
Supramolecular polymer-assisted manipulation of triblock copolymers: understanding the relationships between microphase structures and mechanical properties†
Zhexian
Shentu‡
,
Zhaoming
Zhang‡
*,
Jun
Zhao
 ,
Chuanshuang
Chen
,
Qian
Wu
,
Lei
Wang
and
Xuzhou
Yan
*
,
Chuanshuang
Chen
,
Qian
Wu
,
Lei
Wang
and
Xuzhou
Yan
*
School of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: zhangzhaoming@sjtu.edu.cn; xzyan@sjtu.edu.cn
First published on 27th April 2021
Abstract
Block copolymers with specific microphase-separated structures are a class of important reprocessable elastomers. Numerous approaches have been applied to modify their structures and properties, whereas incorporating supramolecular polymers into the microphase structures has never been achieved. Herein, we report two triblock copolymers in ABA and BAB forms whose hard domains are the A blocks connected by a crown ether-based supramolecular polymer. The ABA triblock copolymer features a regularly distributed hard domain and a tight network, and the rupture of the hard domain is ready to occur upon relatively small deformation. The BAB triblock copolymer shows a hierarchical structure including soft, hard, and secondary hard domains, which results in a relatively tough network. Our work represents a step forward in modifying the phase structures of block copolymers, and would promote the development of block copolymers with emergent properties.
Introduction
Block copolymers having incompatible polymer segments are able to form microphase-separated structures which would endow corresponding materials with elastomeric properties as well as melt reprocessability.1–4 For example, numerous thermoplastic elastomers with high strength and a tunable modulus have been developed by constructing ABA triblock copolymers wherein B is rubbery and the minority A blocks associate to form hard domains as physical cross-links.5–7 Materials based on this kind of copolymer also have inherent defects in mechanical properties, like creep and hysteresis upon deformation.8–10 Discrete supramolecular interactions such as H-bonding and metal-coordination have been utilized to strengthen the hard domain of copolymers, which represents an effective strategy to modify the mechanical properties of materials.11–13 Compared with discrete supramolecular interactions, supramolecular polymers could arrange repeated units into a more ordered and compact form.14–23 Furthermore, in our previous work, we found that the synergistic effect of supramolecular polymers and covalent polymers could endow materials with robust yet dynamic features.24,25 Therefore, we speculate that utilizing supramolecular polymers to connect the A blocks of copolymers would be beneficial to modulate the assembly structure of the hard domains and then result in enhanced strength and toughness. However, as far as we know, incorporating supramolecular polymers into hard domains of copolymers to act as physical cross-links has never been achieved.So far, ABA copolymers have been used widely to develop block copolymer-based elastomers.26–28 By contrast, reports on phase-separated structures and mechanical properties of BAB copolymers in which a hard block is inserted into the middle of the soft block, are limited. It has been reported that well-designed BAB triblock copolymers could also exhibit good mechanical properties.29–31 Besides, a comparison study between ABA and BAB copolymers is helpful to understand the relationships among the polymer structure, phase separation, and mechanical properties of block copolymers.32–34 Nevertheless, in-depth studies on the comparison of the micro-phase structures and mechanical properties of ABA and BAB triblock copolymers are still rare.
Herein, we report two triblock copolymers in forms of ABA and BAB in which the A blocks in hard domains are cross-linked by a supramolecular polymer based on host–guest recognition. Benefiting from the involvement of supramolecular polymers, the two copolymers formed intriguing microphase-separated structures: ABA is composed of regular hard domains to crosslink the soft domains, and BAB presents hierarchical microstructures including soft, hard, and secondary hard domains (Fig. 1). Further, these phase structures endow corresponding materials with different mechanical properties. ABA is relatively rigid due to the tight network structure but the network is also ready to be fractured by stress, and becomes soft after yielding. However, the network of BAB possesses notable elastic properties, showing good stability upon deformation.
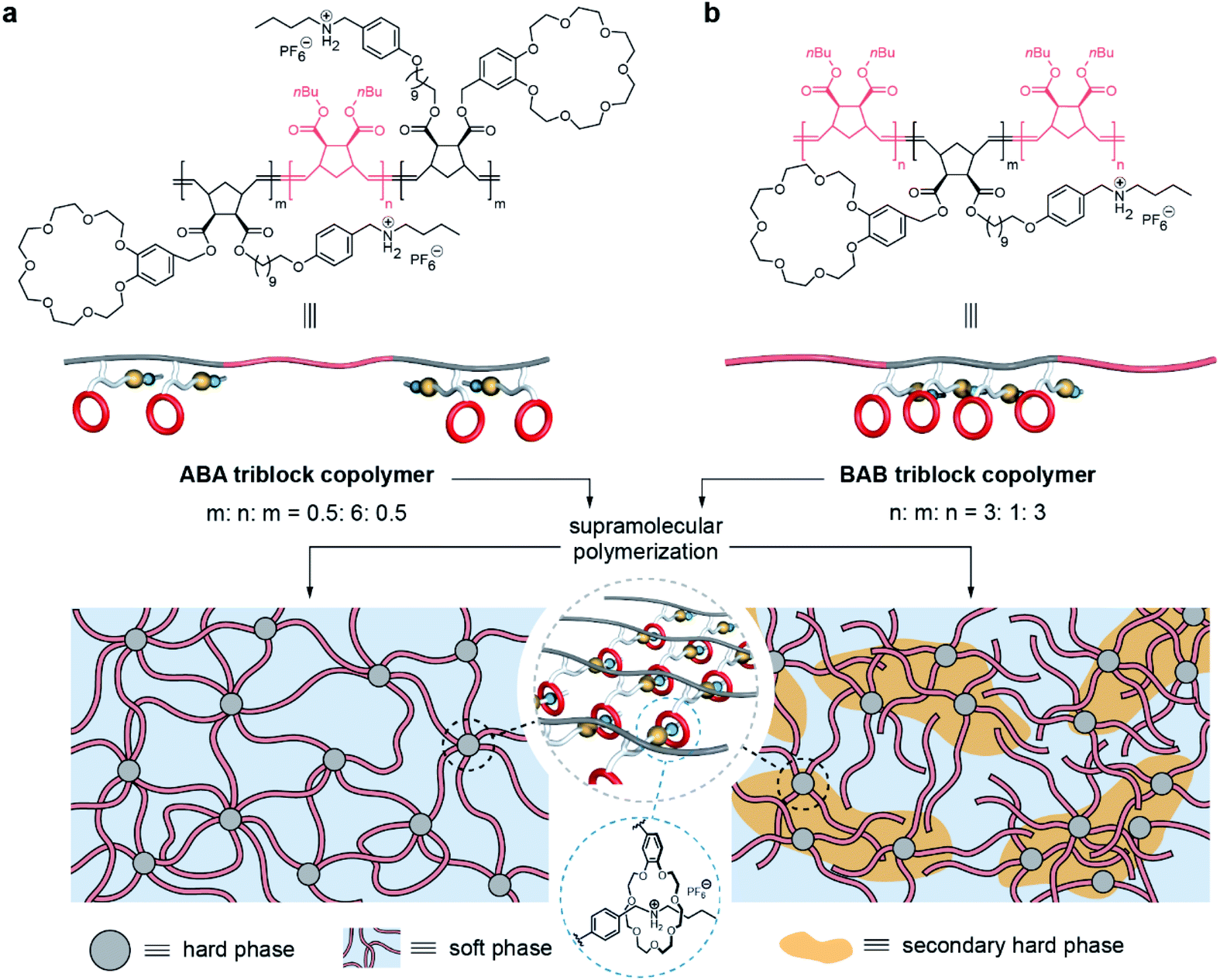 | ||
| Fig. 1 Chemical structures and schematic representation of the two triblock copolymers and their microphase-separated structures. | ||
Results and discussion
The two block segments of the copolymers depend on two monomers: one is norbornenyl exo-di-n-butylester for the soft block (B block), and the other one is a norbornene derivative for the hard block (A block) which bears both a benzo-21-crown-7 (B21C7) moiety and a Boc-protected amino group. Through controlling sequential feeds of the monomers, the ABA and BAB triblock copolymers were precisely prepared by ring opening metathesis polymerization (ROMP). Corresponding 1H NMR spectra and SEC results indicated that the two triblock copolymers had a similar molecular weight, chemical component and content, but differed in the block sequence and block chain length (Figs. S1 and S3†). Table 1 depicts the detailed information about the two copolymers. Notably, host–guest recognition was inactive in the obtained triblock copolymers because the Boc-protected amino group cannot interact with the B21C7 moiety. Hence, acid treatment and anion exchange were further conducted to convert the N-Boc group into a secondary ammonium salt. At this point, a supramolecular polymer was successfully formed based on host–guest recognition, as supported by the 1H NMR spectra (Fig. S1†).| M n (kg mol−1) | M w/Mna | Molar ratio (B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A)b :A)b |
Block sequence (theoretically) | DP of A blockc | DP of B blockc | T g of soft/hard phasesd | |
|---|---|---|---|---|---|---|---|
| a Determined by SEC. b Calculated by peak integration of 1H NMR spectra. c Integrated results of 1H NMR and SEC. d Determined by DSC. *Summation of the two blocks at both ends of the chains. | |||||||
| ABA | 105 | 1.96 | 6.5:1 |
A:B:A = 0.5:6:0.5 |
36* | 219 | −12 °C, 49 °C |
| BAB | 107 | 2.04 | 6.7:1 |
B:A:B = 3:1:3 |
36 | 248* | −20 °C, — |
The thermal properties of the copolymers were firstly characterized. Thermal gravimetric analysis (TGA) results showed that both copolymers had good thermostability below 200 °C. Differential scanning calorimetry (DSC) characterization could differentiate glass transition temperatures (Tg) of different phases. For the soft domains, the Tg of ABA (−12 °C) was slightly higher than that of BAB (−20 °C), which suggested that the movement of B segments in ABA was restricted more tightly. The Tg of hard domains in ABA was observed at 49 °C but that for BAB was fairly inconspicuous, probably indicating a more complexed phase structure in BAB. In addition, similar tendency was also observed in the temperature sweep recorded by dynamic thermomechanical analysis (DMA) (Fig. S6†). Subsequently, tensile tests were carried out to identify the mechanical properties of both samples. As shown in Fig. 2a, the two copolymers showed totally different mechanical behaviors. The ABA copolymer had a much stiffer nature at the initial stage, and then showed a turn-down yield at about 160% strain. In contrast, the BAB copolymer was soft initially but stress strengthening occurred along with the increase of strain. Corresponding Young's modulus and tensile toughness were calculated and are depicted in Fig. 2b. The Young's modulus was calculated to be 1.6 MPa for BAB which was lower than that of ABA (6.0 MPa). The BAB had a relatively superb fracture toughness than ABA (29 vs. 6.1 MJ m−3). The results of frequency sweep were in line with those in tensile tests (Fig. 2c). The storage modulus of ABA was higher than that of BAB, indicative of a stiffer feature of the ABA copolymer.
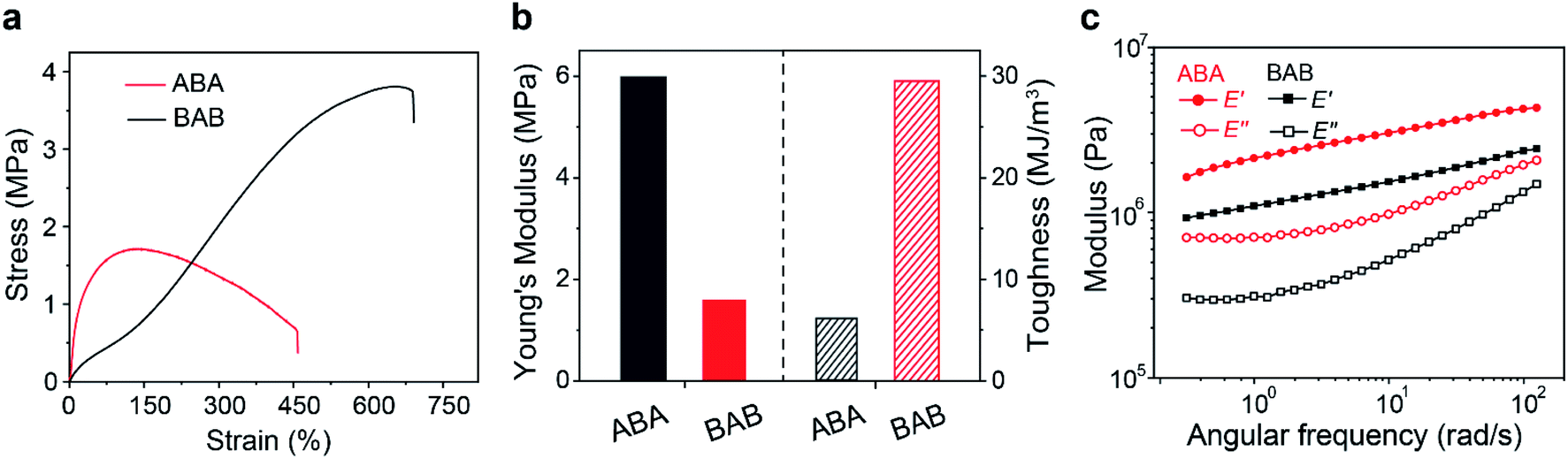 | ||
| Fig. 2 (a) The stress–strain plots of ABA and BAB triblock copolymers. (b) The Young's modulus and toughness of the two copolymers calculated based on their stress–strain curves. (c) Frequency sweep curves of the two copolymers in the range of 0.3–100 rad s−1. | ||
For block copolymers, their mechanical properties are highly dependent on their microphase-separated structures. Hence, the morphologies of the two copolymers were measured by atom force microscopy (AFM). The images showed that ABA formed a typical sea–island dispersed structure (Fig. 3a). Assisted by the modulus distribution image (Fig. S8†), these “islands” could be assigned as the hard domains based on the A blocks connected by the supramolecular polymer. However, for BAB, the system exhibited not only isolated hard domains but also continuous secondary hard phases (Fig. 3b). Moreover, transmission electron microscope (TEM) images also demonstrated similar microphase-separated structures (Fig. S9†). We deduce that the different microphase morphologies of the two copolymers are derived from their peculiar chemical structures and sequences. It is reasonable for the ABA copolymer having shorter supramolecular segments at both ends of the chain to form smaller and discrete hard phases bridging the soft phases. As for the BAB copolymer, the A blocks in the middle of polymer chains are not as flexible as those at the ends of the chain in ABA. Certain B blocks are easy to involve in hard domains and fixed by the supramolecular polymer, forming stable secondary hard-phases. The supramolecular polymer could act as a mediator to stabilize the mixture of the A and B blocks, which is perhaps the reason why the secondary hard-phases are observed in our system rather than other reported BAB triblock copolymers. It has been proved that incorporating extra physical cross-links into the B block could enhance the mechanical properties of ABA copolymers.35–37 In BAB, the B blocks fixed by the supramolecular polymer in the secondary hard-phases are equal to introduce physical cross-links on the B blocks. Therefore, the formation of secondary hard-phases would be beneficial to enhance the mechanical properties of the BAB copolymer.
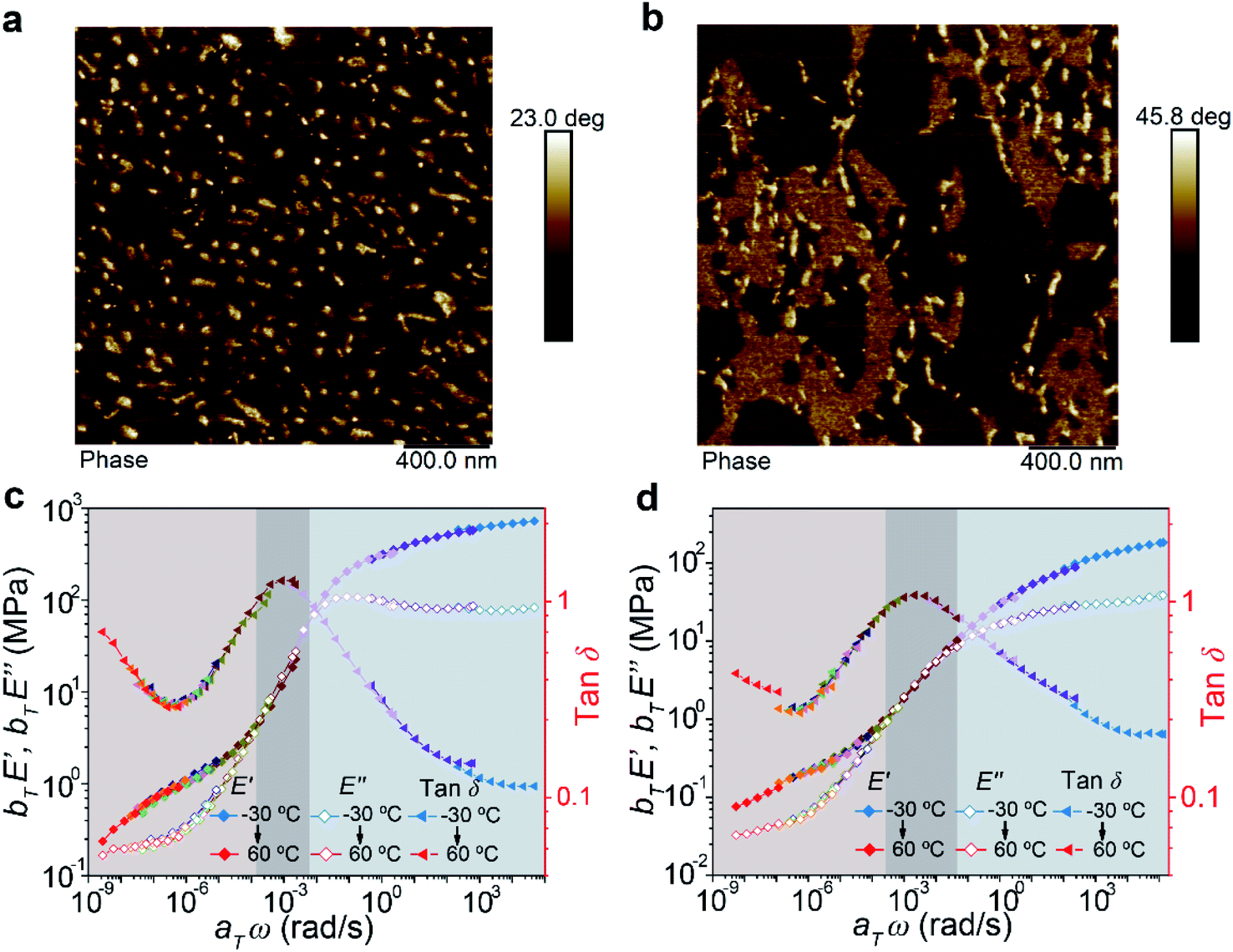 | ||
| Fig. 3 AFM phase images of ABA (a) and BAB (b) triblock copolymers. Master curves of ABA (c) and BAB (d) triblock copolymers at a reference temperature of −20 °C. | ||
Combining the analysis of the viscoelastic behaviors of the two copolymers, basic mechanical characters of their phase structures could be disclosed. Partial master curves exhibiting the rubbery-glassy region of materials are shown in Fig. 3c and d. The low-frequency regions (<10−4) were known as the elastic plateau where the materials exhibited predominately elastic properties. It could be found that the frequency range of the elastic plateau for BAB was broader than that of ABA, and such a phenomenon was also observed in the temperature ramp curves measured by DMA (Fig. S6†). These results implied that the BAB had high flow resistance. In other words, the network of BAB was more stable than that of ABA. Besides, in this region, the E′ modulus of ABA was higher than that of BAB, and was consistent with the tensile behavior (higher strength and stiffness of ABA at the initial stage), which meant that the network of ABA was much tighter. Therefore, the relationships between the phase structures and mechanical properties of the two copolymers could be summarized. The ABA triblock copolymer is prone to form a regular microphase-separated structure which is advantageous to crosslink the polymer chains to form a tight network. But this network is relatively unstable and rupture of the hard domain would be easy to happen upon stress. The BAB triblock copolymer possesses a complicated phase structure including discrete hard domains, continuous secondary hard domains, and soft domains. Such a hierarchical structure could endow the system with high stability to resist large deformation.
To verify the proposed speculation, further investigations on the mechanical behaviors of the two copolymers were performed. The results of cyclic loading–unloading tests are shown in Fig. 4a and b. Continuous tensile tests at 150% strain showed that ABA had a large hysteresis area which became much smaller at the second cycle. This phenomenon as well as the large residual strain reflected that rupture of the network through the dissociation of host–guest recognition may occur under tension. By contrast, BAB displayed a very small hysteresis loop and the loop maintained well at the second cycle, showing a typical elastic energy storage behavior, which was attributed to the superior stability of BAB. Increasing the strain to 300%, a decrease of the hysteresis area at the second cycle for ABA was more distinct due to the rupture of more hard domains. The same phenomenon was also observed for BAB, which implied that structure damage in BAB also took place upon large deformation.
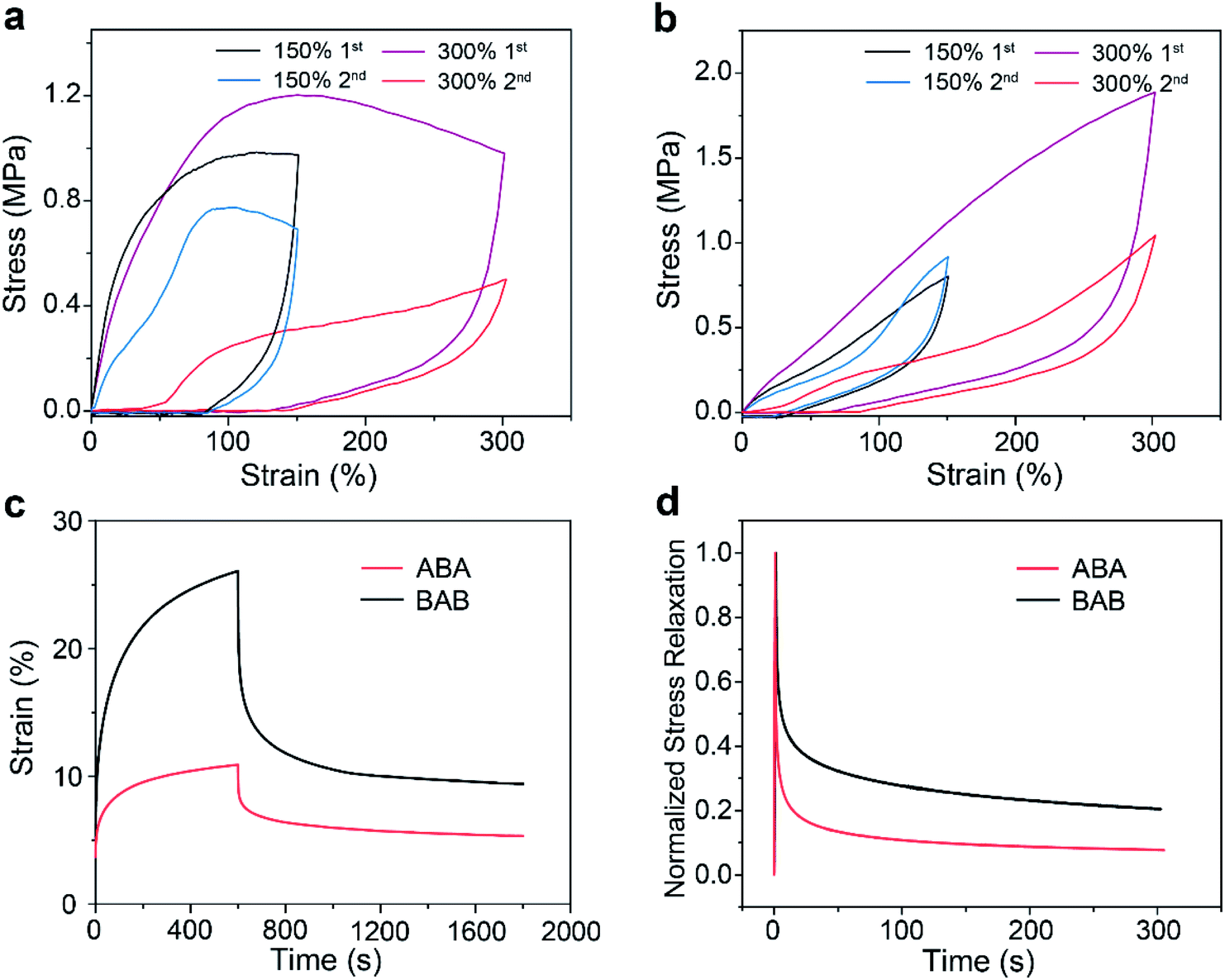 | ||
| Fig. 4 Successive cyclic tensile curves of ABA (a) and BAB (b) at different strains (deformation rate: 100 mm min−1). (c) Creep-recovery response of the two triblock copolymers. (d) Stress relaxation curves of the two triblock copolymers. | ||
Creep-recovery and stress relaxation experiments for the two copolymers were also conducted. For creeping tests with stress at 0.1 MPa for 10 min and then recovery, the ABA samples showed a better creep-resistance performance but a poorer recovery capability (65% for BAB and 51% for ABA) (Fig. 4c). The better creep-resistance of ABA was reasonable because it had higher stiffness compared with that of BAB. However, during the creep process, more hard domains were ruptured in ABA due to the lower stability of its network, which led to its poorer recovery performance. In stress relaxation tests, 50% strain was applied to the two specimens (Fig. 4d). BAB presented a longer elastic relaxation time than ABA, which also supported our speculation that the special hierarchical structures of BAB could maintain a more intact network upon deformation.
Conclusions
In summary, we have synthesized two triblock copolymers in ABA and BAB forms wherein a supramolecular polymer formed in the hard domains driven by the recognition of B21C7 to secondary ammonium salt. The supramolecular polymer played a crucial role in the microphase-separated structures of the copolymers. The ABA triblock copolymer had regular soft-hard domains whereas except for these two phases, secondary hard domains were also observed in the BAB system because some B blocks were mixed with A blocks and fastened by the supramolecular polymer. These unique phase structures endowed the copolymers with different mechanical behaviors. The regular hard phase in ABA gave rise to a tight and stiff network but the hard domains were not so strong to resist large deformation, while the hierarchical phase structures of BAB resulted in a relatively stable network which exhibited good elastic behavior. We believe that using supramolecular polymers as a tool to modulate the microphase structures and properties of block copolymers would facilitate the emergence of more polymeric materials with fascinating properties.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. Y. acknowledges the financial support of the NSFC/China (21901161 and 22071152), Natural Science Foundation of Shanghai (20ZR1429200), and Shanghai Jiao Tong University Scientific and Technological Innovation Funds.References
- G. S. Sulley, G. L. Gregory, T. T. Chen, L. Peña Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 Search PubMed.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps III and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 142, 283–289 Search PubMed.
- J. Hentschel, A. M. Kushner, J. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2012, 51, 10561–10565 Search PubMed.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 Search PubMed.
- T. Isono, S. Nakahira, H. C. Hsieh, S. Katsuhara, H. Mamiya, T. Yamamoto, W.-C. Chen, R. Borsali, K. Tajima and T. Satoh, Macromolecules, 2020, 53, 5408–5417 Search PubMed.
- A. Watts, N. Kurokawa and M. A. Hillmyer, Biomacromolecules, 2017, 18, 1845–1854 Search PubMed.
- Y. Huang, R. Chang, L. Han, G. Shan, Y. Bao and P. Pan, ACS Sustainable Chem. Eng., 2016, 4, 121–128 Search PubMed.
- S. Tomita, I. Wataoka, N. Igarashi, N. Shimizu, H. Takagi, S. Sasaki and S. Sakurai, Macromolecules, 2017, 50, 3404–3410 Search PubMed.
- C. R. Lopez-Barron, A. P. R. Eberle, S. Yakovlev and A. J. Bons, Rheol. Acta, 2016, 55, 103–116 Search PubMed.
- D. Yamaguchi, M. Cloitre, P. Panine and L. Leibler, Macromolecules, 2005, 38, 7798–7806 Search PubMed.
- A. Watts and M. A. Hillmyer, Biomacromolecules, 2019, 20, 2598–2609 Search PubMed.
- M. Nasiri, D. J. Saxon and T. M. Reineke, Macromolecules, 2018, 51, 2456–2465 Search PubMed.
- M. Hayashi, S. Matsushima, A. Noro and Y. Matsushita, Macromolecules, 2015, 48, 421–431 Search PubMed.
- L. Wang, L. Cheng, G. Li, K. Liu, Z. Zhang, P. Li, S. Dong, W. Yu, F. Huang and X. Yan, J. Am. Chem. Soc., 2020, 142, 2051–2058 Search PubMed.
- B. Hua, C. Zhang, W. Zhou, L. Shao, Z. Wang, L. Wang, H. Zhu and F. Huang, J. Am. Chem. Soc., 2020, 142, 16557–16561 Search PubMed.
- B. Qin, S. Zhang, P. Sun, B. Tang, Z. Yin, X. Cao, Q. Chen, J.-F. Xu and X. Zhang, Adv. Mater., 2020, 32, 2000096 Search PubMed.
- Y.-X. Deng, Q. Zhang, B. L. Feringa, H. Tian and D.-H. Qu, Angew. Chem., Int. Ed., 2020, 59, 5278–5283 Search PubMed.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 Search PubMed.
- K. Liu, Y. Jiang, Z. Bao and X. Yan, CCS Chem., 2019, 1, 431–447 Search PubMed.
- S. Wang, Z. Xu, T. Wang, T. Xiao, X. Y. Hu, Y. Z. Shen and L. Wang, Nat. Commun., 2018, 9, 1737 Search PubMed.
- Y. Han, Y. Tian, Z. Li and F. Wang, Chem. Soc. Rev., 2018, 47, 5165–5176 Search PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 Search PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 Search PubMed.
- Z. Zhang, L. Cheng, J. Zhao, L. Wang, K. Liu, W. Yu and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 12139–12146 Search PubMed.
- Z. Zhang, L. Cheng, J. Zhao, H. Zhang, X. Zhao, Y. Liu, R. Bai, H. Pan, W. Yu and X. Yan, J. Am. Chem. Soc., 2021, 143, 902–911 Search PubMed.
- M. Xiong, D. K. Schneiderman, F. S. Bates, M. A. Hillmyer and K. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8357–8362 Search PubMed.
- J. Bai, Z. Shi, J. Yin and M. Tian, Macromolecules, 2014, 47, 2964–2973 Search PubMed.
- A. B. Burns and R. A. Register, Macromolecules, 2016, 49, 269–279 Search PubMed.
- W. Wang, J. Zhang, F. Jiang, X. Wang and Z. Wang, ACS Appl. Polym. Mater., 2019, 1, 571–583 Search PubMed.
- F. Jiang, C. Fang, J. Zhang, W. Wang and Z. Wang, Macromolecules, 2017, 50, 6218–6226 Search PubMed.
- W. Wang, X. Wang, F. Jiang and Z. Wang, Polym. Chem., 2018, 9, 3067–3079 Search PubMed.
- W. Wang, W. Lu, A. Goodwin, H. Wang, P. Yin, N. G. Kang, K. L. Hong and J. W. Mays, Prog. Polym. Sci., 2019, 95, 1–31 Search PubMed.
- A. Zahoranová, M. Mrlík, K. Tomanová, J. Kronek and R. Luxenhofer, Macromol. Chem. Phys., 2017, 218, 1700031 Search PubMed.
- A. Arun, K. Dullaert and R. J. Gaymans, Polym. Eng. Sci., 2010, 50, 756–761 Search PubMed.
- S. Kawana, S. Nakagawa, S. Nakai, M. Sakamoto, Y. Ishii and N. Yoshie, J. Mater. Chem. A, 2019, 7, 21195–21206 Search PubMed.
- Y. Y. Chen and K. R. Shull, Macromolecules, 2017, 50, 3637–3646 Search PubMed.
- M. Hayashi, S. Matsushima, A. Noro and Y. Matsushita, Macromolecules, 2015, 48, 421–431 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02288a |
| ‡ Z. Shentu and Z. Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |