
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCDK4/6 inhibitors: a brief overview and prospective research directions
Tenzin Adon ,
Dhivya Shanmugarajan and
Honnavalli Yogish Kumar*
,
Dhivya Shanmugarajan and
Honnavalli Yogish Kumar*
Department of Pharmaceutical Chemistry, JSS College of Pharmacy, JSS Academy of Higher Education and Research, Sri Shivarathreeshwara Nagar, Mysuru-570015, Karnataka, India. E-mail: yogigcp@gmail.com; Tel: +919726447802
First published on 1st September 2021
Abstract
The discovery of cyclin-dependent kinases (CDK) and their mechanism in regulating the cell cycle process was considered a game-changer in cancer therapy. Cell cycle arrest and apoptosis were both triggered by their inhibition. The CDK4/6 complex acts as a checkpoint during the cell cycle transition from cell growth (G1) to DNA synthesis (S) phase and its deregulation or overexpression induces abnormal cell proliferation and cancer development. Consequently, targeting CDK4/6 has been proposed as a paradigm shift in the anticancer approach. The design and development of effective CDK4/6 inhibitors are increasingly becoming a promising cancer therapy evident with approved drugs such as palbociclib, ribociclib, and abemaciclib, etc. In this article, we explore the biological importance of CDK4/6 in cancer therapy, the development of resistance to monotherapy, and a short overview of PROTAC (Proteolysis Targeting Chimera), a unique and pioneering technique for degrading CDK4/6 enzymes. Overall, our prime focus is to discuss novel CDK4/6 inhibitors with diverse chemical classes and their correlation with computational studies.
1. Introduction
Despite advancements in healthcare technology, an estimated 10 million people worldwide died from cancer in 2020.1 Globally, over the past several decades, cancer continued to be the second prime cause of death owing to various factors such as aging and lifestyle.2 Lung and breast cancer remain the most common cancer, but the mortality rate of lung cancer is highest whereas that of breast cancer is relatively low compared to other types of cancer. Cancer is the uncontrolled proliferation of abnormal cells in a multistage process that eventually spread to other tissues and organs. In the drug design and development of clinical approaches for cancer therapy, understanding the factors that regulate cell division is a key factor. Owing to the complication in specifying target and preventing the development of cancer cells while not harming the proliferation of normal cells, remains a major challenge to date.3The discovery of the catalytic subunit cyclin-dependent kinase (CDKs), which interacts with the regulatory subunit cyclins, serves as a checkpoint in the cell division and growth process.4 As a result, it is extremely important during the cell cycle transition in cancer cell growth. Cyclin is a CDK activating regulatory subunit that is required for enzymatic activity. CDKs that stimulate cell cycle transformation are expected to be key therapeutic targets since several tumorigenic events eventually drive proliferation by impinging on CDK4 or CDK6 complexes in the G1 process of the cell cycle.5
CDKs are proline-binding serine/threonine protein kinases. Based on the sequence of the kinase domain, CDKs are called cyclin-dependent protein kinases, mitogen-activated protein kinases, glycogen synthase kinases, or CDC-like kinases (CMGC).3,6 CDKs are critical regulatory enzymes in the cell cycle playing a major role in controlling cell cycle progression through the different phases and transcriptional regulation in response to several intracellular and extracellular signals. As an outcome of their significance in multiple processes, deregulation of CDKs is a distinctive feature of various diseases, predominantly observed in cancer.6 For instance, in 90 percent of cancers, the cyclin-CDK4/6-Rb-E2F pathway is disrupted with overexpression of cyclin D in most tumour cells.7 Besides, Rb:CDK:cyclin:p16 tumour suppressor pathway undergoes mutational and epigenetic changes in a diverse range of human cancers.8 Targeting CDKs thus serves as an important anti-cancer strategy.
CDK inhibitors are pioneering drug targets and new therapeutic interventions in cancer, for decades as scientists have gained a deeper understanding of CDKs role in transcription control and sustain the oncogenic state in cells. Evidently, resulting in approval of abemaciclib, palbociclib, and ribociclib, (Fig. 1) in treating later stage or stage IV or metastatic breast cancer patients expressing biomarkers like human epidermal growth factor receptor 2-negative (HER2−) and hormone receptor-positive (HR+) and advanced or metastatic breast cancer.4,9–11 Cyclin-CDK4/6 small molecule inhibitors have demonstrated a potential effect compare to traditional steroidal inhibitors in the treatment of breast cancer in ongoing clinical trials (Table 1).
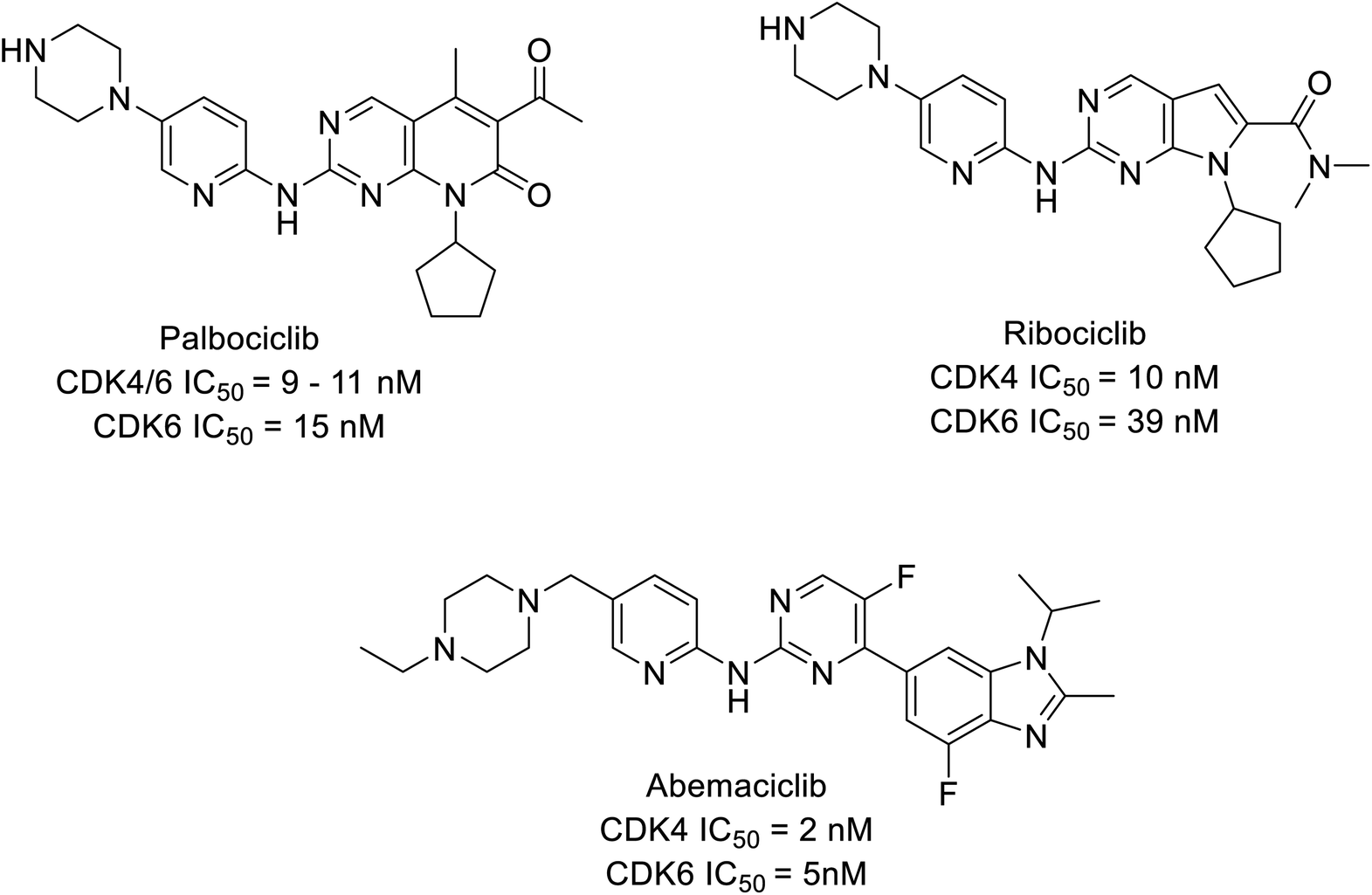 | ||
| Fig. 1 FDA-approved CDK4/6 inhibiting drugs. | ||
| Type of analogues | Compounds | IC50 | -CDOCKER interaction energy | Binding energy (kcal mol−1) | |||
|---|---|---|---|---|---|---|---|
| CDK4 | CDK6 | CDK4 | CDK6 | CDK4 | CDK6 | ||
| Carboxamide | 1a | 0.85 nM | 1.96 nM | 52.9134 | 50.5211 | −22.145 | −20.2216 |
| 2a | 0.77 nM | 0.59 nM | 51.3064 | 50.9082 | −19.0743 | −20.4471 | |
| Pyrimidine | 3a | 30 nM | 4 nM | 53.7621 | 49.8539 | −12.045 | −11.9997 |
| 4a | 115.8 nM | — | 45.9237 | 47.0807 | −15.672 | −14.2252 | |
| 4b | 726.25 nM | — | 47.4417 | 44.4125 | −3.789 | −2.3601 | |
| 5a | 0.8 nM | 2 nM | 58.8961 | 56.0164 | −37.156 | −35.2816 | |
| 5b | 2.7 nM | 4.8 nM | 56.1309 | 57.9118 | −29.7864 | −33.4064 | |
| Quinazoline | 6a | 0.1 μM | 0.026 μM | 48.3002 | 48.1062 | −5.709 | −8.3683 |
| 6b | 0.003 μM | 0.01 μM | 49.0504 | 52.7771 | −12.975 | −11.5884 | |
| Acridone | 7a | 200–800 nM | 53.573 | 51.5219 | −10.197 | −12.9007 | |
| Indole conjugates | 8a | 1.8 μM | — | 35.7233 | 41.4787 | −14.034 | −19.342 |
| 8b | 1.26 μM | — | 38.6651 | 45.6452 | −13.345 | −16.0896 | |
In this review, we have extensively done a literature search and attempted to explain the role of CDK4/6 in cancer, its development of resistance, and the proposed strategy to overcome resistance. Understanding the resistance mechanism and biomarker will contribute to CDK4/6 inhibitor's potential use and its prospect of modification/developments. Furthermore, this review presents insight into the recent design and development of small molecule CDK4/6 inhibitors possessing various chemical pharmacophores or scaffolds exhibiting potential therapy for cancer treatment. Apart from it, we have tried discuss on development of PROTAC, a unique and pioneering technique for degrading CDK4/6 kinases.
2. Cell cycle regulations by cyclin D–CDK4/6 and its biological significance in cancer
The significant role of CDK's is majorly captured during the regulation of the cell cycle in the biological system. The cyclin-D-CDK4/6 complexes control the major phase of cell cycle progression from growth (G1) to DNA replication (S) phase and are thus referred to as cell-cycle controlling kinases (Fig. 2). During the growth (G1) phase of the cell cycle, a distinct family of D-type cyclin act as mitogen sensors12,13 and has latent proto-oncogenic capabilities.12 The level of cyclin D is low in quiescent cells and is up-regulated as cells enter the G1 phase. From an early age, among the D-type cyclins, cyclin D1 received much attention for the reason that it activates cell cycle progression by phosphorylating negative growth regulators.14 For instance, binding of cyclin D1 directly to the retinoblastoma gene (Rb) followed by retinoblastoma protein (pRb) phosphorylation (Fig. 3) by CDK4/6 a family of cyclin D-dependent kinases results in inhibition of tumour suppressor activity of Rb.12,14–16 Subsequently, cyclin D2 gains importance as it promotes the degradation of cyclin-dependent kinase inhibitor p27 (CKI p27Kip1) by translocating it from the nucleus into the cytoplasm at the G0–G1 transition.16,17 Additionally, cell proliferation is controlled by CKI p27Kip1 a family protein of Cip/Kip. However, CKI p27Kip1 has the ability to directly interact to impede the formation of multiple domains like G1 cyclins, p27 and CDKs, which are essential for promoting cell cycle transition from G0–S phase.16,17 The cyclin D types govern the enzymatic activities of the CDK4/6 signalling pathway in the normal and cancerous cells. It was demonstrated neither D type cyclin nor their D-type-dependent kinase CDK4/6 are essential for existing cell proliferation upon mitogenic stimulation.18 But the absence of CDK4/6 kinase can be compensated by CDK2 partially by involving the signalling mechanism associated with D-cyclins-D types.11,13,16 Furthermore, the role of cyclin E–CDK2 was observed more in abundance in the absence of CDK4/6 with pRb kinase activity. Yet the entry mechanism of cell proliferation without cyclin D–CDK4/6 activity that remains poorly characterized before, shown to rely on fluctuating increases in cyclin E–CDK2 activity can be progressed even in the absence of Rb hyperphosphorylation.19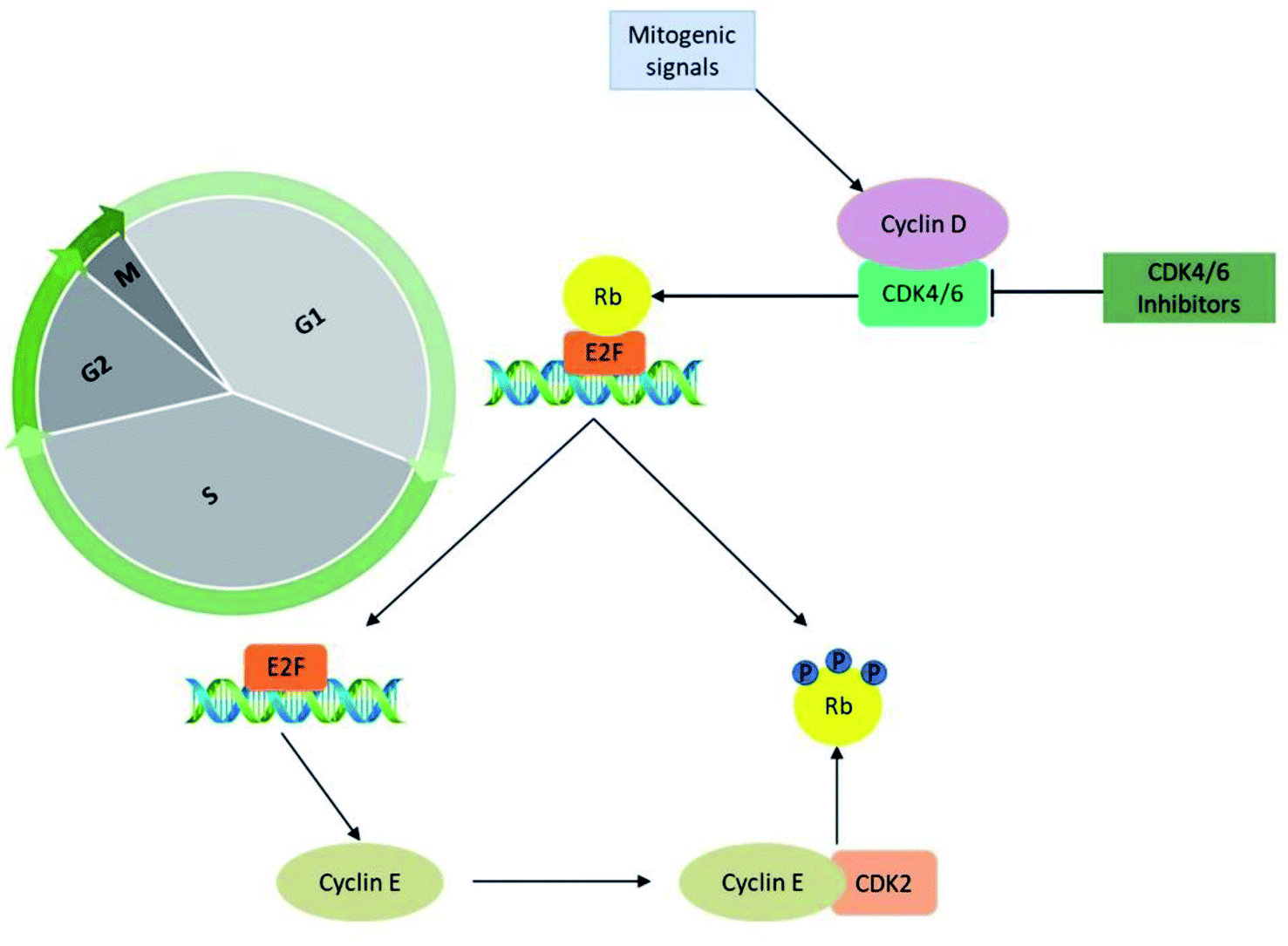 | ||
| Fig. 2 CDK4/6 in cell cycle progression and CDK4/6 inhibitors mechanism of action. | ||
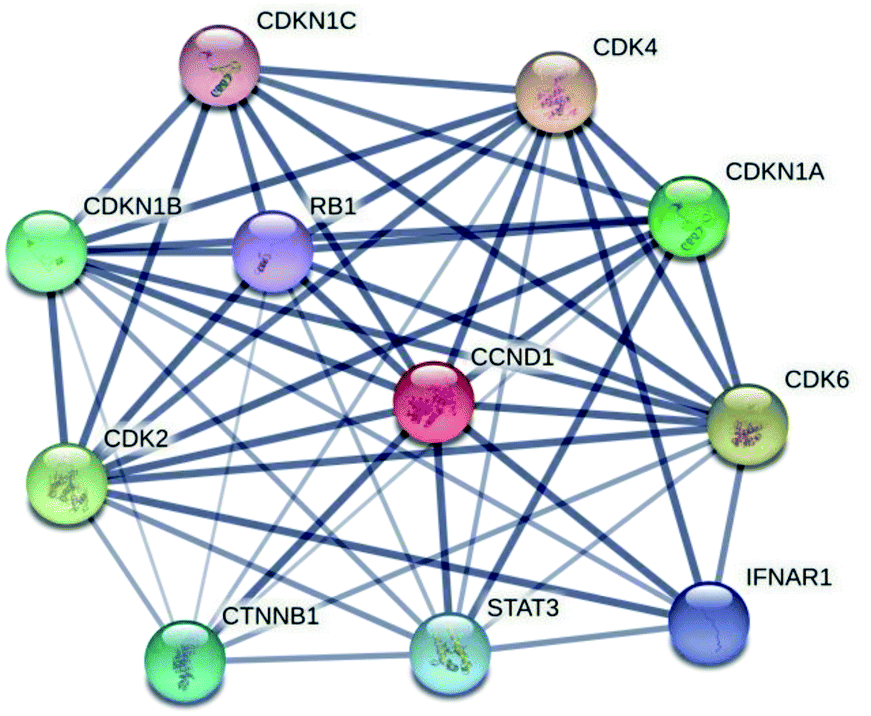 | ||
| Fig. 3 The protein–protein interaction network of cyclin D1 with retinoblastoma and other families of cyclin. A dark line of connection in the network indicates the strong binding interaction vice versa. | ||
CDK4 and CDK6 are structurally related kinases with biochemical and biological similarities. The differential patterns of expression are determined by the specific roles of the catalytic subunits. The role of CDK6 is crucial in the cell proliferation and differentiation of haematological precursors.13,20 CDK4 and CDK6 regulate the mid-G1 phase, a central regulatory checkpoint, after catalytic activation. Mitogenic stimulation causes the synthesis of cyclin D, which leads to the development of cyclin D/CDK4 and cyclin D/CDK6 complexes and their nuclear localization. The pRb is then phosphorylated and inactivated by the activated cyclin D–CDK4/6 complex. The pRb gene is a member of the tumour suppressor gene family. It specifically binds to E2F transcription factors, through its C-terminal region, and in turn, negatively regulates the expression of E2F that is essential for G1 to S progression. The hyperphosphorylation of Rb releases the E2F, induces the expression of the E-type cyclins which binds CDK2. The binding of cyclin E to CDK2 causes its activation that hyper-phosphorylate pRb leading to the liberation of E2F transcription factors. This sequence of functions results in the promotion of transition from growth (G1) to DNA replication (S) phase through the expression of multiple genes thought to be escape mechanisms (Fig. 3). Because of their importance in multiple processes, the deregulated CDK-cyclin-Rb pathway is proposed to be a hallmark of cancer.
3. CDK4/6 inhibitors
3.1 Carboxamide-based analogues
Wang et al. have performed the synthesis of a series of 1-H-pyrazole-3-carboxamide derivatives and evaluated inhibitory activities on CDKs, FLT3 kinases.21 Compound 1a (Fig. 4) exhibited the highest activity to inhibit CDK6 and CDK4 with IC50 values of 1.96 nM and 0.85 nM, respectively. Relevant cytotoxicity effects on NCI60 cancer-cell lines (IC50 < 1 μM) were also found to be significant. In a structure–activity relation (SAR) study, the presence of pyrrolopyrimidine at the 4th position of pyrazole (1a) was necessary for inhibitory activity against FLT3, and CDKs. The replacement of pyrrolopyrimidine moiety of compound 1a by thienopyrimidine exhibited relatively less inhibitory activity against CDK4/6 with IC50 values of 4.24 nM and 5.09 nM respectively.21 Similarly, replacement of pyrrolopyrimidine by furopyrimidine showed inhibitory activity against CDK4/6 with IC50 value of 5.67 nM and 5.09 nM but results in 3–4 folds better CDK4/6 selectivity over CDK2. Compound 1a caused tumour regression and metabolic stability in a liver-microsomal metabolism assay. It's currently in phase I clinical trials in China and the United States, and it may be a potential new therapy for acute myeloid leukaemia (AML).21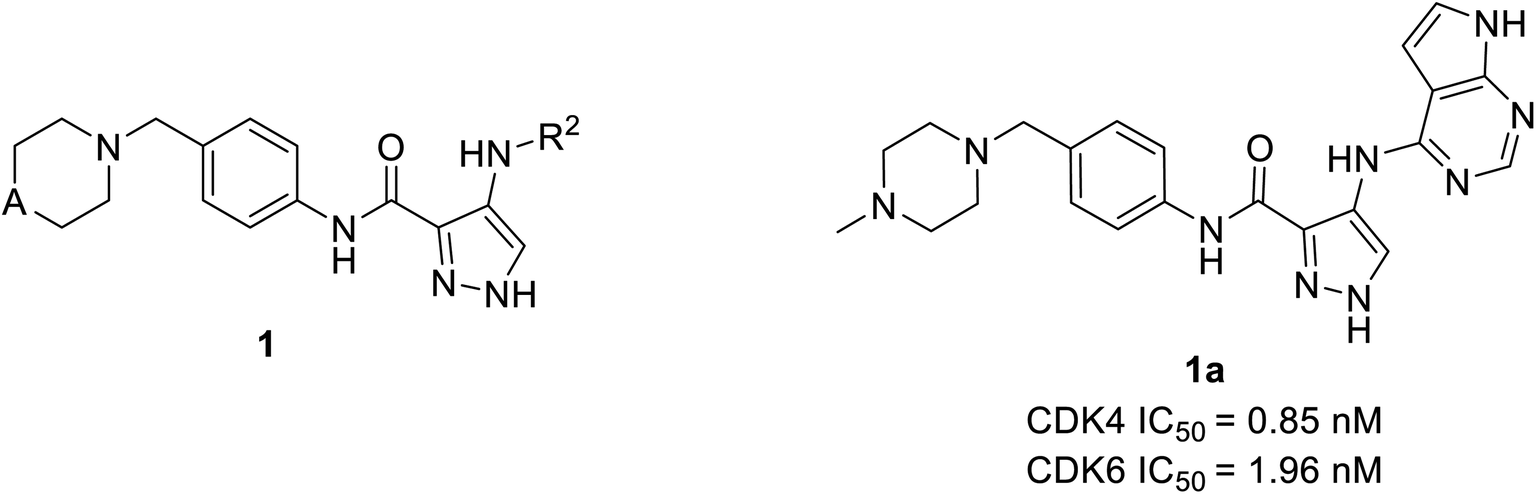 | ||
| Fig. 4 1-H-Pyrazole-3-carboxamide derivatives as potent CDK4/6 and FLT3 inhibitors. | ||
Zhi et al. developed a series of 1H-pyrazole-3-carboxamide derivatives as effective FLT3 and protein kinase inhibitors for AML.22 Inhibitory activity against CDK4/2 and FLT3 was discovered in SAR studies. The combination of piperazine in the hydrophilic pocket, an aromatic linker benzene ring, and a fused bulkier ring in the deep hydrophobic pocket significantly increased anti-proliferative activity against MV4-11 cell line. The compound 2a (Fig. 5) depicted remarkable anti-proliferative bio-efficacy or activities against 60 human cell lines with GI50 < 1 μM for most cell lines. Furthermore, compound 2a exhibited substantial inhibition of CDK4, CDK6, and FLT3 with IC50 values of 0.77 nM, 0.59 nM, and 0.089 nM, respectively, almost 10 times efficacious than FN-1501. Their western blotting analysis report of compound 2a in MV4-11 cell line showed remarkable abolition of phosphorylation of the FLT3 at 1 μM and also inhibited the pRb phosphorylation, in a dose-dependent manner.
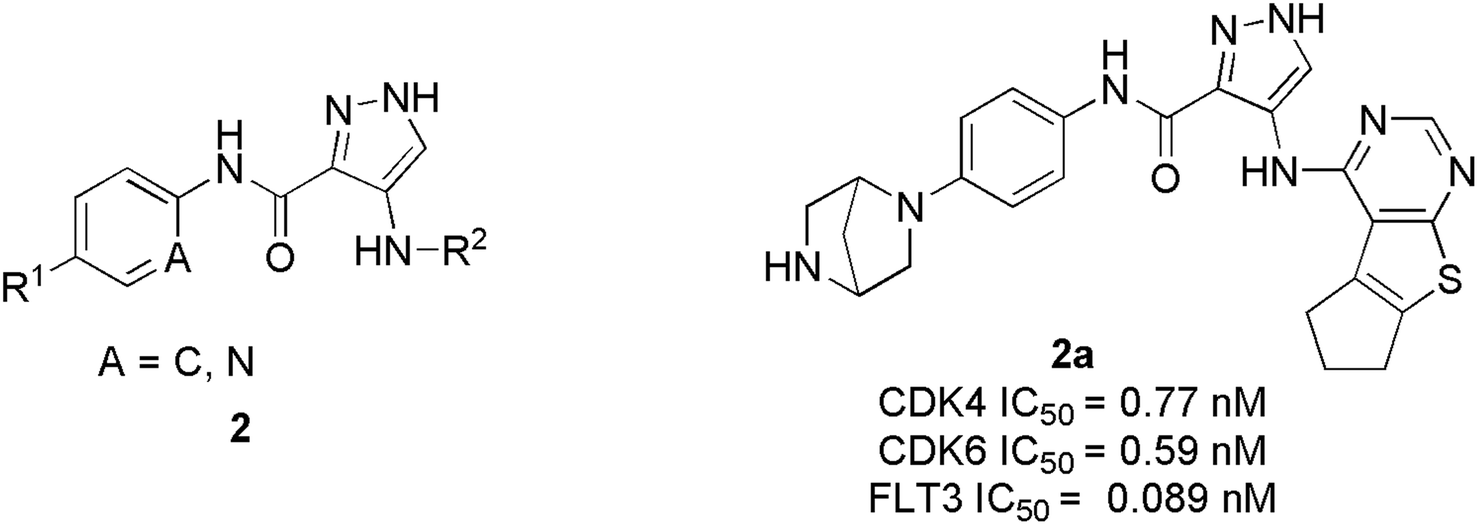 | ||
| Fig. 5 1H-Pyrazole-3-carboxamide derivatives as potent FLT3, CDK4, and CDK6 inhibitors. | ||
3.2 Pyrimidine based analogues
Tadesse et al. synthesized a new series of 4-thiazol-N-(pyridin-2-yl)pyrimidin-2-amine derivatives. Among derivatives synthesized, compound 3a (Fig. 6) was seen to be a highly selective, potent, and orally bioavailable CDK4/6 inhibitor with an IC50 value of 30 nM and 4 nM.23 It also displayed non-proliferation of seven cell lines (M249, PC-3, M229, MOLM13, A2780, NB4, and M249R) with 50% of growth inhibitory (GI50) potency varying from 0.038 μM to 0.681 μM. In the western blot analysis study, compound 3a dose-dependently inhibited pRb phosphorylation at specific amino acid residue Ser780 in MV4-11 cell line treatment at 12 h and 24 h. In an animal model, compound 3a demonstrated a targeted mechanism of action for cellular CDK4/6, appreciable pharmacokinetics, and substantial anticancer efficacy. The SAR analysis shows that substituting a cyclopentyl group at the amino position of the thiazole moiety yielded the optimum combination of enzymatic and cellular potencies, as well as CDK4/6 selectivity.23 The off-target correlation was established when the cyclopentyl group was replaced with an aromatic phenyl group or isopropyl group, resulting in a reduction in both CDK4/6 inhibitory and antiproliferative activities.23 When the methyl group is substituted with electron-withdrawing trifluoromethyl, the toxicity profile increases, resulting in decreased CDK4/6 selectivity. The substitution of the second nitrogen atom of the piperazine with oxygen increase CDK4/6 selectivity where as carbon and the exocyclic primary amino decreases activity and selectivity. Over all, SAR studies indicated that the cyclopentylamino and methyl group at C2 and C4 of the thiazole ring, and morpholine group on the pyridine ring are all essential for CDK4/6 selectivity over other CDKs and optimal cellular activity.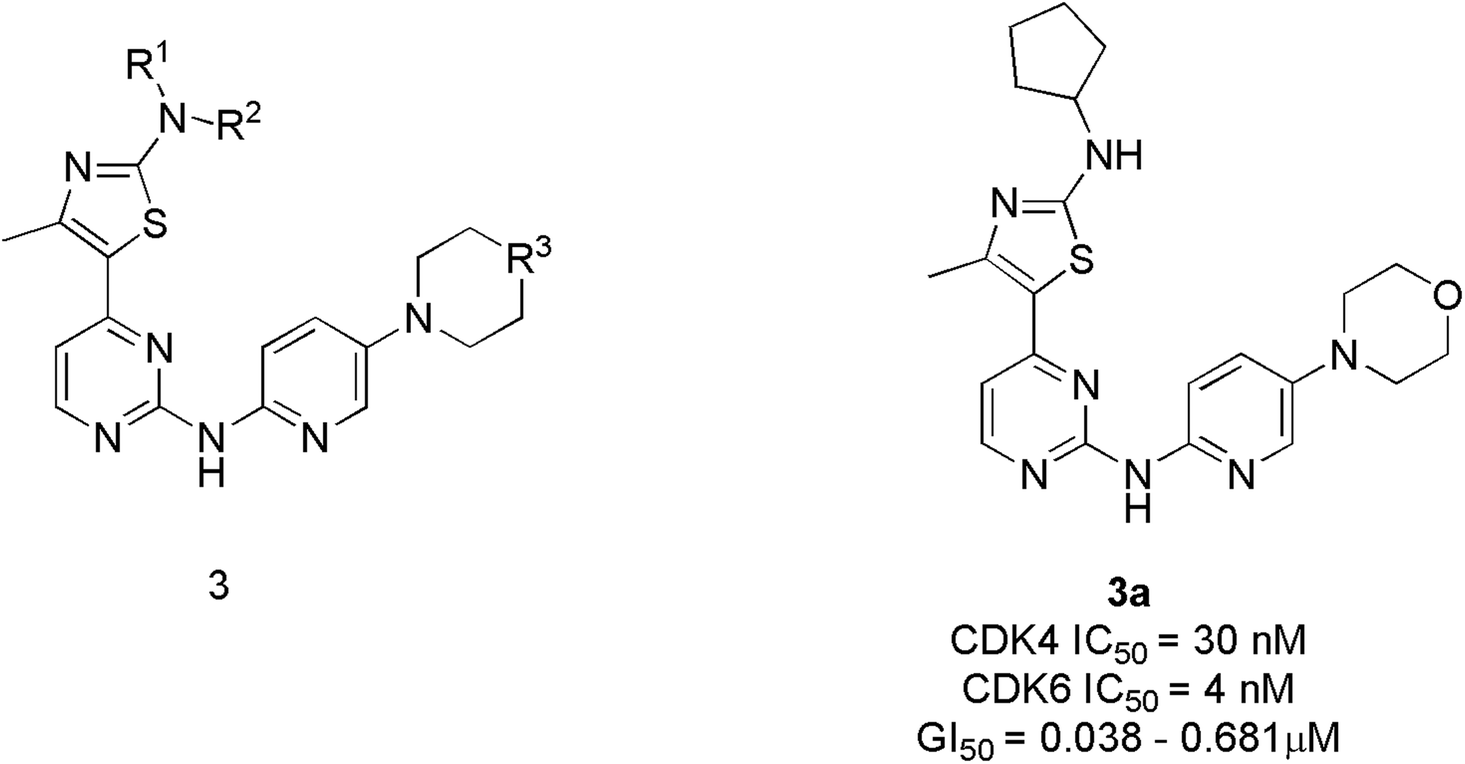 | ||
| Fig. 6 4-Thiazol-N-(pyridin-2-yl)pyrimidin-2-amine derivatives as selective CDK4/6 inhibitors. | ||
Abbas et al. proposed another novel series of pyrido[2,3-d]pyrimidine derivatives as apoptosis inducers and CDK inhibitors.24 From the series, compounds 4a and 4b (Fig. 7) were the most potent direct CDK6 inhibitors with an IC50 value of 115.38 nM and 726.25 nM, respectively. Apoptotic assay of compound 4a in PC-3 cells and compound 4b in MCF-7 cells revealed that they induced apoptosis by 1.9-fold and 1.8-fold, respectively, relative to control. The compound 4a acts as an apoptotic inducer mainly via activation of caspase-3. While compound 4b induces apoptosis mainly through the intrinsic pathway and to a less extent, direct inhibition of CDK6. Furthermore, compounds 4a and 4b cause G1 cell cycle arrest by enhancing the intrinsic pathway of apoptosis via increasing the levels of Bax and p53, and downregulation of Bcl2, and inhibition of CDK4/6. The SAR studies of compound 4a (IC50 = 1.59 μM) revealed that the electronic effect of the substituent for example replacement of chlorine by fluorine (IC50 = 12.2 μM) or 4-methyl (IC50 = 2.8 μM) has no significant effect on the cytotoxic behaviour on MCF-7 breast cancer cell line.24 On the other hand, compound 4b exhibited cytotoxic activity of IC50 = 0.01 μM compared with doxorubicin of IC50 = 0.04 μM and potency of compound 4b is correlated with the presence of electron-donating moieties. Molecular docking analysis of compounds 4a and 4b was also carried out to correlate their ability to interact key amino acids in the binding site with their kinase activity. The compounds 4a and 4b docked with CDK6 kinase with a docking energy score of −12.02 kcal mol−1 and −15.40 kcal mol−1, respectively. The compound 4a forms hydrogen bond interaction with amino acids Val101 and Glu99 by its C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and hydrazine group NH, respectively.24 This hydrogen bonding interaction of compound 4a is correlated with higher CDK6 inhibitory activity compared to compound 4b which failed to bind to the pocket. While its hydrophobicity resulted in its docking score.
O and hydrazine group NH, respectively.24 This hydrogen bonding interaction of compound 4a is correlated with higher CDK6 inhibitory activity compared to compound 4b which failed to bind to the pocket. While its hydrophobicity resulted in its docking score.
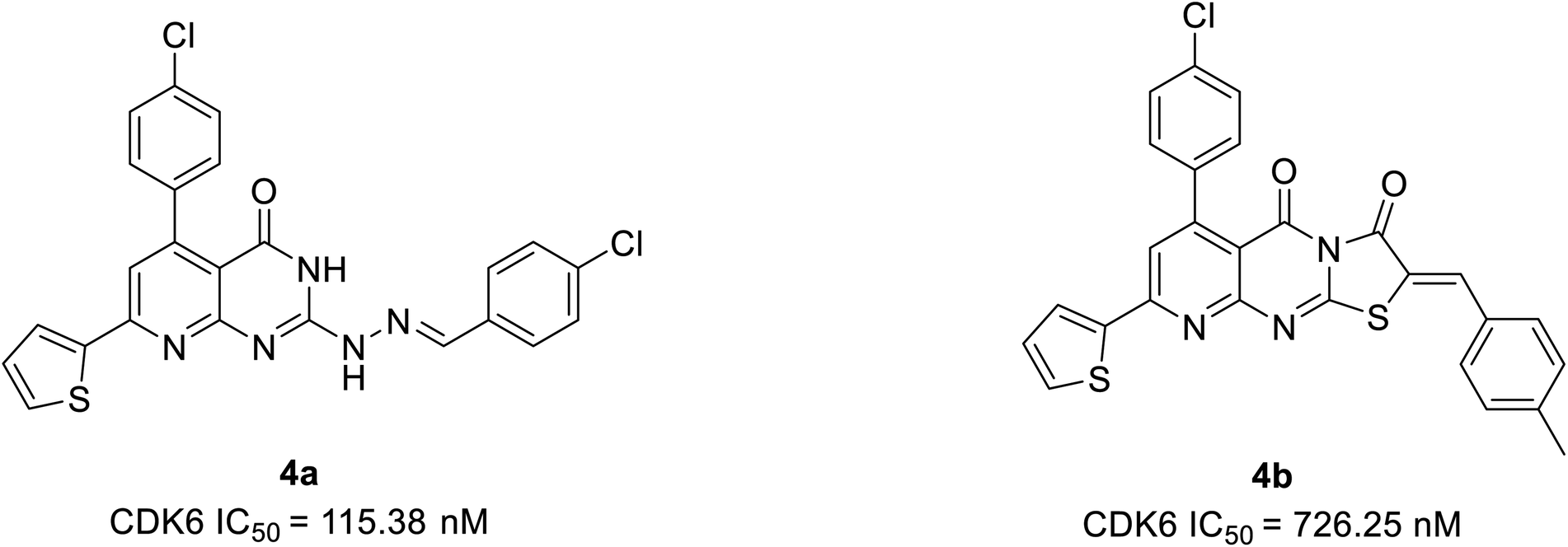 | ||
| Fig. 7 Pyrido[2,3-d]pyrimidine derivatives as potent CDK6 inhibitors and apoptosis inducers. | ||
Shi et al. unveiled a novel class of imidazo[1′,2′:1,6]pyrido(2,3-d)pyrimidine derivatives as CDK4/6 inhibitors.25 Among these derivatives, compounds 5a and 5b (Fig. 8) showed good activities inhibiting CDK4/6 in Colo-205 and U87MG cell lines. Compounds 5a and 5b displayed substantial inhibition of CDK4/D3 (IC50 = 0.8 nM and 2.7 nM, respectively) and CDK6/D3 (IC50 value = 2.0 nM and 4.8 nM, respectively) that are comparable to the IC50 values of palbociclib and abemaciclib. In addition, compounds 5a and 5b showed 23.6-fold and 22.3-fold selectivity against CDK2/CDK4 and slightly higher (29.3-fold and 81.9-fold) selectivity towards CDK4 and CDK9, respectively. The SAR studies reveals the bio-efficacy of the compounds, to explore the pharmacophore of compound 5, the acetyl and isopropyl moieties at C6 and C8 positions (compound 5a) depict 2.5 more potential biological activities for CDK4/cyclinD3 than CDK6/cyclinD3.25 However, the alteration of functional group tert-butyl at C8-substituent (compound 5b) shown to be modest bioactivity for targeting CDK4/6. In Colo-205 and U87MG xenograft models, compounds 5a and 5b displayed favourable antiproliferative properties, outstanding metabolic properties, reasonable pharmacokinetic features, and considerable tumour growth inhibitions with controllable toxicities.
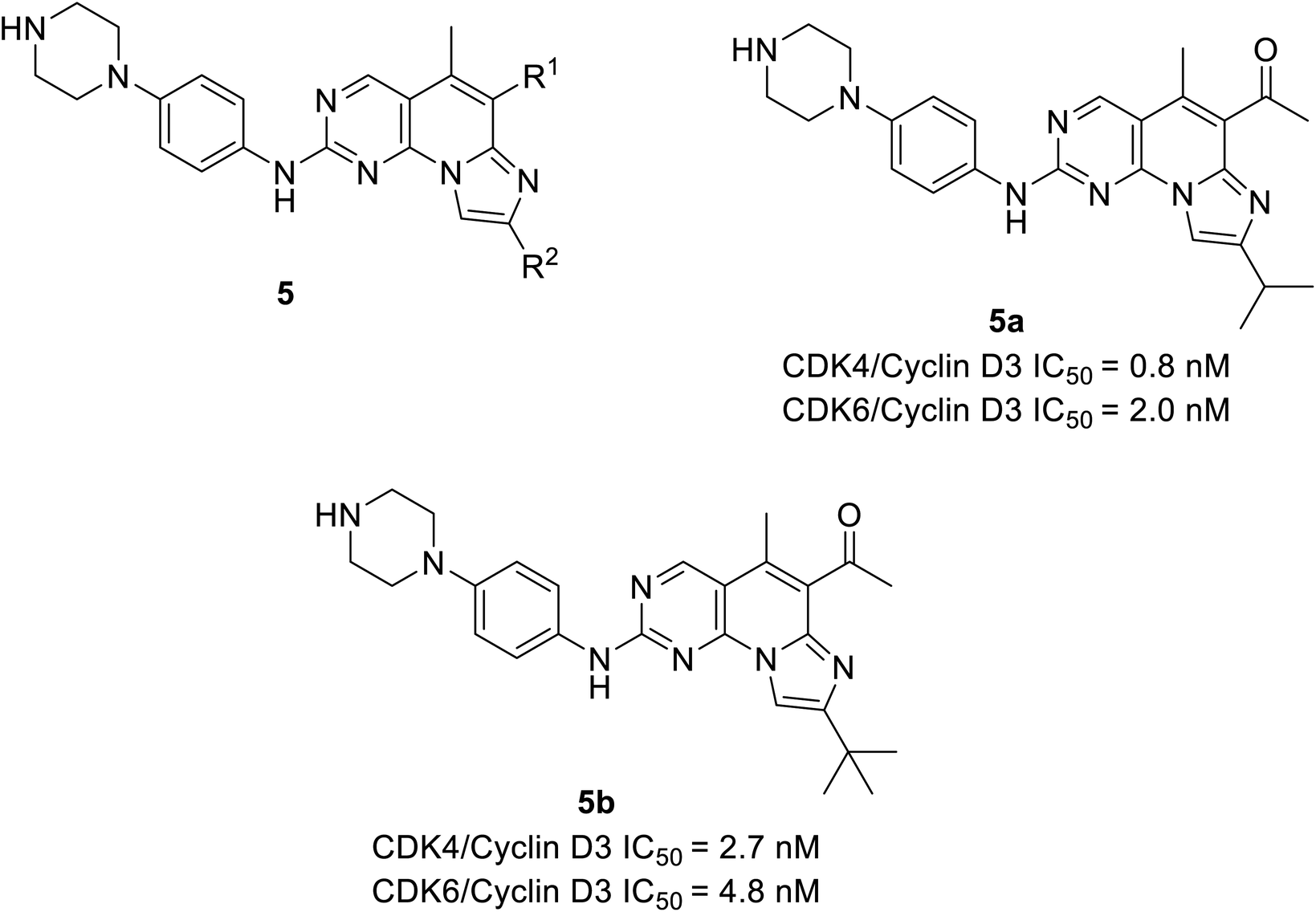 | ||
| Fig. 8 Imidazo[1′,2′:1, 6]pyrido[2,3-d]pyrimidine derivatives as CDK4/6 inhibitors. | ||
3.3 Quinazoline based analogs
Zhao et al. designed and synthesised 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as selective CDK4/6 inhibitors.26 Study investigated compound 6a (Fig. 9) selectively inhibited CDK4/cyclinD3 and CDK6/cyclinD3 with IC50 values of 0.01 μM and 0.026 μM, respectively. Its anti-proliferative activity was higher than that of abemaciclib with an IC50 value of 0.19 μM and 0.13 μM in MCF-7 and HCT116 cell lines, respectively. The SAR studies indicated that the insertion of piperazine at R4 increased selectivity for CDK4 relative to CDK2 by 70-fold, while the exclusion of piperazine decreased potency and selectivity. The presence of piperazine moiety improved pharmacokinetic properties. Although compound 6b (Fig. 9) was more potent than compound 6a in inhibiting CDK4/6 (IC50 values of 0.003 μM and 0.01 μM, respectively), was less selective for CDK4/6 over CDK2.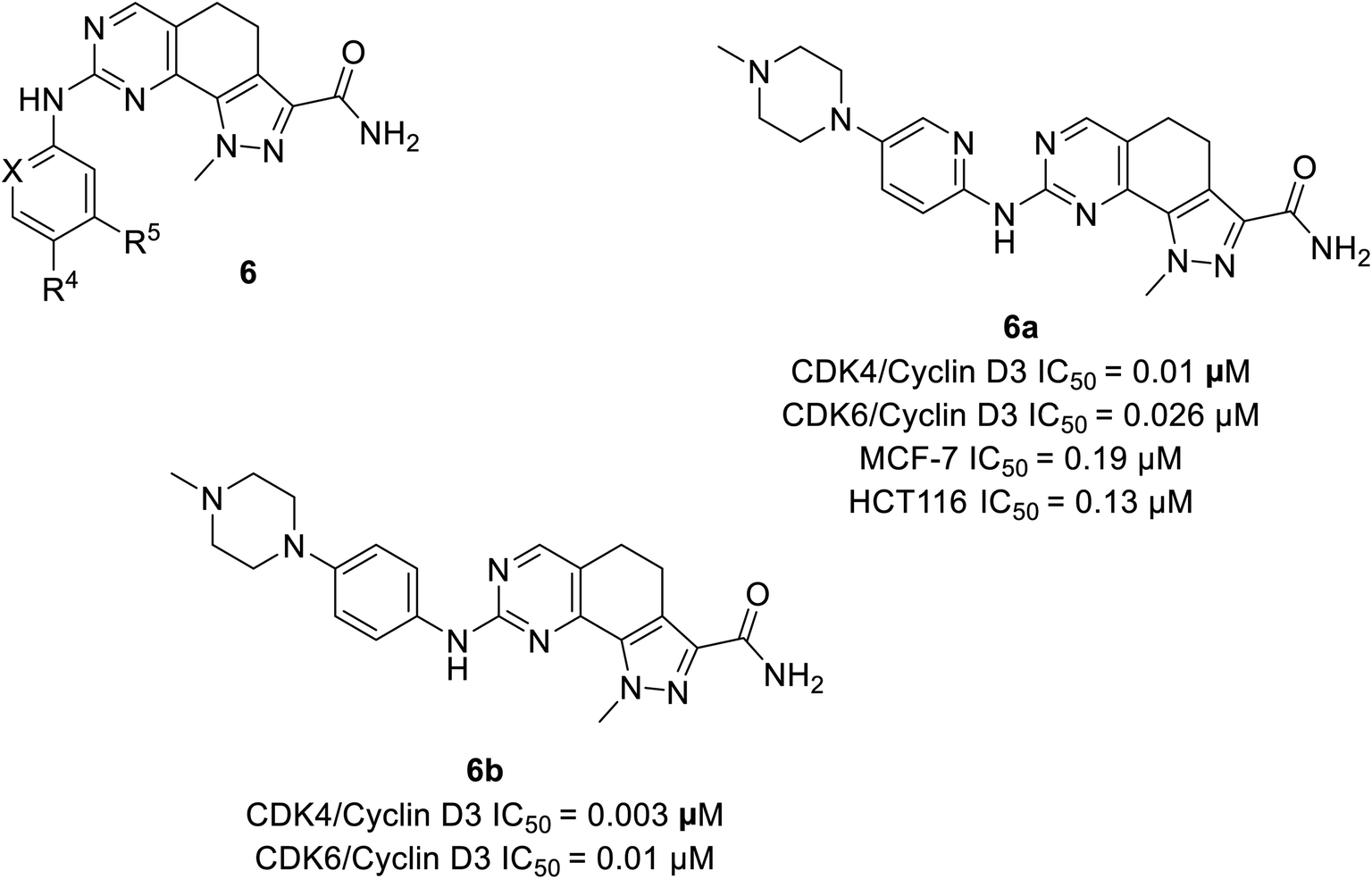 | ||
| Fig. 9 4,5-Dihydro-1H-pyrazolo(4,3-h)quinazoline derivatives as selective CDK4/6 inhibitors. | ||
3.4 Acridone based analogs
Zhang et al. reported the synthesis and the biological activity of methoxybenzyl 5-nitroacridone derivative (7a) (Fig. 10) from an array of novel benzyl acridone derivatives as survivin protein inhibitors for hepatocellular carcinoma treatment.27 In K562 cells, the most potent compound 7a effectively activates G1 phase cell cycle arrest by inhibiting CDK4/6-mediated Rb gene phosphorylation on a nanomolar scale. Furthermore, some compounds showed marked cytotoxicity with IC50 values ranging from 2.92 to 6.84 μM and ≤4 μM on MCF-7 and HepG2 cell lines, respectively. The MTT assay indicated robust anti-proliferative activity against drug-resistant leukaemia cells (K562/ADR cells) and KCL-22 cells. The docking simulations of compound 7a in the active site of CDK4 expressed good complementary interaction involving the significant residues Val196, Asp158, Lys22, Glu144, and Asn145 to form H-bonds.27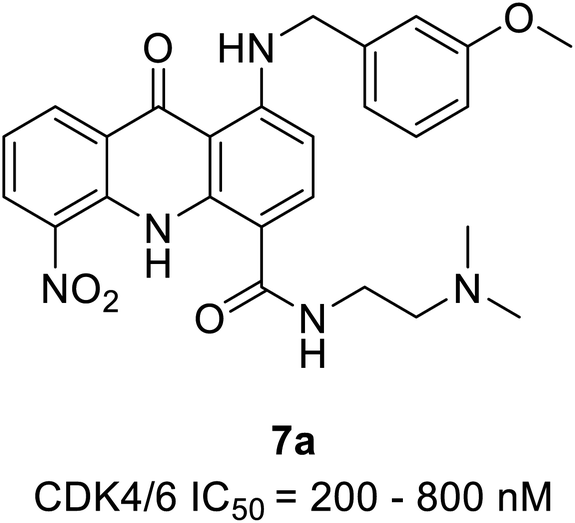 | ||
| Fig. 10 Methoxybenzyl 5-nitroacridone derivative as surviving and CDK4/6 inhibitors. | ||
3.5 Indole conjugates analogues
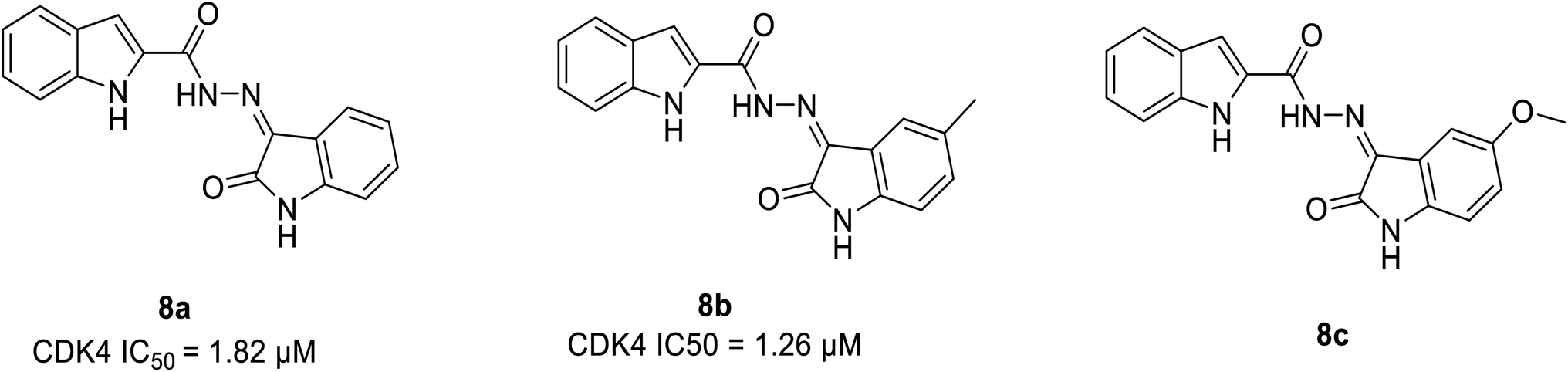
Ayyad et al. developed two series of carbocycle–indole conjugates and oxindole–indole conjugates as effective antitumour agents with potential inhibitory activity against CDK4.28 The most potent conjugates 8a and 8b (Fig. 11) exhibited significant CDK4 inhibitory activity of 92% and 93% having IC50 values of 1.82 μM and 1.26 μM respectively. Compound 8a displayed 78% inhibitory activity against CDK2 depicting the dual inhibitor property on targeting tumor cells. The potent biological activity of conjugates 8b and 8c on the expression levels of the distinct apoptotic markers (Bax, p53 and caspase-3) and the anti-apoptotic marker B-cell lymphoma-2 lead to anti-proliferation activity by accelerating the cell apoptosis process. Similarly, it enhances pro-apoptotic Bax protein by decreasing the expression level of anti-apoptotic Bcl-2 protein by 2.12 folds and 1.7 folds respectively. In the SAR study, the incorporation of an electron-donating substituent on the oxindole ring at 5th position (compound 8a & 8b) increased CDK4 inhibitory activity. Electron-withdrawing substituents such as fluorine, bromine, methoxy, and nitro group, on the other hand, reduced CDK4 inhibitory activity.28 Besides, the molecular docking of CDK4 with oxindole conjugates containing N-substituted derivatives shows similar binding patterns and bonding of bound oxindole ligand. Moreover, it reveals that significant functional moieties (NH) and (CO) are required to form hydrogen bonding interaction with the key amino acids Glu94 and Val96 that present at the active site of the protein. Also, the indole structure is securely situated in a hydrophobic sub-pocket, forming a hydrophobic interaction with the hydrophobic side chains of amino acids Ile12, Val20, and Gln98 which line this sub-pocket. Overall, these findings provided lead hybrid compounds 8a and 8b with good CDK inhibition efficacy, and further these agents could be optimised to identify potential candidate.28
 | ||
| Fig. 11 Oxindole–indole conjugates and carbocycle–indole conjugates as CDK4/6 selective inhibitors. | ||
4. Correlation of activity with computational studies
Cyclin dependent kinases 4 and 6 plays chief role in controlling G1 phase of cell cycle and Rb/E2F transcription with cyclin D. CDK4 and 6 is located in the 12q14.1 and 7q21.2 location of the chromosome involved in many types of cancer including melanoma, stomach cancer, breast cancer, lung cancer, osteosarcoma, bladder cancer, skin cancer.29 There are many drugs which are in approved and few in clinical trials (Table 2) as CDK4/6 inhibitors but met with major limitations with regard to their pharmacokinetic and pharmacodynamics properties.30 Thus, the new pharmacological efficacious inhibitors are required to enhance the bioavailability and to treat disease adverse effects. The synthesis of a range of analogues is described in this study was already evaluated for their inhibitory activity. The pairwise sequence alignment of CDK4 with CDK6 shows sequence identity of 71.6% and similarity of 86% (Fig. 12A). The active site residues of CDK4/6 were depicted in the figure is considered to be crucial for compound binding and interaction. However, 70–80% amino acid residues are highly conserved in the alignment and superimposition of these structure shows <3.1 Å (Fig. 12E) RMSD.| Drug | Combination | Conditions/disease | Clinical phase | Status | Information |
|---|---|---|---|---|---|
| a https://www.ClinicalTrials.gov. | |||||
| Palbociclib | Binimetinib (MEK162) | Advanced KRAS-mutant NSCLC | Phase 1/2 | Active | Possible treatment for lung cancer with a specific change in the KRAS gene |
| Palbociclib | Letrozole | Advanced, ER+, HER2− breast cancer | Phase 2 | Completed | The combination markedly improved PFS in women |
| Palbociclib | Fulvestrant | HR+/HER2− LABC or MBC metastatic, locally advanced breast cancer | Phase 2 | Recruiting | Aim to determine the safety and efficacy of the combination after the failure of a combined treatment of hormonal therapy plus CDK4/6 inhibitors |
| CDK4/6 inhibitors | Non-steroidal aromatase inhibitor (first line) | Breast neoplasm female | Phase 3 | Recruiting | This study aims to see if an aromatase inhibitor plus CDK4/6 in the first line and fulvestrant in the second line is better than an aromatase inhibitor in the first line and fulvestrant plus CDK4/6 in the second line |
| Fulvestrant (second line) | |||||
| Trilaciclib | Gemcitabine and carboplatin | Metastatic triple-negative cancer | Phase 2 | Completed | Assessed the myelopreservation effects of T when combined with GC |
| SHR6390 | Pyrotinib | HER2+ metastatic breast cancer | Phase 2 | Recruiting | To evaluate the efficacy, safety, and tolerability of the combination |
| Lerociclib | Osimertinib | EGFRmut non-small cell lung cancer | Phase 1b | Completed | No clinically relevant drug–drug interaction was observed, well tolerated with only one DLT event to date |
| Ribociclib | Exemestane and everolimus | Men and postmenopausal women with HR+, HER2− ABC refractory to ≥1 line of ET | Phase 1/2 | Completed | An evaluated maximum tolerated dose of triplet combination and clinical benefit rate with centrally-accessed PFS. A preliminary clinical study suggested that this combination can restore sensitivity to inhibitor-based therapy with CDK4/6 |
| Abemaciclib | Anastrozole or letrozole | Breast cancer | Active | To evaluate the nonsteroidal aromatase inhibitors (NSAI) plus abemaciclib combination effectiveness | |
| Ribociclib | Endocrine therapy | Breast cancer | Phase 2 | Active | To evaluate the safety and efficacy of a drug combination as a potential ER+ breast cancer therapy |
| Palbociclib | Fulvestrant and erdafitinib | ER+/HER2−/FGFR-amplified metastatic breast cancer | Phase 1b | Recruiting | To evaluate the safety and tolerability and preliminary anti-tumour activity |
| Ribociclib | Bicalutamide | AR+ triple-negative breast cancer | Phase 1/2 | Active | Safety and investigational study |
| Palbociclib | Gedatolisib | Lung cancer, solid tumors, squamous cell, pancreatic cancer, head & neck cancer | Phase 1 | Recruiting | A combination of drugs as a potential therapy for cancer could have a significant alteration in the PI3K pathway. To study the safety of an investigational drug combination and explore optimal dose(s) for future trials |
| Abemaciclib | Hydroxychloroquine | Breast cancer | Phase 2 | Not yet recruiting | Analyse the combination of drugs to target the disseminated tumour cells in the bone marrow |
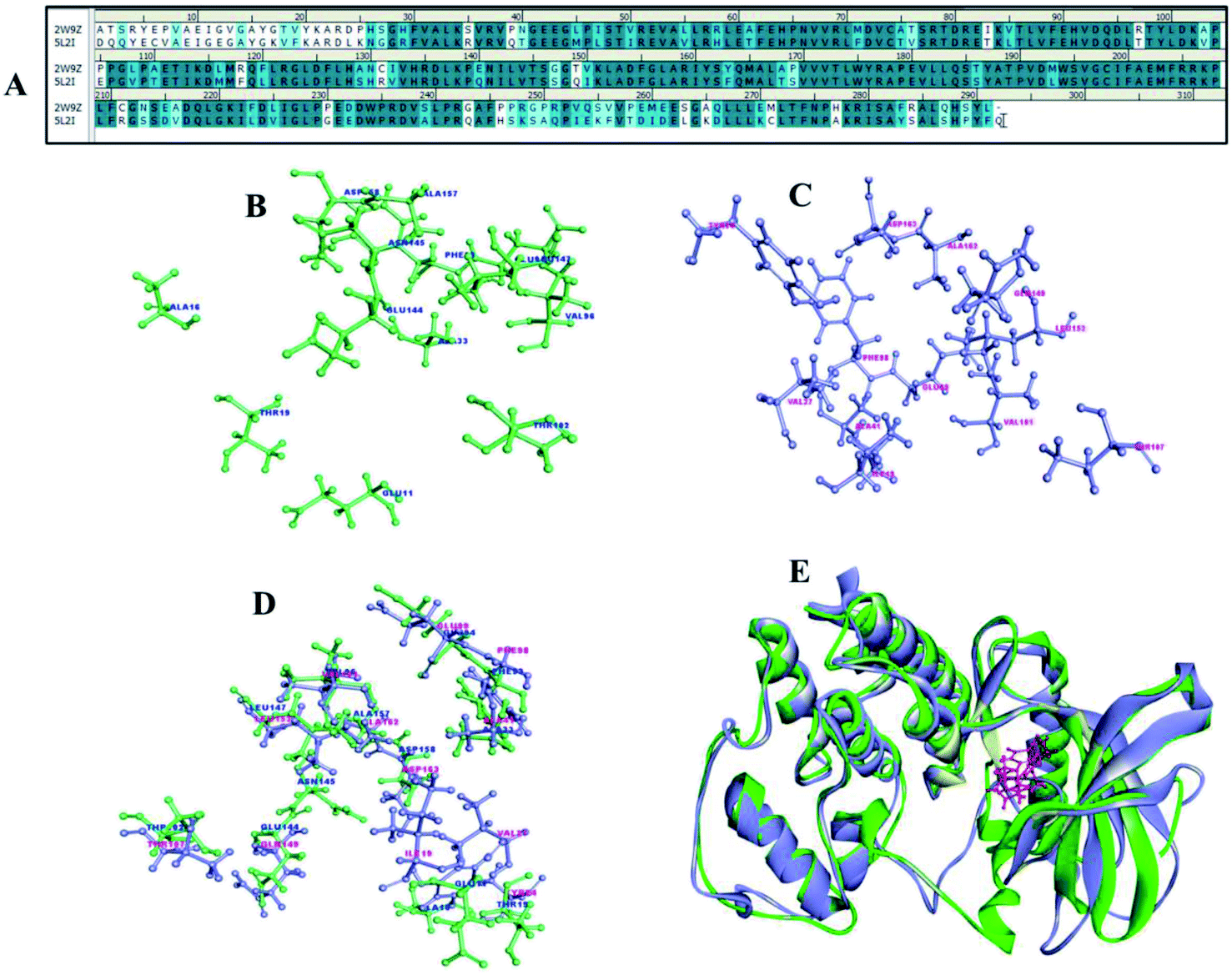 | ||
| Fig. 12 (A) Sequence alignment of CDK4 (2W9Z) and CDK6 (5L2I). (B and C) Active site amino acid residues of CDK4 and CDK6. (D) Overlay representation of CDK4 and CDK6 active site residues. (E) 3D superimposed representation of CDK4 and CDK6. | ||
To concert the importance of pharmacophore of compounds structure-based drug designing approach was implemented to understand the importance of atom–atom interaction. The compounds 1a to 8b were collected to perform molecular docking, non-bonded interaction and free binding energies using PBSA method. The docking was carried out for CDK 4 and CDK6 X-ray crystallography structures, there are totally 9 and 18 resolved structures for CDK4 and CDK6 without and with bound ligands for CDK's. The receptor–ligand interaction analysis of these compounds shows both hydrogen and hydrophobic amino acid interaction. Among 12 compounds docked, at least one of the poses tends to form hydrogen bond interaction with Val101 and Asp163 crucial amino acids in the binding site of CDK6 (Fig. 12C). Similarly, Val 96 and Asp 158 are key amino acid are considered as important amino acid (Fig. 12B) (1) and superimposition of both CDK4 and 6 is shown in the Fig. 12D. The docked results of compound show good binding interaction to form H-bond with CDK4. Fig. 13 depict the interaction compound 5b one of the pyrimidine-based analogues shows favourable hydrogen and hydrophobic interaction with CDK4/6 drug targets. The highest CDocker interaction energy and binding energy was inferred for the compounds IC50 values in nanomolar (Table 1). This implicates the relationship between interaction and biological activity.
 | ||
| Fig. 13 (A and C) shows 2D representation of 5b compound interaction with CDK4 and CDK6. (B and D) is the 3D depict of 5b compound receptor–ligand interaction with CDK4 and CDK6. | ||
5. Cyclin D–CDK4/6-related cancer chemotherapy resistance
Patients with breast cancer gain resistance to CDK4/6 inhibitors after long-term exposure. As a result, the resistance mechanisms involved are extensively studied and the outcome of research on corresponding mechanism aid strategies to counter the development of therapeutic resistance to these agents.5.1 Overexpression of CDK6 and related subunit
Overexpression of CDK6, which is accomplished through the repression of the transforming growth factor (TGF-β) pathway mediated by microRNA (miRNA) expression, is a common cause of acquired resistance to CDK4/6 inhibitors.31 Moreover, CDK6 amplification results in a dramatic rise in its expression and the declined CDK4/6 target sensitivity, pRb to CDK4/6 inhibitors.Yang et al. investigated the potential mechanisms responsible for resistance to CDK4/6 kinase inhibitor in estrogen receptor-positive (ER+) breast cancer using cell line models.32 Their findings suggested that persistent Rb phosphorylation occurs irrespective of high potent CDK4/6 inhibitors in MCF-7 resistant cells (MR). The observed impaired sensitivity to CDK4/6 inhibitors was directly linked to CDK6 mRNA overexpression in MR cells, i.e., a seven-fold increase as compared to parental cells. Their findings also showed that CDK6 overexpression occurs after prolonged exposure to CDK4/6 inhibitors, and that restoring CDK6 expression to levels comparable to those seen in parental cells restored MR cells' sensitivity to LY5219. The acquired CDK6 resistance further promotes hormone resistance and reduces ER/PR expression. The investigators also correlated the inactivity of CDK4/6 with Rb mutation and cyclin E overexpression.32
Genomic analysis of 348 ER+ breast cancer patients treated with CDK4/6 inhibitors in resistant ER+/HER2-breast cancers, Li et al.33 studied loss of FAT1, a tumour suppressor that represses the hippo pathway, led to increased expression of CDK6, which was suppressed, restoring sensitivity to CDK4/6 inhibitors. The hippo pathway that mediates the induction of CDK6 and genomic alterations of other components of the pathway are also found to promote CDK inhibitor resistance. The study suggests that more potent CDK6 inhibitors or dual enzyme inhibitors could be useful strategies to counteract drug resistance because the potential common mechanism of the cell differentiation and resistance of cancer cells to CDK4/6 inhibitors is by the repression of the hippo signalling pathway.
5.2 Loss of pRb and an acquired mutation in the RB1 gene
The involvement of pRb has shown a profound impact on the acute response to palbociclib in the breast cancer cell line, where primary resistance occurs by loss of Rb activity on extended CDK4/6 inhibition.34 Nevertheless, in the absence of Rb, the cell cycle arrest tended to be intrinsic variations in cell-specific and largely correlated with continued inhibition of downstream target genes. For example, in Rb-deficient cases, p107 protein accumulation was observed specifically and associated with E2F-regulated protein repression in the presence of palbociclib.35Early adaptation to CDK4/6 inhibitors restricted the ability of ER+ breast cancer cells to induce complete and stable cell-cycle arrest. The mechanism of acquired resistance to CDK4/6 inhibition in ER+ breast cancers was elucidated by Herrera-Abreu and colleagues,36 who showed that palbociclib-treated breast cancer cell lines T47D that acquired resistance lost pRb expression.
The researchers used a patient-derived tumour xenograft (PDX) made from harvested ER+ metastatic tissue to further investigate an existing acquired resistance to ribociclib (LEE011) and found that the RB1 acquired mutation was acquired through the acquisition of the Rb1 frameshift mutation, which was present at a sub-clonal stage. As a result, inhibiting CDK4/6 contributed to the selection of the RB1 mutant population facilitated the acquisition of palbociclib resistance in ER+ breast cancer.
A retrospective analysis of the pre-treated mutational profile of metastatic breast cancers via whole-exome sequencing demonstrated rare RB1 mutation.37 In the post-treatment ctDNA study, several RB1 mutations were observed, possibly occurring under selective pressure from the CDK4/6 inhibitor that theoretically confers resistance to therapy.38 The latest study by Wander and colleagues of the whole-exome sequence of 59 tumors exposed to CDK4/6 inhibitors demonstrated many candidate resistance mechanisms, including RB1 loss.39
5.3 Cyclin E1 overexpression
Overexpression of cyclin E has been linked to the development or poor prognosis of various human cancers in various studies over the years. It is noted that dysregulated cyclin E has a definite effect on cell cycle regulation and mediated acquired resistance in many tumour cells. For example, through the gain of cyclin E1 amplification, palbociclib acquired resistance and poor prognosis in HR+ breast cancer patients.36,40 The protein expression of cyclin E, for instance, determined the antiproliferative effect of specific inhibitors of CDK4/6. A cohort of 34 HER2+ patients treated with trastuzumab-based therapy analysed by Cortés et al. observed that cyclin E overexpression related to poor clinical benefits along with poor progression-free survival (PFS).41 Cyclin E overexpression promoted trastuzumab resistance both in vitro and in vivo. Knockdown of cyclin E overexpression or treatment with CDK2 inhibitors which resensitized to CDK4/6 inhibitors,36 led to a drastic decrease in proliferation and enhanced apoptosis, thus supporting the concept of cyclin E as a dominant oncoprotein. In a study by Fox et al., phosphorylation of Rb by cyclin E-associated kinase in the absence of cyclin D1-CDK4/6 complexes in tumour cells provided another mechanism that functionally inactivates pRb and reducing the effectiveness of CDK4/6 inhibitors.42The overexpression of cyclin E has also been observed in other types of solid tumors as well as leukaemia. Fingert et al. showed severe quantitative and qualitative alteration in cyclin E protein production by western blot analysis of tissue samples and a blood sample from 42 patients with various malignancies.43 Besides, the alteration of cyclin E expression, particularly in breast cancer is associated with high tumour stage and grade and therefore poor prognosis, hence suggesting its potential use as a prognostic marker for breast cancer.
5.4 Increased lysosomal biomass
CDK4/6 inhibitors failed to treat triple-negative breast cancer after being approved for HR+ breast cancer treatment (TNBCs). Increased lysosomal biogenesis was discovered in a subset of TNBC cell lines, and the resulting lysosomal sequestration of CDK4/6 inhibitors underpins these cells' resistance to these compounds.44 By preventing lysosomal sequestration with lysosomotropic, new CDK4/6 inhibitors have been developed that are effective against resistant TNBC. Eischer et al. reported that the development of lung cancer in genetically engineered mice needs the AMPK. Their results indicated that the AMPK-mediated lysosome biogenesis during the early stages of tumorigenesis promoted cancer cell vitality.45Acidosis-mediated protease activation or release in cancer cell invasion is likely to be mediated by protease-sequestering lysosomes.46–48 Glunde and colleagues investigated that at low extracellular pH, lysosomal burden and diameter decreased in human mammary epithelial cells and poorly metastatic breast cancer cells, but increased in extremely metastatic breast cancer cells.49 Filopodia formation was caused by an acidic pH in highly metastatic breast cancer cells, which could be related to their invasive behaviour. This suggests tumorigenesis and drug resistance are closely associated with the autophagy-lysosomal pathway. An in-depth study of the relationship between tumour drug resistance and the autophagy-lysosomal pathway is significant for the suppression of tumour proliferation.
5.5 Loss of PTEN and increased AKT activation
Earlier studies have shown that loss of PTEN impart resistance to HER2-directed therapies and is associated with the worst outcome in HER2-amplified breast cancer patients.50–52 PTEN is reported, along with other genes (TP53, GATA3, PIK3CA, ESR1, CDH1, MAP3K1, AKT1, MAP2K4, RB1, MAP2K4, CDKN2A, and CBFB), substantially mutated in metastatic breast cancer.37 Costal and colleagues recently investigated that in breast cancer, lack of PTEN mediates therapeutic cross-resistance to CDK4/6 inhibitors.53 They found that in ER+ mBC, both PTEN and Rb loss are resistance pathways to CDK4/6 inhibitors. And their result further reinforced the idea that PTEN absence facilitated the CDK4/cyclin D2 complex's formation and/or stabilization. Gene-editing methods were used to establish an isogenic ER+ breast cancer model to explain how PTEN loss promoted resistance. Overall, their findings showed that loss of PTEN promotes p27 delocalization outside the nucleus by raising AKT activity, which in turn triggers increased CDK4/6 and CDK2 activity, allowing them to overcome the blockade of CDK4/6 inhibitor-induced G1. Also, by activating signalling cascades that induce hyper-activation of CDKs, PTEN deficiency induces reduced immunity to clinically significant doses of CDK4/6 inhibitors.Y. Lu et al. findings are consistent with previous research on PTEN's role in tumour suppression. PTEN overexpression resulted in Akt inactivation and induction of anoikis, a form of apoptosis triggered by cell detachment from the extracellular matrix while restoring PTEN activity resulted in early G1 arrest followed by apoptosis in breast cancer cells.54
6. Strategy to control drug resistance
At present, the number of reports of resistance to approved CDK4/6 inhibitors has steadily increased as a result of compensatory regulation of other corresponding biochemical signalling pathways discussed in the preceding section. Consequently, the selective targeting of one target or signalling pathway may not be beneficial to inhibit the growth of cancerous cells. Considering the role of CDK4/6 inhibitors in cancer treatment, we discuss a few strategies to counteract the resistance.6.1 Major combination therapy with CDK4/6 inhibitors to prevail over resistance
In-depth research has been carried out to develop therapeutic strategies to resolve CDK4/6 inhibitor resistance in ER+/HER2-breast cancer. The multiple clinical/preclinical studies revealed the CDK4/6 inhibitors combination with certain selective inhibitors that overcome acquired resistance to CDK4/6 inhibition and observed synergistic effect. Acquired resistance as a result of CDK4/6 inhibitor monotherapy is associated with a lack of Rb dependency and PI3K/mTOR signalling activation. In ER+/HER2-breast cancer, alpelisib, a p110-selective PI3K inhibitor that blocks PI3K/mTOR signalling, could resolve acquired resistance to CDK4/6-ER-based therapies.55 The CDK4/6:ER:PI3K triple combination therapy prevents the development of resistance to endocrine therapy plus CDK4/6 inhibition. After CDK4/6 inhibition, compensatory induction of cyclin D1 sequestered endogenous p21 and p27 CDK inhibitors. The important role of p21 is to inhibit CDK1/2 activities, while the cyclin E–CDK2 complex, like cyclin D–CDK4/6, can drive cell cycle progression to CDK4/6 inhibitors resistance by phosphorylating Rb and releasing E2F transcriptional activity. Therefore, this sequestration enables CDK2 activity and facilitates CDK4/6 inhibitors resistance.56 Notably, dual inhibition of CDK2 and CDK4/6 produce the synergistic anti-proliferative effect. For instance, cyclin E–CDK2 mediated phosphorylation of c-Myc gene is responsible for CDK4/6 inhibitor resistance by suppressing c-Myc induced senescence. On the contrary, the use of a combination of CDK2 and CDK4/6 inhibitors overcome acquired resistance to palbociclib in MCF27 and MCF7-PR cells with IC50 of 12.5 nM and 11 to 16 nM, respectively.40 MDM2 antagonists suppress CDK4/6 inhibitor resistance by inducing p21 by stabilising the transcriptional activator p53.56Though the inhibition of CDK4/6 arrests the cell cycle, other CDKs compensate and promote the progression of the cell cycle. In a recent cancer research study, selective inhibition of CDK7 with THZ1 and SY-1365 was shown to overcome resistance to CDK4/6 inhibitors in HR+ breast cancer cells in a dose-dependent manner.57
The fibroblast growth factor receptor (FGFR) is often mutated in breast cancers, and overexpression and/or hyperactivation is related to CDK4/6 inhibitor resistance and shorter PFS. The FGFR is found to play a key role in several aspects of cancer development, including cell proliferation, differentiation, and growth. Hence, the FGFR pathway can facilitate tumour progression in CDK4/6 therapy. As a result, it's important to remember that inhibiting aberrant FGFR activity will reverse CDK4/6 inhibitor resistance. In preclinical studies of FGFR1-amplified ER+ breast cancer cells, complete regression of tumors was attributed to a triple combination of ER:CDK4/6:FGFR1 antagonists.58 Thus, a promising therapeutic strategy would be the addition of FGFR inhibitors for delaying or preventing tumour progression in CDK4/6 inhibitors-resistant breast cancer.
The CDK4/6 inhibition-induced feedback regulation of cyclin D1, CDK4, and cyclin E1 lead to the selection of drug-resistant variants in both ER+ and KRAS-mutant NSCLC cells following extended drug therapy. Eukaryotic initiation factor (eIF) 4A inhibitors such as rocaglates suppress this cell-cycle feedback response and synergize with CDK4/6 inhibitors against ER+ breast cancer cells.59 Thus, eIF4A inhibitor can be a potential novel strategy to overcome resistance to CDK4/6 inhibition in cancer.
In glioblastoma (GBM), CDK4/6 inhibition stimulates the NF-kB-mediated TrkA-B and C-Met pathways associated with the clinical resistance to CDK4/6 inhibition of glioblastoma. Therefore, simultaneous inhibition of Trk/C-Met with CDK4/6 has the ability in resolving resistance to cyclin-CDK4/6 inhibitors and demonstrates significant synergy with glioblastoma in combination therapy.60
Many clinical trials are undertaken by various pharma companies incorporating the use of cyclin-CDK4/6 inhibitors in combination therapy (Table 2). Studies are underway to determine their efficacy in other cancers, including mantle cell lymphoma (MCL), non-small-cell lung carcinoma (NSCLC), squamous cell head and neck carcinoma, multiple myeloma (MM), and prostate cancer.
6.2 Proteolysis targeting chimera (PROTAC)
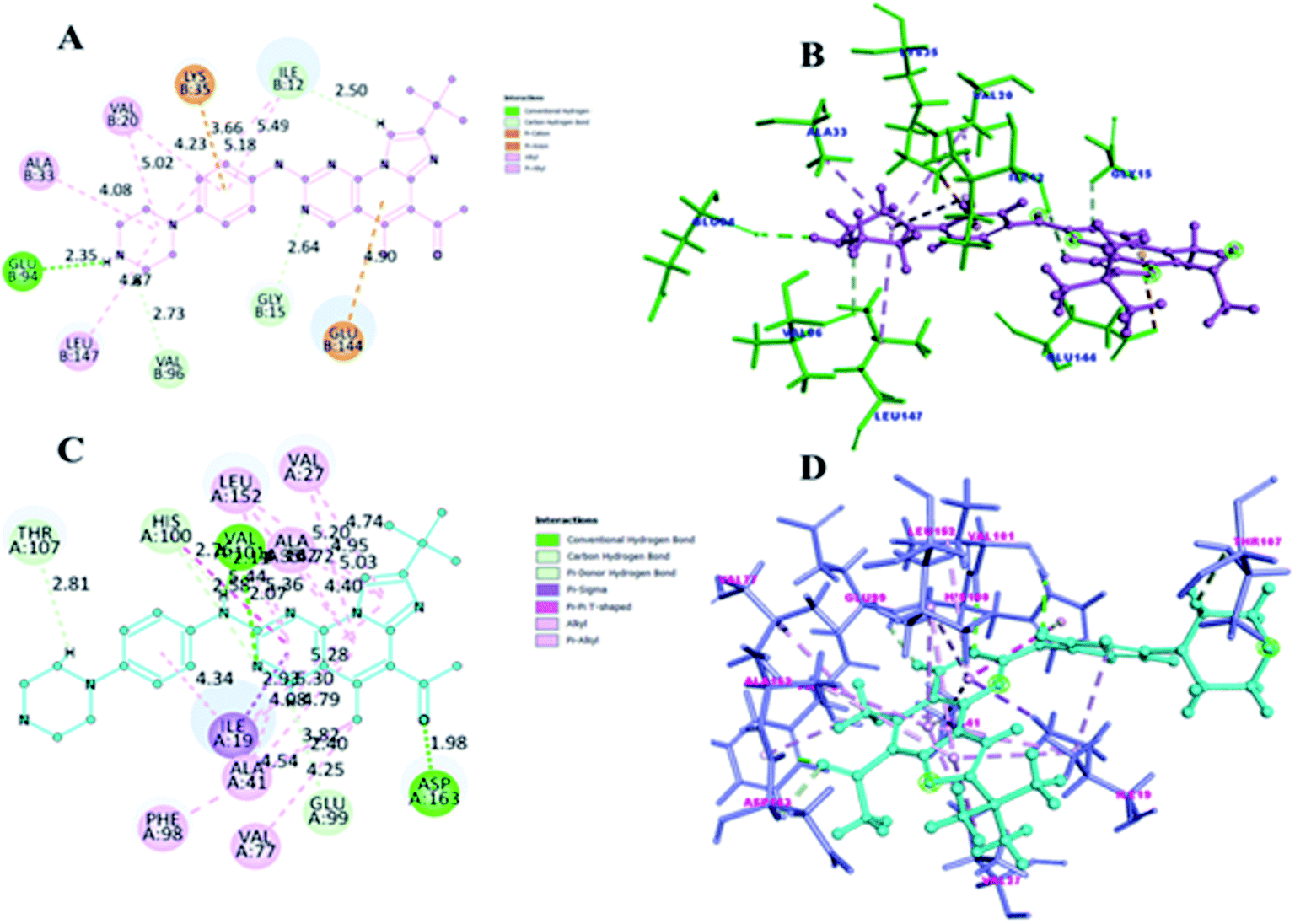
CDK4/6 has validated targets for cancer treatment that have shown a potential therapeutic effect. Acquired resistance has raised concerns with the reduced efficacy of CDK4/6 inhibitors for the treatment of cancer, particularly in breast cancer. The recent development of small molecule CDK4/6 inhibitors are potential therapeutics, however, the off-target activities causing severe adverse effects, making them undruggable.61 Therefore, in order to overcome the limitation of small molecule competitive inhibitors, proteolysis targeting chimeras (PROTACs), a new strategy emerged as a target protein degrader in drug discovery. PROTACs are heterobifunctional molecules comprising a ligand for the target protein of interest and an E3 ligase-recruiting ligand are connected by a linker.62 Thus, the ternary complex formed promotes its catalytic role, where recruitment of E3 ligase to the target protein leads to optimal ubiquitination, resulting in proteasomal degradation, and is released to repeat the cycle, requiring less drug exposure63 (Fig. 14). The composition of PROTACs determines degradation selectivity and influences the stability and conformation of the requisite ternary complex. For example, cereblon-recruiting palbociclib-based PROTACs selectively degrade CDK6 while, cereblon-recruiting abemaciclib-based PROTACs selectively degrades CDK4. PROTACs provide some benefits in comparison with small molecule competitive inhibitors because of their specificity. The key benefits are their catalytic mode of action, high selectivity, the ability to target undruggable proteins, and the ability to resolve resistance by targeting mutant proteins and preventing the feedback-mediated overexpression of target proteins.63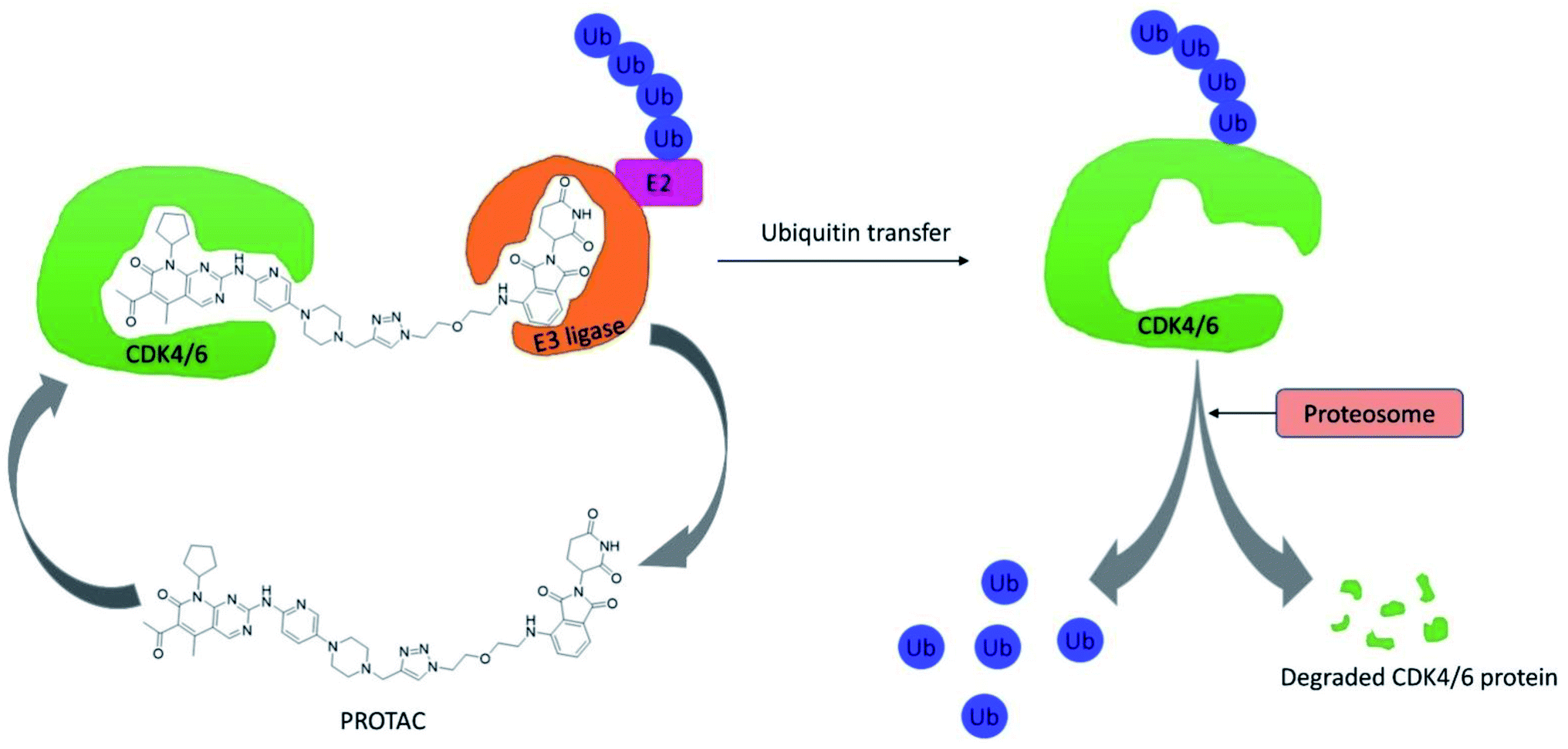 | ||
| Fig. 14 Mechanism of action of PROTACs in the degradation of CDK4/6 protein. | ||
Su et al. established PROTAC that could selectively reduce or degrade CDK6 based on selective inhibitors of CDK4/6 approved by the FDA such as ribociclib, palbociclib, and abemaciclib. The potent PROTAC was developed by coupling four distinct E3 ligases (pomalidomide, VHL, Nutlin-3b, and cIAP) to each of these CDK4/6 inhibitors.64 Palbociclib-derived degraders were reported to be marginally superior to abemaciclib-derived degraders among the CRBN recruiting PROTACs, while ribociclib-derived degraders barely induce CDK6 degradation.64 Palbociclib-based PROTAC 9 (Fig. 15) potentially degraded CDK6 with DC50 (the concentration at which the target is degraded by 50%) values of 2.1 nM and antiproliferation of several hematopoietic cancer cells including multiple myeloma. Their study revealed that PROTAC 9 induced antiproliferation by target protein degradation rather than residual kinase inhibitory action with high CDK6 selectivity and moderate CDK4 degradation at higher concentrations.
 | ||
| Fig. 15 PROTAC 9 based on palbociclib as effective CDK4/6 degrader. | ||
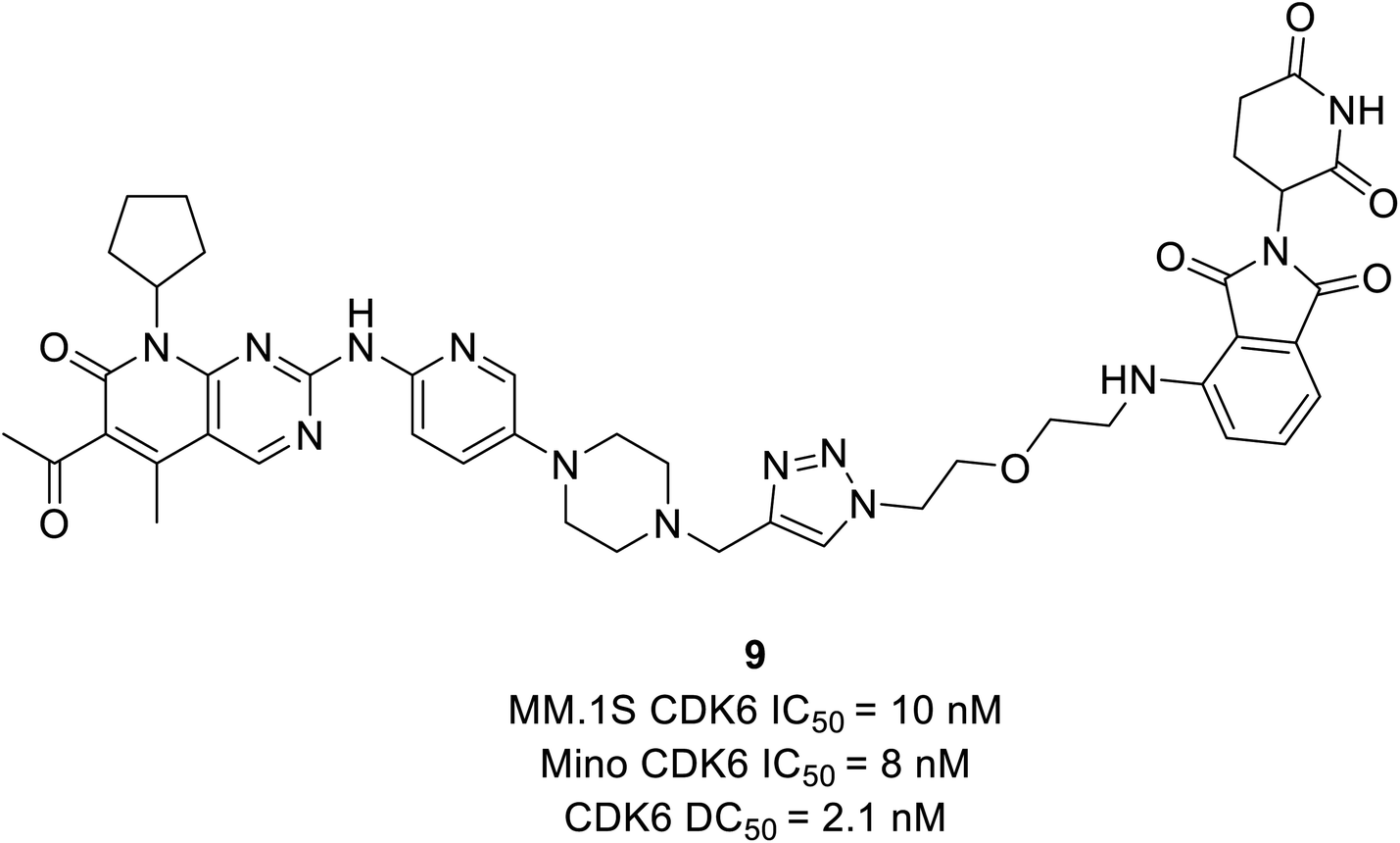
Dominici et al. reported a palbociclib based PROTAC consisting of cereblon (E3 ubiquitin ligase) which selectively degraded CDK6.65 The YX-2-107 (10) (Fig. 16), a cereblon-recruiting PROTAC robustly inhibited both CDK6 and CDK4 kinase activity in vitro with IC50 values of 4.4 nM and 0.69 nM, respectively, and selectively degraded CDK6 in Philadelphia positive (Ph+) BV173 cells with an IC50 of 4 nM. It also distinguished CDK4 from CDK6 by creating a slew of ternary complexes that selectively degraded CDK6. In another study, it inhibited proliferation, phosphor-Rb, FOXM1, and CDK6 but not CDK4 expression in Ph+ ALL.65 On the other hand, YX-2-233 PROTAC consisting of RG7112 as a recruitment group for E3 ligase MDM2 was able to degrade CDK4 and CDK6 with similar efficacy.
 | ||
| Fig. 16 Palbociclib based PROTAC 10 as potent CDK4/6 degrader. | ||
Brand et al. identified a phthalimide-based degrader PROTAC BSJ-03-123 (11) (Fig. 17) that could selectively degrade CDK6 without having an impact on CDK4 by reason of differential ternary complex formation with E3 ligase CRL4CRBN.66 Furthermore, BSJ-03-123 suppressed the pRb proliferation in CDK6 dependent AML cell lines while inducing marked anti-proliferative activity by arresting cells at 200 nM during the growth phase (G1) of the cell cycle. In comparison, no such effect was observed in CDK4 dependent cancer cell lines.
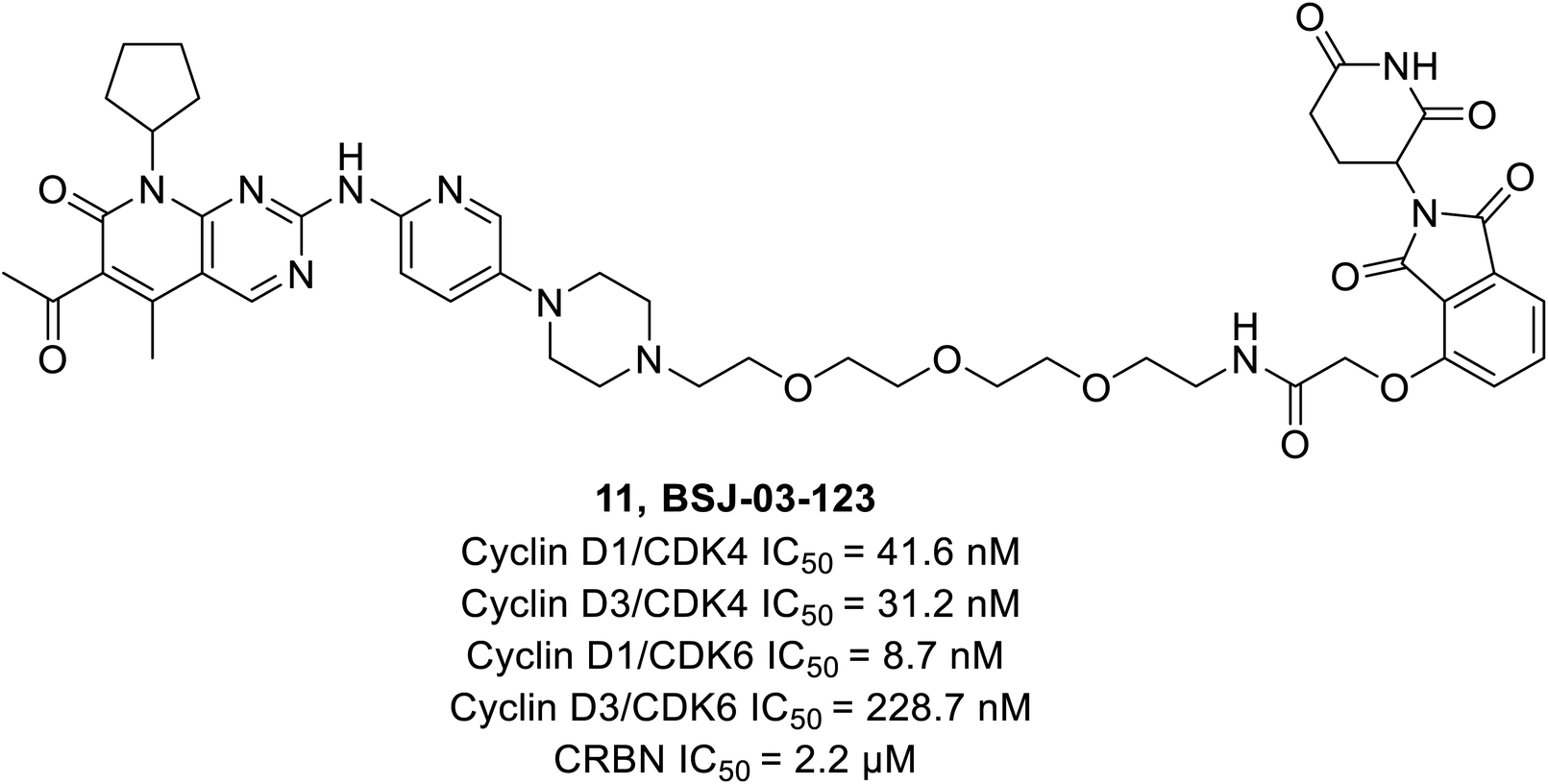 | ||
| Fig. 17 Palbociclib based PROTAC 11 as selective CDK4/6 degrader. | ||
Rana et al. prepared PROTAC compounds from approved drug palbociclib 12 (Fig. 18) as selective CDK6 degrader while sparing the CDK4 in HPNE and MiaPaCa2 pancreatic cell lines.67 This confirms ternary complex formation is a prerequisite for the successful degradation of CDK6. In both cell lines, at 4 and 24 h dose–response tests compound 12 demonstrated quantitative degradation of CDK6 at 100 nM, while CDK4 protein appeared intact.67
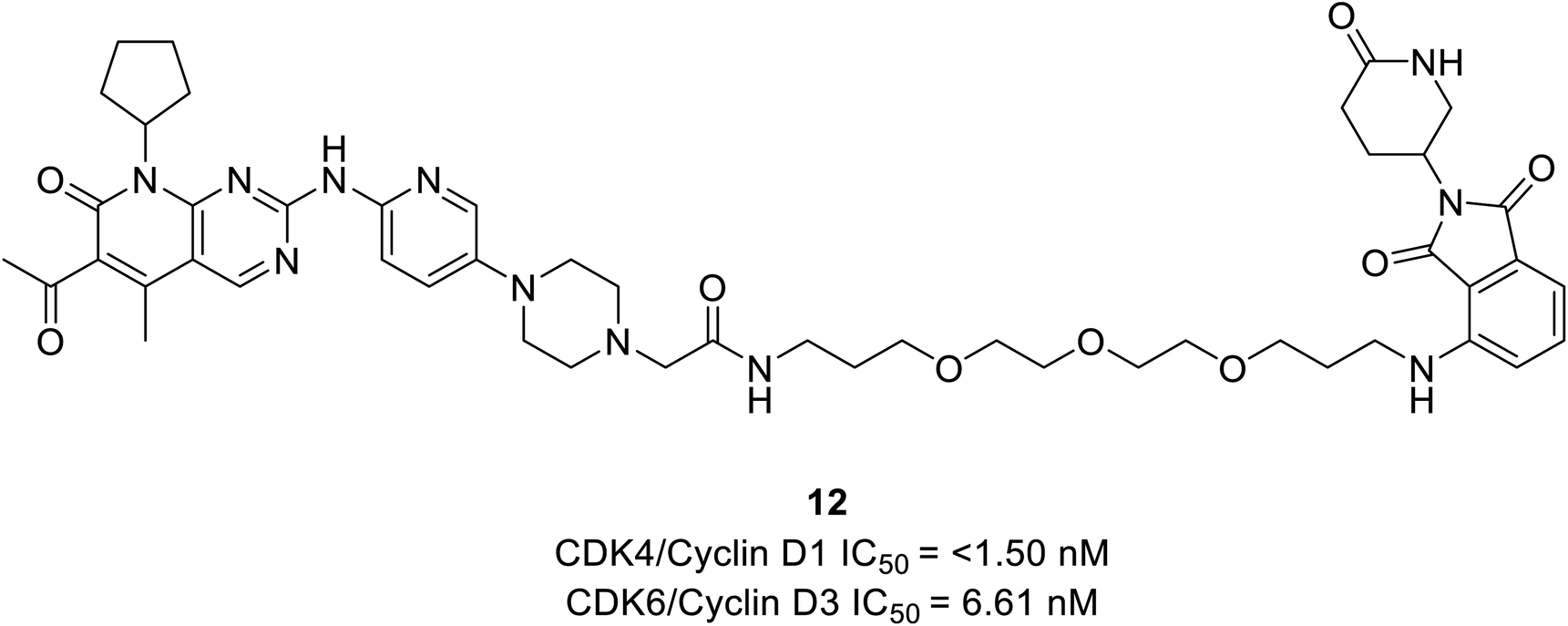 | ||
| Fig. 18 Palbociclib based PROTAC 12 as potent CDK6 degrader. | ||
The PROTAC 13 (Fig. 19) was developed by Zhao et al. proteasome-mediated dual CDK4/6 degrader by conjugating compound pomalidomide, an E3 ligase ligand to both palbociclib and ribociclib.68 The palbociclib-based PROTAC 13 with DC50 values varying from 20 to 50 nM effectively induced the degradation of CDK4/6 in the TNBCs cell line MDA-MB-231 with a dose-dependent reduction in pRb levels. In comparison, degradation caused by ribociclib-based PROTAC was not effective. Their analysis revealed that in MDA-MB-231, palbociclib-based PROTAC acts more efficiently in ER+/HER2− MCF-7 breast cancer cells. They also noticed that both PROTACs selectively degrade CDK4 in the U87 glioblastoma cells, suggesting the scope of the compounds for other cancer types.
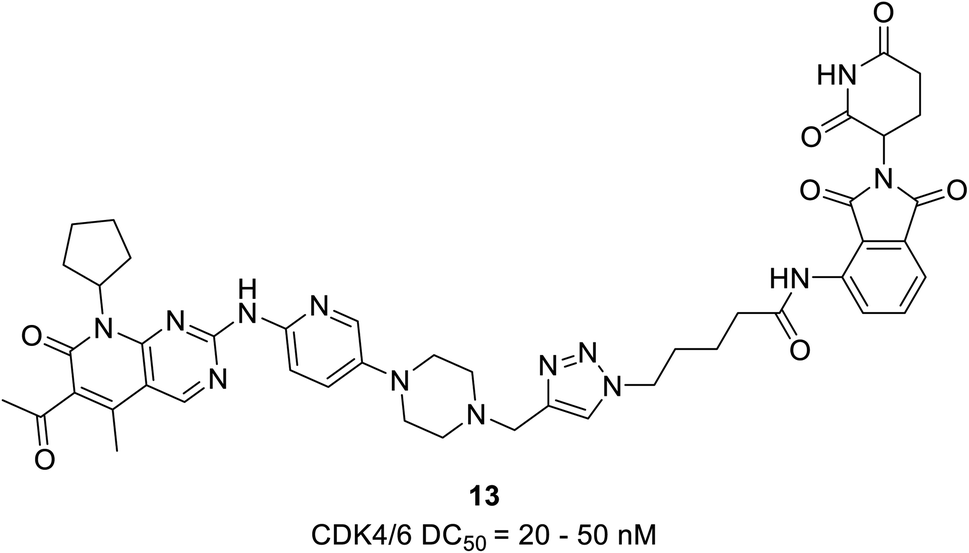 | ||
| Fig. 19 Palbociclib-based PROTAC 13 as dual CDK4/6 degrader. | ||
Jiang et al. developed a series of dual as well as selective CDK4/6 degrader PROTAC comprising FDA-approved CDK4/6 inhibitors and CRBN-recruiting moieties.69 Besides, these degraders displayed decreased pRb levels, an arrest of the G1-phase cell cycle, and increased antiproliferative activity. Palbociclib-based degrader (BSJ-02-162 and BSJ-03-204) (Fig. 20) degraded both CDK4 and CDK6 while, the ribociclib based degrader (BSJ-01-187 and BSJ-04-132) (Fig. 20) selectively degraded CDK4, and abemaciclib based degrader (BSJ-01-184) uniquely exhibited CDK9 degradation. In a panel of MCL cell lines, BSJ-02-162 degrades transcription factors IKZF1/3 with an enhanced anti-proliferative effect, while BSJ-03-204 does not affect IKZF1/3.69 Therefore, the dual inhibitors of CDK4/6 and IKZF1/3 have shown potential for the treatment of MCL.
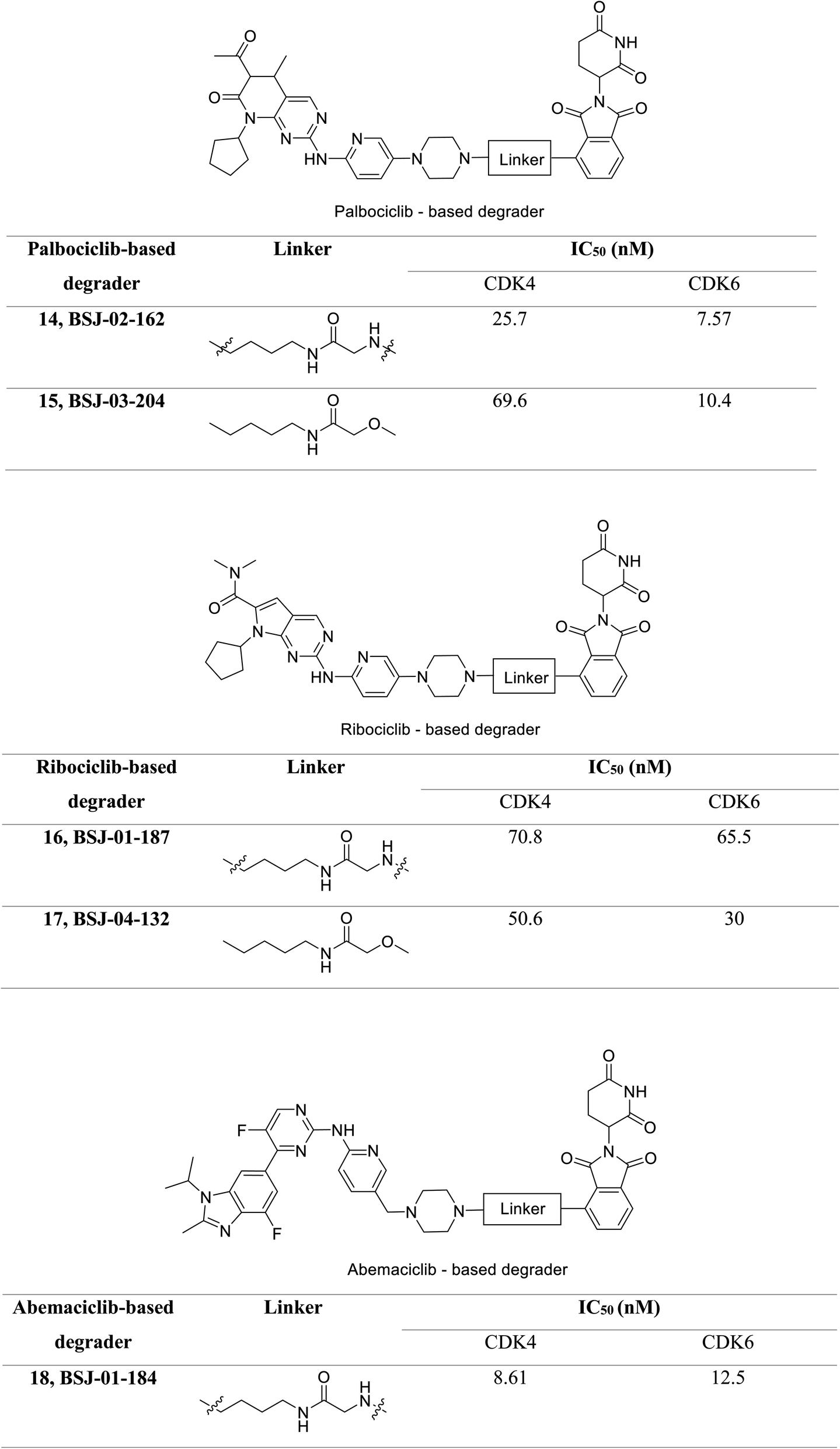 | ||
| Fig. 20 PROTACs 14, 15, 16, 17, 18 as effective CDK4/6 degraders. | ||
Anderson et al. reported the preparation of nine palbociclib-based PROTAC by introducing three separate ligases binder that effectively inhibits CDK4/6 and degrades potency for palbociclib.70 Regardless of the various E3 ligases (VHL, CRBN, and IAP) and linkers, all compounds potently suppress CDK4/6 but exhibited selective degradation of CDK6 with Δ pDC50 ranging from 0.5 nM to 1.5 nM (pDC50 CDK6 to pDC50 CDK4). Regardless of negligible correlation with observed CDK4/6 degradation potency, all compounds showed sub-micromolar inhibition of cyclin D–CDK4/6. Compound 19 (Fig. 21), cereblon-recruiting PROTAC, was the most effective cyclinDK4/6 inhibitor with pDC50 values of 8.0 nM and 9.1 nM against CDK4 and CDK6, respectively. However, with Δ pDC50 of 1.1 CDK6 selectively degraded over CDK4.
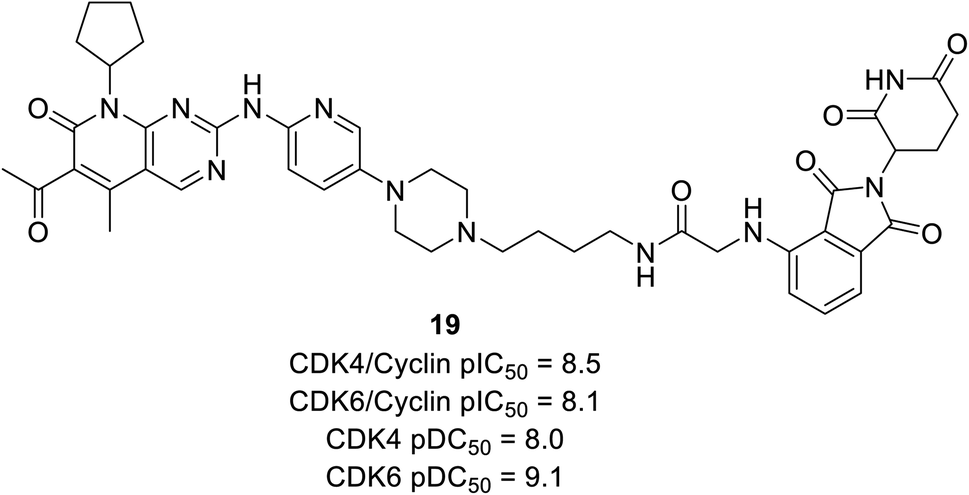 | ||
| Fig. 21 Palbociclib-based PROTAC 19 as potent CDK4/6 degrader. | ||
7. Conclusion
The CDKs are primarily involved in multiple network mechanisms of biology, such as proliferation control and transcription in particular the cyclin D–CDK4/6 complex is a promising therapeutic target for certain types of cancers, particularly breast cancers. The US FDA approved ribociclib, palbociclib, and abemaciclib as potent inhibitors of CDK4/6 for the treatment of ER+, HER2−, and metastatic breast cancer. They are also studies continued to be investigated for use in the treatment of other types of cancers. According to the National Cancer Institute, USA abemaciclib is in phase 2 trials for treating patients with recurrent brain tumors and recurrent glioblastoma. Preclinical and clinical studies suggest CDK4/6 inhibitors may be used to treat a wide range of cancers. But the compensatory regulatory mechanism of other signalling pathways reduced their efficacy due to acquired resistance. In the absence of optimal combination strategies and biomarkers, and prevailing acquired resistance CDK4/6 monotherapy may not be effective. Acquired resistance may render CDK4/6 inhibitors ineffective cytostatic in tumour progression control or absolute tumour cell eradication. As a result, constructing and developing small molecule inhibitors that are more efficient, selective, and resistant to compensatory regulatory mechanisms may have a positive impact on the anticancer drug discovery process. The structural and functional characteristics of recently developed CDK4/6 inhibitors, the evolution of resistance to FDA-approved CDK4/6 inhibitors, and the proposed strategy to overcome resistance are examined and summarized in this review. The inhibitors are classified based on the different chemical scaffolds and a few derivatives showed effectiveness at lower concentrations with favourable anti-proliferative activity. The computational studies carried out at our end are in line with the activity profile showed by various chemical scaffolds discussed.The growing exploration of genetic research and knowledge has offered insight into compensatory mechanisms that control quiescence along with the identification of biomarkers. Overexpression of CDK6, cyclin E, acquired mutations in the pRb and RB1 genes, loss of PTEN, and increased AKT activation, for instance, are signalling mechanisms that compensate for CDK4/6 suppression and are regarded as vital biomarkers. Further studies are needed on these biomarkers that could predict the response and resistance of CDK4/6 inhibitors. Such investigation will allow for the selection of patients who are likely to benefit from selective combination therapies.
To overcome resistance and improve PFS, it is critical to select an efficient combination of therapies with CDK4/6 inhibitors. An array of selective agents is being tested in clinical trials. The development of an optimal CDK4/6 inhibitor-based selective combination therapy would not only pave the way to resolve resistance in breast cancer but could also be extended to other type of cancers. In patients with HR+, HER2− metastatic breast cancer, combining CDK4/6 inhibitors with hormone therapy such as aromatase inhibitors or fulvestrant agents tended to have consistent PFS benefits. In glioblastoma, a combination of CDK4/6 inhibitors and Trk/C-Met therapy showed a synergistic effect. Rational combination therapy is efficient, and CDK4/6 inhibition may be used as a combination agent with a variety of targeted agents.
In a short period, PROTAC is expected to appear as a novel anticancer therapy with the ability to revolutionize drug development and resolve the challenges that conventional small molecule inhibitors face. The ability of PROTACs to induce the degradation of the target protein, rather than only inhibiting it, is one of their distinguishing features. However, their use has been constrained by certain limitations, such as low cell permeability, blood–brain penetration, and poor oral bioavailability. Intriguingly, new PROTAC technology overcomes the permeability concern and is now undergoing clinical development in cancer. Excellent potency, relatively low toxicity, and resistance-free in anticancer therapy, CDK4/6 protein degraders are foreseen to be included in therapeutic strategies. Currently, the PROTACs are mainly represented by four members (VHL, CRBN, cIAP, and MDM2) of the E3 ligase family, even though there are about 600 E3 ligase are encoded in the human genome. Additional studies are needed to explore potential available E3 ligases improving the scope of targeted protein degradation. In future, a thorough structural study of the ligases, their recognition requirements, and their druggability are indeed needed to explore future drug discovery. In summary, the development of potent, selective, and enhanced pharmacokinetics of CDK4/6 inhibitors, PROTACs, and efficient targeted combination therapy, and identification of defined biomarkers would be a major focus in the coming years for a significant impact on anticancer therapy perception.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors have no conflict of interest.Acknowledgements
The authors express their deep sense of gratitude towards the JSS Academy of Higher Education & Research, Mysuru, Karnataka, India, for the support in writing this review article.References
- Cancer, https://www.who.int/news-room/fact-sheets/detail/cancer, accessed March 25, 2021 Search PubMed.
- P. Balakumar, K. Maung-U and G. Jagadeesh, Prevalence and prevention of cardiovascular disease and diabetes mellitus, Pharmacol. Res., 2016, 113, 600–609, DOI:10.1016/j.phrs.2016.09.040.
- R. Roskoski Jr, Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs, Pharmacol. Res., 2018, 139, 471–488, DOI:10.1016/j.phrs.2018.11.035.
- P. Gupta, S. Narayanan and D.-H. Yang, CDK Inhibitors as Sensitizing Agents for Cancer Chemotherapy, in Protein Kinase Inhibitors as Sensitizing Agents for Chemotherapy, Elsevier Inc., 2019, pp. 125–149, DOI:10.1016/b978-0-12-816435-8.00009-2.
- U. Asghar, A. K. Witkiewicz, N. C. Turner and E. S. Knudsen, The history and future of targeting cyclin-dependent kinases in cancer therapy, Nat. Rev. Drug Discovery, 2015, 14(2), 130–146, DOI:10.1038/nrd4504.
- M. Malumbres, Cyclin-dependent kinases, Genome Biol., 2014, 15(6), 1–10, DOI:10.1186/gb4184.
- C. Shi, Q. Wang and X. Liao, et al., Discovery of 6-(2-(dimethylamino)ethyl)-N-(5-fluoro-4-(4-fluoro-1-isopropyl-2-methyl-1H-benzo[d]imidazole-6-yl)pyrimidin-2-yl)-5,6,7,8-tetrahydro-1,6-naphthyridin-2-amine as a highly potent cyclin-dependent kinase 4/6 inhibitor for treatment of cancer, Eur. J. Med. Chem., 2019, 178, 352–364, DOI:10.1016/j.ejmech.2019.06.005.
- F. J. Kaye, Rb and cyclin dependent kinase pathways: defining a distinction between Rb and p16 loss in lung cancer, Oncogene, 2002, 21(45), 6908–6914, DOI:10.1038/sj.onc.1205834.
- S. C. Scott, S. S. Lee and J. Abraham, Mechanisms of therapeutic CDK4/6 inhibition in breast cancer, Semin. Oncol., 2017, 44(6), 385–394, DOI:10.1053/j.seminoncol.2018.01.006.
- R. Jorda, D. Hendrychová, J. Voller, E. Řezníčková, T. Gucký and V. Kryštof, How Selective Are Pharmacological Inhibitors of Cell-Cycle-Regulating Cyclin-Dependent Kinases?, J. Med. Chem., 2018, 61(20), 9105–9120, DOI:10.1021/acs.jmedchem.8b00049.
- M. Poratti and G. Marzaro, Third-generation CDK inhibitors: a review on the synthesis and binding modes of palbociclib, ribociclib and abemaciclib, Eur. J. Med. Chem., 2019, 172, 143–153, DOI:10.1016/j.ejmech.2019.03.064.
- C. J. Sherr, D. Beach and G. I. Shapiro, Targeting CDK4 and CDK6: from discovery to therapy, Cancer Discovery, 2016, 6(4), 353–367, DOI:10.1158/2159-8290.CD-15-0894.
- M. Malumbres, R. Sotillo and D. Santamaría, et al., Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6, Cell, 2004, 118(4), 493–504, DOI:10.1016/j.cell.2004.08.002.
- V. Baldin, J. Lukas, M. J. Marcote, M. Pagano and G. Draetta, Cyclin D1 is a nuclear protein required for cell cycle progression in G1, Genes Dev., 1993, 7(5), 812–821, DOI:10.1101/gad.7.5.812.
- H. Matsushime, M. E. Ewen and D. K. Strom, et al., Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins, Cell, 1992, 71(2), 323–334, DOI:10.1016/0092-8674(92)90360-O.
- J. Kato, H. Matsushime, S. W. Hiebert, M. E. Ewen and C. J. Sherr, Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4, Genes Dev., 1993, 7(3), 331–342, DOI:10.1101/gad.7.3.331.
- E. Susaki, K. Nakayama and K. I. Nakayama, Cyclin D2 Translocates p27 out of the Nucleus and Promotes Its Degradation at the G0-G1 Transition, Mol. Cell. Biol., 2007, 27(13), 4626–4640, DOI:10.1128/mcb.00862-06.
- M. Barbacid, S. Ortega and R. Sotillo, et al., Cell cycle and cancer: genetic analysis of the role of cyclin-dependent kinases, Cold Spring Harbor Symp. Quant. Biol., 2005, 70, 233–240, DOI:10.1101/sqb.2005.70.005.
- C. Liu, Y. Konagaya and M. Chung, et al., Altered G1 signaling order and commitment point in cells proliferating without CDK4/6 activity, Nat. Commun., 2020, 11(1), 1–13, DOI:10.1038/s41467-020-18966-9.
- M. G. Hu, A. Deshpande and M. Enos, et al., A requirement for cyclin-dependent kinase 6 in thymocyte development and tumorigenesis, Cancer Res., 2009, 69(3), 810–818, DOI:10.1158/0008-5472.CAN-08-2473.
- Y. Wang, Y. Zhi and Q. Jin, et al., Discovery of 4-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrazole-3-carboxamide (FN-1501), an FLT3- and CDK-Kinase Inhibitor with Potentially High Efficiency against Acute Myelocytic Leukemia, J. Med. Chem., 2018, 61(4), 1499–1518, DOI:10.1021/acs.jmedchem.7b01261.
- Y. Zhi, Z. Wang and C. Yao, et al., Design and synthesis of 4-(Heterocyclic substituted amino)-1h-pyrazole-3-carboxamide derivatives and their potent activity against acute myeloid leukemia (AML), Int. J. Mol. Sci., 2019, 20(22), 1–15, DOI:10.3390/ijms20225739.
- S. Tadesse, M. Yu and L. B. Mekonnen, et al., Highly Potent, Selective, and Orally Bioavailable 4-Thiazol-N-(pyridin-2-yl)pyrimidin-2-amine Cyclin-Dependent Kinases 4 and 6 Inhibitors as Anticancer Drug Candidates: Design, Synthesis, and Evaluation, J. Med. Chem., 2017, 60(5), 1892–1915, DOI:10.1021/acs.jmedchem.6b01670.
- S. E. Abbas, R. F. George, E. M. Samir, M. M. A. Aref and H. A. Abdel-Aziz, Synthesis and anticancer activity of some pyrido[2,3-d]pyrimidine derivatives as apoptosis inducers and cyclin-dependent kinase inhibitors, Future Med. Chem., 2019, 11(18), 2395–2414, DOI:10.4155/fmc-2019-0050.
- C. Shi, Q. Wang and X. Liao, et al., Discovery of a novel series of imidazo[1,2:1,6]pyrido[2,3-d]pyrimidin derivatives as potent cyclin-dependent kinase 4/6 inhibitors, Eur. J. Med. Chem., 2020, 193, 112239, DOI:10.1016/j.ejmech.2020.112239.
- H. Zhao, X. Hu and K. Cao, et al., Synthesis and SAR of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as potent and selective CDK4/6 inhibitors, Eur. J. Med. Chem., 2018, 157, 935–945, DOI:10.1016/j.ejmech.2018.08.043.
- B. Zhang, T. Zhang and T. Y. Zhang, et al., A novel methoxybenzyl 5-nitroacridone derivative effectively triggers g1 cell cycle arrest in chronic myelogenous leukemia k562 cells by inhibiting cdk4/6-mediated phosphorylation of rb, Int. J. Mol. Sci., 2020, 21(14), 1–16, DOI:10.3390/ijms21145077.
- T. Al-Warhi, A. M. El Kerdawy and N. Aljaeed, et al., Synthesis, biological evaluation and in silico studies of certain oxindole–indole conjugates as anticancer CDK inhibitors, Molecules, 2020, 25(9), 2031, DOI:10.3390/molecules25092031.
- S. Kalra, G. Joshi, A. Munshi and R. Kumar, Structural insights of cyclin dependent kinases: implications in design of selective inhibitors, Eur. J. Med. Chem., 2017, 142, 424–458, DOI:10.1016/j.ejmech.2017.08.071.
- T. W. Miller, N. A. Traphagen and J. Li, et al., Tumor pharmacokinetics and pharmacodynamics of the CDK4/6 inhibitor ribociclib in patients with recurrent glioblastoma, J. Neurooncol., 2019, 144(3), 563–572, DOI:10.1007/s11060-019-03258-0.
- L. Cornell, S. A. Wander, T. Visal, N. Wagle and G. I. Shapiro, MicroRNA-Mediated Suppression of the TGF-β Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance, Cell Rep., 2019, 26(10), 2667–2680.e7, DOI:10.1016/j.celrep.2019.02.023.
- C. Yang, Z. Li and T. Bhatt, et al., Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence, Oncogene, 2017, 36(16), 2255–2264, DOI:10.1038/onc.2016.379.
- Z. Li, P. Razavi and Q. Li, et al., Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway, Cancer Cell, 2018, 34(6), 893–905.e8, DOI:10.1016/j.ccell.2018.11.006.
- J. L. Dean, C. Thangavel, A. K. McClendon, C. A. Reed and E. S. Knudsen, Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure, Oncogene, 2010, 29(28), 4018–4032, DOI:10.1038/onc.2010.154.
- J. Sage, G. J. Mulligan and L. D. Attardi, et al., Targeted disruption of the three Rb-related genes leads to loss of G1 control and immortalization, Genes Dev., 2000, 14(23), 3037–3050, DOI:10.1101/gad.843200.
- M. T. Herrera-Abreu, M. Palafox and U. Asghar, et al., Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer, Cancer Res., 2016, 76(8), 2301–2313, DOI:10.1158/0008-5472.CAN-15-0728.
- C. Lefebvre, T. Bachelot and T. Filleron, et al., Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis, PLoS Med., 2016, 13(12), 1–18, DOI:10.1371/journal.pmed.1002201.
- R. Condorelli, L. Spring and J. OShaughnessy, et al., Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer, Ann. Oncol., 2018, 29(3), 640–645, DOI:10.1093/annonc/mdx784.
- S. A. Wander, O. Cohen and X. Gong, et al., The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer, Cancer Discovery, 2020, 10(8), 1174–1193, DOI:10.1158/2159-8290.CD-19-1390.
- K. Pandey, N. Park and K. S. Park, et al., Combined cdk2 and cdk4/6 inhibition overcomes palbociclib resistance in breast cancer by enhancing senescence, Cancers, 2020, 12(12), 1–17, DOI:10.3390/cancers12123566.
- M. Scaltriti, P. J. Eichhorn and J. Cortés, et al., Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(9), 3761–3766, DOI:10.1073/pnas.1014835108.
- J. Gray-Bablin, J. Zalvide, M. P. Fox, C. J. Knickerbocker, J. A. Decaprio and K. Keyomarsi, Cyclin E, a redundant cyclin in breast cancer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93(26), 15215–15220, DOI:10.1073/pnas.93.26.15215.
- H. J. Fingert, N. OLeary, A. B. Pardee and G. Molnar, Cyclin E, a Potential Prognostic Marker for Breast Cancer, Cancer Res., 1994, 54(2), 380–385 Search PubMed.
- A. Fassl, C. Brain and M. Abu-Remaileh, et al., Increased lysosomal biomass is responsible for the resistance of triple-negative breast cancers to CDK4/6 inhibition, Sci. Adv., 2020, 6(25), 1–16, DOI:10.1126/sciadv.abb2210.
- K. C. Patra, V. K. Weerasekara and N. Bardeesy, AMPK-Mediated Lysosome Biogenesis in Lung Cancer Growth, Cell Metab., 2019, 29(2), 238–240, DOI:10.1016/j.cmet.2018.12.011.
- S. D. Webb, J. A. Sherratt and R. G. Fish, Modelling tumour acidity and invasion, Novartis Found. Symp., 2001, 240, 169–181, DOI:10.1002/0470868716.ch12.
- C. Cuvier, A. Jang and R. P. Hill, Exposure to hypoxia, glucose starvation and acidosis: effect on invasive capacity of murine tumor cells and correlation with cathepsin (L + B) secretion, Clin. Exp. Metastasis, 1997, 15(1), 19–25, DOI:10.1023/A:1018428105463.
- P. Montcourrier, I. Silver, R. Farnoud, I. Bird and H. Rochefort, Breast cancer cells have a high capacity to acidify extracellular milieu by a dual mechanism, Clin. Exp. Metastasis, 1997, 15(4), 382–392, DOI:10.1023/A:1018446104071.
- K. Glunde, S. E. Guggino, M. Solaiyappan, A. P. Pathak, Y. Ichikawa and Z. M. Bhujwalla, Extracellular Acidification Alters Lysosomal Trafficking in Human Breast Cancer Cells, Neoplasia, 2003, 5(6), 533–545, DOI:10.1016/s1476-5586(03)80037-4.
- Y. Nagata, K. H. Lan and X. Zhou, et al., PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients, Cancer Cell, 2004, 6(2), 117–127, DOI:10.1016/j.ccr.2004.06.022.
- H. M. Stern, H. Gardner and T. Burzykowski, et al., PTEN loss is associated with worse outcome in HER2-Amplified breast cancer patients but is not associated with trastuzumab resistance, Clin. Cancer Res., 2015, 21(9), 2065–2074, DOI:10.1158/1078-0432.CCR-14-2993.
- Q. Wang, P. Liu and J. M. Spangle, et al., PI3K-p110α mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers, Oncogene, 2016, 35(27), 3607–3612, DOI:10.1038/onc.2015.406.
- C. Costa, W. Ye and A. Ly, et al., Pten loss mediates clinical cross-resistance to CDK4/6 and PI3Kα inhibitors in breast cancer, Cancer Discovery, 2020, 10(1), 72–85, DOI:10.1158/2159-8290.CD-18-0830.
- Y. Lu, Y. Z. Lin and R. LaPushin, et al., The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells, Oncogene, 1999, 18(50), 7034–7045, DOI:10.1038/sj.onc.1203183.
- N. A. Obrien, M. S. J. McDermott and D. Conklin, et al., Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer, Breast Cancer Res., 2020, 22(1), 1–17, DOI:10.1186/s13058-020-01320-8.
- A. E. Vilgelm, N. Saleh and R. Shattuck-Brandt, et al., MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21, Sci. Transl. Med., 2019, 11(505) DOI:10.1126/scitranslmed.aav7171.
- B. Sun, S. Mason and R. C. Wilson, et al., Inhibition of the transcriptional kinase CDK7 overcomes therapeutic resistance in HER2-positive breast cancers, Oncogene, 2020, 39(1), 50–63, DOI:10.1038/s41388-019-0953-9.
- N. Sobhani, A. Fassl, G. Mondani, D. Generali and T. Otto, Targeting Aberrant FGFR Signaling to Overcome CDK4/6 Inhibitor Resistance in Breast Cancer, Cells, 2021, 10(293), 1–14, DOI:10.3390/cells10020293.
- T. Kong, Y. Xue and R. Cencic, et al., EIF4A inhibitors suppress cell-cycle feedback response and acquired resistance to CDK4/6 inhibition in cancer, Mol. Cancer Ther., 2019, 18(11), 2158–2170, DOI:10.1158/1535-7163.MCT-19-0162.
- I. Olmez, Y. Zhang and L. Manigat, et al., Combined c-Met/Trk inhibition overcomes resistance to CDK4/6 inhibitors in Glioblastoma, Cancer Res., 2018, 78(15), 4360–4369, DOI:10.1158/0008-5472.CAN-17-3124.
- P. Martín-Acosta and X. Xiao, PROTACs to address the challenges facing small molecule inhibitors, Eur. J. Med. Chem., 2021, 210, 112993, DOI:10.1016/j.ejmech.2020.112993.
- M. Konstantinidou, J. Li and B. Zhang, et al., PROTACs– a game-changing technology, Expert Opin. Drug Discovery, 2019, 14(12), 1255–1268, DOI:10.1080/17460441.2019.1659242.
- S. Khan, Y. He and X. Zhang, et al., PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics, Oncogene, 2020, 39(26), 4909–4924, DOI:10.1038/s41388-020-1336-y.
- S. Su, Z. Yang and H. Gao, et al., Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders, J. Med. Chem., 2019, 62(16), 7575–7582, DOI:10.1021/acs.jmedchem.9b00871.
- M. De Dominici, P. Porazzi and Y. Xiao, et al., Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs, Blood, 2020, 135(18), 1560–1573, DOI:10.1182/BLOOD.2019003604.
- M. Brand, B. Jiang and S. Bauer, et al., Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML, Cell Chem. Biol., 2019, 26(2), 300–306.e9, DOI:10.1016/j.chembiol.2018.11.006.
- S. Rana, M. Bendjennat and S. Kour, et al., Selective degradation of CDK6 by a palbociclib based PROTAC, Bioorg. Med. Chem. Lett., 2019, 29(11), 1375–1379, DOI:10.1016/j.bmcl.2019.03.035.
- B. Zhao and K. Burgess, PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer, Chem. Commun., 2019, 55(18), 2704–2707, 10.1039/c9cc00163h.
- B. Jiang, E. S. Wang and K. A. Donovan, et al., Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6, Angew. Chem., Int. Ed., 2019, 58(19), 6321–6326, DOI:10.1002/anie.201901336.
- N. A. Anderson, J. Cryan and A. Ahmed, et al., Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs, Bioorg. Med. Chem. Lett., 2020, 30(9), 127106, DOI:10.1016/j.bmcl.2020.127106.
| This journal is © The Royal Society of Chemistry 2021 |