
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The application of amine-based materials for carbon capture and utilisation: an overarching view†
Louise B.
Hamdy
 *ab,
Chitrakshi
Goel
a,
Jennifer A.
Rudd
ac,
Andrew R.
Barron
ade and
Enrico
Andreoli
*a
*ab,
Chitrakshi
Goel
a,
Jennifer A.
Rudd
ac,
Andrew R.
Barron
ade and
Enrico
Andreoli
*a
aEnergy Safety Research Institute, Swansea University, Bay Campus, Swansea, SA1 8EN, UK. E-mail: e.andreoli@swansea.ac.uk
bCGG, Tyn-y-Coed, Pentywyn Road, Llandudno, LL30 1SA, UK. E-mail: louise.hamdy@cgg.com
cSchool of Management, Swansea University, Bay Campus, Swansea, SA1 8EN, UK
dDepartment of Chemistry and Department of Materials Science and Nanoengineering, Rice University, Houston, TX 77005, USA
eFaculty of Engineering, Universiti Teknologi Brunei, Brunei Darussalam
First published on 1st September 2021
Abstract
In the ongoing research campaign to reduce the global atmospheric CO2 concentration, technologies are being developed to enable the capture of CO2 from dilute sources and conversion into higher-value products. Amine and polyamine-based materials feature widely in the literature as solid CO2 sorbents and as catalyst modifiers for CO2 electrochemical reduction; however, advancing lab-scale research into a pilot or industrial-scale application is fraught with challenges, starting with the definition and identification of an effective adsorbent. This multidisciplinary review serves as an essential introduction to the role of amines in carbon capture and utilisation for scientists entering and advancing the field. The chemical and engineering principles of amine-based CO2 capture are considered to define the parameters required of an adsorbent, describe adsorption testing methods, and introduce the reader to a range of amine-based adsorbents and how they can be specialised to overcome specific issues. Finally, the application of electrocatalysts modified with nitrogen-containing compounds and polymers is reviewed in the context of CO2 utilisation.
 Louise B. Hamdy | Dr Louise Hamdy received her Masters degree in Chemistry from the University of Glasgow, which involved a one year placement with Sasol Technology (UK) Ltd. She was awarded her PhD from the University of Bath, where she studied alkali metal–organic complexes under the supervision of Prof. Chick Wilson. Subsequently, she worked with Dr Enrico Andreoli as a Postdoctoral Researcher at the Energy Safety Research Institute, Swansea University, where she developed new cross-linked polyamine CO2 adsorbents. Louise now works as a Senior Research Scientist at CGG, based in North Wales, where her research interests include carbon capture and storage technologies. |
 Chitrakshi Goel | Dr Chitrakshi Goel obtained her PhD in Chemical Engineering from Thapar University, India, where her research focussed on developing and evaluating carbon adsorbents for carbon dioxide capture. Later, she joined the research group of Prof. Guy Marin at Laboratory for Chemical Technology, Ghent University as a Post-Doctoral Researcher, working on reactor design of a gas–solid vortex reactor. Presently, she is a Research Officer at Energy Safety Research Institute, Swansea University, working with Prof. Andrew Barron and Dr Enrico Andreoli. She is interested in carbon dioxide capture with a focus on reactor design, and process modelling and simulation. |
 Jennifer A. Rudd | Dr Jennifer Rudd has been working on climate mitigation technologies for a decade across Europe and America. Keen to mitigate climate change she has approached it from many angles, working on carbon utilisation, climate change education (http://www.youandco2.org) and lately through the circular economy. |
 Andrew R. Barron | Professor Andrew R. Barron received his PhD from Imperial College (1986), and after post-doctoral research at the University of Texas, Austin, he joined the faculty at Harvard University, from where he moved in 1995 to Rice University as the Charles W. Duncan, Jr – Welch Chair of Chemistry. Since 2013 he is the Sêr Cymru Chair of Low Carbon Energy, as well as the Founder and Director of the Energy Safety Research Institute Barron is the recipient of several awards, including the Star of Asia, Welch Foundation Norman Hackerman Award, and the World Technology Award. His research involves solutions to problems in energy and the environment. He is a visiting professor at the Arizona Institute for Resilience (University of Arizona) and Universiti Technology Brunei. He races on both sides of the Atlantic in his collection of historic race cars. |
 Enrico Andreoli | Dr Enrico Andreoli is an Associate Professor at the Energy Safety Research Institute of Swansea University. His research remit is in low carbon energy and environment particularly on carbon capture and utilisation (CCU) covering both materials and processes for CCU. Currently, he leads research and innovation activities in the field of industrial decarbonisation within flagships research operations in Wales and the UK. His broad experience spans both industry and academia working at Basell Polyolefins (now LyondellBasell) and holding postdoctoral positions at Rice University and the University of Houston. He is author of 45 publications and active reviewer for more that 60 international peer-reviewed journals. |
1 Introduction
Although 2020 saw a reduction on the previous years’ daily global carbon dioxide (CO2) emissions due to the COVID-19 pandemic, the global average atmospheric CO2 concentration continues to increase.1 In 2020 it exceeded 411 ppm, the highest it has been for about the last 5 million years.2 To avoid the irreversible environmental damage caused by climate change, it is necessary to significantly reduce this CO2 concentration to a level commensurate with a global average temperature rise of less than 2 °C, and preferably no more than 1.5 °C, above pre-industrial times.3 The Intergovernmental Panel on Climate Change (IPCC)'s 2018 special report describes the involvement of carbon dioxide removal (CDR) technologies to some extent in all pathways that limit warming to 1.5 °C.4 CO2 capture is considered an economically feasible technology that has been proposed to negate the emission of CO2 produced from fossil fuel combustion, and which can also remove CO2 from the air.5 In order to reduce the absolute amount of CO2 in the atmosphere, it is proposed that the majority of the captured gas will be sequestered by injecting it underground into disused oil and gas reservoirs, where it is expected to be stored on timescales of thousands of years.6 In addition, combining CO2 capture with utilisation offers the potential for a circular economy of carbon itself, with CO2 being converted back into chemicals such as carbon-based fuels to be used again with no net increase in the atmospheric CO2 concentration.1.1 CO2 capture
Carbon dioxide capture is not a new concept. Absorption technologies involving aqueous amines have existed for many decades to capture and release CO2 and H2S, owing to the reversible reaction between amines and acidic gases.7 These technologies have been applied in industrial processes, which primarily use aqueous MEA (monoethanolamine) to capture CO2 from natural gas (pre-combustion purification). Presently, CO2 absorption by such solutions is the most advanced technology available for CO2 capture on an industrial scale.8,9There are currently 51 large scale carbon capture and storage facilities around the world, 19 of which are in operation.10 These include Canada's Boundary Dam facility, the USA's Petra Nova facility in Texas and Norway's Sleipner-T platform in the North Sea, which use mature aqueous amine-based capture systems. However, there is a multitude of drawbacks with aqueous amine solutions: their CO2 uptake needs to be significantly improved upon,8 they are highly corrosive in nature11 and they suffer poisoning and deactivation from SOx and NOx gases.9 Also, they incur a high energy penalty for regeneration, at one point reportedly up to 40% of the energy output of a power plant,12 although costs are reducing as improvements are made.10 Therefore, to expand CO2 capture globally to reduce the hundreds of megatons of post-combustion CO2 emitted from powerplants each year, these technologies need to be adapted and advanced. The UK power station Drax is pilot-testing a new amine-free solvent developed by C-Capture for post-combustion CO2 capture. This technology is superior to traditional amine solutions in that it is less corrosive and has a longer lifetime.13 Evolving beyond solvents altogether however, the materials research community has a growing interest in solid adsorbents. These can have certain advantages over the proven aqueous amines such as their relative ease of handling, higher adsorption capacities, reduced regeneration energy penalties and good recyclability.14 Many solid sorbents are being studied for CO2 capture, including physisorbents such as MOFs (metal–organic frameworks),15,16 COFs (covalent organic frameworks),17 zeolites,18 AC (activated carbon)19 and N-doped carbon black,20 and chemisorbents such as alkali-metal-carbonates,21 and amine-functionalised adsorbents that frequently feature polyamines.22,23 Since amines chemically react with CO2, they are often highly selective and are therefore suited to adsorb CO2 from dilute and ultra-dilute sources such as flue gas or air. Therefore, much research is committed to amine-based adsorbents, and in understanding and optimising their CO2 sorption and separation processes from the lab to an industrial scale.
1.2 CO2 utilisation
To incentivise investment in CO2 capture technologies and find a mechanism by which to exploit a readily available chemical feedstock, CO2 utilisation is now a rapidly growing area, and research in the field has expanded dramatically over the last decade. The reduction of CO2 to higher-value products is an important process for utilising some of the enormous quantity of CO2 that needs to be removed from the atmosphere. Creating a circular economy, where carbon can be recycled again and again, will provide a financial incentive for CO2 capture. Besides, it should mean that traditional fossil fuels, currently removed from the Earth, could be synthesised instead by using CO2 utilisation.Carbon dioxide is already utilised at a commercial scale through Enhanced Oil Recovery. In this process, compressed CO2 is injected into porous rock to displace crude oil and force it into a production well. Some of the utilised CO2 is then stored underground in the rock spaces.24 Several commercial companies capture CO2 directly from the air and utilise it in different ways. Some, including Climeworks, pipe captured CO2 to nearby greenhouses. The CO2-rich atmosphere promotes the growth of food crops and the carbon dioxide is converted into carbohydrates, which stores it temporarily before the food is consumed.25 Other companies, including Carbon Engineering, are creating synthetic fuels, such as gasoline, diesel and jet fuel.26 This is achieved by obtaining hydrogen from water, then combining it with CO2 to make synthetic crude, or ‘syncrude’, then processed into fuels. The company claims that the produced fuels are cleaner-burning than fossil fuels and add ‘no new carbon dioxide to the atmosphere’.26
The electrochemical reduction of CO2 is another promising, but emerging, technique. Rather than making long-chain hydrocarbons (such as diesel), it makes shorter chain hydrocarbons such as methane (the main component of natural gas), ethane and ethanol, which are high-value commodity feedstocks. All of these short-chain molecules are in wide use today but are largely obtained from fossil fuels. Production of such molecules from CO2 would not only financially incentivise carbon capture technology but also has the potential to significantly reduce our dependence on fossil fuels and their high carbon footprint.
1.3 Amine-based materials for CO2 capture and utilisation (CCU)
Amines and polyamines have been identified as a group of compounds that are highly promising for CO2 capture and have found increasing application in methods to enable CO2 utilisation. This review will discuss the role of amine-based materials in CCU, with some focus on polyamines. In Section 2, we consider the parameters that define an ideal adsorbent and introduce the practicalities of analysing new adsorbents during the lab-based development stage. This includes comparing common practices used in CO2 capture performance evaluation and highlighting the factors that are most important in developing new or improving existing CO2 sorbents. In Section 3, we introduce a range of amines and amine-based CO2 adsorbents, and look at how these materials are developed and how their CO2 uptake behaviours are optimised. In Section 4, we look at methods by which the best adsorbents are identified for industrial scale-up. Section 5 considers some of the most successful commercial CO2 capture ventures to date, comparing their sorption technologies and CO2 markets. Finally, in Section 6, we review literature examples in which amines and related nitrogen-containing compounds have been applied in CO2 utilisation via electrochemical reduction.2 Adsorbent: parameters and analysis for CO2 capture
In this section, various parameters and performance indicators that are important for the evaluation of the separation potential of an adsorbent are defined. Also considered are the various methods and operating conditions used for the analysis of proposed CO2 adsorbents. This information is essential to the researcher for the improvement of existing, or discovery of new, carbon capture materials.2.1 Essential parameters of an adsorbent
The separation potential of an adsorbent for a given application is determined using three major parameters, namely: adsorption capacity, selectivity, and heat of adsorption. Adsorption capacity is defined as the amount of the adsorbate taken up by the adsorbent surface per unit mass of the adsorbent. It is the absolute amount of gas adsorbed and can be referred to as equilibrium or breakthrough capacity depending on the method of measurement (as discussed in Section 2.2). Researchers commonly focus on the adsorption capacity of an adsorbent as a screening metric. However, an equally important factor is the material's regenerability for it to be relevant in a real-time separation process. For this purpose, the concept of the working capacity (WC) of an adsorbent has been developed. Adsorbent working capacity is defined as the difference in adsorbate loading at adsorption and desorption conditions. In addition to separation potential, it is an essential parameter for sizing the separation unit and determining the capital cost involved.An important factor for evaluating an adsorbent separation potential is its selectivity. This is the preferential affinity of the adsorbent towards the desired adsorbate (CO2 for example) over other components present in the gas mixture (such as nitrogen) to achieve high product purity. Selectivity is defined as the mole fraction ratio of the desired component in the adsorbed phase to that in the gas phase at a certain condition. The heat of adsorption is another important adsorbent characteristic. This is a measure of the heat generated during the adsorption process and the heat required to carry out the desorption step. It determines the extent of thermal effects and the cost of separation in terms of the energy requirement.
An ideal adsorbent must have a high working capacity and preferential affinity/high selectivity with a low heat of adsorption. However, an adsorbent exhibiting a high heat of adsorption indicates a strong adsorbent–adsorbate interaction, and therefore high selectivity, but potentially high energy-demanding regeneration conditions. Hence, there is always a trade-off between these adsorbent performance parameters and it is challenging to obtain an adsorbent that meets all the desired criteria.
2.2 CO2 adsorption analysis methods and conditions
Carbon capture materials are analysed for their CO2 uptake behaviour by measuring adsorption on exposure to CO2 by different methods including gravimetric, volumetric, and breakthrough analyses. In gravimetric analysis, the sample is placed in a chamber into which a CO2-containing gas flows, and the relative weight increase of the sample is interpreted as CO2 uptake, Fig. 1a. These experiments are frequently carried out under atmospheric pressure by thermogravimetric analysis (TGA), or a custom-built system in which the sample is weighed before and after exposure to CO2. Examples include the CO2-filled balloon set-up used by Xu et al.,27 or the U-shaped glass tube gas delivery system used by Chen and co-workers.28 In volumetric analysis, the sample is placed in a cell and CO2 is introduced at certain increasing partial pressures, with corresponding pressure loss in the cell interpreted as CO2 uptake by the adsorbent, Fig. 1b. A breakthrough analysis is carried out by packing the sample into an adsorption column, or tube, through which pure CO2 or a CO2-containing gas of concentration C0 is passed. The CO2 concentration (C) in the gas exiting the adsorption column is measured using gas chromatography, mass spectrometry or infrared spectroscopy (IR) and this is recorded as a function of time to plot a ‘breakthrough curve’ from which the adsorption capacity is calculated, Fig. 1c. TGA is the most common adsorption analysis method, however, breakthrough analysis is the method most representative of the industrial gas adsorption process.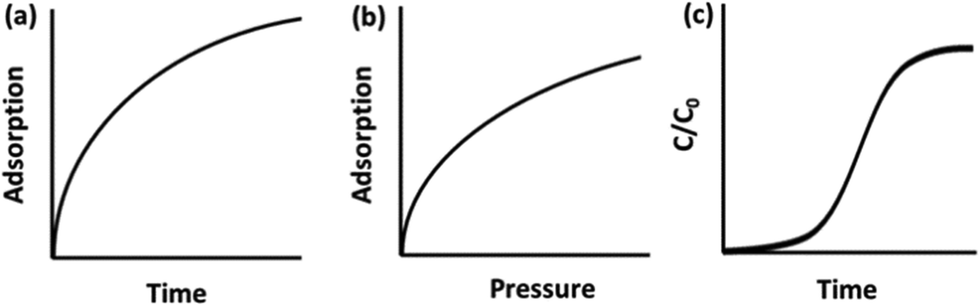 | ||
| Fig. 1 Typical graphs of raw data obtained for CO2 adsorption via (a) gravimetric; (b) volumetric; and (c) breakthrough analyses. | ||
In developing new adsorbents, researchers primarily focus on measuring CO2 capacity for various conditions of temperature, CO2 partial pressure and humidity. Often, CO2 adsorbents are tested under pure CO2 at 1 bar, as they perform better both thermodynamically and kinetically under a higher concentration of CO2.29 However researchers also set up conditions commonly designed to replicate either post-combustion/flue gas or DAC conditions. The relevant adsorption temperatures for DAC are from ambient to around 35 °C, and for post-combustion conditions from 40 °C,30 to above 100 °C.31 Industrial flue gases and air contain a mixture of gases, of which CO2 is a minor component, ranging in concentration from a global average of just above 410 ppm in air, to 5–15% for post-combustion emissions, depending on the fuel source.31 The remaining gas is mainly composed of various amounts of N2, H2O and O2. Therefore, for adsorption tests under a low partial pressure of CO2, a dilute source of 400 ppm or 10% CO2 within a mixed gas system is used. The balance is often composed of an inert gas such as N2 to best represent working conditions, however, Ar or He are also used in some cases. Alternatively, for DAC conditions, Goeppert et al. used ambient air directly from the laboratory, drying over silica gel prior to adsorption tests,32 while Gebald and co-workers used technical grade pressurised air with a CO2 concentration of 400–530 ppm.33 Under a volumetric apparatus set-up, a single component (CO2 only) gas system may be used to test for adsorption under low partial pressures. CO2 is introduced at increasing pressures to measure an adsorption isotherm, and adsorption at 400 ppm or 10% CO2 can be interpolated at 0.0004 bar or around 0.1 bar for DAC and post-combustion conditions respectively.34
Adsorption–desorption experiments are commonly conducted on new adsorbents, sometimes with up to 100 cycles.33,35,36 These are particularly valuable for extended-use stability studies. In designing these experiments, the times set for the adsorption and desorption steps vary widely, with the adsorption step not always sufficiently long enough to allow the adsorbent to achieve its equilibrium capacity, as would be most likely under industrial applications.
2.3 Reporting of CO2 adsorption data
CO2 adsorption capacity is commonly reported in the format of mmol CO2 per g adsorbent but is also reported as g or mg CO2 per g adsorbent or as weight% (wt%) of adsorbent. Studies on polyethyleneimine (PEI)-loaded fibre adsorbents, by Lebrache et al.37,38 and Sujan et al.,39 reported the CO2 capacity of amine-loaded fibres as mmol CO2 per g fibre. The ambiguity of this unit suggests that the reported uptake may not take into account the weight of the polyamine itself, which would be informative as to the overall efficiency of the adsorbent and enable clearer comparison to adsorbents outside of the study. Similarly, Kwon et al. have reported the CO2 adsorption of PEI impregnated silica in terms of mmol CO2 per g SiO2.29Cross-study comparisons of adsorbents’ reported CO2 capacities are not straightforward partly due to widely differing physical analysis conditions, but furthermore, adsorption is a dynamic process and defining boundaries is somewhat arbitrary, therefore, every capacity value comes with certain caveats. Researchers frequently carry out one adsorption event, which is continued until equilibrium capacity, or close to equilibrium capacity, is reached. This may take a matter of minutes or hours depending on the adsorbent and conditions, but the researcher must select an adsorption threshold at which point the capacity is measured. In reporting the results of their breakthrough analyses, Sujan and co-workers39 usefully recorded both a breakthrough and a pseudo-equilibrium capacity and defined these as uptake at the time at which 5%, and 95%, respectively, of C0, is detected in the column eluent (C). It followed that the breakthrough capacity was lower than pseudo-equilibrium uptake, at about two thirds. In contrast, Kwon et al. took the definition for the breakthrough CO2 capacity of PEI impregnated silica as the point at which C was 99% of C0.29 Similarly, Drese et al.40 took the final capacity of a pore-expanded hyperbranched aminosilica adsorbent at the point at which the uptake rate became less than 0.2 μmol CO2 per min, which gave a higher value than for when the breakthrough curve reached 95% of C0.
Although it is informative to determine the maximum uptake potential of an adsorbent, a high equilibrium capacity may not indicate a ‘good’ adsorbent if the CO2 uptake is slow. Knowledge of adsorption kinetics is necessary to fully understand the potential of an adsorbent. Chen et al. considered the CO2 adsorption kinetics of a PEI impregnated resin by looking at its adsorption behaviour over a range of temperatures from 25 °C to 90 °C.28 Building a model to describe adsorption with both a fast step (surface CO2-amine reactions) and a slow step (sub-surface CO2-amine reactions), it was found that 75 °C was the optimum temperature in terms of the adsorption rate and diffusion parameter, so further cyclic adsorption–desorption experiments were conducted at this temperature.
2.4 Adsorbent metrics and process performance indicators
The industrial deployment of CO2 separation presents many challenges to the scientist and engineer. One of these is the identification of suitable candidate adsorbents that perform favourably at industrially relevant conditions.To measure the CO2 uptake of an adsorbent, adsorption–desorption isotherms (primarily single component isotherms) are frequently collected. Isotherms of other gas components such as N2, CH4 and water may also be evaluated, often as single-component isotherms. Experimental single component adsorption data is fitted using isotherm models, e.g., Langmuir, Freundlich, Dual site Langmuir etc. Empirically extended forms of isotherm models, sometimes including the Ideal Adsorbed Solution Theory (IAST), are used to predict the multi-component adsorption isotherms to evaluate the adsorbent CO2 separation potential. The experimental measurement of multi-component adsorption isotherm data is a labour-intensive task. Therefore, very often, the more limited single-component adsorption isotherm data is used to assess adsorbents. However, single-component adsorption data cannot exhibit competitive adsorption between gas components (e.g. for CO2/N2); different gases can compete strongly for the same sites, especially in the case of physisorbents such as zeolites or MOFs. This could lead to uncertainty in the agreement between the experimental multi-component adsorption data and the predicted data based on the isotherm model. Hence, it is imperative to accurately predict the CO2 separation ability of the sorbents in the most industrially relevant conditions, which can be achieved by identifying and calculating the adsorbent screening metrics that are critical to recognising the best adsorbents at the process level. An adsorbent screening metric can assist in the initial screening of multiple adsorbents.
Over the last few decades, several metrics have been defined to determine the efficacy of adsorbents to accurately categorise them based on their equilibrium isotherm data, Table 1. Common metrics are the adsorbent working capacity and selectivity.
| Metric | Definition | Ref. |
|---|---|---|
q
i,ads,pure and qi,des,pure: single-component adsorption capacity for component i with i = 1, 2 under adsorption and desorption conditions, respectively. qi,ads,mix and qi,des,mix: multi-component adsorption capacity for component i with i = 1, 2 under adsorption and desorption conditions respectively. yi: molar gas phase composition (mole fraction) of component i with i = 1, 2. Hi: Henry's law constant for component i with i = 1, 2. WCi,pure: single component working capacity of component i with i = 1, 2. WCi,mix: multi-component working capacity of component i with i = 1, 2. α12,mix![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) atads and α12,mixatdes: equilibrium multi-component selectivity of component 1 over component 2 at adsorption conditions & feed composition and desorption conditions & feed composition, respectively. α12,mix: equilibrium multi-component selectivity of component 1 over component 2. ΔHads,1: heat of adsorption of component 1 (strongly adsorbed component). atads and α12,mixatdes: equilibrium multi-component selectivity of component 1 over component 2 at adsorption conditions & feed composition and desorption conditions & feed composition, respectively. α12,mix: equilibrium multi-component selectivity of component 1 over component 2. ΔHads,1: heat of adsorption of component 1 (strongly adsorbed component). |
||
| Adsorption capacity (mmol g−1) | q i,ads | Present work |
| Single component working capacity, WCi,pure (mmol g−1) | q i,ads,pure − qi,des,pure | Present work |
| Multi-component working capacity, WCi,mix (mmol g−1) | q i,ads,mix − qi,des,mix | Present work |
| Single-component selectivity, α12,pure |
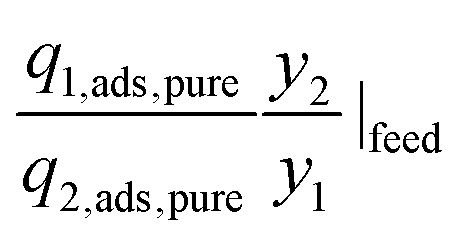
|
Present work |
| Multi-component selectivity, α12,mix |
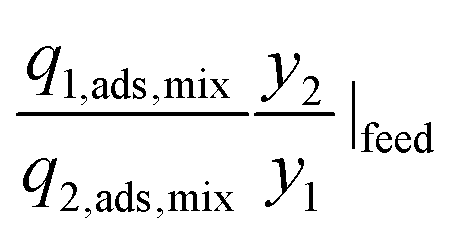
|
Present work |
| Henry selectivity, αH |
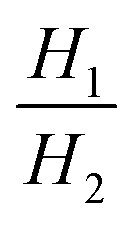
|
41 |
| Adiabatic separation factor, α |
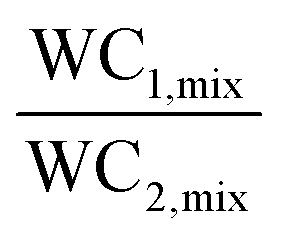
|
42 |
| Adsorption figure of merit (AFM) (mmol g−1) |
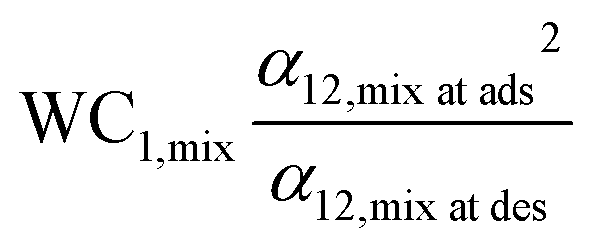
|
43 |
| PSA parameter, S |
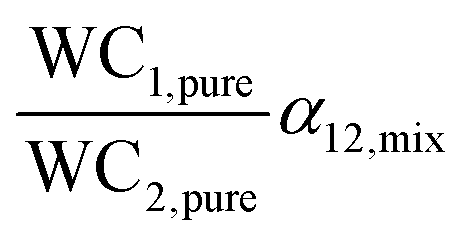
|
44 |
| Adsorbent performance indicator (API) |

|
45 |
It should be noted that absolute capacity is not a suitable measurement of a material's performance; rather working capacity is more informative. The working capacity of an adsorbent can be calculated for the i-th component of both single component (WCi,pure) and multi-component (WCi,mix) adsorption processes, with the multi-component WC being more relevant to CO2 separation processes. Harlick and Tezel used the working capacity to rank 13 zeolites for CO2 separation from flue gas in a pressure swing adsorption (PSA) process.46 In another study, the adsorption capacity of CO2 was used to rank over 120 adsorbents including zeolites, carbons, silica and MOFs under flue gas relevant conditions of 35 °C and 0.1 bar CO2.47
Selectivity is analogous to relative volatility in distillation and is another commonly used metric. It has been defined in different ways in the literature while the most regularly used form is given in Table 1. Again, it can be calculated for both pure-component and mixed-component gas adsorption processes. The difference between these two selectivities is with regards to accounting for competitive adsorption while calculating the equilibrium loading. One of the methods used to calculate an adsorbent selectivity is by simply taking the ratios of Henry's law constants. However, this is a very raw method for adsorbent selection based on single-component data.41
Many researchers have considered either of the metrics (working capacity/selectivity) while evaluating an adsorbent under a given set of operating conditions. Nevertheless, these metrics alone are not sufficient to evaluate the performance of the adsorbents, especially at the process level.48 Also, thermal fluctuations caused by the exothermic adsorption process are not accounted for by either of the parameters.49 Thus, other adsorbent performance metrics have been developed by encompassing both of these parameters with a weighing factor to each42,44 and are usually referred to as figures of merit/performance indicators. These metrics have been defined for a binary system with component 1 and 2 where component 1 is the more strongly adsorbed species than component 2.
Ackley and co-workers proposed the concept of the adiabatic separation factor (α) to evaluate the bulk separation performance of an adsorbent at or near the actual process conditions. This factor was defined as the ratio of the working capacities of two adsorbate components under non-isothermal multi-component conditions. For bulk gas separation, an adsorbent or a range of adsorbents can be selected using the adiabatic separation factor, the adiabatic working capacity of the desired product and/or the product of these two parameters. In this work, two adsorbents NaX and LiX were used for separating air into N2 and O2. Based on the adsorption isotherms, NaX is a weaker adsorbent for N2 adsorption compared to LiX. The adiabatic separation factor was calculated for both the adsorbents at various temperatures (240–340 K) and the weaker NaX turned out to be the preferred adsorbent for low temperature applications of <265 K on account of its higher α value up to 265 K.42
Notaro et al. proposed a lumped parameter called the adsorption figure of merit (AFM) for adsorbent selection for different sections of a bed, containing different adsorbents, to separate N2 from air using a PSA system. It uses the multi-component equilibrium loadings at adsorption and desorption conditions instead of pure component equilibrium data. For O2–N2 separation, the AFM was calculated for four different zeolite adsorbents at various adsorption temperatures (250–320 K). For a given adsorption temperature, the suitable adsorbent was the one with the highest AFM value at that temperature. For instance, NaX having the highest AFM value of 2.5 was the preferred adsorbent at the lowest temperature of 250 K while LiX and CaLiX were the preferred adsorbents for temperatures higher than 270 K.43 The rationale behind this metric is not explained and it is being used as an empirical rule of thumb.45
The PSA parameter (S) was proposed by Rege and Yang for adsorbent selection in a PSA system to separate a binary mixture. For this metric, the working capacity of both components was combined with the equilibrium selectivity of component 1 over component 2. The extended Langmuir isotherm model was used for calculating this metric implying α12,mix is constant for the entire partial pressure range and is equal to the ratio of Henry's constants (also called Henry's selectivity, αH) of the two components, though other models can be used to calculate the selectivity. A direct relationship between the PSA parameter and product recovery, purity and throughput for air separation using molecular sieve zeolites has been demonstrated. The PSA parameter S was calculated for LiX and NaX adsorbents for N2 and O2 separation for pressure ratios (adsorption pressure/desorption pressure) ranging from 2 to 10. An increase in the product (O2) recovery was observed with an increase in the value of the parameter S over the entire pressure ratio. Also, a higher value of S indicated better separation performance of the adsorbent with LiX being superior to NaX for O2 and N2 separation as reported in previous studies;44 however, the PSA parameter does not take into consideration the different objectives of the separation process such as bulk separation (concentration of the desired component in feed stream >10%) or gas purification (concentration of the desired component in feed stream <10%; usually <2%).45,50 This was accounted for by another lumped parameter known as the adsorbent performance indicator (API) proposed by Wiersum et al.45 It accommodates for the relative significance of various adsorbent properties, i.e. working capacity, selectivity and heat of adsorption (as shown in Table 1). The objective of this metric is to provide an initial assessment of adsorbents at an early stage for a given gas separation, thus, identifying the most promising adsorbents for additional assessment at the process level. The heat of adsorption (of the strongly adsorbed component) is in the denominator of the metric as heat produced during the adsorption process is deleterious to adsorption performance. A weighing factor (A, B and C) is assigned to each parameter to adjust their relative significance. For bulk separation, an adsorbent's working capacity is more significant, while for gas purification selectivity has higher significance. The default value of the weighing factors is set to 1 though they can be chosen arbitrarily or from experimental data for a particular gas separation. For all the above-mentioned metrics, the higher the value of the calculated metric, the better the separation performance of the adsorbent under given operating conditions.
Wiersum et al.45 used their API for comparing various adsorbents (zeolite, molecular sieve carbon, MOFs) for CO2/CH4 separation under two different processes (bulk separation and natural gas purification) and compared their results with those obtained by Rege and Yang's PSA parameter.44 Operating conditions involved adsorption and desorption pressures of 80 bar and 1 bar, respectively, with a feed composition of 50% CO2 and 5% CO2 (rest CH4) for bulk separation and natural gas purification, respectively. Both the parameters indicated that the zeolite adsorbent, having the highest selectivity, was the preferred adsorbent for natural gas purification. Values of S and API for the best performing zeolite adsorbent were very high, ca. 40 and ca. 200 respectively, compared to the rest of the adsorbent materials (with values of S and API in the range of 0–8 and 0–40 respectively); however, in the case of bulk separation, the API indicated that two mesoporous MOFs with higher volumetric working capacities were better choices having API values in the range of 8–10 compared to values of <6 for other adsorbents. In contrast, Rege and Yang's PSA parameter suggested the zeolite with a value of S ca. 75 was the preferred adsorbent though the zeolite had a much lower volumetric working capacity than the reported MOFs (the values of S for the MOFs were ca. 30). This could possibly be due to non-inclusion of the energy requirement for the large amount of CO2 in the PSA parameter. Krishna and co-workers defined a new metric called the separation potential by combining the adsorption capacity and IAST-based selectivity to screen and rank MOFs for CO2 separation in a fixed-bed adsorber. Diffusion limitations were ignored while deriving this separation potential and thus it can only be used for preliminary screening purposes without the need to perform detailed breakthrough experiments/simulations at the laboratory scale.51
Although limited research has been conducted on the specific grading of amine-based adsorbents according to their metrics as discussed above, these metrics are entirely applicable to such materials. One commonly asserted advantage of amine-based sorbents over physisorbents is that the strong interaction between CO2 and amine groups, as discussed in Section 3 and illustrated in Fig. 2, results in highly selective CO2 adsorption. Theoretically, this translates into high separation potential, however, this cannot be assumed as many amine-based adsorbents can have physisorbent components or character which may promote the uptake of molecules such as N2, water and CH4, therefore thorough selectivity analysis is essential.52
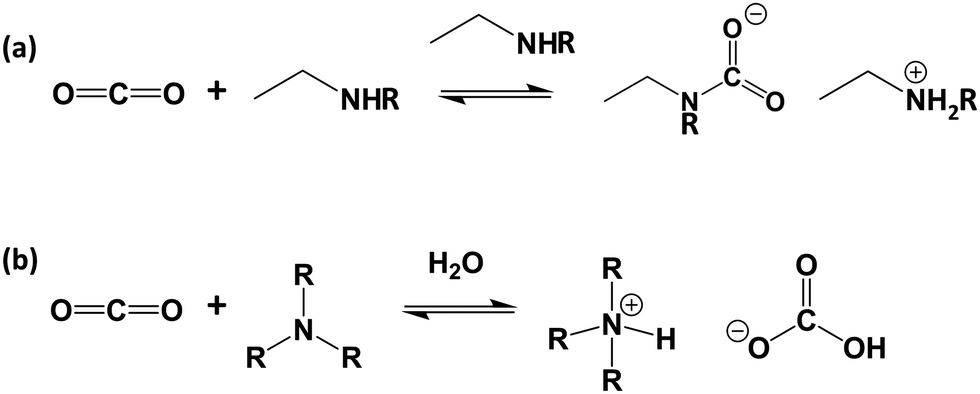 | ||
| Fig. 2 Mechanism of reaction of CO2 with (a) primary- or secondary-amines in the absence of water and (b) tertiary amines in the presence of water. | ||
The above-mentioned adsorbent metrics are employed to evaluate the separation potential of an adsorbent at the material development stage or at the lab scale testing. They are not representative of the technical and economic feasibility of the separation process under industrially relevant conditions. Thus, there is a need to define additional parameters to account for these aspects and to get a more precise idea as to the CO2 separation potential of an adsorbent at the process level. These parameters are product purity, recovery, productivity and parasitic energy. Their definitions are provided in Table 2. Parasitic energy is the amount of energy consumed in all the steps of a PSA or TSA (temperature swing adsorption) cycle per unit of the desired component captured.
| Performance indicator | Definition |
|---|---|
| Product purity (%) |
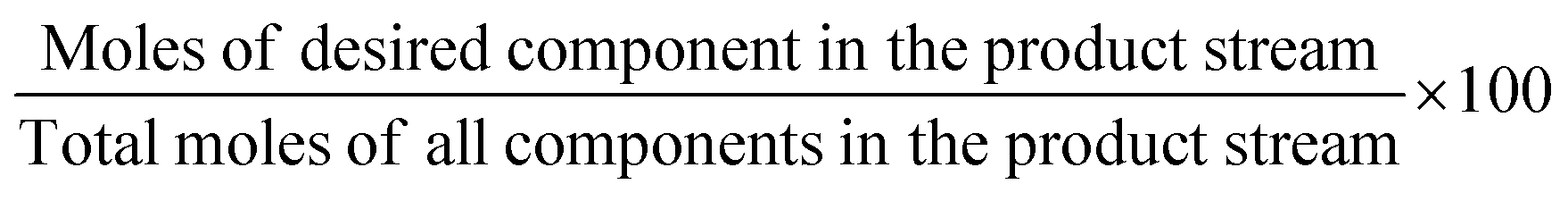
|
| Product recovery (%) |
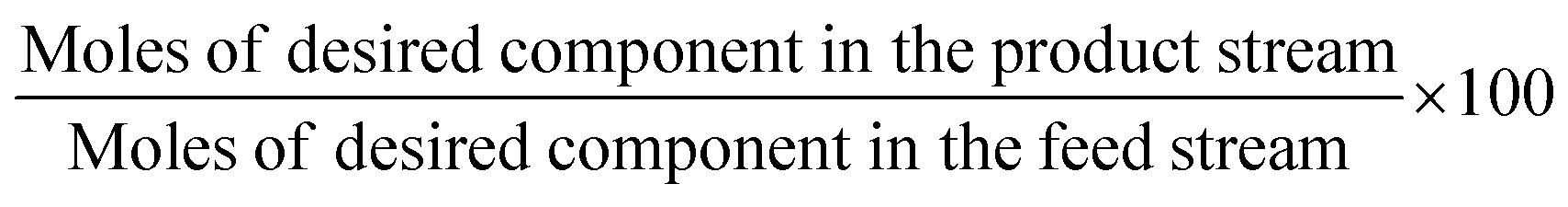
|
| Product productivity (mol kg−1 s−1) |
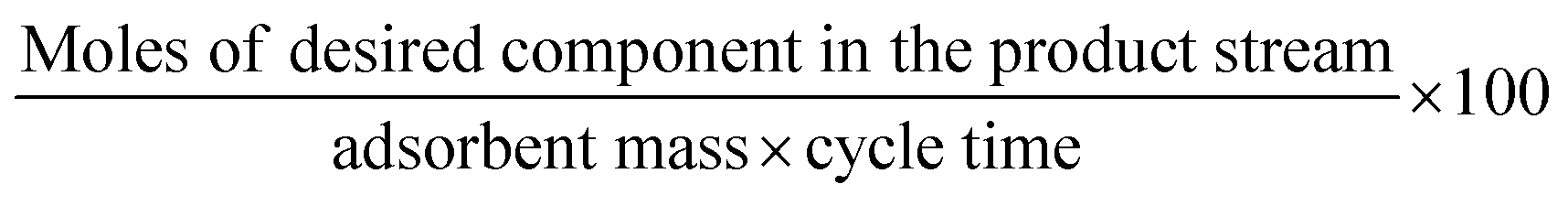
|
| Parasitic energy (kW h per ton) |
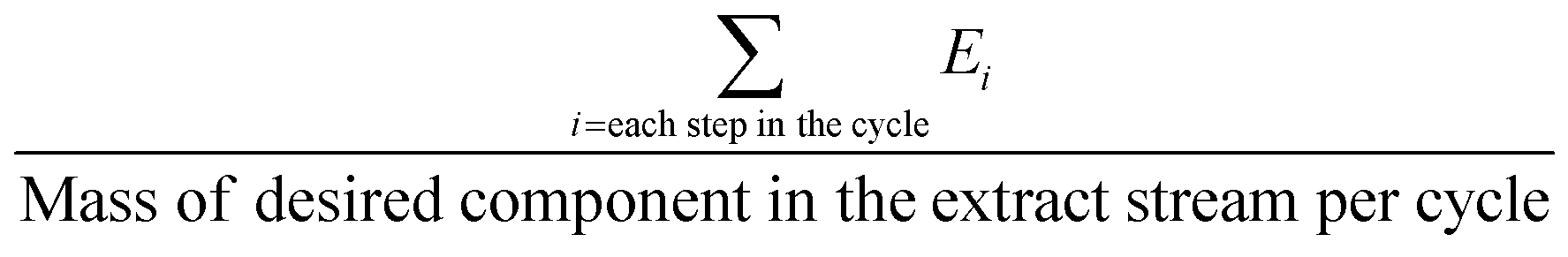
|
Molecular simulations were used to calculate the adsorption isotherms of zeolite and zeolitic imidazolate framework adsorbents and calculate Henry's coefficients, heats of adsorption, etc. as the experimental data is only available for a few materials. This screening study identified a minimum theoretical limit of parasitic energy (1060 kJ kg−1 CO2) and consequently, various zeolite structures meeting this requirement. This study can thus be used to focus efforts in the direction of synthesizing materials that can meet the limits of the minimum energy requirement.54
Where it comes to amine-based adsorbents, purity, recovery, productivity and parasitic energy are highly important and relevant parameters. Due to the strong interaction between CO2 and amines, which affords these sorbents their often-high selectivity, they generally display high heats of adsorption in the chemisorption range of about 50–90 kJ mol−1,55 as discussed further in Section 3. If this can afford these adsorbents a high product purity output and high product recovery it may also potentially result in lower productivity and higher parasitic energy due to the energy required to desorb the captured CO2. It is necessary for all the relevant empirical data to be gathered on amine-based adsorbents in order to get a fuller understanding of their behaviours. The extent to which the balance between purity and recovery, productivity and parasitic energy lies in favour of these adsorbents’ application at the process level ultimately depends on their design and the resulting material properties and characteristics.
Performance targets for CO2 capture systems have been set by the U.S. Department of Energy (DOE) at 95% CO2 purity and 90% recovery. It is more feasible to achieve these targets where the desired product is the weakly adsorbed component (also called the light product) and is collected during the adsorption step, for instance H2 purification. During the adsorption step, the heavy product is adsorbed on the adsorbent surface while the light product exits the bed and is collected. Hence, the purity and recovery of the desired light component will not be impeded here by the heavy component. However, given that CO2 is the heavy product in a given gas mixture, i.e. it adsorbs first on almost all the applicable adsorbents, during regeneration/blowdown (collection of the heavy component), the CO2 is accompanied by some light product (usually N2) present in the gas phase which makes it more difficult to reach these targets.
Productivity and parasitic energy define the economic feasibility of the capture process. A separation cycle must be configured such that maximum productivity is achieved with the minimum parasitic energy. However, there is often a trade-off between these two parameters, i.e. an adsorbent meeting the technical requirements of a CO2 separation process could have high productivity with a high energy requirement or low productivity but with a low energy requirement.
Berger and Bhown developed a simplified model to calculate the parasitic energy of CO2 separation using a TSA process for the in silico screening of a huge number of zeolite-based adsorbents (both physisorbents and chemisorbents).56 Lin et al. also used parasitic energy as a metric for screening thousands of zeolite and zeolitic imidazolate framework adsorbents in a PTSA (pressure–temperature swing adsorption) process for dry flue gas separation.54
3 Carbon dioxide adsorbents
3.1 The role of amine functionality in CO2 capture
In the solution phase, amines react with CO2 by an established acid–base reaction via a zwitterion mechanism, published by Caplow in 1968,57 and outlined in Fig. 2a. In the case of primary or secondary amines, the reaction involves the initial formation of a C–N bond to form carbamic acid, followed by transfer of the amine proton of the carbamic acid to a free base such as a second amine (or water), forming the carbamate salt. Tertiary amines do not have a free proton so cannot form carbamic acid to react with CO2 by the same route taken by primary and secondary amines. Rather, they form highly unstable carbamates due to steric hindrance,58 and as such, tertiary amines react via a different route, only possible in the presence of water, as outlined in Fig. 2b. This mechanism, which was proposed by Donaldson and Nguyen in 1980,59 involves the tertiary amine acting as a base catalyst to deprotonate water, which, during deprotonation, reacts with CO2. The final product of the reaction is ammonium bicarbonate. Generally, the kinetics of the formation of the bicarbonate is slow, therefore tertiary amines have the lowest reaction rates with CO2.60For both mechanisms, the presence of water enables each amine to adsorb one CO2 molecule, therefore giving an amine efficiency (moles of CO2 per moles of amine) of 1. Under dry conditions, two molecules of primary or secondary amines are required to react with one molecule of CO2, giving a lower maximum amine efficiency of 0.5.23 The formation of the bicarbonate ion is also beneficial in the transport of CO2 through an adsorbent, as the bicarbonate ion is able to ‘hop’ efficiently between fixed quaternary ammonium sites.61,62
The products of the adsorbed CO2 on reaction with amines in the solid phase has been elucidated primarily with the application of FTIR studies63 and solid state NMR.64 Under dry conditions, carbamate and carbamic acid species have been identified, and in the presence of moisture, a number of structures can form including monodentate carbonate, bidentate carbonate, monodentate bicarbonate and bidentate bicarbonate.23,65 These different species may be formed under slightly variable conditions depending on the duration of adsorption runs, molar ratios of CO2 and water molecules, and factors specific to the amines such as density and mobility.23 In order to optimise CO2 adsorption, it is desirable to understand and control the adsorption products.
Studies by Zelenak et al.66 and Hahn et al.67 considered and compared the mechanism of CO2 adsorption by (3-aminopropyl)trimethoxysilane (APTMS) and trimethoxy[3-(methylamino)propyl]silane. Zelenak et al. looked at the effect of basicity on CO2 adsorption and found that under dry conditions, the lower reactivity of the secondary amine to CO2, compared to the primary amine, is likely more due to greater steric hindrance and lower accessibility of the lone pair of electrons, rather than due to differences in basicity.66 Hahn et al. reported that primary amines react preferentially to form intermolecular ammonium carbamates, but that secondary amines were able to stabilise carbamic acid at high amine densities due to higher basicity; however, they could also form interactions with Si–OH groups of the support material, preventing interaction with CO2.67
Research by Didas and co-workers compared the CO2 adsorption of primary, secondary and tertiary amines tethered to a silica support. They found primary amines to be the most effective for CO2 capture in both dry and humid conditions, giving higher adsorption capacities and amine efficiencies than secondary or tertiary amines.68 At low loadings they were found to have a higher heat of adsorption. At zero coverage CO2, (on first contact between the adsorbent and CO2), the heats of adsorption for the model primary and secondary amines, (3-aminopropyl)trimethoxysilane and N-methylaminopropyl-trimethoxysilane, were calculated as 130 kJ mol−1, and 88 kJ mol−1, respectively.68 Their higher reactivity to CO2, and the higher heat of adsorption of primary amines can be advantageous in increasing adsorption rate,23 and would explain the higher amine efficiencies of primary amines when exposed to CO2 at ultra-low partial pressures.68 However, since the heat of adsorption represents the energy required to remove CO2 to re-generate the adsorbent,69 the lower heat of adsorption of secondary and tertiary amines would be advantageous in reducing regeneration energy.58,70,71 More recent work by the Jones group, in which heats of adsorption were measured directly by calorimetry, found that the heats of adsorption of strongly basic primary and secondary amines were approximately equal, at around 90 kJ mol−1.72 Considering the CO2 uptakes and heats of adsorption for a range of aminosilanes of varying steric constraints, the authors postulated that the higher CO2 adsorption observed for primary amines under dilute conditions is more likely to be dependent on entropic rather than enthalpic factors.72
Fundamental CO2-reactivity studies are often carried out on adsorbents with discrete amino molecules of defined functionality. Results of these studies do not consistently translate for many polyamine-based adsorbents, which may contain a range of amine functionalities. For example, in contrast to Didas et al.'s study on smaller molecule amine adsorbents,68 a study on two large polymer-based adsorbents featuring either only primary amines or a mixture of primary, secondary and tertiary found that the former was not superior in terms of CO2 capacity especially at higher loading.73 It was suggested this was due to the primary amines occurring on pendent chains of the hydrocarbon backbone, while the mixed amine adsorbent featured primary amines at the ends of the chains, thus being far more accessible to CO2.73 Accessibility is a crucial factor in adsorbent performance, along with stability. These are highly influenced by many elements including different supporting materials or modifiers, which can affect the chemistry of the amine, loading – which is related to density, and molecular size, often affecting mobility. These topics, with respect to polyamine adsorbents in particular, are discussed in Section 3.3 after an introduction to the variety of polyamine-based CO2 adsorbents in Section 3.2.
3.2 Solid polyamine CO2 adsorbents
Solid polyamine sorbents are a broad and varied family of materials. In the most common instances, amines are supported on another material, described as supports, which impart the solid structure. There is a vast range of materials that are used as supports including, but not limited to, inorganic oxides, polymeric fibres, carbon-based materials and organic and coordination polymers. Polyamines may also be cross-linked to form a solid network by reaction with a minor component co-polymer.In this section, we reflect on the diversity of solid polyamine CO2 capture materials. These have been divided according to support material: (i) polyamines supported on inorganic oxides; (ii) polyamines supported on other materials; and (iii) polyamine adsorbents that do not feature a support material. We then consider the major factors affecting their adsorption behaviours and how challenges have been overcome from a materials design perspective. Table S1 (see ESI†) presents the CO2 adsorption data for a range of selected adsorbents where it is reported for low partial pressures relevant to direct air capture (DAC) conditions (∼400 ppm CO2) and post-combustion conditions (5–15% CO2).
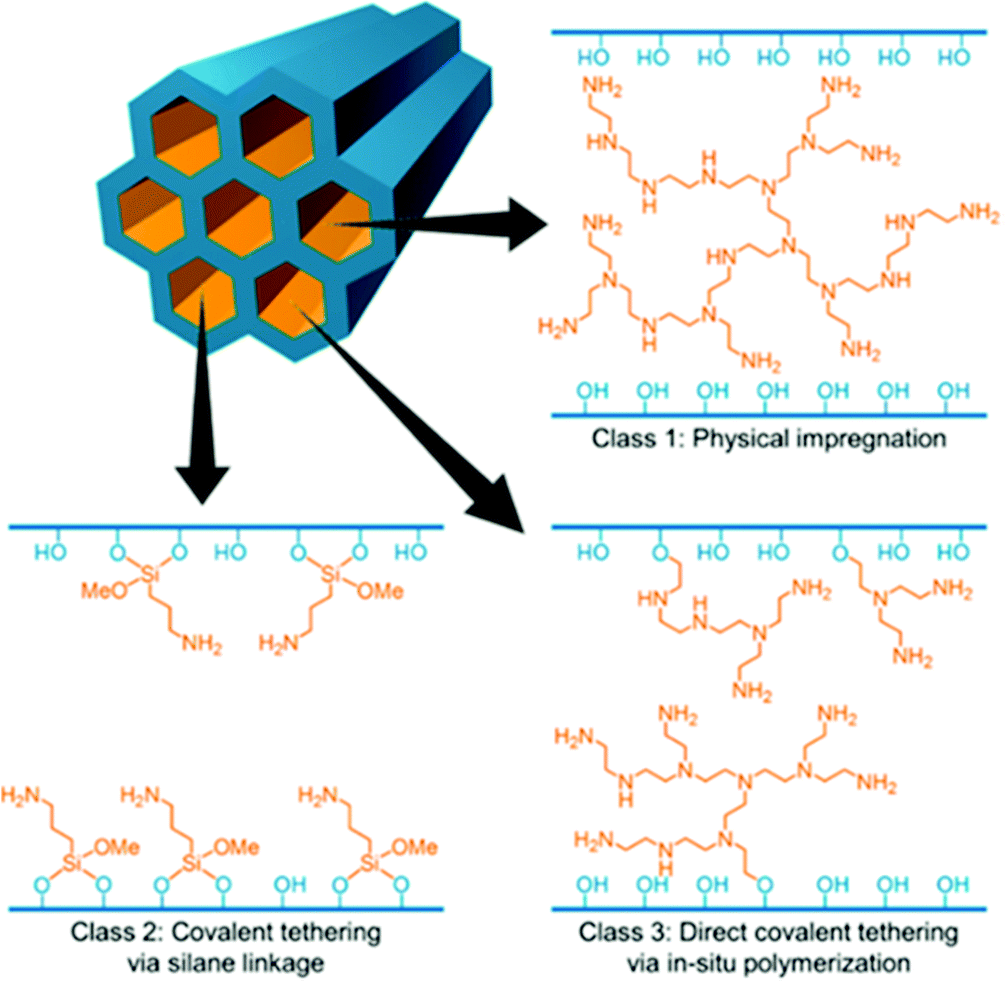 | ||
| Fig. 3 Representation of the three classes of supported amine-based adsorbents. Reprinted with permission from ref. 75. Copyright (2015) American Chemical Society. | ||
Class 1 materials are based on a high surface area porous support physically impregnated with monomeric or polymeric amine species, bonding through hydrogen bonds and/or dipole interactions. There are countless examples of these types of materials, with Xu et al. among the first to report a CO2 sorbent composed of PEI impregnated into the pores of a mesoporous silica, MCM-41, in 2002.76 These materials are generally prepared by conventional wet impregnation routes. One example of this technique is that used by Niu et al. in preparing silica nanotube/PEI nanocomposites; the polyamine is first dissolved in methanol then added to silica and stirred to form a slurry before drying under vacuum and/or under heat.77 The most ubiquitous polyamine used in class 1 adsorbents, and indeed many other types, is polyethyleneimine (PEI), shown in Fig. 4, an easily synthesised, multipurpose polyamine.78 PEI has a high amine content, with one amine for every two carbon atoms and is most often used in the branched rather than linear form, with an average molecular weight from Mw 400 Da to Mw 25000 Da. In the branched form, the ratio of primary:secondary:tertiary amines is 1:1.2:0.76.79 Class 1 PEI adsorbents have proven highly promising candidates for industrial CO2 capture.29
 | ||
| Fig. 4 Polyethyleneimine (PEI) in its (a) branched, and (b) linear form. | ||
Class 2 materials feature low molecular mass amines tethered to a support, commonly as amine-containing organosilanes covalently bonded to silica, or via amine side chains of a polymeric support. These adsorbents were first reported in 1992 by Tsuda et al. In one paper, amino silica gels were prepared by hydrolysis condensation of the products of polyethyleneimines or macrocycle polyamines reacted with trimethoxy(2-phenylethyl)silane.80 In the other paper, adsorbents were prepared by hydrolysis condensation of aminoalkyltrimethoxysilanes.81 Class 2 materials can also be prepared by grafting amino silanes such as 3-[2-(2-aminoethylamino)ethylamino]propyl trimethoxysilane to a silica support under hydro/solvothermal conditions as described in papers by the group of Sayari – particularly recognised for research on class 2 materials.82–85 Within the past decade, a ‘hybrid’ between a class 1 and a class 2 material has emerged in which a porous silica support is both impregnated with PEI and also grafted with (3-aminopropyl)triethoxysilane (APTES).86,87 In 2016, this new type of material was proposed as a ‘Class 4’ sorbent, and research has been focussed particularly on understanding the origins of its high thermal stability (vide infra).88 Class 3 materials feature a porous support within which amine monomers are polymerised in situ within the support. These first emerged in 2008 when Hicks et al. reported a hyperbranched aminosilica material synthesised from the reaction of aziridine with surface silanols of calcined SBA and their subsequent polymerisation in anhydrous toluene.89 In 2013 Chaikittisilp and co-workers reported a novel vapour-phase transport synthesis method to produce a class 3 sorbent using SBA-15 mesoporous silica and alumina supports with aziridine and azetidine monomers.90
000 Da) as an alternative to cellulose acetate.38 PEI (Mw 10000 Da) cross-linked by the epoxy resin bisphenol A,91 or epichlorohydrin92 has also been impregnated onto glass fibres.
Graphene has been employed in a polyamine sorbent in several examples. Yang et al. prepared sandwich-like graphene oxide-based silica sheets on which they impregnated PEI (Mw 800 Da),93 and in 2017, Gadipelli et al. reported an extremely effective CO2 sorbent composed of a highly hierarchical meso and macro-porous graphene-oxide network impregnated with triethylenetetramine (TETA), shown in Fig. 5.94 In exploring other carbon based substrates, Dillon et al. compared the CO2 adsorption of PEI (Mw 25000 Da)-functionalised carbon nanotubes and graphite/graphene composites.95
 | ||
| Fig. 5 SEM micrograph of exfGO-D_7.0TETA, exfoliated graphene-oxide impregnated with 7.0 g g−1 TETA. Reproduced from ref. 94 with permission from the Royal Society of Chemistry. | ||
Versatile CO2 sorbents have been synthesised by impregnating PEI onto resins,28 and polymers of intrinsic microporosity (PIMs), with the benefit of PIMs being the ability to dissolve and process the support into desired geometries appropriate for the adsorption system (Fig. 6).34 There are several examples of polyamines being used in conjunction with MOFs96,97 and MOF/SiO2-hybrids.98
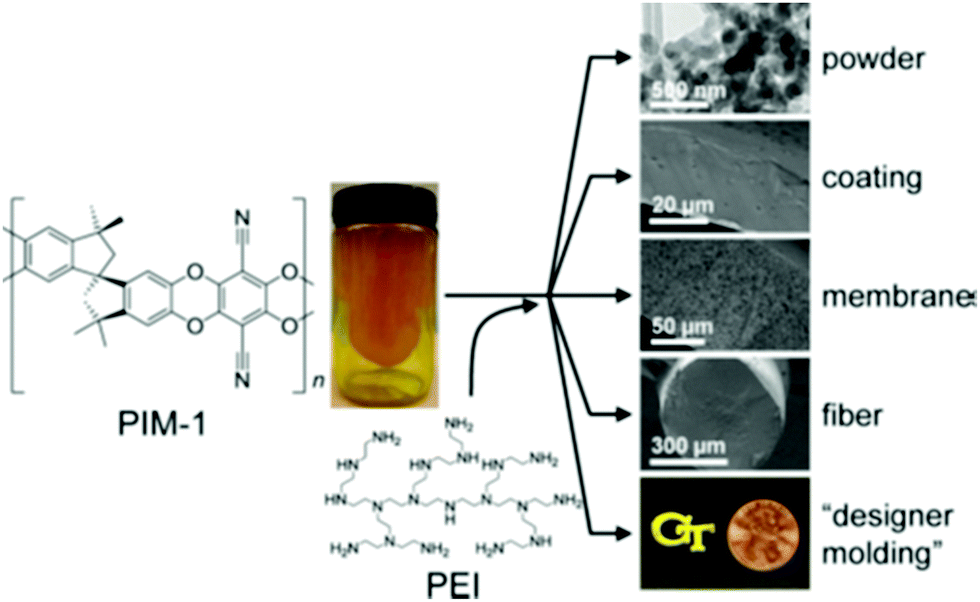 | ||
| Fig. 6 PIM 1 as a solution-processable support for PEI and electron microscopy images show meso-/macroporosity engineered into final structure. Reprinted with permission from ref. 34. Copyright (2015) American Chemical Society. | ||
An amine-functionalised nanofibrilated cellulose sorbent has been prepared by adding N-(2-aminoethyl)-3-aminopropylmethyldimethoxysilane to a cellulose hydrogel,33 and acrylamide modified polypropylene fibres have been grafted with hyperbranched polyamines synthesised from the reaction between pentaethylenehexamine (PEHA) and methyl acrylate.99 In an innovative approach to utilising polyamines in a CO2 physisorbent, PEHA and tetraethylenepentamine (TEPA) and have acted as templating reagents in the synthesis of an inorganic gallium phosphite framework featuring 18-membered-ring channels.100
In 2012, Wang et al. formed porous polyamine particles by the precipitation polymerisation of N-methyl-N-vinylformamide and di[2-(N-vinylformamido)ethyl].101 Later in 2015, Lee et al. reported the synthesis of a CO2-sorbent mesoporous resin by the condensation polymerisation of melamine and phenol resin monomers.102 Also in 2015, Sun, Liu and co-workers published two papers in which polymers were synthesised by nucleophilic substitution between small molecule primary diamines and the alkyl chloride monomer 2,4,6-tris(chloromethyl)-mesitylene.71,103 The resulting covalent organic frameworks had secondary amine functionality. Kumar et al. also explored this synthetic approach in developing the polyamine component for supported adsorbents in 2020, by reacting ethylenediamine and propylenediamine with 1,3,5-tris(bromomethyl)benzene and hexakis(bromomethyl)benzene.104 In a different approach, Huang et al. incorporated amine functionality into existing polymer networks by the post-modification of a divinylbenzene–maleic anhydride (DVB–MAH) copolymer by reaction between the acyl chloride groups of the polymer and a series of small molecule and linear amines.105
Another method to synthesise cross-linked polyamines is via the reaction of an existing polyamine with a cross-linking unit. Varying the relative amounts of reactants can control the extent of cross-linking – and therefore the abundance of primary and secondary amines, influencing the material properties. This technique has given rise to a new and diverse group of unsupported materials, which could be considered a new ‘class’ of polyamine-based adsorbent.
In 2014, Andreoli et al. formed a solid CO2-sorbent by cross-linking PEI (Mw 25000 Da) with fullerene C60.106–108 The material's sorption performance has been investigated after conversion to N-doped graphitic carbon via pyrolysis,109 and improved by spray-drying,110 and C60 has also been used to cross-link polypropylenimine (PPI) dendrimers.111
The reaction of polyamines with aldehyde groups to form imines has been utilised by Hwang et al. in cross-linking PEI (Mw 25000 Da) with glutaraldehyde,112 and by Thompson et al. in cross-linking PEI (Mw 600 Da),113 and TEPA,114 using polyaldehyde phosphorous dendrimers. In 2018, Mane et al. further developed the work of Sun et al. from 2015, using nucleophilic substitution of an aromatic alkyl chloride, by switching the diamines for PEI (Mw 1800 Da) (Fig. 7).115 Epichlorohydrin, used to cross-link PEI supported on glass fibres in 2008,92 was employed in 2018 by Xu and co-workers to form unsupported PEI (Mw 25000 Da) hydrogel beads.27 2019 saw the publication of three studies in which diepoxy crosslinkers yielded effective CO2 adsorbents. The Andreoli group studied a range of materials synthesised from the cross-linking of PEI (Mw 25000 Da) with the epoxy resin, bisphenol-A diglycidyl ether (DER),116 while the Jones group used poly(ethylene glycol)diglycidyl ether to cross-link PEI (Mw 750000 Da) via an ice-templating method, Fig. 8.117 Xu et al., building on their work on hydrogel beads, swapped epichlorohydrin for 1,3-butadiene di-epoxide to cross-link PEI of molecular weights between 800 and 750000 producing a high water content ‘PEI snow’.118 In 2020, the researchers further developed this work by employing the triepoxy–triglycidyl trimethylolpropane ether as the cross-linker which could yield a powdery material.119
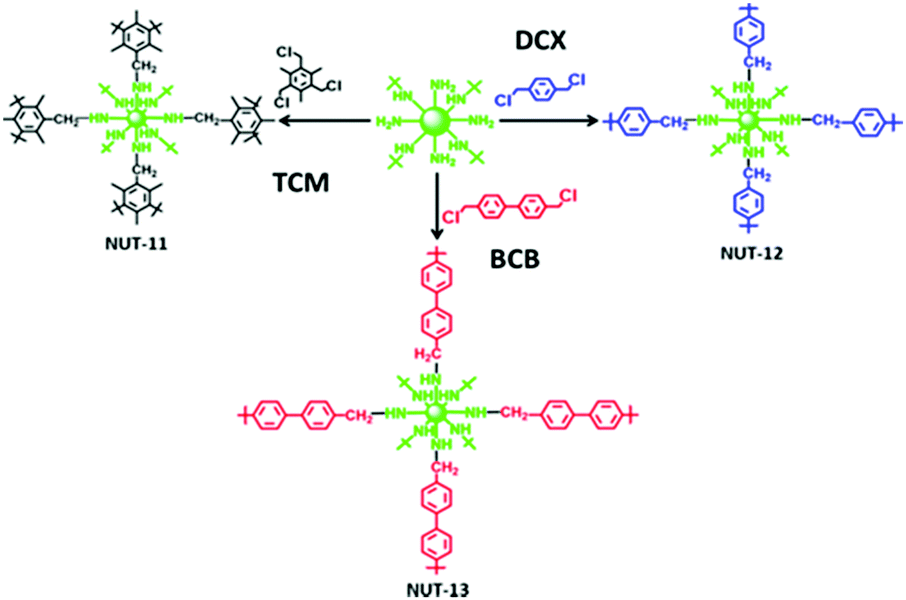 | ||
| Fig. 7 Cross-linking of PEI with 2,4,6-tris(chloromethyl)mesitylene (TCM), 4,4′-bis-(chloromethyl)-1,1′-biphenyl (BCB), and p-dichloroxylene (DCX). Reprinted with permission from ref. 115. Copyright (2018) American Chemical Society. | ||
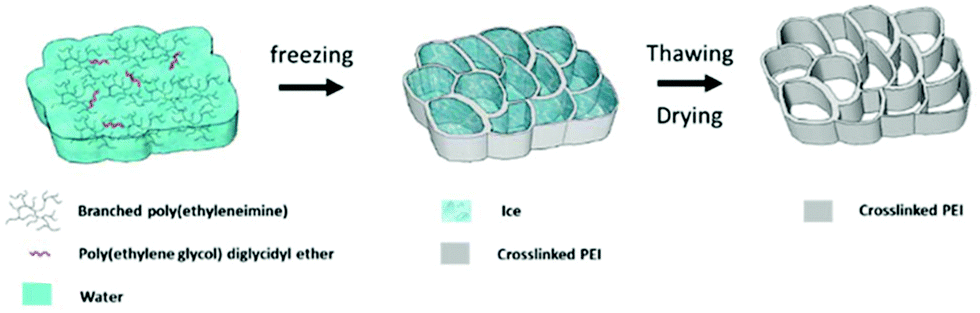 | ||
| Fig. 8 Schematic synthesis of self-supported adsorbent synthesised from cross-linking PEI (Mw 750000 Da) with poly(ethylene glycol). Reproduced from ref. 117 with permission from the Royal Society of Chemistry. | ||
3.3 Factors affecting CO2 adsorption
Water has also been shown to have a beneficial effect on CO2 adsorption capacity and kinetic uptake, to a greater or lesser extent, in many studies in which a polyamine adsorbent is exposed to moisture prior to and/or during adsorption.27,94,99,108,116 Water may promote adsorption in several ways. Firstly, in the presence of water, the theoretical overall amine efficiency is improved as one mole of CO2 can react with one mole of amine to form one mole of ammonium bicarbonate, Fig. 2.23,65 Secondly, in promoting the formation of the bicarbonate ion, water may suppress the formation of ionic cross-linking between alkylammonium carbamate entities, which it has been suggested form a surface diffusion barrier to CO2.29,121 Thirdly, it has been proposed that water can act as a diffusive intermediate, aiding adsorption by acting as a free base for the stabilisation of the zwitterion during carbamate formation, in place of a second amine.122 This adsorption enhancing effect has also been demonstrated in the presence of methanol, which may also act as a diffusive intermediate in the same way as water.108 Fourthly, by disrupting inter/intra molecular interactions within the polyamine layer and weakening hydrogen bonds between polyamine chains, water may have a plasticizing effect and increase polymer flexibility.121
Hyperbranched polyamine grafted acrylamide-modified polypropylene fibres had a high adsorption capacity of 5.64 mmol CO2 per g at 25 °C in 10% CO2/N2 due to low mass transfer resistance after swelling in water.99 Similarly, while dry PEI hydrogel beads adsorbed negligible amounts of CO2 under 15% CO2/N2 at ambient temperature, they adsorbed 1.37 mmol g−1 CO2 when soaked to 70 wt% water content due to better amine accessibility via the water filled pores.27 Glycerol has also been shown to act as a plasticiser; when incorporated into the structure of PAI/silica/PEI hollow fibre adsorbents, breakthrough CO2 capacity was improved by 60%.38 Finally, water can inhibit the formation of urea, which is a common issue in the deactivation of polyamine adsorbents, and a urea-deactivated adsorbent may become fully restored to the active amine adsorbent by hydrolysis under a flow of humid gas.123
3.3.2.1 Polyamine loading. The relationship between polyamine loading and CO2 adsorption has been explored frequently. Increased polyamine does not necessarily translate into higher adsorption capacity, as excess loading causes the pores of a support to become susceptible to blockages due to thick agglomerations, hindering the diffusion of CO2. Ionic cross-linking between alkylammonium carbamate ions at the surface of the polyamine layer may contribute to this diffusion barrier,29 therefore high dispersion of the polyamine is advantageous.
Niu et al. reported adsorption by nanocomposites of mesoporous silica nanotubes impregnated with PEI (Mw 800). At 50 °C under 60% CO2, adsorption increased with PEI loadings up to 50 wt%, but at 60 wt% PEI, CO2 adsorption reduced from 1.83 mmol g−1 to <1.5 mmol g−1.77 Similarly, PEI (Mw 800)-impregnated PIM-1 in powdered form was found to have higher CO2 uptake and amine efficiencies with a 21 wt% PEI loading, compared to 25 wt% loading at 35 °C, under CO2 partial pressures corresponding to 400 ppm CO2 and 10% CO2. The 21 wt% loaded sample adsorbed 0.23 mmol g−1 (0.048 amine efficiency) and 1.15 mmol g−1 (0.24 amine efficiency) at pressures representing 400 ppm and 10% CO2, respectively.34 Goeppert et al. tested the CO2 adsorption of PEI (Mw 25000) deposited on fumed silicas under air at 25 °C.32 Although the sample with 50 wt% polyamine loading displayed a higher dry CO2 capacity of 1.71 mmol g−1, compared to 1.18 mmol g−1 for the sample with 33 wt% loading, the latter completely adsorbed CO2 from the gas flow over a longer period of time (8.3 hours) then had a much faster saturation rate, as shown in Fig. 9. This was likely due to better CO2 diffusion and better dispersion of PEI, reflected in a surface area and pore volume almost three times those of the 50 wt% sample at 79.9 m2 g−1 and 1.057 m2 g−1, respectively.32 Interestingly, it was observed that under humid conditions, while the 33 wt% adsorbent showed increased uptake to 1.77 mmol g−1, the 50 wt% PEI loaded adsorbent had a reduced CO2 uptake of 1.41 mmol g−1. This was attributed to the water blocking the pores of the 55 wt% adsorbent, reducing access to the already least exposed amine groups.
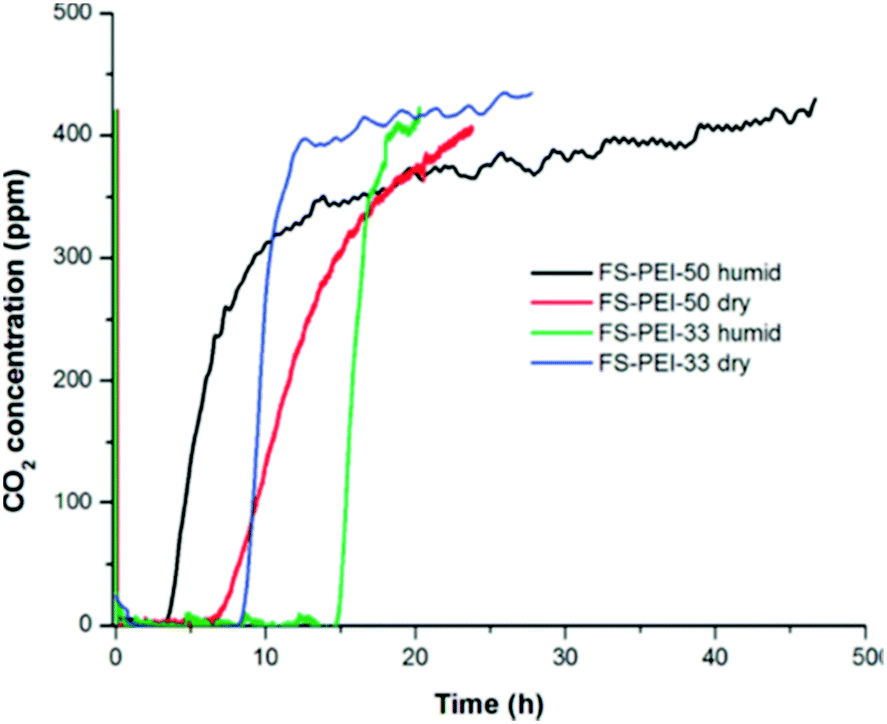 | ||
| Fig. 9 Adsorption of CO2 from the air at 25 °C on FS–PEI-50 and FS–PEI-33 under dry and humid conditions, illustrating longer period before breakthrough for FS–PEI-33 (33 wt% polyamine loading). Reprinted with permission from ref. 32 Copyright (2011) American Chemical Society. | ||
Maresz et al. reported decreased CO2 adsorption (under pure CO2 at 75 °C) with increased PEI loading on hierarchically porous silica monoliths with surface areas of 275 m2 g−1 and 344 m2 g−1.124 Using a larger surface area support of 651 m2 g−1 resulted in improved adsorption with increased polyamine loading, up to 4.16 mmol CO2 per g for the 60 wt% PEI adsorbent. The authors calculated that the lower surface area materials had double the number of PEI layers thus hindering CO2 diffusion.124
Lin et al. compared the CO2 uptake capacity and the amine efficiency of three composite materials consisting of PEI (Mw 600) impregnated into; SBA-15 with a 2-D hexagonal mesostructure, ethane-bridged organosilicas with a 2-D hexagonal mesostructure and ethane-bridged organosilica nanotubes of diameter ∼6 nm.125 Materials of various PEI loadings were tested under 0.1 bar CO2 and at 30 °C, and it was found that the nanotube-based adsorbents displayed the highest adsorption (Fig. 10). Unlike the other adsorbents, increasing the amine loading increased CO2 capacity, and the highest amine efficiency of 0.22 was achieved at a loading of 11.2 mmol N per g for this material. The authors suggest that the high CO2 uptake and amine efficiency is achieved despite the high PEI loading, which resulted in pore blockage for the other adsorbents, due to the hierarchical bimodal porous nanotube structure promoting high dispersal of the PEI, and thus good access to CO2 at low pressure.125
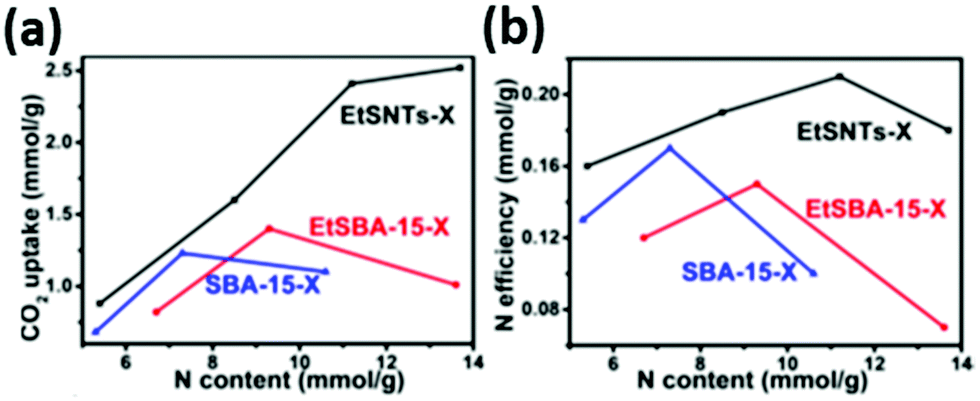 | ||
| Fig. 10 The relationship between (a) CO2 uptake and (b) N efficiency with N content of PEI/silica composites at 30 °C and 0.1 bar CO2. Adapted with permission from ref. 125. Copyright 2020, John Wiley and Sons. | ||
Drese et al. explored the use of pore-expanded, ordered mesoporous silicas of pore diameters 11.4–21.6 nm for the synthesis of class 3 adsorbents in order to increase polyamine loading without causing pore blockage.40 Aziridine polymerisation terminated without causing pore blockage and final amine loadings were lower than expected, therefore the adsorbents captured lower quantities of CO2 compared to the non-pore expanded materials, with adsorption increasing with amine loading. The highest uptake was 0.85 mmol g−1 for PEHAS13, which had an amine loading of 4.49 mmol N per g.40 Higher organic loadings were achieved on a class 3 adsorbent by Chaikittisilp et al. who employed a vapour-phase polymerisation technique to synthesise SBA-15 mesoporous silica-supported PEI adsorbents.90 In a 10% CO2 source at 25 °C, the adsorbent with an organic loading of 31.1 wt% (7.23 mmol N per g) adsorbed 0.93 mmol CO2 per g, with an amine efficiency of 0.13, while increasing PEI loading to 41.5 wt% (9.66 mmol N per g) decreased adsorption to 0.43 mmol CO2 per g, and amine efficiency to 0.04.90
As mentioned in Section 3.3.1, additives to the polyamine can significantly affect adsorption performance. Sakwa-Novak et al. reported that the incorporation of PEG 200 (polyethylene glycol 200) into a silica-supported PEI (Mw 800) adsorbent increased the amine efficiency at any polyamine loading by up to 60%.126 That the highest loaded adsorbents displayed overlapping data suggested that the amount of PEI on the silica wall or in the bulk phase might have been similar. Comparing their CO2 uptakes from 30 to 50 °C, it was found that the adsorbents with PEG 200 showed a more drastic decrease in amine efficiency at higher temperature, suggesting that they were less subject to diffusion resistance and more thermodynamically controlled. It was suggested that this originated from the clustering of the PEI with the PEG, forming smaller aggregates, thus reducing CO2-induced interamine cross-linking and associated diffusional limitations.126
Self-supported cross-linked polyamine adsorbents behaviours are also heavily influenced by the relative polyamine content as demonstrated by Yoo and co-workers in a set of poly(ethylene glycol) diglycidyl ether cross-linked PEI (Mw 750000) materials.117 Adsorption at lower temperatures (25 °C) increased with decreasing relative amine loading. E50-FZ, with the highest amine content at 16.6 mmol N per g, adsorbed 0.25 mmol CO2 g−1, whereas E200-FZ, with 8.2 mmol N per g adsorbed 2.01 mmol g−1. At 75 °C, however, the trend was reversed, with E50-FZ adsorbing up to 3.00 mmol CO2 g−1 (Fig. 11). This was due to the higher amine content, which, at lower temperature, more thermodynamically favourable to adsorption, was mostly inaccessible to CO2 due to diffusion constraints, but at 75 °C, the additional energy enabled the CO2 to overcome this diffusion barrier as kinetic effects became more dominant.117 Interestingly, Sun et al. improved the accessibility of the amines within a self-supported polyamine adsorbent by introducing a surfactant template during polymerisation between monomers 2,4,6-tris(chloromethyl)mesitylene and ethylene diamine.71 The adsorbent synthesised using the template showed mesoporosity and pore size of 4 nm. It displayed CO2 uptake 19% higher than the non-templated adsorbent at 1.93 mmol g−1 at 22 °C and 1 bar CO2.
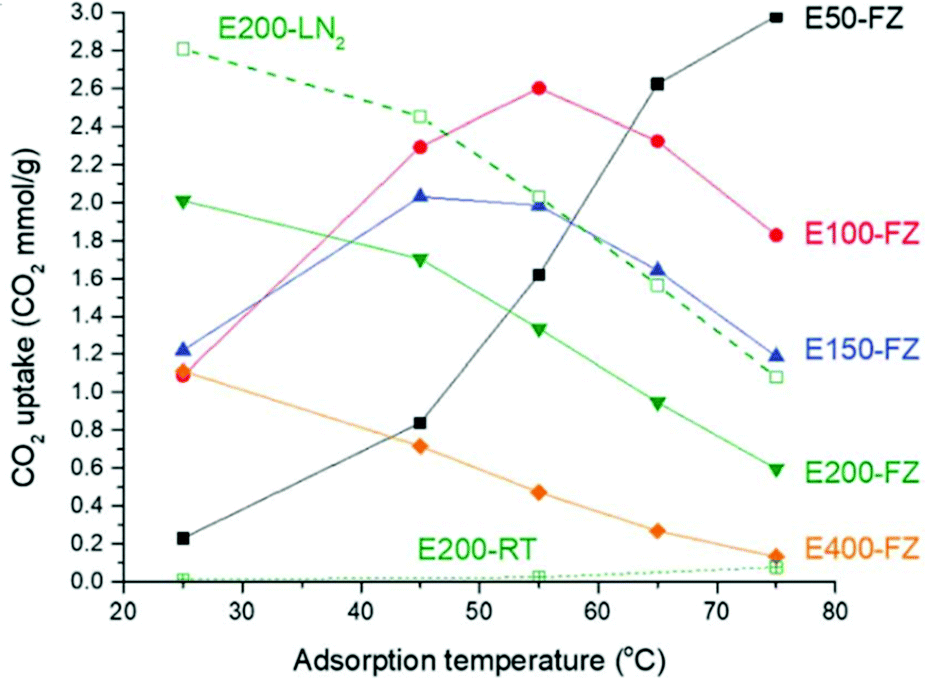 | ||
| Fig. 11 Variation in CO2 capacity with temperature for different amine/cross-linker compositions. Adsorption under 10% CO2 for 3 hours. Reproduced from ref. 117 with permission from the Royal Society of Chemistry. | ||
3.3.2.2 Effect of the support. As described above, polyamine loading is inextricably linked to the morphological character of the support material. However, the chemical modification of supports has been carried out to influence polyamine loading and improve adsorption. Heydari-Gorji et al. modified the surface of pore-expanded MCM-41 mesoporous silica with a layer of long-chain alkyltrimethylammonium cations, onto which they deposited PEI (Mw 423) and TEPA, shown in Fig. 12a.127 At 75 °C under pure CO2, the support-modified material displayed 2.3 times higher CO2 adsorption than the unmodified adsorbent, Fig. 12b. The authors suggested that the higher adsorption was due to decreased diffusion resistance arising from better dispersion of PEI in the modified material by penetration of the hydrophobic alkyl chain network layer. A similar technique was applied by Sanz et al. in optimising the adsorption performance of hybrid class 1/2 adsorbents by double functionalisation of pore-expanded MCM-41 silica supports by impregnation of PEI (Mw 800), PEHA and TEPA over samples grafted with aminopropyl and diethylenetriamino organosilanes.128 Under 1 bar pure CO2 at 45 °C, double functionalisation increased both adsorption capacity and amine efficiency for the 10% PEI loaded samples, and increased capacity for the 10% and 30% PEHA loaded samples. However, higher polyamine loadings led to reduced adsorption, as shown in Fig. 13, due to saturation/pore blockage. Double functionalisation improved adsorption for the TEPA impregnated samples at all loadings, with the highest capacity of 2.37 mmol CO2 per g, and amine efficiency of 0.21, obtained for the 50 wt% TEPA, aminopropyl organosilane-grafted adsorbent. The pronounced benefit for the TEPA adsorbent was due to the lower viscosity of TEPA enabling greater dispersion of the amine without saturation.128
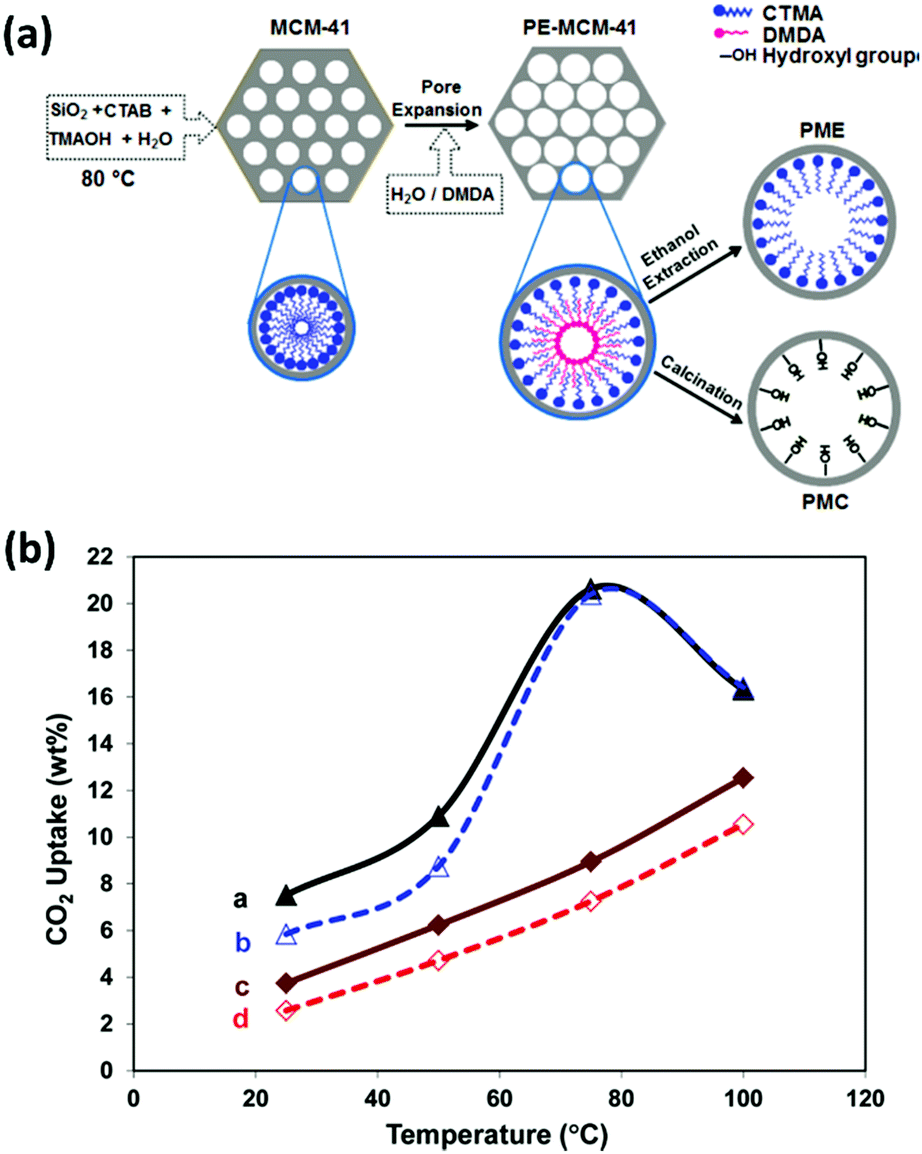 | ||
| Fig. 12 (a) Schematic representation of the syntheses of PMC and PME. (b) CO2 uptake versus temperature for (a) PME–PEI(55) after 180 min, (b) PME–PEI(55) after 30 min, (c) PMC–PEI(55) after 180 min, and (d) PMC–PEI(55) after 30 min of exposure to pure CO2. Adapted with permission from ref. 127. Copyright (2011) American Chemical Society. | ||
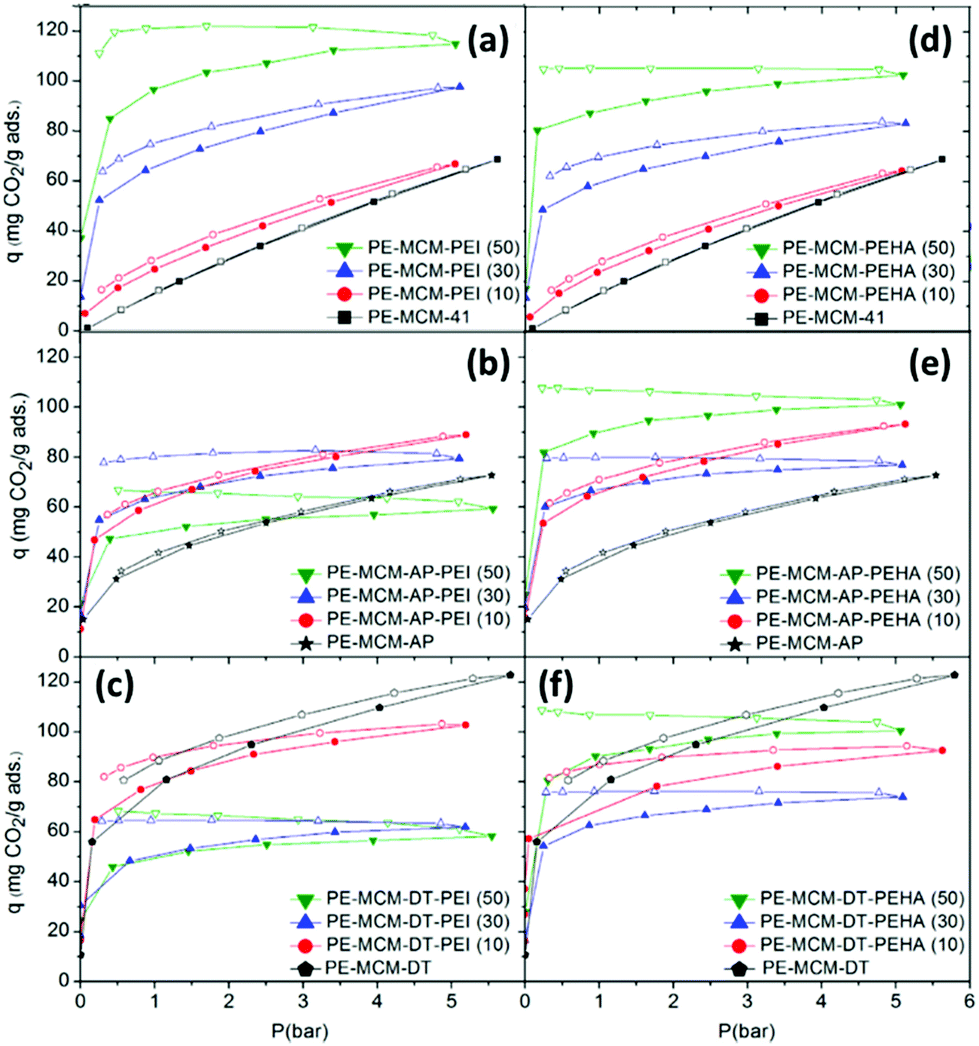 | ||
| Fig. 13 Pure CO2 adsorption–desorption isotherms at 45 °C of pore-expanded-MCM-41 support impregnated with (a) PEI and (b) modified by double functionalisation with AP, or (c) DT. Or impregnated with (d) PEHA and (e) modified by double functionalisation with AP, or (c) DT. Adapted from ref. 128, copyright (2014), with permission from Elsevier. | ||
The chemical nature of the support itself can have a significant effect on the adsorbent behaviour. Chaikittisilp et al. attributed the superior adsorption performance of a γ-alumina-supported PEI (Mw 800) adsorbent relative to a silica supported adsorbent of similar PEI loading to the acid–base properties of the supports.129 The silanol surface groups of silica are slightly more acidic, reducing the basicity of the deposited PEI and weakening its interaction with CO2, while the more basic γ-alumina allows the deposited PEI to interact more strongly with CO2.129 Under 10% CO2 at 25 °C, the γ-alumina supported adsorbent reached 1.73 mmol CO2 per g capacity while the silica-supported adsorbent reached 1.61 mmol CO2 per g. Under 400 ppm CO2, the sorbents showed capacities of 1.33 mmol g−1 and 1.05 mmol g−1 for the γ-alumina and silica-supported adsorbent, respectively.129
Hydrophobicity of the support has been linked to adsorption behaviour in PEI (Mw 600) impregnated resins, HP2MG and HP20.28 Although both supports have similar pore size, volume, and surface area, the latter, non-polar HP20-supported adsorbent, displayed the higher CO2 uptake. It was supposed that adsorption was aided by the hydrophobic nature of HP20, repelling and creating space between the resin microspheres and PEI, and that since PEI occupied the mesopores of <43 nm, good diffusion of CO2 was enabled via the micropores (Fig. 14). The most effective adsorbent with a PEI loading of 50 wt% showed CO2 uptake of 2.29 mmol g−1 under 15% CO2 at 75 °C.28
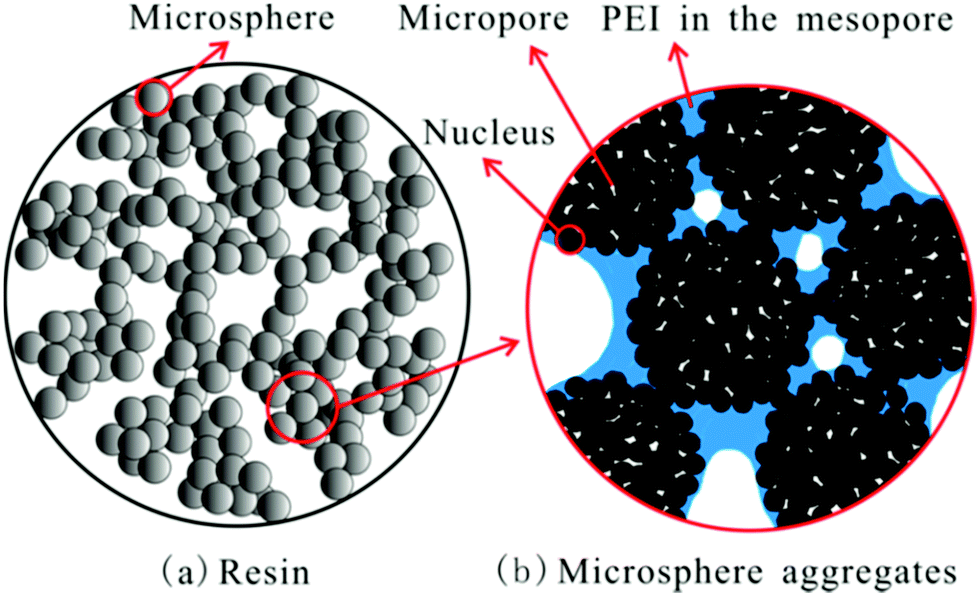 | ||
| Fig. 14 Schematic diagram for PEI distribution in HP20. Reprinted with permission from ref. 28. Copyright (2013) American Chemical Society. | ||
Hydrophobic interactions were also associated with the high CO2 capacity of 4.55 mmol g−1 at 90 °C under 1 bar pure CO2 for unsupported PEI (Mw 25000) cross-linked with C60.106 The adsorption at low partial pressures (<0.2 bar CO2) surpassed that of Mg-MOF-74, and MOF-177 at all temperatures, and reached 3.41 mmol g−1 at 90 °C at 0.15 bar CO2 (Fig. 15). The surface area, at about 2.9 m2 g−1, was considerably smaller than most supported adsorbents, thus it would suggest a very low degree of contact between internal amines and CO2. However, the high CO2 adsorption was attributed to repulsion between the hydrophobic C60 cross-linker and the hydrophilic polyamine, causing the amine groups to be externalised and better exposed for reaction with CO2.106 This hypothesis was further investigated in the study of a set of polypropylenimine dendrimers cross-linked with C60.111 Increasing amounts of C60 caused greater disorder of the amines, likely due to the disruption of hydrogen bonding between the amine groups. This freed the amines to react with CO2via a lower overall energy barrier, confirming the original theory that CO2 enhancement was due to non-affinity repulsive interactions. However, the study also showed that increasing C60 content decreased the probability of successful reactions due to a greater decrease in entropy required to achieve the transition state.111
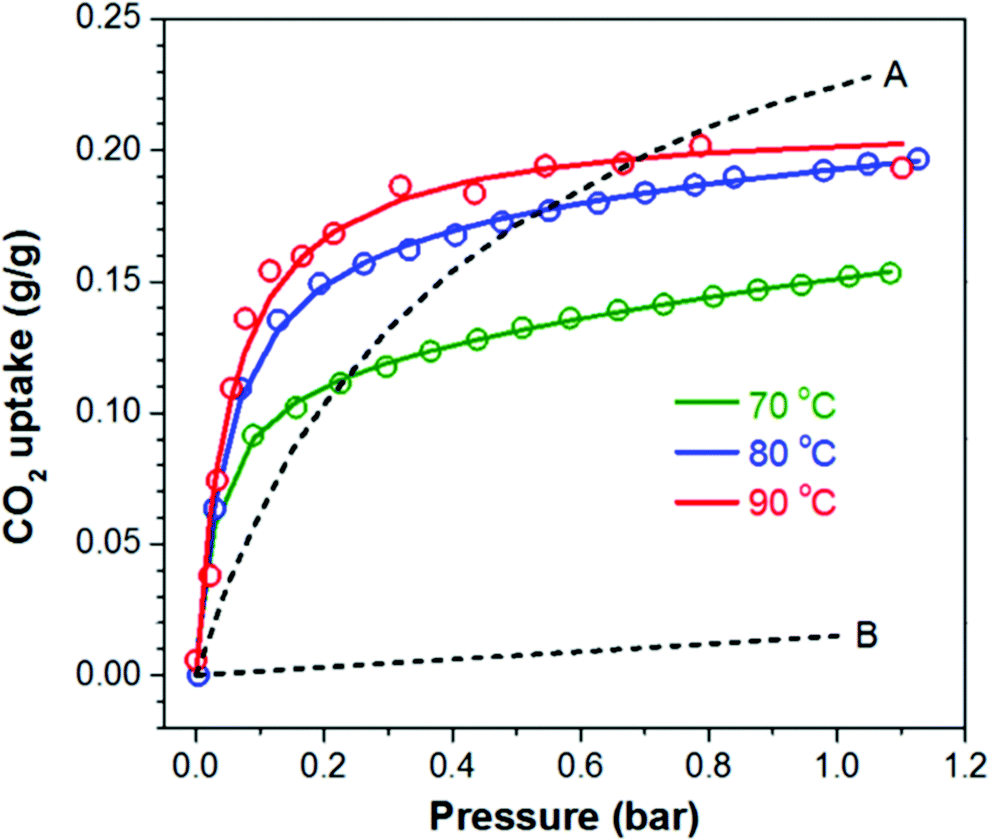 | ||
| Fig. 15 CO2 uptake isotherms of PEI–C60 at 70 °C, 80 °C and 90 °C and compared to the absorption of CO2 on (A) Mg-MOF-74 and (B) MOF-177, both at 90 °C. Reproduced from ref. 106 according to Creative Commons Attribution 4.0 International (CC BY 4.0). | ||
3.3.2.3 Type of polyamine. The type of polyamine has a significant effect on adsorption behaviour, in terms of both uptake capacity, and also stability, as discussed later. Branched chain polyamines containing tertiary as well as primary and secondary amines have reduced adsorption potential due to steric hindrance and the negligible CO2 uptake of tertiary amines under dry conditions. Triethylenetetramine (TETA) supported on mesoporous activated carbon was found to have a higher CO2 capacity and amine efficiency compared to branched chain PEI (Mw 600) at equivalent mass loadings. Meso-AC-TETA(75) adsorbed 1.85 mmol CO2 per g over 5 hours in 75 °C, 0.1 bar CO2, compared to 1.30 mmol CO2 per g for the more sterically hindered PEI adsorbent.130 Reduced polyamine chain length improves dispersion on a support, avoiding saturation of pores and reducing diffusion resistance.128 In a study on the effect of polyamine molecular weight, Zhang et al. compared CO2 adsorption of silica-supported PEI (Mw 800, 1800 and 25
000 Da) at various loadings under 95% CO2 at different temperatures. Generally, due to the lower viscosity and reduced diffusion resistance, adsorption decreased with increasing molecular weight, with the 75 wt% PEI (Mw 800) having the highest capacity of 6.00 mmol CO2 per g at 85 °C. The disparity in adsorption between different molecular weights was most pronounced at a lower temperature, as at higher temperatures, the viscosity of all PEIs was reduced as mobility increased enabling higher adsorption.131
For polyamine chemisorbents, selectivity is dependent on primary and secondary amine abundance and accessibility, therefore, for cross-linked polyamines, where a number of amine groups are reacted with a cross-linker, the conversion of CO2-reactive primary and secondary amines to less reactive tertiary amines can impact selectivity. Mane et al. cross-linked PEI (Mw 1800) with 2,4,6-tris(chloromethyl)mesitylene in a ratio for which there were 0.4 amine groups per alkyl chloride cross-linking reactive site. At the optimum adsorption temperature of 0 °C, CO2/N2 selectivity was 411, based on a partial pressure of 0.15 bar CO2.115 In contrast, at the optimum adsorption temperature of 90 °C, based on a CO2 partial pressure of 0.1 bar, bisphenol A diglycidyl ether cross-linked PEI (Mw 25000) captured 0.101 g g−1 CO2 and showed CO2/N2 selectivity of 2666 (Fig. 16).116 In this case the amine was used in greater excess of the cross-linker, with an amine:epoxy ratio of 40:1, therefore, the higher reactive amine content resulted in higher chemisorbent-activity.
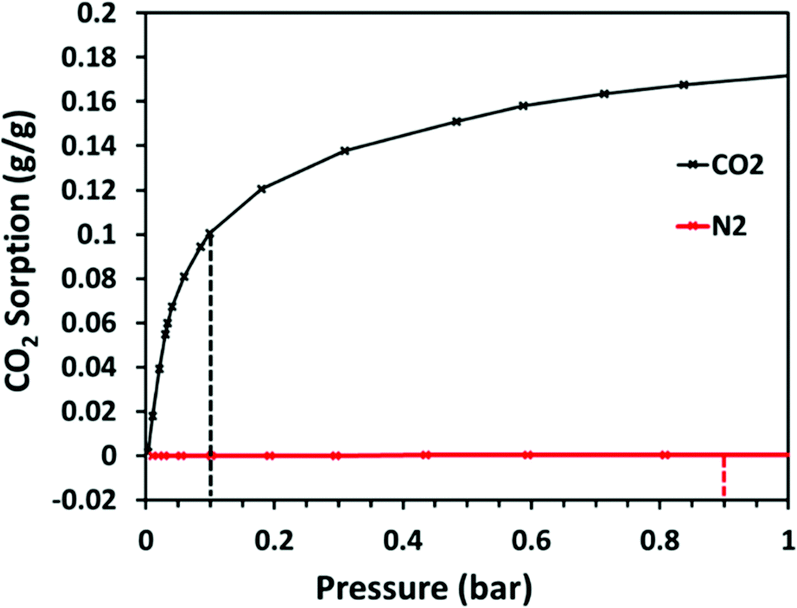 | ||
| Fig. 16 Selectivity of CO2 uptake of BC40. Single-component CO2 and N2 sorption (g g−1) at 90 °C in the low-pressure range. The dashed lines indicate the corresponding interpolated uptakes at the representative pressures of flue gas: 0.10 bar CO2 and 0.90 bar N2. Reprinted with permission from ref. 116. Copyright (2019) American Chemical Society. | ||
3.3.4.1 Thermal stability. High temperatures cause the leaching, i.e. loss or evaporation, of amine content with corresponding reduced adsorption potential over multiple adsorption–desorption cycles, and can be an issue for class 1 supported adsorbents.139 Hydrothermal stability is also an area for consideration as water vapour, present in flue gases, air, and proposed for desorption purposes,74 has also been associated with amine leaching and structural collapse of supports.137 Susceptibility to leaching generally increases as the molecular weight of the amine decreases.140 Yan et al. observed that while PEI (Mw 800) impregnated onto silica mesocellular foam maintained its baseline weight during CO2 adsorption–desorption cycles at 70 °C, with desorption at 100 °C, the TEPA impregnated adsorbent very rapidly lost weight and showed reduced CO2 uptake, shown in Fig. 17.141 In studying amines impregnated on mesoporous activated carbons, Gibson and co-workers observed that a PEI (Mw 600) based adsorbent retained its capacity of 1.22 mmol CO2 per g over four adsorption–desorption cycles at 75 °C in 10% CO2, with desorption under a flow of helium. However, the TETA-based adsorbent leached the more volatile amine under the sustained elevated temperature, resulting in a final CO2 uptake of about 0.85 mmol g−1, down from 0.93 mmol g−1 in the first cycle shown in Fig. 18.130 Similarly, a silica-supported PEI (Mw 1800) adsorbent was found to have greater stability than a poly(propylene guanidine) (PPG) silica-supported adsorbent.142 Over 5 adsorption–desorption cycles, with uptake at 30 °C in 10% CO2, and regeneration at 120 °C in helium, the PEI material's CO2 adsorption reduced from 1.23 to 1.04 mmol g−1 CO2, while for PPG, uptake reduced more drastically from 1.12 to 0.49 mmol g−1 CO2. The thermal analysis did show, however, that the PPG adsorbent was significantly more stable at 100 °C. In each of these examples, the observed reduction in adsorption behaviour was attributed to the instability of the smaller amine during desorption, leading to mass loss due to its lesser affinity for the support surface.
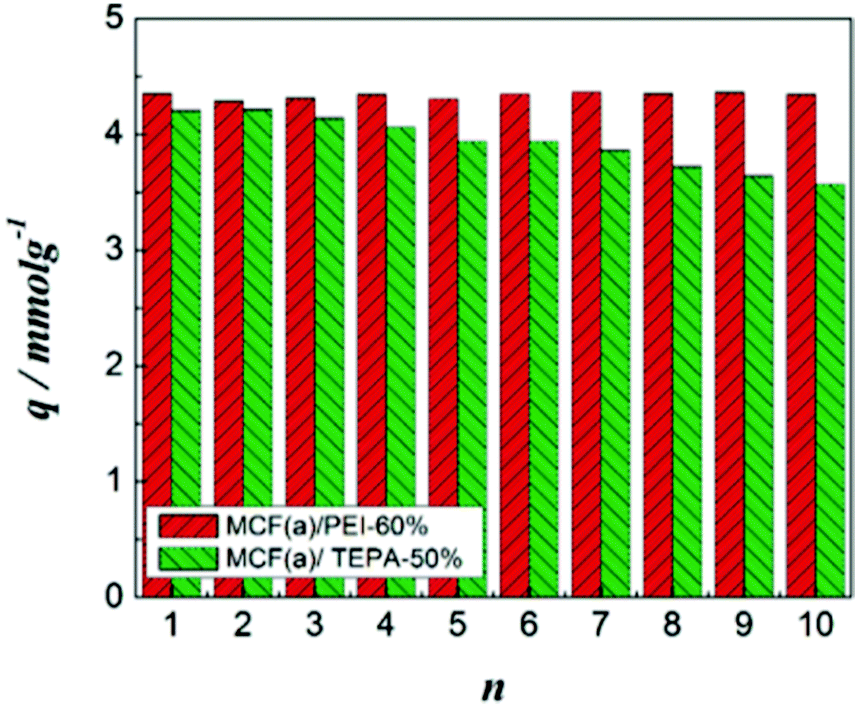 | ||
| Fig. 17 CO2 adsorption capacity of cyclic CO2 adsorption–desorption of MCF(a)/PEI-60% and MCF(a)/TEPA-50%. Reprinted with permission from ref. 141. Copyright (2012) American Chemical Society. | ||
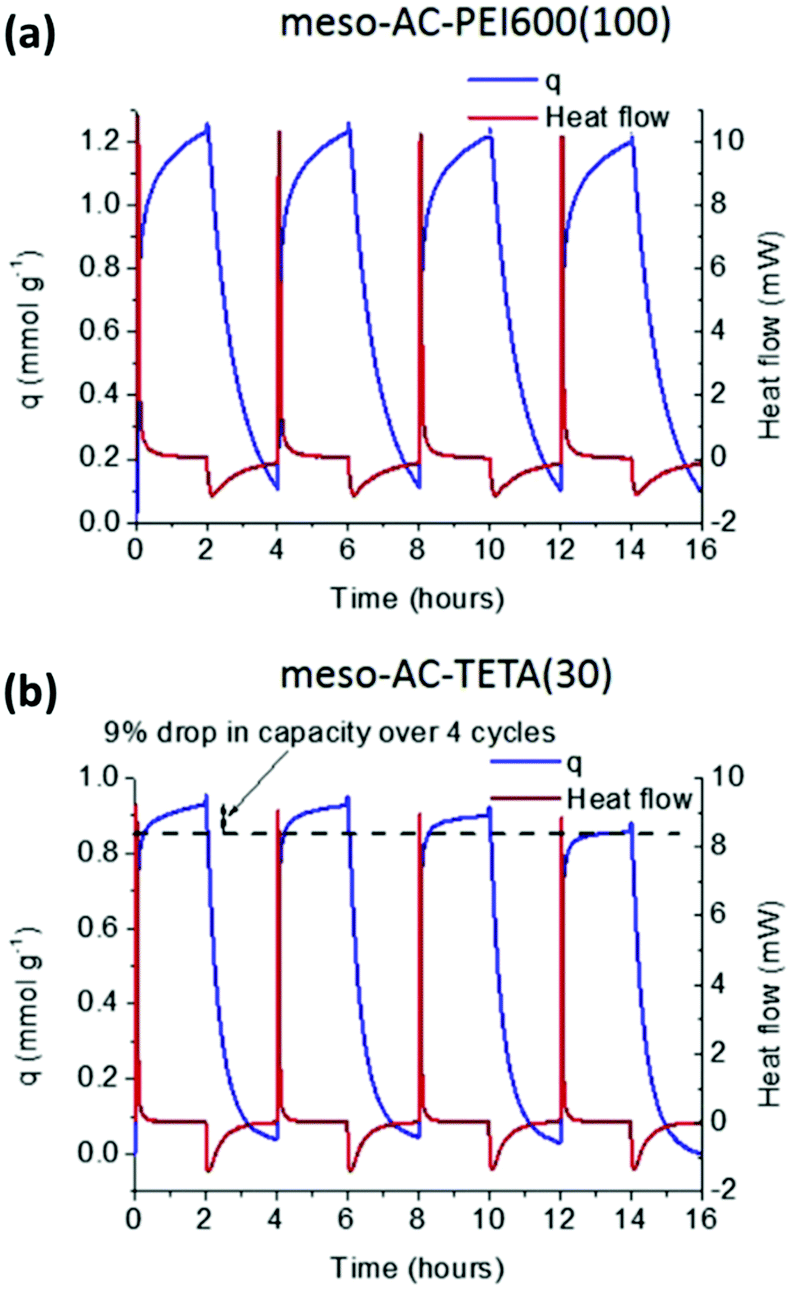 | ||
| Fig. 18 Cyclic CO2 adsorption–desorption experiments at 75 °C on (a) meso-AC-PEI600(100), and (b) meso-AC-TETA(30). Reprinted from ref. 130, Copyright (2015), with permission from Elsevier. | ||
Thermal instability has been mitigated by essentially increasing the organic amine component molecular weight and forming a robust network structure through cross-linking, as demonstrated by Li et al. in preparing cross-linked PEI-impregnated glass fibre matrices.91 Double amine functionalisation has also been shown to improve the thermal stability of supported polyamine adsorbents. The modification of a PEI (Mw 800)-impregnated porous silica with the additives APTES and tetrapropyl orthotitanate to form hybrid adsorbents, A-PEI/silica and T-PEI/silica, were found to improve the thermal stability of PEI by raising its decomposition temperature by 40 °C and 50 °C, respectively (Fig. 19a).87 During adsorption–desorption cycles in 400 ppm CO2, between 25 °C and 110 °C, both the modified adsorbents displayed more stable adsorption capacities reaching 91% and 98% of their original capacities for the APTES and tetrapropyl orthotitanate modified sorbents respectively, compared to just 70% for the unmodified adsorbent (Fig. 19b). It is believed the additives prevented amine leaching through altering the configuration of the PEI and changing the nature of the interactions between the PEI and the support.87 Wilfong et al. carried out NMR studies on similar hybrid or ‘class 4’ adsorbents based on N-(3-(trimethoxysilyl)propyl)ethylenediamine (TMPED)/PEI and APTMS/PEI supported on silica. The hybrid adsorbents’ greater stabilities were attributed to a combination of hydrogen bonding between PEI, aminosilane and Si–OH, and to PEI-catalysed enhanced grafting and polymerisation of the aminosilane.88
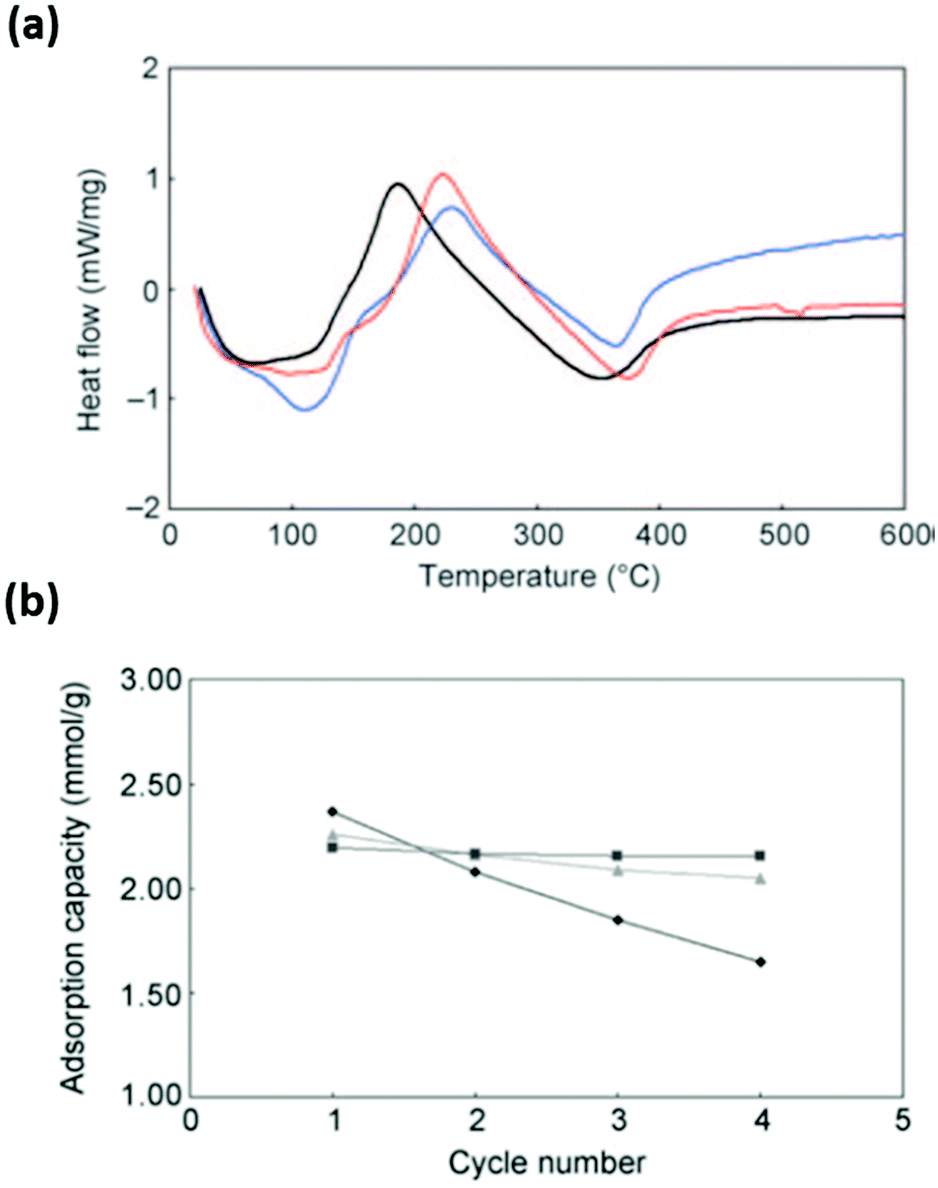 | ||
| Fig. 19 (a) DSC scans showing heat flow versus temperature for PEI/silica (lowest curve at highest temperatures), A-PEI/silica (middle curve at low and high temperature), and T-PEI/silica (upper curve at highest temperatures) samples. Exothermal peaks between 185 °C and 235 °C correspond to PEI decomposition. (b) Changes in CO2 adsorption capacities of hybrid adsorbents over four adsorption/desorption cycles: ♦ PEI/silica; ▲ A-PEI/silica; □ T-PEI/silica. Adapted with permission from ref. 87. Copyright 2011, John Wiley and Sons. | ||
3.3.4.2 Oxidative degradation. Polyamines may undergo oxidative degradation to result in chain cleavage or to form weakly basic species such as amides or imides that are unreactive towards CO2. This is a common problem for polyamine sorbents given that oxygen accounts for about one-fifth of dry air and is present in flue gases at levels of around 5%.8 The inclusion of hydroxyl groups into polyamine adsorbents has been shown to improve oxidative stability. TEPA supported on silica displayed higher oxidative stability when also impregnated with polyethylene glycol. This was found to be due to hydrogen bonding between amine and hydroxyl groups, evidenced by IR spectroscopy, which reduced amine reactivity towards oxygen.143 Similarly, Goeppert et al. modified supported TEPA and pentaethylenehexamine (PEHA) by reaction with propylene oxide and/or a-butylene oxide, thus introducing a secondary hydroxyl group.70 During treatment for 20 hours at 100 °C under 21% O2/N2, the unmodified adsorbents lost up to seven times more weight which was associated with oxidative chain cleavage and material loss. Subsequently, the modified adsorbents’ CO2 capacities were reduced less than the unmodified adsorbents for both the TEPA and PEHA- based adsorbents (Fig. 20). In 95% CO2/N2 at 25, 55, and 85 °C, the CO2 capacity of the TEPA adsorbent decreased by about 90%, while the propylene oxide-modified TEPA adsorbent reduced by a maximum of 18% at 85 °C. Control experiments under pure N2 confirmed that oxidation in air was more significant for capacity reduction than thermal treatment alone.
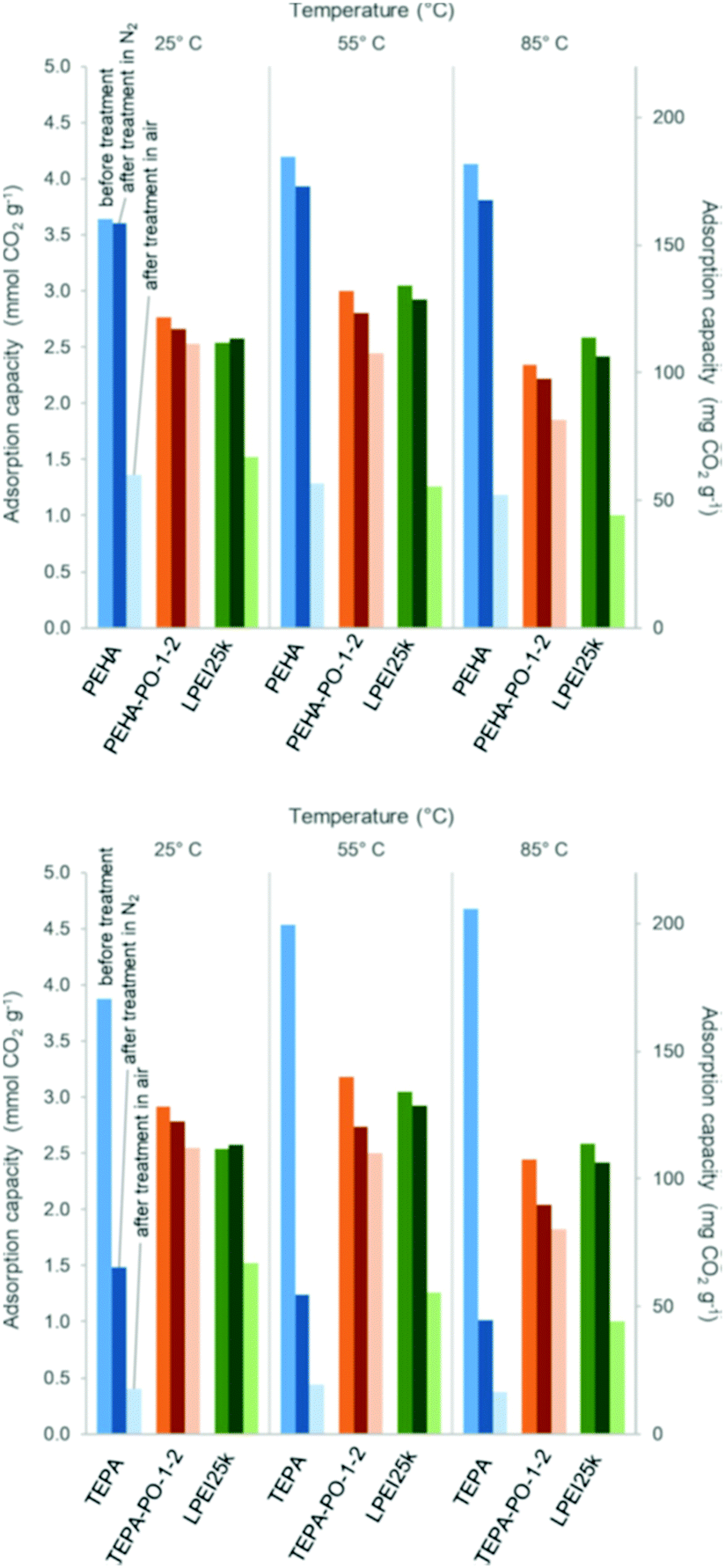 | ||
| Fig. 20 Effect of propylene oxide modification of TEPA- and PEHA-based adsorbents on CO2 adsorption under 95% CO2 in 25, 55, and 85 °C for 3 h before and after treatment under air or N2 at 100 °C for 20 h. Comparison with LPEI25k/50S. Reproduced with permission from ref. 70. Copyright 2019, John Wiley and Sons. | ||
Amine structure is an important factor in susceptibility to oxidative degradation, with secondary amines being less stable than primary or tertiary amines under conditions relevant to flue gas CO2 capture.144 Furthermore, their degradation has been found to simultaneously deactivate neighbouring primary amines, leading to poorer CO2 uptake behaviour.144
One approach to improving oxidative stability is to avoid the use of secondary amines altogether. Poly(allylamine) (PAA) features a linear hydrocarbon backbone and pendant primary amines; by weight, the basic nitrogen content of the polymer is 24%. In 2011, Chaikittisilp et al. demonstrated the use of PAA as an effective alternative to PEI for a class 1 polyamine adsorbent.73 Later, Bali et al. directly compared the oxidative degradation of PAA and PEI (Mw 800) supported on mesoporous γ-alumina under direct air capture conditions, observing that the PAA was far more stable to oxidation than PEI.138 After pre-treatment in a humid flow of 21% O2/N2, followed by CO2 adsorption in 10% CO2/He, the PEI adsorbent's CO2 capacity was reduced by 70.1% and 33.4%, while the PAA adsorbent's CO2 capacity reduced by 10.9% and 7.0% at 110 °C and 70 °C, respectively. The reduction in adsorption of the PEI adsorbent pre-treated at 110 °C was associated with the formation of amide or imide species on oxidation. This was observed in the form of an absorption band at 1693 cm−1 in the IR spectrum and an intense band at 1691 cm−1 in the FT-Raman spectra, both indicative of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency.138
O stretching frequency.138
Although often more prone to oxidation, secondary amines have lower heats of adsorption,70 therefore there is benefit in maintaining their functionality to reduce energy input on desorption. Pan et al. demonstrated that oxidative stability is heavily influenced by the alkyl chain length between amines, with longer linkers associated with greater stability.145 To investigate this, the CO2 uptake performance of pre-oxidised mesoporous silica-supported poly(propylenimine) and poly(ethylenimine) linear polyamines, containing primary and secondary amines, were compared to the non-oxidised equivalents. The tripropylenetetramine (TPTA) adsorbent, with three carbons between amines, had a smaller reduction in CO2 adsorption capacity compared to TETA adsorbent, with only two carbons between amines. Amine efficiency was reduced by 20% for the TPTA adsorbent, while the TETA adsorbent lost 90%. The authors hypothesised that during oxidation the conditions used may have caused oxygen-assisted thermal rearrangement of the amines separated by ethylene linkers.
3.3.4.3 CO2-induced degradation. CO2-induced polyamine degradation by dehydration of the carbamate groups to form urea occurs under dry conditions, and more readily at higher temperature. The presence of moisture suppresses urea formation by directing adsorption towards the ammonium bicarbonate route, (Fig. 21).123 Sayari et al. have carried out extensive studies on the mechanisms of urea formation and have reported that unlike grafted primary monoamines, grafted secondary monoamines are resistant to urea formation in dry CO2 to up to 200 °C;146 however, linear PEI, containing multiple secondary amines, can undergo intramolecular dehydration of ammonium carbamate to form cyclic urea.147
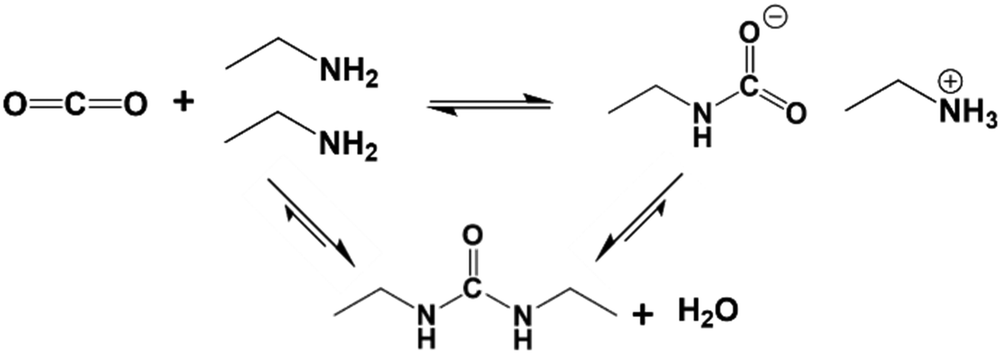 | ||
| Fig. 21 Relationship between formation of carbamate and urea species during CO2 adsorption–desorption under dry conditions. Adapted with permission from ref. 123. Copyright (2010) American Chemical Society. | ||
Didas et al. used density functional theory to study urea formation pathways and proposed an alternative route for urea formation whereby carbamic acid is dehydrated to form an isocyante intermediate.148 It was suggested that the energy barrier for reaction of this intermediate with another amine to form urea decreases with the number of neighbouring amines, thus explaining why adsorbents with a higher amine loading show a greater degree of CO2-induced deactivation. Recently, Jeon et al. reported an innovative way to protect a PEI (Mw 1200)-impregnated silica adsorbent from urea formation by cross-linking PEI with diepoxies of different alkyl chain lengths.149 The adsorbents were subjected to multiple adsorption–desorption cycles under high temperature, high CO2 concentration conditions, with adsorption at 100 °C in 15% CO2 and desorption at 130 °C in 100% CO2. The adsorbent with the longest crosslinker, 1,2,7,8-diepoxyoctane (Si–PEI–O) showed the highest CO2 working capacity at 61.0% of the initial CO2 uptake after 40 cycles, compared to 16.1% for the unmodified PEI adsorbent. FTIR spectra of the samples after 10 cycles displayed peaks matching those for cyclic and open chain urea at 1670 and 1563 cm−1 and 1624 and 1501 cm−1, respectively, in all adsorbents, as shown in Fig. 22. However, the intensity of the peaks was reduced as the diepoxy alkyl chain length increased due to the reduced proximity of neighbouring amines, thus enabling higher CO2 uptake.
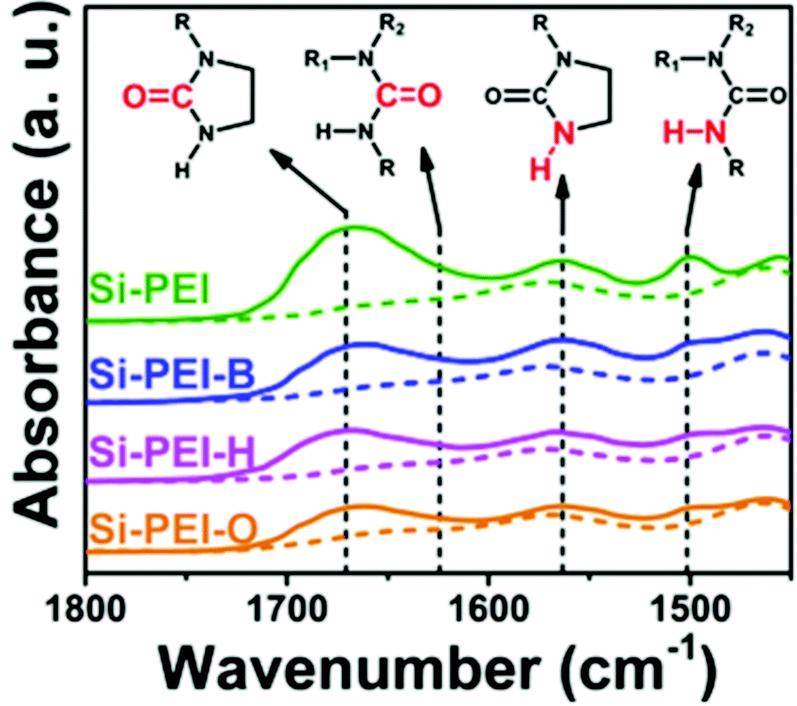 | ||
| Fig. 22 FTIR spectra of diepoxy cross-linked PEI-based adsorbents before (dash line) and after 10 adsorption–desorption cycles (solid line). Reprinted from ref. 149, Copyright (2020), with permission from Elsevier. | ||
3.3.4.4 Poisoning by SOx and NOx. Post-combustion CO2 adsorbents are compromised by the presence of ppm levels of NO, NO2 and SO2, due to their strong interaction with amines, drastically reducing CO2 uptake. Their tendency to compromise adsorption performance is dependent on the type of amine,150 as well as environmental factors of concentration,151 temperature,150 humidity,36,152 and the duration of exposure.151
NO is more abundant in flue gases than NO2, representing 85–95% of the total NOx generated in the combustion process.153 However, a study by Rezaei and Jones has shown that although primary amines have the highest affinity for NO, adsorption is low and the corresponding CO2 capacity loss is less than 5%.150
In terms of adsorbent stability, NO2 is more harmful, as it has a higher affinity for amines due to its higher polarity and acidity.137 Rezaei and Jones investigated the adsorption of NO2 by a class 1 PEI-impregnated silica adsorbent and class 2 adsorbents featuring grafted primary, secondary or tertiary amines.150 Treated under 200 ppm NO2/N2 at 35 °C, for materials with equivalent amine loadings, the adsorbent featuring only secondary amines adsorbed the highest amount of NO2 at approx. 1.9 mmol g−1. After regeneration at 110 °C under N2 and subsequent adsorption under 10% CO2/He, none of the adsorbents re-gained their CO2-sorption properties, adsorbing as little as 7% of their original uptake, indicating irreversible NO2 adsorption. However, the NO2 concentration used in this experiment was higher than that representative of post-combustion conditions. In a later paper, dual component experiments in which CO2 uptake was measured under 20 ppm NO2 in 10% CO2/N2 showed that the NO2 and CO2 breakthrough times for the secondary amine-grafted adsorbent were about equal and that the CO2 working capacity was not significantly reduced in the presence of the lower concentration of NO2 (Fig. 23).151
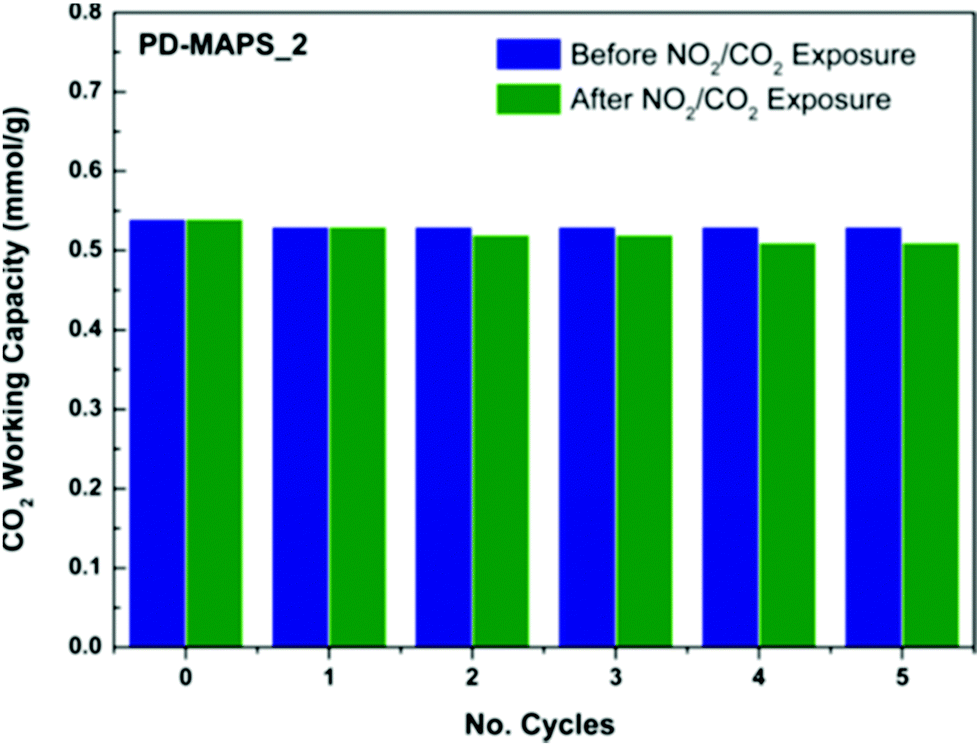 | ||
| Fig. 23 CO2 working capacity of the secondary amine-grafted adsorbent before and after exposure to 20 ppm of NO2 in 10% CO2/N2 at 35 °C. Reprinted with permission from ref. 151. Copyright (2014) American Chemical Society. | ||
The adsorption of SO2 on various amine adsorbents is reported to increase with increasing SO2 concentration and decrease with temperature. The highest capacity of 1.08 mmol SO2 per g was observed for the secondary amine-grafted adsorbent, under the maximum SO2 concentration applied of 200 ppm SO2/N2 and at 35 °C, the lowest temperature investigated in this study. Adsorption performance then decreased in the order: primary amine-grafted > PEI-impregnated > tertiary amine-grafted adsorbents. Subsequent CO2 adsorption analysis after regeneration at 110 °C in N2 indicated that all adsorbents suffered capacity loss, however, this was lowest for the secondary amine-grafted adsorbent.150 Co-adsorption–desorption breakthrough experiments were carried out on the primary amine-grafted, secondary amine-grafted and PEI impregnated adsorbent under 200 ppm SO2 in 10% CO2/N2. These showed that deactivation of the adsorbents could be reduced, and that CO2 capacity could therefore be maintained over multiple cycles, if the adsorption step was continued only until SO2 breakthrough, as opposed to a longer cycle continuing until CO2 breakthrough was observed. During the shorter cycles, there was a slight reduction in CO2 capacity for each adsorbent between the first and second cycle before uptake stabilised – this was attributed to irreversible binding of SO2, which could not be re-gained on desorption at 110 °C in N2. However, the secondary amine-grafted adsorbent showed the best performance with consistent CO2 uptake above 0.5 mmol g−1 over five cycles (without allowing SO2 breakthrough) and the least CO2 capacity loss (when SO2 was allowed to break through).151
SO2 uptake by amine-based adsorbents increases under humid conditions, under which, they can form sulfite and sulfate salts.36,154 Studies by Fan and co-workers on amine-grafted cellulose acetate/silica hollow fibre adsorbents showed that for both the primary amine-grafted and secondary amine-grafted adsorbents, SO2 uptake on exposure to 200 ppm SO2 in 10% CO2/N2 at 35 °C increased when the adsorbent was pre-hydrated under 100% RH (relative humidity) in N2.36 The primary amine-grafted adsorbent's uptake of SO2 increased from 0.15 to 0.28 mmol SO2 per g, while for the secondary amine-grafted adsorbent uptake increased from 0.34 to 0.55 mmol SO2 per g, going from dry to pre-hydrated conditions, respectively. In the presence of SO2, humidity had a detrimental effect on the CO2 adsorption of both adsorbents. However, the secondary amine-grafted adsorbent performed better over 100 CO2 adsorption–desorption cycles, with CO2 uptake decreasing by 38% for the pre-hydrated secondary grafted-amine as compared to a 70% capacity loss for the pre-hydrated primary grafted-amine (Fig. 24). The authors attributed this decline to an increased reaction ratio of SO2/N,36 and it is also likely due to the irreversible adsorption of SO2 in the presence of water, with the formation of heat-stable salts, which pose a major challenge to amine stability.154
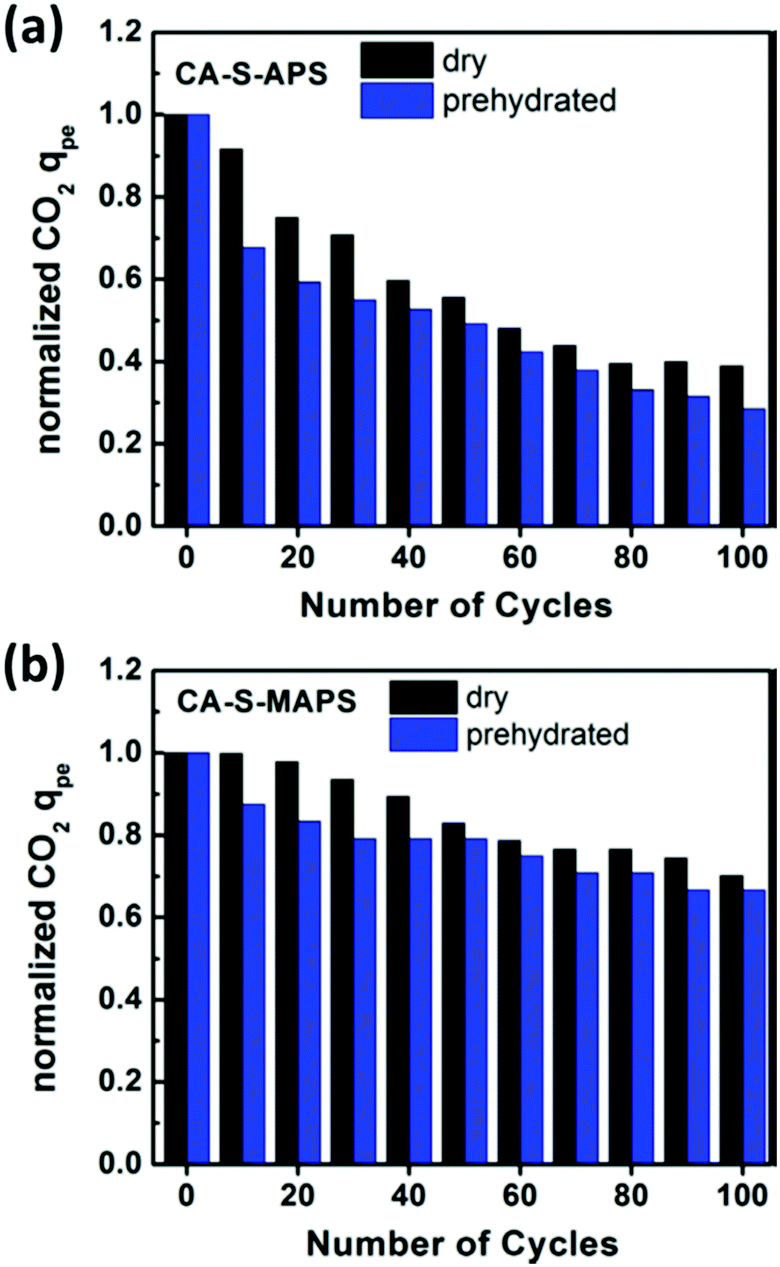 | ||
| Fig. 24 Comparison of the cyclic CO2 capacity of dry fibres and prehydrated fibres in 200 ppm SO2 in 10% CO2/N2 at 35 °C of; (a) primary amine-grafted and; (b) secondary amine-grafted fibres. CO2 capacities normalised to adsorbents’ initial CO2 capacity qpe,0 before exposure degradation. Reprinted from ref. 36 Copyright (2015), with permission from Elsevier. | ||
In agreement with results reported by Fan et al., Luz et al. reported on the stability of a PEI-impregnated MOF/SiO2 hybrid, PEI/(Zn)ZIF-8/SiO2 under a simulated flue gas of 15% CO2, 4.5%, O2, 5.6% water in N2 with 200 ppm SO2. The CO2 uptake capacity was reduced from 12.86 wt% (2.92 mmol g−1) to below 2 wt% over 100 adsorption–desorption cycles, likely due to both the high SO2 concentration and the presence of water; however, under the same flue gas except with 50 ppm SO2, adsorption performance was better maintained with capacity reduced from 12.41% (2.82 mmol g−1) to 8.55%.98
Tertiary amines typically adsorb negligible quantities of CO2; however, they show preferential and reversible adsorption of SO2, it being a stronger Lewis acid. Therefore, the generally proposed solution to SO2 deactivation has been the application of amine scrubbing using tertiary amines to selectively adsorb SO2 to remove it from the flue gas. Wei et al. demonstrated an integrated two-stage removal experiment in which SO2 and CO2 were adsorbed sequentially from a gas mixture of 12% CO2 and 600 ppm SO2 in N2.155 The first adsorption column removed SO2 before the gas entered the second adsorption column. The first column contained SBA-15 impregnated with the tertiary amine triethylolamine which had a breakthrough capacity of 2.28 mmol SO2 gg−1 at 25 °C. The second column contained SBA-15 impregnated with TEPA and adsorbed 2.91 mmol CO2 gg−1 at 80 °C. Tailor and Sayari also demonstrated an effective tertiary amine SO2 adsorbent in synthesising a propyldiethanolamine-grafted pore-expanded mesoporous silica which could adsorb 2.84 mmol SO2 gg−1 under dry 1% SO2/N2 at 23 °C, which was also effective in the presence of CO2. On adsorption–desorption cycles under dry 0.1% SO2/N2, the adsorbent could be completely regenerated by heating to 130 °C under a flow of N2.152 Under humid conditions, adsorption performance was improved, with uptake under 0.05% SO2/N2 at 23 °C increasing from 0.74 mmol g−1 in dry conditions to 1.11 mmol g−1 under 83% RH. However, after regeneration, subsequent adsorption was decreased on exposure to water. The authors inferred that this was due to the irreversible formation of heat-stable sulphites.
3.4 Challenges for industrial application of amine-based adsorbents
There is huge diversity in the make-up of amine-based CO2 adsorbents and while all may fundamentally react with CO2, each is suited to a particular set of working conditions in applications from industrial post-combustion CO2 capture to DAC. However, any scenario comes with a high number of environmental variables, under which, adsorbents must be developed to meet all criteria: high working capacity, recyclability, selectivity, stability, fast kinetics, and low cost.8 How sorbents are analysed in the lab very often only aim to test one or two of the criteria, and for this reason rarely replicate the likely industrial environmental conditions. There are some criteria – particularly regarding selectivity and stability – which studies on new CO2 adsorbents do not necessarily address ‘as standard’ at the initial research stages, since in many instances, adsorbents function as models to test specific characteristics or modifications in isolation.Certain handicaps in analyses need to be addressed more rigorously, a major one of which is regeneration. Regeneration of the adsorbent must be possible under cost-effective, i.e. low energy conditions. In laboratory analyses, amine-based adsorbents are primarily regenerated by a temperature swing, often accompanied by mass displacement via an inert gas such as N2, Ar or He. It has often been commented by leading experts in the field that this technique is irrelevant to an industrial process due to dilution of the CO2 adsorbate, and that practical regeneration is a “critical missing link in the development of supported amine CO2-adsorbents”.74 One proposed solution to this has been to desorb under a flow of pure CO2, however, this can lead to deactivation of the amines due to the formation of urea, as has been shown by Drage et al. on a PEI-impregnated silica.156 The applicability of desorbing under pure CO2 is subject to adsorbent properties, temperature of desorption and time under exposure to pure CO2, as some adsorbents are more resistant to degradation than others. Hamdy et al. reported a good working capacity of 2.14 mmol CO2 gg−1 over 29 cycles in a cyclic adsorption–desorption experiment on epoxy-crosslinked PEI (Mw 25 000), adsorbing under 10% CO2 at 90 °C, and desorbing in pure CO2 at 155 °C. This consistent CO2 uptake, however, was accompanied by a 1.3 wt% sample mass loss over the course of the cycles.116 A promising regeneration approach proposed by the Jones group in 2010, is the use of steam to act as both a thermal and partial pressure driving force to remove CO2, followed by the separation of the water and CO2 by compression and condensation.74 ‘Steam stripping’ has been the focus of many stability-specific studies on adsorbents137 and was explored by Fauth et al. on new class 1/2 polyamine adsorbents, using helium with 90 vol% water vapour for regeneration during cyclic adsorption–desorption experiments.86 Another approach to regenerating a pure CO2 product is by applying the combination of temperature with vacuum as has been demonstrated by Sujan et al. with PEI-functionalised cellulose acetate/silica fibres in a DAC process.39 To investigate the recovery of CO2, the authors carried out a vacuum-assisted desorption experiment in a TSA process at 25 °C, under a gas flow of 380 ppm CO2/397 ppm of He/balance N2. The CO2 concentration at the outlet reached a steady reading after almost 3 hours of adsorption (Fig. 25a). Then, a vacuum was applied reducing the pressure from ambient down to 0.6 mbar, removing the interstitial gas containing He and N2. The system was then sealed before heating the sample to 90 °C to desorb CO2 in the ‘self-sweep’. Mass spectrometry data of the desorbed gas revealed a 88% pure CO2 flow, but on introduction of a back-pressure pulse of N2 in the N2 sweep, CO2 purity increased to 98% over the initial 5 seconds (Fig. 25b).
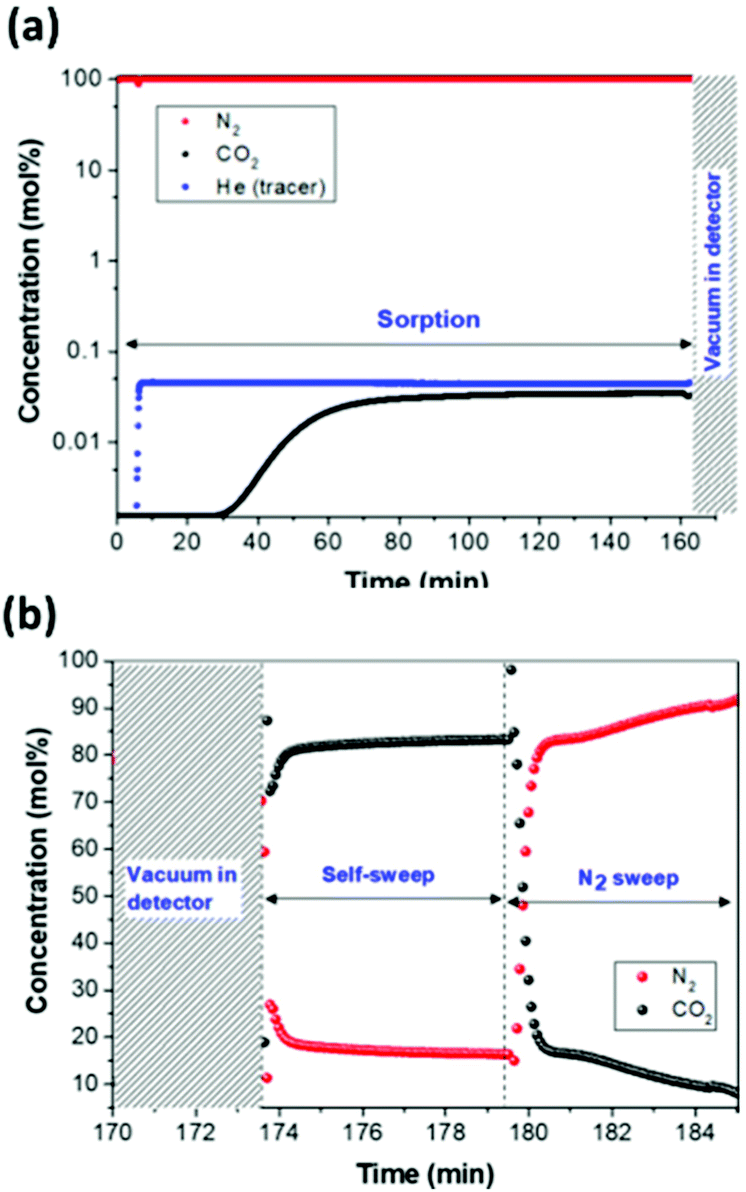 | ||
| Fig. 25 (a) Breakthrough profile of PEI-functionalised cellulose acetate/silica fibres during CO2 adsorption under dry 380 ppm CO2/395 ppm He/balance N2 at 35 °C. (b) Concentration profiles during the ‘self-sweep’ and N2 sweep stages of desorption. Reprinted with permission from ref. 39. Copyright (2019) American Chemical Society. | ||
In designing new adsorbents, researchers are limited to small-scale samples for testing, using in the range of tens of mg to several grams of adsorbent; however, smaller sample sizes mask diffusion limitations of the gas through the bulk sample giving an unclear description of how the sample may behave once scaled up. In large-scale industrial application, several tonnes of adsorbent would be used, often processed into pellet form rather than powder form, which could cause large pressure drops in the adsorption column. Therefore, sorption analysis at the largest scale possible is necessary to determine the potential of an adsorbent.
Economics is also a decisive factor in the development of an adsorbent. Sorbents based on polyamines impregnated into silica, which are the most widely represented in the research literature, are currently portrayed as one of the most economical for industrial scale and leading the way under direct air capture conditions. In 2014, Zhang and co-workers reported on the first scale-up of a 40 wt% PEI (Mw 1800)-impregnated mesoporous silica adsorbent to 1 kg and its application in the ambient air capture of CO2 using a bubbling fluidized bed reactor system.157 More recently Luz et al. demonstrated the scale-up of a PEI-impregnated MOF/SiO2 hybrid to 1 kg. They carried out a cost evaluation study and estimated the price per kilo at approximately $20.98 With further research, the benefits of an increasingly diverse range of adsorbents, utilising better supports, higher capacity and higher stability amines, or the potentially more efficient self-supported adsorbents, will also become realised at a reduced cost.
4. Selection of adsorbents for scale-up
Due to the sheer diversity of amine-based adsorbents and the range of conditions under which they are tested for their separation potential, the evaluation of any adsorbent for application in an industrial process is not straightforward. Consequently, it is crucial to screen this wide range of materials and develop a simplified approach to evaluate and compare their potential for gas separation. The objective of this section is to guide material researchers and engineers in the screening of different candidate adsorbents for larger scale testing (bench/pilot scale). Before we dive into the detailed methodology for screening an adsorbent, it is important to note that this section is applicable to any adsorbent available for any gas separation process. Studies reported in this section are mainly pertaining to adsorbents such as MOFs, zeolites etc. on account of the lack of literature available for amine-based adsorbents. This signifies the gap present in the research on amine-based adsorbents for commercial CO2 capture. The intention of this section is to act as a guiding tool to bridge this gap between lab-based material research and the commercial process implementation of amine-based adsorbents while learning from the progress made with readily available adsorbents like MOFs and zeolites.The task of selecting an adsorbent for a given separation is performed in two steps. The first step is to perform an initial screening of a large number of adsorbents. In terms of separation at lab scale, three main parameters are important at this stage of screening namely CO2 uptake capacity, selectivity, and heat of adsorption. These can be obtained from the adsorption isotherm data from either single component or multi-component conditions and can be achieved with milligrams or grams of adsorbent. There are two ways to carry out the initial screening of adsorbents: using the adsorbent selection metrics (Section 2.4) and/or high-throughput screening of materials (Section 4.1). The adsorbents with the best performance then move to the advanced evaluation step.
4.1 High-throughput screening of materials
High-throughput screening is an easy, fast and cheap method of identifying promising adsorbents from an enormous pool/database of materials for a desired separation process. It can be performed either experimentally or computationally using molecular simulations, genetic algorithms, etc. Most of the research for high-throughput screening has been performed on MOFs and zeolites for various gas separations such as CO2/N2, CO2/CH4, CO2/H2, etc.158 and it is discussed in the following as a guidance for amine-based materials evaluation.Han et al. reported the screening of MOFs for CO2/N2 adsorption and diffusion selectivity using an in-house designed, novel parallel high-throughput (HT) sorption screening system. In this method, the adsorption data were collected at only one pressure value relevant to the partial pressure regime in flue gases, rather than obtaining the isotherm for a complete pressure range. Pressure decay with respect to time was monitored in each of the sample chambers to measure CO2 and N2 adsorption by the adsorbent. The initial pressure of each system was fixed while the gas uptake by the adsorbent determined the exact pressure at which equilibrium adsorption was measured, and the equilibrium adsorption was obtained by pressure sensors at the end of the experiment. This methodology allowed rapid screening of 8 MOF materials, using only 100 mg of adsorbent, and substantially accelerating the process of obtaining the separation data. Additionally, various adsorbate gases could be tested in their HT system and it can thus be implemented for screening various novel materials for a wide range of applications.159 In another study, the parallel evaluation of 6 adsorbents was conducted using a high-throughput gas adsorption apparatus, measuring complete adsorption isotherms up to a pressure of 40 bar. Cyclic adsorption experiments could also be performed with the apparatus, which used four different gases sequentially. With one of the main objectives being to use the minimum amount of adsorbent, reasonably accurate results (within 10% error) were obtained using 60 mg of adsorbent. In this study, optimal thermal activation conditions, adsorbent regenerability and separation properties were determined by obtaining gas uptakes for various MOFs. Adsorbent selectivities and heats of adsorption were obtained from the measured isotherms in combination with isotherm models such as IAST and the Clausius–Clapeyron equation respectively.160 The data were then used for initial adsorbent comparison for a given separation process using the various adsorbent metrics defined in Section 2.4. The same or similar approaches could be applied for the fast screening of amine-based carbon capture materials.
Mason et al. developed a high-throughput instrument for measuring multi-component equilibrium adsorption isotherms of gas mixtures (including water) for up to 28 samples. A set of adsorbents including MOFs, zeolites, activated carbons and amine-functionalised silicas were tested. The volumetric-measured CO2 adsorption under pure-component conditions was compared to that under multi-component conditions (CO2, N2, and H2O) using the HT instrument at the same partial pressure. Only the alkylamine functionalised adsorbents exhibited significant CO2 uptake under multi-component conditions with the highest uptake of 4.2 mmol g−1, at 0.1 bar and 40 °C in the presence of water, by the amine-appended MOF framework mmen-Mg2(dobpdc) (mmen = N,N′-dimethylethylenediamine, dobpdc4− = 4,4-dioxido-3,3′-biphenyldicarboxylate).161
HT adsorption apparatus can be helpful in screening adsorbents experimentally though it would be more impractical to use it to rank several thousands of adsorbents. In such a scenario, high-throughput computational screening can be used for identifying potential adsorbents and identifying structure–property relationships for a given separation process. Wilmer et al.162 used grand canonical Monte Carlo (GCMC) simulations to screen their database of more than hundreds of thousands of hypothetical MOFs. They calculated their pure component adsorption isotherms, followed by calculation of adsorbent metrics including adsorption capacity (both absolute and working), selectivity, regenerability and a modified form of the PSA parameter, S [for non-Langmuir system α12,mix is replaced with (α12,mixatads)2/(α12,mixatdes)].163 The relationship between physical properties of the MOFs and the adsorbent metrics was evaluated providing insight otherwise not possible because of too small a sample set of MOFs.162,163 In another study, GCMC simulations were performed for more than 3800 MOFs in the Cambridge Structural Database (CSD) to predict their binary CO2/H2 mixture adsorption isotherms under PSA and VSA (vacuum swing adsorption) conditions. Subsequently, various adsorbent metrics were calculated to rank the MOFs’ performance, and the top 20 adsorbents for PSA and VSA processes – that surpassed the selectivity of many zeolite adsorbents – were identified. The best MOF adsorbents for CO2/H2 separation were the ones with narrow pore sizes and low porosity as suggested by the structure–performance relation results.164 Computational methods could potentially play a role in the screening of polyamines and amine-based materials and experts in these fields may consider the crucial impact their work would make to decarbonisation targets.
High-throughput screening of a very large number of adsorbents can be computationally expensive depending on the complexity of the simulation conditions. To screen the adsorbents faster, an initial screening can be done using one or more adsorbent metrics thereby reducing the sample set for simulation, which can be followed high-throughput screening for example by GCMC simulations to identify the best adsorbent for the given operating conditions. For instance, using GCMC simulations for estimating the adsorption of water on MOFs is time-consuming. This was avoided by computationally identifying the MOFs that showed high CO2 selectivity over H2O, based on their Henry's law constants, from a database of more than 5000 materials. This initial screening identified 15 high performing MOFs which were then used for GCMC simulations to determine their binary and ternary adsorption isotherms for CO2/H2O and CO2/H2O/N2 mixtures. Subsequently, these top MOF candidates performed well for CO2/N2 separation also.165
Using adsorbent metrics and/or high-throughput screening are useful tools in quickly narrowing down a set of potential adsorbents for a given application. Additionally, high-throughput computational screening can guide the material researcher in synthesising adsorbents with bespoke characteristics. However, experimental studies would still need to be carried out to validate these models or resolve any discrepancies. Also, it should be kept in mind that this approach is to be used only for a preliminary screening and it is not enough to reflect the accurate separation performance in an actual PSA/TSA process. Models do not confirm whether the product purity and recovery targets as defined by the DOE are met. Moreover, adsorbents are ranked for fixed operating conditions, which are not necessarily their best conditions for optimum performance to achieve the purity and recovery targets. Hence, it is essential to account for these additional performance indicators for the selection of adsorbents and implementation at a large scale.
The second step for adsorbent screening also involves two possible approaches, (i) carrying out the testing of the material at the pilot scale, or (ii) performing comprehensive process modelling. For testing the materials at the pilot-scale, one has to synthesise large amounts of adsorbent (100s of grams) and perform process level characterisation i.e. formulation of adsorbent particle (shape and size), binary/multi-component adsorption isotherms at relevant process conditions (lab or bench scale), effect of heat and mass transfer, etc. The evaluation of suitable process performance indicators including product purity, product recovery, throughput, energy requirements, cost, etc. have to be based on the experimental data. Current pilot scale testing for CO2 capture typically uses commercially available adsorbents that can be synthesized in large quantities like zeolites, MOFs and activated carbons. Researchers working in the development of novel adsorbents concentrate mainly on material development with the aim of enhancing material properties and their separation performance. The bulk production of these materials is not contemplated which includes a number of challenges, for example, material availability for large scale production, technical and economic feasibility of production, etc., thereby restricting further exploration of novel and potential materials at bench/pilot scales. However, it is practically not possible to scale-up the production of all the developed materials. Process modelling and simulation acts as a tool to explore the potential of newly developed materials without bulk production.
4.2 Process modelling and simulation
Some researchers have tried to identify the relationship between adsorbent properties and adsorption process targets by screening numerous adsorbents by performing process level modelling. This includes the full process modelling and optimisation of a PSA/TSA unit with the analysis of Pareto fronts giving the process performance. For a multi-objective optimisation problem, there is an optimal trade-off between the two competing objectives, for instance, a trade-off between the product purity and recovery in an adsorption process. A Pareto front represents a range of different sets of operating conditions for these competing objectives. Improvement in one objective leads to a decline in the other objective. Pareto front segregates the section of feasible operating conditions from the unfeasible operating conditions. The optimal performance is obtained by selecting the operating conditions lying on the Pareto front. In the present study, a Pareto front could be a plot between CO2 purity and CO2 recovery or energy consumption versus productivity.Maring and Webley developed a simplified model for a 3-step pressure/vacuum swing adsorption system for rapid adsorbent screening of CO2 adsorbents (zeolite 13X, Mg-MOF-74, activated carbon, PEI/MCF chemisorbent). The three steps included adsorption, blow down (desorption) and re-pressurisation. In this model, the adsorbent bed was simulated as a well-mixed adsorber with no axial gradients, i.e. no temperature, pressure, or concentration gradients either in radial or axial direction throughout the adsorbent bed. This assumption reduced the coupled algebraic partial differential equations of a PSA system to coupled algebraic ordinary differential equations. Furthermore, it was assumed that a cyclic steady state was achieved in a single adsorption–desorption cycle, which significantly reduced the computation time. Heat affects associated with each step were accounted for to capture adiabatic temperature swing associated with pressure swing, in addition to specific work required to pressurise and de-pressurise the adsorbent bed. The PSA process performance was evaluated in terms of CO2 purity, CO2 recovery, working capacity and specific work under both adiabatic and isothermal operating conditions. It was reported that increasing adsorbent selectivity in terms of reducing the adsorbent N2 uptake capacity, and thermal effects concerning the adsorbent heat capacity, impacted both product purity and specific power consumption more substantially than CO2 adsorption capacity.166
The model proposed by Maring and Webley166 was further extended by Balashankar et al. to develop a simplified proxy model for a 4-step isothermal VSA cycle. A set of four adsorbents was used for evaluating this model with a detailed VSA model under optimized conditions, which was followed by illustrating a graphical design approach, and a classification model to rank 79 adsorbents with the constraints of purity and recovery targets of 95% and 90%, respectively. Also, an approach to calculate the parasitic energy was developed for the adsorbents that met those performance targets.167 This model was further applied to screen thousands of adsorbents to classify the potential candidates for post-combustion CO2 capture.167 Adsorbents with the lowest energy requirements demonstrated attributes of low N2 affinity rather than high CO2 affinity with heats of CO2 and N2 adsorption in the ranges of 32 to 42 kJ mol−1 and 8 to 17 kJ mol−1, respectively.
In another study, process optimisation was performed on four adsorbents (2 MOFs: Mg-MOF-74 and UTSA-16; zeolite 13X and a coconut shell-derived activated carbon) in a VSA system to compare the various adsorption metrics reported in the literature.49 A 4-step VSA cycle with a light product pressurisation step was used for post-combustion CO2 capture from dry flue gas and the separation performance was reported in terms of CO2 purity in the final product, recovery, energy requirements and productivity. Two approaches were implemented in this process optimisation study. The first approach was an unconstrained optimisation problem with maximisation of both CO2 purity and recovery while the second approach focussed on maximizing the productivity and minimizing the energy consumption subject to constraints on CO2 recovery and purity. It was found that all the literature reported adsorbent metrics (reported in Table 1) could identify the poorly performing adsorbent (the activated carbon in this case) from the given set of four adsorbents, while for the remaining three, their relative ranking was very different for each of the metrics. Among the reported adsorbent metrics, only the single-component selectivity and Rege and Yang's parameter S predicted the same ranking in terms of the separation performance at the process level. In addition to the four adsorbents under study, three hypothetical adsorbents were also chosen for process optimisation studies to understand the impact of selectivity on process performance. It was found that for a selective CO2 adsorbent, enhancing the CO2 affinity further has a marginal impact on the achievable CO2 purity and recovery. However, lowering the N2 affinity had a huge impact on improving the process performance and decreasing the energy consumption,49 and similar observations have been reported elsewhere.166,168
The working capacity of N2 was reported to be an important factor in post-combustion CO2 capture due to the high CO2 purity requirements. If the amount of N2 adsorbed is high in the feed step, then the regeneration process requires a lower vacuum during counter-current depressurisation thereby increasing the energy requirements. This was deduced from a general evaluation metric (GEM) that was developed for screening MOFs using adsorption data from molecular simulations followed by full-scale process simulations for a PSA unit. The adsorbent ranking was based on the cost of CO2 capture using the Spearman correlation coefficient for the developed GEM and literature reported adsorbent metrics reported in Table 1, subject to 90% CO2 purity and recovery for a given PSA cycle. GEM was the best performing metric with respect to the cost of CO2 capture. In this metric, the working capacity of N2, selectivity at desorption conditions and heat of N2 adsorption were accounted for in addition to the working capacity of CO2. The CO2/N2 selectivity at desorption conditions is more relevant than at adsorption conditions because CO2 is obtained during desorption and it is essential that it desorbs completely during this step – though it is always advantageous to have higher CO2 adsorption than N2 during the feed step. The heat of adsorption exhibits the effect of temperature change on the N2 adsorption on the adsorbent during the PSA cycle.169 As comprehended from the above studies, an adsorbent's N2 loading and affinity for N2 have a huge impact on meeting the purity and recovery targets and economic feasibility of the process. However, while studying/improving the selectivity of amine-based adsorbents, the emphasis is on the interaction of amines with CO2 with no or little attention to N2 loading/affinity on these adsorbents, indicating another missing characteristic required for selecting a suitable adsorbent. It is always beneficial to enhance the CO2 adsorption capacity of a material but N2 loading will strongly influence the achieved purity levels and energy requirements of the process. As amine-based adsorbents are viewed as potential materials for direct air capture, N2 loading becomes a more important factor due to the high N2 concentrations and very dilute CO2 levels in such a separation process.
A neural network model has been developed to screen 74 adsorbents based on product purity and recovery requirements of 95% and 90%, respectively, for post combustion CO2 capture using a 4-step VSA process. This was followed by second stage screening using meta-models with constraints of minimum energy requirement and maximum productivity. This two-step adsorbent screening method identified several promising adsorbents whose performance surpassed that of the current benchmark, zeolite 13X. These adsorbents could then be further tested for stability analysis and cost evaluation.170 Similarly to the previous section, it can be clearly seen that detailed process modelling has been carried out for MOFs, zeolites and activated carbons with studies on amine-based adsorbents again being missing. The above-mentioned simplified process models for a 3 to 4 step adsorption process can be employed for further screening of amine-based carbon capture adsorbents while meeting the product purity and recovery targets and/or optimizing the parasitic energy or cost of CO2 capture involved. An optimization-based approach is essential for accurate adsorbent ranking but it is computationally very expensive and can’t be simply used at the initial screening stage with a large number of adsorbents. Thus, a two step screening is needed starting with a simplified process model with focus on performance targets and then moving on to the full scale modelling with optimization of capture cost.
We know that most of these studies are for a pressure/vacuum swing adsorption system while amine-based adsorbents are more suitable for a temperature swing adsorption process or a hybrid PTSA process. However, for either type of adsorption process, the model equations remain the same, the only difference lies in the boundary conditions being used for solving these model equations. One example of a TSA study is as follows, which included different classes of adsorbents such as zeolites, zeolitic imidazolate frameworks, MOFs, and porous polymer networks. A simplified model for a 3-step TSA cycle was presented with purity, recovery, working capacity and energy demand as the performance metrics and validated using a detailed model of an adsorption bed. In this work, the adsorbent bed is assumed to be mixed well during the TSA cycle, i.e. there were no gradients for the various process variables in the axial or the radial direction. Simplifying the model assumptions thereby significantly reducing the computation time reduced the complexity of the TSA model. Hence, a parametric study can be carried out to identify the optimum operating conditions for an adsorbent in a much faster way. A parametric study was performed for screening 76 real and hypothetical adsorbents, including Zeolite 13X, for CO2 separation from dry flue gas containing 12% CO2, 88% N2, with the TSA cycle operating at 1 bar. Screening was carried out over adsorption temperatures of 278–378 K (5–105 °C) and desorption temperatures of 318–418 K (45–145 °C). 22 out of 76 adsorbents met the CO2 purity and recovery targets for post-combustion CO2 capture. Additional ranking was carried out based on maximum working capacity and minimum energy requirement constraints. This model can be adapted for a PTSA cycle by adding blowdown and pressurisation steps into the TSA cycle and can be further employed for the screening of amine-based adsorbents. The current simplified TSA model can also be coupled with HT computational screening strategies for the fast screening of a large number of adsorbents (real and/or hypothetical).171
It can be concluded that an adsorbent potential for a separation process depends heavily on the process parameters. There is a missing gap in the development of adsorbent materials pertaining to the requirements of the process. Hence, the material researcher and the process engineer must collaborate to perform comprehensive studies on candidate adsorbents.
5. Commercial ventures in direct air capture
The past ten to fifteen years have seen the emergence of several companies that specialise in the direct air capture of CO2 (Fig. 26).172 In each case, the CO2 product is intended for commercial use in various applications such as the atmospheric enrichment of greenhouses, as a feedstock for the synthesis of new fuels, for application in enhanced oil recovery, or it is destined to be stored geologically. In 2011, DAC companies Carbon Engineering, Global Thermostat and Climeworks were announced among the finalists of the Virgin Earth Challenge, recognising them among the top organisations worldwide leading the successful commercialisation in the removal of atmospheric CO2.173 These remain the most prominent DAC companies, and although they utilise – and have patented – various sorption technology processes, it is interesting to note that the latter two use a solid amine-based adsorbent. A Canadian company, Carbon Engineering, founded in 2009, employs a chemical looping system in which atmospheric CO2 is absorbed by an aqueous KOH solution in an air–liquid contactor. The carbonate ions then react with Ca2+ to precipitate CaCO3 which is calcined at 900 °C to release a pure stream of CO2 and regenerate CaO.174 Carbon Engineering's DAC pilot plant, built in 2015 in Squamish, captures about 1 tonne of CO2 per day,175 with a levelised cost ranging from $94 to $232 per tonne of CO2.174 In 2022, the company plan on constructing their first large-scale commercial plant capable of capturing one million tonnes of CO2 per year.175 Carbon Engineering intend on utilising captured CO2 in the production of synthetic fuels via their ‘Air to Fuels’ process and are also interested in geological storage.26 | ||
| Fig. 26 Companies involved in DAC of CO2. Adapted from ref. 188 according to Creative Commons Attribution 4.0 International (CC BY 4.0). | ||
Global Thermostat, a USA-based company formed in 2010 by Eisenberger, targets the capture of CO2 from both the air and from industrial emissions with a view to the commercial sale of CO2.176 The technology involves an adsorbent composed of a porous silica or mesocellular foam substrate with a porous alumina coating, into which an amine-based compound is embedded, comprising 40–60% by volume of the alumina.177 The adsorbent is arranged in modules which can capture 50000 tonnes of CO2 per year and is regenerated by the application of heat from 85–100 °C releasing 98% pure CO2. The heat can be sourced as residual heat from a power plant or from PV solar farms.178 Global Thermostat has been operating a demonstration facility since 2010 at SRI International, Menlo Park, California, and it also has a demonstration plant in Huntsville, Alabama. In 2019, the company partnered with ExxonMobil in order to scale up the technology.179
Arguably the most established DAC venture with the largest portfolio of facilities is Climeworks. The Swiss based company, which was founded in 2009 by ETH Zurich alumni Gebald and Wurzbacher, aims to capture 1% of global CO2 emissions by 2025.180 Their DAC technology centres around modular CO2 collectors that draw air into contact with a porous adsorbent composed of aminosilane-modified cellulose nanofibers.181,182 In 2017, Climeworks launched the world's first commercial DAC plant at Hinwil, Switzerland. The plant, which was projected to cost $3–4m to build, consists of 18 modular CO2 collector units that together can capture 900 tonnes of CO2 per year.183 In each adsorption–desorption cycle, the adsorbent is regenerated by heating at low temperature (<100 °C) using low grade heat provided by a municipal waste incinerator.184 The captured CO2 is sold to a fruit and vegetable company to boost the growth of produce in large greenhouses.183 Together with Carbfix, Climeworks commissioned the world's first negative CO2 emissions facility which couples DAC with CO2 storage in Iceland.185 Heat for regeneration is supplied by Hellisheiði geothermal power station and the captured CO2 is dissolved in water and injected 700 meters underground. Here it reacts with the basaltic rock and within a few years forms calcite and other minerals.186,187 With scaling of the operation, the plant is targeted to capture and permanently store 4000 tonnes of CO2 per year.185 Climeworks has also set up facilities at which captured CO2 is utilised as a feedstock to synthesise fuels demonstrating ‘Power-to-Gas’ technologies. As part of a Horizon 2020 research project, Climeworks launched a DAC plant in Troia, Italy, at which 150 tonnes of CO2 is captured each year. Project partners ATMOSTAT then catalytically combine CO2 with hydrogen – produced from an electrolyser powered by photovoltaic energy – to produce methane, which is liquefied and used as a fuel. Waste heat from the process is utilised in the regeneration of the CO2 adsorbent.189 Climeworks is also involved in the development of a plant to produce 1000 litres of renewable jet fuel per day using atmospheric CO2. The project, initiated by Rotterdam The Hague Airport and involving a consortium coordinated by EDL Anlagenbau Gesellschaft mbH, aims to demonstrate the potential to decarbonise the aviation industry.190
For the operation of their large-scale DAC process, Climeworks quoted typical energy consumption figures of approx. 2000 kW h heat and 650 kW h electricity per tonne of captured CO2.184 The cost per tonne of captured CO2 is estimated to be around $600, but Climeworks aims to reduce this to $100 by cheaper manufacturing on the scaling up of CO2 capture machinery and continued commercialisation of the product.183
Another European-based DAC company is Skytree, founded in 2008 in The Netherlands. A spin-off from the European Space Agency, Skytree use a process originally developed in the 90s as part of the spacecraft onboard life support system. The adsorbent is composed of a high porosity polymer with amino functionality and is regenerated using a temperature swing at 80–100 °C.191 The company aims to make this technology available at the small-scale level for applications including air purification systems in cars and for delivering CO2 to aquariums.192 Oy Hydrocell Ltd., based in Järvenpää, Finland, also developed industrial CO2 filters designed to improve indoor air quality.193 The adsorbent is composed of an amine-functionalised polystyrene resin and is used in the DAC pilot plant operated by the VTT Technical Research Center and Lappeenranta University as part of the Finnish SOLETAIR research project. The DAC plant adsorbs 1.39 tonnes of CO2 per year and uses a regeneration temperature of up to 80 °C. The project aims to utilise the captured CO2 in a renewable-energy fuelled process to produce hydrocarbon fuels.194
A slightly different approach to DAC using a solid adsorbent has been adopted by the USA company, Infinitree, founded in 2014. Infinitree applies the ion exchange adsorption technology developed by Lackner and Wright in which CO2 is adsorbed under dry conditions by a resin featuring ammonium cations with hydroxide or carbonate counter ions, and is then released under humid conditions, utilising a moisture swing desorption process.195,196 Like some of Climeworks’ projects, the company is targeting the application of the technology for CO2 enrichment in commercial agricultural greenhouses.197
DAC companies are an emerging and growing sector and vary substantially depending on their processing systems, locations, partners and business models; however, it is not coincidental that the common denominator of Climeworks, Global Thermostat, Skytree, Oy Hydrocell and Infinitree is their solid amine or ammonium-based adsorption technology. As interest in commercial DAC increases and the direct air capture of CO2 becomes adopted more widely, amine-based chemisorbents may be regarded as advanced materials of the future and further investment and development in amine- and polyamine-based sorbents can be anticipated.
6. Carbon dioxide utilisation
6.1 Overview of carbon dioxide utilisation
During CO2 reduction, an electric current (potentially obtainable from a renewable source) reduces carbon dioxide, which is dissolved in aqueous bicarbonate or alkaline solution. However, carbon dioxide is an extremely stable, linear molecule. Therefore, a catalyst is used for CO2 reduction, decreasing the activation barrier for the reaction of CO2 to ˙CO2. Depending on the catalyst and the number of electrons and protons available, a variety of different reduction products can be formed. The main products discussed in this review are grouped as C1, C2 or C3 products, in line with standard nomenclature198 and are as follows: C1 (carbon monoxide, formic acid, methane), C2 (ethane, ethylene), and C3 (n-propanol).A postulated mechanism for the reduction of CO2 to methane is depicted in Fig. 27.199 The binding mode of the reaction intermediates ultimately determines the end CO2 reduction product. Here, as there is a *HCO intermediate, methane is formed. If instead two *CO intermediates were in close proximity to one another, a C2 product would form.
 | ||
| Fig. 27 A postulated mechanism of methane formation from the electrochemical reduction of CO2. Adapted with permission from ref. 199. Copyright (2015) American Chemical Society. | ||
Hydrogen is also formed from the aqueous solvent medium as a competitive by-product from the CO2 reduction. It is undesirable because it consumes electrons without forming the desired carbon-containing products and can therefore be termed ‘parasitic’. Hydrogen can be made much more efficiently through water oxidation,200,201 which is why the focus of CO2 reduction is on the formation of carbon-containing products. The measure of the success of the CO2 reduction is the faradaic efficiency (FE), eqn (1), where Qexperimental = actual, measured charge passed for a particular molecule, Qtheoretical = expected charge passed for a particular molecule, n = number of electrons passed, F = Faraday constant (96285 C mol−1), QTOT = total charge passed and product = amount of product generated. This describes the efficiency with which electrons (charge) are converted into products.
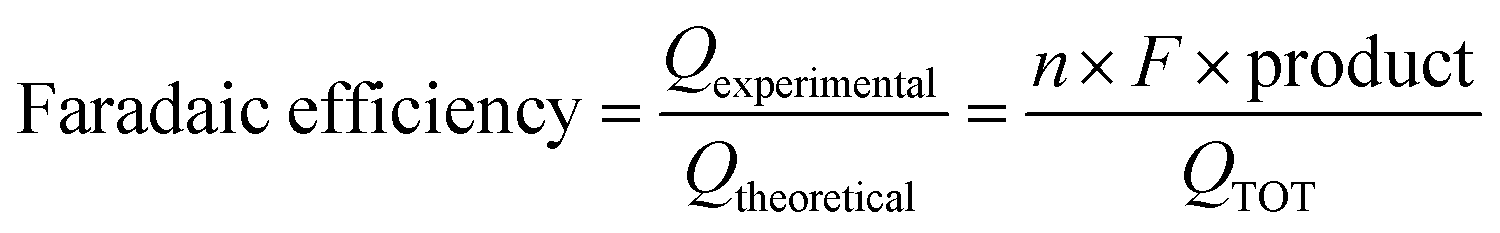 | (1) |
A number of techno-economic studies have been carried out to determine the economic viability of the various CO2 reduction products, taking faradaic efficiency and overpotential into account.202–204 For example, ethylene production requires 89% faradaic efficiency at a 0.7 V overpotential to be commercially viable. This review focuses predominantly on faradaic efficiency because it is a useful standard to compare across different experimental set-ups when referenced to the reversible hydrogen electrode (RHE).
The CO2 reduction catalyst is usually a metal. Additives can be used to change the properties of the catalyst by affecting the surface chemistry and/or reaction energetics. The additive can also be used to interact with carbon dioxide. Polymers are advantageous for this purpose because they can be large and therefore able to cover a large surface area. This is exemplified in the work of Liu and co-workers who used poly(dopamine) to protect the surface of their copper nanowire catalyst from degradation during catalysis.205 They found that the poly(dopamine) wrapped catalyst was active for 14 hours and that there was a 2.4 fold enhancement of methane production.
Polymers are also advantageous because they can feature a number of functional groups and can therefore attach to the catalyst surface as well as interact with carbon dioxide molecules and/or stabilise chemical intermediates. An example of this is the work of Ahn et al., where poly(acrylamide) is added to a porous copper catalyst.206 The poly(acrylamide) binds to the copper surface through the CO bond functionality whilst hydrogen bonding from the NH2 group stabilises the *CO intermediate which is also bound to the copper surface. However, the addition of polymers doesn’t always enhance the production of carbon-containing products. Zhao et al. report the addition of branched PEI (Mw ∼ 800) to gold nanoparticles, supported on reduced graphene oxide (RGO).207 This led to an enhancement in the competitive hydrogen evolution reaction (52% to 80%), with a corresponding decrease in carbon monoxide formation (48% to 20%). The authors suggest that PEI forms a strong interaction with the gold nanoparticles, thereby blocking catalytic sites for CO2 adsorption and reduction. In the same paper, the authors observed that shorter chain linear amines had a positive effect on CO production. Using oleylamine the faradaic efficiency of CO production increased from 52% to 75% with a corresponding decrease of hydrogen production from 48 to 25%. The authors attribute this to the olelyamine's “optimal coverage on the gold nanoparticles”.
In this section of the review, we compare the use of nitrogen-containing polymers and nitrogen-containing small molecules, including amines, pyridines, pyridinic nitrogen, imidazoles and methylcarbamate. The comparisons are categorised according to the products of the CO2 reduction reaction: C1, C2+ products and hydrogen evolution. In each category, two tables are presented summarising the experimental conditions and the faradaic efficiencies of product formation. Wherever possible the potentials are reported versus the reversible hydrogen electrode (RHE). In both tables, the additives are grouped according to the identity of the catalyst (copper, gold, silver, etc.). The categories are (1) nitrogen functionality promoting C1 products, (2) nitrogen functionality promoting C2+ products, and (3) nitrogen functionality promoting H2 evolution.
6.2 Nitrogen functionality promoting C1 products
There are two reports of PEI being combined with a catalyst to promote the formation of C1 products. Chung et al. found the addition of PEI (Mw 2000) to gold nanoparticles increased carbon monoxide production from 40% to 60% faradaic efficiency, at −1.1 V versus RHE.208 The authors state that the addition of PEI increases the surface area of the catalyst, thereby improving current density, and stabilising the catalyst to give 64 hours of stable performance. Zhang et al. used a much larger PEI (Mw 25000) with N-doped multi-walled carbon nanotubes (MWCNTs) to reduce CO2 to formic acid.209 They found that the addition of PEI increased the FE from 59% to 85% and state that PEI acts as a co-catalyst, which in turn stabilises the CO2·- intermediate. The combination of the PEI with the nitrogen-doped MWCNTs was also proven to be important; only 8% FE for formic acid production was achieved with PEI-coated undoped MWCNTs.
Copper catalysts have also been combined with amine-functionalised additives. Buckley and co-workers recently investigated a variety of additives. They found that hydrophilic additives promoted formic acid production, whereas hydrophobic additives enhanced CO selection. They combined molecular dynamic simulations with experimental data, concluding that “aprotic additives are necessary to hone CO2R selectivity”.210 The additives studied included poly(vinylpyrolidone) and didecyldimethylammonium bromide, the results of which are summarised in Tables 3 and 4, below. However, no additive that was combined with the copper catalyst led to the production of methane from CO2. Liu et al. used copper nanowires as the CO2 reduction catalyst, yielding a FE of 13% for CH4 production.205 Addition of the amine-functionalised polymer, poly(dopamine), raised this to 29%. Carbon monoxide was also observed as a minor product, but its FE was unaffected by the addition of the polymer. Interestingly, the authors demonstrated that poly(dopamine) acted as a CO2 reduction catalyst itself, albeit a very inefficient one. It yielded carbon monoxide (FE 6%) and methane (3%) from a CO2 reduction reaction.
| Additive | Metal/support | KHCO3/NaHCO3 (M) | CO2 (mL min−1) | Ref. |
|---|---|---|---|---|
| Poly(ethyleneimine) | Au NPs on C | KHCO3, 0.5 | — | 208 |
| Polyvinyl alcohol | Au NPs on C | KHCO3, 0.5 | 20 | 211 |
| Oleylamine | Au NPs on RGO | KHCO3, 0.1 | 20 | 207 |
| Hexylamine | Au NPs on RGO | KHCO3, 0.1 | 20 | 207 |
| Pyridine | Au NPs on CNTs | KHCO3, 0.1 | NA | 212 |
| Cysteamine | Au foil | KHCO3, 0.1 | 40 | 213 |
| 4-Pyridinylethane mercaptan | Au foil | KHCO3, 0.1 | 40 | 213 |
| Poly(dopamine) (Mw 9–10000) |
Cu nanowires | KHCO3, 0.5 | NA | 205 |
| Poly(vinylpyrolidone) and didecyldimethyl ammonium bromide | Oxide-derived Cu | K2CO3, 0.05 | 0 | 210 |
| Phenethylamine | Cu nanowires | KHCO3, 0.5 | NA | 205 |
| Methyl carbamate | Cu foil | NaHCO3, 0.5 | 60 | 215 |
| Polyethyleneimine (Mw ∼ 25000) |
N-doped MWNTs | KHCO3, 0.1 | NA | 209 |
| 4-Aminomethyl benzene | Pb | KHCO3, 1.0 | 0 | 216 |
| Pyridinic-N | Sn fibres | KHCO3, 0.1 | 20 | 214 |
| Amine functionalised MWCNT | Sn oxide nanosheets | KHCO3, 0.5 | NA | 217 |
| Cysteamine | Ag NPs on CB | KHCO3, 0.5 | 120 | 218 |
| Additive | Add. conc. | Metal/support | V vs. RHE | FE CO (%) | FE HCOOH (%) | FE H2 (%) | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| No add. | Add. | No add. | Add. | No add. | Add. | |||||
| a At −1.0 V the FE of PEM decorated Au foil for HOO-doubles from 12% without to 22.5% with PEM. b The SnOx@MWCNTs were used as gas diffusion electrodes. c Methane production was 85% with methylcarbamate and only 52% without it. d NB. If MWCNTs that weren’t nitrogen-doped were used the formate production FE was 5 and 8% without and with PEI respectively. e Methane production was 30% with poly(dopamine) and 13% without. Methane production was ∼1% with phenethylamine and 13% without it. | ||||||||||
| Oleylamine | 150 mM | Au NPs on RGO | −0.7 | 52 | 75 | — | — | 48 | 25 | 207 |
| Hexylamine | 150 mM | Au NPs on RGO | −0.7 | 52 | 61 | — | — | 48 | 39 | 207 |
| Poly(vinylalcohol) Mw 9000–10000 |
1.0 wt% | Au NPs on carbon | −0.67 | 10 | 90 | — | — | 90 | 10 | 211 |
| Poly(ethyleneimine) Mw 2000 | 1.2 mL | Au NPs on carbon | −1.1 | 40 | 60 | — | — | 60 | 40 | 208 |
| Pyridine | — | Au NPs on CNTs | −0.6 | 85 | 93 | 212 | ||||
| 4-Pyridinyl ethanemercaptana | 20 mM | Au foil | −0.6 | 18 | 3 | 2 | 8 | 45 | 65 | 213 |
| Cysteamine | 20 mM | Au foil | −0.6 | 18 | 35 | 2 | 2.5 | 45 | 55 | 213 |
| Cysteamine | 0.648 mM | Ag NPs on CB | −0.75 | 20 | 80 | — | — | 80 | 20 | 218 |
| Ethylenediamine | 0.648 mM | Ag NPs on carbon | −1.0 | — | 94.2 | — | — | — | 5.8 | 220 |
| −0.75 | 90 | — | — | — | 10 | |||||
| Oleylamine | — | Ag NPs on carbon | −1.0 | — | 45 | — | — | — | 55 | 220 |
| Pyridinic-N | 0.35 g | Sn on CN fibres | −0.5 | — | 62.0 | — | 0 | — | 48 | 214 |
| Pyridinic-N | 0.35 g | Sn on CN fibres | −0.8 | — | 25 | — | 65 | — | 12 | 214 |
| Pyridinic-N | 0.35 g | Atomically dispersed Sn on C–N fibres | −0.6 | — | 91 | — | 0 | — | 9 | 214 |
| Amine functionalised MWCNTb | — | Sn oxide nanosheets | −1.25 vs. SHE | 3 | 35 | 52 | 63 | 23 | 2 | 217 |
| Amine functionalised MWCNTb | — | Sn oxide nanosheets | −1.15 | 41 | 46 | 30 | 43 | 29 | 11 | 217 |
| 4-Aminomethyl-benzene | 50 mM | Pb | −1.29 | — | — | 55 | 80 | 45 | 20 | 216 |
| Polyethyleneimine (Mw ∼ 25000)d |
5 mass% | N-doped MWCTs | −1.8 V vs. SCE | — | — | 59 | 85 | 41 | 15 | 209 |
| Methyl carbamatec | 3.0 mM | Cu | −2.2 | — | — | — | — | 52 | 22 | 215 |
| Poly(dopamine)e | 5 mg | Cu | −0.97 | 1 | 1 | — | — | 10 | 10 | 205 |
| Poly(vinylpyrolidone) | 10 mg + PrOH drop-cast | Oxide derived Cu | −0.7 | 28 | 10 | 34 | 45 | 28 | 43 | 210 |
| Didecyldimethyl ammonium bromide | 0.0274 mM drop-cast | Oxide derived Cu | −0.7 | 28 | 8 | 34 | 62 | 28 | 21 | 210 |
| Phenethylaminee | 100 mM | Cu | −0.97 | 1 | 30 | — | — | 10 | 15 | 205 |
A polymer with no nitrogen functionality, poly(vinylalcohol) (PVA) has also been shown to be an effective additive to gold nanoparticles for CO2 reduction, increasing the faradaic efficiency for carbon monoxide production from 10 to 90%.211 The authors attribute this enhancement to the extensive hydrogen bonding network at the metal–polymer interface, which stabilises the *COOH intermediate. The high faradaic efficiency, low potential (−0.67 V versus RHE) and a 4 hour period of stable CO production is impressive and further research could test PVA with a copper catalyst to see whether it also enhances methane production. There are some small nitrogen-containing molecules, both amines and pyridine derivatives that have been added to a gold catalyst.207,208,211–213 The highest faradaic efficiencies for carbon monoxide production were 75% by adding oleylamine and 93% by adding pyridine.207 Although the increase in FE on the addition of pyridine to carbon nanotubes is insignificant (85% to 93%) the increase in current density from the addition of pyridine is substantial, increasing from only ∼10 A g−1Au to ∼75 A g−1Au (at −0.6 V versus RHE). Therefore, despite a modest difference in faradaic efficiencies, the current density improves dramatically. Supported by density functional theory (DFT) studies, the authors propose that this increase is due to the gold particles and pyridine groups synergistically stabilising the *COOH intermediate. Zhao et al. also find that the addition of amines (namely oleylamine) to gold nanoparticles stabilises the *COOH intermediate. They also propose that using the long-chain, branched, polyethyleneimine results in too high a surface coverage (i.e. all the active sites are blocked) and instead suggest that smaller chain, linear amines, such as oleylamine, cover the surface less effectively therefore leaving active sites free for catalysis.
A cheaper and more effective method for the electroreduction of CO2 to produce CO is the addition of pyridinic nitrogen to atomically dispersed tin nanoparticles on carbon fibres.214 The FE of CO production was 91% at the low potential of −0.6 V versus RHE and the authors suggest that the Sn–N moieties have intrinsically high activity but acknowledge that the reaction kinetics can only be elucidated with further mechanistic studies. The discussed nitrogen-containing additives to catalytic metals are summarised in Tables 3 and 4.
6.3 Nitrogen functionality promoting C2+ products
The addition of nitrogen-containing polymers and smaller molecules to electrocatalysts can also promote the formation of C2+ products. A summary is presented in Tables 5 and 6, summarising the experimental conditions used and comparing the faradaic efficiency of hydrogen production. Until 2018, copper was the only metal catalyst found capable of catalytically reducing carbon dioxide to C2+ products. There has recently been a report of ethylene production from CO2 using a silver catalyst.219| Additive/support | Metal/support | KHCO3/NaHCO3 (M) | CO2 flow rate (mL min−1) | Ref. |
|---|---|---|---|---|
| Glycine | Cu | KHCO3, 0.1 | 0 | 224 |
| N-Tolylpyridinium | Cu | KHCO3, 0.1 | 5 | 225 |
| Benzimidazole | Cu | KHCO3, 0.1 | 10 | 226 |
| Pyridinic-N rich graphene | Cu | KHCO3, 0.5 | 40 | 227 |
| Polyaniline | Cu | KHCO3, 0.1 | 0 | 228 |
| Co-polymerised 2,5-diethynyl-pyridine with 1,3,5-tris(azidomethyl benzene) | Cu | KHCO3, 0.1 | 0 | 229 |
| Co-polymerised 1,3,5-triethynylbenzene with 1,3,5-tris(azidomethyl benzene) | Cu | KHCO3, 0.1 | 0 | 230 |
| Poly(acrylamide) | Cu | NaHCO3, 0.1 | 40 | 206 |
| Additive | Add. conc. (mM) | Metal/support | V vs. RHE | FE C2H4 (%) | FE C2H6 (%) | FE H2 (%) | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| No add. | Add. | No add. | Add. | No add. | Add. | |||||
| a Approximately. Xie et al.224 reported their electrocatalysis vs. Ag/AgCl, without including ohmic drop measurements or corrections. See supporting information of the paper by Ahn et al.206 where Ahn converted the Xie data from Ag/AgCl to RHE. b n-Propanol also produced 2.8% FE without and 7.1% with N-tolylpyridinium, 8.6% with 4-aminophenylpyridinium. Ethanol also produced 7.2% FE without and 30.6% with N-tolylpyridinium, 26.7% with 4-aminophenylpyridinium. c n-Propanol without was 1.75% and with additive was 3.0% ethanol without was 2.0% and with additive was 28.2%. d Data provided by authors at −1.1 V vs. RHE without the additive but not at any other potential. No FE for hydrogen production is reported and so the number has to be extrapolated from data given in the text. Insufficient data is given to extrapolate hydrogen FE at this potential for this material. | ||||||||||
| Glycine | 0.1–10.0 | Cu foil | −1.31a | 9.5 | 24.0 | 0 | 0 | 66.8 | 34.7 | 224 |
| Glycine | 0.1–10.0 | Cu nanowire | −1.31a | 5.9 | 12.7 | 11.7 | 21.1 | 76.2 | 52.2 | 224 |
| N-Tolylpyridiniumb | 10.0 | Cu polycrystalline | −1.1 | 12.4 | 40.5 | — | — | 42.8 | 15.5 | 225 |
| 4-Aminophenylpyridinium | 10.0 | Cu polycrystalline | −1.1 | 12.4 | 40.8 | — | — | 42.8 | 12.4 | 225 |
| Benzimidazolec | 50.0 | Cu Foil | −1.09 | 15 | 41.1 | — | — | 18 | 7.5 | 226 |
| Pyridinic-N rich graphene | 1:1 w/w p-NG:Cu |
Cu NPs (7 nm) | −0.90 | —d | 19.0 | —d | 1.0 | —d | 75.7 | 227 |
| Pyridinic-N rich graphene | 1:1 w/w p-NG:Cu |
Cu NPs (7 nm) | −1.1 | 6.0 | 11 | 3.0 | 4 | 90.0 | —d | 227 |
| Polyaniline (PANI) | 5 g L−1 Nafion PANI–Cu ink | Glassy carbon | −1.2 | 20 | 48.8 | — | — | — | — | 228 |
| Co-polymerised 2,5-diethynyl-pyridine with 1,3,5-triethynylbenzene | 9.9 mM in 5 mL electrolyte | Cu rod | −1.4 | 10 | 21 | — | — | 65 | 45 | 229 |
| 1,3,5-Triethynylbenzene 1,3,5-tris(azidomethyl-benzene) co-polymer | 4.3 mM solution drop cast | Cu rod | −1.4 | 8 | 20 | 0 | 0 | 65 | 48 | 230 |
| Poly(acrylamide) | 10.0 | Cu | −0.96 | 13.0 | 26 | 0.5 | 0.5 | 50 | 50 | 206 |
| Poly(acrylamide) | 10.0 | Cu | −1.05 | 8.0 | 13 | 0 | 0 | 73 | 65 | 206 |
| Amine-functionalised polyamide | 0.012 | Cu on carbon | −0.47 | 27 | 87 | — | — | 12 | 2 | 222 |
The only reported use of a polymer with a copper catalyst to promote C2+ formation was the use of poly(acrylamide) with a copper foam.206 The copper foam alone produced 13% FE for ethylene, this doubled on addition of poly(acrylamide) to 26%, at −0.96 V versus RHE. The current density was also extremely high, 147 mA cm−2, due to the porous nature of the foam giving a high catalytic surface area. Using DFT calculations the authors propose that the poly(acrylamide) binds to the copper metal through the carbonyl moiety of the amide group, leaving the –NH2 protons free to hydrogen bond and stabilise the *CO intermediate (Fig. 28). Concurrently, the polymer encourages multiple *CO intermediates to form nearby leading to CO dimerisation and increased C–C bond formation. Ethylene is the dominant product from this reaction at potentials lower than −0.81 V versus RHE. Additional C2+ products observed are ethane, ethanol and n-propanol. At potentials lower than −0.96 V versus RHE methane (a C1 product) is also observed. Recent studies have shown that modifying the catalyst layer of gas diffusion electrodes221 with nitrogen-containing polymers can indeed enhance CO2-to-ethylene conversion to very high faradaic efficiencies.222 Moreover, the incorporation of phenylpyridinium in porous random copolymers can increase polymer film robustness.223
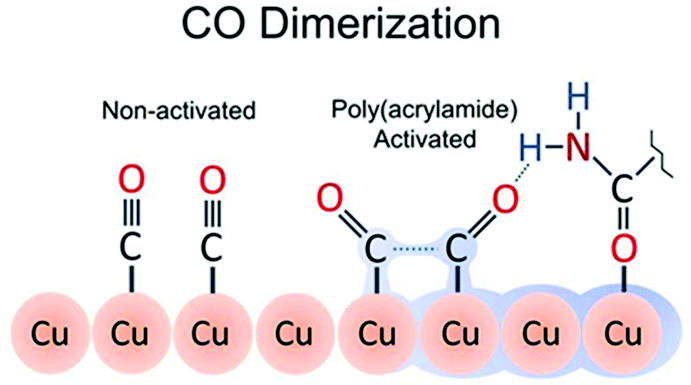 | ||
| Fig. 28 Depiction of the effect of adding poly(acrylamide) to a copper foam, as reported by Ahn et al. Reprinted with permission from ref. 206. Copyright (2018) American Chemical Society. | ||
Xie et al. screened six amino acids as additives to copper nanowire catalysts.224 Glycine led to the greatest improvement, increasing the FE of ethylene production from 9.5% to 24.0%. The authors suggest that the zwitterionic character of the glycine (forming –NH3+) can stabilise the *CHO intermediate in CO2 reduction, thus promoting the formation of C1 and C2 products. However, as a number of the other tested amino acids, e.g., alanine, leucine, arginine, also show zwitterionic character, this theory is worth further investigation to determine whether the electron donating/withdrawing groups attached to the zwitterion also have an effect on catalysis. The N-tolylpyridinium additive added to a copper electrode by Han et al. resulted in an impressive faradaic efficiency for n-propanol production (7.1% at −1.1 V versus RHE).225 Whilst reports of ethylene production are common in the literature,198n-propanol is still a sought after commodity. Han found that some of the N-tolylpyridium reacted during catalysis to form a film upon the catalytic surface. The film was found to be a dimer of the N-tolylpyridinium compound. A conjecture was given that either the film suppressed methane and hydrogen production, or that the film restricted proton diffusion, thereby affecting the local surface pH. Further investigation was carried out by Ovalle and Waegele231 who used surface enhanced infrared absorption spectroscopy and scanning electron microscopy to understand the formation of the aryl pyridinium films on the copper surface. The hypothesis put forward by Han (that the film restricted proton diffusion) was corroborated by Ovalle and Waegele, who found that the N-tolylpyridinium film “leads to an increase in the interfacial pH by limiting the mass transport to/from the interface” and that C2+ products form due to the lack of protons available at the interface. Interestingly, the authors' findings also suggest that nitrogen lone pairs are a disadvantage as they can bind to active copper sites, thus blocking those sites for CO2 reduction.231
Research on arylpyridiniums was expanded by Li et al. to test the same range of pyridinium-containing salts that were reported in Han's paper, but in a liquid-electrolyte flow cell, instead of a static H-cell.232 Through further theoretical and in situ investigation the authors propose that “the nitrogen atoms of the N-aryl-substituted pyridinium ring influences the binding of the *CO” to the copper. Under these conditions the N,N′-(1,4-phenylene)bispyridinium salt was found to be the most active towards ethylene production, achieving 72% efficiency at −0.83 V versus RHE, with an impressively high partial current density of 230 mA cm−2. The authors suggest that the high number of nitrogen atoms in the molecule contributed to the high ethylene efficiency. Overall, the cell was stable for 190 hours of ethylene production during which the faradaic efficiency dropped from 71 to ∼55%. The corresponding hydrogen production increased from ∼10% to ∼25% faradaic efficiency.
Zhong et al. used comparable conditions (electrolyte, bubbling rate, potential) to the original 2017 Han paper, to investigate the addition of benzimidazole to a copper foil for CO2 reduction.226 The addition of benzimidazole (BIM) significantly increases the formation of ethylene (from 15.0% FE to 41.1% FE) and ethanol (28.2% FE versus 2.0% without). These faradaic efficiencies are comparable with those observed by Han when using N-tolylpyridinium and 4-aminomethylpyridinium. The only significant difference is that Han's catalysts achieved double the faradaic efficiency of n-propanol production, compared to Zhong's. Zhong et al. cite (i) the Cu(BIM)x layer restricting proton diffusion (similar to Han and the dimerisation of the N-tolylpyridinium) and (ii) the benzimidazole reacting with water to obtain protons, which leads to the formation of the intermediate [COOH]*, known to lead to C2+ products.
Recently, amine-functionalised polyamides incorporated in copper-based gas diffusion electrodes have achieved high CO2-to-ethylene FE up to 87%.222
6.4 Nitrogen functionality promoting parasitic H2 evolution
Some nitrogen-containing additives promote the competitive hydrogen evolution reaction. A summary is presented in Tables 7 and 8, where Table 7 summarises the experimental conditions used, and Table 8 compares the faradaic efficiency of hydrogen production.| Additive | Metal/support | KHCO3/NaHCO3 (M) | CO2 (mL min−1) | Ref. |
|---|---|---|---|---|
| Poly(allylamine) | Cu | NaHCO3, 0.1 | 40 | 206 |
| Poly(allylamine) | Oxide-derived Cu | K2CO3 (0.5) | 0 | 210 |
| Trimethylamine | Cu | KHCO3, 0.1 | 100 | 233 |
| Pyridinium | Cu | KHCO3, 0.1 | 5 | 225 |
| 1,3-Bis(2,4,6-trimethylphenyl)-1,5-imidazol-1-ylium | Cu | KHCO3, 0.1 | 5 | 225 |
| Methyl carbamate | Cu | NaHCO3, 0.5 | 60 | 215 |
| Poly(ethylene imine) | Au NPs on RGO | KHCO3, 0.1 | 20 | 207 |
| Additive | Add. conc. (mM) | Metal/support | V vs. RHE | FE H2 (%) | Ref. | |
|---|---|---|---|---|---|---|
| No add. | With add. | |||||
| Poly(allylamine) | 10.0 | Cu | −0.96 | 50 | 100 | 206 |
| Poly(allylamine) | 10.0 | Cu | −1.10 | 75 | 100 | 206 |
| Poly(allylamine) | 0.0274 drop cast | Oxide derived Cu | −0.70 | 28 ± 2 | 97 ± 2 | 210 |
| Trimethylamine | 0.2 | Cu | −1.35 (versus SHE) | 15 | 65 | 233 |
| Pyridinium | 10.0 | Cu | −1.1 | 42.8 | 88.5 | 225 |
| 1,3-Bis(2,4,6-trimethylphenyl)-1,5-imidazol-1-ylium | 10.0 | Cu | −1.1 | 42.8 | 91.3 | 225 |
| Methyl carbamate | 3.0 | Cu | −1.10 | 80 | 90 | 215 |
| Poly(ethylene imine) Mw 800 | 150 | Au NPs on RGO | −0.7 | 52 | 80 | 207 |
It is important to understand what effect the nitrogen-containing additive has on the overall catalytic mechanism of hydrogen evolution. Zhao and co-workers proposed that PEI (Mw 800) could block active catalytic sites thereby prohibiting CO2 from interacting with the gold nanoparticles.207 This led to an 80% FE for H2 production at −0.7 V versus RHE. Ahn et al. added poly(allylamine) to a copper foam and found that it completely suppressed CO2 reduction, solely promoting the formation of hydrogen (100% FE for H2 production, up from 50% without additive).206 A similar observation was made by Buckley et al. who added poly(allylamine) to oxide-derived copper – the faradaic efficiency for hydrogen production increased from 28% without poly(allylamine) to 97% with it. Ahn found that the addition of poly(allylamine) affected the morphology of the copper catalyst, leading to a rounded and compact structure.210 Buckley propose that the protic groups in the poly(allylamine) “act as proton shuttles, or that the groups have an electronic effect that promotes the hydrogen evolution reaction”. The promotion of the hydrogen evolution reaction was also observed when trimethylamine was added to a copper catalyst the FE for H2 production increased from 15% to 65%. The strong bonding of amines to copper is cited as the reason for the deactivation of the catalyst.233 Similarly, Han et al. propose that the binding of the pyridine-moieties to the active copper sites causes deactivation of the catalyst.225 Interestingly methyl carbamate was found to have a positive and a negative effect on the CO2 reduction depending on the applied potential. Qiu and co-workers found that at −1.10 V versus RHE the addition of methylcarbamate increased the FE of H2 production from 80% to 90%.215 However, at much more negative potentials (−2.20 V versus RHE) the FE for H2 production was inhibited.
7. Conclusions
For atmospheric CO2 levels to reduce and for global warming to be alleviated, technologies must be developed to capture CO2 at an industrial scale. To help finance this endeavour, and to reduce our dependence on depleting fossil fuel resources, it is widely believed that CO2 utilisation should be adopted and developed across the chemicals, materials and energy sectors, in parallel with carbon capture and storage technology.Amines have been employed as the reactive components within a highly diverse and versatile family of CO2 adsorbents, and these are an intensely researched group of materials. Amines and polyamines have been used heavily in combination with inorganic oxides as cheap and effective support materials, and they have also been applied to a host of other solids to produce novel and effective adsorbents. Polyamines have been supported on hollow fibres and resins, and they have also been used in conjunction with typically researched CO2 physisorbents such as MOFs, carbonaceous materials and microporous polymers. New directions in amine-based adsorbents are evolving too, such as the rise of the ‘unsupported’ polyamine, by way of forming a solid, cross-linked polymer network. In terms of their CO2 capacity, amine-based adsorbents have displayed uptakes in excess of 3 mmol CO2 g−1, even at a low CO2 partial pressure equivalent to that of air.29 In addition to high uptake potentials at low CO2 concentrations, amine-based adsorbents have certain advantages over other CO2 capture technologies, such as their lower regeneration energy penalty compared to aqueous amine solutions, and their high moisture tolerance compared to some of their physisorbent counterparts such as MOFs. Thus, amine-based adsorbent technologies are currently employed by several new ventures commercialising the direct air capture of CO2. With governments worldwide setting increasingly ambitious climate commitments, and the fact that these often rely on atmospheric CO2 removal, it can be anticipated that CO2 capture will receive ever-greater attention and investment. Amine-based adsorbents, much favoured by enterprises specialising in DAC, will very likely be an extremely important feature in the suite of industrial CDR technologies to be deployed in the coming years. However, despite their prominence as some of the leading industrial CO2 capture technologies, challenges remain for the research and development of solid amine adsorbents. To further scale-up CDR operations, adsorbent and operational costs must be significantly reduced. These costs are dependent on a multitude of factors including the price of the component parts of the adsorbent system. As such, cheap support materials such as inorganic oxides are highly favoured, although new methodologies are demonstrating the potential of unsupported polyamine-based adsorbents. For economical adsorption processes, it is necessary for high working capacities to be maintained through optimal amine loading balanced with good availability via favourable dispersion within high surface area adsorbents. Research continues in these directions to improve amine efficiencies through chemical modifications of the supporting materials or amines themselves, or via the introduction of additives to positively influence adsorption behaviour. Amine-based adsorbents may be highly selective for CO2 capture, however, due to this strong affinity, particularly for primary amine functionalities, regeneration of amine-based adsorbents is an issue that requires close attention to ensure the lowest possible energy input for desorption is achieved for economical viability of the process. Stability, and therefore adsorption potential, under the effects of harsh conditions present more trials for amine-based adsorbents. Research is showing how these can be overcome through creative chemical solutions such as altering amine mass or chemical structure to reduce leaching, oxidation and degradation, for example. Alternatively, new processing solutions are proposed such as shortening adsorption steps or introducing pre-CO2 adsorption scrubbing to overcome issues of deactivation by acid–gas pollutants.
When it comes to evaluating an adsorbent for application at the process level, there exist several performance indicators relevant to the adsorbent behaviour, all of which may not consistently be established at laboratory scale, as described, or under the most industrially relevant conditions, as required. These performance indicators include a material's working capacity, selectivity, and heat of adsorption as well as the purity of its product, its recovery, productivity, and the energy input required to regenerate the adsorbent, whether this is via a pressure or temperature swing, or both. These indicators can be applied to provide metrics that can be used to compare multiple adsorbents for a given separation process. Therefore, to enable the most accurate cross-study comparisons, adsorbents’ essential adsorption parameters must be analysed across a wide range of variables to gain a full account of their adsorption behaviours and ensure balance between the improvement of certain metrics over the expense of others. To this end, it is imperative that researchers involved in this area appreciate the full interplay between the fundamental chemical and engineering principles of CO2 capture technologies.
Due to the almost infinite number of adsorbent candidates for separation processes, the identification of the most suitable and highest performing adsorbents may be accelerated by comparison of the adsorbent selection metrics or via high-throughput screening. The latter may entail practical experimentation, potentially using a system in which useful data can be obtained from as small a sample mass as possible, or via a computational model using molecular simulations. The most effective adsorbents can then be subsequently subjected to rigorous pilot scale testing and comprehensive process modelling. To date, cross-study sorbent comparisons have primarily focussed on physisorbents. In order to further the development of amine-based adsorbents for industrial-scale CO2 capture, chemical engineering and processing knowledge must be adapted and advanced to understand their specific issues, especially about their heat of adsorption, product purity and regeneration energy requirements.
As amines gain momentum in developing CO2 capture applications, they also play an increasingly prominent role in CO2 utilisation technology. As components in electrocatalysts for CO2 utilisation, polyamines are highly effective in increasing the faradaic efficiency of product formation. Not only can they increase the proportion of ethylene produced but they can also suppress hydrogen evolution, a parasitic side reaction. In general, polyamines, and nitrogen-containing additives, interact with the surface of an electrocatalyst affecting its morphology, increasing its surface area, and improving its current density. Polyamine and nitrogen-containing additives can also affect electronic properties, influencing the binding of the intermediate products to the metal, and act as a co-catalyst, stabilising and promoting the formation of intermediate products. To this end, amines and polyamines are highly useful to the electrochemist as a tool for the manipulation of an electrocatalytic system to optimise the production of useful carbon compounds.
With the shift in paradigm from viewing CO2 as a waste product, to seeing it as a potentially useful reagent, there is ever-growing momentum and interest in the conversion of CO2 into higher-value chemicals. Solid amine-based materials have proven themselves highly applicable to both CO2 capture and utilisation. It can be expected that they will feature heavily in technologies of the future and play their part in the ‘Green Industrial Revolution’.
Author contributions
Louise B. Hamdy: conceptualisation, writing – original draft, writing – review & editing. Chitrakshi Goel: conceptualisation, writing – original draft, writing – review & editing. Jennifer A. Rudd: conceptualisation, writing – original draft. Andrew R. Barron: funding acquisition, writing – review & editing. Enrico Andreoli: funding acquisition, conceptualisation, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Reducing Industrial Carbon Emissions (RICE) and Flexible Integrated Energy Systems (FLEXIS) research operations part-funded by the EU's European Regional Development Fund through the Welsh Government. Support was also provided by the Engineering and Physical Sciences Research Council through the SUSTAIN Manufacturing Hub EP/S018107/1 and project EP/N009525/1. The authors acknowledge Dr Nada Bjelobrk for valuable discussions.Notes and references
- C. Le Quéré, R. B. Jackson, M. W. Jones, A. J. Smith, S. Abernethy, R. M. Andrew, A. J. De-Gol, D. R. Willis, Y. Shan and J. G. Canadell, Nat. Clim. Change, 2020, 1–7 Search PubMed.
- D. DellaSala and M. Goldstein, Encyclopedia of the Anthropocene, Elsevier, Oxford, England, 1st edn, 2017 Search PubMed.
- M. Meinshausen, in Achieving the Paris Climate Agreement Goals: Global and Regional 100% Renewable Energy Scenarios with Non-energy GHG Pathways for +1.5 °C and +2 °C, ed. S. Teske, Springer International Publishing, Cham, 2019, pp. 459–469 DOI:10.1007/978-3-030-05843-2_12.
- J. Rogelj, D. Shindell, K. Jiang, S. Fifita, P. Forster, V. Ginzburg, C. Handa, H. Kheshgi, S. Kobayashi and E. Kriegler, Global warming of 1.5 °C, Intergovernmental Panel on Climate Change (IPCC), 2018, pp. 93–174.
- O. D. Bert Metz, H. de Coninck, M. Loos and L. Meyer, IPCC Special Report on Carbon Dioxide Capture and Storage, Cambridge, United Kingdom and New York, NY, USA, 2005.
- D. Thomas and S. Benson, Carbon Dioxide Capture for Storage in Deep Geologic Formations-Results from the CO2 Capture Project: Vol 2-Geologic Storage of Carbon Dioxide with Monitoring and Verification, 2015, p. 665 Search PubMed.
- Y.-G. Li and A. E. Mather, Ind. Eng. Chem. Res., 1996, 35, 4804–4809 CrossRef CAS.
- H. A. Patel, J. Byun and C. T. Yavuz, ChemSusChem, 2017, 10, 1303–1317 CrossRef CAS PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- Global CCS Institute, 2019, The Global Status of CCS, 2019, Australia.
- E. E. Ünveren, B. Ö. Monkul, Ş. Sarıoğlan, N. Karademir and E. Alper, Petroleum, 2017, 3, 37–50 CrossRef.
- K. Z. House, C. F. Harvey, M. J. Aziz and D. P. Schrag, Energy Environ. Sci., 2009, 2, 193–205 RSC.
- J. Isles, CCS gets some new chemistry, 2019 Search PubMed.
- A. E. Creamer and B. Gao, Environ. Sci. Technol., 2016, 50, 7276–7289 CrossRef CAS PubMed.
- A. Ö. Yazaydın, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, R. R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425–1430 CrossRef.
- M. Taddei, R. J. Wakeham, A. Koutsianos, E. Andreoli and A. R. Barron, Angew. Chem., 2018, 130, 11880–11884 CrossRef.
- M. Zhang, R. Zheng, Y. Ma, R. Chen, X. Sun and X. Sun, Microporous Mesoporous Mater., 2019, 285, 70–79 CrossRef CAS.
- T. D. Pham, M. R. Hudson, C. M. Brown and R. F. Lobo, ChemSusChem, 2017, 10, 946–957 CrossRef CAS PubMed.
- V. K. Singh and E. Anil Kumar, Appl. Therm. Eng., 2016, 97, 77–86 CrossRef CAS.
- S. Khodabakhshi, P. F. Fulvio and E. Andreoli, Carbon, 2020, 162, 604–649 CrossRef CAS.
- H. Kanoh and H. Luo, Post-combustion Carbon Dioxide Capture Materials, The Royal Society of Chemistry, 2019, pp. 206–258 10.1039/9781788013352-00206.
- A. K. Sekizkardes, S. Hammache, J. S. Hoffman and D. Hopkinson, ACS Appl. Mater. Interfaces, 2019, 11, 30987–30991 CrossRef CAS.
- P. Bollini, S. A. Didas and C. W. Jones, J. Mater. Chem., 2011, 21, 15100–15120 RSC.
- N. E. T. L. NETL, 2010.
- J. Gertner, New York Times, The Tiny Swiss Company That Thinks It Can Help Stop Climate Change, 2019, https://www.nytimes.com/2019/02/12/magazine/climeworks-business-climate-change.html.
- Uses, https://carbonengineering.com/uses, (accessed July 2020).
- X. Xu, B. Pejcic, C. Heath and C. D. Wood, J. Mater. Chem. A, 2018, 6, 21468–21474 RSC.
- Z. Chen, S. Deng, H. Wei, B. Wang, J. Huang and G. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 6937–6945 CrossRef CAS.
- H. T. Kwon, M. A. Sakwa-Novak, S. H. Pang, A. R. Sujan, E. W. Ping and C. W. Jones, Chem. Mater., 2019, 31, 5229–5237 CrossRef CAS.
- P.-Q. Liao, H. Chen, D.-D. Zhou, S.-Y. Liu, C.-T. He, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Energy Environ. Sci., 2015, 8, 1011–1016 RSC.
- S. E. Park, J. S. Chang and K. W. Lee, Carbon Dioxide Utilization for Global Sustainability: Proceedings of the 7th International Conference on Carbon Dioxide Utilization, Seoul, Korea, October 12–16, 2003, Elsevier Science, 2004.
- A. Goeppert, M. Czaun, R. B. May, G. K. S. Prakash, G. A. Olah and S. R. Narayanan, J. Am. Chem. Soc., 2011, 133, 20164–20167 CrossRef CAS.
- C. Gebald, J. A. Wurzbacher, P. Tingaut and A. Steinfeld, Environ. Sci. Technol., 2013, 47, 10063–10070 CrossRef CAS PubMed.
- S. H. Pang, M. L. Jue, J. Leisen, C. W. Jones and R. P. Lively, ACS Macro Lett., 2015, 4, 1415–1419 CrossRef CAS.
- J. C. Fisher II and M. Gray, ChemSusChem, 2015, 8, 452–455 CrossRef PubMed.
- Y. Fan, F. Rezaei, Y. Labreche, R. P. Lively, W. J. Koros and C. W. Jones, Fuel, 2015, 160, 153–164 CrossRef CAS.
- Y. Labreche, R. P. Lively, F. Rezaei, G. Chen, C. W. Jones and W. J. Koros, Chem. Eng. J., 2013, 221, 166–175 CrossRef CAS.
- Y. Labreche, Y. Fan, F. Rezaei, R. P. Lively, C. W. Jones and W. J. Koros, ACS Appl. Mater. Interfaces, 2014, 6, 19336–19346 CrossRef CAS.
- A. R. Sujan, S. H. Pang, G. Zhu, C. W. Jones and R. P. Lively, ACS Sustainable Chem. Eng., 2019, 7, 5264–5273 CrossRef CAS.
- J. H. Drese, S. Choi, S. A. Didas, P. Bollini, M. L. Gray and C. W. Jones, Microporous Mesoporous Mater., 2012, 151, 231–240 CrossRef CAS.
- K. S. Knaebel, Chem. Eng., 1995, 102, 92 CAS.
- M. W. Ackley, A. B. Stewart, G. W. Henzler, F. W. Leavitt, F. Notaro and M. S. Kane, US Pat., US 006027548A, 2000 Search PubMed.
- F. Notaro, J. T. Mullhaupt, F. W. Leavitt and M. W. Ackley, US Pat., 5674311, 1997 Search PubMed.
- S. U. Rege and R. T. Yang, Sep. Sci. Technol., 2001, 36, 3355–3365 CrossRef CAS.
- A. D. Wiersum, J.-S. Chang, C. Serre and P. L. Llewellyn, Langmuir, 2013, 29, 3301–3309 CrossRef CAS.
- P. J. E. Harlick and F. H. Tezel, Microporous Mesoporous Mater., 2004, 76, 71–79 CrossRef CAS.
- E. Mangano, S. Brandani, M. C. Ferrari, H. Ahn, D. Friedrich, M. L. Lozinska, P. A. Wright, J. Kahr, R. Morris, M. Croad, N. McKeown, H. Shamsipour and P. Budd, Energy Procedia, 2013, 37, 40–47 CrossRef CAS.
- E. Braun, A. F. Zurhelle, W. Thijssen, S. K. Schnell, L.-C. Lin, J. Kim, J. A. Thompson and B. Smit, Mol. Syst. Des. Eng., 2016, 1, 175–188 RSC.
- A. K. Rajagopalan, A. M. Avila and A. Rajendran, Int. J. Greenhouse Gas Control, 2016, 46, 76–85 CrossRef CAS.
- R. T. Yang, in Adsorbents: Fundamentals and Applications, ed. R. T. Yang, John Wiley & Sons, Inc. 2003, ch. 3, pp. 17–53 Search PubMed.
- R. Krishna, RSC Adv., 2017, 7, 35724–35737 RSC.
- S. Zohdi, M. Anbia and S. Salehi, Polyhedron, 2019, 166, 175–185 CrossRef CAS.
- J. Park, H. O. Rubiera Landa, Y. Kawajiri, M. J. Realff, R. P. Lively and D. S. Sholl, Ind. Eng. Chem. Res., 2020, 59, 7097–7108 CrossRef CAS.
- L.-C. Lin, A. H. Berger, R. L. Martin, J. Kim, J. A. Swisher, K. Jariwala, C. H. Rycroft, A. S. Bhown, M. W. Deem, M. Haranczyk and B. Smit, Nat. Mater., 2012, 11, 633–641 CrossRef CAS PubMed.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
- A. H. Berger and A. S. Bhown, Energy Procedia, 2013, 37, 25–32 CrossRef CAS.
- M. Caplow, J. Am. Chem. Soc., 1968, 90, 6795–6803 CrossRef CAS.
- F. A. Chowdhury, H. Yamada, T. Higashii, K. Goto and M. Onoda, Ind. Eng. Chem. Res., 2013, 52, 8323–8331 CrossRef CAS.
- T. L. Donaldson and Y. N. Nguyen, Ind. Eng. Chem. Fundam., 1980, 19, 260–266 CrossRef CAS.
- M. Xiao, H. Liu, R. Idem, P. Tontiwachwuthikul and Z. Liang, Appl. Energy, 2016, 184, 219–229 CrossRef CAS.
- J. Wang, S. Wang, Q. Xin and Y. Li, J. Mater. Chem. A, 2017, 5, 6794–6816 CAS.
- Z. Tong and W. S. W. Ho, J. Membr. Sci., 2017, 543, 202–211 CrossRef CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- C.-H. Chen, D. Shimon, J. J. Lee, F. Mentink-Vigier, I. Hung, C. Sievers, C. W. Jones and S. E. Hayes, J. Am. Chem. Soc., 2018, 140, 8648–8651 CrossRef CAS PubMed.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Energy Fuels, 2006, 20, 1514–1520 CrossRef CAS.
- V. Zelenak, D. Halamova, L. Gaberova, E. Bloch and P. Llewellyn, Microporous Mesoporous Mater., 2008, 116, 358–364 CrossRef CAS.
- M. W. Hahn, J. Jelic, E. Berger, K. Reuter, A. Jentys and J. A. Lercher, J. Phys. Chem. B, 2016, 120, 1988–1995 CrossRef CAS PubMed.
- S. A. Didas, A. R. Kulkarni, D. S. Sholl and C. W. Jones, ChemSusChem, 2012, 5, 2058–2064 CrossRef CAS PubMed.
- T. Watabe and K. Yogo, Sep. Purif. Technol., 2013, 120, 20–23 CrossRef CAS.
- A. Goeppert, H. Zhang, R. Sen, H. Dang and G. K. S. Prakash, ChemSusChem, 2019, 12, 1712–1723 CrossRef CAS PubMed.
- L.-B. Sun, A.-G. Li, X.-D. Liu, X.-Q. Liu, D. Feng, W. Lu, D. Yuan and H.-C. Zhou, J. Mater. Chem. A, 2015, 3, 3252–3256 RSC.
- M. A. Alkhabbaz, P. Bollini, G. S. Foo, C. Sievers and C. W. Jones, J. Am. Chem. Soc., 2014, 136, 13170–13173 CrossRef CAS PubMed.
- W. Chaikittisilp, R. Khunsupat, T. T. Chen and C. W. Jones, Ind. Eng. Chem. Res., 2011, 50, 14203–14210 CrossRef CAS.
- W. Li, S. Choi, J. H. Drese, M. Hornbostel, G. Krishnan, P. M. Eisenberger and C. W. Jones, ChemSusChem, 2010, 3, 899–903 CrossRef CAS PubMed.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS PubMed.
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS.
- M. Niu, H. Yang, X. Zhang, Y. Wang and A. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 17312–17320 CrossRef CAS PubMed.
- X. Shen, H. Du, R. H. Mullins and R. R. Kommalapati, Energy Technol., 2017, 5, 822–833 CrossRef CAS.
- Sigma-Aldrich, Polyethylenimine, branched, https://www.sigmaaldrich.com/catalog/product/aldrich/408727?lang=en®ion=GB&gclid=EAIaIQobChMImM6lgKK54QIVw4ayCh1nRgnUEAAYASAAEgJqs_D_BwE, (accessed Dec 2019).
- T. Tsuda and T. Fujiwara, J. Chem. Soc., Chem. Commun., 1992, 1659–1661, 10.1039/C39920001659.
- T. Tsuda, T. Fujiwara, Y. Taketani and T. Saegusa, Chem. Lett., 1992, 2161–2164 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2006, 45, 3248–3255 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2007, 46, 446–458 CrossRef CAS.
- M. Fayaz and A. Sayari, ACS Appl. Mater. Interfaces, 2017, 9, 43747–43754 CrossRef CAS PubMed.
- M. Jahandar Lashaki and A. Sayari, Chem. Eng. J., 2018, 334, 1260–1269 CrossRef CAS.
- D. J. Fauth, M. L. Gray, H. W. Pennline, H. M. Krutka, S. Sjostrom and A. M. Ault, Energy Fuels, 2012, 26, 2483–2496 CrossRef CAS.
- S. Choi, M. Gray and C. Jones, ChemSusChem, 2011, 4, 628–635 CrossRef CAS PubMed.
- W. C. Wilfong, B. W. Kail, C. W. Jones, C. Pacheco and M. L. Gray, ACS Appl. Mater. Interfaces, 2016, 8, 12780–12791 CrossRef CAS PubMed.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS PubMed.
- W. Chaikittisilp, S. A. Didas, H.-J. Kim and C. W. Jones, Chem. Mater., 2013, 25, 613–622 CrossRef CAS.
- P. Li, S. Zhang, S. Chen, Q. Zhang, J. Pan and B. Ge, J. Appl. Polym. Sci., 2008, 108, 3851–3858 CrossRef CAS.
- P. Li, B. Ge, S. Zhang, S. Chen, Q. Zhang and Y. Zhao, Langmuir, 2008, 24, 6567–6574 CrossRef CAS PubMed.
- S. Yang, L. Zhan, X. Xu, Y. Wang, L. Ling and X. Feng, Adv. Mater., 2013, 25, 2130–2134 CrossRef CAS PubMed.
- S. Gadipelli, Y. Lu, N. T. Skipper, T. Yildirim and Z. Guo, J. Mater. Chem. A, 2017, 5, 17833–17840 RSC.
- E. P. Dillon, E. Andreoli, L. Cullum and A. R. Barron, J. Exp. Nanosci., 2015, 10, 746–768 CrossRef CAS.
- Y. Lin, H. Lin, H. Wang, Y. Suo, B. Li, C. Kong and L. Chen, J. Mater. Chem. A, 2014, 2, 14658–14665 RSC.
- Aarti, S. Bhadauria, A. Nanoti, S. Dasgupta, S. Divekar, P. Gupta and R. Chauhan, RSC Adv., 2016, 6, 93003–93009 RSC.
- I. Luz, M. Soukri and M. Lail, Chem. Sci., 2018, 9, 4589–4599 RSC.
- H. He, Y. Hu, S. Chen, L. Zhuang, B. Ma and Q. Wu, Sci. Rep., 2017, 7, 3913 CrossRef PubMed.
- M.-J. Sie, C.-H. Lin and S.-L. Wang, J. Am. Chem. Soc., 2016, 138, 6719–6722 CrossRef CAS PubMed.
- H.-B. Wang, P. G. Jessop and G. Liu, ACS Macro Lett., 2012, 1, 944–948 CrossRef CAS.
- J. H. Lee, H. J. Lee, S. Y. Lim, B. G. Kim and J. W. Choi, J. Am. Chem. Soc., 2015, 137, 7210–7216 CrossRef CAS PubMed.
- L.-B. Sun, Y.-H. Kang, Y.-Q. Shi, Y. Jiang and X.-Q. Liu, ACS Sustainable Chem. Eng., 2015, 3, 3077–3085 CrossRef CAS.
- D. R. Kumar, C. Rosu, A. R. Sujan, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, ACS Sustainable Chem. Eng., 2020, 8, 10971–10982 CAS.
- J. Huang, J. Zhu, S. A. Snyder, A. J. Morris and S. R. Turner, Polymer, 2018, 154, 55–61 CrossRef CAS.
- E. Andreoli, E. P. Dillon, L. Cullum, L. B. Alemany and A. R. Barron, Sci. Rep., 2014, 4, 7304 CrossRef CAS PubMed.
- E. Andreoli, L. Cullum and A. R. Barron, Ind. Eng. Chem. Res., 2015, 54, 878–889 CrossRef CAS.
- A. Koutsianos, A. R. Barron and E. Andreoli, J. Phys. Chem. C, 2017, 121, 21772–21781 CrossRef CAS.
- E. Andreoli and A. R. Barron, Energy Fuels, 2015, 29, 4479–4487 CrossRef CAS.
- E. Andreoli and A. R. Barron, J. Mater. Chem. A, 2015, 3, 4323–4329 RSC.
- E. Andreoli and A. R. Barron, ChemSusChem, 2015, 8, 2635–2644 CrossRef CAS PubMed.
- K.-S. Hwang, H.-Y. Park, J.-H. Kim and J.-Y. Lee, Korean J. Chem. Eng., 2018, 35, 798–804 CrossRef CAS.
- S. J. Thompson, M. Soukri and M. Lail, Chem. Eng. J., 2018, 350, 1056–1065 CrossRef CAS.
- S. J. Thompson, M. Soukri and M. Lail, Energy Fuels, 2018, 32, 8658–8667 CrossRef CAS.
- S. Mane, Y.-X. Li, D.-M. Xue, X.-Q. Liu and L.-B. Sun, Ind. Eng. Chem. Res., 2018, 57, 12926–12934 CrossRef CAS.
- L. B. Hamdy, R. J. Wakeham, M. Taddei, A. R. Barron and E. Andreoli, Chem. Mater., 2019, 31, 4673–4684 CrossRef CAS.
- C.-J. Yoo, P. Narayanan and C. W. Jones, J. Mater. Chem. A, 2019, 7, 19513–19521 RSC.
- X. Xu, B. Pejcic, C. Heath, M. B. Myers, C. Doherty, Y. Gozukara and C. D. Wood, ACS Appl. Mater. Interfaces, 2019, 11, 26770–26780 CrossRef CAS PubMed.
- X. Xu, M. B. Myers, F. G. Versteeg, B. Pejcic, C. Heath and C. D. Wood, Chem. Commun., 2020, 56, 7151–7154 RSC.
- W. Xie, M. Yu and R. Wang, Aerosol Air Qual. Res., 2017, 17, 2715–2725 CrossRef CAS.
- Y. Fan, Y. Labreche, R. P. Lively, C. W. Jones and W. J. Koros, AIChE J., 2014, 60, 3878–3887 CrossRef CAS.
- K. Li, J. D. Kress and D. S. Mebane, J. Phys. Chem. C, 2016, 120, 23683–23691 CrossRef CAS.
- A. Sayari and Y. Belmabkhout, J. Am. Chem. Soc., 2010, 132, 6312–6314 CrossRef CAS PubMed.
- K. Maresz, A. Ciemięga, J. J. Malinowski and J. Mrowiec-Białoń, Chem. Eng. J., 2019, 123175, DOI:10.1016/j.cej.2019.123175.
- L. Liu, J. Chen, L. Tao, H. Li and Q. Yang, ChemNanoMat, 2020, 6, 1096–1103 CrossRef CAS.
- M. A. Sakwa-Novak, S. Tan and C. W. Jones, ACS Appl. Mater. Interfaces, 2015, 7, 24748–24759 CrossRef CAS PubMed.
- A. Heydari-Gorji, Y. Belmabkhout and A. Sayari, Langmuir, 2011, 27, 12411–12416 CrossRef CAS PubMed.
- R. Sanz, G. Calleja, A. Arencibia and E. S. Sanz-Pérez, Microporous Mesoporous Mater., 2015, 209, 165–171 CrossRef CAS.
- W. Chaikittisilp, H.-J. Kim and C. W. Jones, Energy Fuels, 2011, 25, 5528–5537 CrossRef CAS.
- J. A. A. Gibson, A. V. Gromov, S. Brandani and E. E. B. Campbell, Microporous Mesoporous Mater., 2015, 208, 129–139 CrossRef CAS.
- H. Zhang, A. Goeppert, M. Czaun, G. K. S. Prakash and G. A. Olah, RSC Adv., 2014, 4, 19403–19417 RSC.
- K. Pareek, Q. Zhang, R. Rohan and H. Cheng, J. Mater. Chem. A, 2014, 2, 13534–13540 RSC.
- H.-M. Wen, C. Liao, L. Li, A. Alsalme, Z. Alothman, R. Krishna, H. Wu, W. Zhou, J. Hu and B. Chen, J. Mater. Chem. A, 2019, 7, 3128–3134 RSC.
- M. Oschatz and M. Antonietti, Energy Environ. Sci., 2018, 11, 57–70 RSC.
- C. Kim, H. S. Cho, S. Chang, S. J. Cho and M. Choi, Energy Environ. Sci., 2016, 9, 1803–1811 RSC.
- T. H. Nguyen, H. Gong, S. S. Lee and T.-H. Bae, Chem. Phys. Chem., 2016, 17, 3165–3169 CrossRef CAS PubMed.
- M. Jahandar Lashaki, S. Khiavi and A. Sayari, Chem. Soc. Rev., 2019, 48, 3320–3405 RSC.
- S. Bali, T. T. Chen, W. Chaikittisilp and C. W. Jones, Energy Fuels, 2013, 27, 1547–1554 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- Z.-L. Liu, Y. Teng, K. Zhang, Y. Cao and W.-P. Pan, J. Fuel Chem. Technol., 2013, 41, 469–475 CrossRef CAS.
- W. Yan, J. Tang, Z. Bian, J. Hu and H. Liu, Ind. Eng. Chem. Res., 2012, 51, 3653–3662 CrossRef CAS.
- S. J. Park, J. J. Lee, C. B. Hoyt, D. R. Kumar and C. W. Jones, Adsorption, 2020, 26, 89–101 CrossRef CAS.
- C. S. Srikanth and S. S. C. Chuang, ChemSusChem, 2012, 5, 1435–1442 CrossRef CAS PubMed.
- P. Bollini, S. Choi, J. H. Drese and C. W. Jones, Energy Fuels, 2011, 25, 2416–2425 CrossRef CAS.
- S. H. Pang, L.-C. Lee, M. A. Sakwa-Novak, R. P. Lively and C. W. Jones, J. Am. Chem. Soc., 2017, 139, 3627–3630 CrossRef CAS PubMed.
- A. Sayari, Y. Belmabkhout and E. Da’na, Langmuir, 2012, 28, 4241–4247 CrossRef CAS PubMed.
- A. Sayari, A. Heydari-Gorji and Y. Yang, J. Am. Chem. Soc., 2012, 134, 13834–13842 CrossRef CAS PubMed.
- S. A. Didas, R. Zhu, N. A. Brunelli, D. S. Sholl and C. W. Jones, J. Phys. Chem. C, 2014, 118, 12302–12311 CrossRef CAS.
- S. Jeon, J. Min, S. H. Kim and K. B. Lee, Chem. Eng. J., 2020, 398, 125531 CrossRef CAS.
- F. Rezaei and C. W. Jones, Ind. Eng. Chem. Res., 2013, 52, 12192–12201 CrossRef CAS.
- F. Rezaei and C. W. Jones, Ind. Eng. Chem. Res., 2014, 53, 12103–12110 CrossRef CAS.
- R. Tailor and A. Sayari, Chem. Eng. J., 2016, 289, 142–149 CrossRef CAS.
- S. Barman and L. Philip, Environ. Sci. Technol., 2006, 40, 1035–1041 CrossRef CAS PubMed.
- J. Yang, X. Yu, L. An, S.-T. Tu and J. Yan, Appl. Energy, 2017, 194, 9–18 CrossRef CAS.
- L. Wei, Z. Gao and Y. Wang, Asia-Pac. J. Chem. Eng., 2017, 12, 660–670 CrossRef CAS.
- T. C. Drage, A. Arenillas, K. M. Smith and C. E. Snape, Microporous Mesoporous Mater., 2008, 116, 504–512 CrossRef CAS.
- W. Zhang, H. Liu, C. Sun, T. C. Drage and C. E. Snape, Chem. Eng. Sci., 2014, 116, 306–316 CrossRef CAS.
- M. Tong, Y. Lan, Q. Yang and C. Zhong, Green Energy Environ., 2018, 3, 107–119 CrossRef.
- S. Han, Y. Huang, T. Watanabe, Y. Dai, K. S. Walton, S. Nair, D. S. Sholl and J. C. Meredith, ACS Comb. Sci., 2012, 14, 263–267 CrossRef CAS PubMed.
- A. D. Wiersum, C. Giovannangeli, D. Vincent, E. Bloch, H. Reinsch, N. Stock, J.-S. Chang and P. L. Llewellyn, ACS Comb. Sci., 2013, 15, 111–119 CrossRef CAS PubMed.
- J. A. Mason, T. M. McDonald, T.-H. Bae, J. E. Bachman, K. Sumida, J. J. Dutton, S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2015, 137, 4787–4803 CrossRef CAS PubMed.
- C. E. Wilmer, O. K. Farha, Y.-S. Bae, J. T. Hupp and R. Q. Snurr, Energy Environ. Sci., 2012, 5, 9849–9856 RSC.
- Y.-S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- G. Avci, S. Velioglu and S. Keskin, ACS Appl. Mater. Interfaces, 2018, 10, 33693–33706 CrossRef CAS PubMed.
- S. Li, Y. G. Chung and R. Q. Snurr, Langmuir, 2016, 32, 10368–10376 CrossRef CAS PubMed.
- B. J. Maring and P. A. Webley, Int. J. Greenhouse Gas Control, 2013, 15, 16–31 CrossRef CAS.
- V. Subramanian Balashankar, A. K. Rajagopalan, R. de Pauw, A. M. Avila and A. Rajendran, Ind. Eng. Chem. Res., 2019, 58, 3314–3328 CrossRef CAS.
- A. K. Rajagopalan and A. Rajendran, Int. J. Greenhouse Gas Control, 2018, 78, 437–447 CrossRef CAS.
- K. T. Leperi, Y. G. Chung, F. You and R. Q. Snurr, ACS Sustainable Chem. Eng., 2019, 7, 11529–11539 CrossRef CAS.
- M. Khurana and S. Farooq, Ind. Eng. Chem. Res., 2016, 55, 2447–2460 CrossRef CAS.
- A. Ajenifuja, L. Joss and M. Jobson, Ind. Eng. Chem. Res., 2020, 59, 3485–3497 CrossRef CAS.
- AIRMINERS The index of companies and projects mining carbon from the air, http://www.airminers.org/, (accessed Sep 2020).
- Climeworks is a finalist in the Virgin Earth Challenge, https://www.venturekick.ch/Climeworks-is-a-finalist-in-the-Virgin-Earth-Challenge, (accessed Sep 2020).
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- Carbon Engineering Deployment Timeline, https://carbonengineering.com/our-story/, (accessed Aug 2021).
- The GT Solution, https://globalthermostat.com/the-gt-solution/, (accessed Sep 2020).
- P. Eisenberger, M. T. Buelow, M. Durilla, J. Kauffman and P. Tran, US Pat., US 9937461B2, 2018 Search PubMed.
- A Unique Capture Process, https://globalthermostat.com/a-unique-capture-process/, (accessed Sep 2020).
- Inside ExxonMobil's hookup with carbon removal venture Global Thermostat, https://www.greenbiz.com/article/inside-exxonmobils-hookup-carbon-removal-venture-global-thermostat, (accessed Sep 2020).
- Climeworks raises USD 30.8 million to commercialize carbon dioxide removal technology, https://climeworks.com/news/climeworks-raises-usd-30.8-million-to-commercialize, (accessed Sep 2020).
- C. Gebald, T. Zimmermann and P. Tingaut, US Pat., US 2014/0134088 A1, 2014 Search PubMed.
- Direct air capture, https://climeworks.com/page/co2-removal, (accessed Sep 2020).
- The Swiss company hoping to capture 1% of global CO2 emissions by 2025, https://www.carbonbrief.org/swiss-company-hoping-capture-1-global-co2-emissions-2025, (accessed Sep 2020).
- About Climeworks, https://www.climeworks.com/page/faq-about-direct-air-capture, (accessed Sep 2020).
- Carbfix and Climeworks commission the first large-scale permanent removal of carbon dioxide from the atmosphere, https://www.carbfix.com/carbfix-and-climeworks-commission-first-large-scale-permanent-removal-carbon-dioxide-atmosphere, (accessed Sep 2020).
- Underground injections turn carbon dioxide to stone, https://www.sciencemag.org/news/2016/06/underground-injections-turn-carbon-dioxide-stone, (accessed Sep 2020).
- P. Viebahn, A. Scholz and O. Zelt, Energies, 2019, 12, 3443 CrossRef CAS.
- M. Fasihi, O. Efimova and C. Breyer, J. Cleaner Prod., 2019, 224, 957–980 CrossRef CAS.
- Methanation with CO2 captured from air: Climeworks launches DAC-3 plant in Italy, https://climeworkscom.cdn.prismic.io/climeworkscom/5f3617ff-bb86-4e86-a398-7c3c41e4dc05_Climeworks_PressRelease_StoreGo_ENG.pdf, (accessed Sep 2020).
- Renewable Jet Fuel from Air, http://www.edl.poerner.de/en/news-edl/edl-pressemeldung/news/erneuerbarer-flugtreibstoff-aus-luft/, (accessed Sep 2020).
- A. Chanaewa, presented at Chemistry Seminar ‘CO2 from Air - Capture, Technology and Usage’ Swansea, May, 2018.
- Our Technology, https://www.skytree.eu/technology/, (accessed Sep 2020).
- Carbon dioxide filters, air purifiers and DAC equipment, https://hydrocell.fi/suodattimet-puhdistimet-ja-dac/, (accessed Oct 2020).
- F. V. Vázquez, J. Koponen, V. Ruuskanen, C. Bajamundi, A. Kosonen, P. Simell, J. Ahola, C. Frilund, J. Elfving, M. Reinikainen, N. Heikkinen, J. Kauppinen and P. Piermartini, J. CO2 Util., 2018, 28, 235–246 CrossRef.
- T. Wang, K. S. Lackner and A. Wright, Environ. Sci. Technol., 2011, 45, 6670–6675 CrossRef CAS PubMed.
- K. S. Lackner and A. B. Wright, US Pat., US 8133305B2, 2012 Search PubMed.
- Carbon Capture Greenhouse Enrichment, http://www.infinitreellc.com/#about, (accessed Sep 2020).
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- D. Kang, T. W. Kim, S. R. Kubota, A. C. Cardiel, H. G. Cha and K.-S. Choi, Chem. Rev., 2015, 115, 12839–12887 CrossRef CAS PubMed.
- H. Zhou, F. Yu, J. Sun, R. He, S. Chen, C.-W. Chu and Z. Ren, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5607 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti and E. Tzimas, Appl. Energy, 2016, 161, 718–732 CrossRef.
- H. Liu, K. Xiang, Y. Liu, F. Zhu, M. Zou, X. Yan and L. Chai, ChemElectroChem, 2018, 5, 3991–3999 CrossRef CAS.
- S. Ahn, K. Klyukin, R. J. Wakeham, J. A. Rudd, A. R. Lewis, S. Alexander, F. Carla, V. Alexandrov and E. Andreoli, ACS Catal., 2018, 8, 4132–4142 CrossRef CAS.
- Y. Zhao, C. Wang, Y. Liu, D. R. MacFarlane and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1801400 CrossRef.
- Y.-H. Chung, M. G. Ha, Y. Na, H.-Y. Park, H.-J. Kim, D. Henkensmeier, S. J. Yoo, J. Y. Kim, S. Y. Lee, S. W. Lee, H. S. Park, Y.-T. Kim and J. H. Jang, Electroanalysis, 2019, 31, 1401–1408 CrossRef CAS.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
- A. K. Buckley, M. Lee, T. Cheng, R. V. Kazantsev, D. M. Larson, W. A. Goddard Iii, F. D. Toste and F. M. Toma, J. Am. Chem. Soc., 2019, 141, 7355–7364 CrossRef CAS PubMed.
- L. Ma, W. Hu, Q. Pan, L. Zou, Z. Zou, K. Wen and H. Yang, J. CO2 Util., 2019, 34, 108–114 CrossRef CAS.
- Z. Ma, C. Lian, D. Niu, L. Shi, S. Hu, X. Zhang and H. Liu, ChemSusChem, 2019, 12, 1724–1731 CrossRef CAS PubMed.
- Y. Fang and J. C. Flake, J. Am. Chem. Soc., 2017, 139, 3399–3405 CrossRef CAS PubMed.
- Y. Zhao, J. Liang, C. Wang, J. Ma and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1702524 CrossRef.
- Y. Qiu, H. Zhong, W. Xu, T. Zhang, X. Li and H. Zhang, J. Mater. Chem. A, 2019, 7, 5453–5462 RSC.
- N. Zouaoui, B. D. Ossonon, M. Fan, D. Mayilukila, S. Garbarino, G. de Silveira, G. A. Botton, D. Guay and A. C. Tavares, J. Mater. Chem. A, 2019, 7, 11272–11281 RSC.
- Q. Zhang, Y. Zhang, J. Mao, J. Liu, Y. Zhou, D. Guay and J. Qiao, ChemSusChem, 2019, 12, 1443–1450 CrossRef CAS PubMed.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- A. Dutta, C. E. Morstein, M. Rahaman, A. Cedeño López and P. Broekmann, ACS Catal., 2018, 8, 8357–8368 CrossRef CAS.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 CrossRef CAS.
- S. Hernandez-Aldave and E. Andreoli, Catalysts, 2020, 10, 713 CrossRef CAS.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- J. Wang, T. Cheng, A. Q. Fenwick, T. N. Baroud, A. Rosas-Hernández, J. H. Ko, Q. Gan, W. A. Goddard III and R. H. Grubbs, J. Am. Chem. Soc., 2021, 143(7), 2857–2865 CrossRef CAS PubMed.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS PubMed.
- S. Zhong, X. Yang, Z. Cao, X. Dong, S. M. Kozlov, L. Falivene, J.-K. Huang, X. Zhou, M. N. Hedhili, Z. Lai, K.-W. Huang, Y. Han, L. Cavallo and L.-J. Li, Chem. Commun., 2018, 54, 11324–11327 RSC.
- Q. Li, W. Zhu, J. Fu, H. Zhang, G. Wu and S. Sun, Nano Energy, 2016, 24, 1–9 CrossRef CAS.
- X. Wei, Z. Yin, K. Lyu, Z. Li, J. Gong, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Catal., 2020, 10, 4103–4111 CrossRef CAS.
- R. Takeuchi, R. Igarashi, K. Kubo, T. Mizuta and S. Kume, ChemElectroChem, 2020, 7, 2496 CrossRef CAS.
- R. Igarashi, R. Takeuchi, K. Kubo, T. Mizuta and S. Kume, Front. Chem., 2019, 7, 860 CrossRef CAS PubMed.
- V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2019, 123, 24453–24460 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00360g |
| This journal is © The Royal Society of Chemistry 2021 |