
Evaluation of sustainable technologies for the processing of Sargassum muticum: cascade biorefinery schemes
Pablo G.
del Río
 ,
Noelia
Flórez-Fernández
,
Milena
Álvarez-Viñas
,
María Dolores
Torres
,
Aloia
Romaní
*,
Herminia
Domínguez
and
Gil
Garrote
,
Noelia
Flórez-Fernández
,
Milena
Álvarez-Viñas
,
María Dolores
Torres
,
Aloia
Romaní
*,
Herminia
Domínguez
and
Gil
Garrote
Department of Chemical Engineering, Faculty of Science, Universidade de Vigo (Campus Ourense), As Lagoas, 32004 Ourense, Spain. E-mail: aloia@uvigo.es
First published on 13th August 2021
Abstract
The leverage of invasive macroalgae, which represent a serious threat for autochthonous biodiversity, may smooth and mitigate their risk from an environmental point of view while providing an economic benefit. This work proposes the suitability of a closed loop biorefinery employing sustainable technologies (namely autohydrolysis, ultrasound assisted extraction, and microwave hydrodiffusion and gravity) for the complete exploitation of the invasive brown macroalgae Sargassum muticum. In particular, sequential extraction methods allowed recovery up to 85% of fucoidan in the form of sulfated fucooligosaccharides in the liquid phase, besides increasing the phloroglucinol and Trolox equivalent antioxidant capacity (TEAC) content up to 9.73 g per 100 g of extract and 31.1 g per 100 g of extract, respectively. Additionally, extract-free Sargassum muticum presented a high enzymatic susceptibility at a high solid loading, implying a production of 15.6 g ethanol per L (91.9% of ethanol yield) in SHF, while the resulting spent solid provided a calorific power of 10.0 MJ kg−1. This work verified the cascade biorefinery employing sustainable technologies as an appropriate strategy for the integral valorization of Sargassum muticum.
1. Introduction
Seaweed, also referred to as the third generation feedstock, is considered a biomass with high potential to be used in a marine-based biorefinery.1,2 Its rich and varied composition makes it very suitable as a source for the production of biomolecules, platform chemicals, hydrocolloids, biofuels and/or proteins, among others.3–5 In addition, macroalgae have some advantages over first and second generation feedstock (namely, lignocellulosic biomass), for instance: (i) being highly ubiquitous, (ii) presenting a higher photosynthetic efficiency (about 3.5-fold higher) compared to terrestrial biomass, (iii) requiring scarce water consumption, (iv) representing the highest annual CO2 absorption and production of oxygen in the planet, or (v) avoiding the competition for arable land.6,7In this context, Sargassum muticum (Sm) is a brown macroalgae with great potential due to its interesting features, being used as a biosorbent for the treatment of wastewaters,8 as a base for greener antifouling solutions,9 as a source of alginate, fucoidans and phlorotannins10 or for the production of biofuels.11,12 Moreover, since Sm is considered invasive as stated by the Spanish Catalog of Invasive Alien Species, it alters and displaces local native groups with a subsequent environmental impact on the ecosystem. Thus, its exploitation provides a double environmental and economic advantage.11,13
However, the use of pretreatments able to fractionate these kinds of renewable resources is essential to fully leverage macroalgae features and compounds,14,15 emphasizing the use of eco-friendly processes that follow the principles of Green Chemistry, such as using harmless and safe solvents, renewable feedstocks or diminishing derivatives.16 In this context, hydrothermal processing such as autohydrolysis is a sustainable technology based on the employment of high temperatures in a pressurized reactor containing biomass and water.17 It is considered an economical non-toxic pretreatment for the release of important biomolecules10,18 or as a first step for the production of biofuels,19 also with great potential to be used on a larger scale.20 Following this path, ultrasound assisted extraction, using water as the solvent,21,22 and microwave hydrodiffusion and gravity (MHG)23,24 have also been reported as environmentally friendly and energy-efficient techniques of extraction that facilitate the release of biomolecules with antioxidant properties from seaweed biomass.
Within this framework, classic biorefineries are centered on the manufacture of a single product, discarding the residual material.25 In order to avoid the environmental and economic issues regarding this action, a complete valorization of the biomass via sequential steps is highly encouraged following the circular economy concept.25–27 The EU Waste Framework Directive (2018/851) advocates these cascade biorefinery schemes, giving priority to the extraction of high added value biomolecules for chemical or pharmaceutical use and, after that, to the production of biofuels employing residual biomass.28 Consequently, the planning of an appropriate integrated biorefinery with the aim to successfully obtain the target compounds in a cascade approach following green chemistry principles is still a challenge.29,30
Therefore, the aim of this work was the evaluation of sustainable technologies (namely, ultrasound assisted extraction and microwave hydrodiffusion and gravity, combined or not with autohydrolysis) for the integral valorization of S. muticum macroalgae following a biorefinery approach. Firstly, the use of a single or combination of the abovementioned sustainable technologies was evaluated for the extraction of bioactive compounds. Secondly, the effect of different treatments on the enzymatic saccharification of the remaining solid after extraction was also taken into account for the complete exploitation of this brown macroalgae. A mixture of these extract-free seaweeds was blended for bioethanol production using several saccharification and fermentation strategies. Lastly, the spent solid after liquid fuel production was evaluated for heating power by differential scanning calorimetry in order to determine its potential as a solid fuel. As far as the authors know, this is the first work that explores a cascade biorefinery for the production of bioactive compounds and liquid and solid biofuels using Sargassum muticum.
2. Materials and methods
2.1. Raw material and chemical characterization
The renewable resource used in this work was Sargassum muticum (Sm) and was collected at Praia da Mourisca (Pontevedra, NW Spain) in August 2016. The macroalgae was frozen until its use to avoid possible biological contaminations. After that, Sm was washed with tap water and cleaned, cut to a particle size smaller than 8 mm, air dried and stored in plastic bags in a dark, cool and dry place.For the chemical characterization of Sm, the following procedures were used, with a prior mill to a particle size smaller than 1 mm: moisture,31 extractives,32 ash,33 and quantitative acid hydrolysis (QAH).34 The protein content was quantified by the Kjeldahl method, and the result was rectified by the conversion factor 5.38 for brown algae.35
Firstly, Sm was subjected to sequential extractives as stated by del Río and collaborators.11 Subsequently, the proximal composition of the resulting solid was determined by quantitative hydrolysis (QAH) at 121 °C for 60 min and 4% H2SO4. Afterwards, the solid and liquid phases were separated by filtration. The solid insoluble fraction after QAH was gravimetrically quantified and stated as the acid insoluble residue (AIR). The liquid phase was analyzed by HPLC (Agilent) to quantify the concentrations of sugars (namely, glucose, fucose, xylose, galactose, and mannose were quantified as sum, since the retention time for these sugars in the column used is the same), and acetic acid, under the following conditions: detector, refractive index at 40 °C; column, Rezex ROA-Organic Acid H+; mobile phase, 0.03 M H2SO4; flow rate, 0.6 mL min−1; column temperature 60 °C. The liquid after QAH was also employed to quantify the uronic acid concentration as equivalents in galacturonic acid, via a colorimetric procedure.36 All these analyses were performed in triplicate.
The chemical composition of Sargassum muticum measured as g per 100 g Sm in oven dry basis ± standard deviation is as follows:11 glucan 10.2 ± 0.23, xylan + galactan + mannan 6.75 ± 0.19, fucoidan 6.00 ± 0.11, acetyl groups 0.33 ± 0.01, acid insoluble residue (AIR) 25.0 ± 0.40, water extractives 11.3 ± 0.09, ethanol extractives 2.05 ± 0.13, ashes 11.9 ± 0.22, proteins 10.6 ± 0.42, and uronic acids (expressed as equivalents in galacturonic acid) 19.3 ± 0.56.
2.2. Sargassum muticum processing
Three technologies were employed in this work for the processing of S. muticum macroalgae, namely autohydrolysis (AH), ultrasound assisted extraction (US) and microwave hydrodiffusion and gravity (MHG), the combination of which resulted in four biorefinery schemes. Fig. 1 shows a flowchart of seaweed processing including the different cascading technologies proposed in this work and the operational conditions used, which were selected on the basis of previous studies.37–39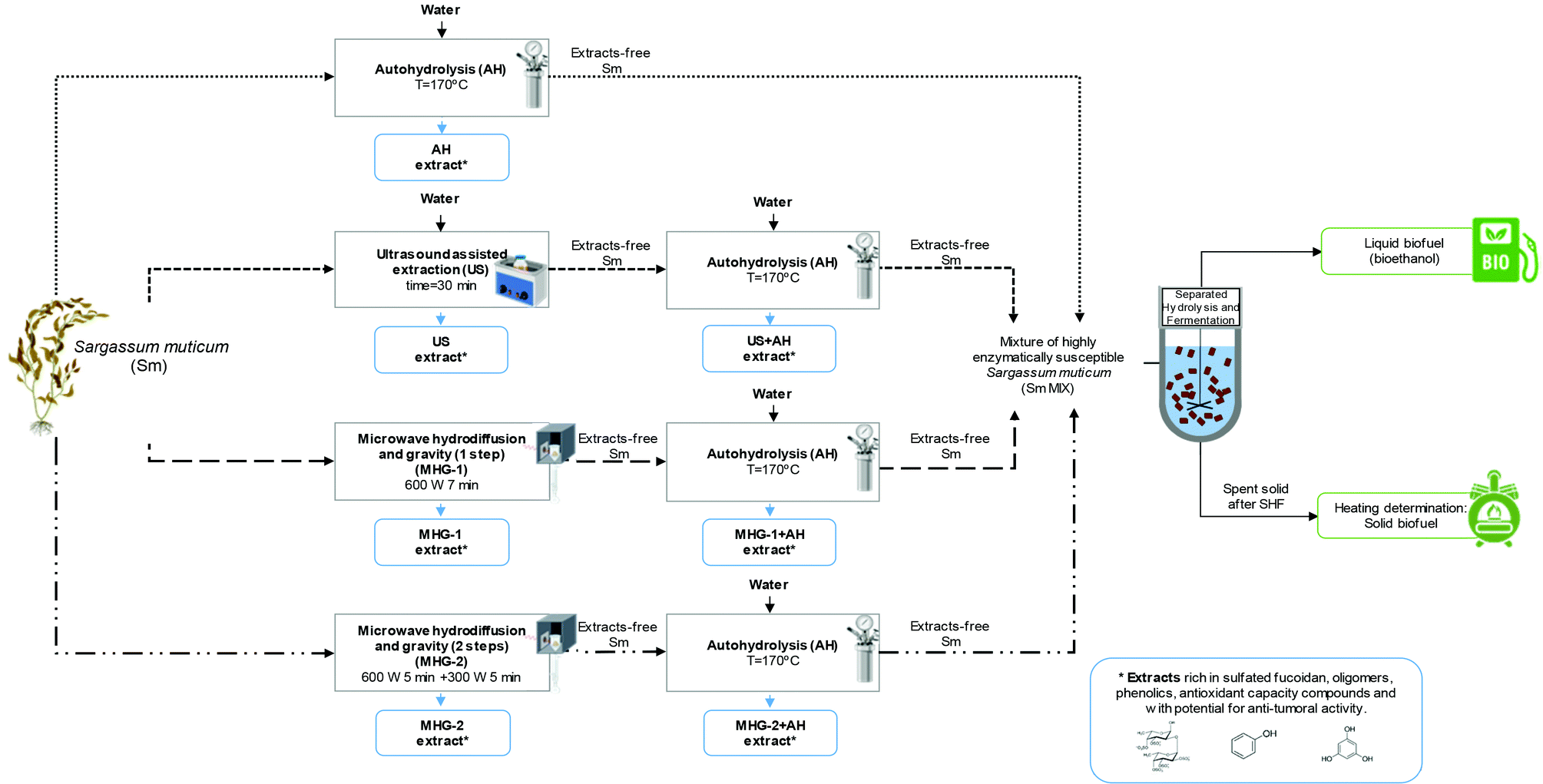 | ||
| Fig. 1 Flow chart of cascading sustainable technologies proposed in this work for Sargassum muticum valorization. Created with BioRender.com. | ||
2.3. Analysis of extracted liquid phases
The phloroglucinol content was determined according to the protocol developed by Koivikko and collaborators.41 The standard curve was done using phloroglucinol (Sigma-Aldrich, USA) as a pattern, in triplicate, then the results were expressed as phloroglucinol equivalents. Briefly, 1 mL of Folin–Ciocalteu (1 N) solution and 2 mL of Na2CO3 (20%) were added to the sample. The mixture was incubated at room temperature in darkness for 30 min. The absorbance was read at 730 nm against a blank (with distilled water). All the measures were performed in triplicate.
2.4. Enzymatic hydrolysis of extract-free Sargassum muticum
Enzymatic hydrolysis of different solid phases obtained from Sm treatments (extracts-free Sm) proposed in this work was performed in an orbital incubator (150 rpm) at 50 °C. These assays were carried out at a LSR (liquid to solid ratio) of 20, 6 and 4 g of liquid per g of solid and an ESR (enzymatic to substrate ratio) of 15 FPU g−1 of Sm and VCR (Viscozyme to Cellic CTec2 ratio) of 5 U FPU−1. The pH was adjusted to 5 with 0.05 N citrate buffer, as explained by NREL.45 Assays were carried out in duplicate.The commercial enzymes employed, kindly provided by Novozymes (Denmark), were “Viscozyme 1.5L” carbohydrases and pectinases from Aspergillus aculeatus and the enzymatic cocktail Cellic CTec2. The polygalacturonase activity for Viscozyme was calculated from the total of D-galacturonic acid formation from 0.5% w/v polygalacturonic acid in a pH 5 solution with 50 mM of sodium acetate buffer, using the DNS method. The unit of enzymatic activity (U) is defined by the quantity of enzymes that catalyzes the formation of D-galacturonic acid per minute at 37 °C and pH 5. The final enzymatic activity was 4206 U mL−1. The cellulase activity of Cellic CTec2 was measured using the filter paper assay and expressed in filter paper units (FPU).46 The final enzymatic activity was 116 FPU mL−1.
At scheduled times samples were taken, centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min, filtered using 0.45 μm pore acetate cellulose membranes and analyzed by HPLC to obtain the glucose concentration.
000 rpm for 10 min, filtered using 0.45 μm pore acetate cellulose membranes and analyzed by HPLC to obtain the glucose concentration.
The results obtained from enzymatic hydrolysis assays were expressed as glucose concentration (g L−1) or as glucan to glucose conversion (GGC, %), which was estimated using the subsequent equation:
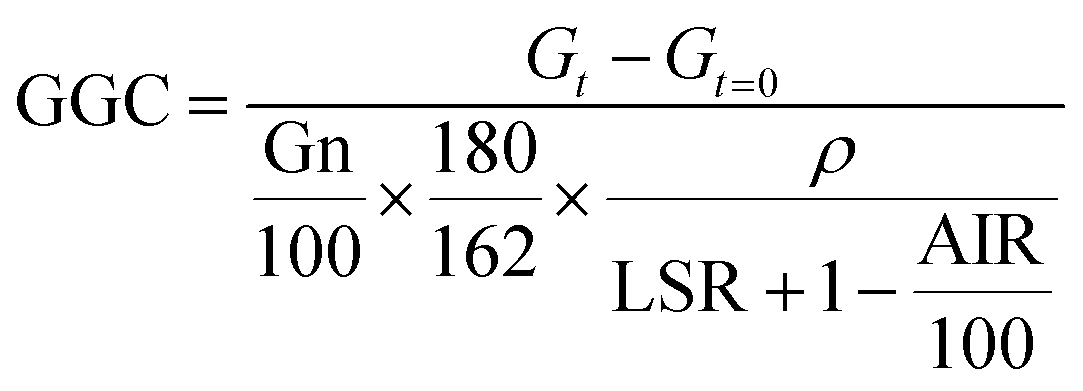 | (1) |
2.5. Yeast and growth conditions
Cells of the industrial Saccharomyces cerevisiae Ethanol Red® strain were grown on yeast peptone dextrose (YPD) agar plates containing 20 g L−1 of glucose and peptone, 15 g L−1 of agar, and 10 g L−1 of yeast extract at 30 °C. Prior to the fermentation assays, cells were incubated in sterilized liquid media of YPD with glucose, peptone and yeast extract (20, 20 and 10 g L−1, respectively) for 15 hours at 30 °C and 200 rpm. Subsequently, the cell suspension was centrifuged for 15 min at 4500 rpm and then resuspended with 0.9% NaCl to achieve a concentration of 200 g fresh yeast per L. The inoculum was added to the fermentation experiments to achieve a final concentration of 1.5 g dry yeast per L.2.6. Saccharification and fermentation assays of processed Sargassum muticum
Different strategies of saccharification and fermentation were employed for the production of third generation bioethanol, namely separated hydrolysis and fermentation (SHF), simultaneous saccharification and fermentation (SSF), and presaccharification and simultaneous saccharification and fermentation (PSSF). The LSR of 6 and 4 g of liquid per g of processed Sm, ESR of 15 FPU g−1 of Sm and VCR of 5 U FPU−1 were employed in these experiments. The presaccharification time was set at 12 hours for the PSSF assays, whereas for the SHF assays it was set at 72 h. The saccharification stage was performed at 50 °C and 150 rpm, while the fermentation stage (either for SHF, SSF or PSSF) was set at 35 °C and 120 rpm, in an orbital shaker with temperature control. After the presaccharification stage, sterilized nutrients (20 g peptone per L, 10 g yeast extract per L) at 100, 50 and 0% concentration and S. cerevisiae cells were added. Assays were carried out in duplicate.The saccharification and fermentation assays were sampled at desired times, centrifuged and the supernatant was analyzed by means of HPLC for monosaccharides, acetic acid and ethanol quantification (after filtration through 0.22 μL cellulose acetate filters). The ethanol yield was calculated as follows:
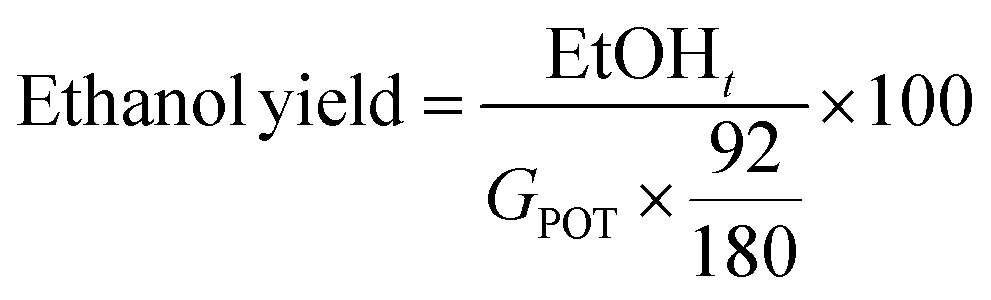 | (2) |
2.7. Differential scanning calorimetry (DSC) of extract-free Sm and spent solid after SHF
Differential scanning calorimetry (DSC) is a thermochemical procedure to evaluate the heat flow features of a material depending on the temperature variation, resulting in a DSC curve that can indicate the reaction path of the assessed biomass.47 The thermal behavior of processed Sm and spent solid after SHF was evaluated in duplicate by differential scanning calorimetry (DSC) assays in a Mettler Toledo thermoanalyzer. In brief, about 10 mg of the air-dried sample was heated up to 600 °C with a constant rate of 10 °C min−1. The resulting data were subjected to further calculations to determine the higher heating value (HHV) of the samples expressed in MJ kg−1 of sample.3. Results and discussion
3.1. Effect of sustainable technologies for the extraction of bioactive compounds
In this work, autohydrolysis, ultrasound and microwave hydrodiffusion and gravity were selected and assessed as alternative, novel, effective and sustainable processes for the extraction of soluble high added value products (for instance, oligomers with prebiotic proprieties) and bioactive compounds (total phenolic compounds with biological activities) from invasive S. muticum.The use of a single extraction is often related to obtaining one specific target compound, and may not contribute significantly to the cell disruption, avoiding the liberation of other interesting biomolecules.25 In this sense, the combination of different technologies was proposed in this work (as shown in Fig. 1) as an alternative to a single extraction. This procedure would allow to evaluate the potential enhancement of the efficiency of the global process and the recovery of a wide range of target compounds.48,49 To evaluate the effect of different extraction processes, the soluble compounds were separated by filtration for a complete characterization.
Therefore, Table 1 displays the non-volatile solid content (NVSC), volatile solid content (VSC), carbohydrate and weak acid content, phloroglucinol, TEAC, protein and sulfate of the extracts after the extractions were accomplished via: autohydrolysis (AH), ultrasound (US), microwave hydrodiffusion and gravity in a single step (MHG-1) and in two sequential steps (MHG-2), and the combination of US + AH, MHG-1 + AH and MHG-2 + AH.
| AH | US | MHG-1 | MHG-2 | US + AH | MHG-1 + AH | MHG-2 + AH | |
|---|---|---|---|---|---|---|---|
| a Data extracted from Álvarez-Viñas et al.44 b Data extracted from Flórez-Fernández et al.38 c Data extracted from Flórez-Fernández et al.37 d Unit of the fractionation data of US + AH, MHG-1 + AH and MHG-2 + AH measured in g per 100 g Sm regarding the Sm resulting from the previous processing. Data represent mean ± standard deviation (n ≥ 3). | |||||||
| Non-volatile solid content (NVSC, g per 100 g Sm) | 45.5 | 10.1 | 0.96 | 0.50 | 38.7d | 42.5d | 41.8d |
| Volatile solid content (VSC, g per 100 g Sm) | 7.70 | 0.00 | 0.00 | 0.00 | 8.30d | 6.80d | 6.20d |
| Liquid phase composition (g per 100 g of extract) (sugars expressed as sum of monomeric and oligomeric compounds) | |||||||
| Glucose | 0.97 ± 0.08 | 3.66 ± 0.20 | 3.94 ± 0.20 | 1.40 ± 0.02 | 1.20 ± 0.24 | 1.62 ± 0.09 | 0.85 ± 0.07 |
| Xylose + Galactose + Mannose | 5.79 ± 0.17 | 5.80 ± 0.20 | 0.57 ± 0.05 | 0.63 ± 0.05 | 7.43 ± 0.26 | 6.06 ± 0.08 | 5.70 ± 0.19 |
| Fucose | 12.3 ± 0.34 | 5.61 ± 0.16 | 2.02 ± 0.09 | 3.21 ± 0.27 | 13.6 ± 0.32 | 10.7 ± 0.10 | 11.4 ± 0.31 |
| Formic acid | 3.66 ± 0.11 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 3.45 ± 0.09 | 3.11 ± 0.09 | 2.95 ± 0.09 |
| Acetic acid | 1.54 ± 0.06 | 1.68 ± 0.09 | 0.04 ± 0.08 | 0.00 ± 0.00 | 1.04 ± 0.11 | 1.13 ± 0.14 | 1.00 ± 0.11 |
| Phloroglucinol | 3.22 ± 0.21a | 2.26 ± 0.18b | 3.52 ± 0.19c | 8.72 ± 0.15c | 8.61 ± 0.05 | 9.73 ± 0.02 | 7.36 ± 0.05 |
| TEAC | 6.47 ± 0.17a | 3.77 ± 0.25b | 13.2 ± 0.21c | 34.4 ± 0.23c | 26.6 ± 0.22 | 31.1 ± 0.20 | 19.9 ± 0.05 |
| Protein | 4.50 ± 0.10a | — | — | — | 4.05 ± 0.09 | 3.02 ± 0.03 | 3.06 ± 0.02 |
| Sulfate | 3.28 ± 0.13a | 3.56 ± 0.02b | — | — | 9.64 ± 0.05 | 12.7 ± 0.10 | 10.2 ± 0.04 |
Firstly, the sum of NVSC and VSC reflected the capacity of solubilization (extraction yield) of different processes evaluated. As seen, the extraction yields for the US (10.1%) and MHG processing (0.50–0.96%) were fairly scarce. On the other hand, the extraction values for AH and its combination with US and MHG ranged from 47.0–53.2%. This fact is related to harsher conditions (temperature and time) used for the AH and its combination with US and MHG. By virtue of these data, the combination of these sustainable technologies allowed to improve the extraction yield when compared with using only US and MHG. This is also related to the liberation of sulfated compounds in the liquid phase, which acted as catalysts of the reaction and resulted in the hydrolysis of compounds from the raw material matrix, incrementing the extraction yield.50 Particularly, values between 9.64–12.7 g of sulfate per 100 g of extract were accomplished when combining US and MHG with AH, whereas 3.28 and 3.56 g of sulfate per 100 g of extract were released in the liquid phase after the extraction of AH and US, respectively.
Regarding the sugar (mostly in the oligomeric form) and weak acid content, their release in the extract seemed to be greatly influenced by the harshness of the technique employed. In general, AH and its combinations with US and MHG resulted in a larger solubilization of these compounds, yielding 21.9–26.7 g of sugars and weak acids per 100 g of extract. Looking closely, fucose represented the major sugar component in the extracts, achieving up to 13.6 g of fucose per 100 g of extract in the combination of US + AH. Slightly lower values were achieved for AH and for the combination of MHG-2 + AH and MHG-1 + AH (12.3, 11.4 and 10.7 g of fucose per 100 g of extract, respectively). On the other hand, extraction techniques of US, MHG-1 and MHG-2 reached up to 5.61 g of fucose per 100 g of extract. Regarding the other sugars and weak acids, a similar behavior was observed, reflecting higher concentrations when autohydrolysis processing was involved.
The extraction of sulfated fucoidan, in this case mostly in the form of fucooligosaccharides, is highly motivated due to its interesting properties such as anti-inflammatory, anti-tumoral, anti-viral, anti-thrombotic, anti-coagulant or contraceptive.50,51 In particular, extracts of MHG at the conditions employed in this work produced a powerful inhibitory effect on the development of glioblastoma cells and pancreatic cancer cells.37 Sm extracts after US also presented anti-proliferative activity for some cancer cells, such as colon carcinoma or pancreatic adenocarcinoma cells.38 Besides that, extracts obtained after autohydrolysis at 170 °C, fractionated by molecular weight via membranes (100–5 kDa), inhibited the growth of cervix cancer cells (10–30 kDa) and ovarian cancer cells (<5 kDa).44
Alternatively, the phenolic compounds can be found in Sm as phlorotannins (polymer of phloroglucinol)52 at different degrees of polymerization, being mainly composed of fuhalols, hydroxyfuhalols and phlorethols.53 In this sense, the total phenolic content (TPC), 2.26–3.52 g of phloroglucinol equivalents per 100 g of extract, was obtained when using AH, US and MHG-1. Conversely, the combination with autohydrolysis or the use of MHG in two steps (MHG-2) caused a rise in the TPC, with values of 7.36–9.73 g of phloroglucinol equivalents per 100 g of extract. Consequently, the antioxidant capacity of the extracts (measured as TEAC) was also affected by the number of steps of extraction, obtaining 3.77–13.20 g per 100 g of extract when using a simple extraction step (AH, US, MHG-1) and 19.9–34.4 g per 100 g of extract when using more than one extraction step. In this sense, these TEAC data can be positively compared to those obtained for the non-isothermal autohydrolysis of Himanthalia elongata at 160 and 180 °C, reaching a value of 2.43–5.18 g eq. Trolox per 100 g of extract.54 Similarly, non-isothermal autohydrolysis at 180 °C was used for the extraction of bioactive compounds from Laminaria ochroleuca, achieving around 1.80 g of Trolox per 100 g of extract.55 On the other hand, TEAC results from the combined processing with autohydrolysis reached similar values to those obtained for Himanthalia elongata by pressurized liquid extraction with ethanol at 50–100 °C (25.9–26.7 g Trolox per 100 g extract),56 which evinces the sequential use of technologies as a suitable approach for the extraction of bioactive compounds, even using water as the solvent. Alternatively, the protein content was fairly similar regardless of the technology employed in this work, ranging from 3.02–4.50 g of protein per 100 g of extract.
In order to complete the physicochemical characterization of the extracts, Fig. 2a shows the molar mass distribution profiles of the samples obtained from the different technologies autohydrolysis, ultrasound assisted extraction and microwave hydrodiffusion and gravity and also with the combination of technologies. This figure displays in all cases the elution profile corresponding to compounds with a molecular mass higher than dextran of 80 kDa, besides the profile obtained for the AH sample in coherence with the profile attained for the combination of technologies US and MHG with AH. In this context, this behavior could be explained according to the harshness of the AH processing, being higher than those in the other technologies. On the other hand, US and MHG-2 profiles exhibit a first peak in the spectra that represent a bigger molar mass. Similar results were found in other studies with results around and superior to 80 kDa for fractions obtained from the brown seaweed Chnoospora minima.57 Other researchers analyzed the main polysaccharide present in brown seaweeds (fucoidan) and the molecular weights were around 105–117 kDa.58 These results were in concordance with those obtained in the present work.
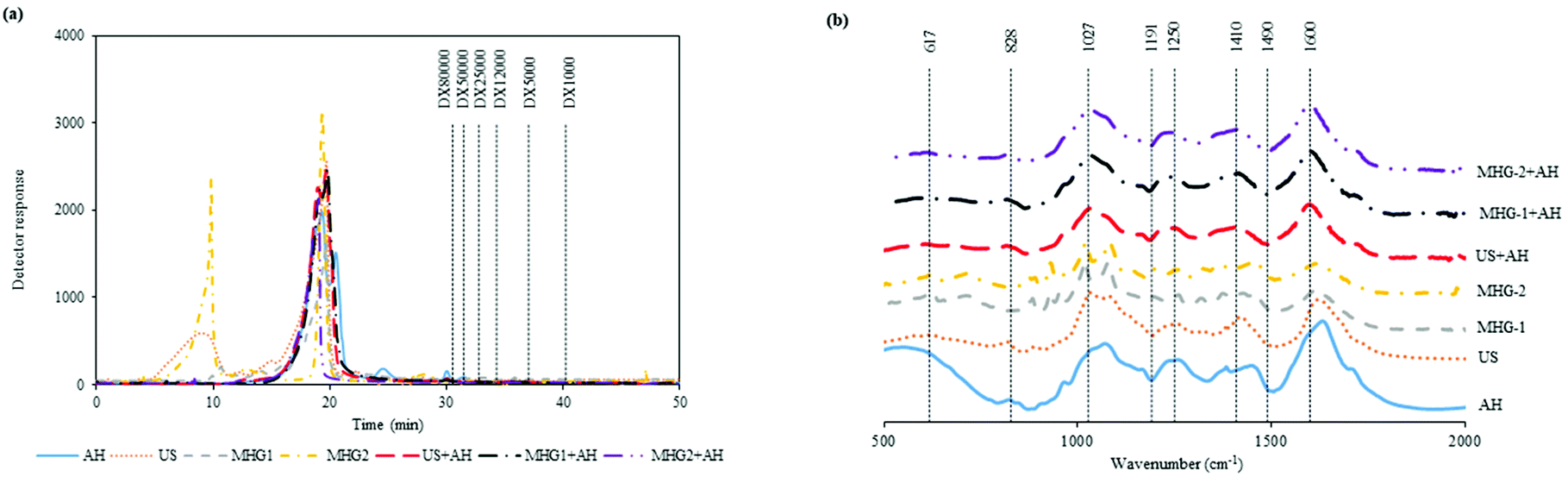 | ||
| Fig. 2 Profiles of (a) the molar mass distribution of the samples eluted by HPSEC, the pattern was represented by dashed lines, dextran (DX) of 80000 Da and (b) FTIR spectra of the extracts. | ||
On the other hand, Fig. 2b represents the FTIR spectra of the extracts obtained by the different configurations of sustainable technologies evaluated. A peak associated with the symmetric and asymmetric deformation of sulfate O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO was represented at 617 cm−1 for the spectra of the AH sample. The band related to sulfation on C2 galactose units (828 cm−1) was found for all the samples except for MHG-1 and MHG-2. It could be because the harshness of this extraction is lower than that of the other processes. The peaks associated with the sulfate ester (1027 and 1250 cm−1) were also observed in all the samples explored. Besides, the carboxylate group and Amide link type II associated with amino sugars and proteins were observed at 1410 and 1600 cm−1. These results were in agreement with other studies where similar bands were obtained and represented in the FTIR spectra results.59–61
SO was represented at 617 cm−1 for the spectra of the AH sample. The band related to sulfation on C2 galactose units (828 cm−1) was found for all the samples except for MHG-1 and MHG-2. It could be because the harshness of this extraction is lower than that of the other processes. The peaks associated with the sulfate ester (1027 and 1250 cm−1) were also observed in all the samples explored. Besides, the carboxylate group and Amide link type II associated with amino sugars and proteins were observed at 1410 and 1600 cm−1. These results were in agreement with other studies where similar bands were obtained and represented in the FTIR spectra results.59–61
3.2. Chemical characterization and enzymatic hydrolysis susceptibility of the processed S. muticum after the biomolecule extraction
In the use of an integrated biorefinery, where the totality of the biomass is exploited, it is essential to follow green chemistry principles, avoiding the waste produced after the extraction of biomolecules. For that, the solid residue after the processing of S. muticum (extracts-free Sm) was analyzed and subjected to enzymatic susceptibility assays to evaluate its potential use as a source of fermentable sugars for bioethanol production, improving their energy revalorization.Table 2 displays the chemical composition of the solid yield and carbohydrate content (as polysaccharide) after the different extraction strategies (Fig. 1).
| AH | US | MHG-1 | MHG-2 | US + AH | MHG-1 + AH | MHG-2 + AH | MIX | |
|---|---|---|---|---|---|---|---|---|
| a Unit of the fractionation data of US + AH, MHG-1 + AH and MHG-2 + AH measured in g per 100 g Sm regarding the Sm resulting from the previous processing. Data represent mean ± standard deviation (n ≥ 3). | ||||||||
| Solid phase composition (g per 100 g processed Sm oven-dry basis, o.d.b.) | ||||||||
| Solid yield (g per 100 g Sm) | 46.8 | 90.4 | 99.3 | 99.6 | 53.0a | 50.7a | 52.0a | — |
| Glucan | 15.8 ± 0.90 | 7.84 ± 0.11 | 10.2 ± 0.36 | 8.06 ± 0.07 | 15.8 ± 0.04 | 14.5 ± 0.06 | 12.0 ± 0.53 | 13.5 ± 0.23 |
| Xylan + galactan + mannan | 2.04 ± 0.08 | 4.51 ± 0.12 | 4.24 ± 0.14 | 4.02 ± 0.08 | 1.61 ± 0.16 | 1.41 ± 0.14 | 1.62 ± 0.02 | 1.78 ± 0.11 |
| Fucoidan | 2.03 ± 0.07 | 6.60 ± 0.17 | 5.99 ± 0.05 | 5.69 ± 0.08 | 1.15 ± 0.10 | 0.96 ± 0.03 | 1.29 ± 0.06 | 1.47 ± 0.09 |
| Acetyl groups | 0.03 ± 0.01 | 0.03 ± 0.01 | 0.03 ± 0.00 | 0.04 ± 0.00 | 0.13 ± 0.03 | 0.16 ± 0.03 | 0.12 ± 0.05 | 0.11 ± 0.04 |
| Acid insoluble residue (AIR) | 52.4 ± 1.02 | 37.0 ± 0.34 | 32.8 ± 0.43 | 34.3 ± 0.43 | 56.6 ± 0.62 | 58.2 ± 0.64 | 59.6 ± 0.38 | 51.2 ± 0.27 |
| Protein (by difference with the extracts) | 18.4 ± 0.09 | 11.7 ± 0.00 | 10.7 ± 0.00 | 10.6 ± 0.00 | 19.2 ± 0.07 | 18.5 ± 0.03 | 18.0 ± 0.02 | 18.5 ± 0.50 |
Firstly, the solid yield reflected the degree of fractionation of the processes, inversely to what was observed for the extraction yield. In this sense, the processing where autohydrolysis was involved implied a lower solid yield (46.8–53.0 g per 100 g of Sm). However, US, MHG-1 and MHG-2 barely affected the macroalgae, reaching solid yields of 90.4, 99.3 and 99.6 g per 100 g of Sm, respectively.
Regarding the chemical composition of the resulting Sm, the acid insoluble residue (AIR) represented the majority of the solid fraction with values ranging from 32.8–59.6 g per 100 g processed Sm. These values implied recoveries of almost or slightly higher than 100% (regarding the initial composition), which can be explained due to the elevated extractive content of Sargassum muticum that can contribute to increase its overall value, or owing to the adhesion of carbohydrates or other compounds to the AIR as already observed for autohydrolyzed Sm11 or to the AIR (Klason lignin) of Paulownia wood after autohydrolysis.62
The second main component of the solid fraction is the glucan, with values between 7.84 and 15.8 g per 100 g of processed Sm. This component mainly remained in the solid phase, with recoveries up to 100% in the case of MHG-1. The selectivity of the technologies evaluated for the totally recovery of glucan in the extract-free Sm is a remarkable result, since this polysaccharide can be used for bioethanol production.
Additionally, other polymers (fucoidan, xylan, mannan and galactan) are also greatly influenced by the harshness of the processing. For instance, fucoidan remained almost quantitatively in the solid fraction after US, MHG-1 and MHG-2 (4.51, 4.24, 4.02 g of fucoidan per 100 g of processed Sm, respectively) with recoveries between 94 and 99% regarding the initial fucoidan content of Sm. On the other hand, AH and its combination with US and MHG implied a recovery of 8–16% of fucoidan regarding initial fucoidan in the solid fraction, representing the great solubilization of this fraction. Similar behavior was observed for other polymers such as xylan, galactan and mannan, with almost quantitative recoveries for US and MHG and recoveries, regarding the initial composition, of about 11–14% in the solid phase for AH, US + AH, MHG-1 + AH, and MHG-2 + AH. Finally, acetyl groups remained marginally in the solid phase in all cases, whereas the protein content remained almost quantitatively in the solid.
In order to evaluate the potential of the processed Sm to obtain bioethanol, enzymatic hydrolysis under favorable conditions (LSR of 20 g of liquid per g of solid, CSR of 15 FPU g−1, VCR of 5 U FPU−1) was carried out. Fig. 3a shows the time course of the glucose concentration while Table 3 displays the maximum glucose concentration and GGC obtained from the saccharification of different residue solids after extraction processes employed in this work. As can be seen, autohydrolysis (and its combination with US and MHG) is the process that enhanced significantly the susceptibility of Sm towards enzymes. As a consequence, a glucan to glucose conversion of (GGC) of 77–100 was achieved within the first 48 h.
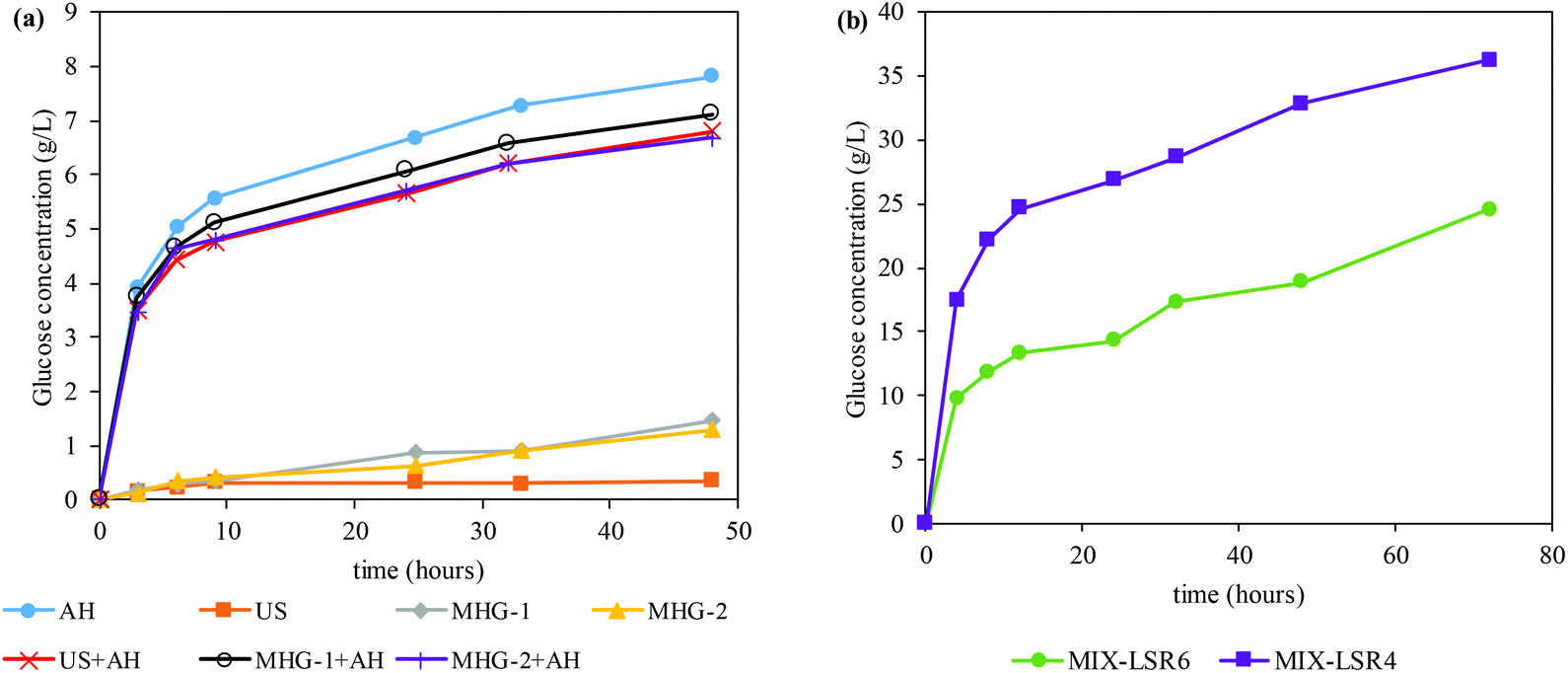 | ||
| Fig. 3 Glucose time course of the enzymatic susceptibility of (a) the solid residues from autohydrolysis (AH), ultrasound (US) and microwave hydrodiffusion and gravity in one step (MHG-1) and two steps (MHG-2), and their combinations with autohydrolysis (US + AH, MHG-1 + AH and MHG-2 + AH) at an LSR of 20 g g−1; (b) mixing the solid residues of AH, US + AH, MHG-1 + AH and MHG-2 + AH at an LSR of 6 and 4 g g−1. | ||
| AH | US | MHG-1 | MHG-2 | US + AH | MHG-1 + AH | MHG-2 + AH | MIX-LSR6 | MIX-LSR4 | |
|---|---|---|---|---|---|---|---|---|---|
| Glucose concentration (g L−1) | 7.82 ± 0.14 | 0.35 ± 0.01 | 1.47 ± 0.03 | 1.30 ± 0.02 | 6.80 ± 0.13 | 7.12 ± 0.13 | 6.68 ± 0.12 | 24.5 ± 0.45 | 36.2 ± 0.67 |
| Glucan to glucose conversion (%) | 89.0 | 7.00 | 24.0 | 21.0 | 77.0 | 89.0 | 100 | 100 | 100 |
Nonetheless, US, MHG-1 and MHG-2 achieved very limited susceptibility towards enzymes, specifically 0.35, 1.47 and 1.30 g glucose per L (analogous to 7, 24 and 21% of GGC). These experimental data illustrated that autohydrolysis was, among the technologies employed, the one that allowed a larger improvement of the enzymatic susceptibility of Sm. Similar results were stated by Gomes-Dias and colleagues in the saccharification of the autohydrolyzed macroalgae Gelidium sesquipedale using non-isothermal autohydrolysis at temperatures between 127.6 and 212.4 °C, reaching a GGC of 72.2–96.2%.3 In addition, as revealed by its time course, the enzymatic hydrolysis of autohydrolyzed Sm (AH, US + AH, MHG-1 + AH, and MHG-2 + AH) was a fast hydrolysis, reaching a GGC of 54–72% at 9 h. In this sense, the use of a sequential subsequent autohydrolysis processing after the US and MHG processing largely augmented the enzymatic susceptibility of the solid residues.
Owing to the fact that the concentrations and GGC of all the experiments after the autohydrolysis procedure were very similar and taking into account the promising results obtained for the all extracts, as bioactive molecules with interesting health-beneficing proprieties (section 3.1), the resulting extract-free Sm samples were mixed in equal quantities in order to validate the suitability of the combination of techniques with autohydrolysis to pretreat Sm for the production of bioethanol.
3.3. Fermentation strategies for third generation bioethanol production from the mixture of extract-free Sm
Firstly, the mixture of extract-free Sm (denominated Sm MIX) was subjected to enzymatic hydrolysis to evaluate its susceptibility. The time course of the Sm MIX saccharification can be observed in Fig. 3b. The LSR was modified to 6 and 4 g g−1 in order to validate the suitability of the blending at more challenging conditions also known as high-solid loadings.63 The data obtained at 72 h of saccharification reflected glucose concentrations up to 24.5 and 36.2 g of glucose per L for the LSR of 6 g g−1 and 4 g g−1, respectively. Both cases achieved a practical GGC of 100%, which is suitable for subsequent bioethanol production. Similar to what was observed in Fig. 3a, a fast conversion of glucan to glucose can be observed within the first 12 h of saccharification, reaching a GGC of 68% for the LSR of 4 g g−1.Three different strategies for the production of third generation bioethanol from Sm MIX were selected in order to validate the most suitable one for the conversion of extract-free Sm into ethanol: (i) simultaneous saccharification and fermentation (SSF), (ii) pre-saccharification and simultaneous saccharification and fermentation (PSSF), and (iii) separate hydrolysis and fermentation (SHF). The pre-saccharification for PSSF was set at 12 h because at that time the fast conversion of glucan to glucose was already accomplished, as stated by previous data (see Fig. 3b). On the other hand, the enzymatic saccharification in the SHF was set at 72 h, when the glucan was completely transformed into glucose (see Fig. 3b). Moreover, due to the high protein content of Sm (that could be used as a nutritional source by the yeast), the addition of different proportions of nutrients (peptone and yeast extract) was tested: (i) 100% of nutrients, i.e. 20 g peptone per L and 10 g yeast extract per L in the fermentation assays, (ii) 50% of nutrients, i.e. 10 g peptone per L and 5 g yeast extract per L in the fermentation assays, and (iii) 0% of nutrients, no commercial supplementation was added. Table 4 displays the main results of the different conditions of saccharification and fermentation evaluated in this study.
| Strategy | LSR (g g−1) | Nutrient supplementation (%) | EtMAX (g L−1) | EtY MAX (%) |
|---|---|---|---|---|
| SSF | 6 | 100 | 6.82 | 54.9 |
| 50 | 7.55 | 60.7 | ||
| 0 | 6.97 | 56.0 | ||
| 4 | 100 | 10.7 | 57.9 | |
| 50 | 10.0 | 54.5 | ||
| 0 | 11.6 | 63.0 | ||
| PSSF | 6 | 100 | 8.69 | 75.8 |
| 50 | 8.22 | 71.7 | ||
| 0 | 6.93 | 60.5 | ||
| 4 | 100 | 13.4 | 79.1 | |
| 50 | 14.5 | 85.2 | ||
| 0 | 11.2 | 65.7 | ||
| SHF | 6 | 50 | 9.77 | 78.6 |
| 0 | 10.4 | 83.6 | ||
| 4 | 50 | 13.9 | 81.8 | |
| 0 | 15.6 | 91.9 | ||
Fig. 4 shows the glucose and ethanol time courses for SSF (Fig. 4a and b), PSSF (Fig. 4c and d) and SHF (Fig. 4e and f).
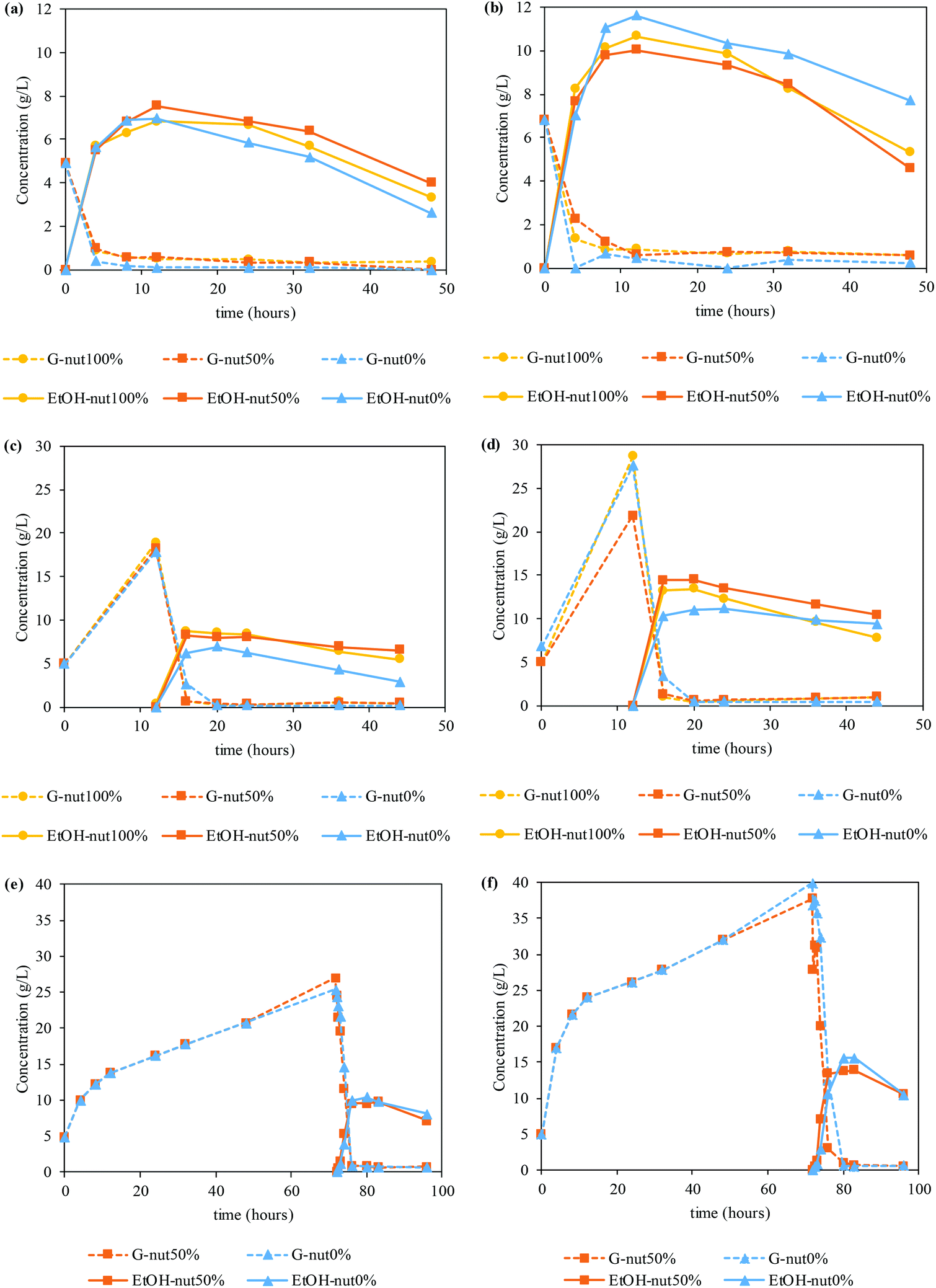 | ||
| Fig. 4 Glucose (G) and ethanol (EtOH) time courses in the fermentation strategies for the mixed solid of Sargassum muticum after autohydrolysis (AH), and sequential processing with the autohydrolysis of ultrasounds (US + AH) and microwave hydrodiffusion and gravity in one step (MHG-1 + AH) and two steps (MHG-2 + AH). (a) SSF at an LSR of 6 g g−1, (b) SSF at an LSR of 4 g g−1, (c) PSSF at an LSR of 6 g g−1, (d) PSSF at an LSR of 4 g g−1, (e) SHF at an LSR of 6 g g−1, and (f) SHF at an LSR of 4 g g−1. The concentration of nutrients in the assays was as follows: nut100%, 20 g peptone per L, 10 g yeast extract per L; nut50%, 10 g peptone per L, 5 g yeast extract per L; and nut0%, no supplementary nutrients were added. | ||
The enzymes employed in this work were stored in a sugar stock, containing glucose. In this sense, the enzymatic hydrolysis graphics only shows the production of glucose, subtracting the initial glucose content (starting at 0 g L−1 at t = 0 h), while saccharification and fermentation assays start with the glucose concentration provided by the sugar stock.64 Firstly, in the case of SSF assays, similar values of ethanol concentration are achieved regardless of the amount of nutrient supplemented. In particular, SSF at a LSR of 6 g g−1 allowed obtained up to 7.55 g of ethanol per L, with an ethanol yield of 61%. In the same way, when employing a LSR of 4 g g−1 the concentration raised up to 11.61 g of ethanol per L, but with a similar ethanol yield (63%). Furthermore, the manufacture of bioethanol is still fast enough to reach the peak of production between 8 and 12 h.
However, PSSF assays exhibit a tendency where the use of nutrients, either the total or the half of it, enhances the production of bioethanol. In these cases, maximum ethanol concentrations are achieved at 16 h of PSSF (4 h of fermentation), with values ranging from 6.97–8.69 g ethanol per L for the LSR of 6 g g−1 and 11.16–14.48 g ethanol per L for the LSR of 4 g g−1. Besides the higher ethanol titer, the ethanol yield was also higher, up to 76% and 85% for an LSR of 6 g g−1 and 4 g g−1, respectively.
In the light of these results, SHF assays were performed at the maximum glucose concentration (72 h) and with 50% and 0% of nutrients to validate whether the final concentration of bioethanol may be increased by a larger amount of glucose in the medium, and whether the nutrients may affect its production.
Accordingly, time courses of glucose and ethanol when performing SHF at an LSR of 6 g g−1 and 4 g g−1 can be found in Fig. 4e and f, respectively. In this case, slightly higher ethanol concentrations are yielded without the use of supplementary nutrients. As a consequence, 10.4 g of ethanol per L (ethanol yield of 84%) at an LSR of 6 g g−1 and 15.6 g of ethanol per L (ethanol yield of 92%) at an LSR of 4 g g−1 are obtained at 80 h of SHF (8 h of fermentation) when no nutrients were added. In contrast, 9.77 and 13.9 g of ethanol per L with an ethanol yield of 79 and 82% (LSR of 6 and 4 g g−1, respectively) are acquired when using 50% nutrient supplementation at 83 h of SHF (11 h of fermentation).
Finally, small amounts of other monomeric sugars were identified during the saccharification and fermentation. In particular, in PSSF 0.94–1.83 g of X + Ga + Ma/L (corresponding to yields of 26.8–40.2%) were detected at 12 h of saccharification, whereas for SHF, 1.37–2.69 g of X + Ga + Ma/L (yield of 45.5–60.2%) were obtained at 72 h of saccharification.
As a summary, autohydrolysis was demonstrated to be an interesting and sustainable processing to increase the digestibility of raw Sm and Sm after US and MHG extractions, reaching up to 15.6 g of ethanol per L with an ethanol yield of 92% using SHF without nutrient supplementation.
Similar ethanol yields (about 89%) were obtained by Borines and collaborators for acid pretreated (4% H2SO4 w/v, 115 °C and 1.5 h) Sargassum spp., but reaching a maximal ethanol concentration of 2.27 g L−1.65 Analogously, Baghel and collaborators achieved ethanol efficiency up to 89% when using the macroalgae Gelidiella acerosa after sequential aqueous, solvent and agar extractions.66 On the other hand, Aparicio and colleagues employed autohydrolyzed Sargassum spp. at 190 °C and 50 minutes in PSSF (24 h of presaccharification) with a solid loading of 13%, reaching 18.1 g of ethanol per L and an ethanol yield of 76.2%.67
3.4. Evaluation of heating power of spent solid after SHF
To evaluate the heating power of processed Sm (Sm MIX) and the spent solid obtained after SHF, samples from those processes were subjected to DSC. Since the solid yield from the SHF was high (69.3 and 75.5 g after SHF per 100 g of Sm MIX used in the SHF, for an LSR of 6 and 4 g g−1 respectively), these data were employed to calculate the higher heating value (HHV) of the solid residues to know the degree of energy recovery during these processes.The DSC curves are shown in Fig. 5. Between 50 and 100 °C (t = 0–5 min) the evaporation of the water in the biomass occurred, represented by a slight fall in the DSC curve (endothermic reaction). In addition, between 300 and 400 °C (t = 25–35 min), there was an exothermic reaction peak owing to the degradation of polysaccharides (such as cellulose or hemicellulose in the lignocellulosic material), reflecting the calorific power of the remaining fucoidan, xylan, galactan or mannan from Sm. Eventually, the largest peak appeared between 450 and 550 °C (t = 40–50 min), being more than likely derived from the degradation of the acid insoluble residue (AIR) from Sargassum muticum, and representing the great majority of the heat flow released from the samples.
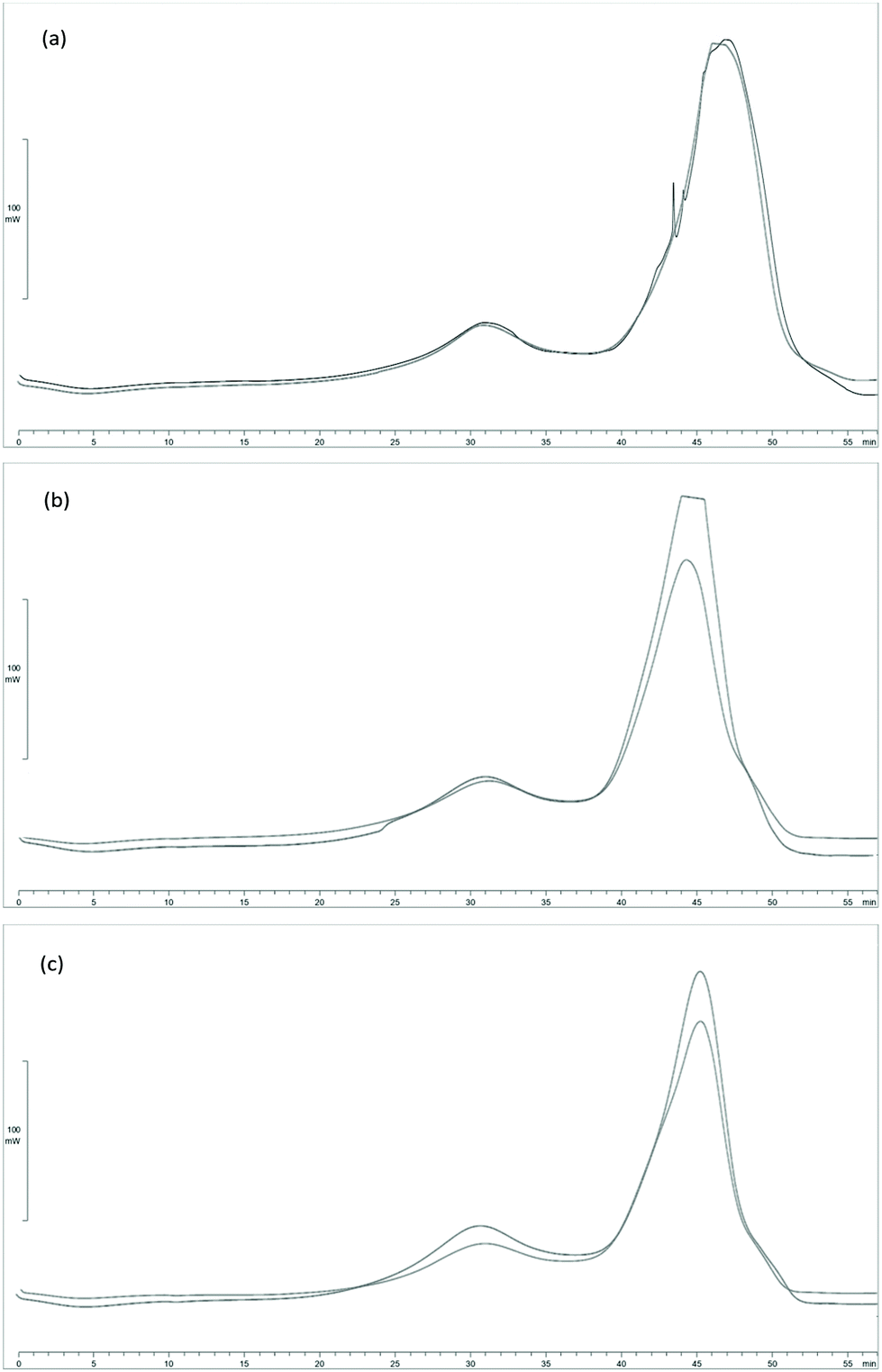 | ||
| Fig. 5 DSC curves for (a) Sm MIX, (b) Sm MIX after SHF at LSR 6 g g−1, and (c) Sm MIX after SHF at LSR 4 g g−1. | ||
On this basis, the obtained results were subjected to further calculation to determine the higher heating value (HHV). Fig. 6 displays the obtained HHV of Sm samples calculated from the DSC data (solid biofuel) and ethanol (liquid biofuel), taking into account the HHV for ethanol (29.6 MJ kg−1). The HHV was calculated using 1 kg of blended Sm after the combined processes with autohydrolysis (Sm MIX). In this context, a HHV of 10.8 MJ kg−1 was achieved for the Sm MIX solid residue. Regarding the spent solid after SHF, 1.71 and 1.87 MJ kg−1 of Sm MIX were obtained from the ethanol, whereas 8.76 and 10.0 MJ kg−1 of Sm MIX were acquired from the solid residues after SHF at LSR 6 and 4 g g−1, respectively.
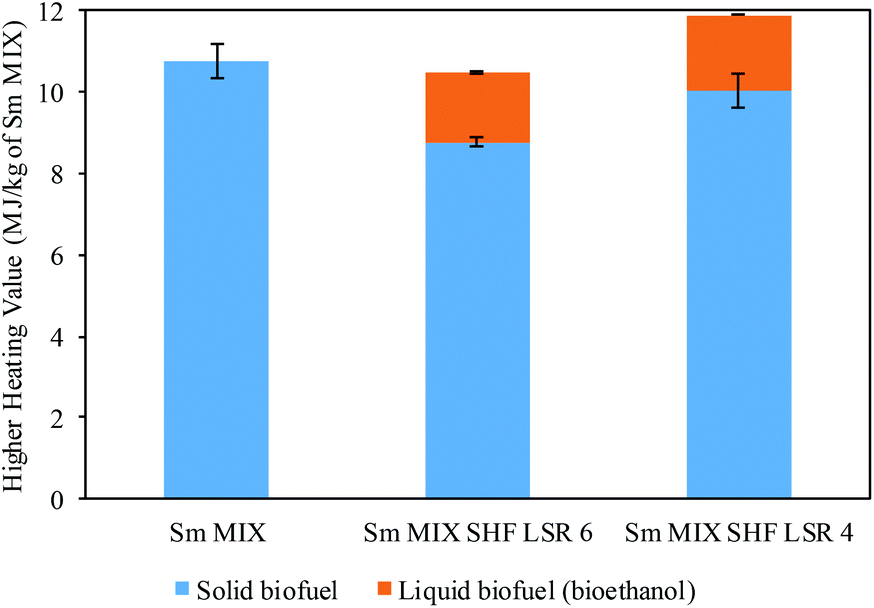 | ||
| Fig. 6 Higher heating value (HHV) and standard deviations calculated from differential scanning calorimetry (DSC) and from ethanol achieved after SHF. | ||
Similar values were obtained by Domínguez et al. for Paulownia elongata × fortunei wood, recovering 14.9 MJ kg−1 of initial wood, calculated using cellulosic and hemicellulosic ethanol and the combustion of residual lignin.68 Similarly, values of 14.2 and 11.9 MJ kg−1 were obtained from the untreated algae P. palmata and L. digitata.69
The results display a practical recovery of 100% of energy concerning the solid residue from the extraction procedures (Sm MIX), regardless of the LSR employed in the SHF process.
3.5. Overall mass balance of the cascading process
Finally, a mass balance provided a better comprehension of the components obtained in the performed cascade biorefinery of Sargassum muticum, as can be seen in Fig. 7. The components were expressed on the basis of 1 ton of initial Sm.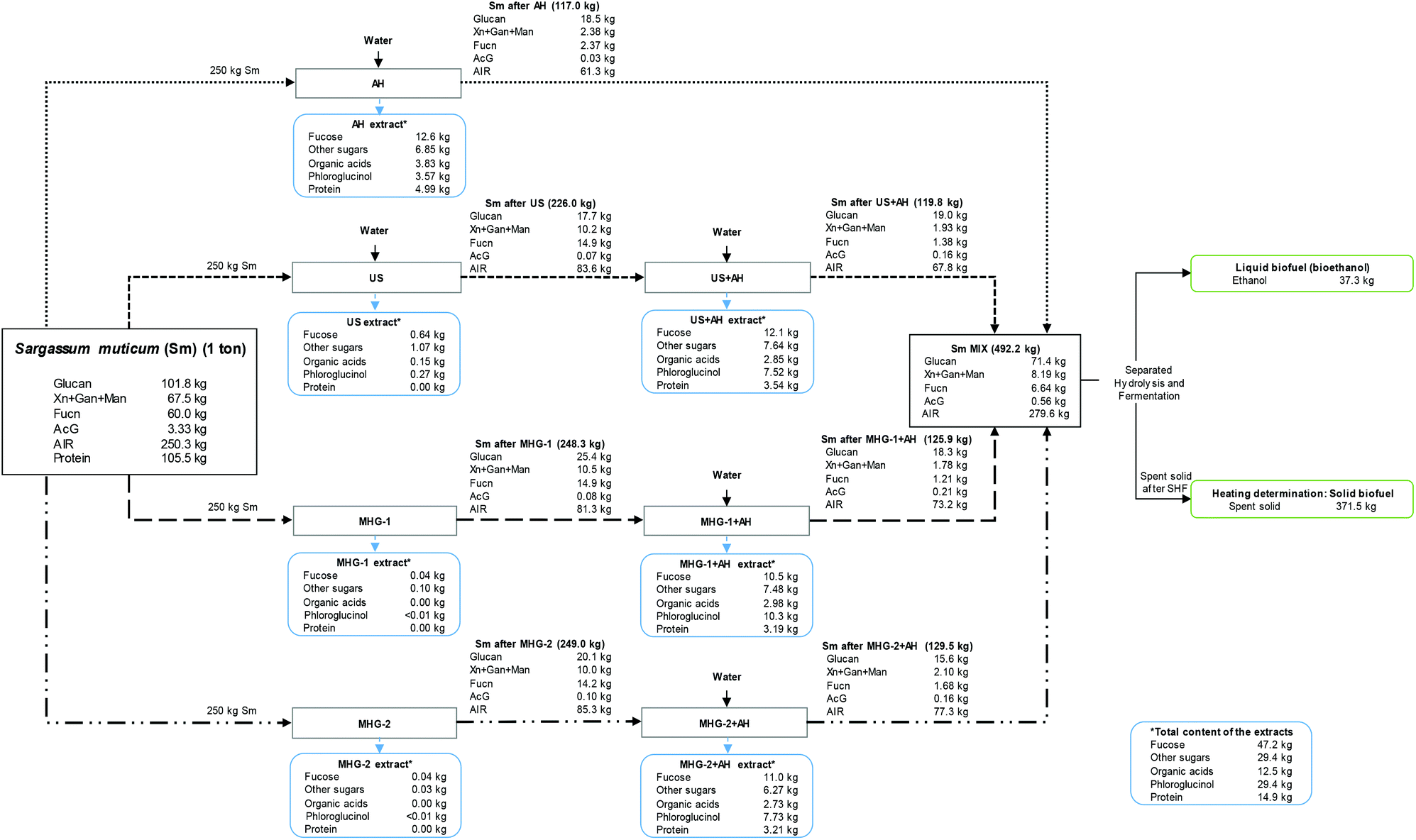 | ||
| Fig. 7 Overall mass balance of the cascading process performed in this study, measured in kg per 1 ton of Sargassum muticum on dry basis. | ||
The total amount of Sm (1 ton) is separated in the four biorefinery schemes studied in the current work (250 kg per biorefinery scheme).
This balance enabled to understand that the use of autohydrolysis allowed a higher recovery of compounds in the extracts, compared with other technologies. However, MHG or US allowed a recovery of other valuable compounds with biological activity, as was explained in a previous section, for instance obtaining a concentrated extract with great inhibitory effect against cancer cells.37 Conversely, autohydrolysis permitted the solubilization of sugars, organic acids, phlorotannins (measured as phloroglucinol) or proteins. In this sense, 10.5–12.6 kg of fucose-derived compounds (mainly sulfated fucooligosaccharides) were obtained after autohydrolysis in the four schemes, summing up to 47.2 kg per 1 ton of Sm, which corresponds to almost an 80% of recovery, regarding the initial fucoidan content of the raw material.
Subsequently, the phloroglucinol content (performing a high antioxidant capacity measured in Trolox equivalents, TEAC) reached 29.4 kg per ton of Sm regarding all the schemes accomplished. Other sugars, organic acids (acetic acid and formic acid) and proteins were also interestingly recovered in values of 29.4, 12.5 and 14.9 kg per ton of Sm.
Concerning the solid fraction, the solid yield from each process is expressed in brackets, demonstrating the high solubilization after autohydrolysis procedures. In this sense, Sm MIX accounted for 492.2 kg per ton of initial Sm, being mainly composed of glucan (71.4 kg) and AIR (279.6 kg). After SHF, 37.3 kg of ethanol and 371.5 kg of spent solid were obtained, reaching an important HHV compared with other biomasses.
Hence, the developed closed loop biorefinery70 proposed in this work using cascading sustainable technologies may propose a new configuration for the sustainable and complete use of Sargassum muticum for biomolecule extraction and for the energy valorization of the resulting solid residue, obtaining both liquid and solid fuels.
4. Conclusions
In summary, this work deals with the production of valuable compounds with application in food and pharmaceutical industries and biofuels following the principles of green chemistry. In this regard, the pretreatments employed are considered as eco-friendly technologies that implicate a non-hazardous and safe solvent (water). In addition, the use of Sargassum muticum as feedstock involves the leverage and revalorization of a problematic and unserviceable material, earning a double environmental and economic profit. Finally, the suitable fractionation of Sm implied the exploitation of a great variety of components: (i) liquid extracts rich in oligomers (specially sulfated fucooligosaccharides), phenolics and antioxidant compounds, (ii) a highly enzymatically susceptible solid, which is key for the manufacture of bioethanol and (iii) a spent solid, after saccharification and fermentation, with high calorific power. However, further work is needed to accomplished a zero-waste biorefinery of Sm, such as employing protease enzymes on the remaining proteins of the Sm MIX to obtain assimilable peptides for the fermentation yeast or performing in-depth characterization of the AIR with the purpose of employing it for a high added value marketable compound, maybe as a source for biomaterials. Additionally, since Sm presents low amounts of glucan, its mixture with other biomasses (as lignocellulosic materials) could provide an interesting tandem to obtain high ethanol titers without the addition of nutrient supplementation during the saccharification and fermentation process. In light of the foregoing, this strategy provides the reduction of derivatives by means of an integral valorization of this invasive macroalgae.Author contributions
Conceptualization, P. G. R., A. R., G. G., formal analysis: P. G. R., N. F. F., M. Á. V., A. R., funding acquisition, H. D., G. G., investigation, P. G. R., N. F. F., M. Á. V., methodology, P. G. R., N. F. F., M. Á. V., A. R., supervision, A. R., validation, P. G. R., N. F. F., M. Á. V., M. D. T., A. R., H. D., G. G., visualization, P. G. R., N. F. F., M. Á. V., writing – original draft, P. G. R., writing – review and editing, P. G. R., N. F. F., M. Á. V., M. D. T., A. R., H. D., G. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Spanish Ministry of Economy and Competitiveness, MINECO (CTM2015-68503-R and PID 2019-110031RB-I00), with partial financial support from the European Regional Development Fund (FEDER). P. G. R. would like to express gratitude to the Ministry of Science, Innovation and Universities of Spain for his FPU research grant (FPU 16/04077). N. F. F. acknowledges Xunta de Galicia for the financial support on her postdoctoral grant (ED481B 2018/071). M. A. V. thanks the Spanish Ministry of Science and Innovation for her predoctoral FPI grant (PRE2019-090567). M. D. T. acknowledges the Ministry of Science, Innovation, and Universities of Spain for her postdoctoral grant (RYC2018-024454-I). The authors acknowledge Prof. Inmaculada Romero from the University of Jaén for help with the DSC analysis.Notes and references
- J. Sadhukhan, S. Gadkari, E. Martinez-Hernandez, K. S. Ng, M. Shemfe, E. Torres-Garcia and J. Lynch, Green Chem., 2019, 21, 2635–2655 RSC.
- F. M. Kerton, Y. Liu, K. W. Omari and K. Hawboldt, Green Chem., 2013, 15, 860–871 RSC.
- J. S. Gomes-Dias, A. Romaní, J. A. Teixeira and C. M. R. Rocha, ACS Sustainable Chem. Eng., 2020, 8, 17143–17153 CrossRef CAS.
- P. Gullón, G. Astray, B. Gullón, D. Franco, P. C. B. Campagnol and J. M. Lorenzo, Curr. Opin. Food Sci., 2021, 40, 20–25 CrossRef.
- D. H. Kim, J. J. Liu, J. W. Lee, J. G. Pelton, E. J. Yun, S. Yu, Y. S. Jin and K. H. Kim, Green Chem., 2020, 22, 1776–1785 RSC.
- P. G. del Río, J. S. Gomes-Dias, C. M. R. Rocha, A. Romaní, G. Garrote and L. Domingues, Bioresour. Technol., 2020, 299, 122613 CrossRef PubMed.
- R. Dickson, B. Brigljevic, H. Lim and J. Liu, Green Chem., 2020, 22, 4174–4186 RSC.
- Y. Hannachi and A. Hafidh, Int. J. Environ. Sci. Technol., 2020, 17, 3875–3890 CrossRef CAS.
- S. Pinteus, M. F. L. Lemos, C. Alves, J. Silva and R. Pedrosa, Sci. Total Environ., 2021, 750, 141372 CrossRef CAS.
- N. Flórez-Fernández, H. Domínguez and M. D. Torres, Ind. Crops Prod., 2019, 138, 111483 CrossRef.
- P. G. del Río, E. Domínguez, V. D. Domínguez, A. Romaní, L. Domingues and G. Garrote, Renewable Energy, 2019, 141, 728–735 CrossRef.
- J. J. Milledge, B. V. Nielsen, M. S. Sadek and P. J. Harvey, Energies, 2018, 11, 1771 CrossRef.
- F. Santos, J. P. Monteiro, D. Duarte, T. Melo, D. Lopes, E. da Costa and M. R. Domingues, Antioxidants, 2020, 9, 1–20 CrossRef PubMed.
- J. Remón, S. H. Danby, J. H. Clark and A. S. Matharu, ACS Sustainable Chem. Eng., 2020, 8, 12493–12510 CrossRef.
- Y. N. Wu, M. Mattsson, M. W. Ding, M. T. Wu, J. Mei and Y. L. Shen, Energy Fuels, 2019, 33, 2278–2284 CrossRef CAS.
- S. L. Y. Tang, R. L. Smith and M. Poliakoff, Green Chem., 2005, 7, 761–762 RSC.
- A. Morales, F. Hernández-Ramos, L. Sillero, R. Fernández-Marín, I. Dávila, P. Gullón, X. Erdocia and J. Labidi, Bioresour. Technol., 2020, 315, 123896 CrossRef CAS PubMed.
- B. Gullón, G. Eibes, I. Dávila, M. T. Moreira, J. Labidi and P. Gullón, Carbohydr. Polym., 2018, 192, 75–83 CrossRef PubMed.
- I. Dávila, B. Gullón, J. Labidi and P. Gullón, Renewable Energy, 2019, 142, 612–623 CrossRef.
- H. A. Ruiz, M. Conrad, S. N. Sun, A. Sanchez, G. J. M. Rocha, A. Romaní, E. Castro, A. Torres, R. M. Rodríguez-Jasso, L. P. Andrade, I. Smirnova, R. C. Sun and A. S. Meyer, Bioresour. Technol., 2020, 299, 122685 CrossRef CAS PubMed.
- C. Jimenez-Lopez, A. G. Pereira, C. Lourenço-Lopes, P. Garcia-Oliveira, L. Cassani, M. Fraga-Corral, M. A. Prieto and J. Simal-Gandara, Food Chem., 2021, 341, 128262 CrossRef CAS PubMed.
- G. Chatel and R. S. Varma, Green Chem., 2019, 21, 6043–6050 RSC.
- L. López-Hortas, L. Gannon, R. Moreira, F. Chenlo, H. Domínguez and M. D. Torres, J. Cleaner Prod., 2018, 197, 1108–1116 CrossRef.
- F. Chemat, M. Abert Vian, A. S. Fabiano-Tixier, M. Nutrizio, A. Režek Jambrak, P. E. S. Munekata, J. M. Lorenzo, F. J. Barba, A. Binello and G. Cravotto, Green Chem., 2020, 22, 2325–2353 RSC.
- M. Álvarez-Viñas, N. Flórez-Fernández, M. D. Torres and H. Domínguez, Mar. Drugs, 2019, 17, 620 CrossRef PubMed.
- A. Cortés, M. T. Moreira and G. Feijoo, Waste Manag., 2019, 95, 70–77 CrossRef.
- J. Trifol, D. C. Marin Quintero and R. Moriana, ACS Sustainable Chem. Eng., 2021, 9, 2180–2190 CrossRef CAS.
- F. Battista, S. Zanzoni, G. Strazzera, M. Andreolli and D. Bolzonella, Renewable Energy, 2020, 157, 1203–1211 CrossRef CAS.
- P. Imbimbo, M. Bueno, L. D'Elia, A. Pollio, E. Ibañez, G. Olivieri and D. M. Monti, ACS Sustainable Chem. Eng., 2020, 8, 2939–2947 CrossRef CAS PubMed.
- C. G. Khoo, Y. K. Dasan, M. K. Lam and K. T. Lee, Bioresour. Technol., 2019, 292, 121964 CrossRef CAS PubMed.
- A. Sluiter, B. Hames, D. Hyman, C. Payne, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and J. Wolfe, Determination of total solids in biomass and total dissolved solids in liquid process samples, Laboratory Anlaytical Procedure (LAP), 2008, vol. 1 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of structural carbohydrates and lignin in biomass, Laboratory Analytical Procedure (LAP) 2008, vol. 1 Search PubMed.
- A. Sluiter, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of Extractives in Biomass, Laboratory Analytical Procedure (LAP), 2008, vol. 1 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of ash in biomass, Laboratory Analytical Procedure (LAP), 2008, vol. 1 Search PubMed.
- S. O. Lourenço, E. Barbarino, J. C. De-Paula, L. O. D. S. Pereira and U. M. Lanfer Marquez, Phycol. Res., 2002, 50, 233–241 CrossRef.
- N. Blumenkrantz and G. Asboe-Hansen, Anal. Biochem., 1973, 54, 484–489 CrossRef CAS.
- N. Flórez-Fernández, M. P. Casas, M. Jesús González-Muñoz and H. Domínguez, J. Chem. Technol. Biotechnol., 2019, 94, 256–264 CrossRef.
- N. Flórez-Fernández, M. López-García, M. J. González-Muñoz, J. M. L. Vilariño and H. Domínguez, J. Appl. Phycol., 2017, 29, 1553–1561 CrossRef.
- E. M. Balboa, S. Rivas, A. Moure, H. Domínguez and J. C. Parajó, Mar. Drugs, 2013, 11, 4612–4627 CrossRef CAS PubMed.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radicals Biol. Med., 1999, 26, 1231–1237 CrossRef CAS.
- R. Koivikko, J. Loponen, T. Honkanen and V. Jormalainen, J. Chem. Ecol., 2005, 31, 195–212 CrossRef CAS PubMed.
- K. S. Dodgson, Biochem. J., 1961, 78, 312–319 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- M. Álvarez-Viñas, N. Flórez-Fernández, M. J. González-Muñoz and H. Domínguez, Algal Res., 2019, 38, 101393 CrossRef.
- B. Adney and J. Baker, Measurement of Cellulase Activities: Laboratory Analytical Procedure (LAP); Technical report: NREL/TP-510-42628, 2008.
- T. K. Ghose, Pure Appl. Chem., 1987, 59, 695–702 Search PubMed.
- M. S. Reza, S. N. Islam, S. Afroze, M. S. A. Bakar, J. Taweekun and A. K. Azad, Data Br., 2020, 30, 105536 CrossRef.
- L. Wen, Z. Zhang, D. W. Sun, S. P. Sivagnanam and B. K. Tiwari, Crit. Rev. Food Sci. Nutr., 2020, 60, 1826–1841 CrossRef CAS PubMed.
- A. D. P. Sánchez-Camargo, L. Montero, V. Stiger-Pouvreau, A. Tanniou, A. Cifuentes, M. Herrero and E. Ibáñez, Food Chem., 2016, 192, 67–74 CrossRef.
- R. M. Rodríguez-Jasso, S. I. Mussatto, L. Pastrana, C. N. Aguilar and J. A. Teixeira, Carbohydr. Polym., 2011, 86, 1137–1144 CrossRef.
- A. S. Silchenko, A. B. Rasin, M. I. Kusaykin, A. I. Kalinovsky, Z. Miansong, L. Changheng, O. Malyarenko, A. O. Zueva, T. N. Zvyagintseva and S. P. Ermakova, Carbohydr. Polym., 2017, 175, 654–660 CrossRef CAS PubMed.
- M. Puspita, M. Déniel, I. Widowati, O. K. Radjasa, P. Douzenel, C. Marty, L. Vandanjon, G. Bedoux and N. Bourgougnon, J. Appl. Phycol., 2017, 29, 2521–2537 CrossRef CAS PubMed.
- L. Montero, A. P. Sánchez-Camargo, V. García-Cañas, A. Tanniou, V. Stiger-Pouvreau, M. Russo, L. Rastrelli, A. Cifuentes, M. Herrero and E. Ibáñez, J. Chromatogr. A, 2016, 1428, 115–125 CrossRef CAS PubMed.
- H. Cernadas, N. Flórez-Fernández, M. J. González-Muñoz, H. Domínguez and M. D. Torres, Food Bioprod. Process., 2019, 117, 275–286 CrossRef CAS.
- N. Flórez-Fernández, M. D. Torres, M. J. González-Muñoz and H. Domínguez, Food Chem., 2019, 277, 353–361 CrossRef PubMed.
- M. Plaza, S. Santoyo, L. Jaime, G. García-Blairsy Reina and E. Ibáñez, J. Pharm. Biomed. Anal., 2010, 51, 450–455 CrossRef CAS PubMed.
- I. P. S. Fernando, K. K. A. Sanjeewa, K. W. Samarakoon, W. W. Lee, H. S. Kim, N. Kang, P. Ranasinghe, H. S. Lee and Y. J. Jeon, Int. J. Biol. Macromol., 2017, 104, 1185–1193 CrossRef CAS PubMed.
- E. Gómez-Ordóñez, A. Jiménez-Escrig and P. Rupérez, Talanta, 2012, 93, 153–159 CrossRef.
- E. Gómez-Ordóñez and P. Rupérez, Food Hydrocolloids, 2011, 25, 1514–1520 CrossRef.
- I. P. S. Fernando, K. K. Asanka Sanjeewa, K. W. Samarakoon, W. W. Lee, H. S. Kim, E. A. Kim, U. K. Gunasekara, D. T. U. Abeytunga, C. Nanayakkara, E. D. De Silva, H. S. Lee and Y. J. Jeon, Algae, 2017, 32, 75–86 CrossRef CAS.
- H. S. A. Koh, J. Lu and W. Zhou, Carbohydr. Polym., 2019, 212, 178–185 CrossRef CAS PubMed.
- P. G. del Río, V. D. Domínguez, E. Domínguez, P. Gullón, B. Gullón, G. Garrote and A. Romaní, Bioresour. Technol., 2020, 314, 123722 CrossRef PubMed.
- P. G. del Río, P. Gullón, F. R. Rebelo, A. Romaní, G. Garrote and B. Gullón, Agronomy, 2020, 10, 1790 CrossRef.
- B. Erdei, B. Franká, M. Galbe and G. Zacchi, Biotechnol. Biofuels, 2012, 5, 12 CrossRef CAS PubMed.
- M. G. Borines, R. L. de Leon and J. L. Cuello, Bioresour. Technol., 2013, 138, 22–29 CrossRef CAS PubMed.
- R. S. Baghel, N. Trivedi, V. Gupta, A. Neori, C. R. K. Reddy, A. Lali and B. Jha, Green Chem., 2015, 17, 2436–2443 RSC.
- E. Aparicio, R. M. Rodríguez-Jasso, C. D. Pinales-Márquez, A. Loredo-Treviño, A. Robledo-Olivo, C. N. Aguilar, E. T. Kostas and H. A. Ruiz, Bioresour. Technol., 2021, 329, 124935 CrossRef CAS.
- E. Domínguez, T. Nóvoa, P. G. del Río, G. Garrote and A. Romaní, Energy Convers. Manage., 2020, 226, 113517 CrossRef.
- H. Redden, J. J. Milledge, H. C. Greenwell, P. W. Dyer and P. J. Harvey, J. Appl. Phycol., 2017, 29, 1037–1046 CrossRef CAS.
- S. Venkata Mohan, M. Hemalatha, D. Chakraborty, S. Chatterjee, P. Ranadheer and R. Kona, Bioresour. Technol., 2020, 295, 122128 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |