
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimal water concentration for aqueous Li+ intercalation in vanadyl phosphate†
Dan
Sun
a,
Masashi
Okubo
 ab and
Atsuo
Yamada
*ab
ab and
Atsuo
Yamada
*ab
aDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, Hongo 7-3-1, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: yamada@chemsys.t.u-tokyo.ac.jp
bElemental Strategy Initiative for Catalysts & Batteries (ESICB), Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan
First published on 11th February 2021
Abstract
Development of high-performance aqueous batteries is an important goal for energy sustainability owing to their environmental benignity and low fabrication costs. Although a layered vanadyl phosphate is one of the most-studied host materials for intercalation electrodes with organic electrolytes, little attention has been paid to its use in aqueous Li+ systems because of its excessive dissolution in water. Herein, by controlling the water concentration, we demonstrate the stable operation of a layered vanadyl phosphate electrode in an aqueous Li+ electrolyte. The combination of experimental analyses and density functional theory calculations reveals that reversible (de)lithiation occurs between dehydrated phases, which can only exist in an optimal water concentration.
Introduction
Lithium-ion batteries (LIBs) dominate the battery market for portable electronics and electric vehicles owing to their long lifetime, high efficiency, and high energy densities. However, LIBs are comprised of flammable and costly organic electrolytes, which unexceptionally accompany both safety hazards and high fabrication costs.1 Batteries that utilize aqueous electrolytes are expected to provide more operational safety, affordability, high power, and environmental benignity, all of which are favorable for large-scale stationary systems and electric vehicle operations.2,3 Although the intrinsically narrow electrochemical stability window for water as an electrolyte solvent (∼1.23 V) imposed severe limitations on any practical applications of aqueous batteries, novel strategies involving highly concentrated aqueous electrolytes have achieved much wider electrochemical potential windows (>3 V), thereby paving a path for the development of more practical aqueous batteries.4–8 Importantly, by exploiting a specialized solution structure without free water,9–14 highly concentrated aqueous electrolytes can also provide unexpected environments for electrode materials that have been considered ‘useless’ in conventional dilute aqueous electrolytes.15The selection criteria of electrode materials for aqueous batteries are (i) suitable redox potential, (ii) durability against water, (iii) no side reactions, (iv) good reversibility, and (v) low cost. Fig. S1† lists selected electrode materials that are potentially compatible with aqueous electrolytes. Among them, for instance, hydrated vanadyl phosphate (VOPO4·nH2O, Fig. 1a) is a versatile layered host for intercalation chemistry, which has been ascertained using various organic electrolyte systems.16–21 However, except for a few very recent reports of its application to aqueous H+/Zn2+ batteries,22,23 VOPO4·nH2O has rarely been considered promising in aqueous systems, due simply to dissolution and decomposition of VOPO4·nH2O in aqueous electrolytes.
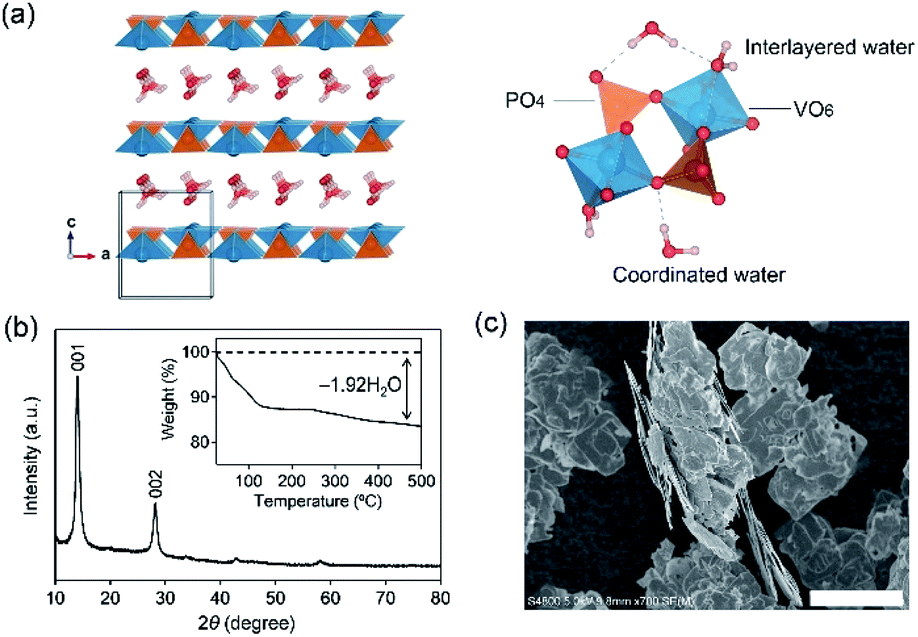 | ||
| Fig. 1 (a) Crystal structure of VOPO4·nH2O. V (dark cyan), P (orange), O (red), and H (gray) atoms are shown. (b) Powder X-ray diffraction pattern and (c) scanning electron microscopy image of VOPO4·nH2O. The inset in (b) is the thermal gravimetric analysis profile of VOPO4·nH2O. | ||
VOPO4·2H2O possesses a bilayer structure where a VOPO4 layer is composed of corner-sharing VO6 octahedra and PO4 tetrahedra, with a layer distance of 7.25 Å along the c axis, as illustrated in Fig. 1a. When VOPO4·nH2O is immersed in liquids that possess high water concentration c(H2O) (e.g., pure water or dilute aqueous solutions), surface vanadium atoms are readily coordinated by the water to form soluble vanadium aquo complexes. Furthermore, high-concentration water is prone to intercalate into an interlayer space to exfoliate VOPO4 layers, accelerating the dissolution process. Alternatively, an extremely low-c(H2O) environment (e.g., highly concentrated aqueous solutions) is expected to desorb interlayer water between VOPO4 layers. Since interlayer water plays an important role in both structural integrity and ion diffusion, the intercalation chemistry may be largely altered via the control of c(H2O) of aqueous electrolytes for achieving reversible lithium-ion (de)intercalation in VOPO4·nH2O. Herein, we demonstrate the stable charge/discharge operation of VOPO4·nH2O through accurate control of c(H2O) in aqueous Li+ electrolytes.
Results and discussion
VOPO4·nH2O was synthesized by refluxing V2O5 and H3PO4,24 and the resulting powder was dried under vacuum overnight at 80 °C. The powder X-ray diffraction (XRD) pattern (Fig. 1b) corresponds to tetragonal VOPO4·nH2O, and a 001 diffraction at 2θ ≈ 14° indicates an interlayer distance of 7.35 Å. The thermogravimetric (TG) analysis (inset of Fig. 1b) reveals a water content per formula unit of n = 1.92. The scanning electron microscopy image (Fig. 1c) shows a lamellar morphology with a lateral size ranging from 10 to 100 μm.The dissolution durability of VOPO4·nH2O was tested using aqueous Li+ electrolytes (Fig. 2a). After immersing VOPO4·nH2O in aqueous electrolytes at various Li+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O ratios for 30 days, the color of the dilute electrolyte (Li+:H2O = 1:50, high c(H2O)) changed to orange, while concentrated electrolytes (e.g., Li+:H2O = 1:4, low c(H2O)) remained transparent, indicating effective suppression of vanadium ion dissolution by lowering c(H2O).
:H2O ratios for 30 days, the color of the dilute electrolyte (Li+:H2O = 1:50, high c(H2O)) changed to orange, while concentrated electrolytes (e.g., Li+:H2O = 1:4, low c(H2O)) remained transparent, indicating effective suppression of vanadium ion dissolution by lowering c(H2O).
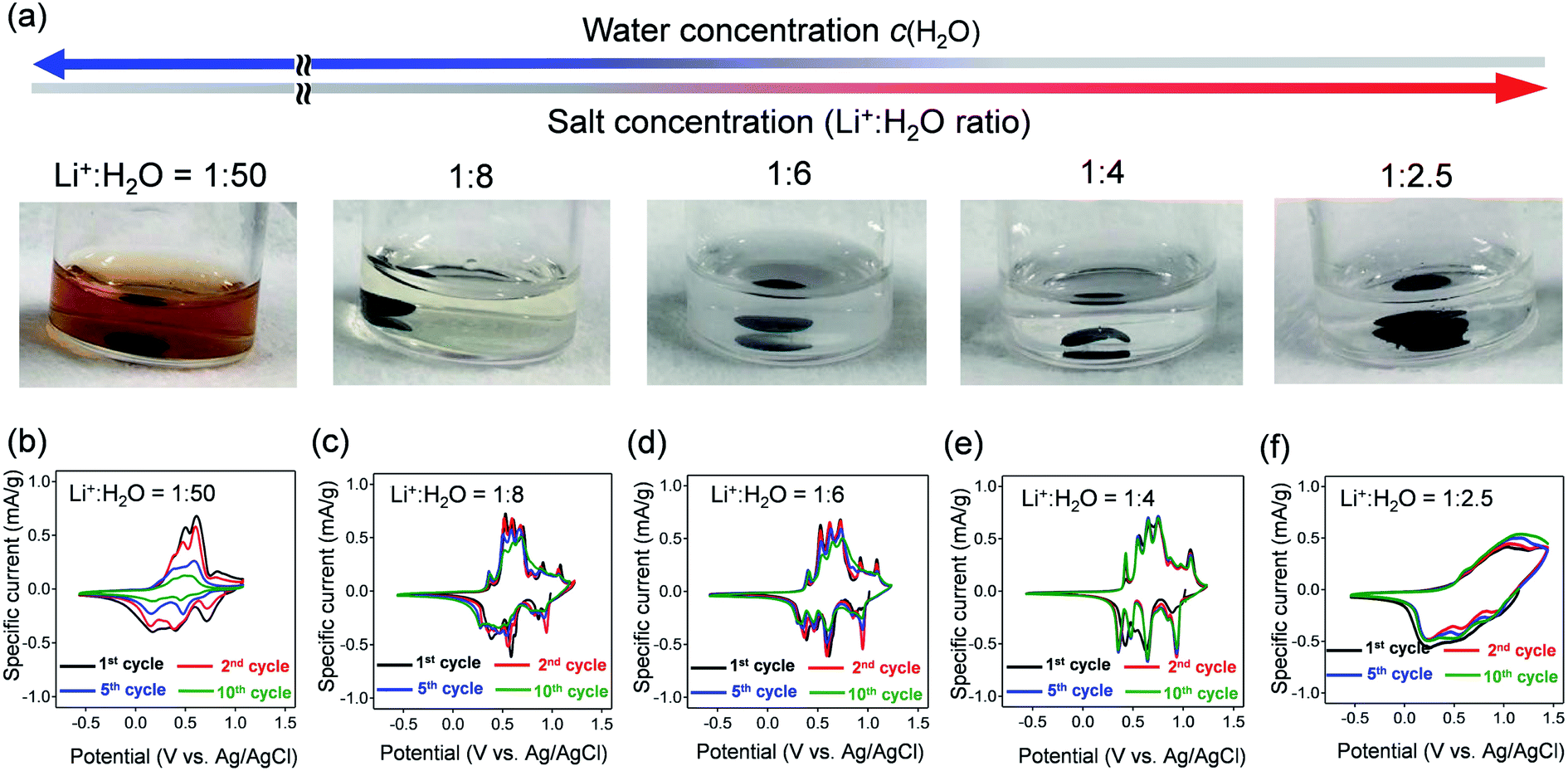 | ||
| Fig. 2 (a) Photographs of aqueous Li+ electrolytes after immersing VOPO4·nH2O for 30 days. The Li+:H2O ratio ranges from 1:50 (LiTFSI/50H2O), 1:8 (LiTFSI/8H2O), 1:6 (LiTFSI/6H2O), 1:4 (LiTFSI/4H2O) to 1:2.5 (water-in-salt, LiTFSI/2.5H2O). (b–f) Cyclic voltammetry curves of VOPO4·nH2O in aqueous Li+ electrolytes at various Li+:H2O ratios at a scan rate of 0.5 mV s−1. | ||
Cyclic voltammetry (CV) was conducted to evaluate the electrochemical properties of VOPO4·nH2O under various c(H2O) conditions (Fig. 2b–f). The CV curves for the dilute aqueous electrolyte (Li+:H2O = 1:50, high c(H2O)) (Fig. 2b) show severe decay of current flow with repeating CV cycles. The decay of current flow coincides with the change in the electrolyte color to orange, arising from the dissolution of vanadium in a high-c(H2O) environment. In contrast, upon increasing the salt concentration (lowering c(H2O)) to a Li+:H2O ratio of 1:4, intense multiple current flows gradually emerge with minimal polarization, indicating reversible (de)lithiation without parasitic reactions/dissolution. However, when further lowering c(H2O) to a Li+:H2O ratio of 1:2.5, the cyclic voltammetry (CV) curve shows broad cathodic/anodic current flows with large polarization (Fig. 2f), which indicates sluggish (de)lithiation of VOPO4·nH2O in an environment of overly low c(H2O). Electrochemical impedance spectroscopy (Fig. S2†) shows the specific increase of both series resistance and charge-transfer resistance specifically when the Li+:H2O ratio changes from 1:4 to 1:2.5. Presumably, the low ionic conductivity of the highly concentrated aqueous electrolyte increases the series resistance while the strong coulombic attraction between Li+ and TFSI− (contact-ion pair) increases the charge-transfer resistance, both of which cause the sluggish (de)lithiation of VOPO4·nH2O.14 Therefore, an optimal-c(H2O) environment (Li+:H2O = 1:4) is required for reversible (de)lithiation of VOPO4·nH2O in aqueous systems.
Fig. 3a–c show the galvanostatic charge/discharge curves of VOPO4·nH2O in various c(H2O) environments at a specific current of 1 A g−1. The delivered capacity with a high-c(H2O) aqueous electrolyte (Fig. 3a) drastically decreases upon cycling, while the potential polarization increases, presumably because of the vanadium dissolution. Upon gradually lowering c(H2O) to an optimal range (Li+:H2O = 1:4), the potential profiles exhibit distinct multiple plateaus without substantial polarization, indicating highly reversible (de)lithiation (Fig. 3b and c). However, when further lowering c(H2O) to Li+:H2O = 1:2.5, the polarization between lithiation/delithiation significantly increases to reduce the reversible capacity to approximately 85 mA h g−1 (Fig. S3†). These results are consistent with the CV results (Fig. 2). The reversible capacity of 118 mA h g−1 in the first cycle (Li+:H2O = 1:4) corresponds to 0.8 Li+ intercalation per formula unit. The reversible reduction/oxidation of vanadium (V5+/V4+) upon charge/discharge, which was confirmed by V K-edge X-ray absorption spectroscopy (Fig. S4†), supports the occurrence of reversible (de)lithiation of VOPO4·nH2O. Furthermore, owing to the optimal-c(H2O) environment, 80% of the initial capacity is retained after 200 cycles (Fig. 3d), which is much higher than that for a high-c(H2O) aqueous electrolyte (7%).
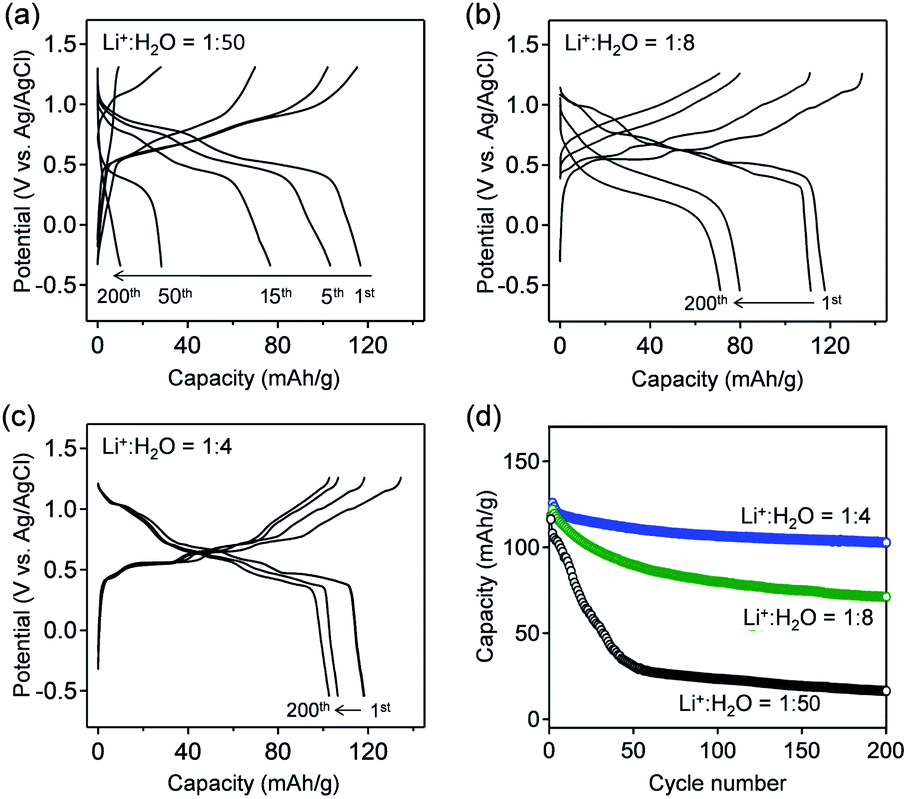 | ||
| Fig. 3 Charge/discharge curves of VOPO4·H2O electrodes with aqueous Li+ electrolytes. The Li+:H2O ratios are (a) 1:50 (LiTFSI/50H2O), (b) 1:8 (LiTFSI/8H2O), and (c) 1:4 (LiTFSI/4H2O), respectively. The specific current is 1 A g−1. (d) Capacity retention during 200 charge/discharge cycles. | ||
To clarify the phase diagram of LixVOPO4·nH2O in the optimal c(H2O) environment, in situ XRD experiments were conducted (Fig. 4a–c). Before immersion in the optimal-c(H2O) electrolyte, VOPO4·nH2O in a composite electrode possesses an interlayer distance of 7.2 Å. The calculated interlayer distance of VOPO4·2H2O (7.25 Å, Fig. S5 and Table S1†) agrees with the experimental value, and the water content of VOPO4·nH2O in a pristine electrode is estimated as n ≈ 2, which is in good agreement with the TG result (the inset of Fig. 1b). However, the in situ XRD pattern for VOPO4·nH2O immersed in a LiTFSI/4H2O electrolyte shows the shift of 001 diffraction to a higher diffraction angle (Fig. 4a), indicating the decrease of the interlayer distance to 6.2 Å. This value is consistent with the calculated interlayer distance for VOPO4·H2O (6.16 Å, Fig. S5 and Table S1†). Therefore, the low-c(H2O) environment dehydrates VOPO4·nH2O to a monohydrate phase (n = 1). Upon lithiation/delithiation, a new 001 diffraction, corresponding to an interlayer distance of 5.3 Å, emerges/diminishes at the expense of the original 001 diffraction (Fig. 4b), suggesting biphasic (de)lithiation. It is noteworthy that the lithiated/delithiated monohydrate phases are stable only in the optimal-c(H2O) environment; for instance, the lithiated phase immediately becomes hydrated to a dihydrate phase (n = 2) after being exposed to the ambient atmosphere (Fig. 4c).
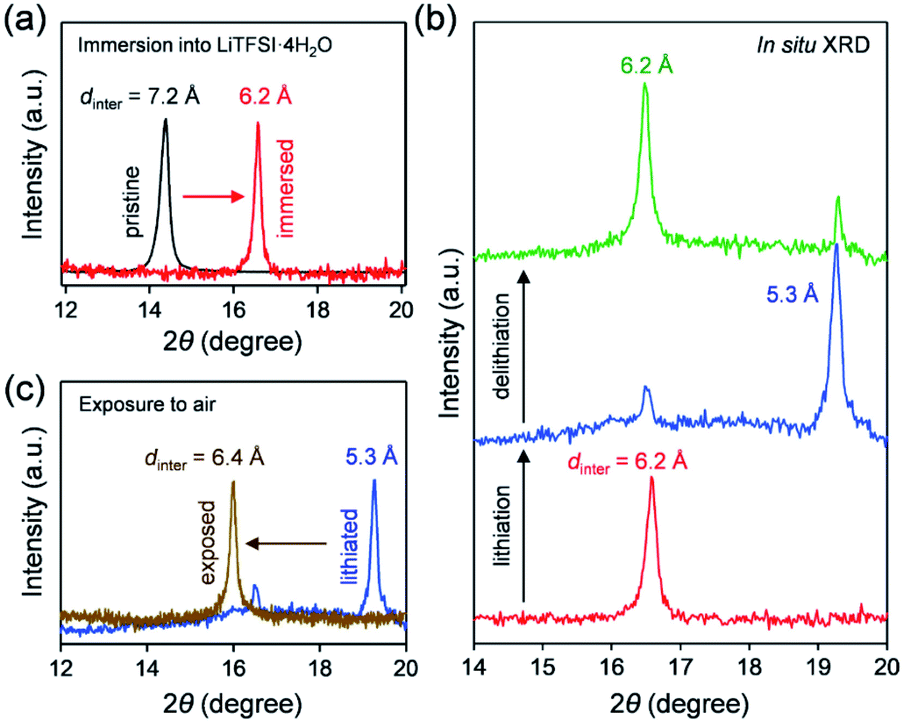 | ||
| Fig. 4 Structural changes of VOPO4·nH2O in LiTFSI/4H2O. The 001 diffraction peak (a) before and after immersion in LiTFSI/4H2O, (b) after lithiation and delithiation in LiTFSI/4H2O, and (c) before and after exposure of a lithiated sample to the ambient atmosphere. dinter denotes the interlayer distance. | ||
A phase diagram of LixVOPO4·nH2O in the optimal-c(H2O) environment is postulated based on the experimental and simulation results as schematized in Fig. 5. The equilibrium between interlayer water in VOPO4·nH2O and low-concentration water in concentrated aqueous electrolytes drives dehydration of VOPO4·nH2O to the monohydrate phase (n = 1). Although exhibiting reversible Li+ (de)intercalation, the monohydrate phases can only be stabilized in a low-c(H2O) environment, and exposure to the ambient atmosphere immediately causes hydration to the dihydrate phase (n = 2).
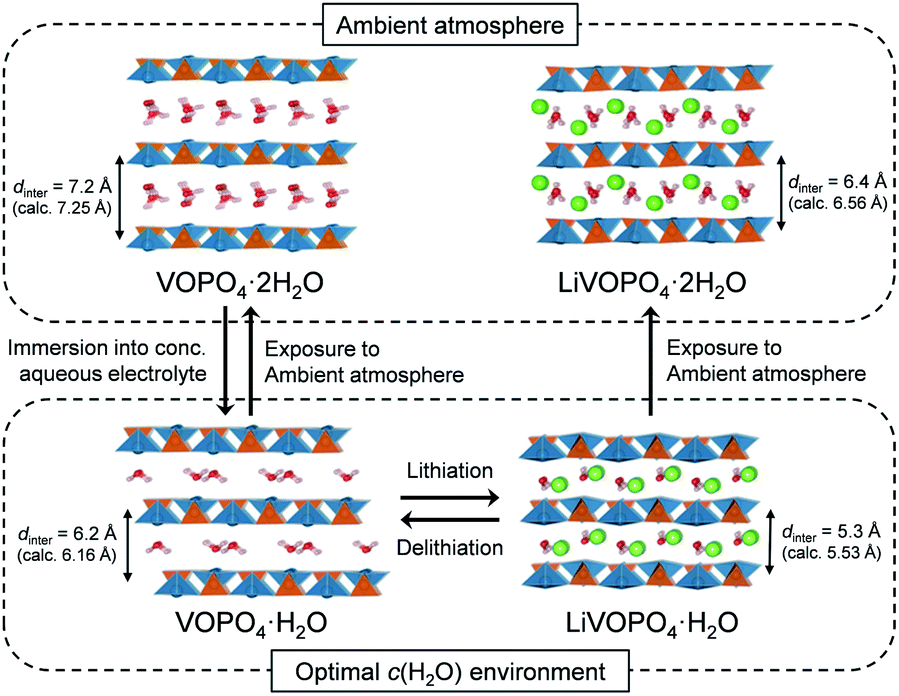 | ||
| Fig. 5 Schematic illustration of the phase diagram of VOPO4·nH2O in the optimal-c(H2O) environment. | ||
Conclusions
In summary, the stable operation of VOPO4·nH2O electrodes in an optimal-c(H2O) environment was successfully demonstrated. The reversible (de)lithiation occurs specifically in monohydrate phases, which are only stable in the optimal-c(H2O) environment. This study provides novel insights into the reaction mechanism and phase diagram of VOPO4·nH2O in a low-c(H2O) environment, and more importantly points to research opportunities to enrich aqueous ion intercalation chemistry by controlling water concentration.Methods
VOPO4·nH2O was synthesized via a reflux method: V2O5 (6 g), H2O (144 mL) and H3PO4 (82%, 57.75 g) at 125 °C for 16 h. After cooling to room temperature, the resulting yellow precipitate was collected by centrifugation and washed three times with water and acetone. Thereafter, the powder was dried under vacuum overnight at 80 °C.X-ray diffraction patterns were recorded on a Bruker AXS D8 Advance X-ray diffractometer using Co Kα radiation. Microstructure analyses were performed using a scanning electron microscope (Hitachi, S-4800) at a beam accelerating voltage of 5 kV. The oxidation states of the samples were measured by V K-edge XANES at the beamline 8B of Photon Factory (PF), High Energy Accelerator Research Organization (KEK) Tsukuba, Japan. The crystal water content of the as-synthesized VOPO4·nH2O sample was confirmed by TG (NETZSCH, STA2500).
VOPO4·nH2O electrodes were fabricated by grinding a mixture containing VOPO4·nH2O, carbon black (super P) and polytetrafluoroethylene (PTFE) in a weight ratio of 75:10:15 for 20 min, and then it was rolled into an electrode film using a rolling machine with a fixed gap of 250 μm. For the preparation of aqueous electrolytes, lithium bis-(trifluoromethanesulfonyl)imide (LiTFSI) was dissolved in ultrapure water as LiTFSI·nH2O (n = 2.5, 4, 6, 8, and 50). The electrochemical performance of VOPO4·nH2O electrodes was evaluated using a three-electrode system (Ag/AgCl and active carbon as the reference and counter electrodes, respectively). CR2032-type coin cells were assembled to measure the galvanostatic charge/discharge properties of the electrodes. The cell consists of a VOPO4·nH2O electrode, active carbon anode and glass fiber separator (GF/F, Whatman). Electrochemical impedance spectroscopy (VMP3 potentiostat, Biologic) was performed at the open circuit potential with an amplitude of 10 mV in the frequency range of 10 mHz to 200 kHz.
First-principles calculations were performed using the Vienna Ab initio Simulation Package (VASP),25 based on density functional theory (DFT).26,27 The exchange–correlation energy is calculated using general gradient approximation (GGA) with the Perdue–Burke–Ernzerhof (PBE) exchange–correlation functional.28 Furthermore, in our calculations, Hubbard U corrections (GGA + U) were adopted with U − J = 3.1 for vanadium. The effect of van der Waals interactions was estimated and implemented in the optimized exchange van der Waals functional B86b of the Becke (optB86b vdW) functional.29,30 The plane wave cutoff energy was 580 eV. The convergence condition for the energy is 10−4 eV, and the structures were relaxed until the force on each atom was less than 0.01 eV Å−1. Spin polarization was considered in all calculations. The k-point meshes of 7 × 7 × 7 and 14 × 14 × 14 in the Monkhorst–Pack sampling scheme were used for geometry optimization and electronic self-consistent computation, respectively.31
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan; Grant-in-Aid for Specially Promoted Research No. 15H05701 and the “Elemental Strategy Initiative for Catalysis and Batteries” (ESICB). M. O. was financially supported by JSPS KAKENHI, Grant Numbers 19H05816, 18K19124, and 18H03924. The structure drawing was generated using VESTA.32Notes and references
- M. R. Palacín and A. de Guibert, Science, 2016, 351, 1253292 CrossRef.
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS.
- W. Tang, Y. S. Zhu, Y. Y. Hou, L. L. Liu, Y. P. Wu, K. P. Loh, H. P. Zhang and K. Zhu, Energy Environ. Sci., 2013, 6, 2093–2104 RSC.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS.
- C. Y. Yang, J. Chen, T. T. Qing, X. L. Fan, W. Sun, A. von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. S. Wang and K. Xu, Joule, 2017, 1, 122–132 CrossRef CAS.
- C. Y. Yang, J. Chen, X. Ji, T. P. Pollard, X. J. Lu, C. J. Sun, S. Hou, Q. Liu, C. M. Liu, T. T. Qing, Y. Q. Wang, O. Borodin, Y. Ren, K. Xu and C. S. Wang, Nature, 2019, 569, 245 CrossRef CAS.
- L. W. Jiang, Y. X. Lu, C. L. Zhao, L. L. Liu, J. N. Zhang, Q. Q. Zhang, X. Shen, J. M. Zhao, X. Q. Yu, H. Li, X. J. Huang, L. Q. Chen and Y. S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- J. Vatamanu and O. Borodin, J. Phys. Chem. Lett., 2017, 8, 4362–4367 CrossRef CAS.
- O. Borodin, L. M. Suo, M. Gobet, X. M. Ren, F. Wang, A. Faraone, J. Peng, M. Olguin, M. Schroeder, M. S. Ding, E. Gobrogge, A. V. Cresce, S. Munoz, J. A. Dura, S. Greenbaum, C. S. Wang and K. Xu, ACS Nano, 2017, 11, 10462–10471 CrossRef CAS.
- K. Miyazaki, N. Takenaka, E. Watanabe, S. Iizuka, Y. Yamada, Y. Tateyama and A. Yamada, J. Phys. Chem. Lett., 2019, 10, 6301–6305 CrossRef CAS.
- N. Takenaka, T. Inagaki, T. Shimada, Y. Yamada, M. Nagaoka and A. Yamada, J. Chem. Phys., 2020, 152, 124706 CrossRef CAS.
- K. Kim, Y. Ando, A. Sugahara, S. Ko, Y. Yamada, M. Otani, M. Okubo and A. Yamada, Chem. Mater., 2019, 31, 5190–5196 CrossRef CAS.
- K. Kim, M. Okubo and A. Yamada, J. Electrochem. Soc., 2019, 166, A3739–A3744 CrossRef CAS.
- A. Yamada, Joule, 2018, 2, 371–372 CrossRef.
- A. J. Jacobson, J. W. Johnson, J. F. Brody, J. C. Scanlon and J. T. Lewandowski, Inorg. Chem., 1985, 24, 1782–1787 CrossRef CAS.
- J. Gaubicher, T. Le Mercier, Y. Chabre, J. Angenault and M. Quarton, J. Electrochem. Soc., 1999, 146, 4375 CrossRef CAS.
- N. Dupre, J. Gaubicher, T. Le Mercier, G. Wallez, J. Angenault and M. Quarton, Solid State Ionics, 2001, 140, 209–221 CrossRef CAS.
- Y. N. Song, P. Y. Zavalij and M. S. Whittingham, J. Electrochem. Soc., 2005, 152, A721–A728 CrossRef CAS.
- M. S. Whittingham, Y. N. Song, S. Lutta, P. Y. Zavalij and N. A. Chernova, J. Mater. Chem., 2005, 15, 3362–3379 RSC.
- Y. Zhu, L. L. Peng, D. H. Chen and G. H. Yu, Nano Lett., 2016, 16, 742–747 CrossRef CAS.
- H. Y. Shi, Y. Song, Z. M. Qin, C. C. Li, D. Guo, X. X. Liu and X. Q. Sun, Angew. Chem., Int. Ed., 2019, 58, 16057–16061 CrossRef CAS.
- F. Wan, Y. Zhang, L. L. Zhang, D. B. Liu, C. D. Wang, L. Song, Z. Q. Niu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 7062–7067 CrossRef CAS.
- J. Hyoung, J. W. Heo, M. S. Chae and S. T. Hong, ChemSusChem, 2019, 12, 1069–1075 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev. B: Solid State, 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. Klimes, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef.
- J. Klimes, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04647g |
| This journal is © The Royal Society of Chemistry 2021 |