
Localized electron density modulation in conjugated polymer nanosheets for boosting photocatalytic H2 evolution†
Yaoyao
Liu
,
Haifeng
Yu
,
Chong
Shi
and
Zhonghua
Xiang
 *
*
State Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: xiangzh@mail.buct.edu.cn
First published on 21st April 2021
Abstract
A localized electron density modulation strategy is proposed to enhance the intrinsic surface catalytic proton reduction reaction by introducing dual pyridinic nitrogen (N) atoms into the polymer skeleton. Theoretical calculations suggest that the increased localized electron density at the N site can enhance the electron density of states around the Fermi level, which would lower the energy barrier to H adsorption and activation during the photocatalytic hydrogen evolution (PHE) process. Moreover, the increased localized electron density also contributes to accelerating the photo-induced charge separation and transportation. Resultingly, an apparent quantum yield of 35.5% of the newly developed conjugated polymer (termed as COP-PB-N2) can be achieved at a polymer concentration of 1 mg mL−1, which is the highest value achieved for conjugated microporous polymers to date. This work provides a new strategy through modulating the electronic properties of catalytically active sites for boosting the intrinsic photocatalytic performance.
Introduction
Photocatalytic hydrogen evolution (PHE) from water has been widely recognized as an ideal strategy to alleviate the energy crisis and environmental pollution.1–3 Recently, conjugated polymers with remarkable advantages, such as versatile chemical composites,4–6 tunable molecular structures,7,8 and high thermal/chemical stability,9,10 have attracted extensive interest for synthesizing efficient hydrogen evolution photocatalysts. The pioneer conjugated polymer photocatalyst poly(p-phenylene) exhibited a low H2 evolution rate (λ ≥ 290 nm) in 1985,11 but it inspired scientists to look further into developing novel polymer-based photocatalysts. Enormous efforts have been devoted to improving PHE performance mainly via three crucial approaches, i.e., by enhancing (i) the visible light absorption, (ii) charge carrier separation, and (iii) surface catalytic reaction.12 Most recently, numerous reported studies mainly focused on the first two approaches. The visible light absorption can be increased via the copolymerization method with large planar conjugated monomers and different substituent-modified cross-linkers.13–16 Meanwhile, the photo-induced charge recombination can be suppressed by fabricating donor–acceptor structures,17–20 building organic heterojunctions,21 or reducing conjugated polymers into polymer dots (Pdots).22–24 However, studies on boosting the intrinsic surface catalytic activity have rarely been performed, and the poor surface catalytic activity is a critical obstacle to further improving the PHE efficiency of conjugated polymers. Loading cocatalysts on the surface of polymer photocatalysts is considered to be the most common way to reduce the energy barrier for boosting the sluggish surface proton reduction reaction,25–30 but how to improve their intrinsic surface catalytic activity is still a great challenge.Alternatively, introducing heteroatoms into the skeleton of conjugated polymers can adjust the electronic configuration of the relevant reactive sites to improve catalytic performance.31–35 For instance, doping N into a metal sulfide is carried out to modulate the electron density of the S site, via the interaction between the 2p orbital of the N atom and 2d orbital of transition metals, to facilitate its surface catalytic proton reduction for electrocatalytic hydrogen evolution.36 After introducing pyridinic N into poly(phenylene) derivatives, the apparent quantum yield (AQY) can be dramatically enhanced from 0.4% to 6.7% at 420 nm.33 However, the surface catalytic activities are still not satisfactory and need to be further improved.
Herein, we newly developed a localized electron density modulation strategy to enhance the intrinsic surface catalytic proton reduction reaction by introducing dual pyridinic N atoms into a conjugated polymer nanosheet (termed as COP-PB-N2). As a control, we also synthesized a fully conjugated polymer nanosheet without N doping (termed as COP-PB) and theoretically developed another polymer doped with only one pyridinic N in the skeleton (denoted as COP-PB-N1). The sp2 occupied orbital of the pyridinic N with lone-pair electrons can interact with the empty 1s orbital of H which makes pyridinic N serve as a reactive site for proton reduction. More importantly, the introduced dual N atoms in COP-PB-N2 can increase the electron density around the N site, which will further strengthen the electron density of states at the Fermi level and thus lower the energy barrier to the formation of intermediate products during the PHE process. Consequently, the newly developed COP-PB-N2 shows a visible-light-driven hydrogen evolution rate of 51.5 μmol h−1 which is 5-fold higher than that of undoped COP-PB, and achieves an apparent quantum yield (AQY) of 35.5% at 400 nm exceeding that of the state-of-the-art conjugated microporous polymer photocatalysts and even being comparable to that of the benchmark inorganic photocatalysts.
Results and discussion
The theoretical models of the three fully conjugated polymers in this work, i.e., COP-PB, COP-PB-N1 and COP-PB-N2, are shown in Fig. S1.† The partial charge density image (Fig. 1a) illustrates that the increased localized electron density is mainly concentrated around pyridinic N. Moreover, the Bader effective charge (Fig. S2 and S3†) clearly reveals that the introduction of dual pyridinic N can induce the redistribution of surface charge and the N atom of COP-PB-N2 at site 1 is more negative than that of COP-PB-N1 at the same position, which would enable the pyridinic N to more easily anchor hydrogen atoms to form intermediate configurations. In order to clearly show the influence of localized electron density change on hydrogen adsorption which is a crucial step to determine the catalytic reaction rate,37,38 we calculate the charge density difference of the optimized adsorption configurations based on density functional theory (DFT), presented in Fig. 1b and S4.† The interaction between adsorbed H and polymers can be strengthened when introducing pyridinic N into conjugated polymers. The COP-PB-N2 has the maximum number of electrons transferred from polymers to adsorbed H, which is beneficial to the H activation.39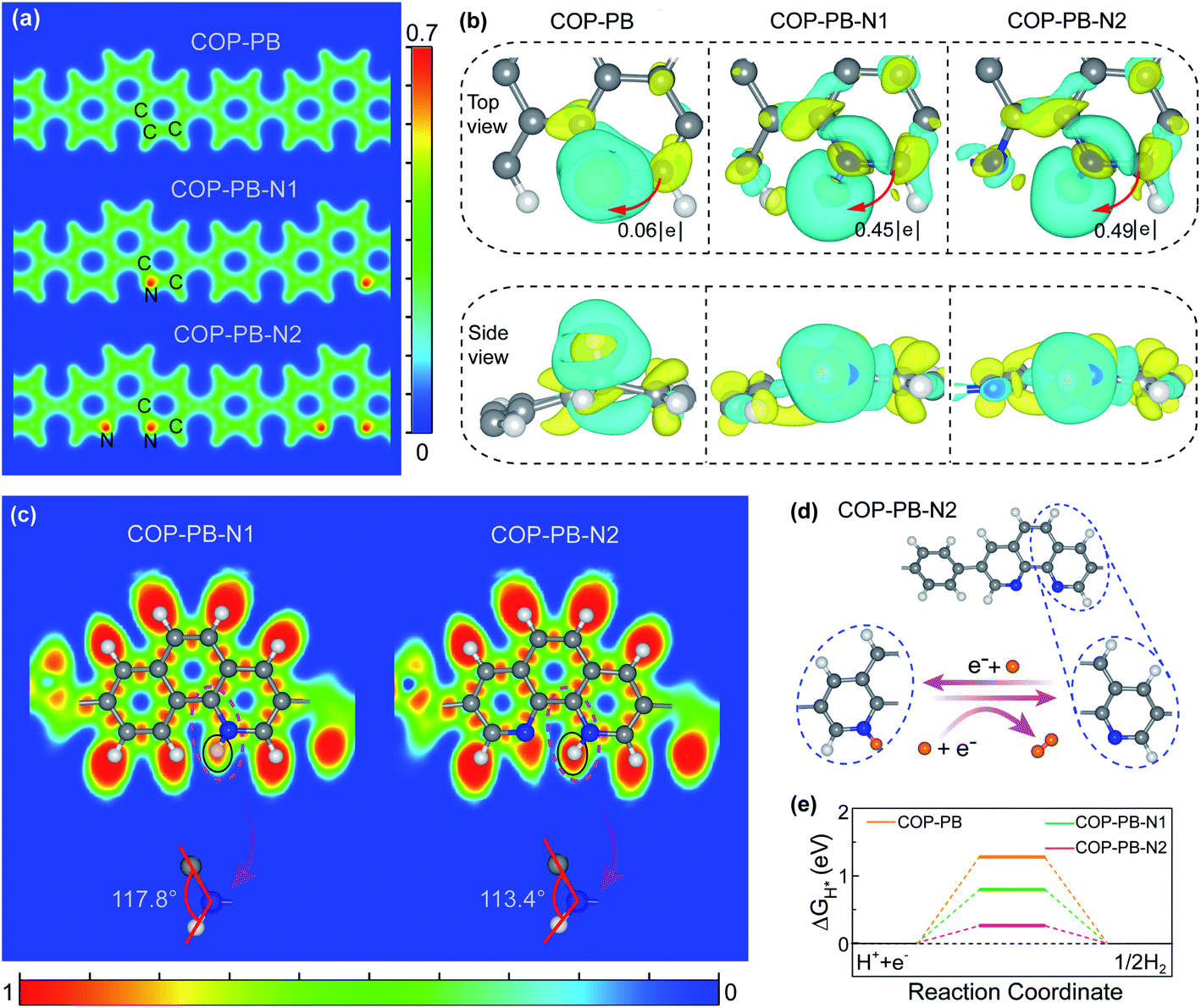 | ||
| Fig. 1 (a) Partial charge density of the polymers. (b) The charge density differences of H chemisorbed on different polymers; cyan and yellow represent charge depletion and accumulation, respectively. The iso-surface value is 0.002 e Å−3 and the Fermi level is set to be 0 eV. (c) The electron localization function (ELF); the bond angles of carbon–nitrogen–H. (d) The H2 evolution process (C grey, N blue, H from COP-PB-N2: white, H from water: orange). (e) The calculated hydrogen evolution reaction energy profile. | ||
As shown in Fig. S5,† the electron density of states of COP-PB-N2 at the Fermi level is the largest among the three samples due to the increased localized electron density on pyridinic N atoms, which contributes to the lower energy barrier to intermediate product formation during the PHE process.40,41 Beyond that, the electron localization function (ELF) also confirms that the interaction of chemisorbed N–H on COP-PB-N2 is stronger than that on COP-PB-N1, which might lead to a more efficient proton reduction (Fig. 1c). Furthermore, due to the absence of one atom in the steric structure, the steric hindrance to chemisorbed N–H in COP-PB-N2 is a little smaller than that in COP-PB-N1 (Fig. 1c), which is favourable for H adsorption.
As mentioned above and emphasized in the reported N-doped polymers,5,17,33 our theoretical screening finds that the pyridinic N in the COP-PB-N2 skeleton is confirmed as the surface catalytic site for PHE (vide infra). The catalytic process of H-adsorption–reduction to H2 is shown in Fig. 1d, i.e., the hydrogen atom from water is first bonded with pyridinic N from COP-PB-N2 to become activated H, which will react with another H from water to produce hydrogen. Furthermore, the calculated Gibbs free energy (ΔGH*) of COP-PB, COP-PB-N1 and COP-PB-N2, which is a good descriptor for evaluating the catalytic activity,42 exhibits different values ranging from 0.26 eV to 1.28 eV (Fig. 1e). The ΔGH* of COP-PB is relatively far from the optimal value of ΔGH* = 0 eV, indicating an inherently low catalytic activity for the hydrogen evolution reaction.43,44 Meanwhile, the ΔGH* of the N site from COP-PB-N2 is only 0.26 eV which is much smaller than the N site (0.80 eV) at the same position in COP-PB-N1 (Fig. 1e). Hence, COP-PB-N2 demonstrates the highest intrinsic catalytic activity for PHE compared with COP-PB and COP-PB-N1.
The as-prepared polymers of COP-PB and COP-PB-N2 were synthesized via a Suzuki cross-coupling reaction with corresponding monomers 1,4-phenylenebisboronic acid, 2,7-dibromophenanthrene and 3,8-dibromophenanthroline (Fig. 2a). The characterization results of FT-IR spectra and 13C solid-state NMR spectra confirm the successful synthesis of COP-PB and COP-PB-N2 (Fig. S10 and S11†).45 Additionally, the powder X-ray diffraction (PXRD) (Fig. S12†) reveals the typical quasi-crystalline properties of both polymers.33 The transmission electron microscope images (Fig. 2b and c) of the two as-synthesized polymers exhibit a nanosheet texture along with atomic force microscopy. The thickness of COP-PB-N2 and COP-PB nanosheets is determined to be 8 and 4 nm, respectively (Fig. 2d and e). The thin sheet-like structure can facilitate charge transfer to the conjugated polymer surface rapidly.46,47
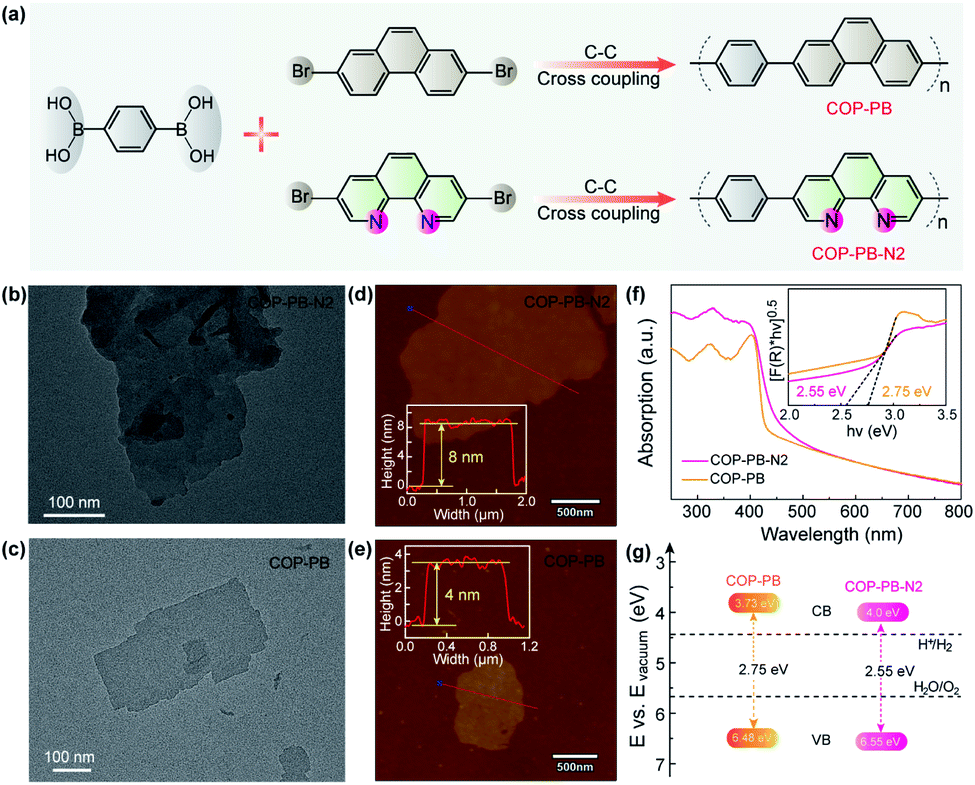 | ||
| Fig. 2 (a) Schematic illustration for the synthesis of COP-PB and COP-PB-N2. (b and c) Transmission electron microscopy (TEM) images of COP-PB-N2 and COP-PB. (d and e) Atomic force microscope (AFM) images of COP-PB-N2 and COP-PB (the insets show the height profiles). (f) UV-vis diffuse reflectance spectra (the inset shows the Tauc plots of COP-PB-N2 and COP-PB). (g) The diagram of electronic band structures for COP-PB and COP-PB-N2. | ||
As shown in Fig. 2f, a broad visible light absorption of COP-PB ranging from 400 to 600 nm was measured by UV-vis reflectance spectra in the solid state. The absorption of COP-PB-N2 is enhanced owing to the extended π-conjugation caused by the increased electron delocalization. Correspondingly, the bandgaps are determined to be 2.75 and 2.55 eV for COP-PB and COP-PB-N2 with the Kubelka–Munk functions from the absorption spectra (Fig. 2g). Meanwhile, ultraviolet photoelectron spectroscopy (UPS) determined the valence band (VB) to be 1.70 eV and 1.85 eV for COP-PB and COP-PB-N2, respectively, below the Fermi level (EF) (Fig. S13†). The work functions (Φ) of COP-PB and COP-PB-N2 are calculated to be 4.78 and 4.70 eV based on the secondary electron cut-off (Fig. S14†). Therefore, the VB of COP-PB and COP-PB-N2 turn out to be 6.48 and 6.55 eV correlated with the vacuum level. Consequently, the bandgaps along with the corresponding conduction band (CB) positions could endow the conjugated polymers with suitable thermodynamic driving force for the proton reduction reaction (Fig. 2g).
The PHE performance of the as-synthesized polymers is studied in a mixture of water, methanol and triethylamine (TEA), where the methanol accelerates the mixing of water and TEA.48 The COP-PB shows a moderate PHE of 11 μmol h−1 under visible light (λ ≥ 400 nm). Encouragingly, after introducing dual pyridinic N into the skeleton of COP-PB, the PHE performance of COP-PB-N2 can be elevated to 51.5 μmol h−1 (Fig. 3a). Significantly, the apparent quantum yield (AQY) of COP-PB-N2 reaches 22.1% at 400 nm and can be further elevated to 35.5% when the polymer concentration is increased from 0.25 mg mL−1 to 1 mg mL−1, which remarkably exceeds the state-of-the-art conjugated microporous polymers (Fig. 3b and g) and is even comparable to the benchmark inorganic photocatalysts.49,50 Although the photocatalytic H2 evolution rate is high in the field of conjugated microporous polymers, it is not enough to meet the practical applications, which needs to be further improved.
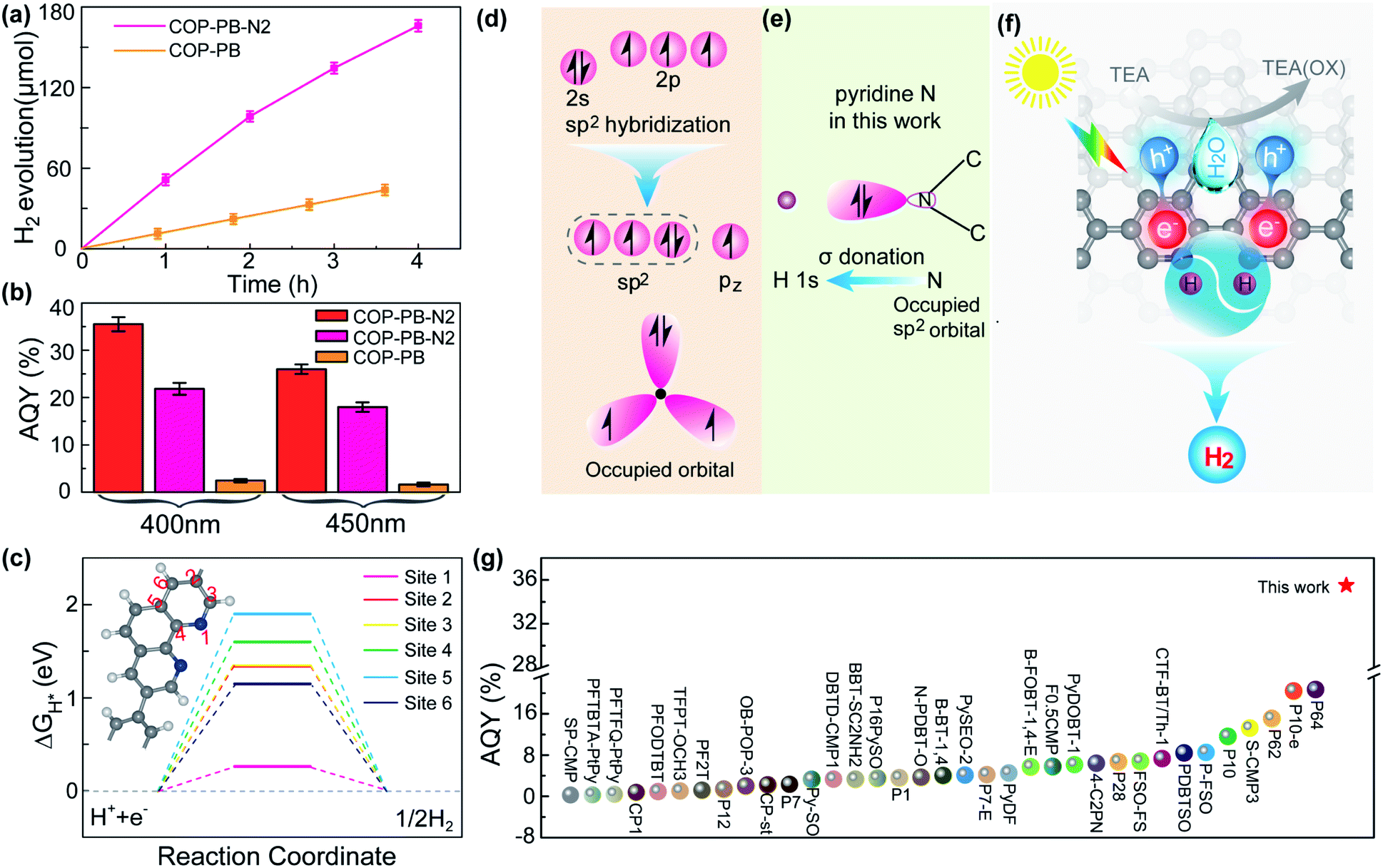 | ||
| Fig. 3 (a) Visible-light-driven hydrogen production of COP-PB-N2 and COP-PB (λ ≥ 400 nm). (b) Apparent quantum yield (AQY) of COP-PB-N2 and COP-PB; red represents a polymer concentration of 1 mg mL−1, pink and orange represent a polymer concentration of 0.25 mg mL−1. (c) The calculated hydrogen evolution reaction energy profile of different sites on COP-PB-N2. The inset is the structure of COP-PB-N2. (d) The electronic configuration of a pyridinic N atom and a N atom with sp2 hybridization. (e) Illustration of H bonding to a pyridinic N atom on COP-PB-N2. (f) The illustration of photocatalytic hydrogen production with COP-PB-N2 under visible light irradiation. (g) The AQY performance comparison between COP-PB-N2 and other reported conjugated polymer photocatalysts; the star represents COP-PB-N2 with a polymer concentration of 1 mg mL−1 at 400 nm (most results are at 400 nm or 420 nm and the detailed irradiation conditions can be found in Table S2†). | ||
The active site screening in COP-PB and COP-PB-N2 confirms the pyridinic N to be the reactive site (Fig. 3c and S15†). The optimal ΔGH* of the carbon site is much larger than that of the N site, demonstrating the pyridinic N to be the reactive site, which is ascribed to the suitable electron configuration of pyridinic N. As illustrated in Fig. 3d and e, the electron structure of the pyridinic N atom is 2s22p3 and the sp2 hybridization of these orbitals will generate three sp2 hybrid orbitals, two of which are half filled and the third one is occupied with a pair of electrons. Both the pz orbital of the pyridinic N atom and the other five pz orbitals of C atoms will result in a large closed π bond. The sp2 hybrid orbital with a pair of electrons will be conducive to bonding with the H 1s orbital and will thus boost the H activation. Therefore, the photocatalytic hydrogen production process of COP-PB-N2 can be illustrated as in Fig. 3f. The photo-excited electrons and holes inside conjugated polymers can rapidly transfer to the surface and subsequently react with adsorbed H and TEA, respectively.
The control experiments are carried out (Fig. S16†) in order to avoid the influence of the above mixed solvent. The pristine H2O/methanol/TEA solvent cannot generate hydrogen with or without visible light irradiation and no hydrogen is measured for the two conjugated polymers in the H2O/methanol/TEA solvent in the dark. Meanwhile, hydrogen cannot be produced with the mixture of water/methanol or pure water, but can be evolved when using a mixture of TEA/water. These results demonstrate that methanol acts as a co-solvent and TEA is a hole scavenger rather than the polymer itself. The FT-IR spectrum indicates that the skeleton of COP-PB-N2 does not fall apart after photocatalysis (Fig. S17†). In addition, we have also synthesized three different batches of samples under the same experimental parameters to ensure the repeatability of the two polymer photocatalysts. Each of the polymers exhibits an almost identical H2 production rate under the same photocatalytic conditions, manifesting the outstanding reproducibility of the as-prepared polymers (Fig. S18†). It is imperative to point out that the COP-PB has more residual palladium than COP-PB-N2 (Table S3†); however, it has much lower photocatalytic performance, which suggests that the residual Pd does not play a crucial role in improving photocatalytic H2 evolution. Meanwhile, we have also carried out photocatalytic cyclic testing of COP-PB-N2, which shows that the photocatalytic H2 evolution rate has been significantly decreased after one cyclic testing (Fig. S19†). To find the reason why the photocatalytic performance is dramatically decreased, we further conducted X-ray photoelectron spectroscopy (XPS) measurements (Fig. S20†). The slight shift of nitrogen peaks indicates that the state of some pyridinic nitrogen atoms may have been changed after photocatalytic H2 evolution. This result confirms that the pyridinic nitrogen atoms are the main catalytic sites. Additionally, the COP-PB-N2 nanosheets tend to aggregate after the photocatalytic cyclic test (Fig. S21†), which might be another reason for the photocatalytic performance decline. Furthermore, previous research has also confirmed that the residual palladium can play a partial catalytic role, but is not the major factor for maximizing the photocatalytic hydrogen evolution rate.5,51–54
Additionally, the photo-excited charge separation and transportation are also fully investigated. The behaviour of photo-induced electrons and holes is detected via electron paramagnetic resonance (EPR) assisted by 2,2,6,6-tetramethyl-L-piperidine-N-oxyl (TEMPO).55 The obvious triplet peak from Fig. 4a in the dark is the characteristic signal of TEMPO, and then the TEMPO will be reacted with photo-induced electrons (e−) or holes (h+) to produce TEMPOH or TEMPO+ in different solutions (Fig. S22†). Subsequently, the decreased intensity of the triplet peaks at intervals of time confirms that the polymers can really produce electrons (e−) and holes (h+) under visible light irradiation.56 Moreover, the time-resolved PL decay plots (Fig. 4b) of the above polymers have been fitted with a multi-exponential kinetic function and the average lifetime has been calculated in the ESI (Table S1†).57,58 A slightly longer lifetime of COP-PB-N2 than COP-PB might reveal the improved separation of photo-excited electron–hole pairs.43,59,60 The transient photocurrent intensity of COP-PB-N2 under on–off visible light irradiation is slightly stronger than that of COP-PB, which confirms the presence of a few more photo-excited charge carriers due to the relatively enhanced visible light absorption.61,62 Additionally, we conducted a contact angle experiment to explore the wettability as shown in Fig. S23.† After introducing pyridinic N into the polymers, the surface affinity of COP-PB-N2 with H2O or H2O/MeOH/TEA is enhanced contributing to boosting the interfacial charge transfer.63 Furthermore, on the basis of electrochemical impedance spectroscopy (Fig. 4d), the smaller Nyquist semicircle curve of COP-PB-N2 than COP-PB also demonstrates the slightly improved interfacial charge transfer.64,65 These results reveal that the separation of photo-induced electron–hole pairs can also be enhanced after introducing dual pyridinic N apart from improving interfacial charge transfer, and both of them synergistically improve PHE performance.
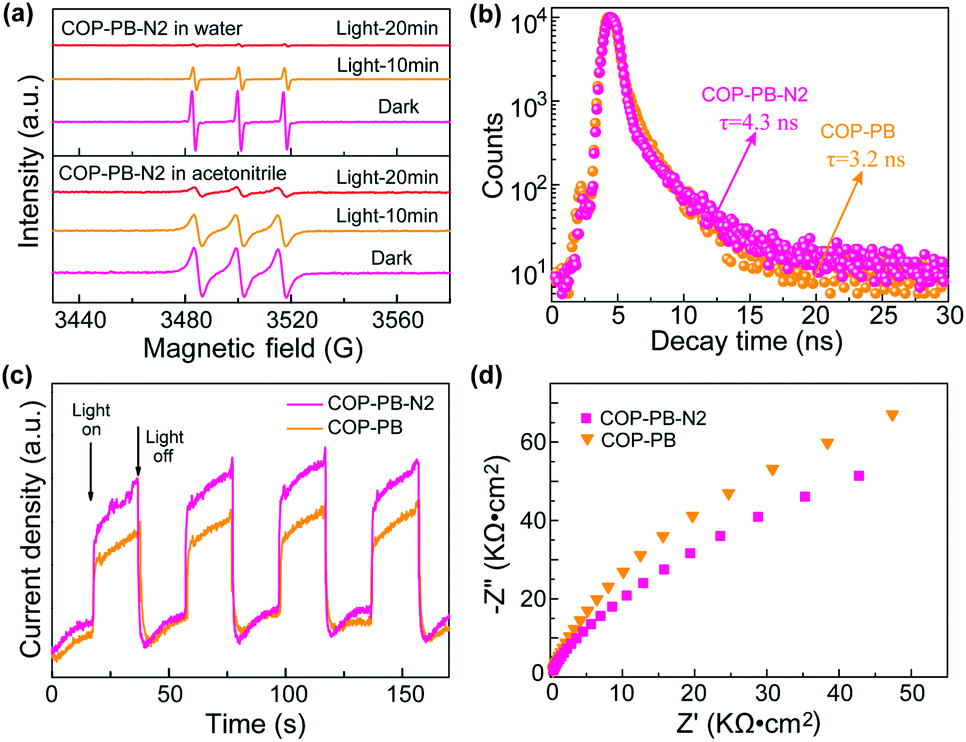 | ||
| Fig. 4 (a) EPR curves for monitoring photo-excited electrons and holes. (b) Time-resolved transient PL decay spectra. (c) On–off photocurrent response under visible light or in the dark. (d) Electrochemical impedance spectroscopy Nyquist plots. | ||
Conclusions
In summary, we have successfully developed a highly efficient conjugated polymer towards PHE via a localized electron density modulation strategy using dual pyridinic N atoms. The newly developed COP-PB-N2 has achieved an excellent photocatalytic H2 production rate of 51.5 μmol h−1. An AQY of 35.5% can be achieved at a polymer concentration of 1 mg mL−1, which exceeds that of the state-of-the-art conjugated microporous polymer photocatalysts. Our theoretical calculations and experiments suggest that the sp2 hybridized orbital of pyridinic N with lone-pair electrons is favourable for facilitating H adsorption and activation. The improved localized electron density of COP-PB-N2 plays a decisive role in increasing the electron density of states at the Fermi level, which significantly lowers the energy barrier to intermediate product formation during the PHE process. Meanwhile, the optimal Gibbs free energy (ΔGH*) of COP-PB-N2 demonstrates its superior intrinsic surface catalytic activity. Moreover, the increased localized electron density is also conducive to enhancing the charge separation and transfer. Therefore, the strategy of localized electron density modulation offers new avenues to develop more highly efficient conjugated polymers for photocatalytic or even other catalytic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Beijing Natural Science Foundation (JQ19007), NSF of China (21676020 and 21922802), the Young Elite Scientists Sponsorship Program by CAST (2017QNRC001), the Talent Cultivation and Open Project (OIC-201801007) of State Key Laboratory of Organic-Inorganic Composites, “Double-First-Class” Construction Projects (XK180301 and XK1804-02), and the Distinguished Scientist Program at BUCT (buctylkxj02).Notes and references
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. W. Tang, Nat. Energy, 2019, 4, 746–760 Search PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050–17067 Search PubMed.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 Search PubMed.
- W. Zhong, J. Liang, S. Hu, X. F. Jiang, L. Ying, F. Huang, W. Yang and Y. Cao, Macromolecules, 2016, 49, 5806–5816 Search PubMed.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 Search PubMed.
- L. Li, R. G. Hadt, S. Yao, Q. Wu, B. Pandit, L. X. Chen and L. Yu, Chem. Mater., 2016, 28, 5394–5399 Search PubMed.
- X. H. Zhang, X. P. Wang, J. Xiao, X. Ding, Y.-G. Xiang and H. Chen, J. Catal., 2017, 350, 64–71 Search PubMed.
- C. Cheng, X. Wang, Y. Lin, L. He, J. X. Jiang, Y. Xu and F. Wang, Polym. Chem., 2018, 9, 4468–4475 Search PubMed.
- C. Yang, B. C. Ma, K. Landfester, K. A. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 Search PubMed.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 Search PubMed.
- A. K. Shozo Yanagida, K. Mizumoto and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 Search PubMed.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 Search PubMed.
- C. Dai, S. Xu, W. Liu, X. Gong, M. Panahandeh-Fard, Z. Liu, D. Zhang, C. Xue, K. P. Loh and B. Liu, Small, 2018, 14, 1801839 Search PubMed.
- Y. Liu and Z. Xiang, ACS Appl. Mater. Interfaces, 2019, 11, 41313–41320 Search PubMed.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 2428–2435 Search PubMed.
- Z. Wang, X. Yang, T. Yang, Y. Chen, J. H. Zeng, C. Yan, F. Huang and J. X. Jiang, ACS Catal., 2018, 8, 8590–8596 Search PubMed.
- Y. Xiang, X. Wang, L. Rao, P. Wang, X. Zhang, S. Wang, H. Chen and Y. Zhu, ACS Energy Lett., 2018, 3, 2544–2549 Search PubMed.
- Y. Liu, Z. Liao, X. Ma and Z. Xiang, ACS Appl. Mater. Interfaces, 2018, 10, 30698–30705 Search PubMed.
- Y. Xu, C. Zhang, F. Wang and J. X. Jiang, Appl. Catal., B, 2018, 228, 1–9 Search PubMed.
- J. Yu, X. Sun, X. Xu, C. Zhang and X. He, Appl. Catal., B, 2019, 257, 117935 Search PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 6597, 1–7 Search PubMed.
- C. M. Aitchison, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2019, 7, 2490–2496 Search PubMed.
- L. Wang, R. Fernandez-Teran, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 Search PubMed.
- P. J. Tseng, C. L. Chang, C. H. Liao, M. L. Tsai and H. H. Chou, ACS Catal., 2018, 8, 7766–7772 Search PubMed.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165–12174 Search PubMed.
- Z. L. Enquan Jin, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 Search PubMed.
- T. Banerjee, F. Haase, G. Savasci, K. Gottschling, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2017, 139, 16228–16234 Search PubMed.
- B. P. Biswal, H. A. Vignolo-Gonzalez, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 Search PubMed.
- W. Liu, W. Cheng, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312–9317 Search PubMed.
- G. Zhao, Y. Sun, W. Zhou, X. Wang, K. Chang, G. Liu, H. Liu, T. Kako and J. Ye, Adv. Mater., 2017, 29, 1703258 Search PubMed.
- Y. Wan, L. Wang, H. Xu, X. Wu and J. Yang, J. Am. Chem. Soc., 2020, 142, 4508–4516 Search PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783–3791 Search PubMed.
- R. S. Sprick, L. Wilbraham, Y. Bai, P. Guiglion, A. Monti, R. Clowes, A. I. Cooper and M. A. Zwijnenburg, Chem. Mater., 2018, 30, 5733–5742 Search PubMed.
- P. Blonski, J. Tucek, Z. Sofer, M. Pumera, M. Otyepka and R. Zboril, J. Am. Chem. Soc., 2017, 139, 3171–3180 Search PubMed.
- I. Choudhuri, G. Bhattacharyya, S. Kumar and B. Pathak, J. Mater. Chem. C, 2016, 4, 11530–11539 Search PubMed.
- Y. Wu, X. Liu, X. Xiao and G. M. Wang, Nat. Commun., 2018, 9, 1–9 Search PubMed.
- C. Ling, L. Shi, Y. Ouyang, Q. Chen and J. Wang, Adv. Sci., 2016, 3, 1600180 Search PubMed.
- Y. Ouyang, Q. Li, L. Shi, C. Ling and J. Wang, J. Mater. Chem. A, 2018, 6, 2289–2294 Search PubMed.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 Search PubMed.
- W. Che, W. Cheng, T. Yao, F. Hu, Z. Pan, Z. Sun and S. Wei, J. Am. Chem. Soc., 2017, 139, 3021–3026 Search PubMed.
- C. F. Fu, X. Wu and J. Yang, Adv. Mater., 2018, 30, 1802106 Search PubMed.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 Search PubMed.
- J. Ran, G. Gao, F. T. Li, T. Y. Ma, A. Du and S. Z. Qiao, Nat. Commun., 2017, 8, 13907–13917 Search PubMed.
- P. Chen, T. Zhou, M. Zhang, Y. Tong, C. Zhong, N. Zhang, L. Zhang, C. Wu and Y. Xie, Adv. Mater., 2017, 29, 1701584 Search PubMed.
- Z. A. Lan, Y. Fang, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 470–474 Search PubMed.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 Search PubMed.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454–3458 Search PubMed.
- R. S. Sprick, C. M. Aitchison, M. A. Zwijnenburg and A. I. Cooper, J. Mater. Chem. A, 2018, 6, 11994–12003 Search PubMed.
- Y. Zhu, J. Chen, L. Shao, X. Xia, Y. Liu and L. Wang, Appl. Catal., B, 2020, 268, 118744 Search PubMed.
- B. Han, S. Liu, N. Zhang, Y. J. Xu and Z. R. Tang, Appl. Catal., B, 2017, 202, 298–304 Search PubMed.
- R. S. Sprick, J. X. Jiang, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 Search PubMed.
- R. S. Sprick, B. Bonillo, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1824–1828 Search PubMed.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017, 7, 1700479 Search PubMed.
- M. Sachs, R. S. Sprick, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968–4979 Search PubMed.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 Search PubMed.
- C. Zhao, Z. Chen, J. Xu, Q. Liu, H. Xu, H. Tang, G. Li, Y. Jiang, F. Qu, Z. Lin and X. Yang, Appl. Catal., B, 2019, 256, 117867 Search PubMed.
- P. Kumar, E. Vahidzadeh, G. M. Bernard, V. K. Michaelis and K. Shankar, J. Am. Chem. Soc., 2019, 141, 5415–5436 Search PubMed.
- H. Huang, H. Yuan, K. P. F. Janssen, J. A. Steele and M. B. J. Roeffaers, ACS Energy Lett., 2018, 3, 755–759 Search PubMed.
- S. Bhattacharyya, F. Ehrat, P. Urban, A. S. Urban and J. K. Stolarczyk, Nat. Commun., 2017, 8, 1401–1410 Search PubMed.
- S. Wei, F. Zhang, W. Zhang, P. Qiang, K. Yu, X. Fu, D. Wu, S. Bi and F. Zhang, J. Am. Chem. Soc., 2019, 141, 14272–14279 Search PubMed.
- X. Gao, C. Shu, C. Zhang, W. Ma, S.-B. Ren, F. Wang, Y. Chen, J. H. Zeng and J. X. Jiang, J. Mater. Chem. A, 2020, 8, 2404–2411 Search PubMed.
- Z. Zhou, P. Peng and Z. Xiang, Sci. Bull., 2018, 63, 369–375 Search PubMed.
- J. Cheng, G. Liao, L. Chen and S. Y. Liu, J. Mater. Chem. A, 2020, 8, 5890–5899 Search PubMed.
- Q. Ruan, J. Xie, Y. Wang, C. J. Carmalt and J. Tang, Angew. Chem., Int. Ed., 2017, 56, 8221–8225 Search PubMed.
- M. Shi, J. Li, X. Jin, E. A. Pidko, R. Li and C. Li, Angew. Chem., Int. Ed., 2019, 59, 6590–6595 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02193a |
| This journal is © The Royal Society of Chemistry 2021 |