluggish multi-step reaction, including tailoring the O–H bond and reproducing the O–O bond to release O2. Improving the overall water splitting efficiency is the main challenge to overcome. As shown in Fig. 4a, E-NiFeOOH-40 min manifests the best performance of 215 mV at 10 mA cm−2, which is not only superior to that of the pure Ni foam, but is also much lower than that of the benchmark RuO2/NF. Furthermore, the Tafel slope of E-NiFeOOH-40 min is only 30.7 mV dec−1 (Fig. 4b). Such a low slope reflects that the reaction rate at the interface between the electrode and the solution is dramatically fast, which benefits from the oxygen defects of E-NiFeOOH. Significantly, as shown in Fig. S8d and f,† under the overpotential of 300 mV, E-NiFeOOH delivers an optimal current density of about 225 mA cm−2, which is about twice that of I-NiFeOOH (102 mA cm−2) and four times that of RuO2/NF (48 mA cm−2). In particular, the Tafel slope value of I-NiFeOOH is 77 mV dec−1 (Fig. S8e†), which is twice that of E-NiFeOOH. The evident difference between these two slopes illustrates that more oxygen defects in E-NiFeOOH can facilitate electron transfer, activate the O–H bond (promoting the formation of the O–O bond to produce O2), and lower the reaction barrier to achieve higher current density with a lower overpotential. Moreover, the OER durability was determined by 1500 CV cycles and continuous electrolysis for 10 hours at a high current density of 110 mA cm−2 under the potential of 1.5 V (vs. RHE), as shown in Fig. 4c. The results manifest that E-NiFeOOH has excellent catalytic performance and impressive OER stability. In addition, the overpotential and Tafel slope value of E-NiFeOOH are compared with those of the related NiFe-based OER electrocatalysts, and both have much better performance than those of most reported state-of-the-art OER electrocatalysts (Fig. 4d and Table S3†). Furthermore, the morphology, composition, and structure of the electrocatalyst after the reaction were revealed. As shown in Fig. S10,† the surface morphology change was revealed by SEM and TEM. It clearly shows that the electrocatalyst undergoes surface reconstruction after the OER. Moreover, the HRTEM image in Fig. S10d† shows clear lattice fringes with an interplanar spacing of 0.25 nm in the crystalline part, which is assigned to the evolution of NiFeOOH. In addition, XRD characterization with a slow scan rate of 2° min−1 was performed to explore the composition of the electrocatalyst. Only a few weak diffraction peaks of NiOOH and FeOOH can be observed in Fig. S10e.† Fig. S10f† shows the high-resolution XPS spectrum of Fe 2p, two peaks at 712.4 eV for Fe 2p3/2 and 725.6 eV for Fe 2p1/2, indicating the Fe3+ oxidation state. In Fig. S10g,† the high-resolution XPS spectrum of Ni 2p shows two spin–orbit peaks at 855.5 (Ni 2p3/2) and 873.4 eV (Ni 2p1/2), along with two satellite peaks, which are characteristic of the Ni2+ oxidation state. Two peaks at 856.8 eV and 874.6 eV correspond to the Ni3+. These results imply that the electrocatalyst delivers incomplete reconstruction. Moreover, the O 1s XPS spectrum displayed in Fig. S10h† shows that the relative intensity of the oxide peak is reduced and the component at 531–532 eV becomes more dominant due to the formation of NiFeOOH.
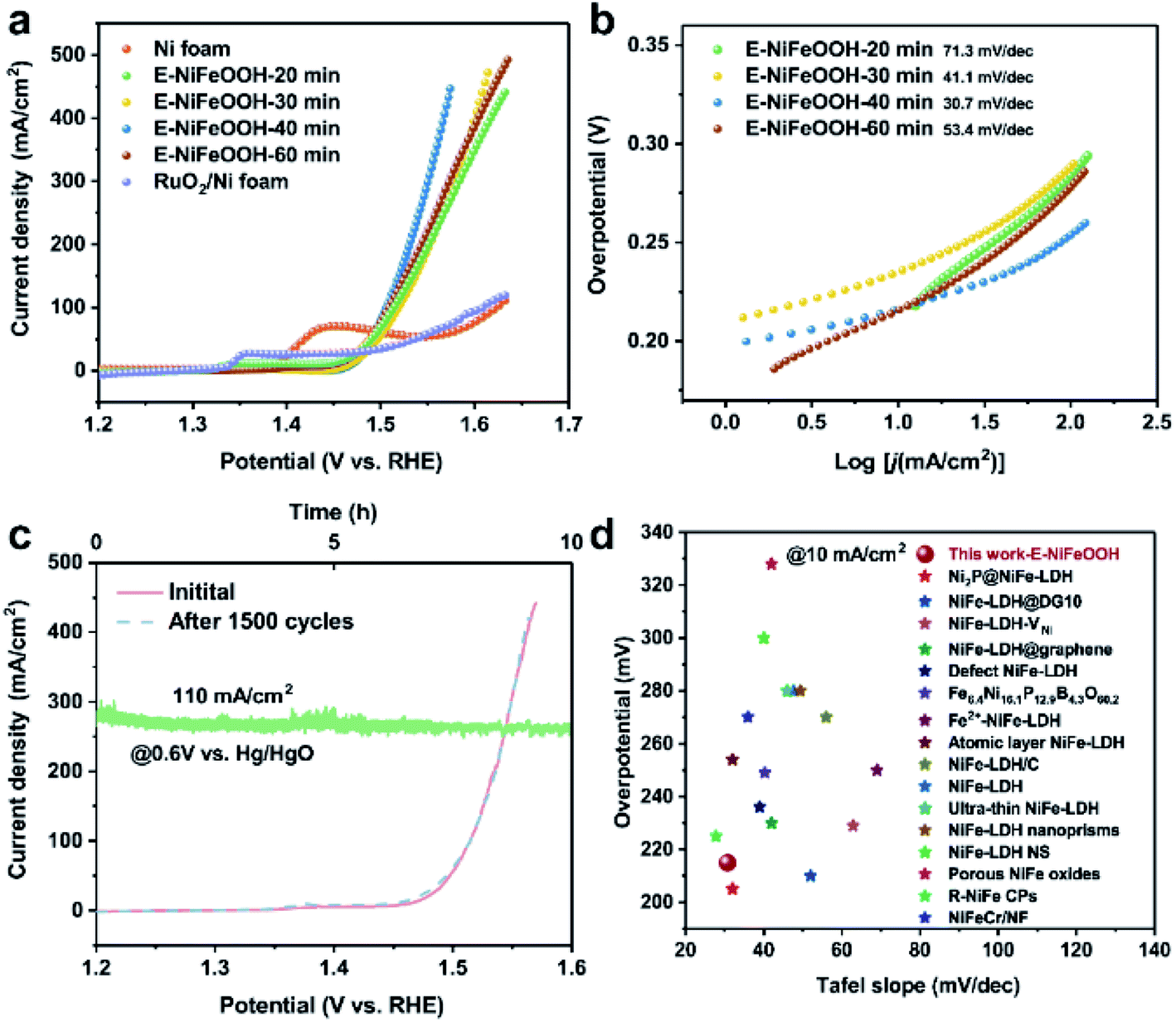 | ||
| Fig. 4 (a and b) Polarization curves and corresponding Tafel plots of the Ni foam, E-NiFeOOH-20, 30, 40, 60 min and RuO2/NF electrodes for the OER. (c) Long-term stability tests and stability measurements of E-NiFeOOH-40 min. (d) Comparison of recent representative studies of NiFe-based OER electrocatalysts with their corresponding overpotentials at 10 mA cm−2 and Tafel slope values (Table S3†). | ||
To gain insight into the catalytic activity of the overall water splitting, we used a home-made two-electrode system to study the electrocatalytic performance. The couple of E-NiFeOOH electrodes required as low as 1.59 V for achieving a current density of 10 mA cm−2 (Fig. 5a), which is much lower than those of the related NiFe LDH/NF (1.7 V), NiFe/NiCo2O4/NF (1.67 V), the state-of-the-art Pt/C–IrO2 counterparts (1.72 V) and other reported electrocatalysts (Fig. 5c). Besides that, we also carried out the cyclic measurement and stability testing to evaluate the practical applications. It is gratifying that the couple of E-NiFeOOH electrodes demonstrated excellent durability in alkaline medium, and there was no obvious change after 1500 cycles and more than 180 hours of long-term operation (Fig. 5b). Moreover, in our perspective, charged droplet mediated corrosion engineering is more suitable for large-scale synthesis by adopting one-step electrospraying operation, compared with immersing the metal-foam in bulk aqueous solution. It can not only greatly reduce the waste of resources, but also realize the control of solution components more conveniently and construct more defects, so as to improve the performance of the electrocatalyst. This concept scheme is shown in Fig. S9.†
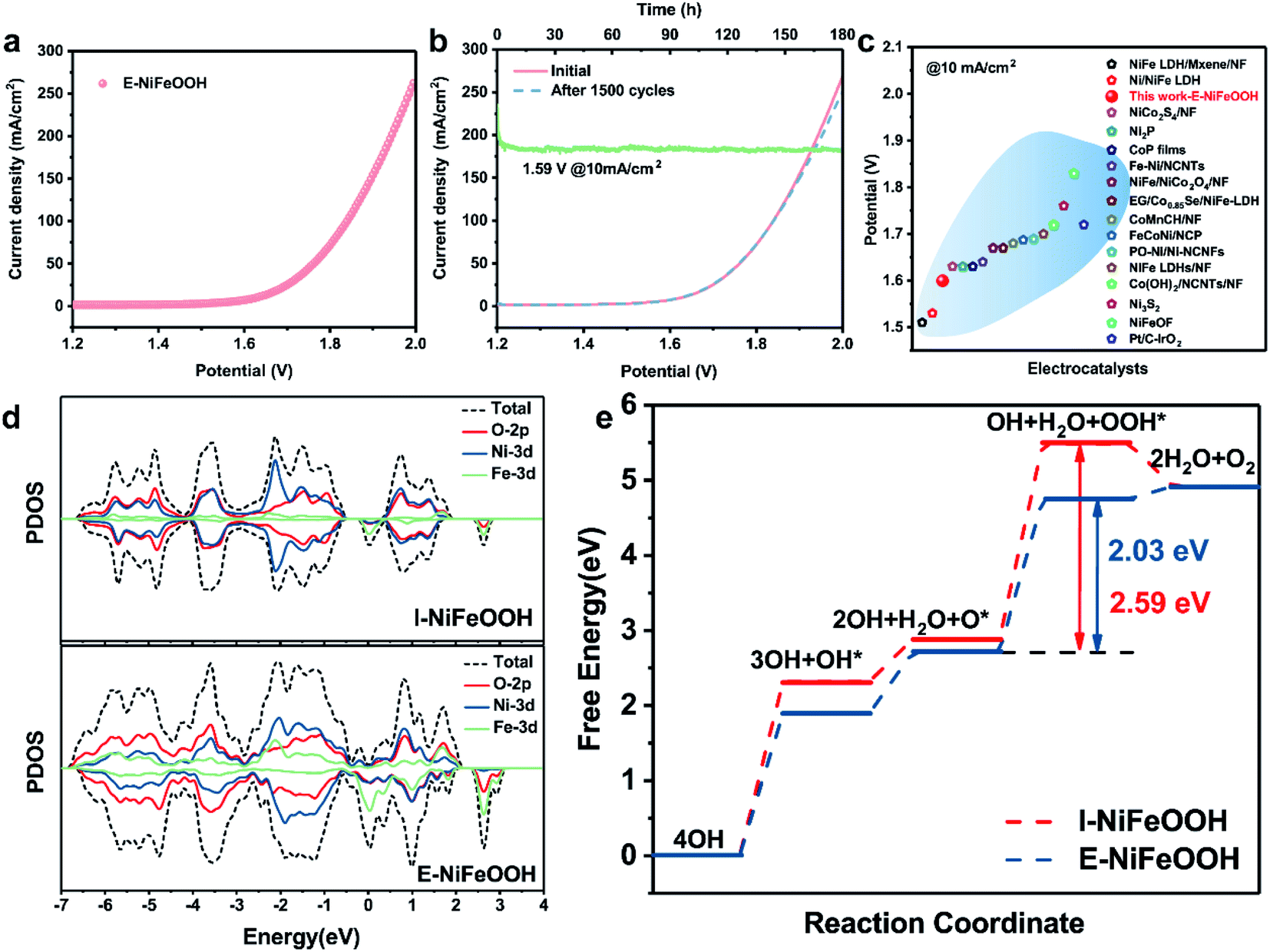 | ||
| Fig. 5 (a) Polarization curves of the E-NiFeOOH-30 min/E-NiFeOOH-40 min electrode couple for overall water splitting. (b) Long-term stability tests and stability measurements for overall water splitting. (c) Comparison of recent representative studies of NiFe-based overall water splitting electrocatalysts at 10 mA cm−2 (Table S4†). (d) Computed partial density of states (pDOS) of I-NiFeOOH (Ni9Fe1-LDH) and E-NiFeOOH (Ni3Fe1-LDH-Ov), respectively. (e) Gibbs free energy profiles of I-NiFeOOH and E-NiFeOOH along the OER reaction pathway. | ||
Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) were performed in 1 M KOH to gain further insight into the electrode kinetics and the effective electrochemically active surface areas (ECSA) of E-NiFeOOH. As shown by the Nyquist plot in Fig. S11a,† compared with E-NiFeOOH-20 min (10.2 Ω) and E-NiFeOOH-40 min (16.1 Ω), E-NiFeOOH-30 min has the smallest charge transfer resistance (Rct) of approximately 8.8 Ω. The small resistance can improve electron transfer between the electrode and the electrolyte, thereby showing striking HER activity. This analysis is consistent with the result of HER measurements. Simultaneously, we calculated the electrochemical double-layer capacitances (Cdl) by CV to reveal the ECSA at various scan rates in a non-faradaic range (Fig. S11b–e†). E-NiFeOOH possesses a larger Cdl value of 6.13 mF, which is about six times higher than that of Ni foam (1.14 mF). The results illustrate that the E-NiFeOOH encompasses more active sites to adsorb and activate water molecules. To evaluate the intrinsic activity of the two electrocatalysts, the ECSA-normalized LSV curves are further recorded. The measured Cdl of I-NiFeOOH is 4.77 mF. As shown in Fig. S11e,† the catalytic performance after ECSA normalization of E-NiFeOOH is still superior to that of I-NiFeOOH, indicating the more efficient intrinsic activity of E-NiFeOOH.
2.4 Electrocatalytic mechanism analysis
Partial density of state (pDOS) calculation was performed to understand the correlations between structural defects, and HER and OER activity. As shown in Fig. 5d, the I-NiFeOOH (Ni9Fe1-LDH) electrode exhibits metallic properties with low occupancy states near the Fermi level. In contrast, E-NiFeOOH (Ni3Fe1-LDH-Ov) shows an increased pDOS in O-2p, Ni-3d and Fe-3d, which benefits from the formation of structural oxygen defects. The increased pDOS of E-NiFeOOH can facilitate the charge transfer of the electrocatalyst, and is also favorable for the adsorption of OER intermediate species. Moreover, the Fe–O bond of E-NiFeOOH was shortened compared with that of I-NiFeOOH, while the Ni–O bond has no change in both the electrodes. The shortened bond and the increased Fe–O hybridization improve the adsorption performance and electron mobility between the intermediate species and the metal cation. The results reveal that the Fe–O bond and oxygen vacancies play important roles in improving the OER kinetics. Additionally, the Gibbs free energy of the OER intermediate species was calculated, as shown in Fig. 5e (detailed DFT calculations are shown in the ESI†). The ideal catalyst requires that the free energy change at each step is appropriate. Compared with the I-NiFeOOH electrode, the E-NiFeOOH electrode displays a lower ΔGOH*, which implies that OH* is more easily adsorbed on the surface with rich Ov and hence favors the subsequent O* and OOH* formation. Notably, the rate-determining steps (RDS) at both I-NiFeOOH and E-NiFeOOH are the ΔGOOH*. The ΔGOOH* energy barrier of E-NiFeOOH electrodes is 2.03 eV, which is superior to that of I-NiFeOOH (2.63 eV). The optimized OER reaction pathway demonstrates that the shortened Fe–O bond and oxygen vacancies can improve the charge mobility between the metal cation and the intermediate species. Moreover, HER activity can also be improved by oxygen vacancies because of the following reasons: (1) rapid transfer of electrons/charges. As shown in Fig. 5d, the oxygen vacancies in E-NiFeOOH can improve the PDOS around the Fermi level, and the enhancement of conductivity in E-NiFeOOH can enable more efficient electron transfer from electrodes to reaction species to realize high HER activity. (2) Optimization of the electronic structure of the catalyst. For the HER in alkaline media, the cleavage of the H–OH bond is very important. It requires appropriate OH− adsorption to improve reaction kinetics, and strong OH− adsorption will poison the electrocatalyst, and weak OH− adsorption will not be favorable for producing Hads. As shown in Fig. S8b,† E-NiFeOOH demonstrates more efficient reaction kinetics and a smaller Tafel slope than I-NiFeOOH. Moreover, both the values are close to 118–120 mV dec−1, which is considered as Volmer-step limited (H2O + e− → H* + OH−). Therefore, the oxygen defects in E-NiFeOOH significantly optimize the adsorption of intermediate species, thereby boosting the HER process.3. Conclusions
In summary, we have developed a facile strategy to construct NiFeOOH with rich oxygen vacancies at the interface between charged droplets and Ni foam. The unique synthesis process of NiFeOOH not only takes less time than classical corrosion engineering, but also can form abundant oxygen vacancies in NiFeOOH based on a non-equilibrium stoichiometric reaction. The electrodes of E-NiFeOOH demonstrate much lower overpotentials of 145 mV (E-NiFeOOH-30 min), 215 mV (E-NiFeOOH-40 min) and 360 mV (the couple electrodes) for the HER, OER and overall water splitting in 1 M KOH at 10 mA cm−2, and have a long-term durability of over 180 h. In this work, the main factors and features of corrosion engineering mediated by charged droplets have been discussed: extreme pH, concentration effect and desolvation state. All these characteristics can contribute to expediting the corrosion reaction in terms of kinetics and thermodynamics. Moreover, thanks to the sub-stoichiometric chemical reaction in the charged droplets, sufficient oxygen vacancies can be rapidly formed in the electrocatalyst. Lastly, we performed a DFT simulation to fundamentally understand the OER mechanism. The calculation results reveal that the shortened Fe–O bonds and oxygen defects synergistically improve the interaction between the metal cations and the intermediate species (OH*, O* and OOH*) and further accelerate the overall reaction kinetics. Our work has greatly accelerated the reaction kinetics of classical corrosion engineering, and offered a new means for the facile formation of oxygen defects by using a unique charged droplet reactor.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully thank La-Jia Yu for help in the EPR experiment, Ye-Chuang Han for help in the experimental facilities, and De-Yin Wu for helpful discussion. This work was supported by the National Natural Science Foundation (2207020940). F. R. Fan acknowledges the Nanqiang Young Top-notch Talent Fellowship from Xiamen University. Z. A. Nan acknowledges the financial support by the China Postdoctoral Science Foundation (2020M671939).References
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC.
- B. C. Steimle, J. L. Fenton and R. E. Schaak, Science, 2020, 367, 418–424 CrossRef CAS PubMed.
- Y. Duan, Z. Y. Yu, S. J. Hu, X. S. Zheng, C. T. Zhang, H. H. Ding, B. C. Hu, Q. Q. Fu, Z. L. Yu, X. Zheng, J. F. Zhu, M. R. Gao and S. H. Yu, Angew. Chem., Int. Ed., 2019, 58, 15772–15777 CrossRef CAS PubMed.
- B. Qiu, L. Cai, Y. Wang, Z. Lin, Y. Zuo, M. Wang and Y. Chai, Adv. Funct. Mater., 2018, 28, 1706008 CrossRef.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- (a) X. Liu, M. Gong, D. Xiao, S. Deng, J. Liang, T. Zhao, Y. Lu, T. Shen, J. Zhang and D. Wang, Small, 2020, e2000663 CrossRef PubMed; (b) Y. Liu, X. Liang, L. Gu, Y. Zhang, G. D. Li, X. Zou and J. S. Chen, Nat. Commun., 2018, 9, 2609 CrossRef PubMed; (c) X. Zou, Y. Liu, G.-D. Li, Y. Wu, D.-P. Liu, W. Li, H.-W. Li, D. Wang, Y. Zhang and X. Zou, Adv. Mater., 2017, 29, 1700404 CrossRef PubMed.
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Gratzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- (a) M. Asnavandi, Y. Yin, Y. Li, C. Sun and C. Zhao, ACS Energy Lett., 2018, 3, 1515–1520 CrossRef CAS; (b) K. Zhu, F. Shi, X. Zhu and W. Yang, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- J. Kim, X. Yin, K.-C. Tsao, S. Fang and H. Yang, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef CAS PubMed.
- (a) R. Liu, Y. Wang, D. Liu, Y. Zou and S. Wang, Adv. Mater., 2017, 29, 1701546 CrossRef PubMed; (b) Z. Wang, Y. Zhang, E. C. Neyts, X. Cao, X. Zhang, B. W. L. Jang and C.-j. Liu, ACS Catal., 2018, 8, 2093–2110 CrossRef CAS.
- C. Meng, M. Lin, X. Sun, X. Chen, X. Chen, X. Du and Y. Zhou, Chem. Commun., 2019, 55, 2904–2907 RSC.
- G. Ou, Y. Xu, B. Wen, R. Lin, B. Ge, Y. Tang, Y. Liang, C. Yang, K. Huang, D. Zu, R. Yu, W. Chen, J. Li, H. Wu, L. M. Liu and Y. Li, Nat. Commun., 2018, 9, 1302 CrossRef PubMed.
- (a) Y. Li, Y. Wang, J. Lu, B. Yang, X. San and Z.-S. Wu, Nano Energy, 2020, 78, 105185 CrossRef CAS; (b) G. Giuffredi, A. Mezzetti, A. Perego, P. Mazzolini, M. Prato, F. Fumagalli, Y. C. Lin, C. Liu, I. N. Ivanov, A. Belianinov, M. Colombo, G. Divitini, C. Ducati, G. Duscher, A. A. Puretzky, D. B. Geohegan and F. Di Fonzo, Small, 2020, e2004047 CrossRef PubMed; (c) Q. Hu, Z. Wang, X. Huang, Y. Qin, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, Energy Environ. Sci., 2020, 13, 5097–5103 RSC.
- (a) K.-L. Yan, J.-Q. Chi, Z.-Z. Liu, B. Dong, S.-S. Lu, X. Shang, W.-K. Gao, Y.-M. Chai and C.-G. Liu, Inorg. Chem. Front., 2017, 4, 1783–1790 RSC; (b) B.-R. Wang, S.-H. Jiao, Z.-S. Wang, M.-J. Lu, D. Chen, Y.-T. Kang, G.-S. Pang and S.-H. Feng, J. Mater. Chem. A, 2020, 8, 17202–17211 RSC; (c) J.-J. Ge, J.-Y. Zheng, J.-W. Zhang, S.-Y. Jiang, L.-L. Zhang, H. Wan, L.-M. Wang, W. Ma, Z. Zhou and R.-Z. Ma, J. Mater. Chem. A, 2021, 9, 14432–14443 RSC; (d) D.-Z. Zhong, L. Zhang, C.-C. Li, D.-D. Li, C.-C. Wei, Q. Zhao, J.-P. Li and J.-L. Gong, J. Mater. Chem. A, 2018, 6, 16810–16817 RSC.
- (a) K. Zhang, G. Zhang, J. Qu and H. Liu, Small, 2018, 14, 1802760 CrossRef PubMed; (b) J. Sun, N. Guo, Z. Shao, K. Huang, Y. Li, F. He and Q. Wang, Adv. Energy Mater., 2018, 8, 1800980 CrossRef.
- (a) H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 7336–7356 CrossRef CAS PubMed; (b) S. Mellouli, L. Bousekkine, A. B. Theberge and W. T. S. Huck, Angew. Chem., Int. Ed., 2012, 51, 7981–7984 CrossRef CAS PubMed.
- R. M. Bain, C. J. Pulliam, F. Thery and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 10478–10482 CrossRef CAS PubMed.
- (a) S. Banerjee and R. N. Zare, Angew. Chem., Int. Ed., 2015, 54, 14795–14799 CrossRef CAS PubMed; (b) X. Yan, R. M. Bain and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 12960–12972 CrossRef CAS PubMed.
- Z. Wei, M. Wleklinski, C. Ferreira and R. G. Cooks, Angew. Chem., Int. Ed., 2017, 56, 9386–9390 CrossRef CAS PubMed.
- (a) J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed; (b) J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19294–19298 CrossRef CAS PubMed; (c) Z. Wei, Y. Li, R. G. Cooks and X. Yan, Annu. Rev. Phys. Chem., 2020, 71, 31–51 CrossRef CAS PubMed.
- (a) A. Gallo, Jr, A. S. F. Farinha, M. Dinis, A. H. Emwas, A. Santana, R. J. Nielsen, W. A. Goddard and H. Mishra, Chem. Sci., 2019, 10, 2566–2577 RSC; (b) S. Banerjee, E. Gnanamani, X. Yan and R. N. Zare, Analyst, 2017, 142, 1399–1402 RSC.
- (a) L. Foti, A. Sionek, E. M. Stori, P. P. Soares, M. M. Pereira, M. A. Krieger, C. L. Petzhold, W. H. Schreiner, M. J. Soares, S. Goldenberg and C. K. Saul, J. Mater. Chem. B, 2015, 3, 2725–2731 RSC; (b) D. Sarkar, R. Singh, A. Som, C. K. Manju, M. A. Ganayee, R. Adhikari and T. Pradeep, J. Phys. Chem. C, 2018, 122, 17777–17783 CrossRef CAS.
- (a) M. Girod, E. Moyano, D. I. Campbell and R. G. Cooks, Chem. Sci., 2011, 2, 501–510 RSC; (b) A. Jana, S. K. Jana, D. Sarkar, T. Ahuja, P. Basuri, B. Mondal, S. Bose, J. Ghosh and T. Pradeep, J. Mater. Chem. A, 2019, 7, 6387–6394 RSC.
- Z. Zhou, X. Yan, Y. H. Lai and R. N. Zare, J. Phys. Chem. Lett., 2018, 9, 2928–2932 CrossRef CAS PubMed.
- X. Liu, M. Gong, S. Deng, T. Zhao, T. Shen, J. Zhang and D. Wang, Adv. Funct. Mater., 2021, 31, 2009032 CrossRef CAS.
- (a) S. Mondal, S. Acharya, R. Biswas, B. Bagchi and R. N. Zare, J. Chem. Phys., 2018, 148, 244704 CrossRef PubMed; (b) M. F. Ruiz-Lopez, J. S. Francisco, M. T. C. Martins-Costa and J. M. Anglada, Nat. Rev. Chem., 2020, 4, 459–475 CrossRef CAS; (c) K. Chingin, V. Frankevich, R. M. Balabin, K. Barylyuk, H. Chen, R. Wang and R. Zenobi, Angew. Chem., Int. Ed., 2010, 49, 2358–2361 CrossRef CAS PubMed; (d) H. Wei, E. P. Vejerano, W. Leng, Q. Huang, M. R. Willner, L. C. Marr and P. J. Vikesland, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7272–7277 CrossRef CAS PubMed.
- S. Niu, W. Jiang, T. Tang, L. Pan Yuan, H. Luo and J. Hu, Adv. Funct. Mater., 2019, 29, 1902180 CrossRef.
- H. X. Lin, Z. C. Lei, Z. Y. Jiang, C. P. Hou, D. Y. Liu, M. M. Xu, Z. Q. Tian and Z. X. Xie, J. Am. Chem. Soc., 2013, 135, 9311–9931 CrossRef CAS PubMed.
- I. Nam, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 36–40 CrossRef CAS PubMed.
- Y. Yao, X. Chen, C. Yan, X. Zhang, W. Cai, J. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4090 CrossRef CAS PubMed.
- W. Guo, D. Li, D. Zhong, S. Chen, G. Hao, G. Liu, J. Li and Q. Zhao, Nanoscale, 2020, 12, 983–990 RSC.
- J. Dong, J. Huang, A. Wang, G. V. Biesold-McGee, X. Zhang, S. Gao, S. Wang, Y. Lai and Z. Lin, Nano Energy, 2020, 71, 104579 CrossRef CAS.
- J. Zhou, L. Yu, Q. Zhu, C. Huang and Y. Yu, J. Mater. Chem. A, 2019, 7, 18118–18125 RSC.
- J. Dong, X. Zhang, J. Huang, J. Hu, C. Zhong and Y. Lai, Chem. Eng. J., 2021, 128556 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Norskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- Y. Zhao, W. Wan, Y. Chen, R. Erni, C. A. Triana, J. Li, C. K. Mavrokefalos, Y. Zhou and G. R. Patzke, Adv. Energy Mater., 2020, 10, 2002228 CrossRef CAS.
- (a) D. Karmakar, S. K. Mandal, R. M. Kadam, P. L. Paulose, A. K. Rajarajan, T. K. Nath, A. K. Das, I. Dasgupta and G. P. Das, Phys. Rev. B, 2007, 75, 144404 CrossRef; (b) O. V. Makhlynets, W. N. Oloo, Y. S. Moroz, I. G. Belaya, T. D. Palluccio, A. S. Filatov, P. Muller, M. A. Cranswick, L. Que, Jr and E. V. Rybak-Akimova, Chem. Commun., 2014, 50, 645–648 RSC.
- A. Alobaid, C. Wang and R. A. Adomaitis, J. Electrochem. Soc., 2018, 165, 3395–3404 CrossRef.
- M. Asnavandi and C. Zhao, Mater. Chem. Front., 2017, 1, 2541–2546 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta05332a |
| This journal is © The Royal Society of Chemistry 2021 |