
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinchpins empower promiscuous electrophiles to enable site-selective modification of histidine and aspartic acid in proteins†
Dattatraya Gautam
Rawale
 a,
Kalyani
Thakur
a,
Pranav
Sreekumar
a,
Sajeev
T. K.
b,
Ramesh
A.
a,
Srinivasa Rao
Adusumalli
a,
Ram Kumar
Mishra
b and
Vishal
Rai
*a
a,
Kalyani
Thakur
a,
Pranav
Sreekumar
a,
Sajeev
T. K.
b,
Ramesh
A.
a,
Srinivasa Rao
Adusumalli
a,
Ram Kumar
Mishra
b and
Vishal
Rai
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal Bypass Road, Bhauri, Bhopal 462 066, India. E-mail: vrai@iiserb.ac.in
bDepartment of Biological Sciences, Indian Institute of Science Education and Research Bhopal, Bhopal Bypass Road, Bhauri, Bhopal 462 066, India
First published on 9th April 2021
Abstract
The conservation of chemoselectivity becomes invalid for multiple electrophilic warheads during protein bioconjugation. Consequently, it leads to unpredictable heterogeneous labeling of proteins. Here, we report that a linchpin can create a unique chemical space to enable site-selectivity for histidine and aspartic acid modifications overcoming the pre-requisite of chemoselectivity.
Introduction
Proteins are natural polypeptides that constitute a key element in the central dogma of molecular biology. Moreover, they are directly involved in diverse chemical processes that are essential for the sustainability of life.1 So, it does not come as a surprise that these biomolecules have been targets for chemical modifications since the emergence of chemical biology.2–5 In this perspective, the tools for attaching probes onto specific proteins provide an opportunity to visualize and track their functions and meet biophysical chemistry and biochemistry demands.6 Besides, the attachment of toxins on protein surfaces has proved promising for protein-based therapeutics.7 Initially, pre-engineered proteins addressed the need for single-site labeling.8,9 However, the growth can be accelerated by several fold if we can achieve the same selectivity with native proteins. The advancement of chemical platforms in recent years offers promise for precision engineering of proteins.10 In this perspective, we and others have developed and established a few chemical technologies that provide single-site labeling of the N-terminus Nα-NH2,11–18 N-Gly,19,20 His,21,22 and Lys,23–26 among others.27–34 However, precise labeling of functional groups with lower reactivity, e.g., His and Asp, presents a monumental challenge.35Here, the knowledge of the principles of organic chemistry unravel a reaction's selectivity and provides the foundation for a chemical platform. As these biomolecules comprise multiple nucleophilic residues, covalent modification would typically require an electrophilic warhead (E1, Fig. 1). As per the general understanding, such a reagent needs to exhibit chemoselectivity for a functional group first. Subsequently, single-site labeling would require such an electrophile to distinguish a unique group from its pool. Such a competition or site-selectivity becomes highly challenging for amino acid residues that display moderate or high-frequency. This perspective ensured that the efforts for single-site modifications of native proteins start by ruling out electrophiles with poor or non-conserved chemoselectivity. However, what if we take such an electrophile with the potential to react with more than one amino acid residue? What if the reactive residues are abundant in the proteome (e.g., His and Asp)?
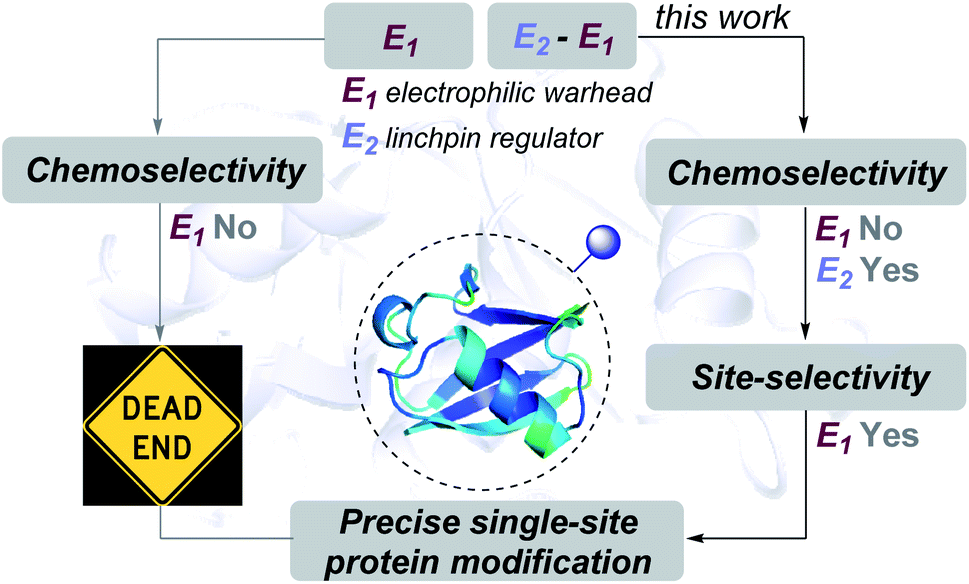 | ||
| Fig. 1 Linchpin (E2) can empower a promiscuous electrophile (E1) to enable site-selectivity and deliver precise modification of proteins. | ||
In this work, we examined whether it is feasible to develop a selectivity regulator for such an electrophile (E1) to get through such a large pool of competitors. Here, we establish that it is possible to enable single-site protein modification even with electrophiles that exhibit non-conserved chemoselectivity. An appropriately designed linchpin regulator (E2, Fig. 1) can limit such a promiscuous electrophile's (E1) localization to reduce the number of competing sites. Besides, chemoselectivity and site-selectivity are deconvoluted; E2 addresses the former in the first step and empowers E1 to enable the latter in the subsequent step. To validate the hypothesis, we used a non-selective alkylating reagent conjugated to 2-hydroxybenzaldehyde with ubiquitin. Further, we demonstrated that the principle extends to a structurally diverse set of proteins, including insulin, α-lactalbumin, and myoglobin. Interestingly, it creates a unique chemical space to label His or Asp, which are otherwise challenging targets for precise labeling. Besides, the proximity regulator delivers ordered single-site immobilization. The methodology is further extended for orthogonal late-stage functionalization and delivers analytically pure tagged proteins.
Results and discussion
Site-selective protein modification
At the onset, we selected aryl sulfonate ester, an alkylating group (E1, Fig. 1), with well-established promiscuity and high reactivity with Glu, Cys, Asp, Tyr, and His in a proteome (Fig. 2a, also see Fig. S88†).35 On the other hand, we derived inspiration from our linchpin-directed modification (LDM®) platform to select the proximity regulator.36,37 We chose 2-hydroxybenzaldehyde (E2, Fig. 1) to enable a rapid, reversible, and chemoselective reaction with Lys residues. Finally, we coupled the two groups through spacers for the multi-step synthesis of electrophilic systems 2b–2f (Fig. 2, and Schemes S2–S6†). In parallel, we screened a range of parameters that kept the intermolecular reaction of aryl sulfonate ester (E1, 2a) with the protein (ubiquitin, 1a) on the fringe. In this pursuit, we established that 25–100 equivalents of 2a do not result in protein labeling at 25–37 °C within 72 hours. In one of the representative examples, the control reaction of 2a (1.25 mM) with ubiquitin (1a, 50 μM) resulted in no noticeable labeling (Table S3, Fig. S1–S6†).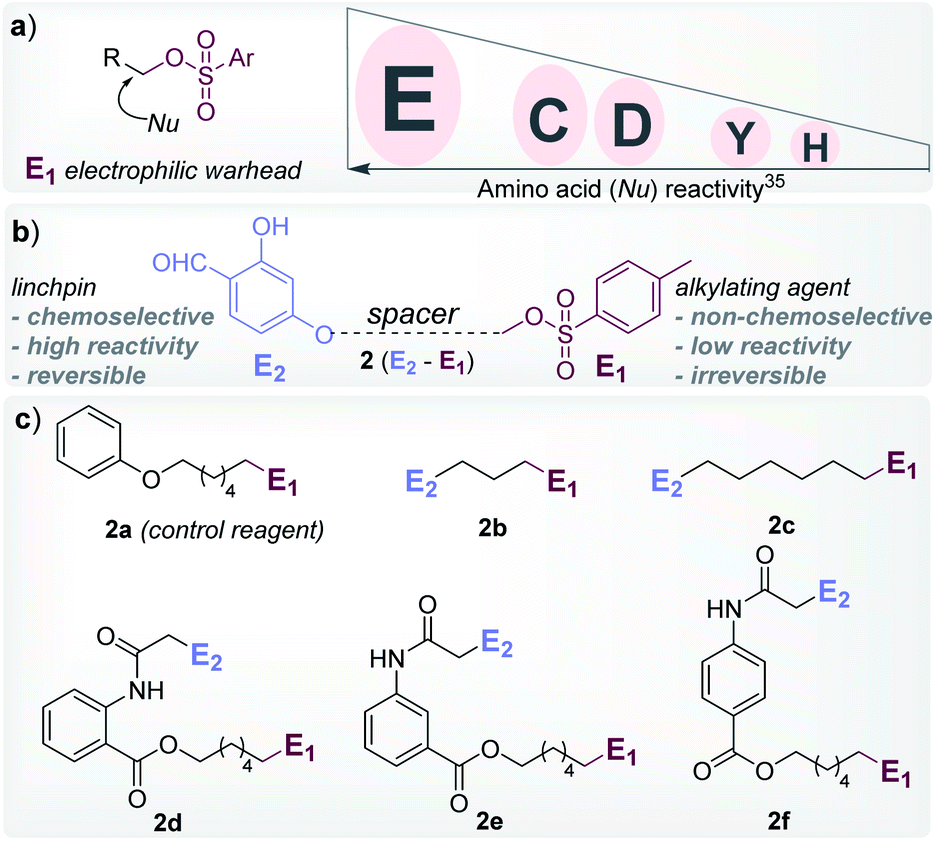 | ||
| Fig. 2 Design elements. (a) Order of proteome reactivity for aryl sulphonate ester: Glu (E) ≫ Cys (C) ≈ Asp (D) > Tyr (Y) > His (H).35 (b) General structure of the chemoselective linchpin-equipped promiscuous electrophile, i.e., aryl sulphonate ester. (c) Control reagent (2a), and other bioconjugation reagents (2b–2f). | ||
To re-validate it, we mixed the aryl sulfonate ester (2a, 1.25 mM) with insulin, lactalbumin, and myoglobin (1b–1d, 50 μM) and observed negligible conversions (Table S4, Fig. S7–S9†). Screening establishes the reaction parameter thresholds below which the background reaction would not occur. With this information in hand, we selected linchpin regulator-empowered electrophiles (E2–E1, 2b–2f) to examine their potential with ubiquitin (1a) as a model protein. Among other nucleophilic residues, it provides six Glu, five Asp, one Tyr, and one His to compete for alkylation. Besides, it provides seven Lys (Nε-NH2) and one N-terminus α-amine (Nα-NH2) to serve as potential linchpins. However, reagents 2d, 2e, and 2f were inefficient within 72 h (8%, 5%, and <3% conversion, respectively, Fig. S18–S20†). The ESI-MS methods facilitated the monitoring of the progress of bioconjugation. These results indicate that the spacer connecting the proximity regulator and the electrophilic warhead has a role in bioconjugation efficiency. The re-designed reagent 2b led to some improvement (15% conversion, Fig. S21†). Finally, reagent 2c delivered 42% mono-labeled ubiquitin (Fig. 3). Interestingly, selectivity remains unperturbed over a range of substrate concentrations and pH of the medium (Tables S5–S7, Fig. S89–91†). The subsequent late-stage installation of benzyloxyamine resulted in >99% conversion to 5a. Tryptic digestion, peptide mapping (IQDK 30–33), and MS/MS confirmed the site of modification (D32, Fig. S10†). Further, circular dichroism spectroscopy confirmed that the structure of ubiquitin remains unperturbed post-labeling (Fig. S34†).
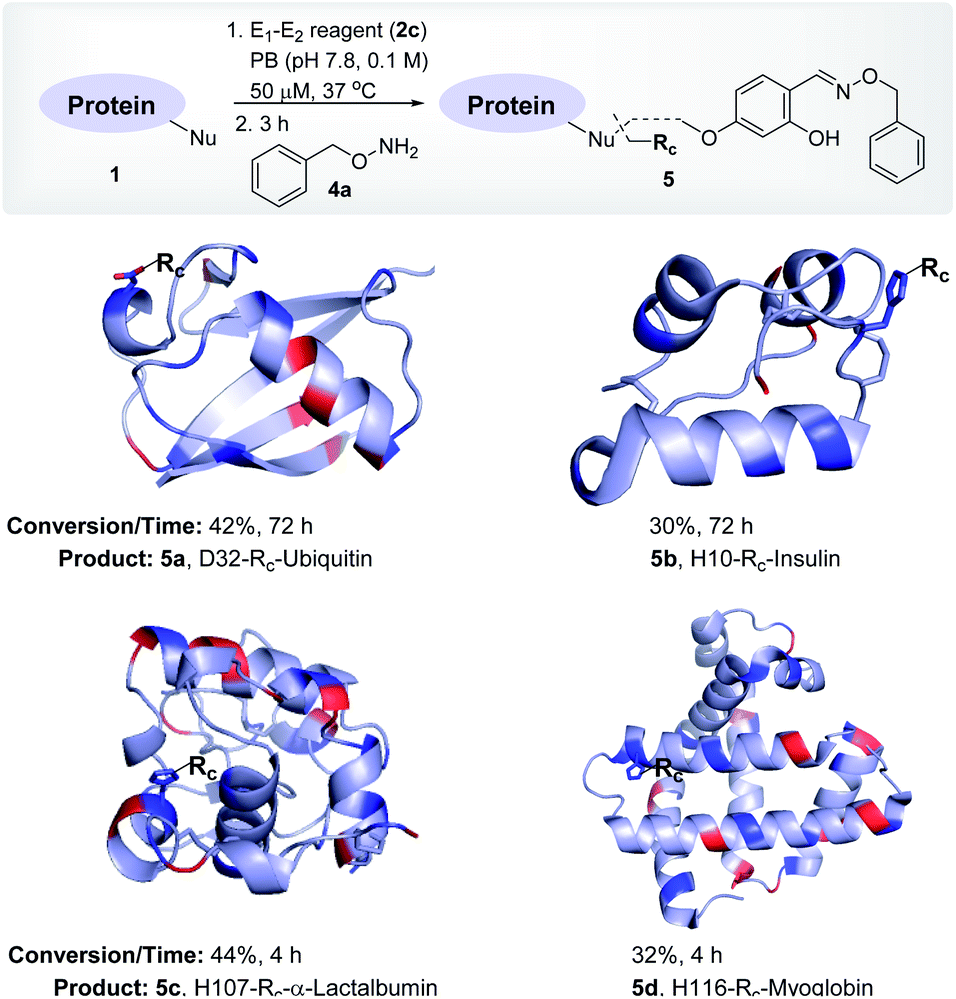 | ||
| Fig. 3 Single-site labeling of proteins. ESI-MS estimates the overall % conversion over two steps (5a–5d). Rc indicates the labels from the oxime derivative of the E1–E2 reagent. The amine residues are highlighted in red and imidazole/carboxylates are highlighted in blue. For data, see Fig. S10–S13.† | ||
To investigate and validate conceptual translation with a structurally diverse protein, we selected insulin. It offers two chains with two Nα-NH2, one Nε-NH2, four Glu, four Tyr, and two His residues. At first, we vortexed insulin (1b) with reagents 2d, 2e, and 2f. The lack of amines to anchor the proximity regulator reflected in poor conversions (0–8%, 72 h, Fig. S22–S24†). Interestingly, reagent 2c resulted in 30% mono-labeled insulin (5b; over two steps, Fig. 3). MS, peptide mapping (FVNQHLCGSHLVEALYLVCGERGFFYTPKT, 1–30), and MS/MS confirmed the modification site to be a His residue (H10, Fig. S11†). It is noteworthy that no Glu or Tyr participated in the alkylation. Amongst the discretely placed primary amines, the N-Phe (Nα-NH2) anchored linchpin is aptly positioned to direct the alkylating electrophile to H10 (Fig. S92†).
After validating that the linchpin can supersede the electrophile's promiscuous chemoselectivity and empower it for site-selective modification, we extended the methodology to α-lactalbumin (1c). It contains twelve Nε-NH2, one Nα-NH2, six Glu, nine Asp, four Tyr, and three His residues. We anticipated that with numerous solvent-accessible amines to serve as linchpins, most of the reagents would facilitate the covalent bioconjugation. As expected, the reaction with reagents 2b, 2d, 2e, and 2f resulted in mono-labeled α-lactalbumin 5c (6–20% conversion over two steps, Fig. S26–S29†). Moreover, reagent 2c delivered 44% conversion within 4 h (Fig. 3). We identified the site-of-conjugation as H107 through a sequence of MS, proteolytic digestion, peptide mapping (AHKAL, 106–110), and MS–MS (Fig. S12†). Finally, we selected myoglobin (1d) that presents nineteen Nε-NH2 and one Nα-NH2 to serve as linchpins. Besides, it possesses thirteen Glu, eight Asp, two Tyr, and eleven His residues to offer a tough selectivity challenge. The treatment of 1d with reagents 2b, 2d, 2e, and 2f led to low conversions (7–13%, respectively, Fig. S30–S33†). Finally, reagent 2c resulted in 32% conversion (over two steps) to mono-labeled myoglobin (5d, Fig. 3). MS, proteolytic digestion of the bioconjugate, peptide mapping (HSKHPGDF, 116–123), and MS–MS confirmed the site of conjugation (H116) (Fig. S13†).
Late-stage modification
Next, we investigated this technology's potential for the late-stage installation of probes (Fig. 4). In this perspective, we selected the ubiquitin bioconjugate (3b) and treated it with hydroxylamine derivatives of the 19F-NMR probe (6a), biotin (6b), and fluorophore (6c) in parallel.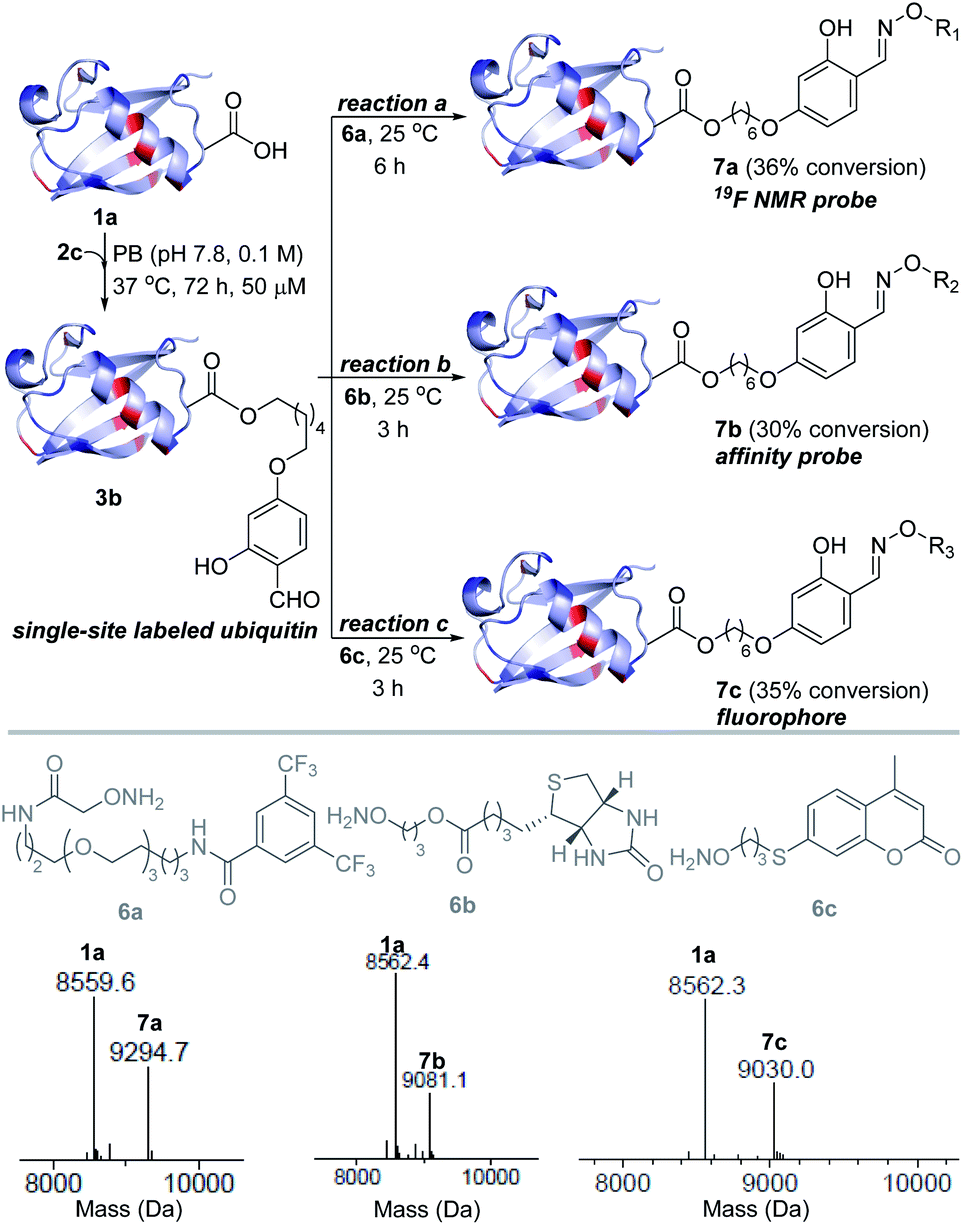 | ||
| Fig. 4 Late-stage modification. Single-site installation of an NMR probe, an affinity probe, and a fluorophore. | ||
We were delighted to note that all three oxime formation reactions exhibit excellent efficiency (>95% conversion, 7a–7c, Fig. 4 and S14–S16†).
Enrichment protocol
Subsequently, we investigated the integration of an enrichment protocol to isolate analytically pure single-site tagged proteins. At first, we incubated D32-labeled ubiquitin (3a/3b, Fig. 3) with acyl hydrazide activated resin (8) in the presence of catalytic p-phenylenediamine (PDA). The hydrazone formation enables bioconjugate capture (3a/3b) through its immobilization within 6 h (9). The process exhibits remarkable capture efficiency (>95%). Besides, the centrifugal spin concentration of the eluted fraction enables excellent recovery (>95%) of the unlabeled ubiquitin (1a). Consequently, the protocol delivers single-site ordered immobilization of the protein. Subsequently, the addition of hydroxylamine (4b) to the immobilized protein enables its release through transoximization (>99% efficiency). The hydrazide activated resin is recovered and recycled up to 5 times. The centrifugal spin concentration delivers the analytically pure single-site tagged ubiquitin bioconjugate (10, Fig. 5 and S17†).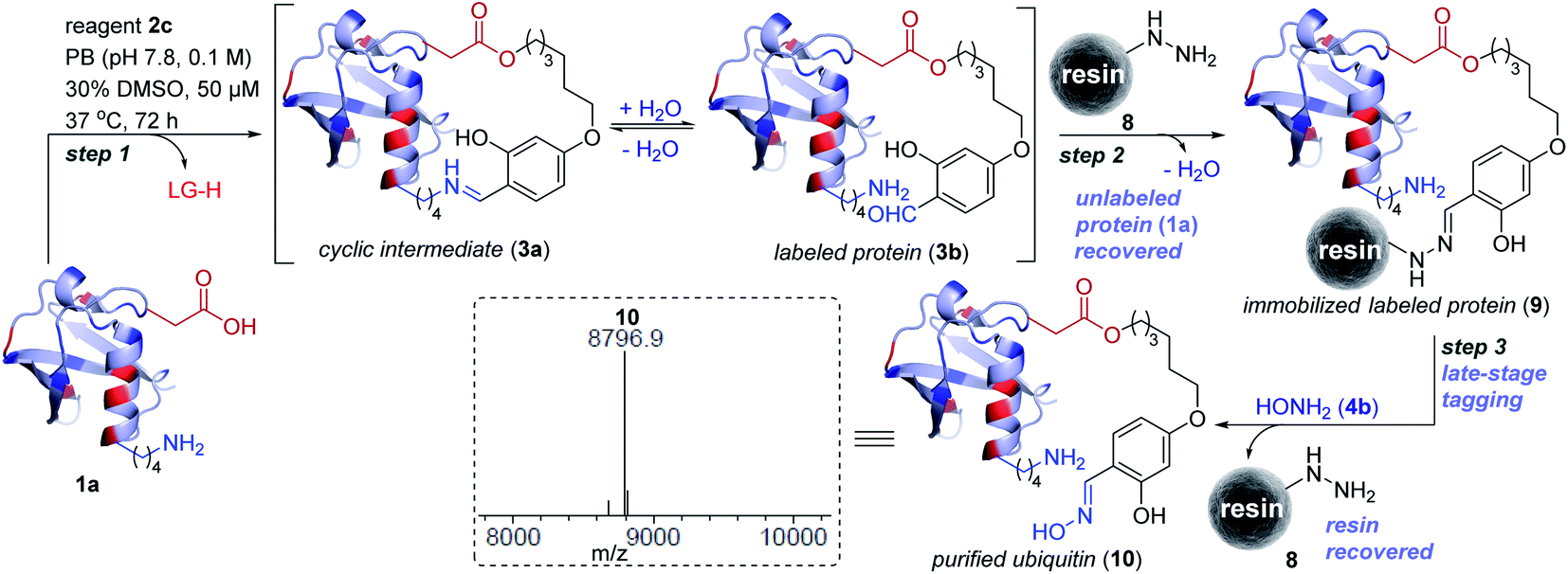 | ||
| Fig. 5 Enrichment protocol. Step 1: linchpin directed alkylation leads to labeled ubiquitin (3a/3b). LG is the leaving group from reagent 2c. Step 2: ordered immobilization of labeled ubiquitin on acyl hydrazide functionalized resin and recovery of the unlabeled protein. Step 3: transoximization delivers single-site tagged analytically pure ubiquitin (10) along with recovery of the resin. ESI-MS spectra of purified labeled ubiquitin (10). | ||
Insulin bioactivity assay
Next, we isolated analytically pure H10-labeled insulin (11, Fig. 6a and S93a†) to test the consequences of bioconjugation. Circular dichroism data confirmed that the labeling and enrichment protocol do not alter insulin's structure (Fig. S93b†). The treatment of cells with purified and native insulin (11 and 1b, respectively) increased the pAkt level as determined by pAkt-S473 detection by western blotting (Fig. 6b). The extent of pAkt-S473 signals in lysates is comparable between native and purified insulin (Fig. 6c). Besides, elevated pAkt levels inside HEK293T cells were detected even after purified insulin treatment and were comparable to those with native insulin (red signals, first vertical panels, Fig. 6d). Together, the enhanced pAkt-S473 levels upon treatment with purified insulin suggest that modification does not affect insulin properties.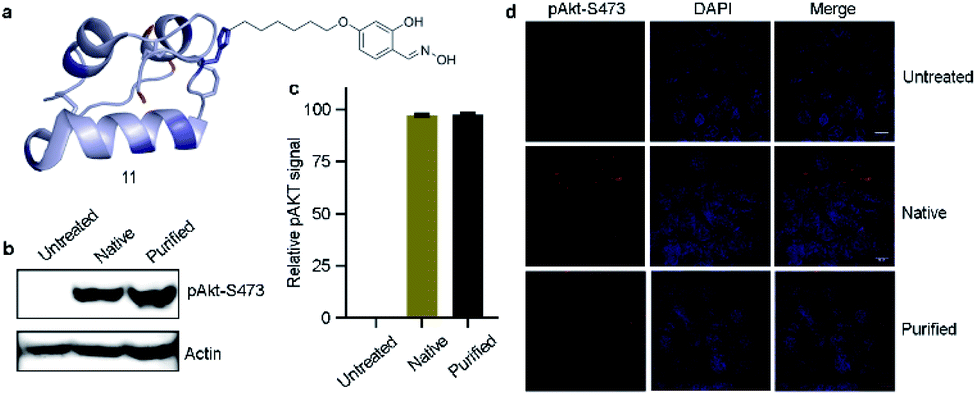 | ||
| Fig. 6 Insulin bioactivity assay. (a) H10-labeled analytically pure insulin. (b) Western blot analysis of pAkt-S473 in HEK293T cell lysate. (c) Quantification of pAkt signals. (d) Comparison of purified H10-labeled insulin and native insulin dependent activation in HEK293T cells (red). Chromatin (blue) (scale bar: 10 μm). | ||
Conclusions
In summary, we demonstrate that the conserved chemoselectivity of an electrophile is not an essential pre-requisite for precise single-site modification of native proteins. A linchpin-based proximity regulator can deconvolute the process of protein labeling into two steps. Subsequently, it can take over the job of chemoselective targeting from a promiscuous electrophile. Also, it regulates the local concentration to empower the latter to deliver site-selectivity. This hypothesis revealed a unique chemical space and enabled single-site modification of Asp in ubiquitin and His in insulin, α-lactalbumin, and myoglobin. It is noteworthy that the examples for precise labeling of these residues are rare due to their low reactivity compared to multiple other residues in a protein. Further, we extended the method for the single-site installation of a probe for NMR, affinity, and fluorescence. Also, we integrated the methodology with a highly efficient enrichment platform to deliver single-site immobilized or analytically pure labeled protein. The mild nature of the overall protocol was highlighted by the insulin bioactivity assay. Besides protein bioconjugation, the findings promise to create a new chemical space for reactive chemical probes to assist the search for efficient covalent inhibitors.Author contributions
V. R. and D. G. R. conceived the research. D. G. R., K. T., P. S., R. A., and S. R. A. designed and performed the bioconjugation experiments and in silico studies. R. K. M. and S. T. K. designed and performed the insulin bioactivity assay. All the authors wrote the manuscript.Conflicts of interest
V. R. is the scientific founder of Plabeltech Private Limited. A patent application has been filed with V. R. and S. R. A. as inventors. D. G. R., K. T., P. S., S. T. K., R. A. and R. K. M. declare no conflicts of interest.Acknowledgements
This work was supported by SERB, DST, DBT, and IISER Bhopal. D. G. R., K. T., P. S., S. T. K., R. A., and S. R. A. are recipients of a research fellowship from CSIR, UGC, DST, and IISER Bhopal. V. R. is a recipient of the Swarnajayanti Fellowship (DST/SJF/CSA-01/2018-19 and SB/SJF/2019-20/01).Notes and references
- D. V. Vranken and G. A. Weiss, Garland Science, Taylor & Francis Group, New York, 2013 Search PubMed.
- N. C. Reddy, M. Kumar, R. Molla and V. Rai, Org. Biomol. Chem., 2020, 18, 4669–4691 RSC.
- D. G. Rawale, K. Thakur, S. R. Adusumalli and V. Rai, Eur. J. Org. Chem., 2019, 6749–6763 CrossRef CAS.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS.
- T. Tomonori and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 2782–2799 CrossRef PubMed.
- E. M. Brustad, E. A. Lemke, P. S. Schultz and A. A. Deniz, J. Am. Chem. Soc., 2008, 130, 17664–17665 CrossRef CAS PubMed.
- A. M. Sochaj, W. S. Karolina and J. Otlewski, Biotechnol. Adv., 2015, 33, 775–784 CrossRef CAS.
- L. Wang, P. G. Schultz and L. Wang, Angew. Chem., Int. Ed., 2005, 44, 34–66 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes and B. G. Davis, Acc. Chem. Res., 2011, 44, 730–741 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- C. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS.
- R. Singudas, S. R. Adusumalli, P. N. Joshi and V. Rai, Chem. Commun., 2015, 51, 473–476 RSC.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS PubMed.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C. H. Weng, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17223–17229 CrossRef CAS PubMed.
- J. I. Macdonald, H. K. Munch, T. Moore and M. B. Francis, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef CAS PubMed.
- D. Chen, M. M. Disotuar, X. Xiong, Y. Wang and D. H. C. Chou, Chem. Sci., 2017, 8, 2717–2722 RSC.
- M. Raj, H. Wu, S. L. Blosser, M. A. Vittoria and P. S. Arora, J. Am. Chem. Soc., 2015, 137, 6932–6940 CrossRef CAS PubMed.
- A. C. Obermeyer, J. B. Jarman and M. B. Francis, J. Am. Chem. Soc., 2014, 136, 9572–9579 CrossRef CAS PubMed.
- L. Purushottam, S. R. Adusumalli, U. Singh, V. B. Unnikrishnan, D. G. Rawale, M. Gujrati, R. K. Mishra and V. Rai, Nat. Commun., 2019, 10, 2539 CrossRef.
- L. Purushottam, V. B. Unnikrishnan, D. G. Rawale, M. Gujrati, S. D. Mishra, T. K. Sajeev, N. C. Reddy, S. R. Adusumalli, R. K. Mishra and V. Rai, Chem. Sci., 2020, 11, 13137–13142 RSC.
- P. N. Joshi and V. Rai, Chem. Commun., 2019, 55, 1100–1103 RSC.
- S. Jia, D. He and C. J. Chang, J. Am. Chem. Soc., 2019, 141, 7294–7301 CrossRef CAS.
- L. Purushottam, S. R. Adusumalli, M. Chilamari and V. Rai, Chem. Commun., 2017, 53, 959–962 RSC.
- M. Chilamari, L. Purushottam and V. Rai, Chem.–Eur. J., 2017, 23, 3819–3823 CrossRef CAS PubMed.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS.
- M. Chilamari, N. Kalra, S. Shukla and V. Rai, Chem. Commun., 2018, 54, 7302–7305 RSC.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS.
- X. Ning, R. P. Temming, J. Dommerholt, J. Guo, D. B. Ania, M. F. Debets, M. A. Wolfert, G.-J. Boons and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 3065–3068 CrossRef CAS.
- H. Ren, F. Xiao, K. Zhan, Y. P. Kim, H. Xie, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 CrossRef CAS.
- G. Casi, N. Huguenin-Dezot, K. Zuberbühler, J. Scheuermann and D. Neri, J. Am. Chem. Soc., 2012, 134, 5887–5892 CrossRef CAS.
- G. J. L. Bernardes, M. Steiner, I. Hartmann, D. Neri and G. Casi, Nat. Protoc., 2013, 8, 2079–2089 CrossRef CAS PubMed.
- D. Bermejo-Velasco, G. N. Nawale, O. P. Oommen, J. Hilborna and O. P. Varghese, Chem. Commun., 2018, 54, 12507–12510 RSC.
- A. Bandyopadhyay, S. Cambray and J. Gao, Chem. Sci., 2016, 7, 4589–4593 RSC.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052–5058 RSC.
- E. Weerapana, G. M. Simon and B. F. Cravatt, Nat. Chem. Biol., 2008, 4, 405–407 CrossRef CAS PubMed.
- S. R. Adusumalli, D. G. Rawale, U. Singh, P. Tripathi, R. Paul, N. Kalra, R. K. Mishra, S. Shukla and V. Rai, J. Am. Chem. Soc., 2018, 140, 15114–15123 CrossRef CAS PubMed.
- S. R. Adusumalli, D. G. Rawale, K. Thakur, L. Purushottam, N. Reddy, N. Kalra, S. Shukla and V. Rai, Angew. Chem., Int. Ed., 2020, 59, 10332–10336 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00335f |
| This journal is © The Royal Society of Chemistry 2021 |